Изобретение относится к органическому синтезу и касается способа получения тетрагидро [1,4]-бензодиазепин-2-тионов.
В журнале Eur. J. Med. Chem, 1978 г., 13, 53-59, описываются три тетрагидроимидазо[4,5,1-jk] [1,4] -бензодиазепины. Соединения согласно изобретению отличаются от них тем, что имидазогруппа замещена тиоксогруппой и тем, что указанные соединения проявляют противовирусное действие.
Изобретение касается получения тетрагидро[1,4]-бензодиазепин-2-тионов, имеющих формулу I
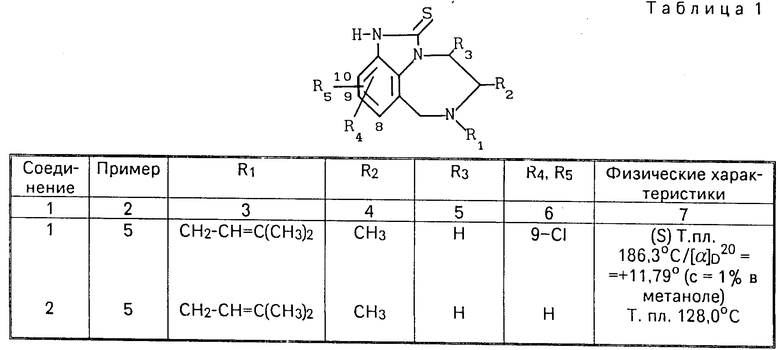
R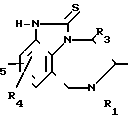 R2 в которой R1 означает алкенил С3-С6 или замещенный циклоалкилом С3-С6алкил С1-С6;
R2 в которой R1 означает алкенил С3-С6 или замещенный циклоалкилом С3-С6алкил С1-С6;
R2 - атом водорода или алкил С1-С6;
R3 - атом водорода или алкил С1-С6;
R4 и R5 независимо друг от друга означают атом водорода, алкил С1-С6, атом галогена, циано-, нитрогруппу или трифторметил,
а также их аддитивных или стереохимических изомеров.
В зависимости от характера различных заместителей, соединения формулы I могут содержать несколько асимметричных атомов углерода. Химическое обозначение соединений охватывает возможные стереохимически изомерные формы, в том числе диастереоизомеры и энантиомеры основной молекулярной структуры. Абсолютная конфигурация каждого хирального центра может быть показана стереохимическими обозначениями R и S, и это R и S обозначение соответствует правилам, описанным в Pure Appl. Chem, 1976, 45, 11-30.
Стереохимически изомерные формы соединений формулы I охватываются объемом изобретения.
Чистые стереохимические изомерные формы соединений формулы I могут быть получены известными способами. Диастереозиомеры могут быть выделены физическими способами разделения, такими как избирательная кристаллизация и хроматография, например противоточным распределением, жидкостной хроматографией и т. д.; и энантиомеры могут быть разделены путем избирательной кристаллизации их диастереоизомерных солей, образуемых с оптически активными кислотами. Чистые стереохимические изомерные формы могут быть также отделены от соответствующих чистых стереохимически изомерных форм подходящих исходных материалов, при условии, что осуществляется стереоспецифическая реакция.
Соединения формулы I обладают основными свойствами и следовательно могут превращаться в их терапевтически активные нетоксичные кислотно-аддитивные соли путем обработки подходящими кислотами, такими как неорганические кислоты, например соляной кислотой, бромистоводородной кислотой и другими, азотной кислотой, фосфорной кислотой и другими; органическими кислотами, например уксусной кислотой, пропановой кислотой, оксиуксусной кислотой, 2-оксипропановой кислотой, этандикислотой, пропандикислотой, бутандикислотой (Z)-2-бутандикислотой, (E)-2-бутандикислотой, 2-оксибу- тандикислотой, 2,3-диоксибутандикислотой, 2,3-диоксибутанкислотой, 2-окси-1,2,3-пропантрикарбоновой кислотой, метансульфокислотой, этансульфокислотой, бензолсульфокислотой, 4-метилбензолсульфокислотой, циклогексансульф- аминовой кислотой, 2-оксибензойной кислотой, 4-амино-2-оксибензойной кислотой, и т.д. И, наоборот, соль может быть превращена в свободное основание в результате обработки ее щелочью. Термин "фармацевтически пригодные кислотно-аддитивные соли" охватывает также сольваты, с которыми могут быть образованы соединения формулы 1, и эти сольваты охватываются объемом изобретения. Примерами таких сольватов являются гидраты, алкоголяты и другие. В случае, когда указанный реагент формулы III представляет собой дисульфид углерода, реакция может также осуществляться в алканоле, таком как метанол, этанол, пропанол, и т. д. , в присутствии основания, такого как гидрат окиси натрия, гидрат окиси калия, и т.д. или в дисульфиде углерода, cлужащем в качестве растворителя, и в присутствии подходящего основания, такого как алкилмагнийгалогенид, например этилмагнийбромид, такого как алкиллитий, например бутиллитий, такого как амин, например N,N-диэтилэтанамин, такого как карбодиимид, например N,N-дициклогексилкарбодиимид, и т.д. Кроме того, как возможный вариант последняя реакция может осущеставляться также в основном растворителе, таком как пиридин и др., в присутствии фосфита, такого как дифенилфосфит.
Соединение формулы I получают путем тионирования 4,5,6,7-тетрагидроимидазо/4,5,1-jk//1,4/-бензодиазепин-2-она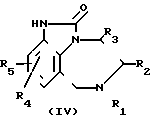
 (1)
(1)
Промежуточные продукты формулы IV могут химически реагировать с галогенирующим реагентом, таким как фосфорилхлорид, трихлорид фосфора, трибромид фосфора, хлористый тионил, оксалилхлорид и др. реагенты, при желании при повышенной температуре, особенно при температуре флегмообразования реакционной смеси, и при желании в присутствии инертного к реагентам растворителя и в присутствии основания, такого как карбонат натрия, бикарбонат натрия, карбонат калия и другие. Полученный таким образом 2-галогено-4,5,6,7-тетрагидроимидазо[4,5,1-jk//1,4/-бензодиазепин может быть далее превращен в соединения формулы I путем реакции с тиомочевиной или с тиосульфатом щелочного металла, например тиосульфатом натрия, в подходящем инертном к реагентам растворителе, таком как вода или алканол, например, метанол, этанол, 1-пропанол, 2-пропанол, бутанол, 1,2-этандиол и т.д., при желании при повышенной температуре, особенно при температуре флегмообразования реакционной смеси.
При желании соединения формулы I могут быть также непосредственно получены из промежуточных продуктов формулы IV путем тионирования с 2,4-бис(4-метоксифенил)-1,3-дитио-2,4-дифосфатан-2,4-дисуль- фидом (реагентом Лавессона) в подходящем инертном к реагентам растворителе, таком как ароматический углеводород, например бензол, метилбензол, диметилбензол, биполярный апротонный растворитель, например, гексаметилфосфотриамид (НМРА) и другие растворители; или путем тионирования с пятисульфидом фосфора.
Затем при желании, превращают соединения формулы I в терапевтически активную нетоксичную кислотно-аддитивную соль путем обработки кислотой, или, наоборот, превращают кислотную соль в свободное основание посредством щелочи, и/или получают его стереохимически изомерные формы.
Продукты реакции могут быть выделены из реакционной смеси и при необходимости дополнительно очищены согласно общеизвестным методикам.
Промежуточные соединения и исходные продукты являются уже известными соединениями, которые могут быть получены согласно принятым методикам получения аналогичных соединений, и некоторые промежуточные соединения являются новыми продуктами.
Промежуточные соединения формулы IV, в которой R1, R2 и R3определены формулой I и (А) R4 и R5 каждый независимо друг от друга представляет собой С1-С6 алкил, галогено, циано, нитро, трифторметил, окси, С1-С6 алкилокси, амино-, или моно- или ди(С1-С6-алкил) амино, или в) R4 представляет собой водород и R5 предствляет собой циано, нитро, трифторметил, окси, С1-С6 алкилокси, амино, или моно- либо ди(С1-С6алкил) амино;
и указанные радикалы R4 и R5 представлены соответственно радикалами R4-a и R5-a; а также указанные промежуточные соединения, описываемые формулой (IV-a), являются новыми соединениями.

