Изобретение относится к новым исходным соединениям, применяемым для получения окситоцина и его аналогов, а именно к защищенным производным окситоцина общей формулы I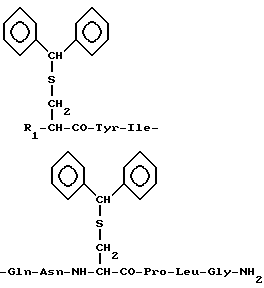
где R1 - трет-бутоксикарбонил-аминогруппа (Ia), аминогруппа (Ib) или атом водорода (Ic).
Окситоцин, нонапептид, содержащий дисульфидный цикл, имеет структуру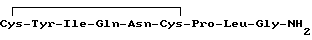
и является одним из важнейших регуляторов репродуктивной сферы млекопитающих.
Окситоцин и его структурные аналоги, получаемые методами химического синтеза, широко применяются в медицине и ветеринарии в качестве лекарственных средств утеротонического действия.
Дисульфидный цикл, образованный двумя остатками цистеина, является существенным элементом структуры, определяющим биологическую активность окситоцина. С точки зрения химического синтеза образование дисульфидного цикла в молекуле окситоцина - очень важная и ответственная стадия, от которой зависит эффективность всего синтеза, а именно выход, химическая чистота и биологическая активность конечного продукта. Дисульфидный цикл химически нестабилен и легко подвергается восстановлению, окислению и изомеризации, поэтому в процессе синтеза окситоцина или его аналогов формирование дисульфидного цикла осуществляют, как правило, на одной из финальных стадий синтеза. С этой целью при сборке пептицидной цепи окситоцина остатки цистеина вводят в синтез с предварительно блокированными сульфгидрильными (SH) группами. Защитные группы для блокирования SH-групп подбирают таким образом, чтобы они, с одной стороны, были устойчивы ко всем реагентам и условиям реакций, применяемым при сборке пептидной цепи, с другой стороны, легко и селективно отщеплялись после сборки пептида с освобождением SH-групп или с непосредственным образованием дисульфидного цикла.
Окситоцин является весьма нестабильным соединением и плохо подается очистке от примесей пептидной природы и солей. Падение его биологической активности может происходить даже при таких операциях, как колоночная хроматография и упаривание растворов; инактивации могут способствовать различные органические и неорганические примеси, присутствующие в растворах. Поэтому очень важно, чтобы после деблокирования и окисления окситоцин был получен в водном растворе с минимальным содержанием солей и нелетучих органических примесей.
На практике при планировании синтеза окситоцина выбор защитных групп и условий их отщепления для остатков цистеина является важнейшим элементом, от которого зависит выбор других защитных групп, реакционных условий и реагентов.
Наиболее известным способом защиты SH-групп остатков цистеина при синтезе окситоцина является S-бензилирование, описанное в J. Amer. Chem. Soc., v. 76, N12, pp. 3115-3121 (1954). S-Бензильная защитная группа отличается большой устойчивостью и совместима практически с любыми типами временных Nα-защитных групп и методами образования пептидной связи. Однако единственным приемлемым способом деблокирования ди-(S-бензил)-окситоцина после синтеза является обработка натрием в жидком аммиаке. В таких условиях происходит частичная деструкция пептида и образуется значительное количество побочных продуктов. В результате после окисления деблокированного пептида получается окситоцин, содержащий большое количество трудноотделимых примесей и имеющий низкую биологическую активность.
В патентах Германии 1038053 и 1059471 описаны способы получения Nα, S, S-трис-трифенилметил-окситоцина и его превращения в окситоцин. Одновременное отщепление всех трифенилметильных (тритильных) защитных групп легко достигается путем обработки защищенного пептида смесью дихлорметана и соляной кислоты. Деблокированный пептид, находящийся в водной фазе, окисляют затем в окситоцин кислородом. По другому способу (патент Швейцарии 498805) ди-(S-тритил)-пептид можно окислять иодом непосредственно в циклический дисульфид. S-Тритильная защита неустойчива в кислых средах, в частности в условиях отщепления трет-бутоксикарбонильной (Boc) и бензилоксикарбонильной (Cbz) Nα-защитных групп. Это создает значительные трудности для синтеза защищенных пептидов, содержащих S-тритилцистеин, поэтому производные ди-(S-тритил)-окситоцина получаются, как правило, с низкими выходами.
