Изобретение относится к фосфорорганическим соединениям, конкретно к способам получения нитрофеноксифосфазенов общей формулы
P3N3Cl(6-n) ˙ (O - Ar)n где n = 1-6, Ar-n - нитрофенил, 2,4-(NO2)2, и может быть использовано для получения полимерных материалов, отвердителей эпоксидных смол, модифицирующих добавок к другим полимерам.
Известен способ получения нитрофеноксифосфазенов, включающий взаимодействие нитрофенолов с тримером фосфонитрилхлорида в присутствии щелочи в ксилоле при 130оС в течение 28-30 ч. Реакция идет благодаря отгонке воды на насадке Дина-Старка в соответствии со следующей схемой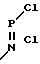 + HO
+ HO  NO2+NaOH __→
NO2+NaOH __→ 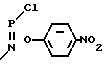 + NaCl+H2O (I)
+ NaCl+H2O (I)
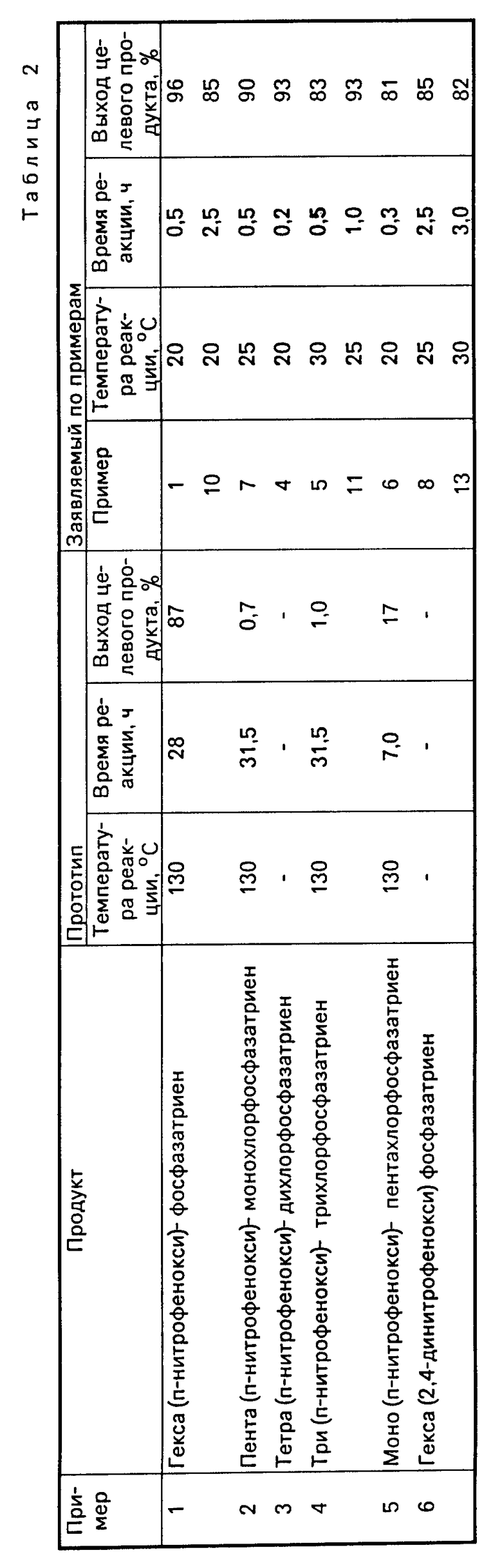
В зависимости от степени замещения хлора в фосфазене на нитрофенокси-группы выход целевых продуктов колеблется от 0,7 до 87%.
Недостатки метода:
длительность процесса; проведение реакции при высокой температуре; низкий выход целевых продуктов.
Известен способ получения замещенных фосфазенов, включающий взаимодействие гексахлорциклотрифосфазена соответствующим нитрофенолом и щелочью в двухфазной системе органический растворитель - вода в присутствии катализатора - четвертичных фосфониевых или аммониевых солей. Реакция протекает при 20-30оС в течение 4-6 ч.
Недостатки метода.
Низкий выход нитрофеноксифосфазенов, так как в указанных условиях в присутствии малоосновных фенолов заметно протекает гидролиз фосфазена, опережающий основной процесс замещения (выход целевого продукта - 3-5%).
Целью изобретения является создание условий, способствующих получению нитрофеноксифосфазенов различной степени замещения с высоким выходом.