Эти промежуточные соединения формулы IV могут быть получены согласно процедурам, описанным в ЕР-А-0336466. Так например, указанные промежуточные соединения формулы IV как известные, так и новые могут быть получены путем N-алкилирования промежуточного соединения формулы IV-в согласно известным процедурам N-алкилирования, как описано выше для превращения промежуточных соединений V в соединения формулы I.
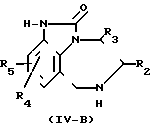
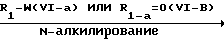 IV
IV
Промежуточные соединения формулы (IV) и (IV-в) могут быть получены из промежуточного соединения формулы (П-Н) путем реакции с химическим реагентом формулы (LVIII), в которой L представляет собой подходящую удаляемую группу.
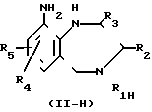
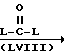
Подходящие реагенты формулы (LVIII) представляют собой, например мочевину, ди(С1-С6 алкил) карбонат, дихлорид угольной кислоты, трихлорметилхлорформат, 1,1'-карбонилбис[1H-имидазол] , этилхлорформат и другие. Данная реакция может легко осуществляться согласно процедурам, описанным выше для превращения промежуточных соединений формулы II в соединения формулы I.
Во всех указанных реакционных схемах химическое обозначение промежуточных соединений охватывает смесь всех возможных стереохимически изомерных форм, смеси ряда возможных стереохимически изомерных форм как таковых, например диастереоизомерные смеси, энантиомерные смеси, например рацемические и обогащенные энантиомерные смеси, и энантиомерно чистые изомерные формы основной молекулярной структуры.
Стереохимически изомерные формы промежуточных соединений, описанных в представленных выше реакционных схемах, и соединения формулы I могут быть получены хорошо известными в данной области способами. Так например, диастереоизомеры могут быть разделены физическими методами разделения, такими как отгонка, избирательная кристаллизация, хроматографические методы разделения, например путем противоточного распределения, жидкостная хроматография и другие.
Энантиомерно чистые промежуточные соединения легко могут быть получены из энантиомерно чистых изомерных форм соответствующих исходных материалов, при условии, что последующие реакции являются стереоспецифическими. Наиболее интересными энантиомерно чистыми исходными материалами для использования в указанных реакционных схемах, являются аминокислоты и/или их замещенные производные, имеющие формулу R1HNH-CHR2-COOR (XLY), и соответствующие аминоалканолы и/или их замещенные производные, имеющие формулу R1HNH-CHR2-CH(R3)OH (XIX).
Как возможный вариант, энантиомерно чистые промежуточные соединения могут быть также получены путем разделения соответствующих рацематов путем, например, избирательной кристаллизации их диастереоизомерных солей оптически активными разделяющими средствами, путем хроматографического разделения диастереизомерных производных, путем хроматографического разделения рацемата с применением хиральной стационарной фазы, и т.д.
Соединения формулы I и промежуточные соединения формулы IV-a обладают противовирусными и особенно противоретровирусными свойствами. До недавнего времени ретровирусы рассматривались как патогенные агенты для ряда заболеваний теплокровных животных, исключая человека, в отличии от вирусов, которые были известны в течение некоторого времени как причина большого числа заболеваний теплокровных животных, включая человека. Однако, после этого было установлено, что ретровирус, вирус человеческого иммунного дефицита, HIV, известный также как LAV, HTLV-III или ARV, является этиологическим агентом синдрома приобретенного иммунного дефицита (СПИДА) у человека, ретровирусных инфекций, и лечение субъектов, страдающих от него, стало заслуживать наибольшего внимания. Вирус HIV заражает предпочтительно клетки человека Т-4 и разрушает их или изменяет их нормальную функцию, особенно координацию иммунной системы. В результате зараженный пациент имеет постоянно уменьшающее число клеток Т-4, которые кроме того ведут себя аномальным образом. Ввиду этого система иммунной защиты не в состоянии бороться против инфекций и неоплазм, и зараженный HIV субъект обычно умирает при заражении условно-патогенными бактериями, в результате таких заболеваний как пневмония, рак, а не в результате прямой инфекции HIV. Другими заболеваниями, связанными с заражением HIV, являются тромбастения, саркома Капози и поражение центральной нервной системы, характеризующееся прогрессирующей демиелинизацией, приводящий в результате к слабоумию и таким симптомам как прогрессирующая дизартрия, атаксия и дизориентировка. Кроме того, инфекционное заражение HIV связано с прогрессирующей невропатией, прогрессирующей распространенной лимфаденопатией (PGL) и связанным со спидом (AIDS) комплексом (ARC).
Противовирусные, в частности противоретровирусные и особенно противо-HIV свойства соединений формулы (I) и промежуточных продуктов формулы IV-a подтверждают, что данные соединения и промежуточные соединения, являются ценными притивовирусными хемотерапевтическими средствами для профилактики или лечения теплокровных животных, страдающих от вирусных инфекций, в частности для лечения людей, зараженных вирусом HIV, особенно HIV-1.
В результате противовирусных и особенно противоретровирусных свойств соединения формулы I и промежуточные соединения формулы IV-a, их фармацевтически пригодные соли и их стереохимически изомерные формы используются для лечения теплокровных животных, зараженных вирусами, в частности ретровирусами, или для профилактики этих теплокровных животных. Примерами ретровирусных инфекций человека являются HIV, в частности HIV-1 и HTLV-1 (человеческий Т-лимфотропный вирус типа I), вызывающие лейкемию и лимфому. В качестве ретровирусной инфекции животных можно назвать FeLV (кошачий вирус лейкемии), который вызывает лейкемию и иммунный дефицит. Состояния, которые могут быть предотвращены или которые могут поддаваться лечению соединениями, отвечающими настоящему изобретению, особенно состояния, связанные с HIV и другими патогенными ретровирусами, включают СПИД (AIDS), связанный со СПИДОМ комплекс (ARS), прогрессирующую распространенную лимфаденопатию (PGL), а также хронические CNS заболевания, вызванные ретровирусами, такие как вызванное HIV слабоумие и рассеянный склероз.
Ввиду противовирусного и особенно противоретровирусного действия соединения согласно изобретению могут быть приготовлены в виде различных фармацевтических препаратов для ввода в организм. Для приготовления фармацевтических композиций, отвечающих данному изобретению, эффективное количество данного соединения, в форме его основной или кислотной соли, служащего в качестве активного ингредиента, тщательно перемешивается с фармацевтически пригодным носителем, который (носитель) может иметь большое разнообразие форм в зависимости от желаемой для ввода в организм формы препарата. Желательно, чтобы эти фармацевтические композиции имели унифицированную дозированную форму, пригодную для ввода в организм, предпочтительно орально, через прямую кишку, подкожно или парэнтерально. Так например, при приготовлении композиций в дозированной форме для орального ввода в организм может использоваться любая обычная фармацевтическая среда, например вода, гликоли, масла, спирты и другие, которые приняты для орального ввода жидких препаратов, таких как суспензии, сиропы, эликсиры и растворы; или могут использоваться твердые носители, такие как крахмал, сахара, каолин, смазки, связующие, дизентегрирующие агенты и другие в случае порошков, пилюль, капсул и таблеток. Ввиду легкости ввода в организм таблетки и капсулы являются самыми предпочтительными дозированными формами для орального ввода и в этом случае используются фармацевтические носители. Для композиций, вводимых парэнтерально, в качестве носителя обычно используется стерильная вода, по меньшей мере в значительной части, хотя для придания растворимости могут быть включены и другие ингредиенты.
