Настоящее изобретение относится к способу модификации формоселективности (конфигурационной, изомерной селективности) цеолитного катализатора и применения модифицированного катализатора в процессах формоселективной конверсии углеводородов, таких как диспропорционирование толуола.
Термин "формоселективный катализ" означает неожиданную каталитическую селективность в цеолитах. Принципы формоселективного катализа подробно рассматриваются, например, в работе N.J.Chen. W.E.Garwood and F.G.Dwyer "Shape Selective Catalysis in Jndustrial Applications", 36 Marcel Dekker Jnc. (1989). Реакции конверсии углеводородов, такие как изомеризация, диспропорционирование, алкилирование и трансалкилирование ароматических соединений, в порах циолита определяется ограничениями, накладываемыми размерами канала (длиной поры). Селективность по реагенту имеет место, когда фракция вводимого потока реагентов является слишком большой для проникновения в поры цеолита для осуществления реакции; тогда как селективность по продукту имеет место, когда некоторое количество продуктов не может выйти из каналов цеолита. Распределение продуктов может также изменяться при переменной селективности, при которой некоторые реакции не могут иметь места, поскольку промежуточные продукты реакции являются слишком большими для образования в цеолитных порах или полостях. Другой тип селективности зависит от конфигурационных ограничений диффузии, где размеры молекулы приближаются к размерам цеолитной пористой системы. Небольшое изменение в размерах молекулы или цеолитной поры может быть большие изменения диффузии, приводящие к различному распределению продукта. Этот тип формоселективного катализа демонстрируется, например, в селективном диспропорционировании алкилзамещенного бензола до парадиалкилзамещенного бензола.
Представителем парадиалкилзамещенного бензола является параксилол. Получение параксилола обычно осуществляется метилированием толуола или диспропорционированием толуола в условиях конверсии. Примеры включают в себя реакцию толуола с метиловым спиртом, как описано в работе Chen et al.J.Amer. Chem. Soc 101 6783 (1979), и диспропорционирование толуола, как описано в работе Pines in "The Chemistry of Catalitic Hydeocarbon Conversions", Academic Precc, N. Y., 1981, p. 72. Такие методы обычно приводят к получению смеси трех изомеров ксилола, т.е. параксилола, ортоксилола и метаксилола. В зависимости от степени селективности катализатора по параксилолу (параселективность) и условий реакции получается различное процентное содержание параксилола. Выход, т.е. отношение количества ксилола к количеству исходного сырья, также зависит от катализатора и условий реакции.
Равновесная реакция конверсии толуола в ксилол и бензол протекает следующим образом: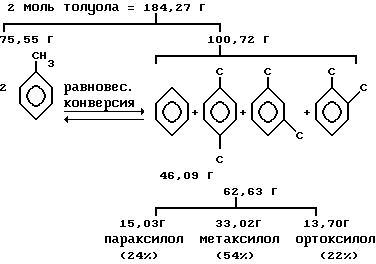
Выход параксилола: 
Селективность по параксилолу: 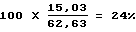
В науке известны различные способы увеличения параселективности цеолитных катализаторов. Один такой способ заключается в модификации катализатора обработкой "агентом, усиливающим селективность". Например, патент США 5173461, 4950835, 4927979, 4465886, 4477583, 4379761, 4145315, 4127616, 4100215, 4090981, 4060568 и 3698157 рассматривают специальные способы контактирования катализатора с агентом, усиливающим селективность, содержащим кремний ("кремнийсодержащее соединение"). Патент США N 4548914 описывает другой способ модификации, включающий пропитку катализаторов оксидами, которые являются трудными для восстановления, такими как оксиды магния, кальция и/или фосфора, с последующей обработкой водным паром для улучшения параселективности.
Европейский патент N 296582 описывает модификацию алюмосиликатных катализаторов пропиткой таких катализаторов фосфорсодержащими соединениями и дополнительную модификацию этих катализаторов введением металлов, таких как марганец, кобальт, кремний и элементы группы ПА. Патент также описывает модификацию цеолитов кремнийсодержащими соединениями.
Традиционно, ex situ предварительная обработка цеолита для усиления его селективности ("селективация"), т.е. селективация вне реакционного сосуда и перед преведением основной реакции, включает в себя единственное применение модифицирующего соединения. Можно отметить, однако, что в патенте США N 4283306 (Herkes) было сделано предположение многократных обработок. Патент рассматривает промотирование кристаллического SiO2-катализатора применением аморфной SiO2, такой как этилортосиликат. Herkes-патент сопоставляет характеристики катализатора, обработанного один раз этилортосиликатным раствором с последующим прокаливанием, с характеристиками катализатора, обработанного дважды этилортосиликатом и прокаленного после каждой обработки. Описание Herkes-патента показывает, что дважды обработанный катализатор является менее активным и менее селективным, чем катализатор, обработанный один раз, в реакции метилирования толуола метанолом, что предполагает, что многократная ex situ селективация не дает выигрыша, и что снижает эффективность катализатора в формоселективных реакциях.
Однако не предполагалось, что селективация алюмосиликатных цеолитов многократной ex situ пропиткой цеолитов кремнийсодержащими соединениями, с последующим прокаливанием после каждой пропитки, будет улучшать селективность и активность катализаторов. Сейчас установлено, что схема многократной пропитки дает неожиданно лучше результаты в формоопределяющих превращениях углеводородов, таких как диспропорционирование толуола, чем схемы предварительной единственной пропитки кремнийсодержащим соединением. Также было установлено, что схема многократной пропитки дает неожиданно более эффективное отложение кремнийсодержащего соединения на катализаторе, чем схемы единственной пропитки кремнийсодержащим соединением.
Обработка острым паром также используется при получении цеолитных катализаторов для модификации их активности или улучшения их стабильности. Например, патент США N 4559314 описывает обработку острым паром композита цеолит/связующее при 200 - 500oC в течение по крайней мере часа для повышения активности, как определяется по их α- величине. Патент США N 4522929 описывает предварительную обработку острым паром свежего цеолитного катализатора, так что α- активность сначала растет, затем падает до уровня не обработанного паром свежего катализатора, с получением стабильного катализатора, который может быть использован для изомеризации ксилола. Патент США N 4443554 описывает обработку острым паром неактивных цеолитов (NaZSM-5) для увеличения α- активности. Патент США N 4487843 описывает контактирование цеолита с острым паром до введения металла группы ШВ.
Также установлено, что схема многократной пропитки кремний-содержащим соединением для селективности цеолитного катализатора с последующей обработкой острым паром в специальных условиях дает дополнительно неожиданно улучшенные результаты, чем только одна многократная обработка пропиткой (без обработки острым паром).
Соответственно изобретение относится к способу модификации формоселективности алюмосиликатного цеолитного катализатора, имеющего молярное отношение SiO2/Al2O3<500, путем обработки катализатора по крайней мере двумя ex situ селективационными обработками, причем каждая ex situ селективационная обработка включает контактирование катализатора с усиливающим селективность агентом в носителе и последующее прокаливание катализатора.
Изобретение также относится к применению модифицированного катализатора в формоселективных превращениях углеводородов, в частности, в параселективных алкилароматических конверсиях, таких как толуольное диспропорционирование и этилбензольное диспропорционирование.
Используемые здесь цеолиты имеют индекс проницаемости (Constraint Index) от 1 до 12 и молярное отношение SiO2:Al2O3<500, обычно более 12, предпочтительно 20 - 100 и наиболее предпочтительно 20 - 60. Соответствующие цеолиты включают в себя ZSM-5, ZSM-11, ZSM-12, ZSM-22, ZSM-35, ZSM-48, ZSM-50 и ZSM-57. Такие цеолиты описываются, например, в патентах США N 3702886 и Re. N 29949, 3709979, 3832449, 4046859, 4556447, 4076842, 4016245, 4229424, 4397827, 4640849, 4046685, 3308069, Re. 28341. Индекс проницаемости и методика его определения описываются, например, в патенте США 4016218.
Помимо одного или более вышеуказанных цеолитов, катализатор, модифицированный по способу изобретения, может содержать связующее, обычно используемое для придания катализатору физической прочности и стойкости к истиранию. Связующее предпочтительно не содержит Al2O3 и наиболее предпочтительно содержит SiO2. Связующее может содержать 2- 70, предпочтительно 20 - 50 мас.% катализатора.
Согласно настоящему изобретению цеолит, используемый либо со связующим, либо без него, контактирует по крайней мере дважды, предпочтительно от 2 до 6 раз, с усиливающим селективность агентом предпочтительно в виде кремнийорганического соединения. Усиливающий селективность агент находится в носителе, который может быть водным или органическим носителем, и после каждой стадии контактирования катализатор прокаливается для удаления носителя и любого органического компонента, усиливающего селективность агента. В каждой фазе селективационной обработки усиливающей селективность агент наносится на наружную поверхность каатализатора любым пригодным способом. Например, усиливающий селективность агент может быть растворен в органическом растворителе, смешан с катализатором и затем высушен выпариванием или вакуумной перегонкой. Этот способ называется "пропиткой". Молекулярное сито может контактировать с соединением кремния при молекулярно-массовом соотношении молекулярное сито/соединение кремния от 100:1 до 1:100.
Предпочтительно кинетический диаметр усиливающего селективность агента превышает диаметр порциолита для того, чтобы избежать попадания усиливающего селективность агента в поры и снижения при этом внутренней активности катализатора.
Используемый усиливающий селективность агент может быть в виде раствора, эмульсии, жидкости или газа в условиях контакта с цеолитом. Нанесенный агент повышения селективности интенсивно распространяется по наружной поверхности цеолита и остается, по существу, исключительно на ней.
Примеры способов нанесения кремния на поверхность цеолита приведены в патентах США NN 4090981, 4127616, 4465886 и 4477583 (Rodewald). Дополнительные примеры нанесения соединения кремния на поверхность цеолитов описываются в работе H. Nakajima, M. Koya, H. Yshida, and M. Kohno. Sekiyu Gakkaishi 35 (2) (1992) и патенте США N 4950835 (Wang et. al.). К тому же, кремнийсодержащий усиливающий селективность агент может контактировать с цеолитом в процессе формирования каталитических частиц, например, при загрузке композиции усиливающего селективность агента/ и носителя вместе с цеолитом и необязательно, связующим, в дробильные вальцы, которые затем используются для формования смеси в экструдированную массу, из которой могут быть экструдированы каталитические частицы.
