Изобретение относится к методике разделения, более конкретно к методике разделения паклитакселя и родственных таксанов.
Паклитаксель является хорошо известным хемотерапевтическим средством против различных метастатических видов рака. Он был испытан FDA и НРB для лечения рака молочной железы и рака яичников.
Данное соединение является природным продуктом, экстрагируемым главным образом из коры тихооокеанского тиса Taxus brevifolia, также он найден в Т. baccata, Т. walichiana, Т. vunnanensis и Т. canadensis.
Концентрация паклитакселя в различных видах сырья, как правило, является низкой, например в коре тихооокеанского тиса содержится порядка от 0,0004 до 0,01 мас.%. Такие низкие концентрации делают экстракцию соединения из сырья и его очистку до фармакопейной чистоты весьма спорной, а поэтому нецелесообразной в промышленном масштабе. Были разработаны различные методики нормально-фазной и обращенно-фазной хроматографии, равно как и колоночной хроматографии низкого и высокого давления для выделения паклитакселя из первичных экстрактов сырья.
Успех применения хроматографии низкого давления в большей мере зависит от устройства колонны. Различные проблемы связаны с использованием силикагеля и окиси алюминия, которые являются классическими носителями в системах разделения с неподвижной фазой. Они образуют неподвижную фазу с большинством систем растворителей, но она является сильным сорбентом и может участвовать в процессе разделения в такой степени, что затрагивает хроматографическое поведение и выход образцов.
Разработаны хроматографические методы определения и выделения паклитакселя из различных видов Taxus в аналитических и препаративных количествах. Эти способы выделения обычно проводятся в ограниченном лабораторном масштабе и обладают низкими селективностью и выходом, высокой себестоимостью производства, поэтому существует насущная и неудовлетворенная потребность в экономически приемлемом способе разделения противоракового соединения паклитакселя от его ближайших аналогов цефаломанина, а также других близкородственных таксанов.
Предшествующий уровень техники описывает использование различных типов хроматографических методик разделения паклитакселя и родственных таксанов, включающих нормально-фазную и обращенно-фазную хроматографию на колоннах с силикагелем или привитым силикагелем. Способы, известные из предшествующего уровня техники, отличаются низким выходом, высокой себестоимостью производства или включают многоступенчатое разделение, которое трудно интенсифицируется до масштабов промышленного производства.
Типичным примером предшествующего уровня техники является патент США 5620875, выданный Хоффману с сотр. 15 апреля 1997 г. В документе указано, что разделение паклитакселя и других таксанов производится многоступенчатой экстракцией гексаном и высокоэффективной жидкостной хроматографией (ВЭЖХ). Данный процесс применяется в лаборатории и дает умеренные выходы целевых соединений.
В патенте США 5670673, выданном Рао 23 сентября 1997 г., изложено разделение и очистка таксола и его аналогов. Способ включает использование обращенно-фазной жидкостной хроматографии на сорбенте C18 с элюированием сорбированных аналогов. Хотя данная методика имеет определенные преимущества, тем не менее существуют ограничения в отношении производительности и чистоты получаемых соединений.
Данное изобретение обеспечивает простой способ разделения, основанный на применении полимерной смолы, который устраняет ограничения существующей методологии.
Объектом данного изобретения является простой и экономически более эффективный способ разделения и очистки важных таксанов, таких как 13-ацетил-9-дигидробаккатин III, 10-деацетилбаккатин III, баккатин III, цефаломанин и паклитаксель.
Обычные методы выделения таксанов, включая паклитаксель, 13-ацетил-9-дигидробаккатин III и баккатин III, обычно включают стадии экстракции таксанов из сырья спиртовым растворителем, обезжиривания экстракта и хроматографического разделения и очистки индивидуального таксана.
Первым объектом данного изобретения является обеспечение способа выделения и очистки таксановых аналогов из источника таксанов.