Могут быть получены, например, инъекционно вводимые растворы, в которых носитель включает солевой раствор, раствор глюкозы или смесь солевого раствора и раствора глюкозы. Могут быть получены также инъекционно вводимые суспензии, в случае чего могут использоваться также подходящие жидкие носители, суспензирующие агенты и другие вещества. В композициях, пригодных для подкожного ввода, носитель при желании включает агент усиления пенетрации и/или подходящий смачивающий агент, при желании в комбинации с подходящими присадками любого типа, вводимыми в незначительных пропорциях, которые (присадки) не вызывают значительного повреждающего воздействия на кожу. Указанные присадки могут облегчать ввод препарата в кожу и/или могут помогать приготовлению желаемых композиций. Эти композиции могут быть введены в организм различными путями, например трансдермально, как локально, так и с нанесением мази. Кислотно аддитивные соли I и IV-a ввиду их повышенной водорастворимости в сравнении с соответствующей основной формой по всей вероятности более пригодны в препарате в виде водной композиции. Особенно желательно приготовление указанных фармацевтических композиций в единичной дозированной форме для облегчения ввода и равномерности дозирования. Единичная дозированная форма, как она определена в описании заявки и в формуле изобретения, относится к физически дискретным дозированным единицам, пригодным в качестве унифицированных дозировок, причем каждая такая единица содержит предварительно заданное количество активного ингредиента, рассчитанное так, чтобы получить желаемый терапевтический эффект в сочетании с требуемым фармацевтическим носителем. Примерами таких дозированных единичных форм являются таблетки (включая простые таблетки или таблетки с покрытием), капсулы, пилюли, пакетики с порошком, вводимые путем инъекции растворы или суспензии, препараты, вводимые по чайной ложке, препараты, вводимые по столовой ложке, их сегрегированные разделенные дозы.
Соединения по изобретению можно использовать для лечения вирусных заболеваний теплокровных животных, страдающих этими вирусными заболеваниями, путем ввода в организм эффективного против вируса количества соединения формулы I или промежуточного продукта формулы IV-a, их фармацевтически пригодной кислотно-аддитивной соли или их стереоизомерной формы. Специалисты по лечению вирусных заболеваний смогут легко определить эффективное противовирусное количество в результате проведения испытания, которое описано в данной заявке. Обычно эффективное количество данного соединения должно составлять 0,01-20 мг/кг массы тела, и особенно 0,1-5 мг/кг массы тела. Может быть желательным ввод требуемой дозы в виде двух, трех, четырех или более разделенных доз и с подходящими интервалами в течение дня. Указанные разделенные дозы могут быть приготовлены в виде единичных дозированных форм, содержащих, например 1-1000 мг, и особенно 5-200 мг активного ингредиента на единичную дозированную форму.
Нижеследующие примеры служат для иллюстрации настоящего изобретения. Все части являются массовыми.
Экспериментальная часть
А. Получение промежуточных продуктов
П р и м е р 1. а) К перемешиваемой охлажденной (-12оС) смеси 9,10 ч. 2-амино-3-нитробензойной кислоты, 6,95 ч. метил-L- α-аланин моногидрохлорида, 13,50 ч. моногидрата 1-окси-1Н-1,2,4-бензотриазола и 178 частей тетрагидрофурана добавляют порциями 5,05 ч N-метилморфолина и затем, через 5 мин в атмосфере аргона 10,3 ч. N,N′-метантетраилбис [циклогексанамина]. Перемешивание продолжают в течение 5,5 ч при -12оС и в течение 15 ч при комнатной температуре. После охлаждения в течение получаса при 0оС реакционную смесь фильтруют. Фильтрат упаривают и осадок распределяют в делительной воронке между этилацетатом и насыщенным раствором NaHCO3. Этилацетатный слой отделяют, промывают насыщенным раствором NaHCO3, высушивают, фильтруют и упаривают. Остаток растирают с гексаном, фильтруют и высушивают, получая в результате 13,08 части (97,9%) (-)-метил-(S)-2-[(2-амино-3-нитробензоил)амино] пропаноата (промежуточное соединение I) с темп. плавл. 132,9оС.
в) Смесь 12,58 ч. промежуточного продукта 1, 3,50 ч. палладиевого катализатора на угле (10%) и 158 ч этанола гидрируют в аппарате Парра в течение 4 ч при комнатной температуре и давлении 3,1˙ 105 Па. Катализатор отфильтровывают на диатомовой земле и фильтрат упаривают. Маслянистый остаток нагревают в вакууме (3,3˙ 103 Па) при 150оС в течение 10 мин и при 202оС в течение 40 мин при перемешивании. После охлаждения измельченный твердый материал растирают с этанолом. Полученный продукт отфильтровывают и промывают этанолом и 1,1'-оксибисэтаном. Выход: 5,58 ч. (57,7%) (+)- (S)-9-амино-3,4-дигидро-3-метил-1Н-бензодиазепин-2,5-она (промежуточное соединение 2).
с) К суспензии 5,55 ч. литийалюминийгидрида в 154,5 ч. 1,4 диоксана в атмосфере аргона при 25оС добавляют 5,00 ч. промежуточного соединения (2). Реакционную смесь кипятят в течение 5 ч. с обратным холодильником. После охлаждения до 10оС к ней последовательно добавляют 5,55 ч. воды, 9,16 ч. 15% -ного раствора NaOH и 16,65 ч. воды. Смесь перемешивают в течение 2 ч и затем фильтруют. Осадок последовательно промывают 178 ч. горячего тетрагидрофурана и 133 ч. горячего дихлорметана. Объединенные фильтраты высушивают, фильтруют и упаривают. Остаток выливают в раствор 3,76 ч. N-метилморфолина в 133 ч. дихлорметана. Полученную смесь добавляют к раствору 4,82 ч. трихлорметилформиата в 160 ч. дихлорметана в течение 15 мин при 0оС, в атмосфере аргона. После перемешивания в течение 10 мин при 0оС реакционную смесь нагревают до комнатной температуры и концентрируют путем упаривания. К остатку добавляют 70 ч. 15%-ного водного раствора 1,4-диоксана и смесь нагревают на паровой бане в атмосфере азота в течение 45 мин, затем охлаждают и подвергают экстракции дихлорметаном (дважды, порциями по 66,5 ч. ). Водный слой фильтруют и подщелачивают концентрированным раствором NH4OH. Выпадающий осадок отфильтровывают, промывают небольшим количеством холодной воды, высушивают и дважды растирают с 6,24 ч. 2-пропанола, получая в результате 1,59 ч. (31,2%) (+)-(S)-4,5,6,7-тетрагидро-5-метилимидазо[4,5,1-jk] [1,4]бензодиазепин-2(1Н)- она.Темп. плавл. 206,5оС (промежуточное соединение 3).
d) К перемешиваемой смеси 0,64 ч. промежуточного соединения (3), 0,5 ч. карбоната натрия, 0,52 ч. иодида калия и 9,4 ч. N,N-диметилформамида добавляют в атмосфере аргона при комнатной температуре 0,56 ч. 1-бром-3-метил-2-бутена. После перемешивания в течение 24 ч растворитель отгоняют при пониженном давлении. Остаток распределяют в делительной воронке между 32 ч. дихлорметана и 35 ч. раствора хлорида натрия. Водную фазу снова подвергают экстракции 32 ч. дихлорметана. Объединенные органические слои промывают 35 ч. раствора хлорида натрия, высушивают, фильтруют и упаривают в вакууме. Остаток перекристаллизовывают из 6,5 ч. ацетонитрила. Кристаллический продукт отфильтровывают и высушивают в течение 16 ч при 81оС в высоком вакууме, получая в результате 0,38 ч. (45%) (+)-(S)-4,5,6,7-тетрагидро-5-метил-6-(3-метил-2-бутенил)имидазо [4,5,1-jk] [1,4] бензодиазепин-2(1Н)-она (промежуточное соединение 4). Т.пл. 136,4оС.
е) Суспензию 38,16 ч. промежуточного соединения (4) и 15 ч карбоната натрия в 578 ч. фосфорилхлорида перемешивают в течение 2 дней при 60оС в атмосфере азота. Избыток фосфорилхлорида отгоняют в вакууме при 30-50оС. Получаемый твердый материал охлаждают на ледяной бане и растворяют в 500 ч. воды. При энергичном перемешивании раствор подщелачивают путем медленного добавления 1000 мл насыщенного раствора NaHCO3. Полученный продукт трижды подвергают экстракции дихлорметаном порциями по 355 ч. Объединенные экстракты промывают насыщенным раствором NaHCO3 и насыщенным раствором NaCl, высушивают, фильтруют и упаривают, получая в результате 27 ч. (66,5%) (S)-2-хлор-4,5,6,7-тетрагидро-5-метил-6-(3-метил-2-бутенил) имидазо[4,5,1-jk] [1,4]-бензодиазепина (промежуточное соединение 5).
