Изобретение относится к новым соединениям, структура которых аналогична 15-дезоксиспергуалину. Оно относится также к способу их получения и их применению в терапии, особенно в качестве иммуносупрессорных агентов.
Известно, что 15-дезоксиспергуалин (ДСГ), который отвечает формуле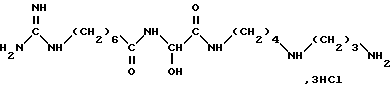
и который известен под общим международным названием Гусперимус, обладает интересной иммуноподавляющей активностью. Многочисленные публикации сообщают об этой активности, в частности она описывается в ряде статей в "Immunomodulating Drugs" - Annals of the New York Academy of Sciences, т. 685, с. 123 - 201.
Однако 15-дезоксиспергуалин не обладает удовлетворительной химической стабильностью, и поэтому предпринимались попытки получить более стабильные соединения, например
(I) заменяя α- гидроксиглициновую группу 15 - дезоксиспергуалина различными α- или ω- аминокислотными группами;
(II) модифицируя структуру центрального звена, или
(III) модифицируя звено, содержащее гуанидиновую функцию. Примеры таких модификаций описываются в европейских патентах A-0181592 и A-0105193 и патенте Франции A-2698628.
Модификации, производимые в отношении гидроксиглицинового остатка, в основном указываются в J. Antibiot. 38: 886 - 898; и J. Antibiot. 41: 1629 - 1643. Согласно этим публикациям, ни одна из предложенных структур не позволяет достичь активности, выше активности ДСГ. Кроме того, изменения, вносимые в спермидиновый остаток, опубликованные например, в J. Antibiot 40: 1303 - 1315, в большинстве случаев приводят к потере активности, и наличие спермидиновой цепи оказывается необходимым для получения активного соединения.
Однако в настоящее время найдено, что соединения, имеющие структуру, родственную ДСГ, но не включающие гидроксиглициновой цепи и спермидиновой цепи, тем не менее, обладают иммуноподавляющей активностью, и эта активность может быть выше активности ДСГ.
В настоящем изобретении предполагаются новые соединения, общая структура которых родствена структуре 15-дезоксиспергуалина, которые химически стабильны и которые обладают активностью выше активности известных продуктов уровня техники в области иммуноподавляющих средств.
Соединения согласно изобретению отличаются от известных продуктов уровня техники по существу значительным изменением центральной части молекулы, которое приводит к группе карбаматного типа. Кроме того, в отличие от всех известных продуктов уровня техники, родственных 15-дезоксиспергуалину, соединения согласно изобретению не содержат полиаминовой цепи типа спермидина, но включают группу типа аминоспирта.
Соединения согласно изобретению отличаются тем, что их выбирают из группы соединений, включающей:
(1) соединения формулы (I)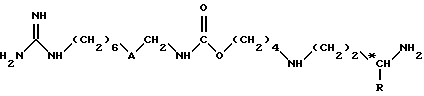
в которой
A обозначает группу -CO-NH или группу -NH-CO-;
R обозначает атом водорода или метильную группу;
*C, когда R не является атомом водорода, представляет собой асимметрический углерод, конфигурация которого может быть неопределенной (R, S) или определенной (R);
и (II) их соли присоединения.
Согласно изобретению предложен также способ получения соединений формулы (I) и их солей присоединения, причем вышеуказанный способ включает удаление защитных групп из соединения формулы (II)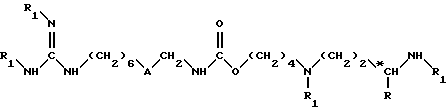
в которой
A обозначает группу -NH-CO- или группу -CO-NH-;
R обозначает атом водорода или метильную группу;
*C, когда R не является атомом водорода, обозначает асимметрический углерод конфигурации (R, S) или конфигурации (R);
причем по крайней мере один из заместителей R1 обозначает защитную группу для аминофункции, а другие обозначают атом водорода;
согласно известной обработке для достижения удаления защитных групп с аминофункций, и замены всех защитных групп R1 атомом водорода.
Изобретение относится также к использованию вещества формулы (I), их нетоксичных солей присоединения и их смесей, для получения лекарственного средства, предназначенного для применения в терапии с целью лечения или профилактики иммунных нарушений или малярии, или для использования в качестве фармакологического реагента.
Под выражением "соли присоединения" понимают соли присоединения кислоты, получаемые путем взаимодействия соединения формулы (I) с неорганической или органической кислотой. Предпочтительными неорганическими кислотами для получения солей являются соляная, бромводородная, фосфорная и серная кислоты. Предпочтительными органическими кислотами для получения солей являются фумаровая, малеиновая, метансульфоновая, щавельная, лимонная и трифторуксусная кислоты.
Как указано в формуле (I), соединения согласно изобретению включают углерод, обозначаемый *C, который, если R не является атомом водорода, представляет собой асимметрический углерод. Если R не является атомом водорода, настоящее изобретение включает рацемические соединения формулы I, где *C имеет конфигурацию (R, S), и энантиомер, где *C имеет конфигурацию (R), согласно правилам определения структуры, описанным Cahn, Ingold и Prelog.
Если R обозначает H, то атом углерода, отмеченный как *C, не является асимметрическим.
На практике из соединений формулы (I), включающих асимметрический углерод *C, предпочитают те, в которых вышеуказанный углерод имеет конфигурацию (R).
Соединения формулы (I) могут быть получены согласно известным способам, основанным на классических реакционных механизмах, в частности с использованием реакций, позволяющих получать группировки карбаматного типа.
Способ получения соединений формулы (I) согласно изобретению включает, как указано выше, удаление защитных групп из соединения формулы (II). На практике группа или группы R1, которые во время реакции должны быть заменены атомами водорода, представляют собой известные защитные группы аминофункций, в частности известные в химии пептидов, которые временно блокируют неполностью замещенные аминофункции.
Из групп, которые пригодны для этой цели, можно использовать:
а) группы оксикарбонильного типа, как, например, алкоксикарбонильные или бензилоксикарбонильные группы:
Boc: трет.-бутилоксикарбонил (или 1,1-диметил-этоксикарбонил);
Fmoc: 9-фторенилметилоксикарбонил;
Z: бензилоксикарбонил (или фенилметоксикарбонил);
Z(p-Cl): 4-хлор-бензилоксикарбонил;
Z(p-OMe): 4-метоксибензилоксикарбонил;
(б) группы бензильного типа, например, фенилметильная группа (Bn).
Из этих защитных групп предпочтительной группой согласно изобретению является группа Boc.
Способ получения соединения формулы (I) или одной из его солей присоединения заключается в том, что он включает следующие стадии:
(1) удаление защитных групп из соединения формулы (II)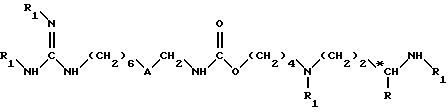
в которой
R обозначает атом водорода или метильную группу;
A обозначает группу -CO-NH- или группу -NH-CO-;
*C, если R не является атомом водорода, обозначает асимметрический углерод неопределенной конфигурации (R, S) или определенной конфигурации (R);
причем по крайней мере один из заместителей R1 обозначает защитную группу аминофункции оксикарбонильного или бензильного типа, а другие заместители R1, если они имеются, обозначают атом водорода;
согласно обработке, зависящей от природы защитной группы, например, если эта защитная группа является группой алкоксикарбонильного типа, осуществляют обработку сильной кислотой, такой как трифторуксусная кислота, или, если эта защитная группа представляет собой группу бензильного типа, осуществляют каталитическое гидрирование в присутствии катализатора на основе палладия, для получения соединения формулы (I) в форме свободного основания или в форме одной из его солей присоединения; и, если необходимо,
(II) получают другие соли присоединения исходя из свободного основания или его соли присоединения, полученных на стадии (I). Так, на стадии (II), из одной соли присоединения получают другие соли присоединения либо с помощью соединения формулы (I) в форме свободного основания, либо за счет обмена противоиона в присутствии избытка соответствующей получаемой соли кислоты.
