Настоящая заявка представляет собой частично продолжающуюся заявку №08/215,203, поданную 21 марта 1994 года, которая является частичным продолжением заявки №08/203, 197, поданной 25 февраля 1994 года, которая является частичным продолжением заявки №08/196,025, поданной 14 февраля 1994 года. Все эти заявки находятся на рассмотрении и используются в данном документе в качестве ссылок.
Предпосылки к созданию изобретения
Гематопоэзис представляет собой процесс, посредством которого клетки крови растут и развиваются в сторону усложнения из ствольных плюрипотентных клеток. Этот процесс включает в себя сложное взаимодействие факторов роста сложных полипептидов (цитокинез), действующих через мембраносвязанные рецепторы на нужные клетки. Действие цитокинеза приводит к пролиферации (быстрому разрастанию) клеток и их дифференциации как реакции на конкретный цитокин, который, как правило, является рядоспецифическим и/или стадиеспецифическим. Развитие клетки одинарного типа, такой как кровяная пластина (тромбоцид), из ствольной клетки может потребовать скоординированного действия множества цитокинов, действующих в нужной последовательности.
Известные цитокины состоят из интерлейкинов, таких как IL-1, IL-2, IL-3, IL-6, IL-8 и т.д.; и из факторов, стимулирующих разрастание колоний, таких как G-CSF, M-CSF, GM-CSF, эритропоэтины (ЕРО) и т.д. Как правило, интерлейкины действуют как переносчик иммуной реакции и реакции воспламенения. Факторы, стимулирующие рост колоний, стимулируют пролиферацию клеток костного мозга, приводят в действие созревшие лейкоциты и другими способами создают единую часть реакции реципиента на контрольные заражения воспламеняемостью и инфекцией и иммунологические заражения.
Разнообразные цитокины были выращены, как терапевтические агенты. Например, эритропоэтин, который стимулирует рост эритроцитов, применяется при лечении анемии, возникшей при почечных нарушениях. Некоторые из факторов, стимулирующих рост колоний, применялись в сочетании с химиотерапией рака для ускорения восстановления иммунных систем пациента. Интерлейкин-2,d-интерферон и γ-интерферон используются при лечении некоторых форм рака. Действия, которые стимулируют мегакариоцитопоэзис и тромбоцитопоээис, были обнаружены в жидкости тела тромбоцитопеничных животных и названы в литературе "тромбопоэтинами" (недавно рассматривались МакДоналдсом в Ехр. Hematol. 16:201-205, 1988 и МакДоналдсом в Am.J.Ped.Hematol.Oncol. 14:8-21, 1992). Несмотря на исследования, проводимые более трех десятилетий, фактор или Факторы, ответственные за эти процессы, не были четко охарактеризованы, частично из-за отсутствия хороших источников, хороших проб для анализов и частично из-за недостатка сведений о месте производства. Слабые нарушения кровотечения (МВР), связанные с дисфункцией тромбоцитов, встречаются сравнительно часто (Бекман, Семинары по Гематологии 17:292-305, 1980), как, например, ряд врожденных нарушений функций тромбоцитов, в том числе синдром бернарда-Сульера (дефицит GРТbтромбоцитов), тромбастения Гланзмана (дефицит GPIIb и GPIIIa), врожденная афибриногенемия (сниженное количество или полное отсутствие фибриногенов в плазме и тромбоцитах) и синдром серых тромбоцитов (отсутствие -гранул). Кроме того, существует еще ряд нарушений, связанных с секрецией тромбоцитов, дефицит пула хранения, аномалии пути аракидоновой тромбоцитной кислоты, дефицит циклооксигеназы тромбоцитов и синтетазы тромбоксана и дефекты активации тромбоцитов (рассматривалось Рао и Холмсеном, Семинар по гематологии 23:102-118, 1986 г.). По сей день молекулярная основа всех этих дефектов не совсем понятна.
