Изобретение относится к хинолинонам и хинолинтионам, композициям, содержащим их, и способам получения этих соединений. Оно относится далее к использованию их в качестве лекарственного средства, в частности, их использованию в качестве лекарственного средства для лечения нарушений кератинизации.
В Европейском патенте описываются (1H-азол-1-илметил)замещенные производные хинолина и хинолинона, которые подавляют удаление ретиноевых кислот из плазмы. Некоторые из этих соединений способны также ингибировать образование андрогенов из прогестинов и/или ингибировать действие ароматазного ферментного комплекса.
Выбранная группа (1H-триазол-1-илметил)замещенных производных хинолинонов и хинолинтионов, которые неизменно замещены 3-(трифторметил)фенильной частью, представляет предмет данного изобретения. Неожиданное превосходство этой выбранной группы соединений над наиболее близкими, известными в данной области соединениями, лежит в их повышенной способности подавлять эффекты кератинизации.
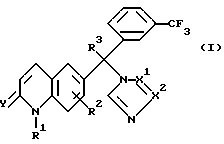
Следовательно, данное изобретение относится к соединениям формулы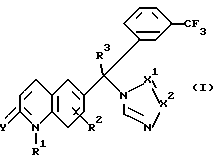
их фармацевтически приемлемым кислотно-аддитивным солям и стереохимически изомерным формам, где:
R1 представляет водород, амино или C1-4 алкил;
R2 представляет водород, галоген или C1-4 алкил;
R3 представляет водород, галоген или C1-4 алкил;
Y представляет O или S и
-X1 = X2 - представляет двухвалентный радикал, имеющий формулу
-N=CH- (а-1)
или
-CH=N- (а-2)
Используемый в предшествующих определениях термин галоген является родовым для фтора, хлора, брома и иода. Термин C1-4 алкил определяет нормальные или разветвленные насыщенные углеводороды, имеющие от 1 до 4 атомов углерода, такие, как, например, метил, этил, пропил, бутил, 1-метилэтил, 1,1-диметилэтил, 2-метилпропил и подобные.
Имеется в виду, что фармацевтически приемлемые кислотно-аддитивные соли, упоминаемые здесь выше, содержат формы кислотно-аддитивных солей, которые можно удобно получить обработкой формы основания соединений формулы (I) подходящими кислотами, такими, как неорганические кислоты, например, галогеноводородная кислота, например, хлористоводородная или бромистоводородная, серная, азотная, фосфорная и подобные кислоты, или органические кислоты, такие, как, например, уксусная, гидроксиуксусная, пропановая, молочная, пировиноградная, щавелевая, малоновая, янтарная, малеиновая, фумаровая, яблочная, винная, лимонная, метансульфокислота, этансульфокислота, бензолсульфокислота, n-толуолсульфокислота, цикламиновая, салициловая, n-аминосалициловая, памоевая и подобные кислоты. Наоборот, эти формы кислотно-аддитивных солей можно превратить в формы свободных оснований обработкой подходящим основанием.
Термин кислотно-аддитивная соль содержит также гидраты и аддитивные формы с растворителем, которые способны образовывать соединения формулы (I). Примерами таких форм являются, например, гидраты, алкоголяты и подобные формы.
Используемый здесь термин стереохимически изомерные формы обозначает все возможные изомерные формы, в которых соединения формулы (I) могут существовать. Если не упоминается или указывается особо, химическое обозначение соединений указывает на смесь всех возможных стереохимически изомерных форм, причем эти смеси содержат все диастереомеры и энантиомеры основной молекулярной структуры. В частности, соединения формулы (I) и некоторые промежуточные продукты, указываемые в дальнейшем, имеют в их структуре по меньшей мере один стереогенный центр. Этот стереогенный центр может присутствовать в R- и S-конфигурации, причем это R- и S-обозначение используют в соответствии с правилами, описанными в Pure Appl. Chem., 1976, 45, 11-30.
Некоторые соединения формулы (I) могут существовать также в их таутомерных формах. Предполагается, что такие формы, хотя они точно не указываются в приведенной выше формуле, включены в объем данного изобретения. Например, соединения формулы (I), где R1 представляет водород, могут существовать в их таутомерной форме.
Подразумевается, что всякий раз, когда его используют здесь позднее, термин соединения формулы (I) включает также фармацевтически приемлемые кислотно-аддитивные соли и все стереоизомерные формы.
Конкретными соединениями данного изобретения являются те соединения формулы (I), где -X1=X2 - представляет двухвалентный радикал формулы (a-1).
Другими конкретными соединениями являются те соединения формулы (I), где R2 замещает (являясь заместителем) положение 5 или 8 хинолиноновой или хинолинтионовой части.
Особенно интересными являются те соединения, у которых -X1=X2- представляет двухвалентный радикал формулы (a-1) и Y представляет O.
Особенно интересными являются также те соединения, у которых Y представляет S и R2 представляет водород.
Далее интересными соединениями являются те соединения формулы (I), у которых R1, R2 и R3 являются водородом.
Другой группой интересных соединений являются те соединения формулы (I), форма основания которых имеет R-конфигурацию.
Предпочтительными соединениями являются те соединения формулы (I), где -X1= X2- представляет двухвалентный радикал формулы (a-1), R1 представляет водород, амино или метил, R2 представляет водород и R3 представляет водород, галоген, метил или этил.
Более предпочтительными соединениями являются те предпочтительные соединения, у которых Y представляет О, R1 представляет водород или метил и R3 представляет водород, метил или этил.
Еще более предпочтительными соединениями являются 6-[1H- 1,2,4-триазол-1-ил-[3-(трифторметил)фенил] метил]-2(1H)-хинолинон, его фармацевтически приемлемые кислотно-аддитивные соли и стереохимически изомерные формы.
Наиболее предпочтительным соединением является соединение (-) -(R)-6-[1 Н-1,2,4-триазол-1- ил-[3-(трифторметил)фенил]-метил]-2-(1H)-хинолинон и его фармацевтически приемлемые кислотно-аддитивные соли.
Соединения формулы (I), где Y представляет О, причем эти соединения представляются формулой (1-b), можно получить в соответствии со способами, описанными в Европейском патенте 0371564.
Соединения формулы (1-b) можно затем превратить в соединения формулы (I), где Y представляет S, причем эти соединения представлены формулой (1-c) с использованием известных в данной области реагентов превращения, таких, как, например, пентасульфид фосфора.
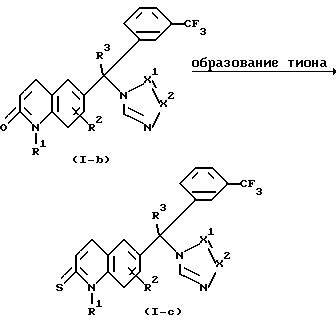
Эти соединения формулы (1-c) можно получить смешиванием реагентов в инертном в реакции растворителе, таком, как, например, пиридин. Реакцию можно подходящим образом проводить при температуре флегмы реакционной смеси.
В этом и следующих получениях продукты реакции можно выделить из реакционной среды и, если необходимо, далее очищать в соответствии с методиками, обычно известными в данной области, таким, как, например, экстракция, кристаллизация, растирание и хроматография.
Соединения формулы (1-b), где R1 представляет водород, причем эти соединения представлены формулой (1-b-1), можно получить реакцией нитрона формулы (II) с подходящим эфирообразующим реагентом, таким, как, например, ангидрид карбоновой кислоты, например, уксусный ангидрид, чтобы таким образом получить соответствующий эфир в положении 2 хинолиновой части. Этот хинолиновый эфир можно гидролизовать in situ в соответствующий хинолинон с использованием основания, такого как, например, карбонат калия. Перемешиванием и повышенными температурами можно повысить скорость реакции.