Для получения соединений формулы (II) можно использовать способ, выбираемый из следующих вариантов его осуществления
(а) вариант А, который включает следующие стадии:
(1) конденсация спирта формулы (III):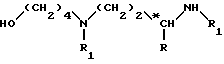
в которой R обозначает атом водорода или метильную группу;
R1 обозначает защитную группу аминофункции, например, группу Boc;
*C обозначает, если R не является атомом водорода, асимметрический углерод конфигурации (R, S) или (R);
с хлорформиатом или симметричным карбонатом, например, с 4-нитрофенил-хлорформиатом, в присутствии основания, например, триэтиламина в инертном растворителе, и при комнатной температуре (15 - 25oC); затем
(II) взаимодействие полученного на стадии (I) соединения с амином формулы (IV)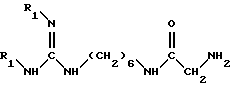
в которой R1 обозначает защитную группу аминофункции, например, группу Boc, в инертном растворителе, при температуре около 25 - 50oC, до получения соединения формулы (II)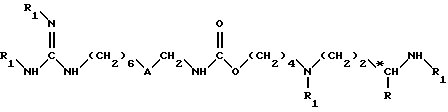
в которой A обозначает группу -NH-CO-;
R обозначает атом водорода или метильную группу;
*C обозначает, если R не является атомом водорода, асимметрический углерод конфигурации (R, S) или (R);
R1 обозначает защитную группу аминофункции, например группу Boc,
б) вариант Б, который включает следующие стадии:
(1) взаимодействие кислоты формулы (V)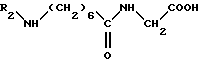
в которой R2 обозначает защитную группу аминофункции, например бензилоксикарбонильную группу, с дифенилфосфорилазидом формулы VI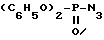
в присутствии основания, такого как триэтиламин, в растворителе, например в тетрагидрофуране, и при комнатной температуре, до получения промежуточного соединения формулы (VII)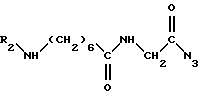
(II) полученное соединение формулы (VII) подвергают перегруппировке, известной под названием реакции Куртиуса, и одновременно таким образом полученный изоцианат вводят во взаимодействие со спиртом формулы (III)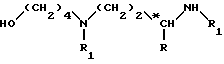
в которой R обозначает атом водорода или метильную группу;
R1 обозначает защитную группу аминофункции, отличную от вышеуказанной группы R2, например группу Boc;
*C обозначает, если R не является атомом водорода, асимметрический углерод конфигурации (R, S) или (R), в растворителе, таком как толуол, при температуре около 80 - 140o, в течение 5 - 50 ч, до получения соединения формулы (VIII)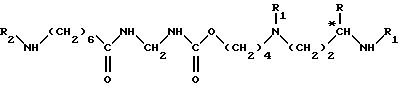
в которой R, R1, R2 и *C имеют вышеуказанные значения;
(III) удаление защитных групп из вышеполученного соединения формулы (VIII), согласно специфической обработке с целью замены защитной для группы R2 атомом водорода, например, если R2 обозначает бензилоксикарбонильную группу, то осуществляют каталитическое гидрирование в присутствии катализатора на основе палладия, до получения соединения формулы (IX)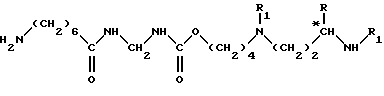
в которой R, R1 и *C сохраняют вышеуказанные значения;
(IV) взаимодействие полученного на стадии (III) соединения формулы (IX) с аминоиминометансульфокислотой, в растворителе, например в метаноле, при комнатной температуре (15 - 25oC) и в течение 8 - 50 ч, до получения соединения формулы (II):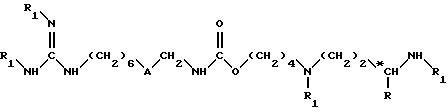
в которой R и *C имеют вышеуказанное значение;
A обозначает группу -CO-NH-;
R1 обозначает защитную группу аминофункции, например группу Boc, за исключением двух групп R1, которые находятся на гуанидиновой функции и каждая из которых обозначаeт атом водорода;
или
(IV') в качестве варианта вышеуказанной стадии (IV), заключающегося во взаимодействии соединения формулы (IX) с соединением формулы (X)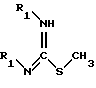
в которой R1 обозначает защитную группу аминофункции, например группу Boc;
в растворителе, например в тетрагидрофуране, в присутствии основания, в частности триэтиламина, при комнатной температуре и в течение 8 - 100 ч, до получения соединения формулы (II)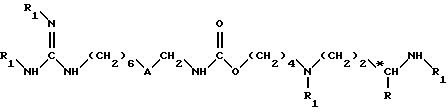
в которой A обозначает группу -CO-NH-;
R обозначает атом водорода или метильную группу;
R1 обозначает защитную группу аминофункции, например группу Boc;
*C обозначает, если R не является атомом водорода, асимметрический углерод конфигурации (R, S) или (R).
Соединение формулы (III)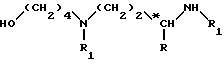
в котором R обозначает метильную группу;
*C обозначает атом углерода конфигурации (R, S) или (R);
R1 обозначает защитную группу аминофункции, например группу Boc; может быть получено: (a) путем взаимодействия 4-аминобутанола с соединением формулы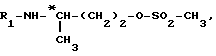
в которой R1 обозначает защитную группу аминофункции, например группу Boc;
до получения соединения формулы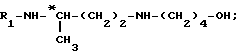
затем путем введения защитной группы свободной аминофункции, например, с помощью трет. -бутилдикарбоната (Boc2O), если R1 обозначает группу Boc, с получением целевого соединения формулы (III); или б) путем взаимодействия галогенида формулы:
X - (CH2)4 - O - R3,
в которой X обозначает галоген, например атом иода или атом брома;
R3 обозначает защитную группу гидроксильной функции, в частности тритильную группу,
с соединением формулы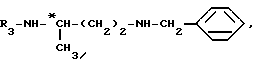
в которой R1 обозначает защитную группу аминофункции, например группу Boc,
до получения соединения формулы (a)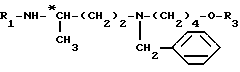
затем путем селективного удаления защитной группы из этого продукта для получения соединения формулы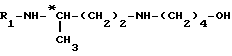
и путем введения в это соединение защитной группы аминофункции R1, например обработкой трет.-бутилдикарбонатом, если R1 обозначает группу Boc, для получения целевого соединения формулы (III).
Соединения формулы (III), в которой R обозначает метильную группу и R1 обозначает группу Boc, являются новыми и составляют один из предметов изобретения.
Соединение формулы (V)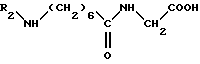
в которой 2 обозначает защитную группу аминофункции, например группу Z (бензилоксикарбонил),
может быть получено исходя из соединения формулы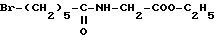
последовательно обрабатывая:
(а) цианидом, который позволяет получить соединение формулы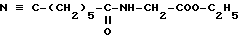
(б) раствором гидроксида натрия, который приводит к кислоте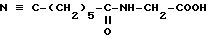
(в) водородом в присутствии катализатора гидрирования для получения амина
(г) наконец, реагентом, вводящим защитную группу аминофункции, например бензилхлорформиатом, для получения целевого соединения формулы (V), в которой R2 обозначает бензилоксикарбонильную группу (Z).
Изобретение будет лучше понятно при ознакомлении с нижеследующими примерами и результатами фармакологических испытаний, полученными при использовании соединений согласно изобретению, по сравнению с результатами, полученными при применении известных продуктов уровня техники. В примерах использована номенклатура, рекомендуемая Chemical Abstracts; также сложный эфир типа "трет.-бутил...оат" описывается в форме "1,1-диметилэтиловый эфир .. . овой кислоты".
В экспериментальной части описания под названием "Приготовления" относятся к промежуточным соединениям, а "Примеры" относятся к продуктам согласно изобретению.
Если соединение содержат в своей структуре асимметрический углерод, то отсутствие особого указания или указание (R, S) обозначает, что речь идет в значительной степени об эквимолекулярной смеси двух энантиомеров (т.е. о "рацемическом" соединении). Если те же самые соединения снабжены знаком (R) или соответственно (S) сразу после идентификации положения заместителя, то это обозначает, что углерод, который содержит этот заместитель, находится в конфигурации (R) или соответственно (S) согласно правилам Cahn, Ingold и Prelog.
Спектральные характеристики сигналов ядерного магнитного резонанса (ЯМР) даются для протона (1H) или для изотопа 13 углерода (13C): указан химсдвиг по отношению к сигналу тетраметилсилана, и в скобках указана форма сигнала (c. для синглета; д. для дублета; т. для триплета; к. для квадруплета; м. для мультиплета; ш. с. - уширенный сигнал) и число протонов, относящихся к сигналу. Для сведения, спектры 1H-ЯМР реализуют при 300 мГц.
