Изобретение относится к процессу последовательного получения алкилпирофосфонатов, алкилпирофосфатов и их мультимеров и, в частности, к получению 4-амино-1-гидрокси- бутилиден-1,1-бисфосфоновой кислоты и ее солей, где конечный продукт получается в практически чистой форме и с высокими выходами в непрерывной реакции.
Из патента США N 4407761 (выдан Henkel Kommandit - gesselschaft) известно получение 4-амино-1-гидроксибутилиден-1,1-бисфосфоновой кислоты с использованием фосфонирующих реагентов и последующим гашением реакционной массы добавлением сильного неокисляющего агента, предпочтительно концентрированной хлороводородной кислоты, при нагревании для гидролиза образовавшихся фосфорных промежуточных продуктов с получением конечного продукта. Однако, эта реакция фосфонирования не является гомогенной, потому что происходит гетерогенное отвердевание реакционной смеси. Отвердевание вызывает различие в выходах и приводит к образованию "горячих точек", которое, отчасти, происходит из-за экзотермической природы реакции. Более того, для получения натриевой соли с использованием известных способов требуется выделение 4-амино-1- гидроксибутилиден-1,1-бисфосфоновой кислоты и дополнительная стадия для превращения ее в мононатриевую соль. Кроме того, требуется использование при гашении концентрированной хлороводородной кислоты, пары которой представляют опасность для окружающей среды.
В патенте США N 4922007 (выдан G.R. Kieczykowsky et al., передан Merck & Co. , Inc. ) описано использование метансульфоновой кислоты для преодоления негомогенности и проблем отвердевания, связанных с образованием промежуточных продуктов на стадии бисфосфонирования. Однако, в этом способе используется водное гашение с неконтролируемым pH, что приводит к сильнокислой и коррозионной гидролизной смеси, которая требует специального оборудования.
В патенте США N 5019615 G.R. (выдан Kieczykowsky et al., передан Merck & Co., Inc.) описано использование стадии гашения с контролируемым pH в диапазоне от 4 до 10 с последующим гидролизом, который исключает концентрированную хлороводородную кислоту, образующуюся на стадии гашения, и необходимость работы с коррозионной смесью кислотного гидролиза продукта.
Известные ранее способы требуют проведения реакции при температурах выше точки кипения PCl3 например при 90oC. Однако известно, что данная температура находится в диапазоне адиабатического саморазогрева, что является небезопасным режимом проведения реакции, поскольку объем загрузки увеличивается, а доступная мощность охлаждения уменьшается. Кроме того, важно поддержание стехиометрических соотношений для получения требуемых промежуточных продуктов. Однако, поддержание стехиометрических соотношений при постоянной температуре, обычно 90oC, с использованием уже известных периодических способов невозможно, поскольку стехиометрические количества PCl3 могут добавляться только при температурах ниже точки кипения. Например, в патенте США N 5019651 стехиометрические соотношения достигаются использованием программирования температуры, поэтому стехиометрическое количество PCl3 может быть добавлено при температурах ниже точки кипения. Альтернативно, в патенте США N 4407761 PCl3 медленно добавляют при постоянной температуре реакции выше точки кипения PCl3. Таким образом, желательно одновременно контролировать как стехиометрию, так и температуру реакции для достижения значительного образования требуемых промежуточных продуктов и гарантированно-безопасных условий проведения реакции. Известные периодические способы проведения реакции делают невозможным контроль за стехиометрическими соотношениями при постоянной температуре.
Настоящее изобретение решает обе эти проблемы путем проведения реакции в емкостном реакторе с непрерывным перемешиванием, что позволяет увеличить теплоперенос для контроля за температурой при поддержании постоянных стехиометрических соотношений реагентов. Более благоприятное соотношение поверхности и объема в настоящем изобретении позволяет увеличить теплоперенос для контроля за температурой. Далее, устойчивое непрерывное перемешивание приводит к фиксированным соотношениям продуктов и промежуточных соединений в небольшом контролируемом объеме путем поддержания как температуры, так и стехиометрического соотношения в течение всего времени. Уменьшение количества реакционной массы уменьшает последствия неожиданного перегрева и позволяет гасить всю реакционную массу.
Настоящее изобретение относится к способу непрерывного получения соединений структурной формулы I:
Z-R1, (I)
где Z выбран из группы, состоящей из
а) H2N-C2-5алкил-,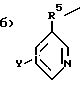
где R5 - это C1-5алкил, и
Y выбран из
(I) водород,
(II) C1-5алкил,
(III) R6O,
(IV) R6S,
(V) R6R6N,
(VI) галоген,
R6 не является H или C1-5алкил; и
в) C2-6алкил-(N-CH3)C2H4-; и
R1 выбран из группы, состоящей из: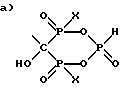
и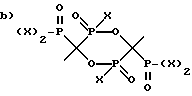
где X - это OH или Cl.