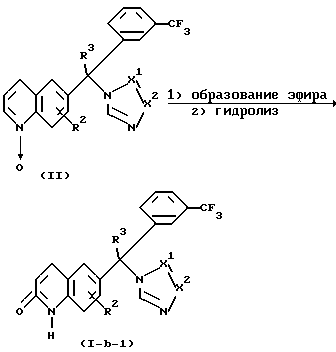
Альтернативно, соединения формулы (1-b-1) можно получить реакцией нитрона формулы (II) с сульфонилсодержащим электрофильным реагентом, таким, как, например, n-толуолсульфонилхлорид, в присутствии основания, такого, как, например, водный карбонат калия. Реакция первоначально включает образование производного 2-гидроксихинолина, который потом таутомеризуют в целевое производное хинолинона. Эту реакцию можно удобно проводить при комнатной температуре в инертном в реакции растворителе, таком, как, например, дихлорметан или толуол. Перемешиванием и применением известных в данной области условий межфазного катализа можно повысить скорость реакции.
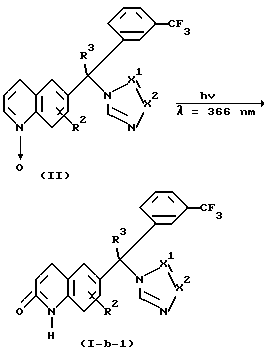
Соединения формулы (1-b-1) можно также получить внутримолекулярной фотохимической перегруппировкой соединений формулы (II). Эту перегруппировку можно проводить растворением реагентов в инертном в реакции растворителе и облучением при длине волны, например, 366 нм. Благоприятно использовать дегазированные растворы и проводить реакцию в инертной атмосфере, такой, как, например, не содержащий кислорода газ аргон или азот, чтобы свести к минимуму нежелательные побочные реакции или снижение количества квантов.
Соединения формулы (1-b-1) можно также превратить в соединения формулы (1-b), где R1 представляет C1-4 алкил, причем эти соединения представлены формулой (1-b-2). Например, соединения формулы (1-b-1) можно N-алкилировать реагентом C1-4 алкил-L, где L - реакционноспособная уходящая группа, такая, как, например, галоген или сульфонилоксигруппа, в присутствии основания, такого, как, например, гидрид натрия.
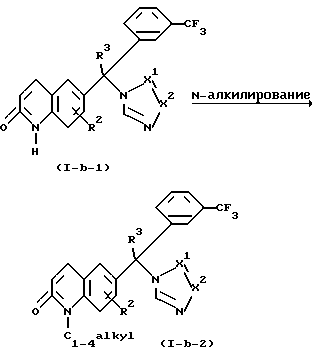
Такое N-алкилирование можно удобно проводить путем смешивания реагентов в инертном в реакции растворителе, таком, как, например, N,N-диметилформамид. Может быть выгодным проведение этого N-алкилирования в инертной атмосфере, такой, как, например, газ аргон или азот.
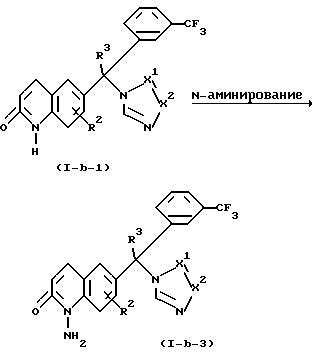
Соединения формулы (1-b-1) можно также превратить в соединения формулы (1-b), где R1 представляет амино, причем эти соединения представлены формулой (1-b-3). Например, соединения формулы (1-b-3) можно получить N-аминированием соединений формулы (1-b-1) агентом аминирования, таким, как, например, гидроксиламин-О-сульфокислота при комнатной температуре в растворителе, таком, как, например, вода, и в присутствии основания, такого, как, например, гидрооксид натрия.
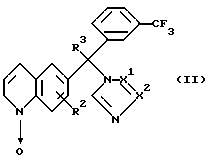
Нитроны формулы (II) можно получить N-окислением хинолинов формулы (III) подходящим окислителем, таким как, например, м-хлорпероксибензойная кислота или фталевый ангидрид в комбинации с пероксидом водорода. Это N-алкилирование можно проводить путем смешивания реагентов при комнатной температуре в инертном в реакции растворителе, таком как, например, дихлорметан. После получения промежуточных продуктов формулы (II) соединения формулы (1-b) можно удобно получить посредством реакции in situ.
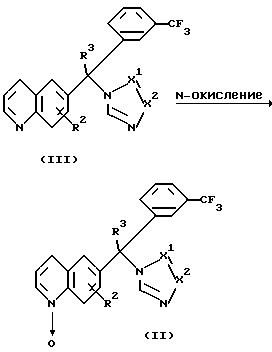
Промежуточные продукты формулы (III) можно получить по способам, описанным в Европейском патенте 0371564.
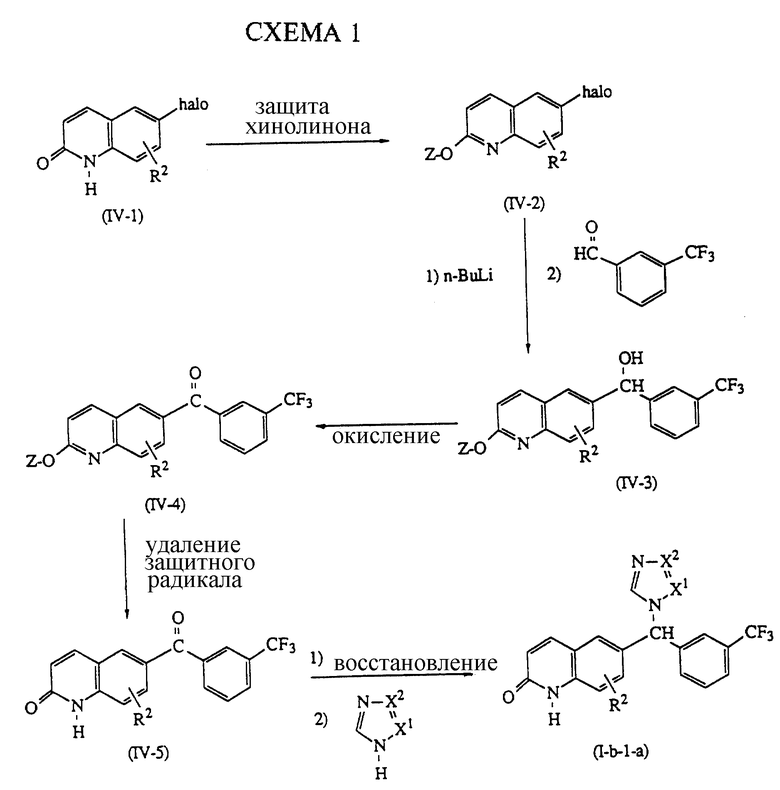
Новый подход к получению соединений формулы (1-b-1), где R3 представляет водород, причем эти соединения представляются формулой (I-b-I-a), включает способ, описанный в схеме 1. Первая стадия включает защиту хинолинона формулы (IV-1), где halo представляет атом галогена, в частности атом брома, для образования таким образом производного хинолина формулы (IV-2), где Z представляет защитную группу, такую как, например, метил. Это производное хинолина реагирует с литийорганическим соединением, таким как, например, н-бутиллитий, в инертной в реакции растворителе, таком как, например, тетрагидрофуран, таким образом замещая атом галогена в положении 6 хинолиновой части в промежуточных продуктах формулы (IV-2) на атом лития. Это литийсодержащий промежуточный продукт реагирует in situ с 3-трифторбензальдегидом или его функциональным производным, таким образом образуя промежуточный продукт формулы (IV-3). Образование промежуточных продуктов формулы (IV-3) из промежуточных продуктов формулы (IV-2) можно удобно проводить при низких температурах, предпочтительно при -78oC. Промежуточные продукты формулы (IV-3) можно окислить в соответствующие кетоны формулы (IV-4) с использованием известных в данной области окислителей. Такие кетоны можно затем освободить от защитных групп, таким образом получая таутомерное производное хинолинона формулы (IV-5), в присутствии кислоты. Повышенные температуры и перемешивание могут повысить скорость реакции превращения.
Соединения формулы (I-b-l-a) можно получить восстановительным алкилированием 1,2,4-триазола или 1,3,4-триазола соединениями формулы (IV-5). Эту реакцию можно удобно проводить перемешиванием и нагреванием реагентов в присутствии муравьиной кислоты или формамидов в качестве восстановителей, необязательно в присутствии кислотного катализатора, такого, как, например, соляная кислота. Если необходимо, соединения формулы (I-b-l-a) можно далее подвергать реакции в соответствии со способами, описанными здесь выше для соединений формул (1-b) и (1-c).