Приготовление 1
Бис-(1,1-диметилэтиловый)эфир 3-{/(1,1-диметилэтокси)карбонил/ амино}-21-/(1,1-диметилэтокси)карбонил/-12,15-диоксо-16-окса-2,4,11, - 14,21,25-гексаазагексакоз-2-ен-диовой кислоты
1 г (2,89 • 10-3 моль) 1,1-Диметилэтилового эфира {3-[/(1,1-диметилэтокси)карбонил/амино] пропил} (4-гидроксибутил)- карбаминовой кислоты растворяют в 20 мл тетрагидрофурана (ТГФ), добавляют 0,45 г (4,5 • 10-3 моль) триэтиламина, затем 0,58 г (2,89 • 10-3 моль) 4-нитрофенилхлорформиата в виде раствор в 5 мл ТГФ. Реакционную смесь перемешивают в течение 15 часов при комнатной температуре, затем добавляют 1,2 г (2,89 • 10-3 моль) 1,1-диметилэтилового эфира 13-амино-3-//(1,1- диметилэтокси)карбонил/амино/-12-оксо-2,4,11-триазатридец-2-еновой кислоты в виде раствора в 6 мл ТГФ, доводят реакционную смесь до температуре 40oC и перемешивают 5 часов. После концентрирования реакционной смеси при пониженном давлении, остаток очищают путем хроматографии среднего давления на силикагеле, элюируя смесью метилциклогексана с этилацетатом (7/3 по объему), затем чистым этилацетатом. Таким образом получают 1 г целевого продукта в форме прозрачного масла (выход = 44%).
1H-ЯМР (CDCl3): 1,3-1,9 (м., 50H); 3,05-3,55 (м., 10H); 3,85 (д., 2H), 4,10 (т. , 2H), 4,7-5,3 (ш.с., 1H); 5,4-5,6 (ш.с., 1H); 6,0-6,2 (ш.с., 1H); 8,3-8,5 (ш.с., 1H); 11,5 (с., 1H).
Пример 1
{ 4-(3-Аминопропил)амино/бутоксикарбониламино} -N-{6-/(аминоиминометил)- амино/гексил}ацетамид-трис(трифторацетат)
Готовят смесь из 1 г (1,277•10-3 моль) полученного в приготовлении 1 соединения в 10 мл дихлорметана и 10 мл трифторуксусной кислоты и реакционную среду перемешивают в течение 15 часов при комнатной температуре. Растворители затем удаляют при пониженном давлении и остаток очищают путем хроматографии среднего давления (ХСД) на силикагеле типа RP18 (гранулометрия 5-20 мкм), элюируя смесью этанол/вода/трифторуксусная кислота (2:7, 5:0,5; по объему). Фракции, содержащие чистый продукт, концентрируют при пониженном давлении, растворяют в воде и лиофилизируют. Таким образом получают 0,78 г целевого продукта в виде аморфного полупрозрачного белого твердого вещества (выход = 84%).
1H-ЯМР (DMCO-d6): 1,2-1,55 (м., 8H); 1,6-1,75 (м., 4H); 1,8-1,95 (м., 2H); 2,8-3,1 (м., 10H); 3,55 (д., 2H); 3,95 (т., 2H); 6,7-7,4 (м., 4H); 7,55 (т., 1H); 7,75-8,00 (м., 4H); 8,5-8,65 (м., 3H).
13C-ЯМР (D2O - диоксан h8): 23,05; 24,55; 26,15; 26,20; 26,30; 28,55; 28,95; 31,05; 37,35; 39,95; 41,88; 45,24; 48,18; 65,71; 158,0; 159,0; 178,5.
Пример 1-бис
{ 4-/(3-Аминопропил)амино/бутоксикарбониламино}-N-{6-/(аминоиминометил)- амино/гексил}ацетил-трис(хлоргидрат)
Готовят раствор из 3,5 г соединения согласно примеру 1 (4,8•10-3 моль) в 7 мл безводного этанола и при 0oC прикапывают 10 мл 1,3 М раствора хлороводорода в этаноле. Выдерживают при перемешивании в течение 30 минут после окончания добавления, затем отфильтровывают выпавший осадок. После промывки твердого вещества безводным этанолом, его высушивают в вакууме при 35 - 40oC. Таким образом получают целевой продукт (1,72 г) в виде белого гигроскопичного порошка (выход = 72%). Т. пл. = 107,5oC.
Приготовление II
1,1-Диметилэтиловый эфир {3-[[4-[трис(фенил)метокси]бутил](фенилметил)- амино]-1-(R)-метилпропил}карбаминовой кислоты
Готовят раствор из 3,77 г (13,5•10-3 моль) 1,1-диметилэтилового эфира [3-(фенилметиламино)-1-(R)- метилпропил] карбаминовой кислоты в 270 мл бутанола, затем добавляют 12 г (27,1•10-3 моль) 1-иод-4-[трис(фенил)метокси] бутана и 3,74 г (27,1•10-3 моль) карбоната калия. Реакционную смесь перемешивают при температуре 95-100oC в течение 48 часов. После охлаждения, реакционную среду отфильтровывают, затем концентрируют при пониженном давлении. Остаток после этого обрабатывают дихлорметаном, и полученный раствор промывают водой. После концентрирования органической фазы при пониженном давлении, продукт очищают путем хроматографии на силикагеле, элюируя смесью гексана с этилацетатом (80:20 по объему). Таким образом получают 6 г целевого продукта в виде масла (выход = 75%).
[α]
1H-ЯМР (CDCl3): 1,02 (д., 3H); 1,40 (с., 9H); 1,59 (м., 6H); 2,38 (м., 3H); 2,55 (м., 1H); 3,0 (ш. с., 2H); 3,40 (д., 1H); 3,6 (д., 2H); 5,70 (с., 1H); 7,21-7,43 (м., 20H).
Приготовление III
1,1-Диметиэтиловый эфир {3-[[4-гидрокси-бутил]амино]-1-(R)- метилпропил} -карбаминовой кислоты (хлоргидрат)
4,94 г (8,33•10-3 моль) полученного в приготовлении II соединения растворяют в 130 мл 95o-ного этанола (т.е. примерно смесь этанол/вода [95/95 по объему]), затем добавляют 4,37 мл 4,3 н раствора HCl в 95o-ном этаноле и 1 г 5%-ного палладия-на-угле. Смесь затем гидрируют при атмосферном давлении в течение 40 часов при комнатной температуре. Катализатор удаляют путем отфильтровывания и фильтрат концентрируют при пониженном давлении. После очистки путем хроматографии на силикагеле, элюируя смесью хлороформа с метанолом (9: 1 по объему), получают 2,09 г целевого продукта. После промывки диэтиловым эфиром получают 1,6 г чистого продукта в виде белого кристаллического твердого вещества (выход = 64,8%). Т. пл. = 135oC [α]
Приготовление IV
1,1-Диметилэтиловый эфир {3-[4(-гидрокси-бутил)(1,1- диметилэтоксикарбонил)амино]-1-(R)-метилпропил}-карбаминовой кислоты
Готовят смесь из 1,54 г (5,18•10-3 моль) полученного согласно приготовлению III соединения, 8 мл диоксана и раствора 0,77 г (7,26•10-3 моль) карбоната натрия и 5,7 мл воды. Эту смесь охлаждают до 5oC, затем добавляют раствор из 1,13 г (5,18•10-3 моль) ди-трет.-бутил-дикарбоната (Boc2O) в 3 мл диоксана, и реакционную смесь выдерживают при перемешивании в течение 15 часов. После добавления 10 мл воды, экстрагируют этилацетатом, после чего органическую фазу концентрируют при пониженном давлении. После очистки путем хроматографии на силикагеле, элюируя смесью гексана с этилацетатом (50:50 по объему), получают 1,82 г целевого продукта в виде вязкого бесцветного масла (выход = 97%) [α]
1H-ЯМР (CDCl3): 0,87 (д., 3H); 1,44-1,46 (2с., 18H); 1,50-1,70 (м., 6H); 3,20 (м., 4H); 3,67 (т., 3H); 4,41-4,56 (2 ш.с., 1H).