Используемые усиливающие селективность агенты представляют собой силоксаны, которые содержат повторяющиеся единицы, характеризующиеся общей формулой
где
R1 - водород, галоген, гидроксил, алкил, алкокси, галогенированный алкил, арил, галогенированный арил, аралкил, галогенированный аралкил, алкарил или галогенированный алкарил. Углеводородные заместители обычно содержат от 1 до 10 углеродных атомов, предпочтительно метил- или этил-группы;
R2 независимо выбирается из той же группы, что и R1;
n - целое число не менее 2 и обычно в пределах от 3 до 1000.
Молекулярная масса используемого кремнийсодержащего соединения обычно составляет от 80 до 20000 и предпочтительно от 150 до 10000. Представители силиконовых соединений включают в себя диметилкремний, диэтилкремний, фенилметилкремний, метилгидрокремний, этилгидрокремний, фенилгидрокремний, метилэтилкремний, фенилэтилкремний, дифенилкремний, метилтрифторпропилкремний, этилтрифторпропилкремний, полидиметилкремний, тетрахлорфенилметилкремний, тетрахлорфенилэтилкремний, тетрахлорфенилгидрокремний, тетрахлорфенилфенилкремний, метилвинилкремний и этилвинилкремний.
Кремнийсодержащее соединение может быть линейным или циклическим, например, гексаметилциклотрисилоксан, октаметилциклотетрасилоксан, гексафенилциклотрисилоксан и октафенилциклотетрасилоксан. Также могут использоваться смеси этих соединений, а также кремнийсодержащие соединения с другими функциональными группами.
Могут быть также использованы другие соединения кремния, включая силаны и алкоксиланы, такие как тетраметоксисилан. Эти используемые кремнийсодержащие усиливающие селективность агенты включают в себя силаны, характеризуемые общей формулой: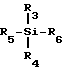
где
R3 - R6 независимо друг от друга выбираются из группы, состоящей из водорода, гидроксила, галогена, алкила, галогенированного алкила, алкокси, алкиламино, арила, галогенированного арила, аралкила, галогенированного аралкила, алкарила, галогенированного алкарила. Алкиламиносиланы благодаря своей амфифильной природе могут растворяться или, по крайней мере, эмульгироваться в водных носителях.
Предпочтительные кремнийсодержащие усиливающие селективность агенты включают в себя диметилфенилметилполисилоксан (например, DOW - 550) и фенилметилполисилоксан (например, DOW - 710). DOW - 550 и DOW - 710 поставляются фирмой DOW Chemical Co., Midland, Mi.
В водных повышающих активность системах по изобретению наиболее предпочтительные усиливающие селективность кремнийсодержащие агенты включают в себя н-пропиламинсилан, поставляемый под маркой Hydrоsil 2627 фирмой Huls America.
В одном варианте настоящего изобретения носителем усиливающего селективного агента является органическая среда. Примерами соответствующих органических носителей являются линейные, разветвленные и циклические алканы, имеющие 5 и более углеродных атомов, предпочтительно 7 или более атомов углерода. В способе настоящего изобретения органический носитель предпочтительно имеет точку кипения выше примерно 70oC. В качестве носителей необязательно могут использоваться смеси низколетучих органических соединений, таких как рециклированное масло гидрокрекинга. Наиболее предпочтительными низколетучими углеводородными носителями являются декан и додекан.
Альтернативно, носителем усиливающего селективность агента является водная среда, которая в случае некоторых описанных выше кремнийсодержащих соединений, например Hydrosil 2627, может быть использована без эмульгатора. Однако в большинстве случаев водная среда требует добавления поверхностно-активного вещества для образования эмульсии между усиливающим селективность кремнийсодержащим агентом и водным носителем. Эмульсии содержат 0,01-5 мас. %, предпочтительно 0,05-2 мас.%, поверхностно-активного вещества и 2,5-50 мас. %, предпочтительно 5-35 мас.%, усиливающего селективность кремнийсодержащего агента.
Несмотря на то, что водные кремнийсодержащие эмульсии являются коммерчески доступными, они могут содержать значительные количества добавленных органических материалов, такие как толуол, для улучшения стабильности. Присутствие таких дополнительных органических веществ увеличивает сложность, опасность и стоимость повышения селективности цеолита. Такие эмульсии являются используемыми, но не предпочтительными согласно способам модификации катализатора настоящего изобретения. С собственной рецептурой изобретателям удалось получить стабильные эмульсии, содержащие силиконовое масло, водный компонент и поверхностно-активное вещество, при этом практически избегая других компонентов.
Предпочтительные стабильные водные эмульсии силиконового масла получаются смешиванием масла и водного компонента в присутствии поверхностно-активного вещества или смеси поверхностно-активных веществ. Используемые для настоящего изобретения поверхностно-активные вещества включают в себя любое из широкого ряда поверхностно-активных веществ, включая ионные и неионные поверхностно-активные вещества. Предпочтительные поверхностно-активные вещества включают в себя неазотистые неионные поверхностно-активные вещества, такие как спирт, алкилфенол и полиалкоксиалканол-производные, глицеролэфиры, полиоксиэтиленэфиры, ангидросорбитолэфиры, этоксилированные ангидросорбитолэфиры, природные жиры, масла, воски и их этоксилированные эфиры, гидрольэфиры, полиалкиленоксид-блоксополимерные поверхностно-активные вещества, поли(оксиэтилен-со-оксипропилен)овые неионные поверхностно-активные вещества и их смеси. Более предпочтительные поверхностно-активные вещества включают в себя поверхностно-активные вещества, имеющие общую формулу α- [4-(1,1,3,3-тетраметилбутил фенил] -w-гидроксиполи(окси-1,2-этандиил)(октоксинолы), наиболее предпочтительно октоксинол-9. Такие предпочтительные поверхностно-активные вещества включают в себя ТРИТОН (TRYTON) серий X, такие как ТРИТОН X-100 и ТРИТОН X-305, поставляемые фирмой Rohm & Haas Co., Philadelphia PA. and the Ydepal CA-серий фирмы GAF Corp., New York, NY.
Водный компонент, пригодный для использования в эмульсии по изобретению, фактически составляет дисперсионную среду эмульсии. Водным компонентом предпочтительно является вода. Наиболее предпочтительно вода дистиллируется перед использованием в эмульсиях. Альтернативно, компонент может включать в себя воду и соединение, выбираемые из группы, включающей неорганические соли, спирты, имеющие 1 -18 углеродов, гликоли, простые эфиры, нейтральные или заряженные сульфоксиды, нейтральные или заряженные амины, альдегиды, кетоны, триофены, фураны, пирролы и их смеси.
Установлено, что схема многократного повышения селективности дает неожиданно увеличенную эффективность нанесения кремнийсодержащего соединения на поверхности катализатора. Эта увеличенная эффективность позволяет использовать относительно небольшие количества кремнийсодержащего соединения, а также относительно небольшие количества носителя.
После каждого нанесения кремнийсодержащего соединения катализатор прокаливается для разложения молекулярных или полимерных частиц до твердого состояния частиц. Катализатор может прокаливаться со скоростью подъема температуры от 0,2oC/мин до 5oC/мин до температуры выше 250oC, но ниже температуры, при которой кристалличность цеолита резко ухудшается. Обычно такая температура ниже 600oC. Предпочтительно температура прокаливания находится в приблизительном интервале от 350oC до 550oC. Продукт выдерживается при температуре прокаливания обычно в течение 1-24 часов, предпочтительно 2-6 часов.
Катализатор может прокаливаться в атмосфере азота, кислородсодержащей атмосфере, предпочтительно на воздухе, в атмосфере азота, за которой следует кислородосодержащая атмосфера, или атмосфере, содержащей смесь азота и воздуха. Прокаливание должно проводиться а атмосфере, практически свободной от водяных паров, для того, чтобы избежать нежелательного неконтролируемого пропаривания покрытого кремнием катализатора. Катализатор может быть прокален один раз и более одного раза после каждого отложения кремния. Прокаливания на каждой стадии обработки не должно быть идентичными, но могут отличаться температурой, скоростью подъема температуры, атмосферой и длительностью прокаливания.
После стадии повышения селективности катализатор может быть подвергнут паровой обработке при температуре 100-600oC, предпочтительно от 175oC до 325oC; от 1 до 100% острого пара, предпочтительно от 50% до 100% пара; при абсолютном давлении от 0,48 Па (0,01 фунт/дюйм2) до 2400 Па (50 фунт/дюйм2) в течение 2-12 часов, предпочтительно от 3 до 6 часов.
Кроме того, подвергнутый ex situ обработке для повышения селективности катализатор по изобретению может быть подвергнут одной или более дополнительным повышающим селективность обработкам in situ в реакторе, в котором катализатор используется для осуществления каталитической конверсии, такой как селективная конверсия алкилбензола в парадиалкилбензол. Эта in situ селективация называется здесь отделочной (окончательной) селективацией.
В одном варианте отделочная селективация осуществляется при подаче второго усиливающего селективность агента и водорода одновременно с исходным алкилбензолом в реакционных условиях до тех пор, пока не достигается необходимая селективность, по параалкилбензолу, например 90%, после чего прекращается одновременная подача усиливающего селективность агента. Реакционные условия для этой стадии in situ отделочной селективации обычно включают в себя температуру от 350oC до 650oC и давление от 100 до 34500 кПа (атмосферное до 5000 фунт/дюйм2). Реакционный поток подается в систему с объемной скоростью от 0,1 до 20 час-1, тогда как водород подается при мольном отношении водород:углеводород от 0,1 до 20.
Второй усиливающий селективность агент для отделочной селективации может содержать кремнийсодержащее соединение, как рассматривалось более подробно выше. Например, пригодными являются такие кремнийорганические соединения, как фенилметилкремний, диметилкремний и их смеси. Согласно одному варианту кремнийсодержащие фенилметил кремниевые и диметилкремниевые группы совместно подаются в систему при соотношении примерно 1:1, тогда как другие компоненты, например, алкилбезол и водород подаются в количествах, установленных выше. Кремний предпочтительно подается в количестве от 0,001 до 10% от массы алкилбензола. В зависимости от процентного содержания используемого агента, усиливающего селективность, отделочная селективация длится в течение по крайней мере одного часа, предпочтительно от 1 до 48 часов, наиболее предпочтительно менее 24 часов.
В этой схеме кремнийсодержащее соединение разлагается с осаждением на катализатор дополнительного количества SiO2. В ходе процесса селективации наблюдается дополнительное увеличение параселективности катализатора. Кремнийсодержание полимерные или молекулярные частицы могут быть растворены в толуоле или другом соответствующем ароматическом или углеводородном носителе.