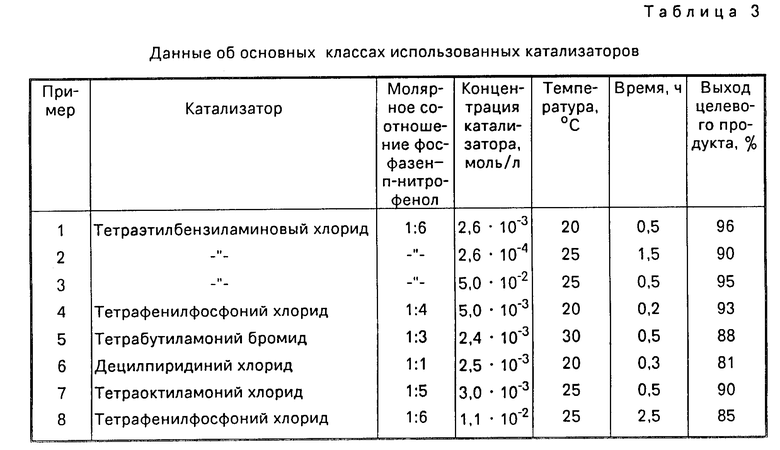
Поставленная цель достигается тем, что исходные продукты (тример фосфонитрилхлорида и нитрофенолы) взаимодействуют в среде органического растворителя, несмешивающегося с водой (хлороформ, хлористый метилен, о-дихлорбензол), тетрахлорэтан, четыреххлористый углерод) и водного раствора щелочи в течение 0,2 ч при 20-30оС в условиях контролируемой рН водной фазы, поддерживаемый в пределах 11,0-12,0 ед., в присутствии катализаторов - четвертичных аммониевых и фосфониевых солей общей формулы:
yx+, где х - галоид,
y - N+(CH2C6H5)(C2H5)3, p+(C6H5)4, N+(C4H9)4, N+(C8H17)4, C10H21N ,
,
Существенными отличиями способа от известного являются применение более эффективных катализаторов, сокращение времени, проведение процесса при поддержании рН водной фазы в заданных пределах.
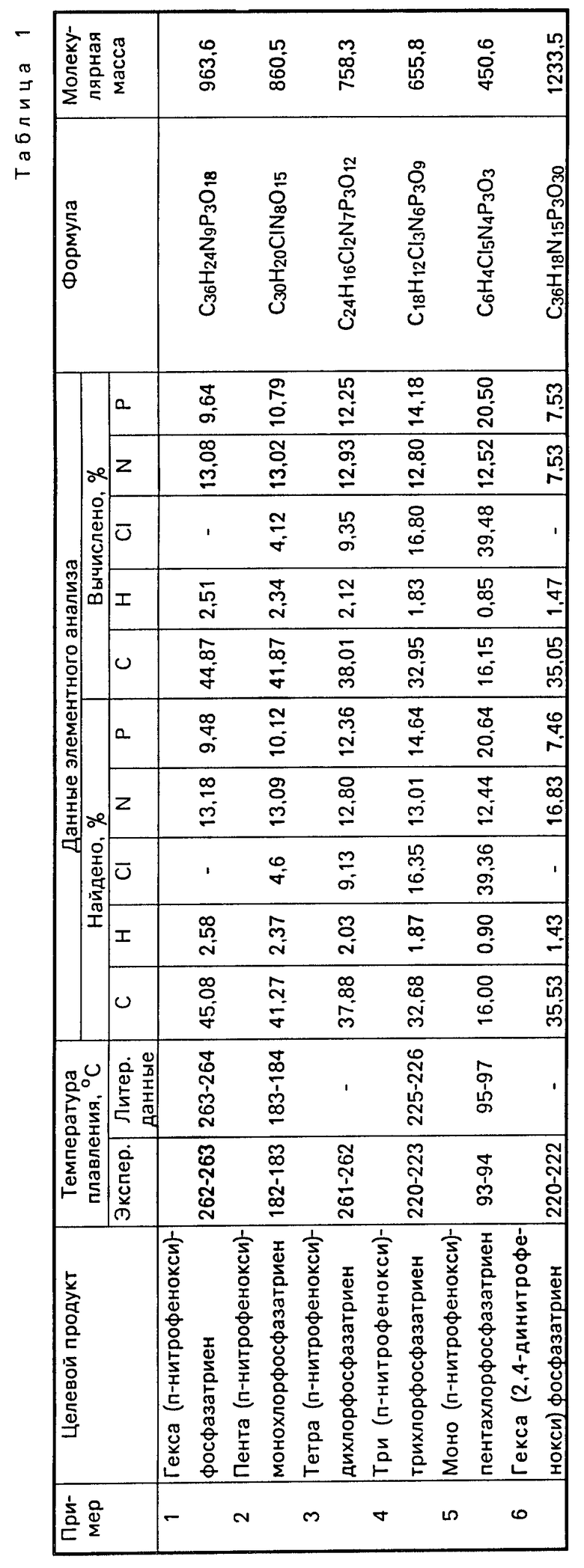
П р и м е р 1. В реакционный сосуд, содержащий 35 мл хлороформа и 25 мл воды, помещают 2,9 г (2,1 ˙ 10-2 моль) п-нитрофенола, 1,2 г (3,5 ˙ 10-3 моль) тримера фосфонитрилхлорида (соотношение реагентов 6:1) и 0,04 г (1,8 ˙ 10-4 моль) триэтилбензиламмоний хлорида. Затем в реакционную смесь добавляют 10 мл 2,5 н. водного раствора NaOH (0,36 моль/л к общему объему раствора) визуально контролируя расход нитрофенолят-иона и поддерживая рН водной фазы в пределах11,0-12,0 ед. После прибавления всего количества щелочи смесь перемешивают при 20оС в течение 30 мин. После окончания реакции органический слой отделяют, высушивают и пропускают через колонку с силикагелем марки L 40/100. Элюент удаляют досуха. Полученный продукт обрабатывают горячим гексаном для удаления следов тримера. Выход гекса(п-нитрофенокси)-фосфазатриена 3,2 г (96%). Т.пл. 262оС (лит. 263-264оС).