Способ включает
- экстракцию источника таксанов органическим экстрагентом;
- обработку рабочей сорбирующей среды экстрагентом и загрузку среды в колонну, содержащую сорбент;
- элюирование на первой стадии смесью органических растворителей под давлением от 68,948 до 137,90 кПа (от 10 до 20 psi) для получения фракций, содержащих таксановые соединения;
- кристаллизацию фракций для получения твердого таксанового соединения и маточного раствора, концентрирование маточного раствора;
- элюирование маточного раствора на второй стадии смесью полярных растворителей через полимерную смолу для выделения по меньшей мере второго таксанового соединения.
Другим объектом данного изобретения является способ выделения и очистки таксанов из источника таксанов.
Способ включает
- получение источника таксанов;
- экстракцию таксанов из источника в органическую экстракционную среду с получением органического слоя, содержащего таксановые соединения;
- обработку носителя органическим слоем;
- применение колонны низкого давления, содержащей сорбент;
- элюирование колонны органическим растворителем на первой стадии для получения очищенных таксановых фракций;
- кристаллизацию таксановых фракций для получения первого таксанового аналога и маточного раствора;
- элюирование маточного раствора на второй стадии через полимерную смолу на хроматографической колонне для очистки и выделения по меньшей мере второго таксанового аналога и третьего таксанового аналога;
- сбор разделенных таксановых аналогов.
Еще одним объектом данного изобретения является способ выделения и очистки таксанов из источника таксанов.
Способ включает
- получение источника таксанов;
- экстракцию таксанов из источника в органическую экстракционную среду с получением органического слоя, содержащего таксановые соединения;
- обработку носителя органическим слоем;
- применение колонны низкого давления, содержащей сорбент;
- элюирование колонны органическим растворителем на первой стадии под давлением 68,948-137,90 кПа (10-20 psi) для получения очищенных таксановых фракций и удаления флавоноидных и лигниновых примесей;
- кристаллизацию таксановых фракций для получения первого таксанового аналога и маточного раствора;
- элюирование маточного раствора на второй стадии через полистирол-ДВБ смолу на хроматографической колонне под давлением 206,84 кПа (30 psi) для очистки и выделения по меньшей мере второго и третьего таксановых аналогов;
- элюирование на третьей стадии второго таксанового аналога и третьего таксанового аналога через нормально-фазную колонну с силикагелем для очистки второго таксанового аналога и третьего таксанового аналога;
- сбор разделенных таксановых аналогов.
Исходным источником таксанов является растительный материал Taxus canadensis, которым изобилует восток Канады. Используются ветки и иглы Taxus canadensis или их смесь. Материал может быть свежим или сушеным.
Сырье замачивают и экстрагируют метанолом при 60oС в течение 5 час, затем фильтруют. Метанольный экстракт смешивают с активированным углем, оставляют на 1 час при комнатной температуре, затем отфильтровывают. Фильтрат концентрируют примерно до 15% от исходного объема путем выпаривания. К концентрату прибавляют смесь вода : дихлорметан 1:1 (v/v) для получения фракции, обогащенной паклитакселем (органический слой).
Органический слой концентрируют для уменьшения объема и наносят на носитель, в данном случае носителем является Celite 545.
Обработанный материал загружают наверху промышленной препаративной колонны, например, размером 150•1500 мм и набитой сорбентом Аl2О3. Колонну элюируют смесью гексан : ацетон (начало 100:0, окончание 45:55) под давлением 68,948- 137,90 кПа (от 10 до 20 psi). Собирают фракции, содержащие таксаны, анализируют тонкослойной хроматографией (ТСХ) с последующим объединением в соответствии с данными ТСХ.
Собранные фракции затем концентрируют в вакууме, полученные вещества растворяют в метаноле и выдерживают в течение суток. Образовавшиеся кристаллы отфильтровывают и промывают метанолом, а затем перекристаллизовывают из ацетона. Получают 13-ацетил-9-дигидробаккатин III в виде белых кристаллов.
