ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
В международной заявке WO 95/19956 (British Biotech Pharmaceuticals Ltd) описан и заявляется класс соединений, являющихся ингибиторами матриксных металлопротеаз (ММП) и ингибиторами выделения фактора, вызывающего некроз опухолевых клеток (TNFa) из клеток. Конкретно, заявка раскрывает соединения общей формулы (I)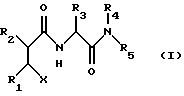
в которой Х обозначает группы -СО2Н или -CONHOH;
R1 обозначает водород; (С1С6)алкил; (С2-С6)алкенил; фенил; замещенный фенил;
фенил(С1-С6)алкил; замещенный фенил(С1-С6)алкил; гетероциклический остаток; замещенный гетероциклический остаток; гетероцикло(С1-С6)алкил; замещенный гетороцикло(С1-С6)алкил; группу ВSОnА, где n обозначает 0, 1 или 2, В обозначает водород или (С1-С6)алкил, фенил, замещенный фенил, остаток гетероцикла, (С1-С6)ацил, фенацильную или замещенную фенацильную группу и А представляет собой (С1-С6)алкил; амино; защищенная аминогруппа; ациламино; ОН; SH; (С1-С6)алкокси; (C1-С6)алкиламино; ди(С1-С6)алкиламино; (С1-С6)алкилтио; арил(С1-С6)алкил; амино(С1- С6)алкил; гидрокси(С1-С6)алкил; меркапто(С1-С6)алкил или карбокси(С1-С6)алкил, где амино-, гидрокси-, меркапто- или карбоксильная группа при необходимости являются защищенными или карбоксильная группа находится в виде амидной; низший карбамоилзамещенный алкил, моно(низший алкил)карбамоил, ди(низший алкил)карбамоил, ди(низший алкил)амино или карбокси-низший-алканоиламино;
R2 обозначает (С1-С6)алкил, (С2-С6)алкенил, (С2-С6)алкинил, фенил(С1-С6)алкил, гетероарил(С1-С6)алкил, циклоалкил(С1-С6)алкильную или циклоалкенил(С1-С6)алкильную группу, любая из которых при необходимости может иметь один или более заместителей, выбранных из (С1С6алкильной, -O(С1-С6)алкильной, -S(C1-С6)алкильной, галоген- или циано (-CN) групп;
R3 обозначает характеристическую группу природной или синтетической α-аминокислоты, в которой любые функциональные группы можно защитить;
R4 обозначает фенильный или 5- или 6-членный гетероарильный цикл, где любой атом азота в цикле может быть окислен до N-оксида, который при необходимости может соединяться с бензольным кольцом или 5-, 6- или 7-членным гетероциклическим кольцом, где любой из циклов может при необходимости быть замещенным с:
(а) одним или более заместителем независимо выбираемым из групп: гидроксил, галоген, -CN, -СО2Н, -СO2(С1-С6)алкил, -(С1-С6)алкил-СO2(С1-С6)алкил,
-CONH2, -СОNН(С1-С6)алкил, -СON(С1-С6)алкил)2, -СНО, -CH2OH, -(С1-C4)перфторалкил, -O(C1-С6)алкил, -SO(С1-С6)алкил, -S(С1-С6)алкил,
SO2(С1-С6)алкил, -NO2, NH, -NH(C1-С6)алкил, -Н(С1-С6)алкил)2 и -NНСО(С1-С6)алкил или
(б) группой, выбираемой из (С1-С6)алкильной, (С2-С6)алкенильной, (С2-С6)алкинильной, (С3-С8)циклоалкильной, (С4-С6)циклоалкенильной, фенильной, бензильной, гетероарильной или гетероарилметильной, причем любая из этих групп может при необходимости иметь один или более заместителей, выбираемых из галогена, гидроксила, амино-, карбоксильной, (С1-С4)-перфторалкильной, (С1-С6)алкильной, -O(С1-С6)алкильной или S(С1-С6)алкильной группы;
R5 обозначает водород или (С1-С6)алкильную группу,
или их соль, гидрат или сольват.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
N1-[2,2-Диметил-1S-пиридин-2-илкарбамоил)пропил] -N4-гидрокси-2R-изобутил-3S-метоксисукцинамид является представителем класса, описываемого и заявляемого в общем в международной заявке WO 95/19956, но ни он, ни его свойства конкретно не охарактеризованы.
В международной заявке WO 95/19956 утверждается, что в описываемом классе соединений ароматический или гетероарильный заместитель R4 вызывает в общем неожиданный и желаемый эффект увеличения активности по отношению к стромелизину по сравнению с известными соединениями практически аналогичного строения, но имеющими другие (обычно метальные) заместители R4, при сохранении активности в отношении коллагеназы и желатиназы. Будучи представителем этого класса, выбранное в данном случае соединение N1-[2,2-диметил-1S-(пиридин-2-илкарбамоил)пропил]- N4-гидрокси-2R-изобутил-3S-метоксисукцинамид обладает этими свойствами.
Однако известно, что некоторые ингибиторы ММП вызывают побочный эффект, мышечную боль (также иногда называемую тендинитом) в суставах, например в плечевом, у некоторых животных и людей, после высоких и/или пролонгированных доз. Хотя считается, что этот эффект является практически обратимым при прекращении приема, он, тем не менее, нежелателен. Механизм возникновения боли до настоящего времени непонятен, и далеко не кажется возможным коррелировать предрасположенность соединения вызывать боль с конкретными особенностями строения молекулы или с ее профилем ингибирования фермента. Соответственно, нельзя заранее предсказать, будет ли какой-либо данный ингибитор ММП вызывать побочный эффект и, в случае его появления, тяжесть этого эффекта, и это нужно определять эмпирически.