Соединения формулы (I) и их промежуточные соединения, получаемые описанными здесь выше способами, обычно являются рацемическими смесями энантиомеров, которые можно отделить друг от друга известными в данной области способами расщепления (на оптические антиподы). Рацемические соединения формулы (I) или их промежуточные соединения можно превратить в формы соответствующих диастереомерных солей реакцией с подходящей хиральной кислотой, такой, как, например, камфорасульфокислота. Такие формы диастереомерных солей затем разделяют, например, селективной или фракционной кристаллизацией и энантиомеры выделяют из них при помощи щелочи. Альтернативный способ разделения энантиомерных форм соединений формулы (I) или их промежуточных соединений включает жидкостную хроматографию с использованием хиральной стационарной фазы. Эти чистые стереохимически изомерные формы можно также получить из соответствующих чистых стереохимически изомерных форм подходящих исходных материалов, при условии, что реакция проходит стереоспецифично. Например, энантиомерно чистые формы соединений формулы (I) можно получить из энантиомерно чистых форм соединений формулы (III). Энантиомерно чистые соединения формулы (III) можно получить с использованием известных в данной области способов разделения.
Соединения данного изобретения имеют превосходство в фармакологических свойствах по сравнению со свойствами самых близких соединений данной области, заключающееся в том, что они более эффективны в подавлении эффектов кератинизации, которые можно показать в "Испытании на вагинальную кератинизацию на крысах с удаленными яичниками", как описывается здесь ниже. С точки зрения их способности подавлять эффекты кератинизации, соединения формулы (I) полезны при лечении и/или предупреждении нарушений кератинизации, таких, как, например, угри, псориаз, тяжелый псориаз, ламеллярный ихтиоз, подошвенные бородавки, мозоли, акантоз, нигрикан, красный плоский лишай, моллюск, меланоз кожи, корнеальная эпителиальная ссадина, десквамативный глоссит, болезнь Фокса-Фордайса, кожная метастатическая меланома и келоиды, эпидермолитический гиперкератоз, фолликулярный дискератоз, pityriasis rubra pilaris (красный отрубевидный волосяной лишай), врожденная ихтиозоформная эритродермия, ладонный и подошвенный гиперкератоз и подобные нарушения.
Соединения формулы (I) подавляют также удаление из плазмы ретиноидов, таких, как, например, все-транс-ретиноевая кислота, 13-цис-ретиноевая кислота и их производные, приводящее к более поддерживаемым тканевым концентрациям ретиноевой кислоты и улучшенному регулированию дифференциации и роста разных типов клеток. Способность задерживать метаболизм ретиноевой кислоты можно доказать в различных экспериментах in vitro и in vivo. Здесь далее описывается конкретный способ in vitro и испытания ингибирующей активности соединений формулы (I) на метаболизм ретиноевой кислоты в раковых клетках молочной железы человека. С точки зрения их способности замедлять метаболизм ретиноевой кислоты, данные соединения полезны для предупреждения и/или лечения нарушений, характеризующихся патологической клеточной пролиферацией и/или дифференциацией, например, рака и, в частности, нарушений кератинизации, таких, как нарушения, упоминаемые здесь ранее (Van Wauwe et al., J. Pharmacol. Exp. Ther., 1992, 261 (2), 773-779).
Кроме того, соединения изобретения полезны при подавлении метаболизма экзогенно введенного и эндогенно образованного 1α,25- дигидроксивитамина D3 (кальцитриол). Ингибирующую активность соединений формулы (I) по метаболической деградации кальцитриола можно доказать путем измерения воздействия этих соединений на деградацию кальцитриола в кератиноцитах крайней плоти человека, клетках свиных почек и клетках гепатомы человека. С точки зрения их ингибирующего действия на метаболизм кальцитриола, соединения формулы (I) можно использовать при лечении состояний недостаточности витамина D. "Классическое" применение соединений ряда витаминов D лежит в области нарушений типа метаболических костей. Описывается также, что кальцитриол влияет на действия и/или продуцирование интерлейкина. Далее, кальцитриал имеет использование при лечении болезней, характеризующихся патологической клеточной пролиферацией и/или дифференциацией, в частности, нарушений кератинизации, например, нарушений, описанных здесь выше (Bouillon et al., Reviews, 1995, 16, 200-257).
Кроме того, соединения формулы (I) ингибируют образование андрогенов из прогестинов и ингибируют действие ароматазного ферментного комплекса, который катализирует образование эстрогенов из андрогенных стероидов у млекопитающих.
С точки зрения указанных выше использований соединений формулы (I) следует, что данное изобретение представляет способ лечения теплокровных животных, страдающих болезнями, которые характеризуются повышенной пролиферацией и/или патологической дифференциацией нормальных клеток, клеток в состоянии злокачественного перерождения или опухолевых клеток, независимо от того, являются ли они эпителиальными или мезенхимальными, независимо от того, имеют ли они эктодермальное, эндодермальное или мезодермальное происхождение или независимо от того, являются ли они эстрогензависимыми, андрогензависимыми или эстроген- и андрогеннезависимыми клетками. Этот способ включает системное или местное введение терапевтического количества соединения формулы (I), эффективного при лечении указанных выше нарушений, в частности, нарушений кератинизации, необязательно в присутствии эффективного количества ретиноевой кислоты, ее производного или стереохимически изомерной формы. Данное изобретение далее относится к способу лечения пациентов, страдающих патологическим состоянием, на которое можно благотворно воздействовать введением кальцитриола или его пролекарства, в частности, нарушениями кератинизации, причем этот способ состоит в введении пациенту (а) эффективного количества кальцитриола или его пролекарства и (б) эффективного количества соединения формулы (I).
Таким образом, данное изобретение относится также к соединениям формулы (I), определяемым здесь выше, для использования в качестве лекарственного средства, в частности, в качестве лекарственного средства для лечения нарушений кератинизации. Данное изобретение относится также к соединениям формулы (I), определяемым здесь выше, в комбинации с ретиноевой кислотой, ее производным или стереохимически изомерной формой или в комбинации с кальцитриолом или его пролекарством для использования в качестве лекарственного средства. Данное изобретение относится также к использованию соединений формулы (I) для производства лекарственного препарата для лечения указанных выше нарушений, в частности нарушений кератинизации.
Для облегчения введения соединения согласно изобретению можно приготовлять в виде различных фармацевтических форм. В качестве подходящих композиций можно сослаться на все композиции, обычно используемые для системного или местного введения лекарственных средств. Для получения фармацевтических композиций этого изобретения терапевтически эффективное количество определенного соединения, необязательно в форме кислотно-аддитивной соли, в качестве активного ингредиента объединяют до получения однородной смеси с фармацевтически приемлемым носителем, который может принимать широкий ряд форм, в зависимости от формы препарата, необходимого для введения. Эти фармацевтические композиции желательны в унифицированной лекарственной форме, подходящей предпочтительно для введения перорально, ректально, подкожно или парентеральной инъекцией. Например, при получении композиций в пероральной лекарственной форме можно использовать любую из обычных фармацевтических сред, таких, как, например, вода, гликоли, масла, спирты и подобные в случае пероральных жидких препаратов, таких как суспензии, сиропы, эликсиры и растворы, или твердые носители, такие как крахмалы, сахара, каолин, смазывающие вещества, связующие, дезинтегрирующие средства и подобные в случае порошков, пилюль, капсул и таблеток. Вследствие легкости их введения, таблетки и капсулы представляют наиболее выгодную пероральную унифицированную лекарственную форму, в этом случае очевидно использование твердых фармацевтических носителей. Для парентеральных композиций носитель обычно содержит стерильную воду, по меньшей мере в большей части, хотя можно включать другие ингредиенты, например, для улучшения растворимости. Можно получить, например, инъецируемые растворы, в которых носитель содержит солевой раствор, раствор глюкозы или смесь солевого раствора и раствора глюкозы. В композициях, подходящих для чрескожного введения, носитель необязательно содержит средство, повышающее проникновение, и/или подходящий смачивающий реагент, необязательно в комбинации с подходящими добавками любой природы в незначительных пропорциях, причем эти добавки не оказывают какое-либо значительное ухудшающее действие на кожу. Эти добавки могут облегчить введение в кожу и/или могут помочь в приготовлении целевых композиций. Эти композиции можно вводить различными способами, например, в виде чрескожного пластыря, в виде наносимых пятен или в виде мази. Кислотно-аддитивные соли соединений формулы (I) вследствие их повышенной растворимости в воде по сравнению с формой соответствующего основания явно более подходящие для получения водных композиций. В качестве подходящих композиций для местного применения можно сослаться на все композиции, обычно используемые для местного введения лекарственных средств, например, кремов, гелей, повязок, шампуней, настоек, паст, мазей, целебных мазей, порошков и подобных. Эти композиции можно применять в виде аэрозолей, например, с пропеллентом, таким как азот, диоксид углерода, фреон, или без пропеллента, например, разбрызгиванием насосом, капель, лосьонов и полутвердой композиции, например, загущенной композиции, которую можно применять при помощи тампона. Особенно удобны для использования полутвердые композиции, например, целебные мази, кремы, гели, мази и подобные.