Маточный раствор упаривают досуха, остаток растворяют в ацетоне. Ацетоновый раствор смешивают с полимерной смолой (смола DowexTM, полистирол-ДВБ). Смесь упаривают в вакууме для удаления ацетона. Полученный порошок подвергают хроматографии низкого давления. Промышленную хроматографическую колонну (150•1500 мм) набивают смолой DowexTM и элюируют смесью ацетон : вода (45:55, v/v) с расходом 150 мл/мин при рабочем давлении 206,84 кПа (30 psi). Собирают фракции объемом порядка 2 л каждая, которые анализируют ТСХ и ВЭЖХ. Фракции, содержащие баккатин III и 10-деацетилбаккатин III, объединяют и упаривают в вакууме до удаления практически всего органического растворителя, разбавляют водой и экстрагируют CH2Cl2. Экстракт упаривают досуха, полученный остаток растворяют в EtOAc и очищают на нормально-фазной колонне с силикагелем, используя в качестве растворителя систему гексан : EtOAc (начало 5: 5, окончание 35:65). Баккатин III и 10-деацетилбаккатин III получают в виде белого порошка со степенью чистоты выше 98%.
Фракции, содержащие паклитаксель и цефаломанин, объединяют и упаривают в вакууме до удаления практически всего ацетона, затем разделяют в системе вода : СН2Сl2. Органический слой отделяют и упаривают досуха в вакууме. Остаток растворяют в метаноле. К метанольному раствору прибавляют примерно 30% (w/v) воды, смесь нагревают до 60oС в течение 5 мин и оставляют на ночь при комнатной температуре. Неочищенный кристаллический осадок отфильтровывают от метанольного раствора и высушивают в вакууме при 70-75oС. Остаток анализируют с помощью ВЭЖХ. Он состоит приблизительно из 70% паклитакселя и 25% цефаломанина и некоторых других таксанов.
Неочищенный паклитаксель может подвергаться обработке в соответствии с тремя альтернативными способами. В первом случае его растворяют в ацетоне и очищают обращенно-фазной колоночной хроматографией. Колонну набивают полимерной смолой (смола Diaion HP2MG) и элюируют смесью ацетонитрил : вода (45: 55). Получают паклитаксель в виде белых кристаллов со степенью чистоты более 99%. Цефаломанин получают в виде белого порошка со степенью чистоты более 98%.
Альтернативно согласно второму способу очистку паклитакселя и цефаломанина проводят с помощью химического превращения, а затем очищают обращенно-фазной колоночной хроматографией на привитом силикагеле. Смесь неочищенных паклитакселя и цефаломанина растворяют в смеси СН2Сl2 : СНСl3 (1:10 w/v) и проводят реакцию с 10 эквивалентами брома в круглодонной колбе, помещенной на ледяную баню (при 0...5oС) в течение приблизительно 40 мин. Полученную смесь паклитакселя и 2",3"-бромоцефаломанина легко разделяют на обращенно-фазной хроматографической колонне (C18 или фенил), элюент СН3СN : Н2O (45:55). Паклитаксель получают в виде белых кристаллов со степенью чистоты более 99%. 2",3"-Бромоцефаломанин получают в виде смеси диастереомеров, которая реагирует с активированным цинком в уксусной кислоте при комнатной температуре, а затем подвергается очистке колоночной флэш-хроматографией. Получают цефаломанин со степенью чистоты более 95%.
Согласно третьему альтернативному способу паклитаксель и бромоцефаломанин очищают на нормально-фазной колонне с силикагелем. Элюирующие системы могут включать гексан : ЕtОАс (начало 6:4, окончание 4:6) или CH2Cl2 : Me2CO (начало 8:2, окончание 65:35) или CH2Cl2 : EtOAc (начало 75:25, окончание 6: 4). Этим способом паклитаксель получают в виде белых кристаллов со степенью чистоты, превосходящей 99%, с содержанием цефаломанина не более 0,2%.
После обсуждения изобретения далее следуют примеры для более точного понимания.