В настоящее время найдено, что выбранный N1-[2,2-диметил-1S-(пиридин-2-илкарбамоил)пропил] -N4-гидрокси-2R-изобутил-3S-метоксисукцинамид обладает резко пониженной тенденцией вызывать побочный эффект - мышечную боль. В этом отношении он отличается от близких по строению аналогов, например N1-[2,2-диметил-1S-(пиpидин-3-илкapбaмoил)-пpoпил] -N4-гидpoкcи-2R-изoбyтил-3S-гидpoкcисукцинамида, более склонного вызывать этот побочный эффект.
В заявке WO 95/19956 также утверждается, что описываемый класс ариламидных ингибиторов ММП включает соединения, которые являются биодоступными при пероральном приеме. Не все соединения, охватываемые заявкой WO 95/19956, являются биодоступными в приемлемой степени при пероральном введении, что доказывается, например, уровнем пика ММП-ингибиторной активности, или уровнем активности во времени ("площадь под кривой"), отнесенным к лекарственному препарату крови животных при пероральном его введении. Было найдено, что соединение, выбранное в соответствии с данным изобретением, N1-[2,2-диметил-1S-(пиридин-2-илкарбамоил)пропил] -N4-гидрокси-2R-изобутил-3S-метоксисукцинамид при пероральном введении является биодоступным в организме человека и в организме других млекопитающих.
Это сочетание пероральной доступности и пониженной склонности вызывать тендинит в качестве побочного эффекта предполагает, что соединение по изобретению должно иметь относительно обширную область применения в терапии для лечения заболеваний, требующих системного введения ингибитора ММП в течение умеренного или более длительного времени. Следовательно, соединение показано для лечения, например, ревматоидного артрита, злокачественных опухолей, рассеянного склероза (PC), синдрома Гийена-Барре-Штроля (ГБС) и псориаза.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Следовательно,данноеизобретениепредставляетсобойN1-[2,2-диметил-1S-(пиридин-2-илкарбамоил)пропил] -N-гидрокси-2R-изобутил-3S-метоксисукцинамид формулы II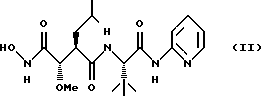
и его фармацевтически приемлемые соли, гидраты и сольваты.
В соединении по изобретению имеются три асимметрических атома углерода, стереохимическая конфигурация которых отражена в названии соединения и проиллюстрирована формулой (II). Однако следует понимать, что изобретение включает смеси энантиомеров соединения (II) при условии, что названный энантиомер преобладает. Предпочтительно, чтобы в такой энантиомерной смеси 90 вес.% или более составлял названный энантиомер.
Соли соединения по изобретению включают физиологически приемлемые соли, получающиеся при присоединении кислот, например гидрохлорид, гидробромид, сульфат, метансульфонат (мезилат), n-толуолсульфонат (тозилат), фосфат, ацетат, цитрат, сукцинат, лактат, тартрат, фумарат и малеат. Соли могут образовываться с основаниями, например натриевые, калиевые, магниевые и кальциевые соли.
Выбранные соединения по изобретению можно получать представленным в примере данного описания способом и можно вводить любым способом, согласующимся с его фармакокинетическими свойствами. Вводимые перорально композиции могут быть в форме таблеток, капсул, порошков, гранул, лепешек, жидких или гелеобразных препаратов, таких как растворы или суспензии для перорального, местного применения или стерильные растворы или суспензии для парентерального введения. Таблетки и капсулы для перорального введения могут быть в виде стандартной дозы и могут содержать подходящие эксципиенты, такие как связующие, наполнители, смазки для таблетирования, расщепляющие вещества или приемлемые увлажнители. Таблетки могут покрываться оболочкой способами, хорошо известными в обычной фармацевтической практике. Жидкие препараты для перорального введения могут быть, например, в виде суспензий в воде или в масле, растворов, эмульсий, сиропов или эликсиров или могут быть в сухом виде и перед употреблением смешиваться ("воссоздаваться") с водой или другим подходящим носителем. Такие жидкие препараты могут содержать соответствующие добавки, такие как суспендирующие агенты; эмульгаторы; неводные носители (которые могут содержать пищевые масла); консерванты и при необходимости соответствующие вкусовые добавки и красители.
Активные ингредиенты могут также вводиться парентерально в стерильной среде. В зависимости от применяемого носителя и концентрации лекарственный препарат может суспендироваться или растворяться в носителе. Адъюванты, например такие, как анестетики для местной анестезии, консерванты и буферизаторы, могут растворяться в носителе.
Доза соединения по изобретению при любом данном клиническом показании и при данном способе введения определяется клиническими испытаниями в соответствии с обычной практикой при условии одобрения соответствующими властями. В целом, в настоящее время полагают, что соединение следует вводить человеку перорально в дозировке 5-100 мг два или три раза в день.
Следующий пример описывает получение соединения по изобретению. Исходные вещества - 2R-(2,2-диметил-5-оксо[1,3]диоксолан-4S-ил)-4-метилпентановую кислоту и L-трет-лейцин-N-(2-пиридил)амид - получают, как описано в международной заявке WO 95/19961.
В примере используются следующие сокращения:
ДМФА - N,N-диметилформамид,
EDC - гидрохлорид N-этил-Nу-(3- диметиламинопропил)карбодиимида,
HOBt - 1-гидроксибензотриазол,
NMM - N-метилморфолин,
ТГФ - тетрагидрофуран.