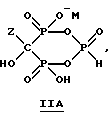
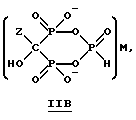
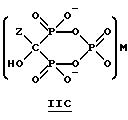
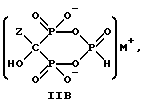
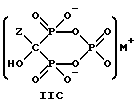
Данное изобретение также относится к способу непрерывного получения промежуточных продуктов формулы IIA, IIB и IIC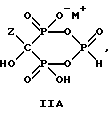
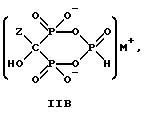
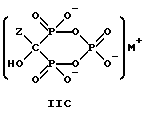
где Z определен выше, и
M - это одновалентный, двухвалентный или трехвалентный катион, такой как Na+, K+, Ca2+, Mg2+.
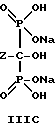
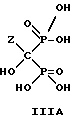
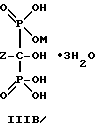
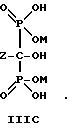
Следует отметить, что все ионные формы этих промежуточных соединений входят в объем данного изобретения. Далее, данное изобретение включает способ непрерывного получения соединений формулы IIIA, IIIB и IIIC.
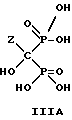
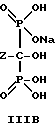
который включает:
а) непрерывное смещение - аминоалканкарбоновой кислоты формулы
Z-COOH,
где Z определен выше с H3PO3 и PCl3 в метансульфоновой кислоте (МСК) или необязательно PCl3 в МСК; и
б) непрерывное прибавление водного основания к переливаемой смеси, содержащей соединение формулы I для получения соединений формулы II; и
в) гидролиз переливаемой смеси, содержащей соединения формулы II для получения соединений формулы III; и
г) выделение продуктов формулы III и их солей.
Отметим, что все возможные гидратированные формы включаются в данное изобретение. Для соединений формулы IIIB тригидрат является предпочтительной формой.
Предпочтительная форма соединений описывается формулой Ia.
Z-R1,
где Z - это группа а) H2N-C2-5алкил.
Предпочтительные промежуточные соединения формулы IIa включают в себя соединения формул IIa(i) и Ia(ii):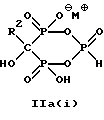
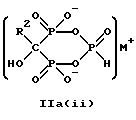
где R2 - это C2-5алкил, замещенный концевым амином или протонированным концевым амином.
Данное изобретение предпочтительно включает способ непрерывного получения соединений формулы IIIa(i), IIIa(ii) и IIIa(iii),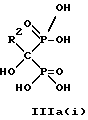
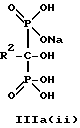
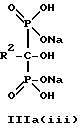
где R2 - это C2-5алкил, замещенный концевым амином, соединения могут находится в любом гидратированном состоянии, или протонированным концевым амином;
и указанный способ включает:
а) непрерывное смешивание аминоалканкарбоновой кислоты формулы
H2N-C2-5алкил-COOH
с H3PO3 и PCl3 в метансульфоновой кислоте (МСК) или необязательно PCl3 в МСК; и
б) непрерывное прибавление водного основания к переливаемой смеси, содержащей соединение формулы Ia для получения соединений формулы IIa; и
в) гидролиз подаваемой смеси, содержащей соединения формулы IIa, для получения соединений формулы IlIa; и
г) выделение продуктов формулы IlIa и их солей.
Настоящее изобретение относится к соединениям структурной формулы I:
Z-R1, (I)
где Z выбран из группы, состоящей из:
a) H2N-C2-5алкил-,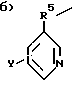
где R5 - это C1-5алкил, и
Y выбран из
(I) водород,
(II) C1-5алкил,
(III) R6O,
(IV) R6S,
(V) R6R6N,
(VI) галоген;
R6 представляет собой H или C1-5алкил; и
в) C2-6алкил-(N-CH3)C2H4-; и
R1 - выбран из группы, состоящей из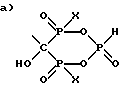
и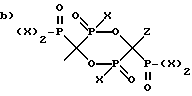
где X - это OH или Cl.
Настоящее изобретение относится также к способу получения указанных соединений и их бисфосфонатных продуктов, включая 4- амино-1-гидрокси-бутилиден-1,1-бисфосфоновую кислоту (АБФ) и ее соли. Конкретно, этот способ может состоять из пяти операций: непрерывной реакции бисфосфонирования, непрерывного или периодического гашения с контролем pH, непрерывного или периодического гидролиза, кристаллизации неочищенного продукта и кристаллизации чистого продукта.
Более конкретно, непрерывная реакция бисфосфонирования состоит из подготовки загрузки карбоновой кислоты и взаимодействия этой загрузки с PCl3 в емкостном реакторе с непрерывным перемешиванием.