В патенте США 3560521 для получения окситоцина использовали производные ди-(S-ацетамидометил)-окситоцина. S-Ацетамидометильные защитные группы отщепляли действием избытка ацетата ртути (II), удаляли ионы ртути из раствора сероводородом, затем окисляли деблокированный дигидроокситоцин кислородом. Способ, описанный в Изв. АН Латв. ССР, Сер.хим., N 4, с.503-505 (1977), позволяет избежать использования соединений ртути. Согласно этому способу удаление S-ацетамидометильных групп с одновременным замыканием дисульфидного цикла проводят путем обработки иодом разбавленного раствора защищенного пептида в смеси диметилформамида и метанола. Недостатком данного способа является сложность выделения целевого продукта из большого объема органического растворителя без потери биологической активности.
В J.Chem. Soc., Chem. Commun., N 20, pp. 1501-1502 (1986), описано получение окситоцина из ди-[S-(9-флуоренил)метил]-окситоцина, синтезированного твердофазным методом. Указанный защищенный пептид в концентрации 0,5 мкмол/мл деблокировали 50% пиперидином в диметилформамиде; при этом происходило одновременное окисление SH-групп в циклический дисульфид. Синтез проведен на микромольном уровне; очевидно, что при увеличении масштаба необходимо пропорционально увеличить объем органических растворителей, используемых на стадии деблокирования-окисления.
Задачей предлагаемого изобретения является синтез новых цистеин-защищенных производных окситоцина, применение которых позволило бы получать окситоцин и его аналоги в виде растворов с минимальным содержанием солей и нелетучих органических примесей.
Задача решается путем получения новых соединений, а именно защищенных производных окситоцина общей формулы I: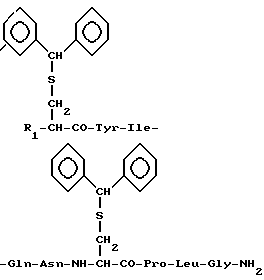
где R1 - трет-бутоксикарбонил-аминогруппа (Ia), аминогруппа (Ib) или атом водорода (Ic).
Соединения формулы I могут быть получены путем синтеза на твердой фазе или в растворе с использованием известных методов и реагентов. Остатки цистеина и 3-меркатопропионовой кислоты в процессе синтеза вводятся в виде S-дифенилметильных (S-Dpm) производных, получаемых известными способами, например, как описано в J. Chtm. Soc. (C), p. 2683-2687 (1970). В качестве временных Nα-защитных групп используются группы, которые можно селективно отщепить, не затрагивая S-дифенилметильную защиту, например, Nα-Boc-группа. При синтезе в растворе для отщепления Nα-Boc-группы могут быть использованы известные методы и реагенты, например растворы хлористого водорода или сульфокислот в органических растворителях, трифторуксусная кислота или ее растворы в дихлорметане или хлороформе. Для наращивания пептидной цепи применяются известные методы конденсации, например конденсация при действии дициклогексилкарбодиимида и 1-гидроксибензотриазола.
Соединения Ia и Ib могут быть использованы для получения окситоцина, соединение Ic - для получения дезаминоокситоцина. Этот процесс проводится в две стадии. На первой стадии проводят отщепление S-Dpm-групп с остатков цистеина и 3-меркаптопропионовой кислоты действием трифторуксусной кислоты в присутствии акцептора дифенилметильных катионов, например фенола (в соединении Ia при этом одновременно отщепляется Nα-Boc-группа). Деблокирование проводят в течение 20 мин при 60-80oC или в течение 18-24 ч при 20-25oC. Затем отгоняют трифторуксусную кислоту и выделяют осаждением деблокированный пептид со свободными SH-группами. На второй стадии полученный пептид растворяют в воде или водно-органическом растворителе, например в водном ДМФА, доводят при необходимости pH раствора до значения в интервале от 6 до 8 и окисляют известным способом, например перекисью водорода или кислородом воздуха.