Пример 1
Приблизительно 200 кг сушеных игл и веток Taxus canadensis экстрагируют 1000 л метанола при 60oС в течение 5 ч в промышленном многоцелевом экстракторе и отфильтровывают. Сырье дополнительно экстрагируют 700 л метанола при температуре от 55 до 60oС в течение 5 ч и отфильтровывают. Фильтраты объединяют и смешивают с 10 кг активированного угля (5% w/w) и выдерживают при комнатной температуре 1 ч, отфильтровывают от активированного угля. Затем фильтрат концентрируют в вакууме приблизительно до 100 л и прибавляют 300 л смеси вода : дихлорметан (1:1). Отделяют органический слой, а водный слой еще дважды экстрагируют 200 л дихлорметана. Дихлорметановые растворы объединяют и упаривают в вакууме до образования суспензии, а затем разбавляют 20 л ацетона.
Ацетоновым раствором пропитывают 20 кг Celite 545. Пропитанный материал высушивают на воздухе, после чего загружают наверху трех промышленных хроматографических колонн низкого давления (размером 150•15 см). Каждая колонна набита 15 кг сорбента (окись алюминия, Аl2О3). Следует отметить, что точный размер колонн не имеет решающего значения, но колонны должны быть достаточно велики, чтобы вместить количество Аl2О3, требуемое для разделения. Использование окиси алюминия эффективно при сорбции флавоноидов и лигнинов, которые также экстрагируются из сырья. Их трудно удалить, поэтому возникает проблема чистоты выпускаемого товарного паклитакселя.
Колонну элюируют системой гексан : ацетон (начало 100:0, окончание 45: 55) под давлением 68,948-103,42 кПа (от 10 до 15 psi) с расходом приблизительно 150 мл/мин.
Фракции, содержащие таксаны, собирают и объединяют согласно результатам тонкослойной хроматографии (ТСХ), после чего концентрируют в вакууме для удаления всех растворителей. Полученное вещество растворяют в метаноле и оставляют на ночь при комнатной температуре до образования игольчатых кристаллов. Кристаллы отфильтровывают и перекристаллизовывают из ацетона. Получают 13-ацетил-9-дигидробаккатин III в виде игольчатых кристаллов со степенью чистоты более 96%, выход составляет 148 г (0,074% в пересчете на сырье).
Маточный раствор после кристаллизации 13-ацетил-9-дигидробаккатина III упаривают досуха в вакууме. Остаток растворяют в 3 л ацетона. Ацетоновый раствор смешивают с 1,5 кг сополимерной смолы полистирола-дивинилбензола. Смесь упаривают для удаления растворителя, полученный порошок загружают наверху промышленной хроматографической колонны низкого давления, набитой сополимерной смолой полистирола-дивинилбензола, и элюируют 45%-ным водным ацетоном с расходом 150 мл/мин при рабочем давлении ниже 206,84 кПа (30 psi). Фракции, объемом 2 л каждая, собирают и анализируют с применением ТСХ и ВЭЖХ.
Фракции, содержащие паклитаксель и цефаломанин, объединяют, упаривают в вакууме до удаления большей части ацетона, затем разбавляют деионизированной водой и трижды экстрагируют 2,5 л дихлорметана. Органический слой упаривают досуха в вакууме, остаток растворяют в 1 л метанола.
К метанольному раствору прибавляют приблизительно 30% воды (w/v), смесь нагревают до 60oС, выдерживают несколько минут, после чего оставляют на ночь при комнатной температуре. Неочищенный кристаллический твердый продукт отфильтровывают от метанола и высушивают в вакуумной сушилке при температуре от 70 до 75oС. Получают 31 г твердого вещества, состоящего приблизительно из 70% паклитакселя и 25% цефаломанина.
Неочищенный паклитаксель (30 г) растворяют в 200 мл ацетонитрила, разбавляют 250 мл деионизированной воды и перекачивают наверх хроматографической колонны низкого давления (размер 10•150 см), набитой полимерной смолой (Diaion, макропористая полиметакрилатная смола). После перекачивания образца колонну подвергают градиентному элюированию водным ацетонитрилом с шагом 35, 40, 45 и 50% ацетонитрила в воде. Изменение состава элюента отмечают по результатам ТСХ и ВЭЖХ фракций.