Загрузку карбоновой кислоты готовят растворением твердой карбоновой кислоты и твердой фосфорноватой кислоты (H3PO3) в метансульфоновой кислоте (МСК). Обычно, на 1 моль карбоновой кислоты используют от 1 до 3, предпочтительно 2 моля, H3PO3, и от 6,3 до 6,4, предпочтительно 6,38 моля, МСК. Для облегчения полного растворения твердых компонентов в жидкой МСК смесь может быть нагрета до температуры от 40 до 90oC, предпочтительно - до 70oC. После растворения твердых компонентов загрузки карбоновой кислоты, эту загрузку можно сохранять при температуре от 10 до 90oC, предпочтительно - при 70oC, используя внешний источник тепла. Альтернативно, добавление H3PO3 может быть исключено из приготовления загрузки карбоновой кислоты. Если выбрана альтернативная методика, то H3PO3 может образовываться in situ из PCl3 в метансульфоновой кислоте (МСК), PCl3 и γ -аминобутановой кислоты (ГАБК) в МСК или H2O в МСК.
Загрузку карбоновой кислоты подают в холодный реакционный сосуд до отметки ниже уровня перелива. Во время заполнения реактора нагревающую среду подают в рубашку и включают перемешивающее устройство сосуда. Температуру доводят до значений от 45 до 100oC, предпочтительно 90oC. Затем вводят в реактор загрузку жидкого PCl3 до тех пор, пока вес PCl3, введенного в реактор (учитывая потери на испарение), деленный на вес загрузки карбоновой кислоты, не будет от 0,22 до 0,33, предпочтительно 0,32. В этот момент загрузку карбоновой кислоты возобновляют при скорости потока, достаточной для обеспечения времени пребывания в реакторе от 1,5 до 2,5 часов, предпочтительно 1,8 часа. Время пребывания выражается как объем реактора в условиях перелива, деленный на скорость потока (объем/мин) подаваемой карбоновой кислоты. Вскоре после возобновления загрузки карбоновой кислоты реакционную массу переливают в сосуд для гашения, который сначала наполняют либо водой, либо разбавленным водным основанием. Карбоновую кислоту и жидкий PCl3 добавляют одновременно при соответствующих скоростях их потоков до тех пор, пока не образуется требуемое количество вещества.
Три времени пребывания обязательны для наступления установившегося режима реакции бисфосфонирования. Известные периодические процессы приводят к неконтролируемому образованию нежелательных промежуточных продуктов. Настоящее изобретение преодолевает эту проблему путем контроля стехиометрии компонентов реакции, минимизирующего образование нежелательных промежуточных продуктов.
Подаваемую реакционную массу нейтрализуют в присоединенном сосуде для гашения добавлением водного основания. Водным основанием может служить любое водное основание формулы MOH, такое как гидроксид натрия, или формулы MHCO3 или MCO3, такие как карбонат натрия или бикарбонат натрия, где M - это любой ион. Отдельно деионизированная вода (ДВ) и загрузки основания используются для поддержания эффективной концентрации основания в гасящем растворе от 15 до 50%, предпочтительно 20%. Водное основание добавляют для поддержания pH, в ответ на колебания pH гасящего раствора. Значение pH в сосуде для гашения поддерживается от 4,0 до 7,0, преимущественно около 5,0. Температура гасимой смеси поддерживается от 0 до 100oC, преимущественно < 50oC.
Смесь бисфосфонирования дает соединение формулы I.
Z-R1, (I)
где Z выбран из группы, состоящей из:
а) H2N-C2-5алкил-,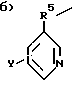
где R5 - это C1-5алкил, и
Y выбран из:
(I) водород,
(II) C1-5алкил,
(III) R6O,
(IV) R6S,
(V) R6R6N,
(VI) галоген,
R6 не является H или C1-5алкил; и
в) C2-6алкил-(N-CH3)C2H4-; и
R1 выбран из группы, состоящей из: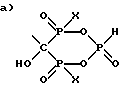
и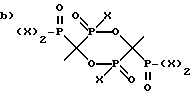
где X - это OH или Cl.
Желательно соединение формулы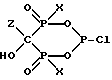
также получать гашением. Предпочтительными соединениями формулы Z-R1 являются те, где Z - это H2N-C2-5алкил-, Z - это предпочтительно, C4алкил, и полученное соединение может использоваться как промежуточное для получения алендроната (тригидрата натриевой соли 4-амино-1-гидроксибутилиден-1,1-бисфосфоновой кислоты).
Для соединений, где Z= б) предпочтительно соединение, в котором R5 - это CH2; полученное соединение может использоваться как промежуточное в производстве ризедроната (1-гидрокси-2-(3-пиридинил)этилиден бисфосфоновой кислоты).
Для соединений, где Z= в), предпочтительно соединение, в котором Z= C4алкил-(N-CH3)C2H4-. Это соединение может использоваться как промежуточное в производстве соединения, обозначаемого ВМ210955 (1-гидрокси-3-(метилпентиламино)пропилиденбисфосфоната).