Изоляция и изучение свойств протеинов тромбоцитов дали бы неоценимую возможность разъяснить наличие дефектов при многих дисфункциях тромбоцитов. Главным препятствием на пути к подробному молекулярному анализу является сложность получения mPHK из тромбоцитов или из их предшественников. Тромбоциты лишены ядер и транскрипций. След mPHK, связанный с тромбоцитами, трудно изолировать и он часто подвергается деградации. Создание библиотек до этого требовало большого количества тромбоцитов, обычно от 25 до 250 единиц цельной крови (Ицуми и др. Рrос. Natl. Acad.Sci.USA 87:7477-7481, 1989: Вики и др.. Thrombosis and Haemostasis 61:448-453, 1989; и Венджер и др. Blood 73:1498-1503, 1989) или форезии (кратковременного использования другого организма для передвижения) пациентов с повышенным содержанием тромбоцитов в крови из-за тромбоцитомии (Рот и др. Biochem.Biophys. Res. Соmm. 160:705-710, 1989). В случаях, когда тромбоцито-специфические сДНК были изолированы целиком от образцов mPHK и, вероятно, представляли собой только малые части от всего спектра кодирования тромбоцитов.
Другим путем создания библиотеки сДНК тромбоцитов является изоляция и создание библиотеки из mPHK, изолированного от мегакариоцитов, непосредственных предшественников тромбоцитов. Мегакариоциты являются полиплоидными клетками и, возможно, содержат mPHK, кодирующий весь хромосомный набор тромбоцитных и мегакариоцитных протеинов. Однако оказалось трудно изолировать мегакариоциты, если они в довольно большом количестве, и сохранить относительную их чистоту.
Недавние успехи молекулярной биологии в значительной степени прояснили гематопоэзис, но в то же время показали, что этот процесс чрезвычайно сложен. Хотя свойства многих цитокин уже определены и многие из них уже нашли клиническое применение, сохраняется необходимость в дополнительных агентах, которые могли бы стимулировать пролиферацию и дифференциацию предшественников милоидов и лимпоидов и производство зрелых кровяных клеток. Особая нужда ощущается в веществах, которые стимулируют развитие и пролиферацию клеток мегакариоцитного ряда, включая тромбоциты. Есть также потребность в веществах, которые могли бы быть использованы в лечении разного вида цитопении, в том числе тромбоцитопении, аномально низкого количества циркулирующих тромбоцитов (менее чем 1×105 тромбоцитов/мм3) и других нарушений в области тромбоцитов. Настоящее изобретение позволяет удовлетворить все эти потребности и предлагает дополнительные преимущества.
Краткое изложение существа изобретения
Задачей настоящего изобретения является обеспечение изолированных протеинов с гематопоэтической активностью.
Другой задачей данного изобретения является предоставление способов производства протеинов с гематопоэтической активностью, а также изолированных молекул ДНК, векторов и клеток, которые можно использовать в осуществлении этих способов.
Другой задачей данного изобретения является обеспечение антителами, которые фиксируют эпитоп (антигенную детерминанту) на гематопоэтическом протеине.
Еще одной задачей данного изобретения является создание способов стимулирования продуцирования мегакариоцитов, тромбоцитов и нейтрофилов у млекопитающих, в том числе и у человека.
Еще одной задачей данного изобретения является обеспечение разноообразных инструментов, которые можно применять при изучении роста клеток костного мозга, дифференциации и пролиферации; и при определении болезней, которые характерны отклонениями в развитии клеток костного мозга, их дифференциации и пролиферации.