Приготовление V
1,1-Диметилэтиловый эфир {3-[N-[4-[(4-нитрофенокси)карбонилокси] бутил] -N-[1,1-диметилэтоксикарбонил]амино]-1-(R)-метилпропил}карбаминовой кислоты
Готовят раствор из 2,73 г (7,57•10-3 моль) полученного в приготовлении IV соединения в 55 мл толуола и 0,62 мл (7,7•10-3 моль) пиридина и добавляют 1,6 г (7,7•10-3 моль) 4-нитрофенил-хлорформиата. Реакционную смесь выдерживают при перемешивании в течение 3-х часов, затем удаляют путем отфильтровывания образовавшиеся соли. Фильтрат концентрируют при пониженном давлении, после чего очищают путем хроматографии на силикагеле, элюируя смесью дихлорметана с этилацетатом (90: 10 по объему). Таким образом получают 3,1 г целевого продукта (выход = 78%).
1H-ЯМР (CDCl3): 1,15 (д., 3H); 1,44 (с., 9H); 1,46 (с., 9H); 1,5-1,8 (м. , 6H); 3,24 (м., 4H); 3,63 (м., 1H); 4,30 (т., 2H); 7,39 (д., 2H); 8,28 (д., 2H).
Приготовление VI
Бис(1,1-диметилэтиловый эфир) 3-{ [(1,1-диметилэтокси)карбонил]амино}- [(1,1-диметилэтокси)карбонил]-24-(R)-метил-12,15-диоксо-16-окса- 2,4,11,14,21,25-гексааза-гексакоз-2-ен-дионовой кислоты
2,8 г (5,33•10-3 моль) соединения согласно приготовлению V растворяют в 300 мл ТГФ, затем добавляют 0,85 мл (6,1•10-3 моль) триэтиламина и раствор 2,54 г (6,1•10-3 моль) 1,1-диметилэтилового эфира 13-амино-3-{[(1,1-диметилэтокси)карбонил] амино} -12-оксо-2,4,11- триазатридец-2-ен-овой кислоты в 50 мл ТГФ. Выдерживают при перемешивании в течение 4-х часов при комнатной температуре, после чего реакционную смесь концентрируют при пониженном давлении. Твердый остаток очищают путем хроматографии на силикагеле, элюируя смесью гексана с этилацетатом (30:70, затем 10:90, по объему). Таким образом получают 3,85 г целевого продукта в виде светло-желтого твердого вещества (выход = 90,2%). [α]
1H-ЯМР (CDCl3): 1,15 (д., 3H); 1,3-1,70 (м., 50H); 3,25 (м., 6H); 3,39 (к., 2H); 3,63 (м., 1H); 3,83 (д., 2H); 4,14 (т., 2H); 4,6 (ш. с., 1H); 5,60 (ш. с., 1H); 6,13 (ш.с., 1H); 8,31 (с., 1H); 11,50 (с., 1H).
Пример 2
{4-[(3-(R)-Амино-бутил)амино]бутоксикарбониламино}-N-[6- (аминоиминометиламино)гексил]ацетамид-трис(трифторацетат)
В колбе, охлаждают до 0oC 3,7 г (4,6•10-3 моль) полученного согласно приготовлению VI соединения и добавляют 37 мл трифторуксусной кислоты. Смесь выдерживают при перемешивании в течение 15 часов при 5 - 10oC, затем концентрируют при пониженном давлении. Сырой продукт очищают путем хроматографии на привитом силикагеле типа RP18, элюируя смесью вода/ацетонитрил/трифторуксусная кислота (70:15:15 по объему). Таким образом получают 3,1 г целевого продукта в виде твердого белого гигроскопичного вещества (выход = 92%). [α]
1H-ЯМР (DMCO-d6): 1,18 (д., 3H); 1,26 (м., 4H); 1,41 (м., 4H); 1,61 (м., 4H); 1,75 (м., 1H); 1,91 (м., 1H); 2,85-3,20 (м., 8H); 3,28 (м., 1H); 3,54 (д. , 2H); 3,96 (ш. с., 2H); 7,25 (т., 1H); 7,64 (с., 1H); 7,86 (т., 1H); 7,98 (с., 3H); 8,62 (с., 2H).
13C-ЯМР (D2O + диоксан): 17,93; 22,98; 26,06; 26,14; 26,23; 28,49; 28,91; 31,13; 39,89; 41,79; 44,31; 44,52; 46,01; 48,08; 65,60; 157,44; 159,38; 172,64.
Пример 2-бис
{4-[(3-(R)-Амино-бутил)амино]бутоксикарбониламино}-N-[6- (аминоиминометиламино)гексил]ацетамид-трис(хлоргидрат)
Готовят раствор из 200 мг (0,27 • 10-3 моль) полученного в примере 2 соединения в 0,5 мл этанола и добавляют 0,235 мл 10,4 н раствора HCl в этаноле. Перемешивают в течение 15 минут при комнатной температуре, затем добавляют 2 мл диизопропилового эфира. Осадившийся продукт отделяют путем декантации, промывают его с помощью диизопропилового эфира, затем поглощают его 3 мл воды и лиофилизируют. Таким образом получают 117 мг целевого продукта в виде аморфного твердого вещества белого цвета (выход = 85%). [α]
1H-ЯМР (DCMO-d6): 1,21 (д., 3H); 1,26 (м., 4H); 1,42 (м., 4H); 1,64 (м., 4H); 1,88 (м., 1H); 2,04 (м., 1H); 2,89-3,10 (м., 8H); 3,35-3,70 (м., 3H); 3,97 (ш.с., 2H); 7,32 (т., 1H); 7,80 (т., 1H); 7,92 (т., 1H); 8,24 (с., 3H); 9,16 (с., 2H).
Приготовление VII
1.1-Диметиловый эфир {3-[N-[4-гидрокси-бутил]амино]-1- (R,S)-метилпропил}карбаминовой кислоты
Готовят раствор из 6,2 г (23,25 • 10-3 моль) 1,1-диметилэтилового эфира { 1-(R, S)-метил-3-[(метилсульфонил)окси]пропил}карбаминовой кислоты в 40 мл 1,2-диметоксиэтана и прикапывают раствор 4,14 г (46,5 • 10-3 моль) 4-аминобутанола в 10 мл 1,2-диметоксиэтана. Реакционную смесь в течение 18 часов кипятят с обратным холодильником, затем концентрируют при пониженном давлении. Сырой продукт очищают путем хроматографии на силикагеле, элюируя смесью этилацетата с этанолом и 33%-ным аммиаком (6:3:0,1 по объему). Таким образом получают 2,5 г целевого продукта в виде масла (выход = 41,5%).
1H-ЯМР (CDCl3): 1,14 (д., 3H); 1,44 (с., 9H); 1,5-1,8 (м., 6H); 2,64 (м. , 4H); 3,58 (т., 2H); 3,71 (м., 1H); 4,50 (д., 1H).
Приготовление VIII
1,1-Диметилэтиловый эфир { 3-[N-(4-гидрокси-бутил)-N-(1,1-диметилэтокси-карбонил)амино] -1-(R, S)-метил-пропил}карбаминовой кислоты
Поступая аналогично способу приготовления IV, исходя из соединения, полученного согласно приготовления VII, получают целевой продукт с выходом 70%.
1H-ЯМР (CDCl3): 1,15 (д., 3H); 1,44-1,46 (2с., 18H); 1,5-1,7 (м., 6H); 3,20 (м., 4H); 3,67 (т., 3H); 4,39-4,56 (2 ш.с., 1H).
Приготовление IX
Бис(1,1-диметилэтиловый эфир) 3-{[(1,1-диметилэтокси)карбонил]амино}-21-[(1,1-диметоксиэтокси) карбонил] -24-(R, S)-метил-12,15-диоксо-16-окса-2,4,11,14,21,25-гексааза- гексакоз-2-ен-дионовой кислоты
1,8-г (5•10-3 моль) Соединения, полученного согласно вышеуказанному приготовлению VII, растворяют в 36 мл ТГФ, затем добавляют 1,1 г (7,8•10-3 моль) триэтиламина и 1,10 г (5,45•10-3 моль) 4-нитрофенилхлорформиата. Реакционную среду выдерживают при перемешивании в течение 15 часов, затем добавляют раствор 2,1 г (5,06•10-3 моль) 1,1-диметилэтилового эфира 13-амино-3-{ [(1,1-диметилэтокси)карбонил] амино}-12- оксо-2,4,11-триаза-тридец-2-ен-овой кислоты в 30 мл ТГФ и выдерживают при перемешивании в течение 18 часов. Образовавшееся за счет реакции твердое вещество удаляют путем отфильтровывания, затем раствор концентрируют при пониженном давлении и остаток очищают путем хроматографии на силикагеле, элюируя смесью метилциклогексана с этилацетатом (50: 50), затем (20:80 по объему). Таким образом получают 0,97 г целевого продукта в виде масла (выход = 24,2%).