П р и м е р 2. В реакционный сосуд, содержащий 35 мл тетрахлорэтана и 25 мл воды, помещают 2,9 г (2,1 ˙ 10-2 моль) п-нитрофенола, 1,2 г (3,5 ˙ 10-3 моль) тримера фосфонитрилхлорида (соотношение реагентов 6:1) и 0,004 г (1,8 ˙ 10-5 моль) триэтилбензиламмоний хлорида. Затем в реакционную смесь добавляют 10 мл 2,5 н. водного раствора NaOH и перемешивают в течение 1,5 ч при 25оС. Контроль за расходованием фенолят-иона и поддержание рН в заданных пределах аналогично примеру 1. Выделение продукта аналогично примеру 1. Выход гекса(п-нитрофенокси)-фосфазена 3 г (90%).
П р и м е р 3. В реакционный сосуд, содержащий 35 мл четыреххлористого углерода и 25 мл воды, помещают 2,9 г (2,1 ˙ 10-2 моль) п-нитрофенола, 1,2 г (3,5 ˙ 10-3 моль) тримера фосфонитрилхлорида (соотношение реагентов 6:1) и 0,08 г (3,5 1,-4 моль) триэтилбензиламмоний хлорида. Затем в реакционную смесь добавляют 10 мл 2,5 н. водного раствора NaOH и перемешивают в течение 30 мин при 25оС. Контроль за расходованием фенолят-иона и поддержание рН в заданных пределах аналогично пр.1. Выделение продукта аналогично пр.1. Выход гекса(п-нитрофенокси)-фосфазатриена 3,1 г (95%).
П р и м е р 4. Проводят реакцию аналогично примеру 1. В качестве катализатора применяют тетрафенилфосфоний хлорид в количестве 0,131 г (3,5 ˙ 10-4 моль). Соотношение реагентов 4:1. Время синтеза 10 мин. Выход тетра(п-нитрофенокси)-дихлорфосфазатриена 2,4 г (93%). Т.пл. 262оС.
П р и м е р 5. В реакционный сосуд, содержащий 35 мл о-дихлорбензола и 25 мл воды, помещают 1,46 г (1,1 ˙ 10-2 моль) п-нитрофенола, 1,2 г (3,5 ˙ 10-3 моль) тримера фосфонитрилхлорида (соотношение реагентов 3:1) и 0,056 г (1,7 ˙ 10-4 моль) тетрабутиламмоний бромида. Затем в реакционную смесь добавляют 10 мл 1 н. водного раствора NaOH и перемешивают в течение 30 мин при 30оС. Контроль за расходованием фенолят-иона и поддержание рН в заданных пределах аналогично примеру 1. После окончания реакции органический слой отделяют, несколько раз (2) промывают равным объемом воды, высушивают и пропускают через колонку с силикагелем марки L 40/100. Элюент удаляют досуха. Полученный продукт обрабатывают горячим гексаном для удаления следов тримера. Выход три (п-нитрофенокси)-трихлорфосфазатриена 2,0 г (88%). Т.пл. 220-223ос (лит. 225-226оС).