Согласно одному аспекту данного изобретения оно предлагает изолированный протеин, отобранный из группы, состоящей из (а) протеинов, содержащих последовательность аминокислот из SEQ BD NO:2 от аминокислотного остатка 45 до аминокислотного остатка 196; (в) протеинов, содержащих последовательность аминокислот из SЕQ ID NO:2 от аминокислотного остатка 45 до аминокислотного остатка 206; (с) протеинов, содержащих последовательность аминокислот из SEQ ID NO:19 от аминокислотного остатка 22 до аминокислотного остатка 173; (d) протеинов, содержащих последовательность аминокислот из SEQ ID NO:19 от аминокислотного остатка 22 до аминокислотного остатка 175; (е) аллельных варинтов (а), (в), (с) и (d); (f) гомологов образцов (а), (в), (с), (d) или (е), в которых протеин стимулирует пролиферацию и дифференциацию предшественников милоидов и липоидов. В определенных вариантах изобретения протеин содержит последовательность аминокислот из SEQ ID NO:2 от аминокислотного остатка 45 до аминокислотного остатка 379 или последовательность аминокислот из SEQ ID NO:19 от аминокислотного остатка 22 до аминокислотного остатка 353.
Другая особенность данного изобретения заключается в том, что оно предлагает изолированную молекулу полинуклеотида, кодирующую протеин, как указывалось выше. В одном из вариантов изобретения молекула полинуклеотида представляет собой молекулу ДНК, содержащую кодирующую цепь, состоящую из последовательности нуклеотидов из SEQ ID NО:1 от нуклеотида 237 до нуклеотида 692 или последовательности нуклеотидов из SEQ ID NO:18 от нуклеотида 64 до нуклеотида 519. В других вариантах изобретения эта молекула содержит нуклеотиды 237-1241, 174-1241, 105-1241, 105-722, 174-722 или 237-722 из SEQ ID NО:1 или соответствующие области из SEQ ID NO:18. Далее данное изобретение предлагает аллельные варианты указанных молекул и молекул ДНК, кодирующих гематопоэтический протеин, молекулы которого кодируют протеин, который, по крайней мере, на 80% идентичен по последовательности аминокислот протеину, кодированному одной из указанных частей из SEQ ID NO:1 или SEQ ID NO:18. Предлагаются также молекулы, комплементарные этим последовательностям.
Относительно другого отличительного признака данное изобретение предлагает изолированную молекулу ДНК, выбираемую из группы, состоящей из (а) вставки Tcj RI-Xho I плазмида pZGmpT-1081 (ATCC 69566), (в) аллельных вариантов молекул ДНК (а) и (с), кодирующих протеин, который, по крайней мере, на 80% идентичен по последовательности аминокислот протеину, закодированному (а) и (в), в которой изолированная молекула ДНК кодирует протеин с гематопоэтической активностью.
Относительно другого отличительного признака данное изобретение предлагает экспрессируюший вектор, состоящий из следующих связанных в действии элементов: стимулятор (промотор) транскрипции; сегмент ДНК, отобранный из группы, состоящей из (а) сегментов ДНК, кодирующих гематопоэтический протеин и содержащих последовательность нуклеотидов, как показано в SEQ ID NO:1 от нуклеотида 237 до нуклеотида 692, (в) сегменты ДНК, кодирующие гематопоэтический протеин и содержащие последовательность нуклеотидов, как показано в SEQ ID NO:18 от нуклеотитда 64 до нуклеотида 519; (с) аллельных вариантов (а) или (в) и (d) сегментов ДНК, кодирующих гематопоэтический протеин, который, по крайней мере, на 80% идентичен последовательности по аминокислотам протеину, закодированному (а), (в) или (с); и терминатор транскрипции.
Относительно другого отличительного признака данное изобретение предлагает культивируемую клетку, в которую был введен экспрессирующий вектор, как указывалось выше, в который эта клетка экспрессирует гематопоэтический протеин, кодируемый сегментом ДНК. В определенных вариантах изобретения эта клетка представляет собой грибную (грибковую) клетку, клетку млекопитающего или бактериальную клетку.
Относительно другого отличительного признака данное изобретение предлагает введение клетки млекопитающего (не человека) в зародышевую линию, в которую был введен гетерологовый сегмент ДНК, кодирующий гематопоэтический протеин, как описано выше, в которой клетка млекопитающего продуцирует гематопоэтический протеин, кодируемый указанным сегментом ДНК.