Фракции, объемом приблизительно 1 л каждая, собирают и анализируют с применением ТСХ и ВЭЖХ. Расход 75 мл/мин. Фракции с колонны, содержащие паклитаксель или цефаломанин, объединяют по отдельности. Два раствора, полученных объединением таких фракций, оставляют при температуре приблизительно 5oС до окончания кристаллизации индивидуальных соединений.
Кристаллы раздельно отфильтровывают, оба вещества перекристаллизовывают при 65oС из смеси метанол-вода.
Паклитаксель получают в виде белых игольчатых кристаллов со степенью чистоты выше 99%. Выход 18,5 г (0,009%).
Цефаломанин получают в виде белых игл с чистотой выше 98%. Выход 6,5 г (0,003%).
Фракции с предыдущей стадии, содержащие баккатин III и 10-деацетилбаккатин III, объединяют и упаривают в вакууме до по существу удаления ацетона, после чего экстрагируют 3 л смеси вода-дихлорметан (1:1 v/v). Органический слой упаривают досуха, остаток подвергают нормально-фазной хроматографии. Размеры применяемой колонны, набитой силикагелем (ситовый состав 200-300), равны 4"•4'. Состав элюента гексан : этилацетат (начало 50:50, окончание 35: 65) изменяют шаговым градиентным способом. Фракции, объемом приблизительно 1000 мл каждая, собирают и анализируют с применением ТСХ.
Фракции, содержащие баккатин III или 10-деацетилбаккатин III, собирают по отдельности и объединяют согласно результатам ТСХ, после чего упаривают досуха.
Часть продукта, содержащую баккатин III, растворяют в 200 мл метанола и оставляют на ночь в холодильнике. Образовавшиеся белые кристаллы отфильтровывают и перекристаллизовывают из метанола. Получают баккатин III в виде белых кристаллов. Выход 7,5 г (0,003%).
Неочищенный 10-деацетилбаккатин III растворяют в 150 мл ацетона, затем разбавляют 150 мл гексана. Смесь оставляют на ночь при комнатной температуре. Образовавшиеся белые кристаллы отфильтровывают и перекристаллизовывают из того же растворителя. Получают 10-деацетилбаккатин III в виде белых кристаллов. Выход 15 г (0,007%).
Пример 2
С использованием аппаратов и методики, подобных приведенным в примере 1, получают неочищенный паклитаксель. Неочищенный паклитаксель растворяют в 300 мл дихлорметана, раствор переносят в круглодонную трехгорлую колбу емкостью 1000 мл. Колбу помещают на ледяную баню, раствор перемешивают магнитной мешалкой. По достижении температуры около 5oС при перемешивании медленно добавляют раствор 10 эквивалентов брома в дихлорметане (1:1 v/v). Мольное соотношение цефаломанина и брома составляет 1:10.
За ходом бромирования следят с помощью ТСХ, отбирая пробы с периодичностью от 5 до 10 мин. По окончании реакции реакционную массу разбавляют 300 мл дихлорметана и переносят в делительную воронку (для завершения реакции требуется от 40 до 50 мин). В реакционную смесь добавляют 350 мл 10%-ного водного раствора тиосульфата натрия для удаления избытка брома. Слой дихлорметана отделяют и промывают водой и насыщенным раствором поваренной соли, упаривают досуха в вакууме. Получают легкий коричневый порошок.
Пример 3
Неочищенный продукт из примера 2 растворяют в 120 мл ацетонитрила, разбавленного 150 мл деионизированной воды. Раствор перекачивают наверх обращенно-фазной хроматографической колонны (5•100 см), набитой силикагелем, привитым C18.
Колонну элюируют смесью ацетонитрил : вода (45:55) при рабочем давлении 206,84-275,79 кПа (от 30 до 40 psi). Расход 30 мл/мин. Собирают фракции по 250 мл каждая, которые затем анализируют с помощью ТСХ и объединяют.