ПРИМЕР
N1-[2,2-Диметил-1S-пиридин-2-илкарбамоил)пропил]-N4-гидрокси-2R-изобутил-3S-метоксисукцинамид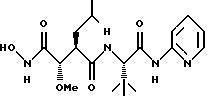
Стадия А. Диметиловый эфир 2S-гидрокси-3R-изобутилсукциновой кислоты.
2R-(2,2-Диметил-5-оксо[1,3] диоксолан-4S-ил)-4-метилпентановую кислоту (75.0 г, 0.326 ммоль) растворяют в метаноле (500 мл) и охлаждают до 0oС и полученный раствор насыщают газообразным хлористым водородом. Реакционную смесь доводят до комнатной температуры и перемешивают в течение ночи. Растворитель удаляют при пониженном давлении, остаток растворяют в хлористом метилене и промывают последовательно насыщенным раствором бикарбоната натрия и рассолом. Органическую фазу сушат над безводным сульфатом магния, отфильтровывают и упаривают досуха при пониженном давлении с получением вышеназванного соединения в виде желтого масла (53 г, 75%).
1Н-ЯМР, δ (СDС13): 4.10 (1Н, д, J=4.0 Гц), 3.60 (3Н, с), 3.50 (3Н, с), 3.18 (ш.с.), 2.78 (1Н, м), 1.61-1.40 (2Н, м), 1.28 (1Н, м) и 0.76-0.73 (6Н, м).
Стадия В. Диметиловый эфир 2R-изобутил-3S-метоксисукциновой кислоты.
Диметиловый эфир 2S-гидрокси-3R-изобутилсукциновой кислоты (23.9 г, 110 ммоль) растворяют в ДМФА (200 мл) и добавляют перегнанный иодистый метил (8.2 мл, 132 ммоль), а затем окись серебра (I) (27.95 г, 121 ммоль). Реакционную массу перемешивают в течение 7 дней при комнатной температуре в темноте. Растворитель удаляют при пониженном давлении и остаток очищают с помощью колоночной хроматографии (силикагель, хлористый метилен в качестве растворителя) с получением вышеназванного соединения в виде вязкого желтого масла (19.16 г, 75%).
1Н-ЯМР, δ (CDCl3): 3.83 (1Н, д, J=7.5 Гц), 3.71 (3Н, с), 3.62 (3Н, с), 3.30 (3Н, с), 2.85 (1Н, м), 1.65-1.39 (2Н, м), 1.10 (1Н, м) и 0.83-0.81(6Н, м).
Стадия С. Дилитиевая соль 2R-изобутил-3S-метоксисукциновой кислоты.
Гидроксид лития (1.76 г, 42.0 ммоль) добавляют к раствору диметилового эфира 2R-изобутил-3S-метоксисукциновой кислоты (4.70 г, 20.0 ммоль) в метаноле (30 мл) и воде (30 мл). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов, затем удаляют растворитель при пониженном давлении с получением вышеназванного соединения в виде твердого вещества желтого цвета (4.40 г, количественный выход).
1Н-ЯМР, δ (СD3ОD): 3.52 (1Н, д, J=5.1 Гц), 3.27 (3Н, с), 2.65 (1Н, м), 1.56-1.53 (2Н, м), 1.31 (1Н, м) и 0.82-0.78 (6Н, м).
Стадия D. 4-Метиловый эфир 2R-изобутил-3S-метоксисукциновой кислоты.
Дилитиевую соль 2R-изобутил-3S-метоксисукциновой кислоты (25.14 г, 116 ммоль) растворяют в сухом ТГФ (150 мл) и охлаждают раствор до 0oС. Добавляют трифторуксусный ангидрид (30 мл) и смесь перемешивают при 0oС дополнительно 4 часа. Растворитель удаляют при пониженном давлении, остаток растворяют в безводном метаноле (200 мл) при 0oС и раствор перемешивают в течение ночи при комнатной температуре. Растворитель удаляют при пониженном давлении с получением вышеназванного соединения в виде желтого масла (54.3 г, включая 2 эквивалента литиевой соли трифторуксусной кислоты), которое используют в стадии Е без дополнительной очистки.
1Н-ЯМР, δ (CD3OD): 7.71 (1H, д, J=7.5 Гц), 3.65 (3Н, с), 3.24 (3Н, с), 2.72 (1H, м), 1.56-1.42 (2Н, м), 1.06 (1H, м) и 0.81-0.79 (6Н, м).
Стадия Е. Метиловый эфир 3R-[2,2-диметил-1S-(пиридилкарбамоил)пропилкарбамоил]-2S-метокси-5-метилгексановой кислоты.
Продукт из стадии D (25.06 г, эквивалентны 53,7 ммоль 4-метилового эфира 2R-изобутил-3S-метоксисукциновой кислоты) растворяют в ДМФА (200 мл) и раствор охлаждают до 0oС, добавляя HOBt (8.70 г, 53.7 ммоль) и затем EDC (12.35 г, 64.4 ммоль). Смесь перемешивают и спустя 2 часа доводят до комнатной температуры. Раствор активного эфира, полученный таким образом, охлаждают до 0oС, добавляют L-трет-лейцин-N-(2-пиридил)амид (11.11 г, 53.7 ммоль) и смесь перемешивают в течение ночи при комнатной температуре. Растворитель удаляют при пониженном давлении, остаток растворяют в этилацетате. Промывают раствор последовательно 1М раствором карбоната натрия и рассолом, сушат над безводным сульфатом магния, фильтруют и упаривают досуха. Остаток очищают с помощью колоночной хроматографии (силикагель, от 0 до 5% метанола в хлористом метилене) с получением вышеназванного соединения в виде белого твердого вещества (13.41 г, 61%).