Относительно другого отличительного признака данное изобретение предлагает способы для стимулирования продуцирования тромбоцитов в клетке млекопитающего. Эти способы состоят во введении в клетку млекопитающего терапевтическим путем эффективного количества гематопоэтического протеина, отобранного из группы, состоящей из (а) протеинов, содержащих последовательность аминокислот SEQ ID NО:1 от аминокислотного остатка 45 до аминокислотного остатка 196; (в) протеины, состоящие из последовательности аминокислот из SEQ ID NO:19 от аминокислотного остатка 22 до аминокислотного остатка 173; (с) аллельные варианты (а) и (в); и (d) гомологи образцов (а), (в) или (с), в которых протеин стимулирует пролиферацию или дифференциацию предшествеников милоидов или лимпоидов, в сочетании с фармацевтически приемлемым вектром.
Эти и другие отличительные признаки данного изобретения станут очевидными после того, как мы обратимся к приведенному ниже подробному описанию и прилагаемым чертежам.
Краткое описание чертежей
Фиг.1 представляет собой частично суженную карту вектора pDX. Используемые символы SV40 ori, начало репликации из SV40: SV40 Е, SV40 усилитель (участок ДНК вне гена, усиливающий экспрессию этого гена); MLP, основной поздний стимулятор (промотор) аденовируса; L1-3, трехраздельная лидерная последовательность; ss, сигналы сплайсинга; рА, участок полиаденилирования.
Фиг.2 показывает конструкцию плазмида pBJ3. Используемые символы промоторы TPIp, ТРII; терминатор TPIt, ТРII; ААТ, α-1 сДНК антитрипсин; альфа, лидерная последовательность альфа-фактора; mТРО, кодирующая последовательность ТРО мыши.
Подробное описание изобретения
Перед подробным описанием настоящего изобретения полезно определиться с терминологией.
Аллельный вариант: Альтернативная форма гена, которая возникает благодаря мутации, или измененный полипептид, кодируемый мутированным геном. Генные мутации могут быть молчащими (без изменений в кодированном полипептиде) или могут закодировать полипептиды с измененной аминокислотной последовательностью.
сДНК: Комплементарная ДНК, приготовленная по обратной транскрипции РНК матрицы посредника или клон или увеличенная копия такой молекулы. Комплементарная ДНК может состоять из одной нити или из двух нитей.
Экспрессирующий вектор: Молекула ДНК линейная или круговая, которая содержит сегмент, кодирующий полипептид, в действии связанный с дополнительными сегментами, которые дают ему транскрипцию. Такие дополнительные сегменты включают в себя промоторную и терминаторную последовательности и также могут содержать один или более источники репликации, один или более селектируемые маркеры, усилитель, сигнал полиадениляции и т.д., экспрессирующие векторы обычно ответвляются от плазмида или вирусного ДНК или могут содержать элементы того и другого. Термин "в действии связанный" указывает на то, что эти сегменты расположены так, что могут действовать сообща, то есть транскрипция начинается в промоторе и проходит через кодирующий сегмент до терминатора.
Ген: Сегмент хромосомной ДНК, который кодирует полипептидную цепь. Ген содержит одну или более области, кодирующие аминокислоты, которые в некоторых случаях перемежаются с не кодирующими "вставочными последовательностями" (интронами), вместе с фланкирующими, некодирующими областями, которые обеспечивают транскрипцию кодирующей последовательности.
Молекулы, комплементарные: Молекулы полинуклеотидов с комплементарной последовательностью оснований и с обратной ориентацией по сравнению с эталонной последовательностью. Например, последовательность 5’ATGCACGGG 3’ комплементарна 5’ CCCGTGCAT 3’’.
Промотор: Часть гена, на которой начинаются полимеразовые РНК связи и синтез mPHK.