1H-ЯМР (CDCl3): 1,15 (д., 3H); 1,20-1,80 (м., 50H); 3,26 (м., 6H); (к., 2H); 3,62 (м., 1H); 3,83 (д., 2H); 4,12 (м., 2H); 4,60 (ш.с., 1H); 5,60 (ш. с., 1H); 6,13 (ш.с., 1H); 8,30 (т., 1H); 11,49 (с., 1H).
Пример 3
{ 4-[(3-(R,S)-Амино-бутил)амино]бутоксикарбониламино}- N-[6-амино-иминометиламино)гексил]ацетамид-трис(трифторацетат).
0,95 (1,18 • 10-3 моль) Соединения, полученного согласно приготовлению IX, растворяют в 10 мл дихлорметана, затем добавляют 10 мл трифторуксусной кислоты. После перемешивания в течение 24 часов при комнатной температуре реакционную смесь концентрируют при пониженном давлении и остаток очищают путем хроматографии на привитом силикагеле типа RP18, элюируя смесью ацетонитрил/вода/трифторуксусная кислота (15:80:5 по объему). После лиофилизации чистых фракций получают 580 мг целевого продукта в виде твердого вещества белого цвета (выход = 66%).
1H-ЯМР (DMCO-d6): 1,18 (д., 3H); 1,26 (м., 4H); 1,41 (м., 4H); 1,61 (м., 4H); 1,74 (м., 1H); 1,91 (м., 1H); 2,85-3,10 (м., 8H); 3,28 (м., 1H); 3,54 (д. , 2H); 3,96 (т., 2H); 7,23 (т., 1H); 7,58 (т., 1H); 7,93 (с., 3H); 8,56 (м., 2H).
13C-ЯМР (D2O + диоксан h8): 18,02; 23,09; 26,17; 26,22; 26,30; 28,58; 29,00; 31,24; 40,00; 41,88; 44,40; 44,63; 46,10; 48,19; 65,72; 157,55; 159,55; 172,82.
Приготовление X
Сложный этиловый эфир N-(6-цианогексаноил)глицина
23,73 г (84,7 • 10-3 моль) Сложного этилового эфира N-(6-бромгексаноил)глицина растворяют в 200 мл этанола и добавляют 6,5 г (0,1 моль) цианида калия в виде порошка. Реакционную смесь доводят до температуры рефлюкса и таким образом перемешивают в течение 15 часов. После концентрирования смеси при пониженном давлении остаток обрабатывают дихлорметаном и органическую фазу промывают водным раствором хлорида натрия. Органическую фазу сушат и концентрируют при пониженном давлении. Таким образом получают 19 г целевого продукта (выход = 99%).
1H-ЯМР (CDCl3): 1,29 (т. , 3H); 1,45-1,55 (м., 2H); 1,6-1,8 (м., 4H); 1,27 (т., 2H); 2,36 (т., 2H); 4,03 (д., 2H); 4,22 (к., 2H); 5,35-6,05 (ш.с., 1H).
Приготовление XI
N-(6-Цианогексаноил)глицин
Готовят смесь из 19 г (84 • 10-3 моль) соединения, полученного согласно приготовлению X, 120 мл 1,2-диметоксиэтана и 120 мл 1 М раствора гидроксида натрия и перемешивают в течение 15 минут при комнатной температуре. После этого добавляют 100 мл воды и 200 мл дихлорметана, и подкисляют до pH=1, охлаждая на ледяной бане. Водную фазу экстрагируют несколько раз дихлорметаном, и объединенные органические фазы сушат и концентрируют при пониженном давлении. Таким образом получают 10,2 г целевого продукта в виде масла бледно-желтого цвета (выход = 61%).
1H-ЯМР (CDCl3): 1,45-1,55 (м., 2H); 1,6-1,8 (м., 4H); 2,3 (т., 2H); 2,36 (т., 2H); 4,08 (д., 2H); 6,15-6,25 (ш., с., 1H).
Приготовление XII
N-(7-Амино-гептаноил)глицин (натриевая соль)
8,2 г (41,4 • 10-3 моль) N-(6-цианогексаноил) глицина растворяют в 100 мл этанола, добавляют 60 мл 1М раствора гидроксида натрия и 800 мг никеля Ренея. Смесь затем перемешивают в атмосфере водорода при комнатной температуре и давлении 3,5 • 105 Па, в течение 8 часов. По окончании реакции добавляют 20 мл 1М соляной кислоты и концентрируют при пониженном давлении. Tаким образом получают 10,2 г твердого пастообразного вещества белого цвета, которое содержит целевую соль и хлорид натрия и которое может быть использовано без другой очистки в следующей стадии.
1H-ЯМР (DMCO-d6): 1,15-1,6 (м., 8H); 2,08 (т., 2H); 2,56 (т., 2H); 3,32 (д., 2H); 7,1-7,25 (ш.с., 1H).
Приготовление XIII
N-{7-[(Фенилметоксикарбонил)амино]гептаноил}глицин
2 г Соединения, полученного согласно приготовлению XII, растворяют в смеси из 50 мл воды и 50 мл этанола, добавляют 1,06 г (10-2 моль) карбоната натрия, затем 2,34 г (19,8 • 10-3 моль) бензилхлорформиата. После перемешивания в течение 15 часов при комнатной температуре реакционную среду доводят до pH=1 с помощью 1М соляной кислоты, после чего экстрагируют дихлорметаном. Органическую фазу сушат и концентрируют при пониженном давлении, и сырой продукт очищают путем хроматографии на силикагеле, элюируя смесью этилацетат/этанол/гидрат окиси аммония (6:3:0,5 по объему). Таким образом получают 1,3 г целевого продукта в виде твердого пастообразного вещества.
1H-ЯМР (DMCO-d6):1,2-1,6 (м., 8H); 2,09 (т., 2H); 2,98 (т.д., 2H); 3,64 (д., 2H); 5,01 (с., 2H); 7,25 (т., 1H); 7,3-7,4 (м., 5H); 7,94 (т., 1H).
Приготовление XIV
1-(Фенилметил)-24-(1,1-диметилэтиловый) эфир 19-[(1,1-диметилэтокси)-карбонил] -9,13-диоксо-14-окса-2,10,12,19,23- пентааза-тетракозандиовой кислоты
0,5 г (1,49 • 10-3 моль) Кислоты, полученной согласно приготовлению XIII, растворяют в 25 мл ТГФ, добавляют 0,18 г (1,8 • 10-3 моль) триэтиламина и смесь охлаждают до 0oC. Затем прикапывают 0,46 г (1,67 • 10-3 моль) дифенилфосфорилазида в виде раствора в 5 мл ТГФ и оставляют при перемешивании при комнатной температуре в течение 45 минут. Растворитель удаляют при пониженном давлении, остаток обрабатывают в 2 мл толуола и добавляют 0,18 г (1,8 • 10-3 моль) триэтиламина и 1,04 г (3 • 10-3 моль) 1,1-диметилэтилового эфира { 3-[[(1,1-диметилэтокси)карбонил]амино]пропил}(4-гидроксибутил) карбаминовой кислоты. Реакционную смесь кипятят с обратным холодильником в течение 20 часов, после чего концентрируют при пониженном давлении. Сырой продукт очищают путем хроматографии на силикагеле, элюируя смесью метилциклогесана с этилацетатом при градиенте (8/2 ---> 1/9 по объему). Таким образом получают 0,4 г целевого продукта в виде твердого вещества белого цвета (выход = 39,6%).
Т.пл. = 115oC.
1H-ЯМР (CDCl3): 1,2 - 1,7 (м., 32H); 2,15 (т., 2H); 3,05 - 3,3 (м., 8H); 4,05 (т., 2H); 4,5 (т., 2H); 4,7 - 4,9 (ш.с., 1H); 5,08 (с., 2H); 5,7 - 5,9 (ш.с., 1H); 6,4 - 6,6 (ш.с., 1H); 7,3 - 7,4 (м., 6H).