Данная реакция и/или сама бисфосфонирующая смесь проявляют значительные экзотермические характеристики. Поэтому следует предпринять существенные меры безопасности для обеспечения нормального протекания процесса. С этой точки зрения для заданной производительности меньший объем в непрерывной реакции обеспечивает скорейшее гашение в случае разгона реакции, чем периодическая система равной производительности. Сосуд, в который в нормальном режиме поступает реакционная масса реакции бисфосфонирования, также применяется для быстрого гашения. Минимальный объем быстрого гашения примерно в два раза больше реакционного объема реакционного сосуда. Это позволяет, в случае нежелательного перегрева, быстро погасить весь реакционный объем.
Соединение формулы IIA, IIB и IIC: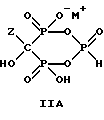
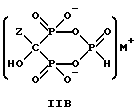
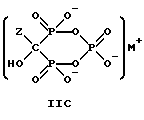
где Z определен выше, и, предпочтительно, является C2-5алкилом, замещенным концевым амином или протонированным концевым амином;
M+ - это катион одновалентного металла или двухвалентного металла, такой как Na+, K+, Ca2+, Mg2+,
могут накапливаться или непрерывно удаляться из сосуда для гашения посредством перетока в новый реактор для гидролиза. Следует отметить, что другие анионные формы соединений формулы II, например трехионная, образуются в соответствующих условиях pH: (структуры во всем данном описании следует понимать как включающие все возможные ионные формы в зависимости от pH среды). Значение pH гашеного материала проверяется и устанавливается, при необходимости, в интервале от 3,3 до 12,6, преимущественно от 4,6 до 5,0. Реакционную массу нагревают в тонкостенном сосуде, изготовленном из пирекса (PYREXTM), или в случае возможности разрушения сосуда - в аппарате, покрытом изнутри HastalloyTM C-276, примерно до 100-175oC, предпочтительно до 140oC, при 4,22 кг/см2 и выдерживают около 20 часов до разложения соединений IIA и IIB с получением соединения IIIC.
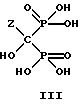
где Z определен выше, и предпочтительно является C2-5алкилом, замещенным концевым амином,
и его солей, в частности, мононатриевой или динатриевой солей.
Затем реакционную массу охлаждают до 85oC и берут пробу для подтверждения pH и завершения гидролиза. Однако, гидролиз пирофосфоната может проводиться и при комнатной температуре, и выделение целевого конечного продукта является возможным. Объем реакционной загрузки может устанавливаться до или после гидролиза как упариванием, так и добавлением воды. Чистые маточные растворы могут быть возвращены в реакцию до гидролиза, а избыточный объем - удален упариванием для обеспечения выполнения общих требований при первичной кристаллизации твердых веществ.
Значение pH теплого раствора при необходимости корректируется добавлением подходящей кислоты или основания. После установления pH при 85oC, в гидролизованную реакционную массу можно вносить затравки в виде неочищенного или чистого соединения формулы III или его формы моно- или ди-соли, которые могут присутствовать при соответствующих pH.
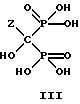
Реакционную массу охлаждают до 0-25oC. Раствор выдерживают более б часов и отфильтровывают кристаллы. Вещество на фильтре можно промыть холодной деионизированной водой. Неочищенный осадок с фильтра может быть высушен или непосредственно подан на стадию очистки.
Неочищенный влажный продукт и деионизированную воду подают в сосуд, в котором проводится очистка. Температуру в сосуде поднимают до 40-100oC, предпочтительно до 50oC, и смесь выдерживают до завершения растворения. Выделение конечного продукта зависит от pH в интервале от 3,0 до 12,0. Предпочтительно устанавливают pH 4,3 для получения моно-соли. Полученную реакционную массу фильтруют и затем концентрируют упариванием. Полученную массу охлаждают до 0-5oC и выдерживают более 2 часов. Охлажденную массу фильтруют и влажный осадок промывают на фильтре холодной деионизированной водой (0-5oC) и затем высушивают в вакууме. Данным способом получают соединение формулы III.
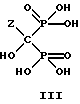
где Z определен выше, и, предпочтительно, является C2-5алкилом, замещенным концевым амином;
и его соли, в частности мононатриевую или динатриевую соли.
Реакция представлена нижеследующей схемой для случая, если основанием является NaOH: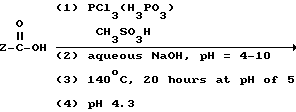
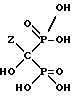
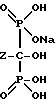
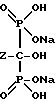
где Z определен выше, и предпочтительно является C2-5алкилом, замещенным концевым амином.