Аналогичным примеру Id образом получают (+)-(S)-4,5,6,7-тетрагидро-5-метил-6-(2-метил-2-пропенил)имидазо [4,5,1-jk][1,4]бензодиазепин 2(1Н)-он (промежуточное соединение 6). Т.пл. 152,4оС.
П р и м е р 2. а) Раствор 2,6 ч. метил-2-бром-3-нитробензоата, 1,75 ч. N-[(2-амино-1-метил)этил] бензометанамина и 1,06 ч. карбоната натрия в 8 ч. 1-бутанола перемешивают и кипятят в течение 30 мин с обратным холодильником. После этого растворитель отгоняют, к остатку добавляют 20 ч. воды и полученный продукт дважды подвергают экстракции 30 ч. трихлометана. Объединенные экстракты высушивают, фильтруют и упаривают. Из маслянистого свободного основания получают хлористоводородную соль обычным способом. Полученную соль отфильтровывают, промывают 2-пропанолом и высушивают. Выход 3,4 ч. (89,5%) гидрохлорида 3-нитро-2-[[2-метил-2-[(фенилметил)амино]этил]ами- но]бензоата (промежуточное соединение 7). Т.пл. 204оС.
в) Смесь 3,8 ч. промежуточного соединения (7), 15 ч. 2 н. раствора гидроксида натрия и 4 ч. 2-пропанола перемешивают и кипятят в течение часа с обратным холодильником. К кипящей реакционной смеси добавляют затем раствор 3 ч. концентрированной соляной кислоты и 5 ч. воды. После охлаждения отфильтровывают, промывают водой и перекристаллизовывают из 80 ч. ледяной уксусной кислоты, получая в результате 3 ч. (82%) 3-нитро-2-[[[2-[(фенилметил)амино] -2-метил] этил] амино]-бензойной кислоты (промежуточное соединение 8). Т.пл. 227оС.
с) Смесь 189,3 ч. промежуточного соединения 8,400 ч. тионилхлорида и 400 ч. метилбензола перемешивают и кипятят в течение 2 ч. с обратным холодильником. После этого растворитель отгоняют и остаток растворяют в 600 ч. метилбензола. Полученный раствор обрабатывают раствором гидрокарбоната натрия. Органический слой после отделения высушивают над безводным карбонатом натрия, фильтруют и концентрируют до объема примерно 500 ч. При стоянии при комнатной температуре продукт частично выпадает в осадок. Его отфильтровывают (фильтрат отбрасывают), промывают последовательно 2-пропанолом и 1,1'-оксибисэтаном и высушивают, получая в результате первую фракцию 123,5 ч. сырого 2,3,4,5-тетрагидро-3-метил-9-нитро-4-(фенилметил)-1Н-1,4-бензодиазепин- 5-она. Из маточного раствора отгоняют растворитель. Остаток растворяют в 160 ч. кипящего 2-пропанола и проводят кристаллизацию при комнатной температуре. Выпадающий в осадок продукт от- фильтровывают, промывают последовательно 2-пропанолом и 1,1'-оксибисэтаном и высушивают, получая в результате вторую, менее чистую фракцию 28 ч. 2,3,4,5-тетрагидро-3-метил-9-нитро-4- (фенилметил)-1Н-1,4-бензодиазепин-5-она. Обе сырые фракции перекристаллизовывают из этанола, получая в результате 137 ч. (85%) 2,3,4,5-тетрагидро-3-метил-9-нитро-4-(фенилме- тил)-1Н-1,4- бензодиазепин-5-она (промежуточное соединение 9). Т.пл. 125оС.
d) К перемешиваемой и кипятящейся с обратным холодильником суспензии 14 ч. литийалюминийгидрида в 40 ч. бензола и 50 ч. тетрагидрофурана добавляют раствор 20,2 ч. промежуточного соединения (9) в 200 ч. тетрагидрофурана и смесь продолжают перемешивать и кипятить с обратным холодильником в течение 2,5 ч. Реакционную смесь охлаждают, затем измельченным льдом и разлагают путем последовательного добавления воды, 15%-ного раствора гидроксида натрия и снова воды. Неорганический продукт отфильтровывают и фильтрат упаривают. К остатку добавляют 40 ч. метилбензола и полученный раствор упаривают досуха, получая в результате 19,8 ч. (87,6%) 9-амино-2,3,4,5-тетрагидро-3-метил-4-) (фенилметил)-1Н-1,4-бензодиазепин-5-она в виде красной маслянистой жидкости (промежуточное соединение 10), которую без дополнительной очистки используют на последующей стадии.
е) Смесь 19,8 ч. промежуточного соединения (10) и 7,2 ч. мочевины нагревают при 210-220оС до прекращения вспенивания и образования газообразного аммиака (примерно в течение 10 мин). Реакционную смесь охлаждают до примерно 100оС и кипятят со 120 ч. 1 н. раствора соляной кислоты. Раствор отделяют декантированием от маслянистого остатка, который обрабатывают активированным углем и фильтруют. Фильтрат охлаждают, подщелачивают гидроксидом аммония и подвергают экстракции один раз 75 и один раз 150 ч. трихлорметана. Объединенные экстракты высушивают и упаривают. Остаток растирают в 24 ч. 2-пропанола, фильтруют и перекристаллизовывают из этанола, а затем из 4-метил-2-пентатнона, получая в результате 2,5 ч. (11,5%) 4,5,6,7-тетрагидро-5-метил-6-(фенилметил)имидазо-[4,5,1-jk][1,4] бензодиазепин-2(1Н)-она (промежуточное соединение II). Темп. плавл. 205оС.
f) Смесь 8 ч. промежуточного соединения (II), 1 ч. 10%-ного палладиевого катализатора на угле в 80 ч. ледяной уксусной кислоты гидрируют при примерно 38оС. После поглощения расчетного количества водорода катализатор отфильтровывают и уксусную кислоту отгоняют. Остаток растворяют в 75 ч. воды и раствор подщелачивают 30 ч. концентрированного раствора гидроксида аммония. Продукт кристаллизуют при комнатной температуре, отфильтровывают, промывают водой и перекристаллизовывают из 20 ч. 2-пропанола, получая в результате 3,7 ч. (66,8% ) 4,5,6,7-тетрагидро- 5-метилимидазо-[4,5,1-jk] [1,4] бензодиазепин-2- (1Н)-она (промежуточное соединение 12). Темп. плавл. 190,5оС.
д) К перемешиваемому раствору 1,0 ч. промежуточного соединения (12), 0,816 ч. иодида калия и 0,782 ч. карбоната натрия в 56,4 ч. N,N-диметилформамида добавляют по каплям раствор 0,88 ч. 1-бром-3-метил-2-бутена в 14 ч. N, N-диметилформамида. После перемешивания в течение 22,5 ч при комнатной температуре реакционную смесь концентрируют в вакууме при примерно 70оС. Остаток дважды распределяют в делительной воронке между 130 ч. дихлорметана и 100 ч. смеси воды и насыщенного водного раствора гидрокарбоната натрия (объемное соотношение 50:50). Объединенные водные слои подвергают экстракции 78 ч. дихлорметана. Дихлорметановые слои объединяют и проводят из них экстракцию 100 ч. насыщенного раствора хлорида натрия. Экстракт высушивают и концентрируют в вакууме при температуре около 40оС. Остаток дважды перекристаллизовывают из 16 ч. ацетонитрила. Полученный продукт охлаждают в течение 45 мин при 0-5оС, кристаллы отфильтровывают, промывают 4 ч. холодного (0-5оС) ацетонитрила и сушат в течение ночи в вакууме при 78оС. В результате получают 0,805 ч. (60,3% ) (±)-4,5,6,7-тетрагидро-5-метил-6-(3-метил-2-бутенил)-имидазо- [4,5,1-jk] [1,4]-бензодиазепин-2(1Н)-она (промежуточное соединение 13). Т.пл. 158,0оС.
h) Суспензию 1,0 ч. промежуточного соединения 13 в 8,25 ч. фосфорилхлорида нагревают в течение 15 ч при 90оС в атмосфере азота. Реакционную смесь затем упаривают и остаток распределяют в делительной воронке между насыщенным раствором NaHCO3 и дихлорметаном. Водный слой подвергают реэкстракции дихлорметаном. Объединенные органические слои промывают последовательно насыщенным раствором NaHCO3 и NaCl, высушивают, фильтруют и упаривают, получая в результате 1,05 ч. (98,3%) 2-хлор-4,5,6,7-тетрагидро-5-метил-6-(3-метил-2-бутенил)имидазо [4,5,1-jk] [1,4]-бензодиазепина (промежуточное соединение 14).