1Н-ЯМР, δ (CDCl3): 9.61 (1H, с), 8.24 (1H, д, J=8.4 Гц), 7.74 (1H, м), 7.07 (1H, м), 6.97 (1H, д, J=8.9 Гц), 4.64 (1H, д, J=8.9 Гц), 4.01 (1H, д, J=7.6 Гц), 3.76 (3Н, с), 3.41 (3Н, с), 2.75 (1H, м), 1.79 (1H, м), 1.51 (1Н, м), 1.11(1Н, м), 1.02 (9Н, с), 0.84 (3Н, д, J=6.3Гц) и 0.82 (3Н, д, J=6.3 Гц).
Стадия F. Литиевая соль 3R-[2,2-диметил-1S-(пиридин-2-илкарбамоил)пропилкарбамоил]-2S-метокси-5-метилгексановой кислоты.
Метиловый эфир 3R-[2,2-диметил-1S-(пиридилкарбамоил)пропилкарбамоил]-2S-метокси-5-метилгексановой кислоты (13.4 г, 32.9 ммоль) растворяют в ТГФ (265 мл) и воде (65 мл) и добавляют моногидрат гидроксида лития (1.396 г, 33.3 ммоль). Раствор перемешивают при комнатной температуре в течение 2 часов, затем упариванием при пониженном давлении и получают желтое масло, которое сушат при помощи азеотропной отгонки с толуолом. Продукт (13.4 г, содержащий остатки растворителя) немедленно используют без дополнительной очистки.
1Н-ЯМР, δ (CD3OD, смесь диастереомеров 3.5:1): 8.31 (1Н, м), 7.99 (1H, д, J=8.3 Гц), 7.66 (1H, м), 7.00 (1Н, м), 4.49 (0.23Н, с; минорный диастереомер), 4.37 (0.77Н, с; основной диастереомер), 3.68 (0.23Н, д, J=6.7 Гц; минорный диастереомер), 3.52 (0.77Н, д, J=6.7 Гц; основной диастереомер), 3.21 (0.69Н, с; минорный диастереомер), 3.20 (2.31Н, с; основной диастереомер), 2.64 (1H, м), 1.53 (2Н, ш.с.), 1.18 (1H, м), 1.01 (9Н, с), 0.85 (3Н, д, J= 6.4 Гц) и 0.81 (3Н, д, J=6.3 Гц).
Стадия G. N4-Бензилокси-N1-[2,2-диметил-1S-пиридин-2-илкарбамоил]пропил-2R-изобутил-3S-метоксисукцинамид.
Продукт cтадии F (13.4 г, 33 ммоль) растворяют в сухом ДМФА (250 мл), помещают в атмосферу аргона и охлаждают до -10oС с перемешиванием. Добавляют по каплям этилхлорформиат (3.47 г, 36 ммоль), затем NMM (1.8 мл, 16.5 ммоль). Смесь перемешивают в течение 30 минут и затем по каплям добавляют O-бензилгидроксиламин (6 г, 49 ммоль) в ДМФА (10 мл). Реакционную смесь доводят до комнатной температуры и перемешивают в течение ночи. Растворитель удаляют при пониженном давлении, а остаток распределяют между этилацетатом и рассолом. Органический слой промывают 1М раствором карбоната натрия, сушат над сульфатом магния, фильтруют и упаривают. Остаток очищают флеш-хроматографией (силикагель, 5% метанола в хлористом метилене). Фракции, содержащие нужный продукт, собирают и упаривают. Продукт насыщают диэтиловым эфиром, чтобы удалить слабо окрашенные примеси. Выход: 9.44 г (58%, смесь диастереомеров >10:1).
1Н-ЯМР, δ (СDС13, основной диастереомер): 10.26 (1Н, с), 9.93 (1Н, с), 8.32 (1Н, м), 8.23 (1Н, д, J=8.2 Гц), 7.63 (1Н, м), 7.25 (5Н, м), 7.12 (1Н, д, J=9.2 Гц), 7.02 (1Н, м), 4.94 (1Н, д, J=10.8 Гц), 4.76 (2Н, д, J=3.8 Гц), 3.88 (1Н, д, J=5.5 Гц), 3.32 (3Н, с), 2.91 (1Н, м), 1.72 (1Н, м), 1.55 (1Н, м), 1.35 (1Н, м), 1.02 (9Н, с), 0.89 (3Н, д, J=6.5 Гц) и 0.85 (3Н, д, J=6.5 Гц).
Стадия Н. N1-[2,2-Диметил-1S-(пиридин-2-илкарбамоил)пропил]-N4гидрокси-2R-изобутил-3S-метоксисукцинамид.
N4-Бензилокси-N1-[2,2-диметил-1S-(пиридин-2-илкарбамоил)пропил]-2R-изобутил-3S-метоксисукцинамид растворяют в смеси метанола (75 мл) и этанола (75 мл) и помещают в атмосферу аргона. Добавляют 10%-ный палладий на угле и через раствор пропускают водород в течение 2 часов, после чего ТСХ-анализ показывает, что весь исходный продукт прореагировал. Систему продувают аргоном и катализатор удаляют фильтрованием. Растворитель удаляют при пониженном давлении с получением вышеназванного соединения в виде белого твердого вещества (8.8 г, количественный выход; смесь диастереомеров 12:1). Т. пл. 163-164oС.