Приготовление XV
1,1-Диметилэтиловый эфир 22-амино-6-[(1,1-диметилэтокси)карбонил] -12,16-диоксо-11-окса-2,6,13,15- тетрааза-докозановой кислоты
Раствор из 0,33 г (0,48 • 10-3 моль) соединения, полученного согласно приготовлению XIV в 15 мл этанола подвергают каталитическому гидрированию в присутствии 30 мг 5%-ного палладия на угле при атмосферном давлении и при комнатной температуре, в течение 5 часов. После удаления катализатора путем отфильтровывания растворитель удаляют при пониженном давлении и получают 0,26 г целевого продукта в форме масла (выход 0,99%).
1H-ЯМР (CDCl3): 1,2 - 1,75 (м., 32H); 2,2 (т., 2H); 2,8 (т., 2H); 2,85 - 3,3 (м., 8H); 4,07 (т., 2H); 4,53 (т., 2H); 4,7 - 5,4 (ш.с., 1H); 5,7 - 6,1 (ш.с., 1H); 6,4 - 7,1 (ш.с., 1H).
Приготовление XVI
Бис-(1,1-диметилэтиловый) эфир 3-{[(1,1- диметилэтокси)карбонил]амино} -21-[(1,1-диметилэтокси)карбонил] -11,15-диоксо-16-окса-2,4,12,14,21,25- гексааза-гексакоз-2-ен-диовой кислоты
Готовят смесь из 0,26 г (0,477 • 10-3 моль) соединения, полученного согласно приготовлению XV, 0,276 г (0,95 • 10-3 моль) 1,1-диметилэтилового эфира {[[(1,1- диметилэтокси)карбонил]амино](метилтио)метилен}-карбаминовой кислоты и 200 мкл триэтиламина в 12 мл ТГФ и реакционную среду выдерживают при перемешивании в течение 4-х дней при комнатной температуре. После выпаривания растворителя при пониженном давлении сырой продукт очищают путем хроматографии на силикагеле, элюируя смесью метилциклогексана с этилацетатом при градиенте (7/3 ---> 2/8 по объему). Таким образом получают 0,10 г целевого продукта в виде масла (выход = 26,7%).
1H-ЯМР (CDCl3): 1,0-1,8 (м., 50H); 2,16 (т., 2H); 3,05 - 3,3 (м., 6H); 3,4 (т. д. , 2H); 4,07 (т., 2H); 4,55 (т., 3H); 5,2 - 5,3 (ш.с., 1H); 5,7 - 5,8 (ш.с., 1H); 6,45 - 6,55 (ш.с., 1H); 8,3 (т., 1H); 11,5 (с., 1H).
Пример 4
7-[(Аминоиминометил)амино] -N-{ [4-[(3-аминопропил)амино]бутокси] карбониламинометил}гептанамид-трис(трифторацетат)
Поступая аналогично способу примера 3 и исходя из соединения, полученного согласно приготовлению XVI, получают целевой продукт с выходом = 54%.
1H-ЯМР (DMCO-d6): 1,2 - 1,35 (м., 4H); 1,4 - 1,7 (м., 8H); 1,8 - 1,95 (м. , 2H); 2,06 (т., 2H); 2,8 - 3,1 (м., 8H); 3,96 (т., 2H); 4,32 (т., 2H); 6,85 - 7,4 (ш.с., 3H); 7,55 - 7,7 (м., 2H); 7,8 - 8,0 (м., 4H); 8,33 (т., 1H); 8,55 - 8,7 (м., 2H).
Приготовление XVII
1-(Фенилметил)-24-(1,1-диметилэтиловый) эфир 19-[(1,1-диметилэтокси)-карбонил] -22-(R)-метил-9,13-диоксо-14-окса- 2,10,12,19,23-пентааза-тетракозандиовой кислоты
Поступая аналогично способу приготовления XIV, исходя из кислоты, полученной согласно приготовлению XIII, и спирта, полученного согласно приготовлению IV, получают целевой продукт в виде твердого аморфного вещества с выходом 46%. [α]
1H-ЯМР (CDCl3): 1,15 (д., 3H); 1,2 - 1,7 (м., 32H); 2,15 (т., 2H); 3,05 - 3,35 (м. , 6H); 3,55 - 3,7 (м., 1H); 4,05 (т., 2H); 4,55 (т., 2H); 4,8 - 4,9 (ш. , ст., 1H); 5,09 (c., 2H); 5,01 - 5,2 (ш.с., 1H); 5,7 - 5,9 (ш.с., 1H); 6,45 - 6,6 (ш.с., 1H); 7,3 - 7,4 (м., 5H).
Приготовление XVIII
1,1-Диметилэтиловый эфир 22-амино-6-[(1,1-диметилэтокси)карбонил] -3-(R)-метил-12,16-диоксо-11- окса-2,6,13,15-тетрааза-докозановой кислоты
Поступая аналогично способу приготовления XV и исходя из соединения, полученного согласно приготовлению XVII, получают целевой продукт в виде масла с выходом 99%. [α]
1H-ЯМР (CDCl3): 1,15 (д., 3H); 1,2 - 1,7 (м., 32H); 2,19 (т., 2H); 2,77 (т, 2H); 2,85 - 3,3 (м., 6H); 3,55 - 3,7 (м., 1H); 4,07 (т., 2H); 4,52 (т., 2H); 5,7 - 6,3 (ш.с., 2H); 6,8 - 7,0 (ш.с., 1H).
Приготовление XIX
1,1-Диметилэтиловый эфир 22[(аминоиминометил)амино]-6- [(1,1-диметилэтокси)карбонил] -3(R)-метил-12,16-диоксо-11-окса- 2,6,13,15-тетраазоадокозановой кислоты
0,4 г (0,716 • 10-3 моль) Соединения, полученного согласно приготовлению XVIII, растворяют в 10 мл метанола, добавляют 0,186 г (1,5 • 10-3 моль) аминоиминометансульфокислоты и реакционную смесь перемешивают при комнатной температуре в течение 3-х дней. После удаления растворителя при пониженном давлении, сырой продукт очищают путем хроматографии на силикагеле, элюируя смесью этилацетат/этанол/аммиак (6: 3: 0,1, затем 6:3:1; по объему). Таким образом получают 0,37 г целевого продукта в виде масла (выход = 86%). [α]
1H-ЯМР (CDCl3): 1,15 (д., 3H); 1,2 - 1,7 (м., 32H); 2,2 (т., 2H); 2,9 - 3,3 (м. , 6H); 3,5 - 3,75 (м., 1H); 4,05 (т., 2H); 4,52 (т., 2H); 5,4 - 5,7 (ш. с. , 2H); 6,3 - 6,5 (ш.с., 1H); 7,05 - 7,3 (ш.с., 4H); 7,8 - 8,0 (ш.с., 1H).
Пример 5
7-[(Аминоиминометил)амино] -N-[[4-[(3-(R)-аминобутил)амино] -бутокси] - карбониламиноэтил]гептанамид-трис(трифторацетат)
Поступая аналогично способу примера 3, и исходя из соединения, полученного согласно приготовлению XIX, получают целевой продукт в виде аморфного твердого вещества с выходом 42%. [α]
1H-ЯМР (DMCO-d6): 1,18 (д., 3H); 1,2-1,35 (м., 4H); 1,4-1,55 (м., 4H); 1,55-1,7 (м. , 4H); 1,7-1,85 (м., 1H); 1,85-2,0 (м., 1H); 2,05 (т., 2H); 2,85-3,1 (м., 6H); 3,2-3,85 (м., 1H); 3,96 (т., 2H); 4,32 (т., 2H); 6,8-7,5 (м. с. , 3H); 7,60 (т., 1H); 7,68 (т., 1H); 7,95 (с., 4H); 8,35 (т., 1H); 8,5-8,7 (м., 2H).
13C-ЯМР (D2O + диоксан h8): 18,0; 23,06; 25,79; 26,12; 26,25; 28,45; 28,50; 31,22; 36,27; 41,86; 44,61; 46,08; 46,55; 48,17; 65,48; 157,52; 159,06; 178,36.