П р и м е р 3. а) Смесь 41,49 ч. 6-хлор-2Н-3,1-бензоксазин-2,4(1Н)-диона и 31,40 ч. метил-L-α-аланин моногидрохлорида в 108 ч. пиридина кипятят в течение 10 ч. с обратным холодильником в атмосфере аргона. Реакционную смесь затем охлаждают и перемешивают в течение 12 ч при комнатной температуре. Выпадающий осадок отфильтровывают и промывают этанолом, получая в результате 24,77 ч. (52,5%) (S)-7-хлор-3,4-дигидро-3-метил-1Н-1,4-бензодиазепин 2,5-диона (промежуточное соединение 15).
в) 24,55 ч. промежуточного соединения 15 добавляют порциями при 0оС в атмосфере аргона к 142 ч. азотной кислоты. После выдержки в течение 3,5 ч при 0оС раствор медленно при перемешивании, добавляют к 450 ч льда. Выпадающий осадок отфильтровывают, промывают водой и высушивают при комнатной температуре, получая в результате 27,84 ч. (93,9%) (S)-7-хлор-3,4-дигидро-3-метил-9-нитро-1Н-бензодиазепин-2,5-диона (промежуточное соединение 16).
с) К охлажденной (0оС) суспензии 18,2 ч. литийалюминийгидрида в 261 ч. 1,2-диметоксиэтана добавляют порциями 16,14 ч. промежуточного соединения 16 в атмосфере аргона. Смесь перемешивают в течение 2 ч при 0оС и затем в течение еще 40 ч при температуре кипения. После охлаждения до 0оС к ней добавляют смесь 18,2 ч. воды и 48,1 ч. тетрагидрофурана, 21,1 ч. 15%-ного раствора NaOH и 54,6 ч. воды. Смесь перемешивают в течение часа при комнатной температуре и фильтруют. Осадок кипятят в течение 5 мин в тетрагидрофуране и смесь снова фильтруют. Объединенные фильтры высушивают, фильтруют и упаривают. Остаток растворяют в 399 ч. дихлорметана. После высушивания и фильтрования раствор смешивают с 18,2 ч. N-метилморфолина и всю массу добавляют по каплям при 0оС в атмосфере аргона к смеси 11,9 ч. трихлорметилхлорформиата и 665 ч. дихлорметана. Смесь упаривают и остаток растворяют в 150 мл смеси воды и 1,4-диоксана, взятых в соотношение 85:15. Смесь нагревают в течение 2 ч на паровой бане в атмосфере азота. После охлаждения твердый материал отфильтровывают и растворяют в 80 ч. воды. Раствор подщелачивают NH4OH и перемешивают в течение 45 мин. Образующийся продукт отфильтровывают и последовательно перекристаллизовывают из ацетонитрила и 2-пропанола, получая в результате 2,28 ч. (16%) (+)-(S)-9-хлор-4,5,6,7-тетрагидро-5-метилимидазо[4,5,1-jk] [1,4] бензодиазепин-2(1Н)-она (промежуточное соединение 17). Темп. плавл. 202,2оС [α]D20 = +72,6о (с = = 0,98% в метаноле).
d) К перемешиваемой смеси 2,99 ч. промежуточного соединения (17), 2,00 ч. карбоната натрия, 2,08 ч. иодида калия и 37,6 ч. N,N-диметилформамида добавляют 2,24 ч. 1-бром-3-метил-2-бутена в атмосфере аргона. После перемешивания в течение 4 дней при комнатной температуре реакционную смесь упаривают и остаток распределяют в делительной воронке между водой и дихлорметаном. Органический слой промывают насыщенным раствором NaCl, высушивают, фильтруют и упаривают. Остаток дважды перекристаллизовывают из ацетонитрила. Продукт отфильтровывают, промывают холодным ацетонитрилом и высушивают, получая в результате 1,74 ч. (45,2%) (+)-(S)-9-хлор-4,5,6,7-тетрагидро-5-метил-6(3-метил-2-бутенил) имидазо[4,5,1-jk] [1,4] бензодиазепин-2(1Н)-она (промежуточное соединение 18). Т.пл. 135,6оС.
е) Суспензию 2,5 ч. промежуточного соединения 18 и 0,87 ч. карбоната натрия в 33 ч. фосфорилхлорида перемешивают в течение 24 ч при 90оС в атмосфере азота. Избыток фосфорилхлорида отгоняют в вакууме. Образующееся в результате твердое вещество охлаждают на ледяной бане и растворяют в воде. При энергичном перемешивании смесь подщелачивают, медленно добавляя к ней насыщенный раствор NaHCO3. Продукт экстрагируют дихлорметаном (трижды порциями по 44,3 ч.) и объединенные экстракты промывают последовательно насыщенными растворами NaHCO3 и NaCl, высушивают, фильтруют и упаривают, получая в результате 2,57 ч. (97,0%) (S)-2,9-дихлор-4,5,6,7-тетрагидро-5-метил-6-(3-метил-2-бутенил)имидазо [4,5,1-jk] [1,4] бензодиазепина (промежуточное соединение 19).
Аналогичным образом получают (S)-2,8-дихлор-4,5,6,7-тетрагидро-5-метил-6-(3-ме-тил-2-бутенил)имидазо [4,5,1-jk] [1,4]бензодиазепина (промежуточное соединение 23).
П р и м е р 4. а) К охлажденной (-35оС) смеси 7,4 ч. промежуточного соединения 3 и 395 ч. ацетонитрила добавляют 24,8 ч. ангидрида (1,1-диметилэтокси) муравьиной кислоты и 0,45 ч. N,N-диметил-4-пиридинамина. Смесь оставляют стоять при комнатной температуре в течение 12 ч, послье чего упаривают. Остаток подвергают очистке с помощью флеш-хроматографии на колонке, заполненной силикагелем, с использованием в качестве подвижной фазы смеси CH2Cl2 и CH3OH в соотношении 99:1. Нужную фракцию элюата упаривают и остаток перемешивают в течение 3 ч со смесью 79 ч. метанола и 1,5 ч. карбоната калия. Растворитель затем отгоняют и остаток распределяют в делительной воронке между этилацетатом и водой. Органический слой отделяют, высушивают, фильтруют и упаривают, получая в результате 6,4 ч. (57,5%) (1,1-диметилэтил)-(S)-1,2,4,5,6,7-гексагидро-5-метил-2-оксоимидазо [4,5,1-jk] [1,4]бензодиазепин-6-карбоксилата (промежуточное соединение 20).
е) Смешивают при комнатной температуре в атмосфере аргона 5,43 ч. промежуточного соединения 20 и 224 ч. трихлорметана. После охлаждения до -35оС к смеси добавляют 2,29 ч. N-бромсукцинимида, выдерживают всю массу в течение 6 ч при этой температуре, затем дают ей нагреться до комнатной температуры и оставляют стоять в течение 12 ч. Реакционную смесь затем упаривают и остаток подвергают очистке с помощью колоночной хроматографии LC; RP). Нужную фракцию элюента упаривают, получая в результате 2,10 ч. (30,7%) (1,1-диметил)-(-)-(S)-8-бром-1,2,4,5,6,7-гексагидро-5- метил-2-оксоимидазо[4,5,1-jk] [1,4] бензодиазепин-6-карбоксилата (промежуточное соединение 21). Т.пл. 228,6оС; [α]D25 = -60,0о (конц. = 0,30 в метаноле).
f) К 2,96 ч. охлажденной (0оС) трифторуксусной кислоты добавляют 2,0 ч. промежуточного соединения 21 в атмосфере аргона. После перемешивания в течение 2 ч при 0оС смесь упаривают. К остатку добавляют последовательно 18,8 ч. N,N-диметилформамида, 0,86 ч. 1-бром-3-метил-2-бутена, 3,0 ч. карбоната натрия и 0,96 ч. иодида калия, смесь перемешивают в течение ночи при комнатной температуре и упаривают. Остаток распределяют в делительной воронке между водой и этилацетатом. Органический слой отделяют, промывают последовательно насыщенными растворами NaHCO3 и NaCl, высушивают, фильтруют и упаривают. Остаток перекристаллизовывают из ацетонитрила. Продукт отфильтровывают и высушивают в течение ночи в вакууме при 60оС, получая в результате 1,5 ч. (81,7%) (+)-(S)-8-бром-4,5,6,7-тетрагидро-5-метил-6-(3-ме- тил-2-бутенил)имидазо [4,5,1-jk] [1,4]-бензодиазепин-2(1Н)-она (промежуточное соединение 22). Т.пл. 125,2оС; [α]D25= +12,6о (конц. =1% в СН3О).