1Н-ЯМР, δ ((CD3)2SO): 10.67 (1H, с), 10.13 (1H, с), 8.90 (1H, с), 8.15 (1H, м), 7.89 (1H, д, J=8.4 Гц), 7.81 (1H, д, J=8.8 Гц), 7.60 (1H, м), 6.93 (1H, м), 4.53 (0.12Н, д, J=9.4 Гц), 4.43 (0.88Н, д, J=8.8 Гц), 3.74 (0.12Н, д, J= 10.0 Гц), 3.32 (0.88Н, д, J=9.8 Гц), 2.98 (0,3Н, с), 2.96 (2.64Н, с), 2.78 (1H, м), 1.23 (2Н, м), 0.84 (10Н, с и м), 0.65 (3Н, д, J=6.4 Гц) и 0.56 (3Н, д, J= 6.3 Гц). 13С-ЯМР, δ (СD3OD): 172.6, 170.0, 166.0, 151.5, 147.9, 136.0, 119.5, 113.6, 61.2, 60.6, 56.7, 46.2, 37.0, 34.0, 26.5, 25.2, 23.7 и 21.7. ИК, nмакс (KBr): 3255, 2957, 1700, 1645, 1524, 1467, 1435, 1370, 1301, 1213 и 1152 см-1.
Найдено, %: С 58.40; Н 7.92; N 13,61. С20Н32N4O5•0.2 Н2O. Найдено,%: С 58.29; Н 7.92; N 13.60.
БИОЛОГИЧЕСКИЙ ПРИМЕР А
Эффективность соединения по изобретению в качестве ингибитора интерстициальной коллагеназы определяют по методу Cawston and Barrett (Anal. Biochem. , 99, 340-345, 1979), в соответствии с которым 1 мМ раствор испытуемого соединения или его разведение термостатируют при 37oС в течение 16 часов с коллагеном и коллагеназой (буферированными 25 мМ Hepes, рН 7.5, содержащей 5 мМ CaCL2, 0.05% Brij 35 и 0.02% NаN3). Коллаген представляет собой ацетилированный 14С коллаген, приготовленный по методу Cawston and Murphy (Methods in Enzvmology, 80, 711, 1981), вводимому в данное описание в качестве ссылки. Образцы центрифугируют для седиментации негидролизованного коллагена и отбирают аликвотную часть надосадочной жидкости для определения числа сцинтилляций (с помощью сцинтилляционного счетчика) как степени (меры) гидролиза. Активность коллагеназы в присутствии 1 мМ испытуемого соединения или его разведения сравнивают с активностью контрольного (образца), без ингибитора, и результат, представленный ниже, дан как концентрация ингибитора, вызывающая 50%-ное ингибирование активности коллагеназы (IС50).
Эффективность соединения по изобретению в качестве ингибитора стромелизина-1 определяют по методике Cawston et al. (Biochem. J., 195, 159-165, 1981), в соответствии с которой 1 мМ раствор испытуемого соединения или его разведение термостатируют при 37oС в течение 16 часов со стромелизином и 14С-ацетилированным казеином (буферированными 25 мМ Hepes, рН 7.5, содержащей 5 мМ СаС2, 0.05% Brij 35 и 0.02% NaN3). Казеин представляет собой ацетилированный 14С-казеин, приготовленный по методу Cawston et al (ibid). Активность стромелизина в присутствии 1 мМ испытуемого соединения или его разведения сравнивают с активностью в контрольном образце, не содержащем ингибитора, и результаты, представленные ниже, выражены как концентрация ингибитора, вызывающая 50%-ное ингибирование активности стромелизина (IC50).
Эффективность соединения по изобретению в качестве ингибитора 72 kDa-желатиназы определяют по методике, основанной на методе Sllers et al., Biochem. J. , 171, 493-496 (1979). kDa-желатиназу, полученную из клеток RPMI-7951, очищают с помощью хроматографии на желатин-агарозном геле. Фермент активируют термостатированием с аминофенилртутьацетатом и примерно 0.05 единиц термостатируют с 50 мкг меченого [14С]-желатина в соответствующем буфере в течение 16 часов при 37oС. В конце термостатирования добавляют 50 мкг бычьего сывороточного альбумина вместе с трихлоруксусной кислотой (конечная концентрация 16%) для прекращения реакции и осаждения нерасщепленного субстрата. Реакционные пробирки помещают в лед на 15 минут перед тем, как центрифугировать в течение 15 минут при 10 000 об/мин для седиментации осаждающегося субстрата. Берут аликвотную часть 200 мкл надосадочной реакционной жидкости и определяют радиактивность с помощью сцинтилляционного счетчика для жидкостей. Действие ингибитора определяют по кривой эффекта дозы. IC50 (концентрация ингибитора, требующаяся для 50%-ного уменьшения активности фермента) определяют, вычерчивая кривую по данным и вычисляя концентрацию ингибитора, требуемую для достижения 50%-ного ингибирования фермента. Для каждого определения IС50 определяют действие ингибитора на активность желатиназы при примерно 8 концентрациях ингибитора. Ингибиторы растворяют и разводят (разбавляют) в ДМСО.