Альтернативно, ex situ селективированный катализатор до контактирования с исходным алкил бензолом может быть подвергнут отделочной селективации термически разлагаемым органическим соединением при повышенной температуре, которая выше температуры разложения указанного соединения, но ниже температуры, при которой резко снижается кристалличность цеолита. Обычно эта температура ниже примерно 650oC. При этом варианте отделочной селективации образуется кокс на поверхности катализатора.
Органические материалы, термически разлагающиеся при указанных выше температурных условиях с образованием коксовых отложений, охватывают широкий ряд соединений, включая, например, углеводороды, такие как парафиновые, циклопарафиновые, олефиновые, циклоолефиновые и ароматические; кислородсодержащие органические соединения, такие как спирты, альдегиды, простые эфиры, кетоны и фенолы; гетероциклические соединения, такие как фураны, тиофены, пирролы и пиридины. Обычно, предполагается, что термически разлагающийся термоводород, такой как алкилзамещенная ароматика, является источником кокса, причем наиболее предпочтительно подвергается превращению на катализаторе по изобретению. В последнем случае алкилбензол сначала приводится в контакт с катализатором в температурных условиях и при концентрации водорода, вызывающих быстрое коксообразование. Обычно образование кокса происходит в условиях, более жестких, чем рабочие параметры, использованные в течение основного промежутка времени каталитического цикла. Когда желаемое отложение кокса уже произошло, продолжается подача алкилбензола на кокссодержащий катализатор в условиях температуры и концентрации водорода, способствующих требуемой конверсии, с значительно сниженной скоростью коксования.
Не претендуя на теоретические обобщения, можно предположить, что преимущества настоящего изобретения отчасти являются результатом превращения кислотных участков на наружных поверхностях катализатора в практически недоступные для реагентов, пори увеличении извилистости катализатора. В случае селективной конверсии алкилбензола в парадиалкилбензол, предполагается, что на кислотных участках, имеющихся на наружной поверхности катализатора, происходит изомеризация в растворе парадиалкилбензола обратно в равновесное состояние с другими диалкилбензольными изомерами, в результате чего снижается процент селективности по парапродукту. Снижением доступности этих кислотных участков для растворенного диалкилбензола может поддерживаться относительно высокое содержание параизомера. Предполагается, что усиливающие селективность агенты настоящего изобретения блокируют, или иными словами, делают эти наружные кислотные участки недоступными для парадиалкилбензола в результате химической модификации указанных участков.
Вообще каталитические конверсии на модифицированном цеолитном катализаторе по изобретению проводятся при температуре от 100oC до 760oC, давлении от 10 до 20000 кПа (0,1-200 ати), среднечасовой скорости подачи сырья от 0,08 до 2000 час-1 и мольном соотношении водород/органическое, например углеводородное, соединение от 0 до 100.
Цеолиты, модифицированные в соответствии с изобретением, обычно используются в качестве катализаторов в формоселективных способах конверсии углеводородов, в частности, формоселективном диспропорционировании алкилзамещенных бензолов с выходом диалкилзамещенных бензолов. Модифицированные цеолитные катализаторы по изобретению, преимущественно, используются для конверсии алкилбензольных соединений с получением диалкилбензольных продуктов, которые являются высокообогащенными парадиалкилбензольны изомером. Реакции превращения этого типа включают в себя трансалкилирование (переалкилирование) и диспропорционирование алкилбензолов. Алкилирование ароматических соединений, в котором могут быть использованы катализаторы изобретения, описывается, например в патентах США NN 3755483, 4086287, 4117024 и 4117026.
Было установлено, что модифицированные катализаторы настоящего изобретения, в частности, используются для селективного получения парадиалкилзамещенных бензолов, содержащих алкильные группы с 1 - 4 углеродными атомами, таких как параксилол. Такие процессы типизируются диспропорционированием, в присутствии модифицированного катализатора, углеводородного предшественника, обычно моноалкилзамещенного бензола, имеющего 1 - 4 атомов углерода в алкальном заместителе.
Диспропорционирование алкилбензолов
Катализатор настоящего изобретения предпочтительно применяется для диспропорционирования алкилзамещенных бензолов, таких как толуол и этилбензол. Обычно однократное превращение алкилбензольного потока дает поток продукта, который содержит диалкилбензолы, имеющие алкильные группы во всех положениях, т. е. орто-, мета- и парадиалкилбензолы. Катализатор, обработанный описанным здесь образом, имеет желаемый показатель скорости сорбции ортодиалкилбензола и дает при алкилбензольном диспропорционировании продукт с значительной параселективностью. Например, константа скорости диффузии в толуольном диспропорционировании рассматривается в работе D. H. Olcon and W.O. Haag. "Structure-Selectivity Relationship in Xylene Ysomerization and Selective Tolyene Disproportionation". Catalytic Materials: Relationship Between Structure and Reactivity, ACS Symposium Ser. N 248 (1984).
При диспропорционировании толуол диффундирует в цеолит с коэффициентом диффузии Dr. Толуол претерпевает диспропорционирование на пара-, мета- и ортоксилол и бензол при константе общей скорости KD. Для высокой селективности и каталитической эффективности желательно, чтобы выдерживалось неравенство
Степень параселективности зависит от активности и диффузионных характеристик катализатора. Главный продукт является обогащенным параизомером, если вначале полученные мета- и ортоксилолы диффундируют из цеолитного кристалла со скоростью (Dm,o/r2, которая ниже скорости из превращения в параксилол (Kr), а также ниже скорости диффузии параксилола (Dp/r2) из катализатора, где Dm - диффузия метаксилола, Do - диффузия ортоксилола, Dp - диффузия параксилола, r - длина диффузионного пути (размер кристалла), Kr - скорость взаимопревращения через изомеризацию изомеров ксилола с получением вторично метаксилол и ортоксилол.
Желательно увеличить параселективность катализатора. Практически это включает в себя снижение коэффициентов диффузии орто- и метаксилола, так что
В таком случае скорость превращения мета- и ортоксилолов в параксилол превышает коэффициенты диффузии мета- и параксилолов. В результате выхода пара-ксилола увеличивается. Специалистам должно быть понятно, что подобные положения применимы к коэффициентам диффузии других алкилбензолов.
Катализатор по изобретению достигает очень высокой парадиалкилбензольной селективности, обычно более 80% и предпочтительно более 90%, диспропорционировании алкилбензолов при условиях однократного превращения не менее 10%, предпочтительно не менее 15-23%. Используемый здесь термин "парадиалкилбензольная селективность" означает процентное содержание во всех диалкилбензольных продуктах, т. е. парадиалкилбензола, ортодиалкилбензола, метадиалкилбензола. Специалистам должно быть понятно, что относительная близость точек кипения этих изомеров обуславливает неизбежно относительно дорогую стоимость процессов разделения для выделения парадиалкилбензола. С другой стороны, парадиалкилбензолы легче отделяются от других компонентов в потоке продукта, таких как бензол, моноалкилбензолы и другие алкилзамещенные бензолы.
Алкилбензольное сырье предпочтительно содержит 50-100% алкилбензола, более предпочтительно не менее 80% алкилбензола. В толуольном сырье могут также присутствовать другие соединения, такие как бензол и другие алкилзамещенные бензолы.
Алкилбензольное сырье может также высушиваться при необходимости таким образом, чтобы минимизировать количество влаги, поступающее в реакционную зону. Многочисленные известные в науке методы являются пригодными для сушки исходного алкилбензола для способа изобретения. Эти методы включают в себя процеживание через любой осушитель, например силикагель, активированный Al2O3, молекулярные сита или другие подходящие вещества, или применение сушилок жидких загрузок.
При использовании для диспропорционирования алкилбензолов цеолит, селективированный способом изобретения, предпочтительно имеет размер кристаллов более 0,1 мкм, более предпочтительно более 0,2 мкм. Точное измерение размеров кристаллов цеолитных материалов является часто очень трудным. Часто используются методы микроскопии, такие как СЭМ (сканирующая электронная микроскопия) и ПЭМ (просвечивающая электронная микроскопия), но эти методы требуют измерений на большом числе кристаллов для каждого измеренного кристалла могут быть необходимы значения до трех измерений. Для ZSM-5-цеолитов, описанных ниже в примерах, были сделаны определения эффективного размера кристалла по измерению скорости сорбции 2,2-диметилбутана при 90oC и давлении углеводорода 60 мм рт.ст. Размер кристалла рассчитывается с применением уравнения диффузии, приведенного в работе J. Crank "The Mathemamatics of Diffusion". Oxford at the Clarendon Press 1957, p. 52-56, для скорости поглощения сорбата твердым веществом, диффузионные свойства которого могут быть апроксимированы плоской листовой моделью. К тому же, константа диффузии, D, 2,2-диметилбутана в этих условиях берется равным 1,5•1--14 см2/сек. Связь между размером кристалла α, измеренным в микронах, и временем диффузии t0,3, измеренным в минутах, временем, требуемым для потребления 30% объема углеводорода, является следующей
В настоящем случае эти измерения делаются на контролируемых компьютером термогравиметрических электровесах, но имеется множество известных путей получения данных. Более крупный используемый здесь кристаллический материал имеет время сорбции t0,3 497 мин, что дает расчетный размер кристалла 1,6 мкм. Более мелкий кристаллический материал имеет время сорбции 7,8 мин и расчетный размер кристалла 0,20 мкм.
Цеолит, селективированный для использования в реакции диспропорционирования алкилбензолов, предпочтительно имеет конечную α- величину не менее 50 и предпочтительно не менее 100. Альфа-величина является апроксимированным показателем каталитической активности катализатора в реакции крекинга по сравнению с стандартным аморфным расщепляющим SiO2 - Al2O3 - катализатором. Определение α описано в патенте США 3354078 и в The Journal of Catalycis т. 4, стр. 522-529 (август 1965), т. 6, стр. 278 (1966) и т. 61, стр. 395 (1980). Отмечается, что истинные константы скорости для многих кислотно-катализированных реакций являются пропорциональными α- величине для частично кристаллического силикатного катализатора (смотри "The Aktive Site of Acidic Aluminosilicate Catalysts" Nature т. 309, N 5959, стр. 589-591, 14 June 1984).
Рабочими условиями способа алкилбензольного диспропорционирования по настоящему изобретению является температура на входе реактора 200-600oC, предпочтительно 350-540oC; давление от 100 до 34500 кПа (атмосферное давление 5000 фунт/дюйм2), предпочтительно 700-7000 кПа (100-1000 фунт/дюйм2); объемная скорость от 0,1 до 20 час-1, предпочтительно 2-10 час-1; и мольное соотношение H2:углерод 0,05 до 20, предпочтительно 0,5-6. Этот способ может осуществляться либо периодически, либо псевдоожиженном слое с легко достигаемыми преимуществами любого способа. Выходящий поток может быть отделен и дистиллирован с удалением нужного продукта, т.е. параизомера, а также других побочных продуктов. Альтернативно соответствующая фракция может быть подвергнута дополнительной сепарации, как в случае ксилолов, подвергнутых кристаллизацией или PAPEX-процессу с получением параксилола.