Частным примером данной реакции, если Z представляет собой H2N-CH2-CH2-CH2, является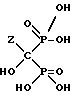
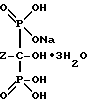
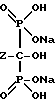
Описанные здесь бисфосфоновые кислоты находят применение благодаря своей способности изолировать ионы поливалентных металлов и комплексообразованию с ионами щелочноземельных металлов, предпочтительно кальция. Поэтому, замещенные бисфосфоновые кислоты могут использоваться для смягчения воды, водоочистки и при приготовлении низкотоксичных фармакологических препаратов.
В частности, описанный здесь тригидрат мононатриевой соли 4-амино-1-гидроксибутилиден-1,1-бисфосфоновой кислоты используется в фармацевтической композиции для лечения или профилактики болезней, включающих костную резорбцию. Такие болезни, как злокачественная гиперкальцемия, болезнь Пагета (Paget) и остеопороз успешно лечатся тригидратом мононатриевой соли 4-амино-1-гидроксибутилиден-1,1-бисфосфоновой кислоты, полученным способом по данному изобретению.
Другие фармацевтически приемлемые соли, такие как, например, калиевые соли, могут быть получены в соответствии со способом по настоящему изобретению и входят в объем изобретения. Другими бисфосфонатами, которые могут быть получены по данному непрерывному способу, являются:
(а) 2-амино-1-гидроксиизо-бутилиден-1,1-бисфосфоновая кислота,
(б) 3-амино-1- гидроксипропилиден-1,1-бисфосфоновая кислота,
(в) 5-амино-1- гидроксипентилиден-1,1-бисфосфоновая кислота,
(г) 6-амино-1- гидроксигексилиден-1,1-бисфосфоновая кислота,
(д) ризедронат (1-гидрокси-2-(3-пиридинил)этилен-1,1-бисфосфоновая кислота,
(е) BM 210955 N-бутил-N-метил-3-амино-1-гидроксипропилиден- 1,1-бисфосфоновая кислота.
Нижеследующие примеры иллюстрируют изобретение, не ограничивая его.
Пример 1. Непрерывное производство 4-амино-1- гидроксибутилиден-1,1-бисфосфоновой кислоты
В реакционную колбу загружают 2,6 кг МСК. При перемешивании в колбу прибавляют 0,545 кг ГАБК, после чего загружают 0,865 кг H3PO3. В дальнейшем смесь МСК, ГАБК и H3PO3 именуется загрузкой ГАБК. Во время растворения смесь выдерживают при 70oC. Оставшиеся 0,645 кг МСК добавляют в качестве промывной жидкости и перемешивают раствор при 70oC до растворения ГАБК и H3PO3.
Реактор для бисфосфонирования снабжен рубашкой и механической мешалкой, вводными штуцерами, датчиком температуры, обратным холодильником и нижним спуском. При разработке реактора использовалась стандартная конфигурация аппарата для гидрирования. Аппарат включает четыре полуперегородки, отходящие от дна реактора и расположенные под углом 90o друг к другу. Турбинная мешалка Раштона (Rushton) расположена внизу вала мешалки. Над мешалкой Раштона расположена пропеллерная мешалка, также прикрепленная к валу мешалки. Пропеллерная мешалка имеет больший диаметр, чем турбинная мешалка Раштона. Рубашка, окружающая реактор, расположена ниже смоченной стенки. Нагреватель, используемый для обогрева среды в рубашке, установлен на 97-105oC, в зависимости от требований реакционной массы к тепловой нагрузке для поддержания температуры в 90oC. Конденсатор и среда установлены для достижения температуры отходящих газов -10oC.
До достижения установившегося режима непрерывной реакции бисфосфонирования используется полупериодический режим. Нагреватель реактора устанавливают на 97oC для поддержания температуры реакционной массы 90oC. Циркуляцию в рубашке реактора не производят до загрузки в реактор ГАБК. Температуру нагревателя непрерывно регулируют, как это требуется для поддержания температуры загрузки 90oC. Резервуар с PCl3 наполняют и, если требуется, наполняют повторно. Также резервуар с загрузкой ГАБК наполняют и, если требуется, наполняют повторно. Реакционный сосуд заполняют 400 мл нагретого ГАБК. Одновременно продолжают перемешивание и циркуляцию теплоносителя в рубашке реактора. Загрузку ГАБК в реакторе нагревают до 90oC. 50 мл загрузки ГАБК сливают из реактора через дренаж. PCl3 вводят в реактор со скоростью 0,95 мл/мин. Через 95 минут загрузку ГАБК вводят со скоростью 3,7 мл/мин. Это время соответствует 90 мл PCl3, поданным в реактор, и соотношению PCl3/ГАБК 0,33 г/г. На этой стадии полупериодический режим завершается и устанавливается режим непрерывного производства.
Подачу PCl3 и ГАБК продолжают при 0,95 мл/мин и 3,7 мл/мин, соответственно, для требуемого времени превращения. Скорости потоков выбраны, чтобы обеспечить время пребывания 1,8 часа на основе скорости потока подачи ГАБК. В течение всего процесса из реактора осуществляется перелив в сосуд для гашения. Выход промежуточных продуктов, которые образуются после гидролиза и могут быть выделены, составляет 60-72%, как правило - 70%, в установившемся режиме. Это на 10% больше, чем выход, ожидаемый от прямой замены периодического способа непрерывным.