В. Получение целевых соединений
П р и м е р 5. К раствору 2,57 ч. промежуточного соединения 19 в 27,7 ч. этанола добавляют 1,21 ч. мочевины. После кипячения в течение 24 ч с обратным холодильником реакционную смесь упаривают и остаток распределяют в делительной воронке между насыщенным раствором NaHCO3 и дихлорметаном. Органический слой последовательно промывают насыщенным раствором NaHCO3, водой и насыщенным раствором NaCl, высушивают, фильтруют и упаривают. Остаток дважды подвергают очистке с помощью колоночной хроматографии (флеш-хроматография; силикагель; CH2Cl2/CH3OH = 30:1; высокопроизводительная жидкостная хроматография; силикагель; СН3СООС2Н5 (гексан = 4:6). Нужную фракцию элюата упаривают, получая в результате 0,34 ч. (13,3%) (+)-(S)-9-хлор-4,5,6,7-тетрагидро-5-метил-6-(3-метил-2-бутенил)-имидазо [4,5,1,-jk] [1,4] бензодиазепин-2(1Н)-тиона (соединение I). Т.пл. 180,3оС; [α]D20= +8,3о (с = 0,96% в метаноле).
Аналогичным образом получают (±)-4,5,6,7-тетрагидро-5-метил-6-(3- метил-2-бутенил)имидазо[4,5,1-jk][1,4]-бензодиазе- пин-2(1Н)-тион (соединение 2). Темп. плавл. 128,0оС (с разложением);
(+)-(S)-4,5,6,7-тетрагидро-5-метил-6-(3-метил-2-бутенил)имидазо- [4,5,1-jk] [1,4] -2(1Н)-тион (соединение 3); темп. плавл. 174,5оС; [α]D20= +15,95оС (с = 1% в этаноле).
П р и м е р 6. а) К раствору 2,5 ч. промежуточного соединения 23 в 12,6 ч. этанола при 20оС добавляют 0,53 ч. тиомочевины. После кипячения в течение 1,5 ч с обратным холодильником при перемешивании реакционную смесь охлаждают до 0-5оС и фильтруют при 0оС. Остаток высушивают в вакууме при 30оС, получая в результате 1,70 ч. (75,4%) (-)-(S)-8-хлор-4,5,6,7-тетрагидро-5-метил-6-(3-метил-2-бутенил) имидазо/4,5,1-jk)/1,4 бензодиазепин-2(1Н)-тион моно- гидрохлорида (соединение 17). Температура плавления 230оС [α]D20 = -76,61о (с = 1% в СН3ОН).
в) 0,5 ч. соединения 17 распределяют в делительной воронке между дихлорметаном и водой. Всю смесь подщелачивают, органический слой отделяют, высушивают, фильтруют и упаривают. Продукт высушивают в вакууме при 40оС, получая в результате 0,43 ч. (95,7%) (+)-(S)-8-хлор-4,5,6,7-тетрагидро-5-метил-6-(3-метил-2-бутенил) имидазо[4,5,1-jk] [1,4]бензодиазепин-2(1Н)-тиона (соединение 22). Темп. плавл. 163,3оС; [α]D25 = +9,37о (с = 1% в метаноле).
П р и м е р 7. Смешивают в атмосфере аргона 1,16 ч. промежуточного соединения 6 и 1,65 ч. фосфорилхлорида и кипятят смесь в течение 5,5 ч с обратным холодильником. Избыток фосфорилхлорида отгоняют, добавляют 10 ч. воды и нейтрализуют смесь насыщенным водным раствором NaHCO3. Образующийся продукт экстрагируют дихлорметаном и экстракт высушивают, фильтруют и упаривают. К остатку добавляют 7,9 ч. этанола и 1,00 ч. тиомочевины и кипятят смесь в течение 2 ч с обратным холодильником. Затем ее упаривают и остаток распределяют в делительной воронке между водой и дихлорметаном. Органический слой отделяют, высушивают, фильтруют и упаривают. Остаток подвергают очистке с помощью колоночной хроматографии (силикагель; CH2Cl2/C2H5OH = 96:5). Нужную фракцию элюата упаривают, получая в результате 0,29 части (+)-(S)-4,5,6,7-тетрагидро-5-метил-6-(2-метил-2-пропенил)имидазо [4,5,1-jk][1,4]бензодиазепин-2(1Н)-тиона (соединение 6). Темп. плавл. 137,6оС; [α]D25 = +26,2оС (конц. = 1,09% в СН3ОН).
П р и м е р 8. К охлажденному (-78оС) раствору 1,18 ч. промежуточного соединения 22 в 100 ч. дихлорметана добавляют в атмосфере аргона 1,26 ч. ангидрида трифторуксусной кислоты, через 15 мин 0,61 ч. 2,6-диметилпиридина и еще через 15 мин 21,1 мл 1,1'-оксибисэтана, насыщенного HCl. Смесь перемешивают в течение по- лучаса и затем нейтрализуют насыщенным раствором NaHCO3. Образующийся продукт дважды экстрагируют дихлорметаном, порциями на 66,5 ч. и объединенные экстракты высушивают, фильтруют и упаривают. Остаток подвергают очистке с помощью колоночной флеш-хроматографии (силикагель; CH2Cl2/CH3OH/NH4OH = 98:2:0,1). Содержащую целевой продукт фракцию элюата упаривают и остаток растворяют в 7,9 ч. этанола. К раствору добавляют 2,0 ч. тиомочевины и смесь кипятят в течение 6 ч с обратным холодильником, а затем упаривают. Остаток распределяют в делительной воронке между водой и дихлорметаном. Органический слой отделяют, последовательно промывают насыщенными растворами NaHCO3 и NaCl, высушивают, фильтруют и упаривают. Остаток подвергают очистке с помощью колоночной флеш-хроматографии (силикагель; CH3COOC2H5/гексан = 25:75). Фракцию элюата, содержащую целевой продукт, упаривают и остаток перекристаллизовывают из ацетонитрила. Продукт отфильтровывают и высушивают в вакууме при 60оС, получая в результате 0,284 ч. (20,2%) (+)-(S)-8-бром-4,5,6,7-тетрагидро-5-метил-6-(3-метил-2-бутенил) имидазо [4,5,1-jk] [1,4] бензодиазепин-2(1Н)-тиона (соединение 19). Темп. плавл. 161,1оС; [α]D25 = +4,6о (конц. = 1% в метаноле).
Все другие соединения, перечисленные в табл.1, могут быть получены по способу в соответствии с указанным примером.