Порцию, содержащую паклитаксель, оставляют в холодильнике на ночь, образовавшиеся кристаллы отфильтровывают и перекристаллизовывают из 70% метанола. Получают паклитаксель в виде белых игольчатых кристаллов со степенью чистоты выше 99%. Выход 6,1 г. Фракции, содержащие 2",3"-бромоцефаломанин, объединяют, экстрагируют смесью воды и дихлорметана. Органический слой отделяют и упаривают в вакууме досуха. Остаток растворяют в 150 мл метанола, разбавляют 50 мл воды для получения игольчатых кристаллов. Кристаллы грязно-белого цвета перекристаллизовывают из смеси метанол-вода. Получают 2", 3"-бромоцефаломанин в виде белых кристаллов.
В 30 мл АсОН растворяют 2 г 1",3"-бромоцефаломанина и прибавляют 2 г свежеактивированного цинка. Смесь перемешивают 3 ч при комнатной температуре, за ходом реакции следят с помощью ТСХ. После того как результаты ТСХ укажут на завершение реакции, смесь экстрагируют 300 мл смеси дихлорметана и 10% NaHCO3 (2:1). Органический слой промывают водой и упаривают досуха. Неочищенный продукт очищают флэш-хроматографией, получают цефаломанин в виде белого порошка.
Пример 4
Неочищенное вещество из примера 2 растворяют в 120 мл дихлорметана. Органический слой упаривают досуха, остаток подвергают нормально-фазной хроматографии. Размеры применяемой колонны, набитой силикагелем (ситовый состав между 200 и 300), равны 4"•4'. Проводят градиентное элюирование системой дихлорметан : этилацетат (начало 75:25, окончание 6:4), для выделения паклитакселя и 2",3"-бромоцефаломанина также могут быть использованы системы гексан : этилацетат (начало 6:4, окончание 4:6) и дихлорметан : ацетон (начало 8: 2, окончание 65:35). Фракции объемом 500 мл каждая собирают и анализируют с помощью ТСХ.
Фракции, содержащие паклитаксель и 2",3"-бромоцефаломанин, собирают по отдельности и объединяют согласно результатам ТСХ, а затем упаривают досуха.
Часть, содержащую паклитаксель, растворяют в 100 мл метанола и разбавляют 35 мл воды, после чего выдерживают при охлаждении. Образовавшиеся белые кристаллы отфильтровывают и перекристаллизовывают из смеси метанол-вода. Получают 5,5 г паклитакселя в виде белых кристаллов.
Неочищенный 1", 3"-бромоцефаломанин растворяют в 50 мл ацетона, затем разбавляют 50 мл гексана. Смесь оставляют на ночь при комнатной температуре. Образовавшиеся белые кристаллы отфильтровывают и перекристаллизовывают из того же растворителя. Получают 2",3"-бромоцефаломанин в виде белых кристаллов.
Структуры I и II иллюстрируют строение соединений.
Несмотря на то, что выше уже были описаны осуществления данного изобретения, это никак не ограничивает его, и любому специалисту в данной области очевидно, что многочисленные модификации являются частью данного изобретения, в той мере, в какой они не противоречат идее, содержанию и рамкам заявленного и описанного изобретения.