Как указывалось выше, настоящее изобретение предлагает вещества и способы для применения в продуцировании протеинов с гематопоэтической активностью. В настоящем документе термин "гематопоэтический" означает способность стимулировать пролиферацию и/или дифференциацию предшественников милоидов и лимпоидов, что имеет место в стандартных образцах. См., например, Меткалф. Proc.natl.Acad. USA 77:5327-53306, 1980, Меткалф и др. J. Cell.Physiol. 116: 198-206, 1983; и Меткалф и др. Exp Hematol.15:288-295, 1987. Обычно клетки костного мозга инкубируются в присутствии контрольного образца и испытательного образца. Затем эти культуры выбираются для пролиферации и дифференциации клетки путем визуального обследования и/или окрашивания. Особенно предпочтительным образцом является МТТ калориметрический образец Мосмана (J. Immunol.Meth. 65; 55-63, 1983; упоминается в данном документе для справки), более подробно описанный в приведенном ниже примере.
Настоящее изобретение основано частично на обнаружении активности, которая стимулирует рост клеток через рецептор MPL. Этот рецептор (Союри и др. Cell. 73: 1137-1147, 1990) был до этого открытия "сиротским" рецептором, естественный лиганд которого был неизвестен. Посредством процессов клона и мутагенеза, подробно описанного в примерах, приведенных ниже, авторы изобретения вырастили клеточную линию, которая зависит от стимулирования пути MPL, связанного через рецепторы, на выживание и рост и которая была способна к автокринному стимулированию рецептора. Была найдена кондиционированная среда из этих независимых от интерлейкина-3 (IL-3) клеток, которая поддерживает рост клеток, экспрессированных MPL рецептором и зависимых от IL-3. Эксперименты по нейтрализации антитела продемонстрировали, что эта активность возникала не благодаря IL-3 или IL-4, а что нейтрализация могла происходить посредством растворимого типа рецептора. Затем создавалась сДНК библиотека из клеточной линии, независимой от IL-3. ДНК применялась для трансфекции клеток почки детеныша хомяка (ВНК), и среда трансфектантов была исследована на способность к стимуляции пролиферации клетки, независимой от MPL. Положительный клон был изолирован, и продуцировался рекомбинантный лиганд MPL. Было обнаружено, что рекомбинантный протеин стимулирует пролиферацию широкого спектра предшественников милоидов и лимпоидов и, в частности, стимулирует продуцирование мегакариоцитов и нейтрофилов из недифференцированных клеток-предшественников в клетках костного мозга. Кроме того, оказалось, что рекомбинантный протеин стимулирует продуцирование тромбоцитов в испытуемых животных. В свете сказанного, протеин был назван тромбопоэтином (ТРО).
Настоящее изобретение предлагает изолированные молекулы полинуклеотида, кодирующие тромбопоэтин. Полезные для этого молекулы полинуклеотидов содержат mРНК, геномный ДНК, сДНК, синтетический ДНК и молекулы ДНК, вырабатываемые благодаря связи фрагментов от разных источников. Для продуцирования рекомбикантных ТРО предпочтительны молекулы ДНК с недостатком интронов в наиболее экспрессирующих системах. Под словом "изолированный" подразумевается, что молекулы удаляются из своей естественной генетической среды. Таким образом, настоящее изобретение позволяет получить молекулы ДНК, свободные от других генов, с которыми они обычно связаны. В частности, молекулы свободны от периферических или нежелательных кодирующих последовательностей и представлены в виде, удобном для использования внутри созданных генетической инженерией систем, продуцирующих протеин.
Последовательности клонов сДНК, кодирующие типичные протеины мышиного или человеческого ТРО, показаны в SEQ ID NO:1 и в SEQ TD NO:18 соответственно, а соответствующие последовательности аминокислот показаны в SEQ ID NO:2 и SEQ ID NO:19 соответственно. Специалистам в данной области науки будет понятно, что последовательности, показанные в SEQ ID NO:1, 2, 18 и 19 и геномные последовательности, показанные в SEQ ID NO:28 и 29, соответствуют одиночным аллелям мышиного или человеческого гена, и что предполагается существование аллельной вариации. Аллельные вариации последовательностей ДНК, показанные в SEQ ID NO:1, SEQ ID NO:18 и SEQ ID NO:28, в том числе и те, которые содержат молчащие мутации, и те, в которых мутации приводят к изменению последовательности аминокислоты, попадают в объем данного изобретения, так же, как и протеины, которые представляют собой аллельные вариации SEQ ID NO:2 и SEQ ID NO:19. Также будет очевидно, что специалистам в данной области будет нетрудно сконструировать сайты, которые облегчат и упростят манипулирование нуклеотидной последовательностью при помощи альтернативных кодонов.