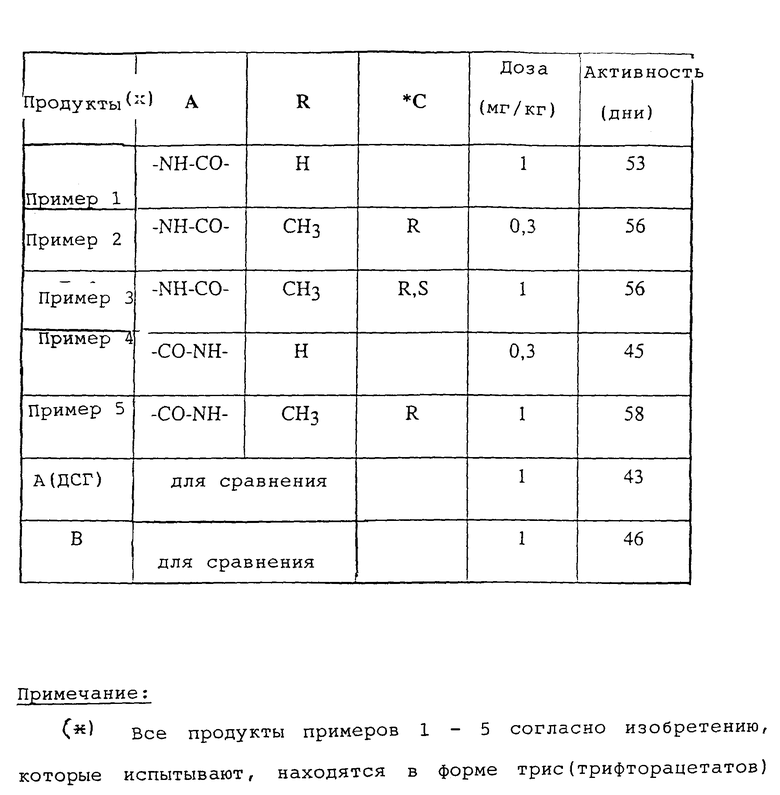
Иммуноподавляющую активность продуктов согласно изобретению выявляют с помощью так называемого теста реакции имплантата против хозяина. Самцов мышей B6D2F1 (гибриды первой генерации C57B1/6 • DBA/2) иммуноугнетают путем интраперитональной инъекции циклофосфамида. Тремя днями позже (день 0 эксперимента: J0) они получают внутривенно 4 • 107 спленоцитов мыши C57B1/6. Животных затем распределяют на партии минимально по 8 особей и они получают ежедневное лечение в дни J1 - J5 и J7 - J10 интраперитонально. Контрольная группа получает один эксципиент. За гибелью наблюдают в течение 60 дней (J60). Результаты, выраженные средней величиной выживания в днях при указанной дозе, представлены в таблице, где указанные величины значительно согласно тесту Логранка (вероятность менее или равно 5%). Для сравнения в таблице указаны значения, полученные с известными продуктами (или родственной структуры) уровня техники: (А) 15-дезоксипергуалин (в виде трис/хлоргидрата/) и соединение (B), которое соответствует примеру 16 европейского патента A-0600762.
Продукт B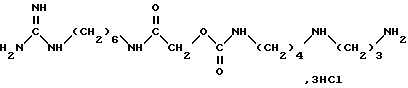
Из этого сравнения следует, что продукты согласно изобретению обладают более высокой активностью, чем продукты уровня техники, или требуют более незначительной дозировки для достижения эквивалентного эффекта.
Продукты согласно изобретению пригодны в терапии в качестве лечебных или профилактических иммунодепрессирующих агентов, особенно для профилактики отторжения аллогенных или ксеногенных сосудистых или несосудистых органов; для предупреждения реакции трансплантата против хозяина в результате пересадки сосудов, при лечении аутоиммунных генетических зависимостей или приобретенных заболеваний (например, диссеминированная эритематозная волчанка, рассеянный склероз, ревматоидный полиартрит); хронических воспалительных заболеваний, таких как, например, суставные ревматизмы, а также в случае любых патологий, при которых иммунные нарушения являются причиной или фактором из-за которых сохраняются ухудшенное клиническое состояние.
Продукты согласно изобретению также можно вводить в качестве добавки к антираковым цитотоксическим лекарственным препаратам, чтобы ограничивать их вторичные эффекты, и в качестве добавки при приеме продуктов биологического происхождения, в частности рекомбинантных цитокининов, моно- или поликлональных антител, чтобы уменьшить появление защитных антител, продуцируемых пациентом.
Продукты согласно изобретению можно использовать при лечебной обработке паразитов, в особенности в соответствии в случае малярии.
Продукты согласно изобретению можно вводить перорально, путем инъекций (в частности внутримышечной или внутривенной), топическим путем (например, в виде крема для локального нанесения глазных капель), чрескожно, ректально с помощью свечей или путем ингаляции.
Продукты согласно изобретению также находят свое применение в качестве фармакологических агентов, в частности при исследовании аутоиммунных заболеваний.
Пример A.
Препарат для перфузии.
Готовят маточный раствор из раствора 200 мг соединения примера 1 в 5 мл чистой воды, который доводят до pH 4,0 добавлением 0,01 N раствора щелочи натрия и добавляют к нему 30 мг хлорида натрия, а затем очищенную воду для инъекций в количестве, достаточном до объема 10 мл.
Раствор, который хранят при 4oC, вводят в перфузионную жидкость из расчета дозировки 2 мл/кг/день.
Пример B.
Препарат для подкожного введения.
Готовят раствор из 12 мг соединения по примеру 2, растворенных в 0,5 мл чистой воды для инъекций, и доводят этот раствор до pH 3,0 с помощью очень разбавленного раствора соляной кислоты. Добавляют к раствору чистую воду для доведения до 1 мл. Раствор хранят при температуре +4oC.
Инъецируемый раствор получают добавлением 1 мл разбавляющего растворителя. Этот разбавитель получают из фосфатного буфера с pH 7 (фармакопейного), который разбавляют до 4%-ной концентрации чистой водой для инъекций.
Получают таким образом 2 мл инъецируемого раствора для подкожного введения, который содержит 12 мг активного начала. Рекомендуемая доза 2 - 4 инъекций в день в зависимости от веса пациента.
Пример C.
Лиофилизат для подкожных инъекций.
Получают раствор, соответствующий 12 мг соединения примера 2 и 120 мг маннита, растворенных в 2 мг чистой воды. Раствор фильтруют, затем лиофилизуют во флаконе. Лиофилизат хранят при 4oC.
Инъецируемый раствор готовят перед употреблением путем разведения лиофилизата в 4 мл чистой воды для инъекций. Рекомендуемая доза составляет 1-4 инъекций в день при подкожном введении.
Пример D.
Таблетка, содержащая 50 мг активного начала.
Активное начало - 50 мг
Микрокристаллическая целлюлоза - 400 мг
Преджелитинированный крахмал - 48 мг
Стеарат магния - 25 мги
| название | год | авторы | номер документа |
|---|---|---|---|
| АНАЛОГИ 15-ДЕЗОКСИСПЕРГУАЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ (ВАРИАНТЫ), АМИНОЗАЩИЩЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ОСНОВЕ АНАЛОГОВ 15-ДЕЗОКСИСПЕРГУАЛИНА | 1995 |
|
RU2113431C1 |
| АНАЛОГИ 15-ДЕОКСИСПЕРГУАЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1993 |
|
RU2114823C1 |
| СПОСОБ ПОЛУЧЕНИЯ СУЛЬФОНИЛ- β -D-ТИОКСИЛОЗИДА | 1991 |
|
RU2033995C1 |
| СПИРОПИПЕРИДИНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 1993 |
|
RU2168512C2 |
| Новое тетерагидропиримидиновое соединение или его соль | 2015 |
|
RU2636310C1 |
| СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ ПРОТЕАЗЫ ВИЧ | 1994 |
|
RU2139280C1 |
| ПРОИЗВОДНЫЕ 2,6-ДИАМИНОПУРИН-β-D-РИБОФУРАНУРОНАМИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМСОСТАВ ДЛЯ ПОДАВЛЕНИЯ АКТИВНОСТИ ЛЕЙКОЦИТОВ, СПОСОБ ЕГО ПОЛУЧЕНИЯ, СПОСОБ ПОДАВЛЕНИЯ АКТИВНОСТИ ЛЕЙКОЦИТОВ | 1994 |
|
RU2129561C1 |
| ПРОИЗВОДНЫЕ 3-Н-1,2,3-ТРИАЗОЛО-[4,5-D]ПИРИМИДИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1996 |
|
RU2174518C2 |
| ЦИКЛОГЕКСАНОВОЕ ПРОИЗВОДНОЕ, СПОСОБ РАССЛАБЛЕНИЯ ГЛАДКОЙ МЫШЦЫ МЛЕКОПИТАЮЩЕГО | 1994 |
|
RU2129113C1 |
| АЗОТСОДЕРЖАЩЕЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ | 2013 |
|
RU2632253C2 |
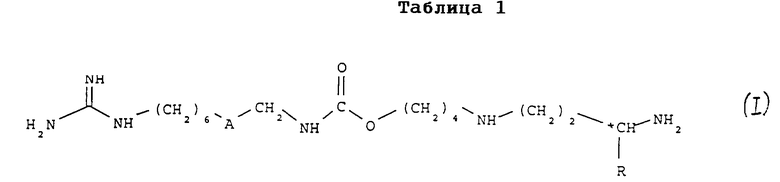
Раскрыты аналоги 15-дезоксиспергуалина общей формулы I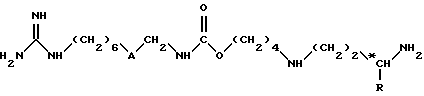
или их нетоксичные соли присоединения, где А обозначает группу -CO-NH- или группу -NH-CO-; R обозначает атом водорода или группу СН3; *С обозначает, если R не является атомом водорода, асимметрический углерод конфигурации (R, S) или конфигурации (R), а также способ их получения, промежуточные соединения и фармацевтическая композиция на их основе. Изобретение может быть использовано для получения лекарственного средства, предназначенного для применения в терапии с целью лечения или профилактики иммунных нарушений или малярии. 2 c. и 6 з.п. ф-лы, 1 табл.