Отщепление S-Dpm-групп проходит гладко и с высоким выходом. Благодаря осаждению и тщательной промывке первой стадии деблокированный пептид практически не содержит органических примесей непептидного характера и солей и имеет высокое содержание основного вещества. При растворении деблокированного пептида в воде или водном ДМФА раствор имеет кислую реакцию, что позволяет минимизировать неконтролируемое окисление на стадии растворения. В результате окисления получается окситоцин или дезаминоокситоцин с высокой биологической активностью в растворе, не содержащем солей и органических примесей. Такой раствор может быть сконцентрирован известными способами, например упариванием при пониженном давлении или лиофилизацией, без потери биологической активности целевого продукта.
Таким образом, предлагаемые защищенные производные окситоцина можно эффективно использовать для получения окситоцина и дезаминоокситоцина, причем целевые продукты получаются с высокими выходами и в форме, удобной для концентрирования и очистки без потери биологической активности.
Сущность предлагаемого изобретения иллюстрируется примерами. При описании примеров используются следующие сокращения и условные обозначения:
ДМФА - диметилформамид
ДЦГК - дициклогексилкарбодиимид
Сокращенные обозначения аминокислот и защитных групп используются в соответствии с рекомендациями Комиссии по биологической номенклатуре при IUPACIUB, опубликованными в Eur, J. Biochem, v.138, N 1, pp. 9-37 (1984). Оптически активные аминокислоты, приведенные в описаниях примеров, по умолчанию имеют L-конфигурацию.
Значения хроматографических подвижностей Rf приведены для пластинок для тонкослойной хроматографии Alufolien Kieselgel 60 F254 (Merck, ФРГ) в системах хлороформ-метанол-уксусная кислота 90:10:3 (А) и ацетонитрил-уксусная кислота-вода 40: 1:10 (Б). Обнаружение пятен на пластинках проводили в УФ-свете и нингидриновым реактивом после прогревания. Массы молекулярных ионов (M + H)+ измерены на времяпролетном масс-спектрометре МСБХ-1 (НПО "Электрон", Украина).
Пример 1. Получение трифторацетата (S-дифенилметил)цистеинил-пролил-лейцил-глицинамида [H-Cys(Dpm)-Pro-Leu-Gly-NH2•TFA].
К раствору 9.12 г пролил-лейцил-глицинамида, 7.6 г Boc- (S-дифенилметил)- цистеина и 1.35 г 1-гидроксибензотриазола в 30 мл ДМФА при охлаждении в ледяной бане и перемешивании прибавляют 4.3 г ДЦГК. Смесь перемешивают 1 ч при охлаждении и еще 4 ч при комнатной температуре. Выпавший осадок фильтруют, фильтрат упаривают при пониженном давлении, остаток растворяют в 250 мл этилацетата. Раствор промывают в делительной воронке 3 х 100 мл 5% водного раствора бикарбоната натрия, 2 х 70 мл 1 н. соляной кислоты и 100 мл насыщенного водного раствора хлорида натрия, сушат над безводным сульфатом натрия и упаривают при пониженном давлении. Остаток растворяют в 30 мл дихлорметана, добавляют 20 мл трифторуксусной кислоты и выдерживают 40 мин при комнатной температуре. Раствор упаривают до вязкого масла, к остатку добавляют 60 мл толуола и вновь упаривают. Остаток растворяют в 30 мл этанола, добавляют 150 мл диэтилового эфира и оставляют на ночь в холодильнике. Выпавший осадок растирают, фильтруют, промывают на фильтре эфиром и сушат в вакуум-эксикаторе. Получают 11 г целевого тетрапептида в виде белого мелкокристаллического порошка; Rf(A) 0.17; масс-спектр: (M+H)+ 556.1 (вычислено: 554.775).
Пример 2. Получение Boc-аспарагинил-(S-дифенилметил)цистеинилпролил-лейцил-глицинамида [Boc-Asn-Cys(Dpm)-Pro-Leu-Gly-NH2].