Катализатор может быть дополнительно модифицирован для того, чтобы снизить количество нежелательных побочных продуктов, таких как этилбензол в случае ксилолов, вытекающий из реактора,
Продукт обычного толуольного диспропорционирования обычно содержит около 0,5% этилбензольного побочного продукта. При дистилляции реакционных продуктов содержание этилбензола в C8-фракции часто увеличивается до 3 - 4%. Это содержание этиленбензола является неприемлемым для параксилола качества для полимеров, так как этилбензол в параксилоле, если он не удален, ухудшает качество волокон, получаемых в конечном счете из параксилольного продукта. Следовательно, содержание этилбензола в параксилольном продукте должно поддерживаться низким. Технические условия на промышленный параксилол определяют допустимое в нем содержание этилбензола как не более 0,3%. Этилбензол может быть практически удален способами кристаллизации или суперфракционирования.
Для того, чтобы избежать необходимость удаления падающего вниз этилбензола, содержание этилбезольного побочного продукта благоприятно снижается введением функции гидрирования/дегидрирования в катализатор так, как при добавлении соединения металла, такого как платина. В то время как платина является предпочтительным металлом, могут быть использованы другие металлы от группы IB до группы VIII периодической системы элементов, такие как палладий, никель, медь, кобальт, молибден, родий, рутений, серебро, золото, ртуть, осмий, железо, цинк, кадмий и их смеси. Металл может быть добавлен катионообменом в количествах от 0,001% до 2%, обычно около 0,5% от массы катализатора. Например, модифицированный пластиной катализатор может быть получен сначала добавлением катализатора к раствору нитрата аммония для того, чтобы перевести катализатор в аммониевую форму. Катализатор последовательно контактирует с водным раствором нитрата тетрааминплатины (II) или хлорида тетраминплатины (II). Катализатор затем может быть отфильтрован, промыт водой и прокален при температурах от 250oC до 500oC.
Нижеследующие неограничивающие примеры иллюстрируют изобретение по диспропорционированию толуола и этилбензола.
В примерах определяется параметр скорости сорбции ортоксилола Do/r2 при 120oC и 505 Па (3,8 мм рт.ст.). Do - коэффициент диффузии ортоксилола, r - размер кристалла, Do/r2 - параметр скорости диффузии, который является мерой скорости движения ортоксилола в и из кристаллов катализатора.
Кроме того, в примерах определение атмосферного диспропорционирования толуола (ДПТ) проводится при 482oC, I атмосфере при 4% конверсии. Константа скорости ДПТ катализатора получается при тех же самых условиях.
Пример 1 (Сравнительный)
Атмосферное отборочное испытание по каталитической активности и селективности в процессе ДПТ проводится следующим образом с использованием образца HXSM-5/SiO2 (65% HZSM-5/35% SiO2) с размером кристалла 1,6 мкм. Осуществляется контактирование необработанного образца катализатора с толуолом при атмосферном давлении при 482oC, и конверсия толуола варьируется регулированием объемной скорости реакционного потока. Параселективность необработанного катализатора равняется 37% при 40 конверсии толуола с константной скорости ДПТ 167. Коэффициент ортоксилольной диффузии в необработанном катализаторе равняется 4,7•10-6, и сорбция н-гексана равняется 69 мг/г.
Пример 2 (Сравнительный).
К 8,0 г необработанного катализатора (пример 1) добавляется 1,55 г диметилфенилметилполисилоксана (DOW-550), растворенных в 40 см3 гексана. Катализатор перемешивается в растворе кремнийсодержащего соединения в течение нескольких минут, и гексан отгоняется высоковакуумной перегонкой. Высушенный образец катализатора охлаждается до комнатной температуры и затем прокаливается на воздухе при подъеме температуры со скоростью 1oC/мин до 538oC и выдержке при этой температуре в течение 3 часов. Модифицированный SiO2 катализатор взвешивается, причем его привес оказывается равным 3,7 мас.%, вероятно, в виде SiO2.
Атмосферное ДПТ-испытание проводится на катализаторе, обработанном один раз, как описывается в примере 1. Параселективность катализатора равняется 67,3% при 4% конверсии толуол с константой скорости ДПТ 226. Ортоксилольный коэффициент диффузии снижается до 1,1•10-6, а затем H-гексана равняется 68 мг/г.
Пример 3.
К 5,75 г катализатора, обработанного один раз (пример 2), добавляется 1,12 г диметилфенилметилполисилоксана (DOW-550), растворенных в 40 см3 гексана. Катализатор перемешивается в силиконовом растворе в течение нескольких минут, и гексан отгоняется высоковакуумной перегонкой. Высушенный образец катализатора охлаждается до комнатной температуры, а затем прокаливается на воздухе со скоростью 1oC/мин до 538oC и выдерживается при этой температуре в течение 3 часов. Модифицированный SiO2 катализатор взвешивается, причем привес оказывается равным 5,0 мас.%, вероятно, в виде SiO2.
Атмосферное ДПТ-испытание проводится на дважды обработанном катализаторе, как описано в примере 1. Параселективность катализатора увеличивается до 92,9% при 4% конверсии толуола с константой скорости ДПТ 251. Коэффициент ортоксилольной диффузии снижается до 0,29•10-6, и сорбция H-гексана равняется 65 мг/г.
Пример 4.
К 4,18 г дважды обработанного катализатора (пример 3) добавляется 0,81 г диметилфенилметилполисилоксана (DOW-550), растворенных в 40 см3 гексана. Катализатор перемешивается в кремнийсодержащем растворе в течение нескольких минут, и гексан отгоняется высоковакуумной перегонкой. После охлаждения высушенного катализатора до комнатной температуры образец прокаливается на воздухе при подъеме температуры со скоростью 1oC/мин до 538oC и выдерживается при этой температуре в течение 3 часов. SiO2-модифицированный катализатор взвешивается, причем привес равняется 0,8 мас.%, по-видимому, в виде SiO2.
Атмосферное ДПТ-испытание проводится на дважды обработанном катализаторе, как описано в примере 1. Параселективность катализатора равняется 99,1% при 4% конверсии толуола с константой скорости ДПТ 249. Коэффициент ортоксилольной диффузии дополнительно снижается до 0,073•10-6, и сорбция Н-гексана равняется 64 мг/г.
Сравнение характеристик необработанного катализатора и трех обработанных кремнийсодержащим соединением катализаторов, описанных в примерах 1-4, приводится ниже в таблице 1.
Данными таблицами 1 (примеры 1 - 4) показано, что в результате многократного нанесения кремниевых покрытий на цеолитный катализатор значительно меняются характеристики катализатора. У трижды обработанного катализатора наблюдается чрезвычайно высокая параселективность (99,1%) по сравнению с необработанным образом (37,0%). Необходимо также отметить, что сорбция Н-гексана является идентичной во всех примерах 1 - 4; это подтверждает, что SiO2 наносится практически только на наружную поверхность кристалла. Наконец, коэффициент диффузии трижды обработанного катализатора является примерно в 5 - 6 раз меньше, чем у необработанного материала. Это наблюдение, в основном, согласуется с определенным количеством SiO2, добавленным к катализатору, указывая на то, что при нанесении SiO2 вводится значительный диффузионный барьер.
Пример 5.
Оценка каталитической активности селективированного катализатора проводится на автоматизированной установке (приборе) с отбором проб на линии. Один грамм трижды покрытого материала (пример 4) загружается в трубчатый реактор диаметром 6,35 мм (0,25 дюйма) из нержавеющей стали. Образец нагревается до 538oC в 200 см3/мин воздуха при скорости нагревания 2,0oC/мин. Затем вводится чистый толуол при 485oC, объемной скорости 4 час-1 H2/HC=2 и 3550 кПа (500 фунт/дюйм2). После 20 часов работы продукт содержит 80% параксила при 37%-конверсии. Варьированием объемной скорости демонстрируется возможность реализации очень высокой параксилольной селективности, например 96% параксилола при 19% конверсии с объемной скоростью 16 час-1.
Для определения активности/селективности селективированного катализатора варьируется температура реакции (в реакторе) с получением профиля конверсии толуола как функции температуры. Например, при 465oC, 4 час-1 H2/HC=2 и 3550 кПа (500 фунт/дюйм2) катализатор дает 93% параксилола при 29% конверсии.
Пример 6. Для определения степени постоянства селективации используемый катализатор из примера 5 прокаливается на воздухе. После 24 часов при 466oC, 4 час-1 H2/HC=2 и 3550 кПа (500 фунт/дюйм2) на чисто толуольном сырье профиль катализатора изменяется до 93% параксилола при 30% конверсии. Таким образом, превосходные характеристики катализатора сохраняются после регенерации.
Пример 7. Для того, чтобы дополнительно исследовать свойства модифицированного катализатора из примера 5, инициируется in situ отделочная селективация с помощью 0,1 мас.% диметилфенилметилполисилоксана (DOW-550) в толуоле в течение четырехчасового периода (485oC, 4 час-1 H2/HC=2 и 3550 кПа). В результате этой отделки достигается увеличенная параксилольная селективность, т.е. 91% параксилола при 32% конверсии против 86% параксилола при 35% конверсии. В результате непрерывной отделки в тех же самых реакционных условиях достигается очень высокая параксилольная селективность.
Пример 8.
HZSM-5/SiO2-катализатор с размером кристалла приблизительно 1,6 мкм подвергается 4-м последовательным (подряд) обработкам диметилфенилметилполисилоксаном (DOW-550), как описано выше и иллюстрируется примерами 2 - 4. Образец этого модифицированного материала массой 1 г загружается в трубчатый реактор диаметром 6,35 (0,25 дюйма) из нержавеющей стали. Образец предварительно сушится при 300oC в течение нескольких часов. Затем с использованием чистого толуольного сырья при 484oC, 4 час-1 H2/HC= 2 и 3550 кПа (500 фунт/дюйм2 ) инициируется цикл селективной реакции толуольного диспропорционирования. Результаты приводятся в таблице 2.
Пример 9.
Несколько граммов многократно обработанного катализатора, описанного в примере 8, обрабатывается острым паром (100% пар) в течение 4 часов при 204oC при 1 атмосфере. Затем активность и селективность пропаренного катализатора оценивается при осуществлении цикла селективного ДПТ, как описывается в примере 8. Результаты приводятся в таблице 2.