П р и м е р 6. В реакционный сосуд, содержащий 50 мл хлористого метилена и 30 мл воды, помещают 0,70 г (5,0 ˙ 10-3 моль) п-нитрофенола, 1,74 г (5,0 ˙ 10-3 моль) тримера фосфонитрилхлорида (соотношение реагентов 1:1) и 0,064 г (2,5 ˙ 10-4 моль) децилпиридиний хлорида. Затем в реакционную смесь добавляют 20 мл 0,35 н. раствора К2СО3 и перемешивают в течение 20 мин при 20оС. Контроль за расходованием фенолят-иона и поддержание рН в заданных пределах аналогично примеру 1. Выделение продукта аналогично примеру 5. Выход моно(п-нитрофенокси)-пентахлорфосфазатри- ена 1,8 г (81%). Т.пл. 93оС (лит. 95-97оС).
П р и м е р 7. В реакционный сосуд, содержащий 50 мл хлороформа и 30 мл воды, помещают 3,5 г (2,5 ˙ 10-3 моль) п-нитрофенола, 1,74 г (5 ˙ 10-3 моль) тримера фосфонитрилхлорида (соотношение реагентов 5:1) и 0,15 г (3 ˙ 10-4 моль) тетраоктиламмоний хлорида. Затем в реакционную смесь добавляют 20 мл 1 н. водного раствора NaOH и перемешивают в течение 30 мин при 25оС. Контроль за расходованием фенолят-иона и поддержание рН в заданных пределах аналогично примеру 1. Выделение продукта аналогично примеру 5. Выход пента(п-нитрофенокси)-монохлорфосфазатри- ена 3,9 г (90%). Т.пл. 182оС.
П р и м е р 8. В реакционный сосуд, содержащий 50 мл хлороформа, помещают 3,48 г (1 ˙ 10-2 моль) тримера фосфонитрилхлорида и 11 г (6 ˙ 10-2 моль) 2,4-динитрофенола (соотношение 6:1). К раствору добавляют 40 мл воды, содержащей 0,393 г (1,05 ˙ 10-3 моль) тетрафенилфосфонийхлорида. Затем в реакционную смесь при интенсивном перемешивании добавляют 10 мл 6 н.раствора NaOH и затем продолжают перемешивание еще в течение 2,5-3 ч при 25оС. Контроль за расходованием фенолят-иона и поддержание рН в заданных пределах аналогично примеру 1. После окончания реакции органический слой вместе с осадком отделяют, промывают равным объемом воды и фильтруют. Полученный осадок промывают бензолом и высушивают. Выделение целевого продукта проводят с помощью колоночной хроматографии на силикагеле марки L 40/100 (элюент - ацетон). Выход гекса(2,4-динитрофенокси)-фосфазатриена 10,4 г (85%). Т.пл. 220-222оС.
В табл. 1 приведены данные элементного анализа и точки плавления продуктов, полученных по примерам.
В табл. 2 приведены сравнительные данные по получению нитрофеноксифосфазенов.