В особенности выгодно приготовлять вышеупомянутые фармацевтические композиции в унифицированной лекарственной форме для облегчения введения и постоянства дозировки. Термин унифицированная лекарственная форма, используемый здесь в описании и формуле изобретения, относится к физически дискретным единицам, подходящим в качестве унифицированных доз, причем каждая единица содержит заданное количество активного ингредиента, рассчитанное для достижения желаемого терапевтического действия, в сочетании с требуемым фармацевтическим носителем. Примерами таких унифицированных лекарственных форм являются таблетки (включая таблетки с покрытием и бороздками), капсулы, пилюли, пакетики с порошком, облатки, инъецируемые растворы или суспензии, полные чайные ложки, полные столовые ложки и подобные и разделенные части их.
Другие такие композиции представляют препараты косметического типа, например, туалетную воду, тампоны, лосьоны, молочко для кожи или молочные лосьоны. Эти препараты содержат кроме активного ингредиента компоненты, обычно применяемые в таких препаратах. Примерами таких компонентов являются масла, жиры, воски, поверхностно-активные вещества, увлажнители, загустители, антиоксиданты, стабилизаторы вязкости, хелатообразователи, буферы, консерванты, отдушки, красители, низшие алканолы и подобные. При желании в композиции можно вводить другие ингредиенты, например, противовоспалительные средства, антибактериальные средства, антигрибковые средства, дезинфицирующие средства, витамины, солнцезащитные средства, антибиотики или другие средства против угрей.
Еще одной особенностью данного изобретения является представление определенных фармацевтических или косметических композиций, которые содержат инертный носитель, эффективное количество соединения формулы (I) и эффективное количество ретиноевой кислоты, ее производного или стереохимически изомерной формы. Такие содержащие ретиноевую кислоту композиции в частности полезны для лечения угрей или для замедления действия старения на кожу и обычно улучшают качество кожи, в частности кожу лица человека.
Кроме того, данное изобретение относится также к конкретным фармацевтическим или косметическим композициям, которые содержат инертный носитель, эффективное количество соединения формулы (I) и эффективное количество кальцитриола или его пролекарства. Последние композиции в частности полезны при лечении нарушений кератинизации.
Конкретное воплощение изобретения относится к продукту, содержащему ретиноевую кислоту или ее производное и соединение формулы (I) в качестве комбинированного препарата для одновременного, отдельного или последовательного использования в дерматологических нарушениях. Изобретение относится также к продукту, содержащему кальцитриол или его пролекарство и соединение формулы (I), как комбинированному препарату для одновременного, раздельного или последовательного использования при нарушениях, на которые благотворное влияние оказывает кальцитриол. Такие продукты могут содержать, например, набор, содержащий контейнер с подходящей композицией, содержащей соединение формулы (I), и другой контейнер с композицией, содержащей кальцитриол или ретиноид. Такой продукт может иметь преимущество, заключающееся в том, что врач может выбрать на основе диагноза пациента, которого нужно лечить, подходящие количества каждого компонента и последовательность и время введения их.
Специалисты в данной области при лечении нарушений кератинизации могут определить эффективное терапевтическое суточное количество на основе результатов испытаний, представленных здесь ниже. Эффективное терапевтическое суточное количество должно быть от около 0,1 мг/кг до около 40 мг/кг массы тела, более предпочтительно от около 0,3 мг/кг до около 10 мг/кг массы тела. Может быть подходящим назначение введения терапевтически эффективной дозы один раз в сутки или назначение введения субдоз два, три, четыре или более раз в сутки. Эти субдозы можно приготовить в виде унифицированной лекарственной формы, например, содержащей от 0,1 мг до 500 мг, в частности от 0,5 мг до 50 мг активного ингредиента на унифицированную лекарственную форму.
Точная дозировка и частота введения зависит от конкретного используемого соединения формулы (I), конкретного состояния, которое лечат, тяжести состояния, которое лечат, возраста, массы и общего физического состояния конкретного пациента, а также другого лекарственного средства, которое может принимать пациент, как хорошо известно специалистам данной области. Кроме того, очевидно, что это эффективное суточное количество можно снизить или повысить в зависимости от ответной реакции пациента и/или в зависимости от оценки врача, прописывающего соединения данного изобретения. Диапазон эффективного суточного количества упоминался здесь ранее и поэтому представляет только рекомендации.
Следующие примеры предназначены для иллюстрации, а не для ограничения объема данного изобретения.
Экспериментальная часть
А. Получение промежуточных продуктов
Пример 1
а) Смесь этилового эфира 4-амино-2-хлорбензойной кислоты (20 г), глицерина (32,17 г) и 3-нитробензолсульфоната натрия (46,73 г) в серной кислоте (75%) (160 мл) перемешивали в течение 3 часов при 100oC и в течение 1 часа при 140oC. После охлаждения смеси до 60oC и добавления этанола (200 мл) смесь перемешивали в течение 16 часов при 60oC. Этанол выпаривали и остаток выливали в ледяную воду, нейтрализовали NH4OH и экстрагировали этилацетатом. Отделенный органический слой сушили над MgSO4, фильтровали и фильтрат выпаривали. Остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/этилацетат, 97,5/2,5). Целевые чистые фракции собирали и выпаривали, получая 7,56 г (28%) этил 5-хлор-6-хинолинкарбоксилата (промежуточный продукт 1).
б) Литийалюминийгидрид (1,66 г) добавляли по частям в раствор промежуточного продукта 1 (10,11 г) в тетрагидрофуране (325 мл) при 0oC в атмосфере N2 и смесь перемешивали в течение 1 часа. В смесь добавляли этилацетат (70 мл) и воду (3 мл), которую затем фильтровали.
Фильтрат сушили над MgSO4, фильтровали и выпаривали, получая 7,9 г (93%) 5-хлор-6-хинолинметанола (промежуточный продукт 2).
в) Смесь промежуточного продукта 2 (7,9 г) и диоксида марганца (10,64 г) в CH2Cl2 (165 мл) перемешивали в течение 6 часов при комнатной температуре. Смесь фильтровали через целит и фильтрат перемешивали снова с диоксидом марганца (10,64 г) в течение 24 часов. Смесь фильтровали через целит и фильтрат выпаривали, получая 7,82 г (100%) 5-хлор-6-хинолинкарбоксальдегида (промежуточный продукт 3). Аналогичным образом получали 8-хлор-6- хинолинкарбоксальдегид (промежуточный продукт 4).
Пример 2
а) Раствор 1-бром-3-(трифторметил)бензола (10,67 г) в тетрагидрофуране (15 мл) добавляли по каплям в суспензию магниевой стружки (1,15 г) в тетрагидрофуране (15 мл). Смесь охлаждали до 0oC и добавляли в нее по каплям раствор промежуточного продукта 3 (7,57 г) в тетрагидрофуране (90 мл). Смесь распределяли между этилацетатом и насыщенным водным раствором NH4Cl. Органический слой отделяли, сушили над MgSO4, фильтровали и фильтрат выпаривали. Остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/CH3OH, 98/2). Чистые фракции собирали и выпаривали, получая 10,9 г (81%) (±)-5-хлор -α- [3-(трифторметил)фенил]-6-хинолинметанол (промежуточный продукт 5).