Пример 10.
Несколько граммов многократно обработанного катализатора, описанного в примере 8, обрабатывается паром (100% пар) в течение 4 часов при 316oC при 1 атм. Затем активность и селективность пропаренного катализатора оценивается путем проведения цикла селективного ДПТ, как описывается в примере 8. Результаты приводятся в таблице 2.
Пример 11.
Несколько граммов многократно обработанного катализатора, описанного в примере 8, обрабатывается паром (100% пар) в течение 4 часов при 1 атм. Затем активность и селективность пропаренного катализатора оценивается путем проведения цикла селективного ДПТ, как описывается в примере 8. Результаты приводятся в таблице 2.
Пример 12.
Несколько граммов многократно обработанного катализатора, описанного в примере 8, обрабатывается паром (100% пар) в течение 4 часов при 500oC при 1 атм. Затем активность и селективность пропаренного катализатора оценивается путем проведения цикла селективного ДПТ, как описывается в примере 8.
В таблице 2 ниже приводятся данные, сравнивающие значения активности и селективности для необработанного паром (пример 8) и различно обработанных паром (примеры 9 - 12) катализаторов с многократным покрытием.
Образцы катализаторов, обработанные паром в течение 4 часов при 204oC (пример 9) или 316oC (пример 10), являются как более активными, так и более селективными, чем необработанный паром материал, тогда как при более высокой температуре обработки паром (500oC, 4 часа) достигается значительно меньшая активность и снижается селективность (пример 12). У материала, обработанного паром при 371oC (пример 11), наблюдается активность и селективность, соразмерная с активностью и селективностью необработанного паром материала (пример 8).
Важно, что с умеренно пропаренными материалами примера 9 (204oC) и примера 10 (316oC) достигается 25% конверсии при 9 и 10 час-1 соответственно, тогда как для непропаренного материала примера 8 требуется 4 час-1. Таким образом, при низкотемпературной обработке паром достигается 2-3-кратное увеличение активности. Для материала, обработанного паром при высокой температуре, пример 12, требуется 2 час-1 для того уровня конверсии, и, таким образом, только половина такого активного, как необработанной паром материал.
Пример 13.
Катализатор данного примера получается посредством процедуры многократного покрытия. 5,38 г необработанного HZSM-5/SiO2-материала с размером кристалла 1,6 мкм подвергается 3-м последовательным обработкам диметилфенилметилполисилаксаном (DOW-550) в додекане. Для каждой обработки примерно 1,9 г диметилфенилметилполисилексана (DOW-550) растворяется в 10 г додекана и используется в качестве пропитывающего раствора. После каждой обработки катализатор прокаливается на воздухе со скоростью нагревания 5oC/мин до 538oC и выдерживается при этой температуре в течение 0,5 часа. Общий привес катализатора равняется примерно 8 мас.%.
Толуольное диспропорционирование проводится на автоматизированной установке с отбором образцов на линии. Приблизительно 1 г экструдата катализатора загружается в трубчатый реактор диаметром 6,35 мм (0,25 дюйма) из нержавеющей стали.
Каталитический цикл инициируется подачей чистого толуола при 486oC, 4 час-1, H2/HC= 2 и 3550 кПа (500 фунт/дюйм2). Первоначально на катализатор получают 89% параксилола при 30% конверсии. Через 20 часов активность катализатора слегка улучшается до 92% параксилола при 28% конверсии. При 8 час-1 получается 96% параксилола при 21% конверсии. Таким образом, замена растворителя на более низко летучий не оказывает отрицательного влияния на селективность катализатора.
Варьирование температуры в пределах 80oC оказывает незначительное влияние на результаты, в которых поддерживается высокое значение параселективности (более 90%). Например, 95% параксилола получается при 23% конверсии при 465oC.
Пример 14.
После каталитического цикла, описанного в примере 13, катализатор удаляется из установки. Для подтверждения степени устойчивости селективации катализатор регенерируется быстрым прокаливанием на воздухе со скоростью нагревания 5oC/мин до 538oC в муфельной печи. После прокаливания исходного образца, полученного в процессе каталитического цикла при 486oC, 4 час-1, H2/HC= 2 и 3550 кПа (500 фунт/дюйм2) на чисто толуольном сырье, получается 86% параксилола при 24% конверсии. Через несколько часов селективность катализатора увеличивается до 90% параксилола при 22% конверсии. Общая потеря селективности регенерированного катализатора (примерно 25%) по сравнению с исходным модифицированным катализатором может относиться к возможной непреднамеренной обработке паром, происходящей от быстрого воздушного прокаливания регенератора.
Пример 15.
К 20,0 г необработанного HZSM-5/SiO2-катализатора, имеющего размер кристалла 1,6 мкм, добавляется 3,88 г диметилфенилметилполисилоксана (DOW-550), растворенного в 60 см3 гексана. Катализатор перемешивается в кремнийсодержащем растворе в течение нескольких минут, и гексан отгоняется высоковакуумной перегонкой. Сухой катализатор затем прокаливается со скоростью нагревания 1oC/мин в азоте до 538oC. После охлаждения до комнатной температуры образец затем прокаливается на воздухе со скоростью нагревания 1oC/мин до 538oC и выдерживается при этой температуре в течение 3 часов. Привес SiO2-модифицированного катализатора равняется 1,4 мас.%, вероятно, в виде SiO2. Катализатор затем обрабатывается подобным образом еще 3 раза с использованием 6,77 г, 6,82 г, 6,78 г DOW-550 соответственно. Дополнительные привесы равняются 3,54 мас.%, 1,67 мас.% и 1,39 мас.%, соответственно, при общем привесе около 8,23 мас.% после четырех силиконовых обработок.
Каталитическая активность и селективность оцениваются путем осуществления цикла ДПТ в автоматизированной установке с отбором образцов на линии. Приблизительно 1 г модифицированного катализатора загружается в трубчатый реактор диаметром 6,35 мм (0,25 дюйма) из нержавеющей стали. Образец нагревается до 538oC в 200 см3/мин воздуха со скоростью 20oC/мин. Каталитический цикл инициируется подачей чистого толуола при 445oC, 4 час-1, H2/HC=2 и 3550 кПа (500 фунт/дюйм2). Температурным сканированием подтверждается, что катализатор является активным и селективным. Например, при 485oC с другими идентичными условиями и при 22 час на потоке с данным катализатором получается 88% параксилола при 32% конверсии. При 465oC с другими неизменными условиями через несколько часов с данным катализатором получается 88% параксилола при 26% конверсии.
При прокаливании для регенерирования катализатора при 485oC, 4 час.-1, H2/HC= 2 и 3550 кПа (500 фунт/дюйм2) с данным катализатором получается 91% параксилола при 30% конверсии. Таким образом, характеристики катализатора при регенерации сохраняются.
Пример 16.
Для определения влияния транспортировки катализатора, например, загрузки, выгрузки и т.д., образец экструдата из примера 15 измельчается до 14/30 меш и испытывается на каталитическую активность и селективность. Загружается образец массой 1 г, и проводится каталитический цикл как описано в примере 15.
Каталитический цикл инициируется подачей чистого толуола при 445oC, 4 час-1, H2/HC=2 и 3550 кПа (500 фунт/дюйм2). Сканирование температуры подтверждает, что катализатор является активным и селективным. Через 8 часов (485oC, 4 час-1, H2/HC= 2 и 3550 кПа) получается 76% параксилола при 37% конверсии. Через 19 часов на потоке (485oC, 6 час-1, H2/HC=2 и 3500 кПа) получается 85% параксилола при 32% конверсии.
Таким образом, через эквивалентное количество времени измельченный экструдат демонстрирует приблизительно аналогичные оригинальному модифицированному материалу профили активности/селектиности. Эти результаты предполагают, что физическое повреждение экструдера катализатора, ex situ обработанного усиливающим селективность агентом, приводит только к минимальным потерям (например, 3%) каталитической селективности; однако, часть этих потерь может быть восстановлена в ходе процесса.
Для определения устойчивости селективации и влияния на него процесса измельчения катализатор регенерируется прокаливанием на воздухе, как описывается в примере 15. На катализаторе получают 87% параксилола при 32% конверсии (485oC, 6 час-1, H2/HC=2 и 500 фунт/дюйм2), что приблизительно эквивалентно селективности и активности измельченного катализатора до регенерации. Не наблюдается значительного влияния измельчения на регенерационное поведение катализатора, многократно покрытого силиконом.
Пример 17.
105,0 г необработанного HZSM-5/SiO2-катализатора (1,6 мм экструдата, высушенный при 130oC) с размером кристалла 0,2 мкм добавляется к раствору 10,0 г диметилфенилметилсилоксана (DOW-500), растворенного в 92 г додекана. Катализатор смешивается с кремнийсодержащим раствор при комнатной температуре в течение 2 часов. Затем избыток растворителя удаляется фильтрацией, после чего проводится двухстадийная процедура прокаливания. Экструдат нагревается в азоте до 140oC и выдерживается при этой температуре в течение 2 часов, затем нагревается в азоте со скоростью 2oC/мин до 538oC и выдерживается при этой температуре в течение 2 часов. Образец затем охлаждается в азоте до температуры 300oC, при котором вводится воздух, с последующим нагреванием со скоростью 2oC/мин до 538oC, и выдерживается при этой температуре в течение 4 часов. После охлаждения в азоте до комнатной температуры определяется, что привес катализатора равняется 5,7 мас.%.
111,0 г однократно обработанного катализатора добавляется к раствору 10,0 г диметилфенилметилполисилоксана (DOW-550), растворенных в 92 г додекана. Катализатор смешивается с кремнийсодержащим раствором при комнатной температуре в течение 2 часов. Затем избыток растворителя удаляется фильтрацией, после чего проводится двухстадийная процедура прокаливания. Экструдат нагревается в азоте до 140oC и выдерживается при этой температуре в течение 2 часов, затем нагревается в азоте со скоростью 2oC/мин до 538oC и выдерживается в течение 2 часов при этой температуре. Образец затем охлаждается в азоте до температуры 300oC, при которой вводится воздух, с последующим нагреванием со скоростью 2oC/мин до 538oC и выдержкой в течение 4 часов при этой температуре.