Количество требуемого материала является лимитирующим фактором продолжительности операции. В конце операции загрузки PCl3 и ГАБК извлекают. Реактор опорожняется, поскольку PCl3 более не возвращается в него.
Непрерывное гашение производится в цилиндрическом реакционном сосуде объемом 500 мл, снабженном рубашкой, переточной трубой и перемешивающим механизмом с тефлоновой лопастной мешалкой. Датчик pH калиброван по буферным растворам с pH 4,0 и 7,0. Нижнее значение установлено при 5,0. Заполняют резервуар 47% NaOH. Заполняют резервуар деионизированной водой (ДВ) или чистым маточным раствором. Во время полупериодического режима устанавливают скорость потока водного NaOH 12,2 мл/мин. Устанавливают скорость потока ДВ или чистого маточного раствора 18,75 мл/мин. В сосуд для гашения подают начальную загрузку 700 мл ДВ. Как только реакционная масса подается в сосуд для гашения, устанавливают значение pH 5,0 включением насоса NaOH через pH- метр. По достижении достаточной загрузки реакицонной смеси и NaOH, что приводит к общей концентрации твердых веществ >550 г/л, включают насос ДВ или чистого маточного раствора. В это время масса из сосуда для гашения переливается через переливную трубу и полупериодический период завершается.
В непрерывном режиме сосуд гашения работает с pH-метром и перетоком до сбора требуемой массы продукта. По завершении реакции pH в сосуде для гашения контролируется до погашения всей массы. Через 20 минут после завершения гашения массы насосы и pH-метр выключают, а сосуд для гашения опорожняют.
Получают соединение формулы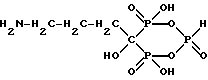
имеющее следующие характеристики:
а) Молекулярная масса 295;
б) ЯМР 31P при 161,98 МГц с H3PO4 ( δ 0,00) в качестве внешнего стандарта: δ 3,8 (т, Jрр = 13,5, Jpн = 669,4), δ 15,9 (д, Jрр = 13,5);
в) ЯМР 13C при 100,61 МГц с диоксаном ( δ 67,4) в качестве внешнего стандарта: δ 83,2 (тд, Jср = 134,9, 10,4), δ 41,2, δ 31,8 (д, Jср = 3,2), δ 23,8 (д, Jср = 6,4).
Аналогично получают соединение формулы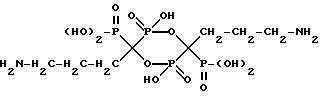
имеющее следующие характеристики:
а) Молекулярная масса 462;
б) ЯМР 31P при 161,98 МГц с H3PO4 ( δ 0,00) в качестве внешнего стандарта: δ 12,9 (т, Jрр = 17,1), 8,0 (т, Jрр = 17,1);
в) ЯМР 13С при 100,61 МГц с диоксаном ( δ 67,4) в качестве внешнего стандарта: δ 86,4 (ддд, Jср = 139,7, 129,3, 15,3), δ 41,0, δ 33,3, δ 23,0 (м).
Гидролиз проводят в толстостенной бутыли Ace glass с защитным покрытием объемом 250 мл, снабженной магнитной мешалкой с якорем в тефлоновой оболочке, усовершенствованной тефлоновой крышкой с термопарой в тефлоновой изоляции для контроля температуры. Сосуд нагревают на силиконовой бане. Загружают в сосуд для гидролиза 200 мл массы со стадии гашения, Измеряют pH массы и соответствующим образом устанавливают в пределах от 4,6 до 5,5. Содержимое сосуда для гидролиза нагревают до 140oC. После достижения требуемой температуры гидролиз проводят в течение 20 часов при 140oC. После выдержки содержимому сосуда дают охладиться до 85oC, проверяют значение pH и устанавливают pH 4,3 добавлением 50% NaOH или HCl (37%).
Первичную кристаллизацию проводят в трехгорлой круглодонной колбе объемом 250 мл, снабженной тефлоновой мешалкой. При перемешивании в трехгорлую колбу объемом 250 мл загружают при температуре 85oC 200 мл раствора из сосуда для гидролиза. Измеряют значение pH и устанавливают соответствующим образом. Однако, если pH ниже 4,0, раствор выгружают и проводят гидролиз заново. Раствору дают охладиться до 20-25oC; в это время вещество кристаллизуется. Осадок выдерживают в течение > 15 часов при комнатной температуре при перемешивании и отфильтровывают под вакуумом. Кристаллы промывают 2х15 мл ледяной ДВ. Продукт оставляют сушиться на ночь под вакуумом при 45-50oC.