К раствору 10.8 г трифторацетата (S-дифенилметил)цистеинил-пролил-лейцил-глицинамида (пример 1), 4.2 г Boc- аспарагина и 2.5 г 1-гидроксибензотриазола в 30 мл ДМФА при охлаждении в ледяной бане и перемешивании прибавляют 2.8 мл триэтиламина, затем 4.1 г ДЦГК. Смесь перемешивают 1 ч при охлаждении и еще 3 ч при комнатной температуре. Выпавший осадок фильтруют, фильтрат упаривают при пониженном давлении, остаток обрабатывают 300 мл воды. Образовавшийся осадок растирают, фильтруют, промывают на фильтре 100 мл 5% водного раствора бикарбоната натрия и 100 мл воды и сушат на воздухе. Полученный сырой продукт растворяют в 30 мл ДМФА и осаждают диэтиловым эфиром, промывают на фильтре эфиром и сушат на воздухе. Получают 10.8 г Boc-пентапептида; Rf(A) 0.35; масс-спектр: (M+H)+768.3(вычислено: 769,008).
Пример 3. Получение Boc-глутаминил-аспарагинил-(S-дифенилметил)цистеинил-пропил-лейцил-глицинамида [Boc-Gln-Asn-Cys(Dpm)-Pro-Leu-Gly-NH2].
15.4 г Boc-аспарагинил-(S-дифенилметил)цистеинил-пролил-лейцил-глицинамида (пример 2) растворяют в 40 мл дихлорметана и 30 мл трифторуксусной кислоты и выдерживают раствор 1 ч при комнатной температуре. Раствор упаривают до вязкого масла, к остатку добавляют 60 мл толуола и вновь упаривают. Остаток растирают с 200 мл диэтилового эфира, осадок фильтруют, промывают на фильтре эфиром и сушат в вакуум-эксикаторе. Полученный трифторацетат пентапептида растворяют в 50 мл ДМФА и добавляют 5.5 г Boc-глутамина и 2.8 г 1-гидроксибензотриазола. При охлаждении в ледяной бане и перемешивании к раствору прибавляют 3.3 мл N-метилморфолина, затем 4.6. г ДЦГК. Смесь перемешивают 1 ч при охлаждении и еще 6 ч при комнатной температуре. Выпавший осадок фильтруют, фильтрат упаривают при пониженном давлении, остаток обрабатывают 50 мл этилацетата и 100 мл диэтилового эфира. Образовавшийся осадок растирают, фильтруют, промывают на фильтре 100 мл эфира и сушат на воздухе. Получают 15.6 г Boc-гексапептида; Rf(A) 0.22, Rf(Б) 0.45; масс-спектр: (M+H)+ 895.5 (вычислено: 897.141).
Пример 4. Получение Boc-изолейцил-глутаминил-аспарагинил-(S-дифенилметил)цистеинил-пролил-лейцил-глицинамида [Boc-Ile-Gln-Asn-Cys(Dpm)-Pro-Leu-Gly-NH2].
15.4 Boс-глутаминил-аспарагинил-(S-дифенилметил)цистеинил-пролил-лейцил-глицинамида (пример 3) растворяют в 40 мл дихлорметана и 40 мл трифторуксусной кислоты и выдерживают раствор 1 ч при комнатной температуре. Раствор упаривают до вязкого масла, к остатку добавляют 80 мл толуола и вновь упаривают. Остаток растирают с 250 мл диэтилового эфира, осадок фильтруют, промывают на фильтре эфиром и сушат в вакуум-эксикаторе. Полученный трифторацетат гексапептида растворяют в 50 мл ДМФА и добавляют 4.8 г полугидрата Boc-изолейцина и 2.7 г 1-гидроксибензотриазола. При охлаждении в ледяной бане и перемешивании к раствору прибавляют 3.3 мл N-метилморфолина, затем 4.8 г ДЦГК. Смесь перемешивают 1 ч при охлаждении и еще 4 ч при комнатной температуре. Выпавший осадок фильтруют, фильтрат упаривают при пониженном давлении, остаток обрабатывают 100 мл этилацетата и 150 мл диэтилового эфира. Образовавшийся осадок растирают, фильтруют, промывают на фильтре 150 мл эфира и сушат на воздухе. Получают 15.2 г Boc-гептапептида; Rf(Б) 0.50; масс-спектр: (M+H)+1009.1 (вычислено: 1010.314)
Пример 5. Получение Boc-тирозил-изолейцил-глутаминил-аспарагинил-(S-дифенилметил)цистеинил-пролил-лейцил-глицинамида [Boc-Tyr-Ile-Gln-Asn-Cys(Dpm)-Pro-Leu-Gly-NH2].