98,0 г дважды обработанного катализатора добавляется к раствору 9,6 г диметилфенилметилполисилоксана (DOW-550), растворенных в 88 г додекана. Катализатор смешивается с кремний-содержащим раствором при комнатной температуре в течение 2 час. Затем избыток растворителя удаляется фильтрацией, после чего проводится двухстадийное прокаливание. Экструдер нагревается в азоте до 140oC и выдерживается при этой температуре 2 час, затем нагревается в азоте со скоростью 2oC/мин до 538oC и выдерживается при этой температуре в течение 2 часов. Образец затем охлаждается в азоте до температуре 300oC, при которой вводится воздух, и затем нагревается со скоростью 2oC/мин до 538oC и выдерживается при этой температуре в течение 4 часов. После охлаждения в азоте до комнатной температуры определяется привес образца, который оказывается равным 2,0 мас.%.
96,0 г трижды обработанного катализатора добавляется к раствору 9,4 г диметилфенилметилполисилоксана (DOW-550), растворенных в 86 г додекана. Катализатор смешивается с силиконовым раствором при комнатной температуре в течение 2 часов. Затем избыток растворителя удаляется фильтрацией, после чего проводится двухстадийное прокаливание. Экструдат нагревается в азоте до 140oC и выдерживается при этой температуре в течение 2 часов, затем нагревается в азоте со скоростью 2oC/мин до 538oC и выдерживается при этой температуре в течение 2 часов. Образец затем охлаждается в азоте до температуре 300oC, при которой вводится воздух, и нагревается со скоростью 2oC/мин до температуры 538oC, при которой выдерживается в течение 4 часов. После охлаждения в азоте до комнатной температуры определяется привес катализатора, который равняется 2,0 мас.%.
Образец 4-кратно обработанного катализатора массой 2 г испытывается в автоматизированной установке с отбором проб на линии. Образец катализатора загружается в трубчатый реактор диаметром 7,6 мм (0,305 дюйма) из нержавеющей стали и затем нагревается в водороде со скоростью 3,5oC/мин до 425oC в потоке водорода 40 см3/мин. Чистый толуол вводится при 425oC при 4 час-1, H2/HC= 1,5-2 и 2170 кПа (300 фунт/дюйм2). Характеристики приводятся ниже в таблице 3.
Пример 18.
50,0 г необработанного HZSM-5/SiO2-катализатора (1,6 мм (1/16 дюйма) экструдат, высушенный при 105oC), имеющего размер кристалла 1,6 мкм, добавляется к раствору 4,6 диметилфенилметилполисилоксана (DOW-550), растворенных в 49 г додекана. Обеспечивается контактирование/абсорбирование катализатора с силиконовым раствором в течение нескольких минут, после чего додекан удаляется в атмосфере азота при 210-220oC. Катализатор затем помещается в металлический ящик и прокаливается в муфельной печи при нагревании со скоростью 2oC/мин в 80% / 20% смеси N2/воздух (общий расход 60 см3/мин) до 538oC. Катализатор выдерживается при температуре 538oC в течение примерно 4 часов. После охлаждения в атмосфере определяется, что масса SiO2-модифицированного катализатора увеличивается на 1,3 мас.%.
50,7 однократно обработанного катализатора добавляется к раствору 4,6 г диметилфенилметилполисилоксана (DOW-550), растворенных в 50 г додекана. Обеспечивается контактирование катализатора с кремнийсодержащим раствором в течение нескольких минут, после чего додекан удаляется в атмосфере азота при 210-220oC. Катализатор затем помещается в металлический ящик и прокаливается в муфельной печи при нагревании до 538oC со скоростью 2oC/мин в 80% /20% смеси N2/воздух (общий расход 60 см3/мин). Катализатор выдерживается при температуре 538oC в течение примерно 4 часов. После охлаждения в атмосфере азота определяется, что масса SiO2-модифицированного катализатора увеличивается на 3,1 мас.%.
51,2 г дважды обработанного катализатора добавляется к 2,4 г диметилфенилметилполисилоксана (DOW-550), растворенных в 51 г додекана. Обеспечивается контактирование катализатора с кремнийсодержащим раствором в течение нескольких минут, после чего додекан удаляется в атмосфере азота при 210-220oC. Катализатор затем помещается в металлический ящик и прокаливается в муфельной печи с нагреванием до 538oC со скоростью 2oC/мин и 80% /20% смеси N2/воздух (общий расход 60 см3/мин). Катализатор выдерживается при температуре 538oC в течение примерно 4 часов. После чего катализатор охлаждается в атмосфере азота, взвешивается, и определяется, что масса катализатора увеличивается на 2,1 мас.%.
Катализатор делится на две порции по 25 г. Одна порция обрабатывается паром при 316oC в течение 4 часов в 100% паре, и испытывается с использованием реакции толуольного диспропорционирования, как описывается ниже. Результаты сравниваются с характеристиками непропаренной порции многократно обработанного усиливающим селективность агентом катализатора, а также с катализатором, обработанным коксом.
Характеристики ex situ селективированного катализатора оказываются подобными характеристикам селективированного коксом катализатора. Например, трижды обработанный кремнием катализатор проявляет только слегка сниженную активность, что определяется по требуемой более высокой средней температуре (471oC против 464oC). Выход в случае ex situ селективированного катализатора, однако, является обычно лучшим, чем в случае коксом селективированного образца. Например, выход параксилола является более высоким для ex situ катализатора, чем для катализатора, модифицированного коксом.
Пример 19.
Проводится сравнение активности и селективности катализатора, модифицированного посредством единичного SiO2-покрытия, и катализатора, модифицированного способом многократного покрытия в соответствии с настоящим изобретением.
50,2 г необработанного HZM-5/SiO2-катализатора (1,6 мм (1/16 дюйма) экструдат, высушенный при 105oC), имеющего размер кристалла 1,6 мкм, добавляется к раствору 11,6 г диметилфенилметилсилоксана (DOW-550), растворенных в 50 г додекана. Обеспечивается контактирование катализатора с кремнийсодержащим раствором в течение нескольких минут, после чего додекан удаляется в атмосферу азота при 210-220oC. Катализатор затем помещается в металлический ящик и прокаливается в муфельной печи при нагревании до 538oC со скоростью 2oC/мин и 80% /20% смеси N2/воздух (общая подача 60 см3/мин). Катализатор выдерживается при температуре 538oC в течение примерно 4 часов. После охлаждения в атмосфере азота определяется привес SiO2-модифицированного катализатора, который оказывается равным 5,2 мас.%.
Характеристики катализатора, обработанного только один раз, оцениваются с использованием реакции диспропорционирования толуола, как описано ниже. Результаты сравниваются с характеристиками трижды селективированного катализатора из примера 16. Отмечается, что при одинаковой степени конверсии толуола трижды обработанный катализатор является более селективным, чем катализатор, обработанный один раз.
Пример 20
Исследуемый в данном примере катализатор получается способом многократного нанесения покрытия. Необработанный образец HZHM-5/SiO2 подвергается двум последовательным обработкам диметилметилполисилоксаном (DOW-550), предназначенным для добавления примерно 5 мас.% SiO2. Подробная обработка описывается ниже. 6,9 г необработанного HZSM-5/ SiO2-катализатора с размером кристалла 1,6 мкм (1,6 мм(1/16 дюйма) экструдат, высушенный при 300oC) добавляется к раствору 0,65 г диметилфенилметилсилоксана (DOW-550), растворенных в 3 г додекана. Катализатор затем помещается в вертикальную трубчатую печь и прокаливается с нагреванием до 538oC со скоростью 2oC/мин в 80%/20% смеси N2/воздух. Катализатор выдерживается при температуре 538oC в течение примерно 3 часов. После охлаждения в атмосфере азота определяется прирост массы SiO2-модифицированного катализатора, который оказывается равным 2,9 мас.%.
7,1 г модифицированного катализатора добавляется к раствору 0,66 г диметилфенилметилсилоксана (DOW-550), растворенных в 3 г додекана. Катализатор затем помещается в вертикальную трубчатую печь и прокаливается при температуре до 583oC, которую поднимают со скоростью 2oC/мин в 80% /20% смеси N2/ воздух (общая подача 60 см3/мин). Катализатор выдерживается при температуре 538oC в течение 3 часов. После охлаждения в атмосфере азота определяется прирост массы катализатора, который оказывается равным 2,7 мас.%.
Характеристики дважды модифицированного катализатора оцениваются при проведении цикла селективного этилбензольного диспропорционирования (СЭБДП) в автоматизированной установке с отбором образцов на линии. Приблизительного 1 г модифицированного катализатора загружается в трубчатый реактор диаметром 6,625 мм (0,25 дюйма) из нержавеющей стали.
Каталитический цикл инициируется подачей этилбензола при 6 час-1, 213 кПа (28 фунт/дюйм2) и H2/HC=0. Данные получаются в течение 20-дневного периода времени на потоке в интервале температур от примерно 314oC до примерно 337oC. Скорость старения 0,6oC/день наблюдается примерно 7 дней. На всем цикле наблюдается усредненная парадиэтилбензольная селективность около 97,4% при 13-14% этилбензольной конверсии.
Пример 21.
Характеристики модифицированного катализатора из примера 20 оцениваются дополнительно при проведении цикла СЭБДП с использованием модифицированного катализатора в присутствии одновременно подаваемого водородного разбавителя.
Каталитический цикл инициируется подачей этилбензола при 10 час-1, 690-790 кПа (85-100 фунт/дюйм2) и H2/HC=1,0. Активность и селективность катализатора контролируется в течение 11 дн. при различных температурах.
После достижения видимого устойчивого состояния конверсия этилбензола остается при 20% в течение приблизительно 170 часов (H2/HC=1,0, 350oC, 690 кПа, 10 час-1). При отсутствии одновременной подачи водорода в этих условиях ожидается скорость старения катализатора около 4oC/день. Совсем напротив, в присутствии водорода наблюдается незначительное старение или вообще его отсутствие в течение исследуемого периода времени.
Пример 22 (сравнительный)
20 г полимера Н-пропиламинсилана (Hydrosil 2627) разбавляется 20 г деионизованной воды. 7 г NaZSM-5/SiO2 добавляется к этому раствору и остается стоять до равновесия на 16 часов, после чего остаточная вода выпаривается при 130oC. Образец затем прокаливается в N2 до 538oC со скоростью 2oC/мин в течение 2 часов, с последующим прокаливанием на воздухе при 538oC в течение 2 часов. Затем определяется увеличение массы экструдата, которое оказывается равным 16,8% SiO2.
Оценка каталитической способности селективированного катализатора проводится при атмосферном давлении, 482oC и 4% конверсии толуола. В этих условиях параксилольная селективность оказывается равной 31%.
Пример 23.