Мышиные и человеческие последовательности, рассматриваемые в данной заявке, представляют собой очень важные средства для подготовки изолированных молекул полинуклеотидов, кодирующих ТРО протеины из других образцов ("образцов гомологов"). Предпочтительно, чтобы такие образцы гомологов содержали гомологи млекопитающих, такие как протеины коровы, собаки, свиньи, овцы, лошади и, особенно, приматов. В этой области знаний уже известны способы использования последовательной информации от первого образца для клонирования соответствующей полинуклеотидной последовательности из второго образца. См., например, Озабел и др. Current Protocols in Molecular Bioligy, John Wiley and Sons, Inc., NY, 1987. Молекулы ДНК по настоящему изобретению, кодирующие ТРО, как правило, на 60%, а желательно на 80%, а могут на 90-95% и более быть идентичными последовательностям из SEQ ID NO:1 и SEQ ID NO:18 и их аллельным вариантам.
Тромбопоэтиновые молекулы характеризуются способностью специфически связываться с рецептором MPL из того же образца и стимулировать продуцирование тромбоцитов in vivo. В обычном испытуемом животном ТРО способен увеличить уровень тромбоцитов на 100% и более за 10 суток, считая от начала ежедневного введения препарата.
Анализ распределения mРНК показал, что кодирующий ТРО присутствует в некоторых тканях человека и мыши, и особенно его много в легких, печени, сердце, скелетных мышцах и в почках. Таким образом, для того чтобы изолировать гомологи от других образцов, создается библиотека сДНК, желательно из одной из тканей, которая обнаружила свойства продуцировать особенно много mРНК. В данной области науки существуют хорошо известные способы создания библиотеки сДНК. См., например, Самбрук и др., Molecular Cloning: A Laboratory Manual, 2-е издание. Cold Spring Harbor Laboratory Press, 1989 и приведенные там ссылки. Для того чтобы определить ТРО, кодирующий молекулы, эта библиотека опробуется на сДНК мыши или человека, рассмотренной в данной заявке, или на его фрагменте, или на одном или более зондах ДНК на основе рассмотренных последовательностей. Особенно полезным являются зонды, содержащие полигонуклиотиды из, по крайней мере, 14 или более нуклеотидов и по длине до 25 и более нуклеотидов, которые, по крайней мере, на 80% идентичны части SEQ ID NO:1, SEQ ID NO:18, SEQ ID NO:28 или им комплеметнарным последовательностям той же длины. Желательно зондировать библиотеку с небольшими ограничениями по гибридизации, т.е. при примерно 2×SSC (раствор хлорида и нитрата натрия) и при температуре гибридизации примерно 50%. Молекулы, с которыми зонд скрещивается, обнаруживаются по стандартной процедуре обнаружения. Позитивные клоны подтверждаются анализом последовательности и активности, такой как способность фиксировать рецептор гомологового MPL (т.е. рецептора MPL из того же образца, что и сДНК) или стимулировать гематопоэзис из гомологовых клеток костного мозга. Специалисту будет очевидно, что можно использовать и другие способы клонирования.