Результаты вышеописанных испытаний соединения по изобретению следующие:
Фермент - IC50(нM)
Коллагеназа - 10
kDa-желатиназа - 70
Стромелизин - 30
БИОЛОГИЧЕСКИЙ ПРИМЕР Б
Измеряют концентрацию соединения по изобретению в крови лабораторных животных во времени после введения испытуемого соединения. Соединение вводят через зонд группе из 6 подопытных самцов крыс (вес 300 г). Образцы крови отбирают венепункцией хвостовой вены через 0.5, 1.0, 2.0, 6.0 и 24 часа после введения. 0.4 мл крови помещают в пробирки на 4.5 мл, содержащие 0.1 мл кислого цитрата декстрозы (ACD) в качестве антикоагулянта. Для экстракции добавляют 3 мл метанола и осаждающаяся кровь гранулируется при центрифугировании (30 мин, 3000 об/мин). Отбирают аликвотную часть 2 мл надосадочной жидкости и концентрируют лиофилизацией. Экстракт снова растворяют в 200 мкл ДМСО и отбирают аликвоту 10 мкл для анализа активности ингибирования коллагеназы. Ингибиторную активность экстрактов определяют, используя метод анализа коллагеназы, описанный выше, в Биологическом Примере А, и концентрацию ингибитора (т. е. лекарственного препарата плюс любых активных метаболитов) получают сравнением со стандартной кривой. Результаты выражаются в виде максимальной концентрации в нг/мл х часы через 0.5-24 часа и в виде AUC в числе IC50 x часы. Результаты приведены в табл.1.
Соединение по изобретению также испытывают, вводя его перорально игрункам и людям-добровольцам, при этом отмечается высокая степень ингибиторной активности в крови испытуемых после такого введения.
БИОЛОГИЧЕСКИЙ ПРИМЕР В
Действие соединения по изобретению при злокачественных новообразованиях испытано на двух видах подопытных животных, при этом обнаружена его активность.
Модель: меланома у мышей В16
В этом случае испытуемым мышам C57/BL6J вводят подкожно 105 клеток меланомы мыши В16. Опухоль растет в течение 18 дней и вес опухоли вычисляют с помощью микрометра (штангельциркуля) по следующей формуле: вес (мг) - длина (мм) х ширина (мм) ÷ 4. Группа мышей (n=19) получает соединение, вводимое с помощью осмотического мини-насоса, имплантированного подкожно на стороне, противоположной той, где находится В16-опухоль. Насос имплантируют за день до введения (посева) опухолевых клеток, а доза вводимого соединения составляет 360 мкг/день. Контрольная группа (n=17) получает соответствующий объем носителя с помощью такого же имплантированного насоса.
Средний вес контрольной опухоли через 18 дней составляет 1145±97 мг. Вес опухоли у мышей, получавших соединение по изобретению, составляет 774±50 мг. Это снижение веса (32% в случае испытуемого соединения) является значительным (р<0.005). Оценка образцов в конце эксперимента показывает, что уровень плазмы составляет 26.8±3.2 нг/мл в случае испытуемого соединения.
БИОЛОГИЧЕСКИЙ ПРИМЕР Г
Соединение по изобретению испытывают на его предрасположенность вызывать видимые признаки тендинита у крыс. Способность соединения по изобретению (Соединение А) вызывать тендинит у крыс ("тендинит-эффект") сравнивают с таковой близкого по строению изомера, а именно N1-[2,2-диметил-1S-пиридин-3-илкарбамоил)пропил]N4гидрокси-2R-изобутил-3S-гидроксисукцинамида (Соединение Б).
Берут самцов крыс Льюиса. В каждом эксперименте до 30 крыс взвешивают и произвольно распределяют на группы вне зависимости от веса тела, n=6/группа. В каждом эксперименте одной группе дают соответствующий носитель для сравнения с каждой группой, получающей лекарственное средство.
Соединение А берут в виде мезилата (соль метансульфоновой кислоты), тогда как Соединение Б находится в виде свободного основания. В случае Соединения А @ 15 мг/мл носитель представляет собой: 50% ДМСО, 37% 0.1 М метансульфокислота и 13% стерилизованная вода; и @ 30 мг/мл носитель: 50% ДМСО, 37% 0.2 М метансульфокислота и 13% стерилизованная вода. Для соединения Б @ 15 мг/мл носитель представляет собой 50% ДМСО/вода.
Alzet (торговая марка) осмотические мининасосы (2ML2, 14 дней, 5 мкл/час от Akza Corp., Palo Alto CA 94303) взвешивают пустыми и заполненными, чтобы быть уверенными в правильности заполнения объема. Перед имплантацией заполненные насосы помещают в термостат при 37oС. Всех крыс анестезируют с помощью галотана. После анестезии загривок выбривают и дезинфицируют. На подготовленной стороне делают продольный разрез, примерно 2 см длиной, и с помощью пары кровоостанавливающих щипцов под кожей делают подкожный карман. Мининасосы помещают так, чтобы выходное отверстие насоса было обращено наружу от раны. На разрез накладывают швы и снова дезинфицируют область вокруг раны. Сразу после операции всем крысам дают анестетик (Temgesic-trade mark-Reckitt and Colman), 0.1 мг/кг подкожно в бок. После того, как кончится действие анестетика, животных возвращают в клетки с сухой подстилкой, чтобы гарантировать, что рана остается чистой до заживления. На следующий день после хирургического вмешательства всех крыс помещают назад на стандартную подстилку и размещают по группам из 3, чтобы можно было более четко наблюдать за признаками тендинита.