Очистку проводят в трехгорлой круглодонной колбе объемом 250 мл, снабженной тефлоновой мешалкой. Высушенный неочищенный продукт массой 10 г загружают в трехгорлую колбу. В колбу приливают 150 мл ДВ. Колбу нагревают до 50oC и выдерживают при этой температуре до растворения твердого вещества. Прекращают нагревание и содержимое фильтруют под вакуумом. Фильтрат помещают в трехгорлую колбу и перегоняют под атмосферным давлением до объема остатка 44 мл. Прекращают нагревание и дают колбе остыть до комнатной температуры. Содержимое колбы выдерживают в течение 2 часов. Осадок охлаждают до 0-5oC, выдерживают 2 часа и отфильтровывают под вакуумом. Кристаллы промывают 2х15 мл воды при температуре 0-5oC.
Пример 2. Непрерывное получение (а) 2-амино-1-гидроксиизо- бутилиден-1,1-бисфосфоновой кислоты, (б) 3-амино-1-гидроксипропилиден- 1,1-бисфосфоновой кислоты, (в) 5-амино-1- гидроксипентилиден-1,1-бисфосфоновой кислоты, (г) 6-амино-1- гидроксигексилиден-1,1-бисфосфоновой кислоты
Используя соответствующие аминокарбоновые кислоты в количествах, эквивалентных по отношению к 4-аминобутановой кислоте, можно получить указанные в заголовке бисфосфоновые кислоты, пользуясь способом примера 1. Соответствующие аминокарбоновые кислоты включают, но не ограничиваются:
2-аминоизобутановую кислоту,
3-аминопропионовую кислоту,
5-аминовалериановую кислоту и
6-аминокапроновую кислоту.
Пример 3. Непрерывное получение ризедроната (а) и BM 210955 (б)
Используя соответствующие исходные материалы, можно получить указанные в заголовке соединения, пользуясь способом примера 1. Исходные материалы включают, но не ограничиваются:
3-пиридилуксусную кислоту,
N-бутил-N-метил-3-амино-пропионовую кислоту.
Изобретение относится к циклическим фосфорсодержащим соединениям ф-лы Z-R1 (I), где Z выбран из группы состоящей из: а) H2N-C2-5алкилен, б)пиридил-3-С1-5 алкилен, в) С2-6 алкил (N-CH3)C2H4; R1 выбран из структур (а) и (в), где Х представляет ОН и Cl, которые являются промежуточными продуктами для получения γ-амино-1-гидроксиалкилиден-1,1-бисфосфоновых кислот формул IIIA, IIIB и IIIC, где Z имеет вышеуказанные значения, М - катион основания. Способ получения указанных соединений включает: а) непрерывное смешивание аминоалканкарбоновой к-ты ф-лы Z-COOH с H3PO3 и PCl в метансульфокислоте (МСК) или, необязательно, с PCl3 в МСК с получением и выделением соединений ф-лы Z-R1 (I), б) непрерывное прибавление водного основания с получением соединений ф-лы IIA, IIB или IIC, в) гидролиз вышеуказанной смеси с получением соединений ф-лы IIIA, IIIB или IIIC в практически чистой форме и с высоким выходом в непрерывном процессе. 3 с. и 20 з.п.ф-лы.
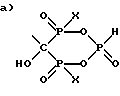
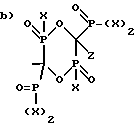
Z-R1 (1),
где Z выбран из группы, состоящей из: а) H2N-C2-5алкилен б)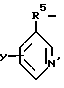
где R5 представляет C1-5алкилен;
Y выбран из группы: водород, C1-5алкил, R6O, R6S, R6R6N, галоген;
R6 представляет водород или C1-5алкил,
в) C2-6алкил-(N-CH3)C2H4 и R1 выбран из группы, состоящей из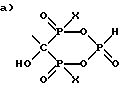
и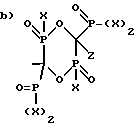 ,
,
где X представляет OH или Cl.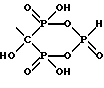
и характеризующееся: а) молекулярной массой 295; б) ЯМР 31Р при 161,98 МГц с H3PO4 (δ 0,00) в качестве внешнего стандарта: δ 3,8 (т, Jрр = 13.5, Jрн = 669,4), δ 15,9 (д, Jрр = 13,5); в) ЯМР 13C при 100,61 МГц с диоксаном (δ 67,4) в качестве внешнего стандарта: δ 83,2 (тд, Jсp = 134.9, 10.4), δ 41.2, δ 31.8 (д, Jср = 3,2), δ 23,8 (д, Jср = 6,4).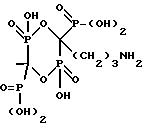
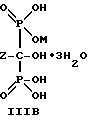