15.1 г Boc-изолейцил-глутаминил-аспарагинил-(S-дифенилметил)цистеинил-пролил- лейцил-глицинамида (пример 4) растворяют в 50 мл дихлорметана и 40 мл трифторуксусной кислоты и выдерживают раствор 1 ч при комнатной температуре. Раствор упаривают до вязкого масла, к остатку добавляют 80 мл толуола и вновь упаривают. Остаток растирают с 200 мл диэтилового эфира, осадок фильтруют, промывают на фильтре эфиром и сушат в вакуум-эксикаторе. Трифторацетат гептапептида растворяют в 50 мл ДМФА и добавляют 4.8 г Boc-тирозина и 2.6 г 1-гидроксибензотриазола. При охлаждении в ледяной бане и перемешивании к раствору прибавляют 2,9 мл N-метилморфолина, затем 3.6 г ДЦГК. Смесь перемешивают 1 ч при охлаждении и еще 8 ч при комнатной температуре. Выпавший осадок фильтруют, фильтрат упаривают при пониженном давлении, остаток обрабатывают 300 мл воды. Образовавшийся осадок растирают, фильтруют, промывают на фильтре 100 мл 5% водного раствора бикарбоната натрия и 100 мл воды и сушат на воздухе. Полупродукт растворяют в 30 мл ДМФА, упаривают до масла и растирают с диэтиловым эфиром, промывают на фильтре эфиром и сушат на воздухе. Получают 16.4 г Boc-октапептида; Rf(Б) 0.55; масс-спектр: (M+H)+1172.1 (вычислено: 1173.497).
Пример 6. Получение Boc-(S-дифенилметил)цистеинил-тирозил-изолейцил-глутаминил-аспарагинил-(S-дифенилметил)цистеинил-пролил-лейцил-глицинамида [Boc-Cys(Dpm)-Tyr-Ile-Gln-Asn-Cys(Dpm)-Pro-Leu-Gly-NH2, соединение Ia].
16.2 г Boc-тирозил-изолейцил-глутаминил-аспарагинил-(S-дифенилметил)цистеинил-пролил-лейцил-глицинамида (пример 5) растворяют в 50 мл дихлорметана и 50 мл трифторуксусной кислоты и выдерживают раствор 1 ч при комнатной температуре. Раствор упаривают до вязкого масла, к остатку добавляют 100 мл толуола и вновь упаривают. Остаток растирают с 200 мл диэтилового эфира, осадок фильтруют, промывают на фильтре эфиром и сушат в вакуум-эксикаторе. Трифторацетат октапептида растворяют в 50 мл ДМФА и добавляют 5.2 г Boc-(S-дифенилметил)цистеина и 2.2 г 1-гидроксибензотриазола. При охлаждении в ледяной бане и перемешивании к раствору прибавляют 2.9 мл N-метилморфолина, затем 3.3 г ДЦГК. Смесь перемешивают 1 ч при охлаждении и еще 5 ч при комнатной температуре. Выпавший осадок фильтруют, фильтрат упаривают при пониженном давлении, остаток обрабатывают 300 мл воды. Образовавшийся осадок растирают, фильтруют, промывают на фильтре 100 мл 5% водного раствора бикарбоната натрия и 100 мл воды и сушат на воздухе. Полупродукт растворяют в 30 мл ДМФА, упаривают до масла, затем добавляют 70 мл ацетонитрила и оставляют на ночь в холодильнике. Гелеобразную массу разбавляют 50 мл диэтилового эфира, осадок фильтруют, промывают на фильтре эфиром и сушат на воздухе. Получают 17.3 г соединения Ia; Rf(Б) 0.60; масс-спектр: (M+H)+1441.1 (вычислено: 1442.88).