В табл. 3 приведены данные об основных классах использованных катализаторов.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИЗОТИУРОНИОАЗИНЫ, ПРОЯВЛЯЮЩИЕ ПРОТИВООПУХОЛЕВУЮ АКТИВНОСТЬ | 1991 |
|
RU2032676C1 |
| 1,3-БИС-(2,3-ДИАКРИЛОИЛОКСИПРОПИЛ) -5,5-ДИМЕТИЛ-2,4-ИМИДАЗОЛИДИНДИОН В КАЧЕСТВЕ МОДИФИКАТОРА ФОТОПОЛИМЕРИЗУЮЩИХСЯ АКРИЛАТНЫХ КОМПОЗИЦИЙ | 1991 |
|
RU2032672C1 |
| 7-АЛКОКСИ-4-ОКСА-1,2,6-ТРИНИТРАТГЕПТАНТРИОЛЫ, ОБЛАДАЮЩИЕ КАРДИОТОНИЧЕСКОЙ АКТИВНОСТЬЮ | 1991 |
|
RU2027700C1 |
| Способ получения аренселеноновых кислот | 1985 |
|
SU1293176A1 |
| @ , @ -Бис-(4-оксифенокси)фторзамещенные арилы в качестве мономеров для термостойких полимеров | 1981 |
|
SU1048679A1 |
| 1,2-БИС-(1-ГЛИЦИДИЛБЕНЗИМИДАЗОЛ-2-ИЛТИО)ЭТАН В КАЧЕСТВЕ СООТВЕРДИТЕЛЯ ЭПОКСИДНЫХ СМОЛ И 1,2-БИС-(БЕНЗИМИДАЗОЛ-2-ИЛТИО)ЭТАН КАК ПРОМЕЖУТОЧНЫЙ ПРОДУКТ В СИНТЕЗЕ 1,2-БИС-(1-ГЛИЦИДИЛБЕНЗИМИДАЗОЛ-2-ИЛТИО)ЭТАНА В КАЧЕСТВЕ СООТВЕРДИТЕЛЯ ЭПОКСИДНЫХ СМОЛ | 1990 |
|
SU1743160A1 |
| Способ очистки фенольных сточных вод | 1990 |
|
SU1745694A1 |
| Способ получения 4-(метиламино)-бифенила | 1987 |
|
SU1512966A1 |
| СОСТАВ ПЛЕНОЧНОГО ПОКРЫТИЯ ДЛЯ ВРЕМЕННОЙ ПРОТИВОКОРРОЗИОННОЙ ЗАЩИТЫ | 1990 |
|
RU2034885C1 |
| 1-ГЛИЦИДИЛ-2-МЕТАЛЛИЛТИО-4,5-ДИФЕНИЛМИДАЗОЛ В КАЧЕСТВЕ СООТВЕРДИТЕЛЯ ЭПОКСИДНЫХ СМОЛ | 1990 |
|
SU1743169A1 |
Сущность изобретения: продукт P3N3Cl6-4(OAr)u где n = 1 - 6; Ar = n-NO2C6H4i;2,4-C6H3(NO3)2i; БФ C36H24N9P3O18 , БФ C30H20ClN8O15 , БФ C24H16Cl2N7P3O12 , БФ C18H12Cl3N6P3O9 , БФ C6H4Cl5N4P3O3 , БФ C36H18N15P3O30. Выход 85 - 95%. Реагент 1: HO - Ar, где Ar - как указано выше; Реагент 2: P3N3Cl6. Условия реакции: в присутствии  , где X - галоид, Y -
, где X - галоид, Y - 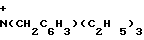 ;
; 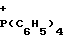 ;
;  ,
, 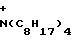 ,
, 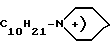 , в двухфазной системе, органический растворитель - вода, щелочь при pH 11,0 - 12,0 ед. при 20 - 30*03С. 3 табл.
, в двухфазной системе, органический растворитель - вода, щелочь при pH 11,0 - 12,0 ед. при 20 - 30*03С. 3 табл.
СПОСОБ ПОЛУЧЕНИЯ НИТРОФЕНОКСИФОСФАЗЕНОВ формулы
P3 N3 Cl6-n · (O - Ar)n,
где n = 1 - 6;
Ar - п-нитрофенил, 2,4-динитрофенил,
взаимодействием гексахлорциклотрифосфазена с соответствующим нитрофенолом и щелочью в двухфазной системе органический растворитель - вода в присутствии в качестве катализатора четвертичных аммониевых или фосфониевых солей при 20 - 30oС, отличающийся тем, что в качестве четвертичных аммониевых или фосфониевых солей используют соединения формулы
Y+ X-,
где X - галоид;
Y -  (CH2C6H5)(c2H5)3,
(CH2C6H5)(c2H5)3,  (C6H5)4,
(C6H5)4, (C4H9)4,
(C4H9)4,  (C8H17)4,
(C8H17)4,
C10H21-N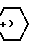 ,
,
в количестве 2,6 · 10-4 - 1,1 · 10-2 моль/л и процесс ведут при рН водной фазы 11 - 12 ед. в течение 0,2 - 3,0 ч.
| Способ получения ди- и триарилоксизамещенных производных гексахлорциклотрифосфазена | 1984 |
|
SU1225844A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