и характеризующееся: а) молекулярной массой 462; б) ЯМР 31Р при 161,98 МГц с H3PO4 (δ 0,00) в качестве внешнего стандарта: δ 12,9 (т, Jрр = 17,1), 8,0 (т, Jрр = 17,1); в) ЯМР 13C при 100,61 МГц с диоксаном (δ 67,4) в качестве внешнего стандарта: δ 86,4 (ддд, Jср = 139.7, 129.3, 15.3), δ 41.0, δ 33.3, δ 23.0 (м).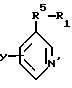
где R5 представляет C1-5алкилен;
R1 и Y имеют значения, указанные в п.1.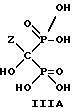
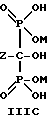 ,
,
где Z выбран из группы, состоящей из: а) H2N-C2-5алкилен, б)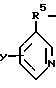
где R5 представляет C1-5алкилен;
Y выбран из группы: водород, C1-5алкил, R6O, R6S, R6R6N, галоген;
R6 представляет водород или C1-5алкил,
в) C2-6алкил-(N-CH3)C2H4,
М представляет одновалентный, двухвалентный или трехвалентный катион основания, включающий: а) непрерывное смешивание аминоалканкарбоновой кислоты формулы
Z-COOH,
где Z имеет значения, определенные выше, с H3PO3 и PCl3 в метансульфоновой кислоте (МСК) или, необязательно, с PCl3 в МСК с получением и выделением соединений формулы 1
Z-R1 (1),
где R1 выбран из группы, состоящей из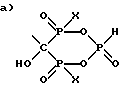
и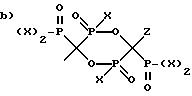
где X представляет OH или Cl,
б) непрерывное прибавление водного основания к соединению формулы 1 с получением соединений формулы IIA, IIB или IIC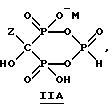
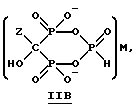
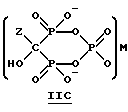
где Z и M имеют вышеуказанные значения,
в) гидролиз подаваемой смеси, содержащей соединения формулы IIA, IIB или IIC, с получением целевых соединений формулы IIIA, IIIB или IIIC.
H2N-C2-5алкилен-R1;
где R1 выбран из группы, состоящей из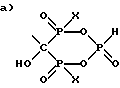
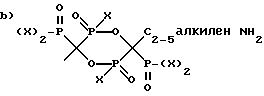
X представляет OH или Cl,
включающий а) непрерывное смешение аминоалканкарбоновой кислоты формулы H2N-C2-5алкилен - COOH с H3PO3 и PCl3 в метансульфоновой кислоте (МСК), или необязательно, с PCl3 в MCK и б) непрерывное удаление смеси, содержащей соединение формулы I.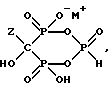
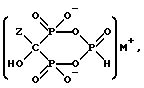
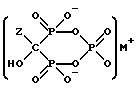
где Z представляет C2-5алкил, замещенный концевым амином или протонированным амином;
M представляет одновалентный, двухвалентный или трехвалентный катион основания, б) непрерывное удаление смеси, содержащей соединение формулы IIA, IIB или IIC.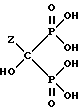
или его соли.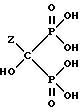
и их солей,
где Z представляет C2-5 алкил, замещенный концевым амином,
который включает: а) непрерывное смешение аминоалканкарбоновой кислоты формулы H2N-C2-5алкилен - COOH с H3PO3 и PCl3 в метансульфоновой кислоте (МСК) или, необязательно, с PCl3 в MCK и б) непрерывное прибавление водного основания к подаваемому соединению формулы I
H2N-C2-5алкилен-R1,
где R1 выбран из группы, состоящей из :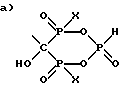
и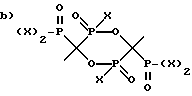
где X представляет OH или Cl,
с получением соединения формулы II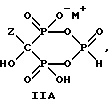
где Z имеет вышеуказанные значения;
M представляет одновалентный, двухвалентный или трехвалентный катион основания,
и в) гидролиз подаваемой смеси, содержащей соединения формулы II, с получением соединений формулы III.
Приоритет по признакам
25.08.93 при Z-H2N-C2-5алкилен;
04.08.94 при Z-пиридил-3-C1-5алкилен и C2-6алкил-(N-CH3)C2H4.
| US 5019651 A, 28.05.1991 | |||
| US 4922007 A, 01.05.1990 | |||
| Способ получения производных дифосфоновой кислоты или их натриевых солей | 1985 |
|
SU1475487A3 |
| Способ получения @ -аминозамещенных- @ -оксипропилидендифосфоновых кислот | 1981 |
|
SU1002300A1 |
| ЛЕНТОЧНЫЙ ЭЛЕКТРОМАГНИТНЫЙ СЕПАРАТОР ПЕРЕМЕННОГО ТОКА | 1933 |
|
SU39033A1 |
| ПАССИВНЫЙ УСПОКОИТЕЛЬ КАЧКИ СУДНА | 1966 |
|
SU216465A1 |