15 г NaZSM-5A/SiO2 добавляется к 15 г раствора полимера эминосилана/вода и все остается стоять до сухости. Катализатор затем нагревается в азоте до 538oC со скоростью 2oC/мин и выдерживается при температуре 538oC в течение 2 часов. Образец затем охлаждается до 300oC, вводится воздух, образец нагревается со скоростью 2oC/мин до 538oC и выдерживается при этой температуре в течение 2 часов. По отношению к известному силиконовому содержанию в полимере H-пропиламинсилана (Hydrosil 2627) определяется, что привес экструдата равняется 4,5 SiO2.
Однократно модифицированный катализатор обрабатывается второй раз с использованием методики, описанной для первой модификации.
13 г дважды модифицированного катализатора добавляется к 13 г раствора 27% полимера аминосилан/вода и остается стоять до сухости. Катализатор затем нагревается в азоте до 538oC со скоростью 2oC/мин и выдерживается при температуре в течение 2-х часов. Образец охлаждается до 300oC, вводится воздух, образец нагревается со скоростью 2oC/мин до 538oC и выдерживается при этой температуре в течение 2 часов.
10 г трижды обработанного катализатора добавляется к 10 г 27% раствора полимера аминосилана в воде и остается стоять до сухости. Катализатор затем нагревается в азоте со скоростью 2oC/мин до температуры 538oC, при которой катализатор выдерживается в течение 2 часов. Образец охлаждается до температуры 300oC, вводится воздух, образец нагревается со скоростью 2oC/мин до температуры 538oC и выдерживается при этой температуре в течение 2 часов.
После четвертой модификации образец обрабатывается для обмена 1 М NH4NO3 при комнатной температуре в течение 1 часа для общих 3-х обменов.
Оценка каталитической активности четырежды обработанного катализатора (0,77 г масса золы) проводится в автоматизированной установке с отбором образцов на линии. Образец загружается в трубчатый реактор диаметром 7,75 мм (0,305 дюйма из нержавеющей стали и затем прокаливается на воздухе при 538oC в течение двух часов для превращения аммониевой формы в водородную форму М-5. Образец охлаждается до 300oC в атмосфере азота для удаления избыточного кислорода. Образец затем нагревается в атмосфере азота со скоростью 3,3oC/мин до 425oC в потоку водорода 40 см3/мин. Вводится чистый толуол при 425oC при 4 час-1, H2/HC= 2 и 3550 кПа (500 фунт/дюйм2). Для определения активности/селективности модифицированного (селективированного) катализатора реакционная температура и скорость подачи толуола варьируется для измерения толуольной конверсии. В таблице 6 приводятся данные некоторых примеров толуольной конверсии, параксилольной селективности и выхода продукта.
80,7% параксилольной активности наблюдается при 14,1% конверсии толуола для четырехкратно обработанного катализатора по сравнению с 31% параксилольной селективностью при 4% конверсии толуола для однократно обработанного катализатора из примера 22.
Пример 24.
Процесс многократной селективации для водной системы описывается ниже. Описанный в данном примере катализатор модифицируется в натриевой форме с использованием Hidrosil 2627, водорастворимого полимера H-пропиламинсилана.
10 г экструдата NaZSM-5/SiO2 с размером кристалла 0,20 мкм добавляется к 11 г 16% раствора полимера H-пропиламинсиламина (Hylrosil 2627) в воде и остается стоять до сухости. Катализатор затем нагревается в азоте со скоростью 2oC/мин до температуры 538oC, затем выдерживается при этой температуре в течение 2 часов. Образец затем охлаждается до 300oC, вводится воздух, образец нагревается со скоростью 2oC/мин до 538oC и выдерживается при этой температуре в течение 2 часов. К экструдату добавляется 2,9 мас.% SiO2 в расчете по содержанию кремния в полимере силана.
10 г однократно модифицированного катализатора добавляют к 11 г 16% раствора полимера H-пропиламинсилана (Hydrosil 2627) в воде и выдерживается до сухости. Катализатор затем нагревается в азоте со скоростью 2oC/мин до 538oC и выдерживается при этой температуре в течение 2 часов. Затем образец охлаждается до 300oC, вводится воздух, образец нагревается со скоростью 2oC/мин до температуры 538oC и выдерживается при ней в течение 2 часов. В расчете на содержание кремния в полимере силана к экструдату добавляется 2,9 мас.% SiO2 до общего количества добавленного SiO2 5,9 мас.%.
2 г дважды модифицированного катализатора добавляют к 25 г 10% раствора полимера H-пропиламинсилана (Hydrosil 2627) в воде и выдерживают до сухости. Катализатор затем нагревается в азоте со скоростью 2oC/мин до 538oC и выдерживается при этой температуре в течение 2 часов. Образец затем охлаждается до 300oC, вводится воздух, образец нагревается со скоростью 2oC/мин до 538oC и выдерживается при этой температуре в течение 2 часов. В расчете на содержание кремния в полимере силана к экструдату добавляется 3,3 мас.% SiO2 до общего добавленного количества SiO2 9,4 мас.%.
2 г трижды модифицированного катализатора добавляется к 2,5 г 16% раствора полимера H-пропиламинсилана (Hydrosil 2627) в воде и остается стоять до сухости. Катализатор затем нагревается в азоте со скоростью 2oC/мин до 538oC и выдерживается при этой температуре в течение 2 часов. Образец охлаждается до 300oC, вводится воздух, образец нагревается со скоростью 2oC/мин до 538oC и выдерживается в течение 2 часов. В расчете на содержание кремния в полимере силана к экструдату добавляется 3,3 мас.% SiO2 до общего добавленного количества SiO2 13 мас.%.
После четвертой модификации образец обрабатывается для обмена 1 М NH4NO3 при комнатной температуре в течение 1 часа для общих трех обменов.
Оценка каталитической активности модифицированного катализатора (0,738 г масса золы) проводится, как описывается в примере 23. С использованием данного модифицированного катализатора получается 92% параксилола при 14,2% конверсии при 446oC, 16 час-1, H2/HC=2, 3550 кПа (500 фунт/дюйм2).
Пример 25.
11 г NaZSM-5/SiO2 с размером кристалла 0,20 мкм добавляется к 11 г 21% раствора полимера H-пропиламинсилана (Hydrosil 2627) в воде и остается стоять до сухости. Катализатор затем нагревается в азоте со скоростью 2oC/мин до 538oC и затем выдерживается при этой температуре в течение 2 часов. По отношению к силиконовому содержанию Hydrosil 2627 к экструдату добавляется 3,6 мас.% SiO2.
Повторяется процедура, описанная для первой модификации.
8 г дважды обработанного катализатора добавляют к 8 г 21% раствора полимера H-пропиламинсилана (Hydrosil 2627) в воде и выдерживают до сухости. Катализатор затем нагревается со скоростью 2oC/мин до 538oC и затем выдерживается при этой температуре в течение 2 часов. Образец охлаждается до 300oC, вводится воздух, образец нагревается со скоростью 2oC/мин до 538oC и выдерживается при этой температуре в течение 2 часов.
6 г трижды обработанного катализатора добавляют к 6 г 21% раствора полимера H-пропиламинсилана (Hydrosil 2627) в воде и выдерживают до сухости. Катализатор затем нагревается в азоте со скоростью 2oC/мин до 538oC и выдерживается при этой температуре в течение 2 часов. Образец охлаждается до 300oC, вводится воздух, образец нагревается со скоростью 2oC/мин до 538oC и выдерживается при этой температуре в течение 2 часов. После четвертой модификации образец обрабатывается для обмена IM NH4NO3 при комнатной температуре в течение 1 часа для суммы трех обменов. Затем образцы прокаливаются на воздухе при 538oC в течение 2 часов с последующим пропариванием при 430oC в течение 4 часов (100% H2O-пар).
Указанный выше катализатор испытывается для этилбензольного диспропорционирования при 6 час-1, 310 кПа (30 фунт/дюйм2), 315 - 330oC и 14% этилбензольной конверсии. Парадиэтилбензольная селективность является выше 98%, что является более высоким показателем, чем возможная селективность традиционных катализаторов.
Пример 26
10 г NaZSM-5/SiO2 добавляют к 10 г 50% раствора полимера H-пропиламиносилана (Hydrosil 2627) в воде и выдерживают до сухости. Катализатор затем нагревается в азоте со скоростью 2oC в минуту до 538oC и выдерживается при этой температуре в течение 2 часов. Образец затем охлаждается до 300oC, вводится воздух, затем образец нагревается со скоростью 2oC/мин до 538oC и выдерживается при этой температуре в течение 2 часов.
9,5 г однократно модифицированного катализатора добавляется к 9,5 г 70% раствора полимера H-пропиламиносилана (Hydzosil 2627) в воде и выдерживается до сухости. Катализатор затем нагревается в азоте со скоростью 2oC/мин до 538oC и выдерживается при этой температуре в течение 2 часов. Образец охлаждается до 300oC, вводится воздух, образец нагревается со скоростью 2oC/мин до 538oC и выдерживается при этой температуре в течение 2 часов. После второй модификации образец обрабатывается для обмена 1 M NH4NO3 при комнатной температуре в течение 1 часа для суммы трех обменов, затем прокаливается на воздухе при 538oC в течение 2 часов.
Оценка каталитической активности дважды модифицированного катализатора в диспропорционировании толуола проводится, как описывается в примере 23. Представляющие селективность/конверсию данные приводятся в таблице 7.
Затем проводится отделочное коксование образца для определения возможности более высокой параксилольной селективности. Реакционная температура повышается до 540oC, скорость подачи сырья до 6,5 час-1, H2/HC снижается до 0,5, азот добавляется при 3,5 H2/HC, давление снижается до 2860 кПа (400 фунт/кв. дюйм). После 15 часов в этих условиях в реакторе устанавливается температура 456oC, 4 час-1, H2/HC=2 и 3550 кПа (500 фунт/дюйм2). Представляющие селективность/конверсию данные приводятся в таблице 7. При сравнении данных до и после модификации коксом более высокая параксилольная селективность наблюдается для катализатора с коксовой отделкой при данной толуольной конверсии.
Пример 27
Смесь 70% /30% (по массе) поверхностно-активных веществ получается смешением 14,0 г Тритона X-100 и 6,0 г Тритона X-305. 17,0 г полученной смеси поверхностно-активных веществ затем смешивается с 983,0 г дистиллированной воды с получением раствора, содержащего 1,7% поверхностно-активного вещества в воде. 60 г водного раствора поверхностно-активного вещества затем смешивается с 111,42 г DC-510 (DOW-фенилметилсиликоновое масло), затем смесь эмульгируется в смесителе в течение 1 мин с получением 65% эмульсии силиконовое масло/вода. 6,5 г 65% эмульсии силиконового масла разбавляется до 65 г раствором вода/поверхностно-активное вещество. Разбавленная эмульсия (6,5% масло/вода) рециркулируется через неподвижный слой, содержащий 20,0 г 80/20 NaZSM, причем всего в течение 12 час трижды прокаливают на воздухе до 538oC между пропитками. Полученный катализатор три раза H+ 4-обменивается обработкой 1 M NH4NO3 по одному часу каждый раз при комнатной температуре. Изменяется увеличение массы готового катализатора, которое оказывается равным 6%.