б) В раствор промежуточного продукта 5 (8 г) в CH2Cl2 (400 мл) при 0oC по каплям добавляли тионилхлорид (8 мл) и смесь перемешивали при комнатной температуре в течение ночи. Растворитель выпаривали и остаток распределяли между CH2Cl2 и водой, которую подщелачивали насыщенным раствором K2CO3. Отделенный органический слой сушили над MgSO4, фильтровали и фильтрат выпаривали, получая 8,4 г (100%) (±)-5-хлор-6-[хлор- [3-(трифторметил)фенил]-метил]хинолина (промежуточный продукт 6).
в) Смесь промежуточного продукта 6 (8,4 г), 1,2,4-триазола (4,89 г) и карбоната калия (9,78 г) в ацетонитриле (300 мл) перемешивали и кипятили с обратным холодильником в течение 12 часов. Растворитель выпаривали и остаток распределяли между CH2Cl2 и водой. Отделенный органический слой сушили над MgSO4, фильтровали и фильтрат выпаривали. Остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/CH3OH/-NH4OH, 98,5/1,5/0,1). Чистые фракции собирали и растворитель выпаривали. Остаток кристаллизовали из смеси диизопропиловый эфир/петролейный эфир, получая 1,51 г (32%) (±)-5-хлор-6-[1Н-1,2,4-триазол-1-ил-[3- (трифторметил)фенил]метил]хинолин(промежуточный продукт 7; т. пл. 144,5oC).
Аналогичным образом были получены:
(±)-8-метил-6-[1Н-1,2,4-триазол-1-ил-[3- (трифторметил)фенил]метил]хинолин (промежуточный продукт 8; т. пл. 108,9oC и (±)-8-фтор-6-[1Н-1,2,4-триазол-1-ил-[3- (трифторметил)фенил]метил]хинолин (промежуточный продукт 9; т. пл. 146,6oC.
Пример 3
Рацемическую смесь 6-[1H-1,2,4-триазол-1-ил-[3- (трифторметил)-фенил] метил] хинолина (10 г), которая описана и приведена в качестве примера в Европейском патенте 371564, разделяли на его чистые энантиомеры на колонке с Chiracell OD® (элюент: этанол/гексан, 1/1). Чистые фракции первого элюированного пика собирали, объединяли и растворитель выпаривали, получая 3,6 г (±)-(S)-6-[1H-1,2,4-триазол-1-ил- [3-(трифторметил)фенил] метил] хинолина (промежуточный продукт 10). Чистые фракции второго элюированного пика собирали, объединяли и разделяли диэтиловым эфиром. Объединенный органический слой фильтровали и растворитель выпаривали, получая 3,48 г (-)-(R)-6-[1H-1,2,4-триазол-1-ил-[3- (трифторметил)фенил] метил] хинолина (промежуточный продукт 11).
Пример 4
Иодметан (8,8 мл) добавляли в перемешиваемую смесь (±)-6-[1H-1,2,4-триазол-1-ил-[3-(трифторметил)фенил] метил] хинолина (10 г) в N,N-диметилформамиде (100 мл) при 0oC в атмосфере N2. По частям добавляли трет-бутоксид калия (9,5 г) и смесь перемешивали в течение 2 часов при комнатной температуре. Смесь распределяли между ледяной водой и CH2Cl2. Отделенный органический слой промывали водой, сушили над MgSO4, фильтровали и фильтрат выпаривали. Маслянистый остаток очищали колоночной хроматографией на силикагеле (элюент: толуол/2-пропанол, 94/6). Чистые фракции собирали и выпаривали. Остаток превращали в соль азотной кислоты (1:2) в CH3OH и кристаллизовали из смеси 2-пропанон/диэтиловый эфир, получая 1,15 г (9%) динитрата (±)-6-11H-1,2,4-триазол-1-ил-[3- (трифторметил)фенил]этил]хинолина (промежуточный продукт 12; т. пл. 116,4o).
Аналогичным образом были получены:
(±)-6-[1H-1,2,4-триазол-1-ил-[3-(трифторметил)фенил] пропил] хинолин (промежуточный продукт 13); (±)-6-[1H-1,2,4-триазол-1-ил-[3-(трифторметил)фенил] -2-метилпропил] хинолин (промежуточный продукт 14) и (±)-6-[1H-1,2,4-триазол-1-ил-[3- (трифторметил)фенил] пентил] хинолин (промежуточный продукт 15).
Пример 5
Трет-бутоксид калия (0,945 г) добавляли по частям в перемешиваемую смесь (±)-6-[1H-1,2,4-триазол-1-ил-[3- (трифторметил)фенил]метил]хинолина (2,7 г) в N,N-диметилформамиде (80 мл) при 0oC в атмосфере N2. Смесь перемешивали в течение 30 минут при 0oC и добавляли N-фторсультам (2,43 г), описанный в Helv. Chim. Acta 72 p., 1248 (1989). Затем смесь перемешивали при 0oC в течение 1 часа и при комнатной температуре в течение 1 часа. Добавляли несколько миллилитров воды и растворитель выпаривали. Остаток распределяли между водой и этилацетатом. Отделенный органический слой промывали водой, сушили над MgSO4, фильтровали и фильтрат выпаривали. Масляный остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/CH3OH, 98/2). Чистые фракции собирали и выпаривали. Остаток превращали в соль этандиовой (щавелевой) кислоты (1:1) в смеси 2-пропанон/диэтиловый эфир, получая этандиоат (1: 1) (±)-6-[фтор-(1H-1,2,4-триазол-1-ил)-[3- (трифторметил)фенил]метил]хинолина (промежуточный продукт 16; т. пл. 142,6oC.
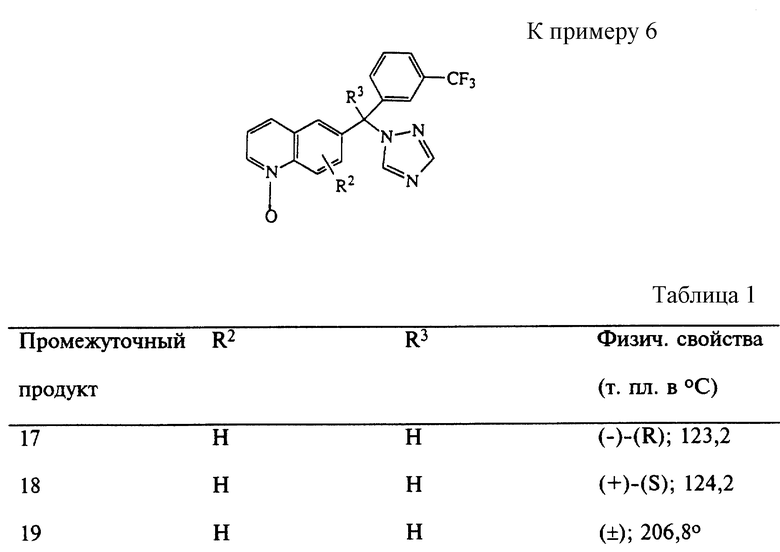
Пример 6
3-Хлорпероксибензойную кислоту (49 г) добавляли по частям в раствор промежуточного продукта 11 (50,3 г) в CH2Cl2 (500 мл) и смесь перемешивали в течение 1 часа при комнатной температуре. Смесь разделяли 10% раствором NaHCO3. Отделенный органический слой промывали насыщенным раствором NaCl, сушили над MgSO4, фильтровали и фильтрат выпаривали, получая 1-оксид (-)-(R)-6-[1H-1,2,4-триазол-1-ил-[3- (трифторметил)фенил] метил] хинолина (промежуточный продукт 17; т. пл. 123,2oC).
Аналогичным образом были получены промежуточные продукты, показанные в табл. 1.