Пример 7. Получение трифторацетата (S-дифенилметил)цистеинилтирозил-изолейцил-глутаминил-аспарагинил- (S-дифенилметил)цистеинил-пролил-лейцил-глицинамида [H-Cys(Dpm)-Tyr-Ile-Gln-Asn-Cys(Dpm)-Pro-Leu-Gly-NH2 x TFA, соединение 1b].
17.2 г соединения Ia (пример 6) растворяют в 50 мл дихлорметана и 50 мл трифторуксусной кислоты и выдерживают раствор 1 ч при комнатной температуре. Раствор упаривают до густого масла, к остатку добавляют 100 мл толуола и вновь упаривают. Остаток растирают с 250 мл диэтилового эфира, осадок фильтруют, промывают на фильтре эфиром и сушат в вакуум-эксикаторе. Получают 17.1 г трифторацетата соединения Ib; Rf(Б) 4.40; масс-спектр: (M+H)+ 1341.2 (вычислено: 1342.75).
Пример 8. Получение 3-(дифенилметилтио)пропионил-тирозил-изолейцил-глутаминил-аспарагинил-(S-дифенилметил)цистеинил-пролил-лейцил-глицинамида [Mpr(Dpm)-Tyr-Ile-Gln-Asn-Cys(Dpm)-Pro-Leu-Gly-NH2(Ic)].
4.2 г Трифторацетата тирозил-изолейцил-глутаминил-аспарагинил-(S-дифенилметил)цистеинил-пролил-лейцил-глицинамида, полученного как описано в примере 6, растворяют в 15 мл ДМФА и добавляют 1.1 г 3-(дифенилметилтио)пропионовой кислоты и 0.7 г 1-гидлроксибензотриазола. При охлаждении в ледяной банен и перемешивании к раствору прибавляют 0.60 мл N-метилморфолина, затем 0.9 г ДЦГК. Смесь перемешивают 1 ч при охлаждении и еще 2 ч при комнатной температуре. Выпавший осадок фильтруют, фильтрат упаривают при пониженном давлении, остаток обрабатывают 100 мл воды. Образовавшийся осадок растирают, фильтруют, промывают на фильтре 30 мл 5% водного раствора бикарбоната натрия и 50 мл воды и сушат на воздухе. Полупродукт растворяют в 10 мл ДМФА, упаривают до масла, затем добавляют 50 мл этилацетата и оставляют на ночь в холодильнике. Осадок фильтруют, промывают на фильтре диэтиловым эфиром и сушат на воздухе. Получают 4.3 г соединения Ic; Rf(Б) 0.62; масс-спектр: (M+H)+ 1327.1 (вычислено: 1327.74).
Пример 9. Получение окситоцина.
14.4 г соединения Ia, 60 г перегнанного фенола и 320 мл трифторуксусной кислоты перемешивают в атмосфере аргона 20 ч при комнатной температуре, затем трифторуксусную кислоту отгоняют при пониженном давлении. Остаток встряхивают с 200 мл петролейного эфира, после расслаивания петролейный эфир декантируют, эту операцию повторяют еще дважды. Остаток растворяют в 50 мл ледяной уксусной кислоты и к раствору при перемешивании добавляют 300 мл диэтилового эфира. Выпавший белый осадок быстро фильтруют, промывают на фильтре 3 • 50 мл эфира и сушат в вакуум-эксикаторе. Полученный трифторацетат дигидроокситоцина (11г) при перемешивании растворяют в 10 л бидистиллированной воды, затем добавлением концентрированного водного аммиака доводят pH раствора до 7.5. Раствор интенсивно перемешивают в открытом сосуде 2 суток при комнатной температуре до отрицательной реакции с нитропруссидом натрия. Получают раствор окситоцина с биологической активностью 290 ± 15 ME/мл. К раствору добавляют 500 мл уксусной кислоты и упаривают его при пониженном давлении и температуре бани 35-40oC до объема 1 л; измеренная удельная активность окситоцина составляет 3010 ± 160 ME/мл.
Пример 10. Получение дезаминоокситоцина.