После имплантации мини-насосов крыс взвешивают и контролируют возможное начало тендинита. Начало и тяжесть тендинита измеряют с помощью наблюдений, основанных на системе оценок (см. ниже). Крыс наблюдают ежедневно в течение 16 дней. Средние оценки >2 считаются значительными. Применяемая система оценок такова:
Положение лежа
Нормальное - 0
Опирается на одну лапу - 1
Не опирается на лапы - 2
При стимуляции движения животные
Двигаются нормально - 0
Неохотно двигаются - 1
Двигаются с умеренным нежеланием - 2
Очень неохотно двигаются - 3
Походка
Нормальная - 0
Избегают пользоваться одной задней лапой - 1
Избегают пользоваться обеими задними лапами - 2
Результаты показывают, что крысы, получающие дозу Соединения А (как 15 мг/мл, так и 30 мг/мл) и только носитель, не проявляют заметных признаков тендинита, тогда как крысы, получающие дозу Соединения Б, проявляют заметные признаки, начиная с 8 дня и далее (средние оценки >2 на 8 день увеличиваются до 6 на 15 и 16 день).
Было подтверждено, что крысы, получающие соединения А и Б в вышеописанных тестах, подвергаются воздействию соединений из мини-насосов на плазму. Отбирают образцы крови на 3 и 10 дни после имплантации мини-насосов после слабой анестезии крыс голотаном из хвостовой вены. Образцы крови (0.5 мл) помещают в пробирки, содержащие 3.0 мл метанола для экстракции свободного и связанного соединения. Концентрацию в крови Соединения А и Соединения Б определяют методом флуорометрии с применением кумарин-пептидного субстрата Mca-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2 (Mca=(7-метоксикумарин-4-ил)ацетил, Dpa= Н-3-(2,4-динитрофенил)-L-2,3-диаминопропил) (см. Knight et al., FEBS Lett., 1992, 296, 263-266). На 16 или 17 день после имплантации эксперимент заканчивают, крыс окончательно усыпляют и берут образцы крови (0.5 мл) сердечной пукцией и определяют концентрацию Соединения А и Соединения Б.
БИОЛОГИЧЕСКИЙ ПРИМЕР Д
На подопытных животных ГБС (GBS) испытывают действие соединения по изобретению. Экспериментальный аутоиммунный неврит (ЭАН, EAN) представляет собой опосредуемое Т-клетками аутоиммунное расстройство периферической нервной системы. У подопытных животных проявляются многие патологические особенности ГБС (GBS) с симптомами атаксии, слабости и паралича. EAN можно вызвать у животных, вводя им инъекцию миелина периферических нервов или белковых компонентов миелина, например протеин 0, в адъюванте. EAN-повреждения происходят в спинальных корешках и периферических нервах и характеризуются инфильтрацией периваскулярных мононуклеарных клеток, демиелинизацией аксонов и нарушением нервной проводимости. В патологии GBS и EAN участвует TNFa; уровни TNFa в крови у больных GBS поднимаются и антитела к TNFa уменьшают тяжесть заболевания при EAN (Hartung HP. Annals of Neurology, 1993, 33, 563-567).
Соединение по изобретению испытывают на крысах с EAN, симптомы заболевания вызваны инъекцией миелина периферических нервов в адъюванте. Соединение вводится с помощью имплантированных хирургическим путем мини-насосов с концентрацией 15 или 30 мг/мл со скоростью 5 мкл/час (см. Биологический пример Г), доставка соответственно 7 и 14 мг/кг в день. Терапия соединением в течение всего эксперимента, начиная с 1 до 15 дня, значительно ослабляет проявление клинических симптомов в зависимости от дозы. Соединение также ослабляет, в зависимости от дозы, клинические симптомы в случае EAN-модели при терапевтическом введении с 8 по 15 день при концентрации 15 или 30 мг/мл со скоростью 10 мкл/час и доставке 14 и 28 мг/кг в день соответственно.
БИОЛОГИЧЕСКИЙ ПРИМЕР Е
Активность соединения по изобретению в случае экспериментальной модели PC (МС).
Метод.
Применяется Модель PC с гиперчувствительностью замедленного типа, описанная Matyszak and Perry (Matyszak M.K. and Perry V.H., 1995. Demyelination in the central nervous system following a delayed type hypersensitivity response to bacillus Calmette-Guerin. Neuroscience 64, 967-977). Анестезируют самцов крыс Льюиса, вводя инъекции 1 мкл физиологического раствора с фосфатньм буфером, содержащего 105 убитых термически бацилл Calmette Guerin (BCG) в полосатое тело левого полушария. BCG вводят стереотаксически с помощью шприца 27G на 10 мкл. Координаты для инъекции: брегма +1.2 мм, латерально (в бок)+3.0 мм и глубина 4.5 мм. Через 4 недели вводят внутрикожно (интрадермально) в заднюю лапу 200 мкл раствора, содержащего 107 термически убитых BCG-организмов в полном адъюванте Фрейнда. Еще через 15 дней крысам вводят внутривенно инъекцию 2750 Ед. пероксидазы хрена тип II (HRP). Через 30 минут крыс более глубоко анестезируют с помощью пентобарбитала натрия и осуществляют транскардиально перфузию 100 мл 0.9% (вес/объем) NaCI, содержащего 5000 Ед. /л гепарина, а затем 200 мл параформальдегид-лизин-периодатного фиксатора (PLP), содержащего 2% параформальдегида и 0.05% глутаральдегида. Мозг удаляют, вторично фиксируют еще в течение 4 часов в PLP и подвергают криозащите, погрузив в 30% раствор сахарозы на ночь при 4oС перед тем, как поместить в Tissue-Tek О.С.Т. (miles inc Elkhart USA) и заморозить в жидком азоте. Для HRP-локализации по методу Hanker-Yates (Perry V.H. et al. , 1992) делают подвижные венечные срезы толщиной 50 мкм. Эта процедура вызывает гиперчувствительность замедленного типа в месте стереотаксической инъекции BCG, характеризующуюся локальным нарушением гематоэнцефалического барьера, на что указывает окрашивание за счет внесосудистого присутствия HRP; инфильтрацией лимфоцитов, на что указывает окрашивание антителами ОХ-22, специфическими к высокомолекулярным формам общих лейкоцитарных антигенов; активацией лейкоцитов, на что указывает окрашивание антителами ОХ-6, специфическими к МНС-антигенам и расщеплением миелина, на что указывает окрашивание с помощью антител к основному миелиновому белку. Все эти особенности являются признаками активных патологических изменений или бляшек, наблюдаемых в центральной нервной системе больных PC (MS). Число лимфоцитов определяют, подсчитывая количество ОХ-22-позитивных клеток на участке, где окрашивание наиболее интенсивно. Число клеток на единичном участке в поле зрения записывают и выражают как клетки/мм2. Области экспрессии МНС класс II, утечки через гематоэнцефалический барьер и демиелинизации рассчитывают, применяя автоматизированный (компьютерный) визуальный анализ и выходные данные в мм2.