Пример 7
а) Бутиллитий добавляли по каплям при -78oC в раствор 6-бром-2-метоксихинолина (4 г) в тетрагидрофуране (160 мл). После окончания добавления смесь перемешивали при -78oC в течение 15 минут. По каплям добавляли раствор 3-(трифторметил)- бензальдегида (3,51 г) в тетрагидрофуране (40 мл) и смесь перемешивали при -78oC в течение 30 минут, затем гасили водой (50 мл) и экстрагировали этилацетатом. Органический слой отделяли, промывали солевым раствором, сушили над MgSO4, фильтровали и выпаривали растворитель. Остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/CH3OH, 97/3). Чистые фракции собирали и растворитель выпаривали, получая 3,7 г (66%) (±)-2-метокси -α- [3-(трифторметил)фенил]-6-хинолинметанола (промежуточный продукт 28).
б) Смесь промежуточного продукта 28 (1 г) и HCl (25 мл, 3 N) перемешивали и кипятили с обратным холодильником в течение 2 часов. Раствор подщелачивали NaOH и экстрагировали CH2Cl2. Органический слой отделяли, промывали солевым раствором, сушили над MgSO4, фильтровали и растворитель выпаривали, получая 1,8 г (±)-6-[гидрокси-[3-(трифторметил)фенил]метил]-2(1H)хинолинона (промежуточный продукт 29).
в) Тионилхлорид (12,3 мл) добавляли по каплям при 0oC в раствор промежуточного продукта 29 (12,3 г) в CH2Cl2 (900 мл) и смесь перемешивали в течение 1 часа. Тионилхлорид (12,3 мл) снова добавляли по каплям при 0oC и смесь перемешивали при комнатной температуре в течение 1 часа. Смесь выливали в лед и экстрагировали. Органический слой сушили над MgSO4, фильтровали и растворитель выпаривали, получая 13 г (100%) (±)-6-[хлор-[3-(трифторметил)фенил]метил]-2(1H)хинолинона (промежуточный продукт 30).
Б. Получение соединений формулы (I)
Пример 8
10%-ный раствор K2CO3 (700 мл) и п-толуолсульфонилхлорид (36,8 г) добавляли в раствор промежуточного продукта 17 (52,5 г) в CH2Cl2 (700 мл) и смесь перемешивали при комнатной температуре в течение 1 часа. Смесь промывали насыщенным раствором NaCl и отделенный органический слой сушили над MgSO4, фильтровали и фильтрат выпаривали. Остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/CH3OH, 96,5/3,5). Чистые фракции собирали и выпаривали. Остаток кристаллизовали из смеси метилэтилкетон/диизопропиловый эфир, получая 15,3 г (29%) (-)-(R)-(±)-6-[1H-1,2,4-триазол-1-ил-[3-(трифторметил)фенил]метил]- 2(1H)- хинолинона (соединение 1; т. пл. 192,8oC; [α]
Пример 9
Аналогично соединение 1 было получено перемешиванием раствора промежуточного продукта 17 (1,2 г) в уксусном ангидриде (10 мл) в течение 12 часов при 140oC. Избыточный уксусный ангидрид выпаривали, смесь подщелачивали 10% раствором K2CO3 и экстрагировали CH2Cl2. Отделенный органический слой промывали насыщенным раствором NaCI, сушили над MgSO4, фильтровали и фильтрат выпаривали. Остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/CH3OH, 95/5). Чистые фракции собирали, выпаривали и далее очищали колоночной хроматографией на хиракеле AD® (элюент: н-гексан/-этанол, 90/10). Чистые фракции собирали и выпаривали, получая 0,235 г (33,7%) соединения 1 (т. пл. 176,0oC; [α]
Указанную выше методику реакции повторяли несколько раз и смесь получаемых фракций растворяли в метилэтилкетоне (300 мл) и нагревали до полного растворения. Смесь фильтровали теплой. Фильтрат оставляли на 18 часов для перекристаллизации соединения. Осадок фильтровали, промывали диизопропиловым эфиром (100 мл) и сушили, получая 118 г (90,8%) соединения 1. Эту фракцию снова сушили, получая 109,5 г (64,2%) соединения 1 (т. пл. 195oC; [α]
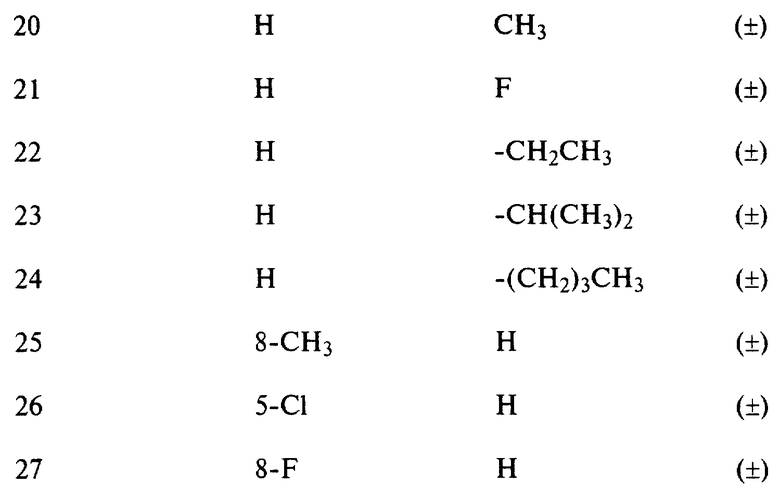
Пример 10
Смесь промежуточного продукта 30 (0,846 г), 1,2,4-триазола (0,346 г) и карбоната калия (0,7 г) в CH3CN (30 мл) перемешивали и кипятили с обратным холодильником в течение 2 часов. Растворитель выпаривали и остаток растворяли в воде и экстрагировали CH2Cl2. Органический слой сушили над MgSO4, фильтровали и выпаривали. Остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/CH3OH/NH4OH, 98/2/0,1) и растворитель целевых фракций выпаривали, получая 0,25 г (27%) (±)-6-[1H-1,2,4-триазол-1-ил-[3-(трифторметил)фенил]метил]-2 (1H)-хинолинона (соединение 3).
В табл. 2 перечисляются соединения, которые были получены способом, аналогичным способу одного из вышеупомянутых здесь примеров.
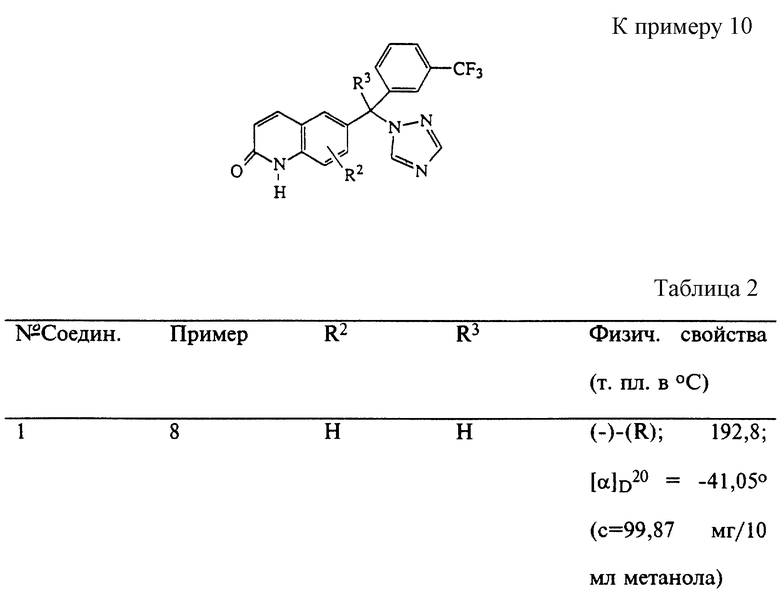
Пример 11
Смесь соединения 3 (10 г) N,N-диметилформамиде (100 мл) перемешивали при комнатной температуре в атмосфере N2. По частям добавляли гидрид натрия (0,51 г) и смесь перемешивали в течение 15 минут. Смесь охлаждали, добавляли по каплям иодметан и реакционную смесь перемешивали в течение 12 часов. Растворитель выпаривали и остаток распределяли между CH2Cl2 и водой. Отделенный органический слой сушили над MgSO4, фильтровали и фильтрат выпаривали. Масляный остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/CH3OH/NH3OH, 98/2/0,1). Чистые фракции собирали и растворитель выпаривали. Остаток кристаллизовали из смеси метилэтилкетон/диэтиловый эфир, получая 2,9 г (28%) (±)-1-метил-6-[1H-1,2,4-триазол-1-ил-[3- (трифторметил)фенил] метил] -2(1H)-хинолинона (соединение 12; т. пл. 186,7oC).