4 г соединения Ic, 20 г перегнанного фенола и 120 мл трифторуксусной кислоты нагревают в атмосфере аргона 20 мин в водяной бане с температурой 70oC, затем раствор быстро охлаждают и трифторуксусную кислоту отгоняют при пониженном давлении. Остаток встряхивают со 100 мл петролейного эфира, после расслаивания петролейный эфир декантируют, эту операцию повторяют еще дважды. Остаток растирают со 100 мл диэтилового эфира, образовавшийся белый осадок быстро фильтруют, промывают на фильтре 3 х 20 мл эфира и сушат в вакуум-эксикаторе. Полученный дигидро-дезаминоокситоцин (2.85 г) при перемешивании растворяют в 10 мл ДМФА и при интенсивном перемешивании по каплям приливают к раствору 1 мл 25%-ного водного аммиака и 0.5 мл 32%-ной перекиси водорода в 3 л бидистиллированной воды. Раствор перемешивают 12 ч при комнатной температуре до отрицательной реакции с нитропруссидом натрия. Получают раствор дезаминоокситоцина с биологической активностью (350±15) ME/мл. К раствору добавляют 300 мл уксусной кислоты и упаривают его при пониженном давлении и температуре бани 35-40oC до объема 150 мл, измеренная удельная активность дезаминоокситоцина составляет (6900±200) ME/мл.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАЩИЩЕННЫЕ ПРОИЗВОДНЫЕ ВАЗОПРЕССИНА | 1997 |
|
RU2123498C1 |
| RGD-СОДЕРЖАЩИЕ ПЕПТИДЫ | 2001 |
|
RU2214416C2 |
| Способ получения окситоцина и дезаминоокситоцина | 1976 |
|
SU586165A1 |
| ЗАЩИЩЕННЫЕ ПЕПТИДЫ КАЛЬЦИТОНИНА | 2000 |
|
RU2193567C2 |
| Способ отщепления сульфенильных групп от -сульфениламинокислот и сульфенилпептидов | 1978 |
|
SU767090A1 |
| Цистеинсодержащие пептиды для синтеза пептидных гормонов окситоцина и вазопрессина | 1974 |
|
SU523083A1 |
| Способ получения -сульфоната в-цепи инсулина человека | 1977 |
|
SU696011A1 |
| СПОСОБ СТАБИЛИЗАЦИИ ОЛИГОНУКЛЕОТИДНЫХ ДУПЛЕКСОВ | 1996 |
|
RU2124523C1 |
| Способ получения трипептидов | 1980 |
|
SU1085505A3 |
| Способ получения амида N -трет.бутоксикарбонилпролил-лейцил-глицина | 1979 |
|
SU857117A1 |
Производные окситоцина формулы I, где R1 - трет-бутоксикарбонил-амино, амино или водород, пригодны для использования в качестве исходных при получении окситоцина и дезаминоокситоцина.
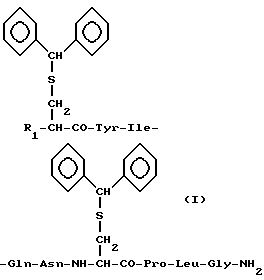
Производные окситоцина общей формулы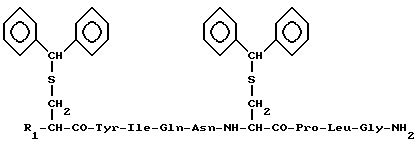
где R1 - трет-бутоксикарбонил-аминогруппа, аминогруппа или атом водорода,
в качестве исходных соединений для получения окситоцина и дезаминоокситоцина.
| Цистеинсодержащие пептиды для синтеза пептидных гормонов окситоцина и вазопрессина | 1974 |
|
SU523083A1 |
| УСТРОЙСТВО ДЛЯ ПНЕВМОПЕРЕПУТЫВАНИЯ КОМПЛЕКСНЫХ ХИМИЧЕСКИХ НИТЕЙ | 1994 |
|
RU2074582C1 |
| СПОСОБ ПОЛУЧЕНИЯ АРОМАТИЧЕСКИХ ОКСИАЛЬДЕГИДОВ | 1994 |
|
RU2078755C1 |
| СИСТЕМА ТЕРМОСТАТИРОВАНИЯ РАДИОЭЛЕКТРОННОЙ АППАРАТУРЫ | 1974 |
|
SU498805A1 |