С. Пример фармакологического испытания
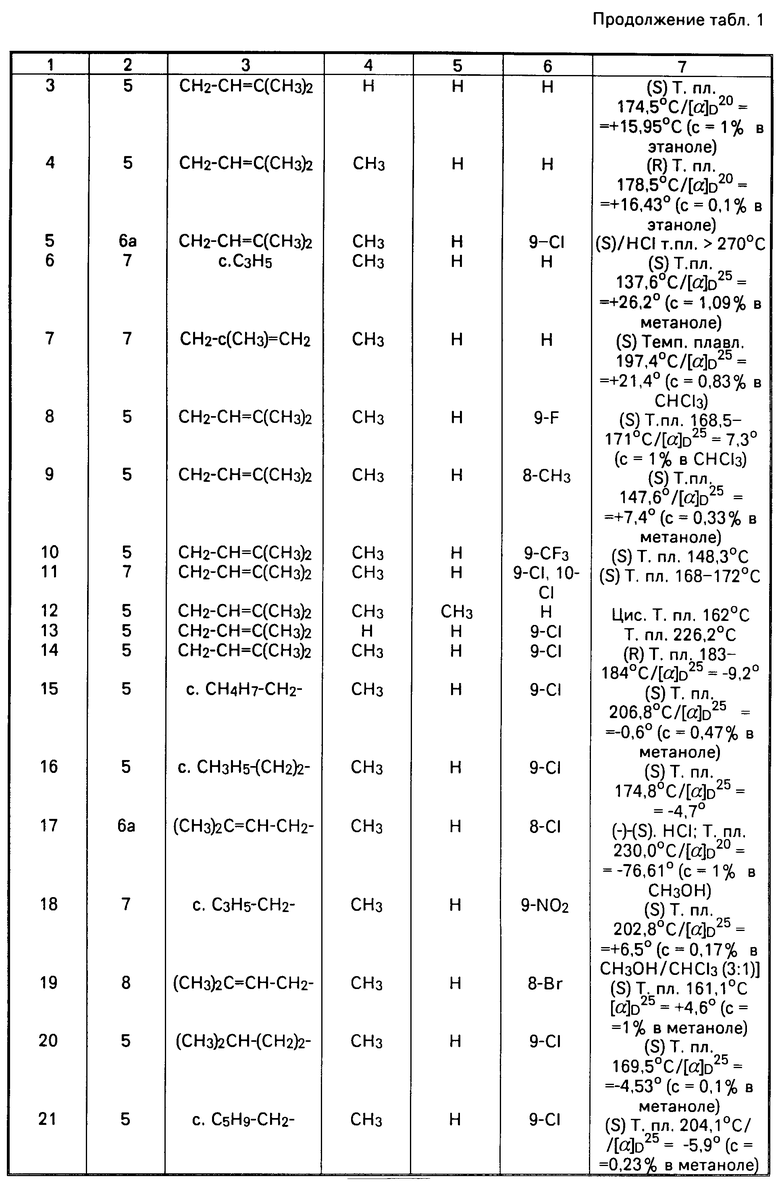
П р и м е р 9. Для оценки в условиях "ин витро" противо-HIV агентов осуществляется быстрая чувствительная и автоматизированная процедура анализа. В качестве испытываемой клеточной линии служит трансформированная HIV-1 клеточная линия Т-4, то есть МТ-4, которая была описана ранее (Koganagi и др. , Int. J. Caneer, 36. 445-451, 1985 г.) как очень высоко чувствительная и возможная для инфекционного заражения. Конечной целью является ингибирование индуцированного HIV цитопатического (связанного с заболеванием клеток) эффекта. Жизнеспособность зараженных как HIV, так и Мок (Mock) клеток определяется спектрофотометрически путем восстановления непосредственно в данных условиях 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолбромида (МТТ). 50% Цитотоксическая доза (СД50 в мкг/мл) определяется как концентрация соединения, которая снижает поглотительную способность зараженного мок (Mock) контрольного образца на 50%. Процент защиты, обеспечиваемый данным соединением в зараженных HIV клетках рассчитывается по следующей формуле: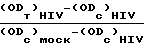 выражено в %. где (ODт)HIV - оптическая плотность, измеренная в зараженных HIV клетках при заданной концентрации испытываемого соединения: (ODc)HIV - оптическая плотность контрольных необработанных зараженных HIV клеток; (ODc)мocк - оптическая плотность контрольных необработанных зараженных мок клеток; все значения оптической плотности определены при 540 нм. Доза, обеспечивающая 50% защиты согласно указанной выше формуле определяется как 50% эффективная доза (ЕД50 в мкг/мл). Отношение CД50 к ЕД50 определяется как показатель избирательности (SI).
выражено в %. где (ODт)HIV - оптическая плотность, измеренная в зараженных HIV клетках при заданной концентрации испытываемого соединения: (ODc)HIV - оптическая плотность контрольных необработанных зараженных HIV клеток; (ODc)мocк - оптическая плотность контрольных необработанных зараженных мок клеток; все значения оптической плотности определены при 540 нм. Доза, обеспечивающая 50% защиты согласно указанной выше формуле определяется как 50% эффективная доза (ЕД50 в мкг/мл). Отношение CД50 к ЕД50 определяется как показатель избирательности (SI).
50% цитотоксическая (СД50), 50% эффективная доза (ЕД50) и показатель избирательности SI приведены в табл.2.
Д. Примеры композиции
П р и м е р 9. Вводимые орально капли.
500 ч. активного ингредиента растворяется в 0,5 л 2-оксипропановой кислоты и 1,5 л полиэтиленгликоля при 60-80оС. После охлаждения до 30-40оС вводится 35 л полиэтиленгликоля и смесь тщательно перемешивается. Затем вводится раствор 1750 ч. натрийсахарина в 2,5 л очищенной воды и при перемешивании вводится 2,5 л порошка какао и полиэтиленгликоль до объема 50 л, в результате чего получается раствор для капельного орального ввода, включающий 10 мг/мл активного ингредиента. Полученный раствор вводится в соответствующие емкости.
П р и м е р 10. Раствор для орального ввода.
9 ч. метил-4-оксибензоата и 1 ч. пропил-4-оксибензоата растворяются в 4 л кипящей очищенной воды. В 3 л этого раствора растворяют сначала 10 ч. 2,3-диоксибутандионовой кислоты и затем 20 ч. активного ингредиента. Последний раствор смешивается с остальной частью первого раствора, и в него вводят 12 л раствора 1,2,3-пропантриола и 3 л сорбита концентрацией 70%. 40 ч. натрийсахарина растворяют в 0,5 л воды и вводят малиновую (2 мл) и крыжовниковую (2 мл) эссенцию. Последний раствор смешивается с первым, вводится вода до объема 20 л, и получается раствор для орального ввода, содержащий 5 мг активного ингредиента на чайную ложку (5 мл). Полученный раствор вводят в соответствующие емкости.
П р и м е р 11. Капсулы.
20 ч. активного ингредиента, 6 ч. натрийлаурилсульфата, 56 ч. крахмала, 56 ч. лактозы. 0,8 ч. коллоидной двуокиси кремния и 1,2 ч. стеарата магния тщательно перемешивают друг с другом. Затем полученной смесью заполняются 1000 подходящих отвержденных желатиновых капсул, каждая из которых будет содержать по 20 мг активного ингредиента.
П р и м е р 12. Покрытые пленкой таблетки
Приготовление ядра таблетки.
Смесь 100 ч. активного ингредиента, 570 ч. лактозы и 200 ч. крахмала тщательно перемешивается и затем увлажняется раствором 5 ч. натрийдодецилсульфата и 10 ч. поливинилпирролидона (Kollidon-K 90R) примерно в 200 мл воды. Смоченная порошкообразная смесь просеивается, высушивается и снова просеивается. Затем вводится 100 ч. микрокристаллической целлюлозы (Avice ) и 15 ч. гидрогенизированного растительного масла (Sterotex
) и 15 ч. гидрогенизированного растительного масла (Sterotex ). Весь объем смеси тщательно перемешивается и прессуется в таблетки, и получается 10000 таблеток, каждая из которых содержит 10 мг активного ингредиента.
). Весь объем смеси тщательно перемешивается и прессуется в таблетки, и получается 10000 таблеток, каждая из которых содержит 10 мг активного ингредиента.
Покрытие.
В раствор 10 ч. метилцеллюлозы (Metgael60 HG ) в 75 мл денатурированного этанола вводят раствор 5 ч. этилцеллюлозы (Ethocel 22 cps
) в 75 мл денатурированного этанола вводят раствор 5 ч. этилцеллюлозы (Ethocel 22 cps ) в 150 мл дихлорметана. Затем в него вводится 75 мл дихлорметана и 2,5 мл 1,2,3-пропантриола, 10 ч. полиэтиленгликоля расплавляются и растворяются в 75 мл дихлорметана. Последний раствор вводится в первый и затем вводится 2,5 ч. октадеканоата магния. 5 ч. поливинилпирролидона и 30 мл концентрированной суспензии красителя (Opaspray K-1-2109
) в 150 мл дихлорметана. Затем в него вводится 75 мл дихлорметана и 2,5 мл 1,2,3-пропантриола, 10 ч. полиэтиленгликоля расплавляются и растворяются в 75 мл дихлорметана. Последний раствор вводится в первый и затем вводится 2,5 ч. октадеканоата магния. 5 ч. поливинилпирролидона и 30 мл концентрированной суспензии красителя (Opaspray K-1-2109 ) и вся смесь гомогенизируется. Эти ядра таблеток покрываются полученной таким путем смесью в аппаратуре для нанесения покрытия.