Модифицированный (селективированный) катализатор используется для превращения толуола при 446oC, 2170 кПа (300 фунт/дюйм2) и мольном соотношении водород/углеводород= 2. Результаты приводятся в таблице 8, причем параксилольная селективность равняется 55,4% при 29,6% конверсии толуола.
Селективированный катализатор подвергается 4-м последовательным обработкам водной силиконовой эмульсией/ с промежуточным воздушным прокаливанием, после чего определяется увеличение массы катализатора, которое оказывается равным 1% SiO2. Полученный катализатор используется для превращения толуола при 425oC, 2170 кПа (300 фунт/дюйм2) и молярном отношении водород/углеводород= 2. Результаты приводятся в таблице 8, причем параксилольная селективность равняется 88,4% при 30,9% конверсии толуола.
Необработанный 80/20 HZSM-5/ SiO2-катализатор с размером кристалла 0,2 мкм используется для превращения толуола при 446oC, 2170 кПа (300 фунт/дюйм2) и молярном отношении водород/углеводород=2. Результаты приводятся в таблице 8, причем параксилольная селективность равняется 27,2% при 61,6% конверсии толуола.
Пример 28.
SiO2-модифицированный HZSM-5 получается двухстадийным способом пропитки водной силиконовой эмульсией. К 1,13 г фенилметилсиликоновой эмульсии (65% масла/0,6% поверхностно-активного вещества) и 7,20 г дистиллированной воды добавляется 4,5 г HZSM-5 с размером кристалла 0,2 мкм. Вода отгоняется с использованием ротационного испарителя. Продукт прокаливается запрограммированно на воздухе со скоростью нагревания 1oC/мин до 538oC, затем выдерживается при этой температуре в течение 6 часов. Прокаленный катализатор взвешивается, привес оказывается равным 4,89 г (7,98% добавочного SiO2).
Указанная выше процедура повторяется с использованием 1,23 г фенилметилсиликоновой эмульсии, 3,30 г дистиллированной воды и 2,46 г Si)2-модифицированного HZSM-5. После отгонки воды и воздушного прокаливания катализатор взвешивается, привес оказывается равным 2,80 г (19,6% добавочного SiO2).
Коксовая отделочная селективация этого SiO2-модифицированного HZSM-5-катализатора (2,00 г) выполняется при 579oC, 790 кПа (100 фунт/дюйм2) и 0,5 час-1 с использованием толуольного сырья. Смесь азота и водорода (N2/H2=8) пропускается через катализатор с расходом 19,9 см3/мин. Через 72 часа оценивается эффективность селективации. В нижеследующей таблице (таблица 9) приводится конверсия толуола и параксилольная селективность после отделочной селективации.
После отделочной селективации катализатор испытывается в условиях процесса: 465oC, 3550 кПа (500 фунт/дюйм2), 3 час-1 и отношение водород/углерод = 2. При 23% конверсии толуола параксилольная селективность является высокой 95,4%. Температура увеличивается до 485oC. Конверсия толуола увеличивается до 28% при еще высокой параксилольной селективности при 93,2%.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФОРМОСЕЛЕКТИВНЫЙ ЦЕОЛИТОВЫЙ КАТАЛИЗАТОР И СПОСОБ КОНВЕРСИИ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ | 1996 |
|
RU2163506C2 |
| СПОСОБ ДИСПРОПОРЦИОНИРОВАНИЯ ТОЛУОЛА | 1993 |
|
RU2131862C1 |
| СПОСОБ КАТАЛИТИЧЕСКОЙ КОНВЕРСИИ УГЛЕВОДОРОДНОГО СЫРЬЯ, ВКЛЮЧАЮЩЕГО ПО КРАЙНЕЙ МЕРЕ ОДНО АРОМАТИЧЕСКОЕ СОЕДИНЕНИЕ, СОДЕРЖАЩЕЕ ПО КРАЙНЕЙ МЕРЕ 9 АТОМОВ УГЛЕРОДА, В ПРОДУКТ, СОДЕРЖАЩИЙ C-C-АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ | 1991 |
|
RU2011650C1 |
| СЕЛЕКТИВНОЕ ПОЛУЧЕНИЕ ПАРА-КСИЛОЛА ПОСРЕДСТВОМ МЕТИЛИРОВАНИЯ ТОЛУОЛА | 1997 |
|
RU2179964C2 |
| СИНТЕТИЧЕСКИЙ СЛОИСТЫЙ МАТЕРИАЛ МСМ-56, ЕГО ПОЛУЧЕНИЕ И ИСПОЛЬЗОВАНИЕ | 1994 |
|
RU2140962C1 |
| СПОСОБ КОНВЕРТИРОВАНИЯ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ | 1996 |
|
RU2148573C1 |
| Способ получения смесей изомеров этилтолуола или диэтилбензола | 1978 |
|
SU1181532A3 |
| СПОСОБ СНИЖЕНИЯ СОДЕРЖАНИЯ БРОМ-РЕАКЦИОННОСПОСОБНЫХ ЗАГРЯЗНЯЮЩИХ ПРИМЕСЕЙ В АРОМАТИЧЕСКИХ МАТЕРИАЛАХ | 1999 |
|
RU2204584C2 |
| СПОСОБ АЛКИЛИРОВАНИЯ АРОМАТИЧЕСКОГО СОЕДИНЕНИЯ | 1991 |
|
RU2014316C1 |
| СПОСОБ АЛКИЛИРОВАНИЯ ИЗОПАРАФИНА ОЛЕФИНОМ | 1991 |
|
RU2031900C1 |
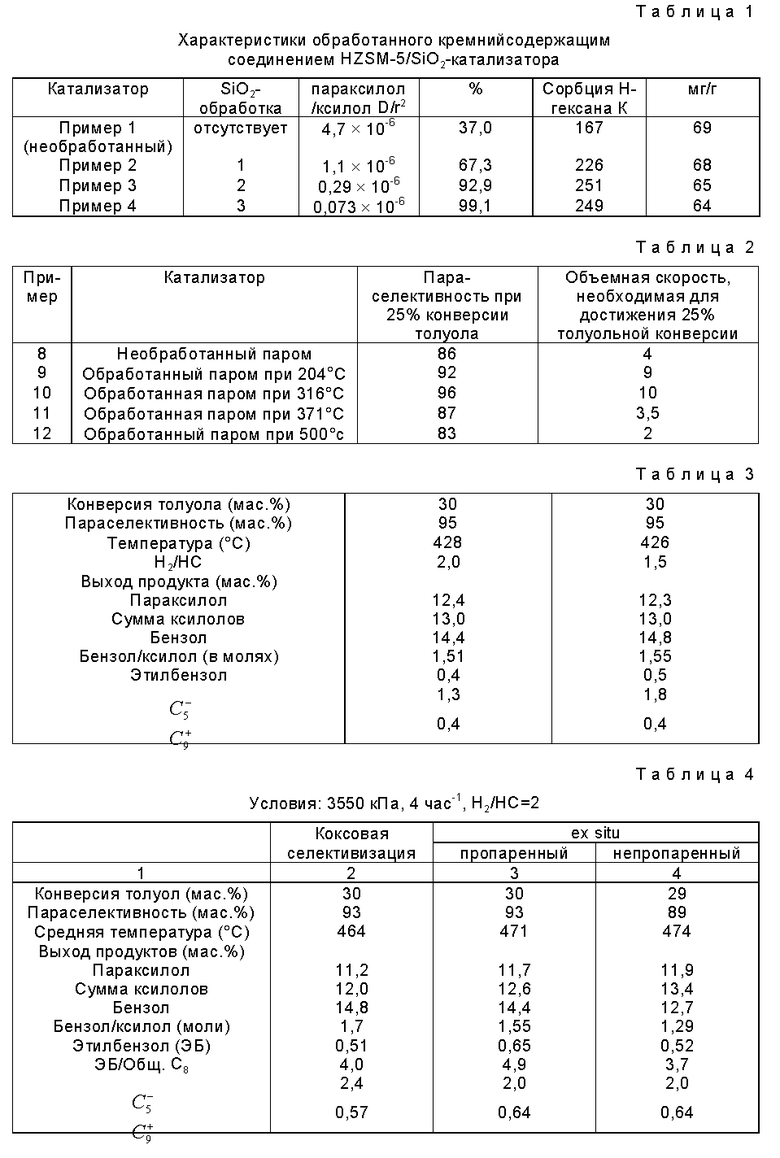


Изобретение относится к способу модификации формоселективности цеолитного катализатора и применения модифицированного катализатора в процессах формоселективной конверсии углеводородов, таких как диспропорционирование толуола. В способе модификации формоселективности алюмосиликатного цеолитного катализатора, имеющего молярное отношение SiO2: AI2O3<500, катализатор подвергается по крайней мере двум ex situ последовательным усиливающим селективность обработкам (селективациям), в котором каждая ex situ последовательная селективация включает в себя стадию контактирования катализатора с усиливающим селективность агентом, обычно кремнийорганическим соединением, в носителе и стадию последующего прокаливания катализатора. Катализатор по изобретению достигает очень высокой парадиалкилбензольной селективности, обычно более 80%. 2 с. и 20 з.п.ф-лы, 9 табл.

где R1 и R2 независимо друг от друга выбирают из группы, состоящей из водорода, галогена, гидроксила, алкила, алкокси, галогенированного алкила, арила, галогенированного арила, аралкила, галогенированного аралкила, алкарила и галогенированного алкарила;
n = 2 - 1000,
и органосиланы общей формулы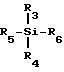
где R1 - R6 независимо друг от друга выбирают из группы, состоящей из водорода, галогена, гидроксила, алкила, аминоалкила, галогенированного алкила, арила, галогенированного арила, аралкила, галогенированного аралкила, алкарила и галогенированного алкарила.
15 Способ по пп.1 - 14, отличающийся тем, что дополнительно осуществляют стадию in situ отделочной селективации модифицированного цеолита.
| US 4477583 A, 16.10.84 | |||
| Способ активации катализатора | 1985 |
|
SU1409321A1 |
| US 4402867 A, 06.09.83 | |||
| US 4559314 A, 17.12.85. | |||