Аналогичным образом были получены соединения, показанные в табл. 3.
Пример 12
Смесь соединения 3 (7,3 г) и NaOH (4,12 г) в воде (200 мл) перемешивали при комнатной температуре в течение 15 минут. По частям добавляли гидроксиламин-O-сульфокислоту (6,3 г) и смесь перемешивали в течение 12 часов. Осадок фильтровали и кристаллизовали из 2-пропанола, получая 2,3 г (±)-1-амино-6-[1H-1,2,4-триазол-1-ил-13-(трифторметил)фенил] метил] - 2(1H)-хинолинона (соединение 20; т. пл. 222,3oC).
Пример 13
Смесь соединения 12 (7 г) и пентасульфида фосфора (8,1 г) в пиридине (100 мл) перемешивали и кипятили с обратным холодильником в течение 4 часов. Растворитель выпаривали, остаток распределяли между водой и CH2Cl2. Отделенный органический слой сушили над MgSO4, фильтровали и фильтрат выпаривали. Остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/CH3OH/NH4OH, 99/1/0,1). Чистые фракции собирали и выпаривали. Остаток кристаллизовали из смеси диизопропиловый эфир/метилэтилкетон/2-пропанон, получая 1,4 г (20%) (±)-1- метил-6-[1H-1,2,4-триазол-1 -ил-[3-(трифторметил)фенил] метил] -2(1 Н)-хинолинтиона (соединение 21; т. пл. 143,7oC).
Аналогичным образом был получен (±)-6-[1H-1,2,4-триазол-1-ил-(3- (трифторметил)фенил]метил]-2(1H)-хинолинтиол (соединение 22).
Пример 14
Соединение 1 (3,7 г) растворяли в этаноле (50 мл). Через смесь в течение 2 минут барботировали HBr (газ). Растворитель выпаривали и остаток растворяли в смеси CH3CN/C2H5OH, 80/20. Растворитель выпаривали и остаток растирали в метилизобутилкетоне. Осадок фильтровали и сушили. Эту фракцию перекристаллизовали из смеси 2-пропанон/2-пропанол, (35 мл/15 мл). Осадок фильтровали и сушили. Эту фракцию перекристаллизовали из этанола в закрытой камере, насыщенной 2-пропаноном. Жидкость декантировали и кристаллы сушили, получая 0,74 г (16,4%) моногидробромида (1:1) (R)-6-[1H-1,2,4-триазол-1-ил-[3-(трифторметил)фенил] метил] -2(1H)- хинолинона (соединение 23, т. пл. 220oC).
B. Фармакологические примеры
Пример 15: Ингибирование метаболизма ретиноевой кислоты (РК)
Раковые клетки MCF-7 молочной железы человека выращивали в качестве исходной культуры в соответствии с известным в данной области протоколом. За один день до экспериментов в исходные культуры добавляли РК для стимулирования РК-метаболизма. В начале эксперимента клеточные суспензии инкубировали в среде из тканевой культуры, содержащей 3H-РК, в качестве субстрата. В смеси инкубирования добавляли разные концентрации испытуемого соединения (растворенного в ДМСО) и в конце инкубирования неметаболизированную РК отделяли от ее полярных метаболитов. Фракции, содержащие полярные 3H-меченые метаболиты, собирали и подсчитывали в счетчике сцинтилляции. Для каждого эксперимента параллельно проводили контрольное и слепое инкубирование. Величины IC50, перечисленные в табл. 4, представляют концентрации, необходимые для снижения количества метаболитов до 50% от контрольного.
Пример 16: "Испытание на вагинальную кератинизацию на крысах с удаленными яичниками"
Крысам с удаленными яичниками подкожно инъецировали раствор кунжутного масла, содержащий 100 мкг ундецилата эстрадиола в объеме 0,1 мл на 100 г массы тела, контрольным животным инъецировали кунжутное масло. В первый, второй и третий дни испытуемым животным вводили один раз в день принимаемую дозу испытуемого соединения, контрольным животным вводили наполнитель для лекарственного средства (ПЭГ 200). Через день после последнего введения животных умерщвляли и их влагалища обрабатывали для гистологической оценки в соответствии со способом, описанным в J. Pharmacol. Exp. Ther. 261(2), 773-779 (1992). Доза, при которой 50% испытуемых крыс показывают полное подавление индуцированного ундецилатом эстратриола эффектов кератинизации, определяется как активная доза. Самая низкая активная доза (СHАД) для соединений данного изобретения перечисляется в табл. 4.
Неожиданное превосходство соединений данного изобретения над самыми близкими соединениями, известными в данной области, демонстрируется в табл. 5. В этой таблице влияние данного соединения под номером 3 на кератинизацию сравнивается с наиболее близкими по структуре, известными в данной области соединениями, причем последние описываются в Европейском патенте 0371564.
Г. Примеры композиций
Следующие готовые лекарственные формы приводятся в качестве примеров типичных фармацевтических композиций, пригодных для системного или местного введения животным или людям в соответствии с данным изобретением. Термин "активный ингредиент" (А.И.), используемый на всем протяжении этих примеров, относится к соединению формулы (I) или его фармацевтически приемлемой кислотно-аддитивной соли.
Пример 17: пероральный раствор
9 г Метил-4-гидроксибензоата и 1 г пропил-4-гидроксибензоата растворяли в 4 л кипящей очищенной воды. В 3 л этого раствора растворяли сначала 10 г 2,3-дигидроксибутандиовой кислоты и затем 20 г А.И. 1 и/или 0,2 г А.И. 2. Последний раствор объединяли с оставшейся частью первого раствора и в него добавляли 12 л 1,2,3-пропантриола и 3 л 70% раствора сорбита. 40 г натриевой соли сахарина растворяли в 0,5 л воды и добавляли 2 мл эссенции малины и 2 мл эссенции крыжовника. Последний раствор объединяли с первым, добавляли воду (сколько нужно) до объема 20 л, обеспечивая получение перорального раствора, содержащего 5 мг А.И. 1 и/или 0,05 А.И. 2 на полную чайную ложку (5 мл). Подходящие контейнеры заполняют получаемым раствором.
Пример 18: пероральные капли
500 г А.И. растворяли в 0,5 л 2-гидроксипропановой кислоты и 1,5 л полиэтиленгликоля при 60-80oC. После охлаждения до 30-40oC туда добавляли 35 л полиэтиленгликоля и смесь хорошо перемешивали. Затем туда добавляли раствор 1750 г натриевой соли сахарина в 2,5 л очищенной воды и при перемешивании добавляли 2,5 л добавки с запахом и вкусом какао и полиэтиленгликоль (сколько нужно) до объема 50 л, обеспечивая получение раствора для перорального введения каплями, содержащего 10 мг/мл А.И. Подходящие контейнеры заполняли полученным раствором.
Пример 19: капсулы
20 г А. И. 1 и/или 0,2 г А.И. 2, 6 г лаурилсульфата натрия, 56 г крахмала, 56 г лактозы, 0,8 г коллоидного диоксида кремния и 1,2 г стеарата магния энергично вместе перемешивали. 1000 подходящих отвержденных желатиновых капсул потом заполняли получаемой смесью, причем каждая капсула содержала 20 мг А.И. 1 и/или 0,2 мг А.И. 2.
Пример 20: инъецируемый раствор
0,5 г А. И. 1 и/или 0,05 мг А.И. 2, 50 мг безводной глюкозы и 0,332 мл концентрированной соляной кислоты смешивали с 0,8 мл воды для инъекции. Для установления pH 3,2±0,1 добавляли едкий натр и воду добавляли до объема 1 мл. Раствор стерилизовали и заполняли в стерильные контейнеры.
Пример 21: таблетки, покрытые пленкой
Получение сердцевины таблетки
Смесь 100 г А.И., 570 г лактозы и 200 г крахмала хорошо смешивали и затем увлажняли раствором 5 г додецилсульфата натрия и 10 г поливинилпирролидона Kollidon-K 90® приблизительно в 200 мл воды. Смесь смоченного порошка просеивали, сушили и просеивали снова. Затем туда добавляли 100 г микрокристаллической целлюлозы Avicel® и 15 г гидрогенизированного масла Sterotex® Всю массу хорошо перемешивали и прессовали в 10000 таблеток, причем каждая содержала 10 мг активного ингредиента.