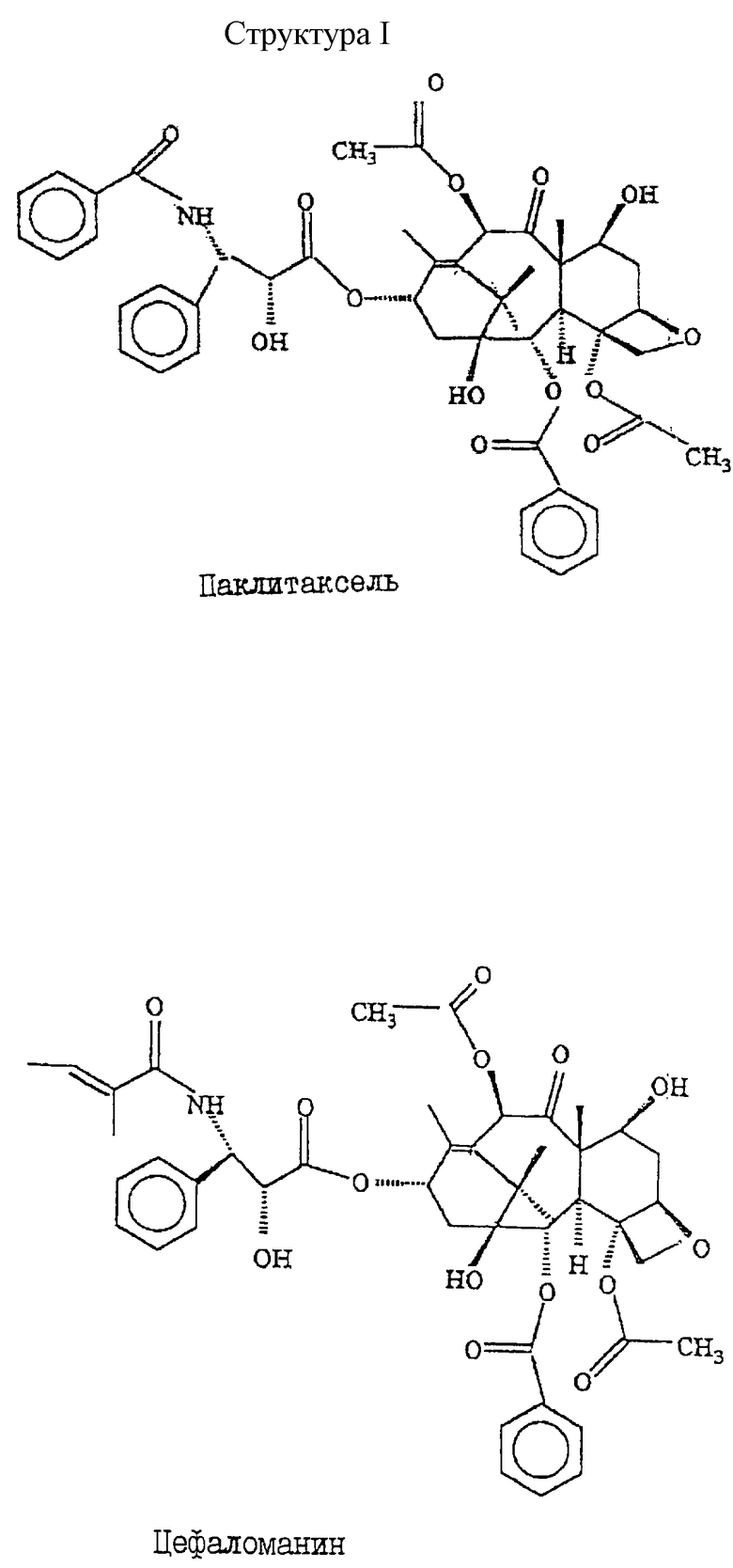
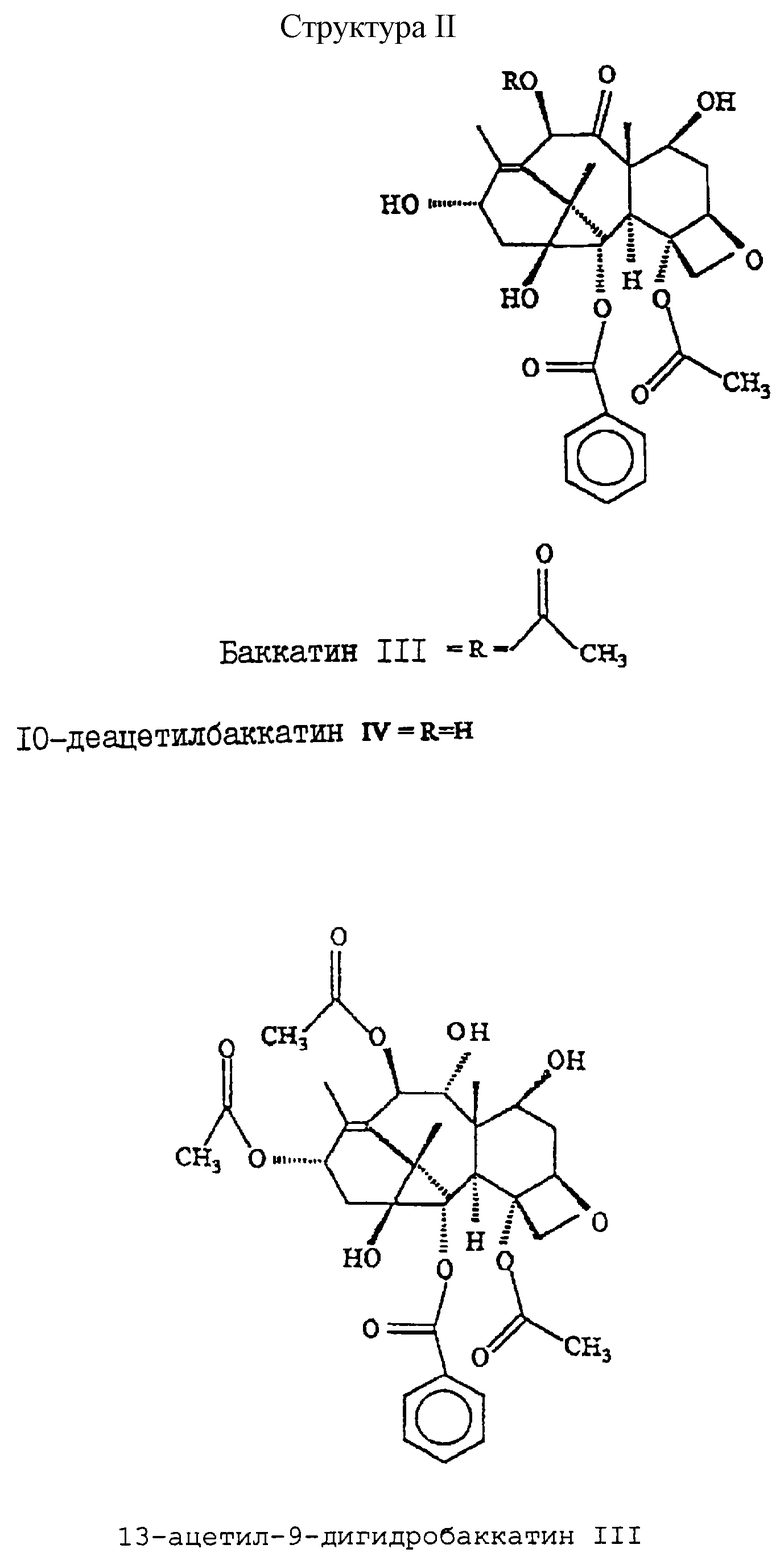
Способ получения таксановых аналогов из источника, содержащего таксаны. Способ включает экстракцию таксанов, использование полимерной смолы для выделения таксанов при низком давлении без применения сложных и дорогостоящих стадий разделения/очистки, кристаллизацию фракций, содержащих таксановые соединения, концентрирование маточного раствора и использование полимерной смолы для выделения второго таксанового соединения из маточного раствора. Способ обеспечивает высокий выход и чистоту. 3 с. и 17 з.п. ф-лы.
| US 5670673 А, 25.03.1997 | |||
| US 5620875 А, 15.04.1997 | |||
| УСТРОЙСТВО ДЛЯ СКРИНИНГОВОЙ ДИАГНОСТИКИ ЗРЕНИЯ | 2001 |
|
RU2210972C1 |
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| ТАКСАНЫ С БОКОВОЙ ЦЕПЬЮ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ | 1993 |
|
RU2125042C1 |