) и вся смесь гомогенизируется. Эти ядра таблеток покрываются полученной таким путем смесью в аппаратуре для нанесения покрытия.
П р и м е р 13. Инъекционный раствор
1,8 ч. метил-4-оксибензоата и 0,2 ч. пропил-4-оксибензоата растворяются примерно в 0,5 л кипящей воды для инъекции. После охлаждения до температуры примерно 50оС вводят с одновременным перемешиванием 4 ч. молочной кислоты, 0,05 ч. пропиленгликоля и 4 ч. активного ингредиента.
Раствор охлаждается до комнатной температуры и дополняется водой для инъекции до объема 1 л и в результате получается раствор, содержащий 4 мг/мл активного ингредиента. Этот раствор стерилизуется путем фильтрации (Фармакопея США, ХУП, стр. 811) и вводится в стерильные емкости.
П р и м е р 14. Свечи.
3 ч. активного ингредиента растворяются в растворе 3 ч. 2,3-диоксибутандионовой кислоты в 25 мл полиэтиленгликоля 400. 12 ч. поверхностно-активного вещества (SPAN ) и триглицериды (Witepsol 555
) и триглицериды (Witepsol 555 ) дополненные до объема 300 ч. , расплавляются вместе друг с другом. Последняя смесь тщательно перемешивается с первым раствором. Полученная таким путем смесь вливается в формы при 37-38оС и в результате получается 100 свечей, каждая из которых содержит по 30 мг/мл активного ингредиента.
) дополненные до объема 300 ч. , расплавляются вместе друг с другом. Последняя смесь тщательно перемешивается с первым раствором. Полученная таким путем смесь вливается в формы при 37-38оС и в результате получается 100 свечей, каждая из которых содержит по 30 мг/мл активного ингредиента.
П р и м е р 15. Инъекционный раствор.
60 ч. активного ингредиента и 12 ч. бензилового спирта тщательно перемешиваются и добавляется кунжутное масло до объема 1 л, и получается раствор, содержащий 60 мг/мл активного ингредиента. Этот раствор стерилизуется и вводится в стерильные емкости.
Сравнительные биологические данные.
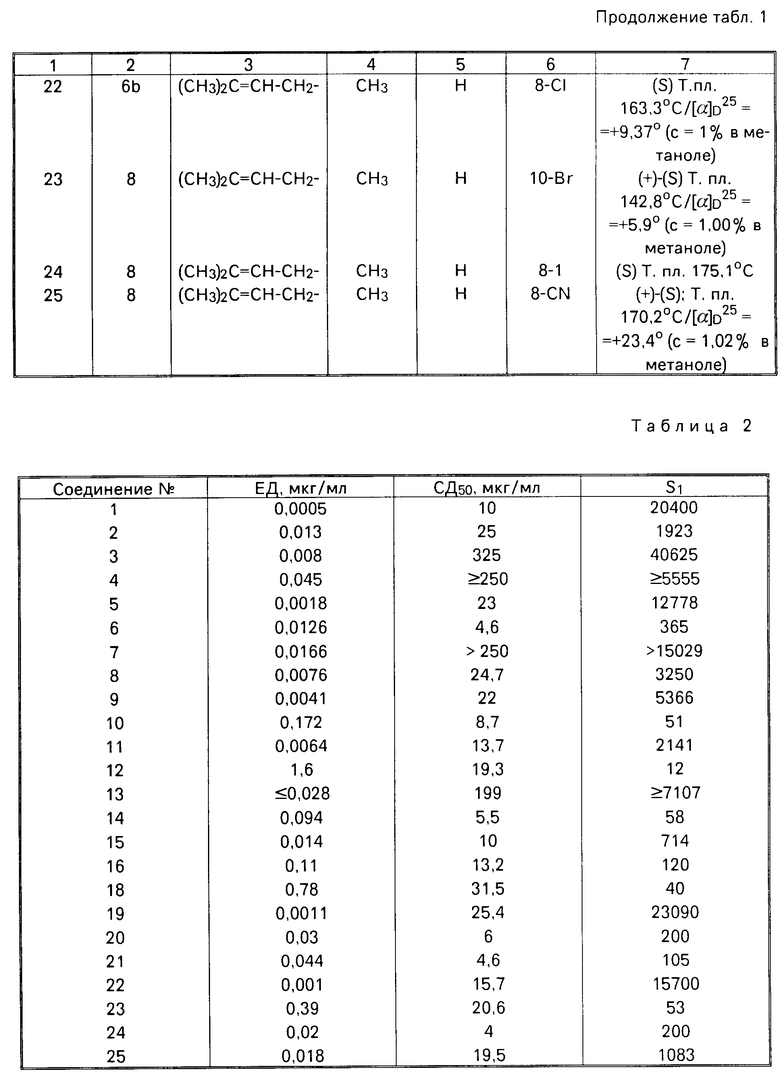
Соединения 1,3,4,8 и 9, полученные согласно изобретению, и их структурные аналоги (соединения А-Е), испытывали по описанному тесту. Полученные данные по биологической активности соединений по изобретению и известных соединений представлены в табл.3.
Индекс СД50 означает величину 50% цитотоксичной дозы.
Индекс ЕД50 означает величину 50% эффективной антивирусной зоны.
Индекс селективности SJ =  - показатель относительной безопасности лекарства.
- показатель относительной безопасности лекарства.
Сущность изобретения: продукт- ф-лы 1, где R1 - C3-C6 -алкил, замещенный C3-C6 -циклоалкилом; R2 и R3 -независимо означают водород или C1-C6 -алкил; R4 и R5 - независимо означают водород, C1-C6 -алкил, галоид, циано, нитро или трифторметил; или фармацевтически приемлемые соли. Реагент 1: соответствующее 4, 5, 6, 7-тетрагидроимидазо-[4, 5-ik][1,4]-бензодиазепин-2-он. Реагент 2: галоид. Реагент 3: тиомочевина. Условия реакции: в среде инертного органического растворителя. Структура соединения 1 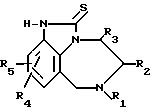 . 3 табл.
. 3 табл.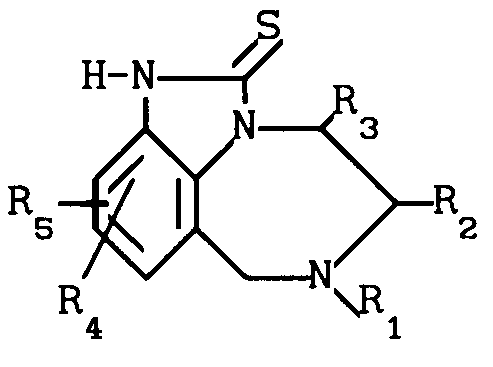
Способ получения тетрагидро [1,4]-бензодиазепин-2-тионов общей формулы
R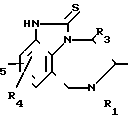 R2
R2
где R1-C3-C6-алкенил или замещенный C3-C6-циклоалкилом C1-C6-алкил;
R2 и R3 - независимо водород или C1-C6-алкил;
R4 и R5 - независимо друг от друга водород, C1-C6-алкил, галоген, циано-, нитро- или трифторметилрадикал,
или их фармацевтически приемлемых кислых аддитивных солей или стереохимических изомеров, отличающийся тем, что 4,5,6,7-тетрагидроимидазо-[4,5-jk] [1,4]бензодиазепин-2-он общей формулы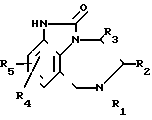
где R1-R5 имеют указанные значения,
галоидируют и полученный 2-галоид-4,5,6,7-тетрагидроимидазо-[4,5-jk] [1,4] -бензодиазепин подвергают взаимодействию с тиомочевиной в среде инертного органического растворителя и, в случае необходимости, полученный целевой продукт путем обработки его кислотой переводят в терапевтически активную нетоксичную кислую аддитивную соль или кислую соль, путем обработки ее щелочью переводят в свободное основание и/или получают его стереохимические изомеры.
| P | |||
| Geneste | |||
| L | |||
| M | |||
| Kamenka | |||
| Y | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Chem | |||
| Чугунный экономайзер с вертикально-расположенными трубами с поперечными ребрами | 1911 |
|
SU1978A1 |