Покрытие
В раствор 10 г метилцеллюлозы (Methocel 60 HG® ) в 75 мл денатурированного этанола добавляли раствор 5 г этилцеллюлозы Ethocel 22 cps® в 150 мл дихлорметана. Затем туда добавляли 75 мл дихлорметана и 2,5 мл 1,2,3-пропантриола. 10 г полиэтиленгликоля расплавляли и растворяли в 75 мл дихлорметана. Последний раствор добавляли в первый и затем туда добавляли 2,5 г октадеканоата магния, 5 г поливинилпирролидона и 30 мл концентрированной суспензии красителя Opaspray K-1-2109® и всю массу гомогенизировали. Сердцевины таблеток покрывали таким образом полученной смесью в устройстве для покрытия.
Пример 22:2% крем
75 мг стеарилового спирта, 2 мг цетилового спирта, 20 мг моностеарата сорбитана (ангидросорбита) и 10 мг изопропилмиристата вводят в сосуд с рубашкой и нагревают до тех пор, пока смесь полностью не расплавится. Эту смесь добавляют в отдельно приготовленную смесь очищенной воды, 200 мг пропиленгликоля и 15 мг полисорбата 6, имеющую температуру 70-75oC, при использовании гомогенизатора для жидкостей. Получаемую эмульсию оставляют для охлаждения ниже 25oC при непрерывном перемешивании. Раствор 20 мг А.И., 1 мг полисорбата 80 и очищенной воды и раствор 2 мг безводного сульфита натрия в очищенной воде затем добавляют в эмульсию при непрерывном перемешивании. Крем, 1 г А.И. гомогенизируют и наполняют в подходящие тюбики.
Пример 23: гель для местного введения
В раствор 200 мг гидроксипропил-β-циклодекстрина в очищенной воде при перемешивании добавляют 20 мг А.И. До полного растворения добавляют хлористоводородную кислоту и затем добавляют едкий натр до установления pH 6,0. Этот раствор добавляют в дисперсию 10 мг каррагенана PJ в 50 мл пропиленгликоля при перемешивании. При медленном перемешивании смесь нагревают до 50oC и оставляют охлаждаться приблизительно до 35oC, после чего добавляют 50 мг 95% (об. /об. ) этилового спирта. Добавляют остальную очищенную воду (сколько нужно) до 1 г и смесь перемешивают до достижения гомогенности.
Пример 24: крем для местного введения
В раствор 200 мг гидроксипропил-β-циклодекстрина в очищенной воде при перемешивании добавляют 20 мг А.И. До полного растворения добавляют хлористоводородную кислоту и затем добавляют едкий натр до установления pH 6,0. При перемешивании добавляют 50 мг глицерина и 35 мг полисорбата 60 и смесь нагревают до 70oC. Получаемую смесь добавляют в смесь 100 мг минерального масла, 20 мг стеарилового спирта, 20 мг цетилового спирта, 20 мг моностеарата глицерина и 15 мг сорбата 60, имеющую температуру 70oC, при медленном перемешивании. После охлаждения до ниже 25oC добавляют остальную очищенную воду (сколько нужно) до 1 г и смесь перемешивают до достижения гомогенности.
Пример 25: 2% липосомная готовая лекарственная форма
Смесь 2 г А.И., 20 г фосфатидилхолина, 5 г холестерина и 10 г этилового спирта перемешивают и нагревают при 55-60oC до полного растворения и добавляют в раствор 0,2 г метилпарабена, 0,02 г пропилпарабена, 0,15 г двунатриевой соли этилендиаминтетрауксусной кислоты и 0,3 г хлористого натрия в очищенной воде при гомогенизации. Добавляют 0,15 г гидроксипропилметилцеллюлозы в очищенной воде до достижения 100 г, и перемешивание продолжают до достижения полного набухания.
Пример 26: липосомная готовая лекарственная форма
Смесь 10 г фосфатадилхолина, 1 г холестерина и 7,5 г этилового спирта перемешивают и нагревают при 40oC до полного растворения. 2 г тонкоизмельченного А. И. растворяют в очищенной воде смешиванием их при нагревании при 40oC. В водный раствор медленно добавляют спиртовой раствор при гомогенизации в течение 10 минут. При перемешивании до полного набухания добавляют 1,5 г гидроксипропилметилцеллюлозы в очищенной воде. pH получаемого раствора устанавливают 5,0 при помощи 1 N едкого натра и разбавляют остальной (сколько нужно) очищенной водой до достижения 100 г.

Настоящее изобретение относится к соединениям общей формулы I и их стереохимически изомерным формам, где R представляет собой водород, амино, С1-4 алкил, R2 и R3 - водород, галоген, С1-4 алкил, Y - О или S, -X1 = Х2 - представляет двухвалентный радикал, имеющий формулу -N= CH- или -CH=N-. Данное изобретение относится также к способу получения композиции и к самой композиции, которая содержит соединение формулы I, обладающее способностью подавлять эффекты кератинизации. Изобретение касается нового способа получения соединений формулы I путем взаимодействия нитрона формулы II с эфирообразующим реагентом или сульфонилсодержащим электрофильным реагентом, получая соединение формулы I-b-I и затем, если необходимо, превращают соединение формулы I в кислотно-аддитивную соль. 5 c. и 6 з.п. ф-лы, 5 табл.
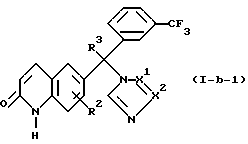
его фармацевтически приемлемая кислотно-аддитивная соль или стереохимически изомерная форма,
где R1 представляет водород, амино или C1-4 алкил;
R2 представляет водород, галоген или C1-4 алкил;
R3 представляет водород, галоген или C1-4 алкил;
Y представляет O или S;
-X1= X2 - представляет двухвалентный радикал, имеющий формулу -N=CH- (a-1) или -CH=N- (a-2).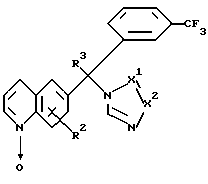
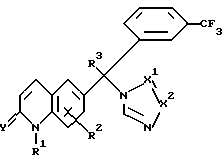
где R2 представляет водород, галоген или C1-4 алкил;
R3 представляет водород, галоген или C1-4 алкил;
-X1= X2 - представляет двухвалентный радикал, имеющий формулу -N=CH- (a-1) или -CH= N- (a-2), его кислотно-аддитивная соль или стереоизомерная форма.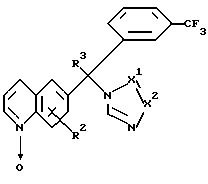
где R2, R3 и -X1=X2- определяется как в п.1,
подвергают реакции с подходящим эфирообразующим реагентом или с сульфонилсодержащим электрофильным реагентом, получая таким образом соединение формулы I-b-1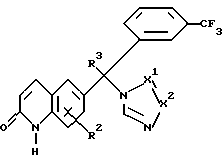
и затем, если необходимо, превращают соединение формулы I в кислотно-аддитивную соль обработкой подходящей кислотой, или превращают кислотно-аддитивную соль в свободное основание обработкой щелочью, и/или получают его стереохимически изомерные формы.
| Способ получения производных (1Н-азол-1-илметил)-замещенных хинолина, хиназолина или хиноксалина или их терапевтически активных нетоксичных кислотно-аддитивных солей или стереохимически изомерных форм | 1989 |
|
SU1780536A3 |
| СПОСОБ УСТРАНЕНИЯ ВЛИЯНИЯ ОСТАТОЧНОЙ | 0 |
|
SU355583A1 |
| КЛИНОВОЙ ЗАЖИМ ДЛЯ ЗАКРЕПЛЕНИЯ РОЛИКОВ И ДРУГИХ ВРАЩАЮЩИХСЯ МАШИННЫХ ЧАСТЕЙ | 1932 |
|
SU30044A1 |
| ГШЕВМО ГИДРАВЛИЧЕСКИ И АККУМУЛЯТОР''и ^^•• | 0 |
|
SU399394A1 |