Действие соединения по изобретению оценивают, давая его крысам перорально 30 мг/кг дважды в день, начиная с 5 дня после второй инъекции BCG и продолжая давать до 15 дня. Соединение по изобретению дают в физиологическом растворе с фосфатньм буфером в качестве носителя, содержащем 0.01% Tween 80. Контрольным животным дают только носитель.
Области утечки через гематоэнцефалический барьер, экспрессии МНС класс II, демиелинизации и число Т-клеток у животных, принимающих соединение по изобретению, сравнивают с контрольными, получающими носитель, применяя критерий Т Стьюдента.
Результаты. DTH-ответ (замедленная гиперчувствительность) у животных, получавших носитель, характеризуется разрушением гематоэнцефалического барьера, инфильтрацией лимфоцитов, экспрессией МНС класс II и демиелинизацией. У животных, получавших соединение по изобретению, наблюдается заметное снижение площади утечки через гематоэнцефалический барьер (р<0.05) и показателя инфильтрации Т-клеток (р<0.0001). Уменьшение демиелинизации и экспрессии МНС класс II наблюдается, но не достигают статистически значимой величины.
Действие соединения по изобретению на DTH-модели PC см. в табл.2.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЦИТОСТАТИЧЕСКИЕ АГЕНТЫ | 1998 |
|
RU2187499C2 |
| ПРОИЗВОДНЫЕ ГИДРОКСАМОВОЙ ИЛИ КАРБОНОВОЙ КИСЛОТЫ И ФАРМАЦЕВТИЧЕСКАЯ ИЛИ ВЕТЕРИНАРНАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1995 |
|
RU2136657C1 |
| АНТИБАКТЕРИАЛЬНЫЕ АГЕНТЫ | 2000 |
|
RU2269525C2 |
| АНТИБАКТЕРИАЛЬНЫЕ АГЕНТЫ | 1999 |
|
RU2246941C2 |
| ПРОИЗВОДНЫЕ ГИДРОКСАМОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ ИЛИ ВЕТЕРИНАРНАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1993 |
|
RU2126791C1 |
| ПИРРОЛОПИРИДАЗИНОВЫЕ ПРОИЗВОДНЫЕ | 2001 |
|
RU2254335C2 |
| ИЗОСЕРИНОВЫЕ ПРОИЗВОДНЫЕ ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ФАКТОРА СВЕРТЫВАНИЯ КРОВИ IXA | 2007 |
|
RU2446157C2 |
| МОДУЛЯТОРЫ MAS-СВЯЗАННОГО РЕЦЕПТОРА G-БЕЛКА X2 И СВЯЗАННЫЕ С НИМИ ПРОДУКТЫ И СПОСОБЫ | 2021 |
|
RU2841536C1 |
| ПРОИЗВОДНЫЕ ЭТИЛЕНДИАМИНА И СОДЕРЖАЩИЕ ИХ ИНГИБИТОР FXa И АНТИКОАГУЛЯНТ | 2001 |
|
RU2268259C2 |
| СОЕДИНЕНИЯ, ПРИГОДНЫЕ ДЛЯ ТЕРАПИИ ПРОТИВ ВИЧ | 2019 |
|
RU2806030C2 |


Изобретение относится к новому N1-[2,2-диметил-1S-(пиридин-2-илкapбaмoил)пропил] -N4-гидpoкcи-2R-изoбyтил-3S-метоксисукцинамиду или его фармацевтически приемлемым солям, гидратам или сольватам. Указанные соединения обладают биологической активностью и могут быть использованы в медицине для лечения ревматоидного артрита, злокачественных новообразований, рассеянного склероза или псориаза. 2 табл.
N1-[2,2-Диметил-1S-(пиридин-2-илкарбамоил)пропил]-N4-гидрокси-2R-изобутил-3S-метоксисукцинамид формулы (II)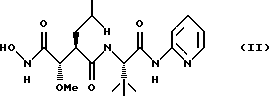
или его фармацевтически приемлемая соль, гидрат или сольват.
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| 5-ХЛОР-2-ПИРИДИЛАМИД-4-НИТРО-N-(КАРБОКСИМЕТИЛ)АНТРАНИЛОВОЙ КИСЛОТЫ, ПРОЯВЛЯЮЩИЙ ПРОТИВОВОСПАЛИТЕЛЬНУЮ АКТИВНОСТЬ | 1991 |
|
RU2024507C1 |