в которой A обозначает группу -CO-NH- или группу -NH-CO-;
R обозначает атом водорода или группу CH3;
*C обозначает, если R не является атомом водорода, асимметрический углерод конфигурации (R,S) или конфигурации (R);
или их соли присоединения.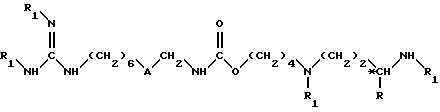
в которой
R обозначает атом водорода или метильную группу;
A обозначает группу -CO-NH- или группу -NH-CO-;
*C обозначает, если R не является атомом водорода, асимметрический углерод конфигурации (R, S) или (R), причем по крайней мере один из заместителей R1 обозначает защитную группу аминофункции оксикарбонильного или бензильного типа, а другие заместители R1, если они имеются, обозначают атом водорода, согласно обработке, соответствующей природе защитной группы аминофункции с получением соединения формулы (I) в виде свободного основания или одной из его солей присоединения; и, если необходимо, полученное свободное основание или его соль присоединения переводят в другую аддитивную соль.
(а) либо (вариант А):
конденсируют спирт формулы III: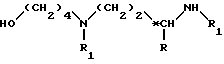
в которой R обозначает атом водорода или метильную группу;
R1 обозначает защитную группу аминофункции
*C обозначает, если R не является атомом водорода, асимметрический углерод конфигурации (R,S) или (R),
с хлорформиатом или симметричным карбонатом в присутствии основания, в инертном растворителе и при комнатной температуре (15 - 25oC);
полученное соединение подвергают взаимодействию с амином формулы IV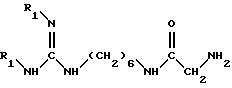
в которой R1 обозначает защитную группу аминофункции,
в инертном растворителе, при температуре около 25 - 50oC, до получения соединения формулы II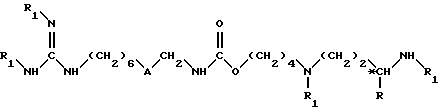
в которой A обозначает группу -NH-CO-;
R обозначает атом водорода или метильную группу;
*C обозначает, если R не является атомом водорода, асимметрический углерод конфигурации (R,S) или (R);
R1 обозначает защитную для аминной функции группу;
(б) либо (вариант В):
кислоту общей формулы V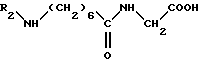
в которой R2 обозначает защитную группу аминофункции, подвергают взаимодействию с дифенилфосфорилазидом формулы VI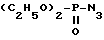
в присутствии основания в растворителе при комнатной температуре для получения промежуточного соединения формулы VII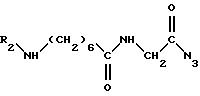
полученное соединение формулы VII подвергают перегруппировке, известной под названием реакции Куртиуса, и одновременно таким образом полученный изоцианат вводят во взаимодействие со спиртом формулы III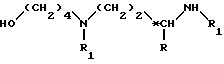
в которой R обозначает атом водорода или метильную группу;
R1 обозначает защитную группу аминофункции, отличную от вышеуказанной группы R2;
*C обозначает, если R не является атомом водорода, асимметрический углерод конфигурации (R,S) или (R); в растворителе, при температуре около 80 - 140oC, в течение 5 - 50 ч, для получения соединения формулы VIII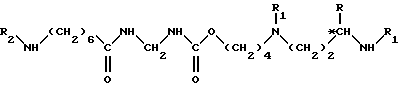
в которой R, R1, R2 и *C имеют вышеуказанные значения;
удаляют защитные группы из полученного соединения формулы VIII согласно специфической обработке замены защитной группы R2 атомом водорода, для получения соединения формулы IX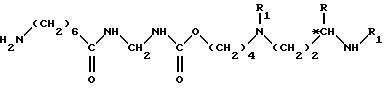
в которой R, R1 и *C имеют вышеуказанные значения;
и затем полученное соединение формулы IX подвергают взаимодействию с аминоиминометансульфокислотой, в растворителе, при комнатной температуре (15 - 25oC) и в течение 8 - 50 ч, до получения соединения формулы II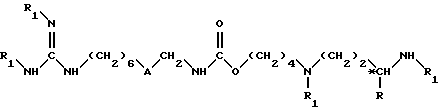
в которой R и *C имеют вышеуказанное значение;
A обозначает группы -CO-NH-;
R1 обозначает защитную группу аминофункции, за исключением двух групп R1, которые находятся на гуанидиновой функции и которые обозначают каждый атом водорода:
или, в качестве варианта, соединение формулы IX подвергают взаимодействию с соединением формулы X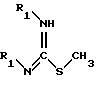
в которой R1 обозначает защитную группу аминофункции в инертном растворителе, в частности в тетрагидрофуране, в присутствии основания, при комнатной температуре (15 - 25oC), в течение 8 - 100 ч, до получения соединения формулы II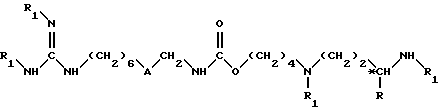
в которой A обозначает группы -CO-NH-;
R обозначает атом водорода или метильную группу;
R1 обозначает защитную для аминной функции группы;
*C обозначает, если R не является атомом водорода, асимметрический углерод конфигурации (R,S) или (R),
и далее удаляют защитные группы как указано в п.3 для получения целевого продукта формулы I.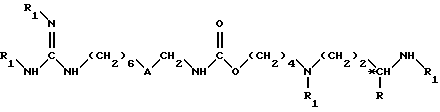
в которой A обозначает группу -CO-NH- или группу -NH-CO-;
R обозначает атом водорода или метильную группу;
*C обозначает, если R не является атомом водорода, асимметрический углерод конфигурации (R,S) или конфигурации (R),
причем по крайней мере один из заместителей R1 обозначает защитную группу аминофункции оксикарбонильного или бензильного типа, а другие заместители R1, если они имеются, обозначают атом водорода.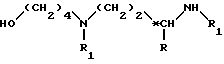
в которой R обозначает метильную группу;
R1 обозначает защитную группу аминофункции;
*C обозначает асимметрический углерод конфигурации (R,S) или (R).
| RU 95102479 A1, 10.05.97 | |||
| Устройство для экспонирования печатных плат | 1977 |
|
SU669316A1 |
| EP 0600762 A1, 1994 | |||
| Прессоточка для круговой затяжки заготовки обуви с предварительно пришитым к ней рантом | 1955 |
|
SU105193A1 |
| СПОСОБ ОБРАБОТКИ ВОДНО-СПИРТОВЫХ РАСТВОРОВ (СОРТИРОВОК) В водочном ПРОИЗВОДСТВЕ | 0 |
|
SU181592A1 |