Полинуклеотидные молекулы, кодирующие ТЗО (включая аллельные варианты и образцы гомологов молекул, рассматриваемых в данном изобретении) можно также изолировать клонированием из клеточной линии, которая продуцирует лиганд MPL и стимулирует рост отокрина. Короче говоря, зависимая от факторов клеточная линия, трансфексируется для экспрессии рецептора MPL (Вигон и др., Proc.Natl.Acad.Sci. USA 89:5640-5644, 1992; Шкода и др., EMBO J. 12:2645-2653, 1993; и SEQ ID NО:17), затем мутагенизируется и отбираются фактор-независимые клетки. Затем эти клетки используются в качестве источника ТРО mРНК. Соответствующие линии фактор-независимых клеток содержат линию IL-3-независимых BaF3 клеток (Паласиус и Стейнмец, Cell 41%727-734, 1985; Матей-Прево и др., Mol.Cell.Biol.6: 4133-4135, 1986), FDC-P1 (Хапел и др. Blood 64:786-790, 1984) и МО7Е (Кисc и др. Leukemia 7:235-240, 1993). Линии клеток, зависимых от фактора роста, можно организовать по опубликованным способам (т.е. Гринберг и др., Leukemia Res., 8:363-375, 1984; Декстер и др. и Баум и др. Experimental Hematology Today, 8-th Ann.Mtg. Int. Soc.Exp.Hematol. 1979, 145-156, 1980). Типичная процедура предусматривает извлечение клеток из ткани (костного мозга, селезенки, печени) и культивирование в обычной сывороточной среде, такой как RPMI 1640 с 10% коровьей сывороткой, 15% лошадиной сывороткой и 10-6 М гидрокортизона. С интервалом в 1-2 недели получаются неприкрепленные клетки и культуры питаются свежей средой. Выращенные, неприкрепленные клетки промываются и культивируются в среде с дополнительным источником фактора роста (т.е. RPMI 1640+5-20% WEHI-3 кондиционная среда в качестве источника IL-3). Эти клетки питаются свежей средой с интервалом в 1-2 недели и увеличиваются по мере роста культуры. По прошествии нескольких недель или нескольких месяцев индивидуальные клоны изолируются высеванием клеток на полутвердую среду (т.е. среду, содержащую метилцеллулозу) или путем ограничения расширения. Фактор-зависимость клонов подтверждается культивированием отдельных клонов при отсутствии фактора роста. Для достижения более частого выращивания фактор-зависимых клеток можно использовать ретровирусную инфекцию или химический мутагенез. Эти фактор-зависимые клетки трансфектируются для экспрессирования рецептора MPL, затем мутагенизируются, скажем, путем химической обработки, облучением УФ-лучами, облучением рентгеновскими лучами или ретровирусным мутагенезом. Мутагенезированные клетки затем культивируются в условиях, в которых выживание клетки зависит от продуцирования фактора роста отокрина, то есть в отсутствии экзогенного фактора роста, который требуется для родительской клетки. Предуцирование ТРО подтверждается результатами скрининг (массовой проверки), например проверкой кондиционированной среды на клетках, экспрессируощих и неэкспрессирующих рецепторы MPL, или проверкой деятельности кондиционной среды в присутствии растворимого рецептора MPL или антител в отличие от известных цитокинов.
Настоящее изобретение также предлагает изолированные протеины, которые по существу гомологичны протеинам из SEQ ID NO:2 или SEQ ID NO:19 и их образцов последовательностей. Под "изолированный" следует понимать протеин, который оказывается в условиях, отличных от своей естественной жизненной среды, от крови или животной ткани. Предпочтительно, чтобы изолированный протеин был по существу свободен от других протеинов, особенно от других протеинов животного происхождения. Желательно получать протеины высокого уровня чистоты, т.е. с более чем 95% чистоты, и еще более предпочтительно с уровнем чистоты более 99%. Термин "по существу гомологичны" используется здесь для указания на то, что протеины с 50% последовательной идентичностью, а более предпочтительно с 60% и еще более предпочтительно с 80% последовательной идентичностью последовательности, показанной в SEQ ID NO:2 или SEQ ID NO:19 или образцам их гомологов. Последовательная идентичность, выраженная в процентах, определяется известными способами. СМ., например, Алтшул и др. Bull. Math. Bio.48:603-616, 1986 и Хеникоф и Хеникоф, Proc.Natl.Acad.Sci.USA 89: 10915-10919, 1992. Короче говоря, две аминокислотные последовательности выравниваются для опитимизации выравнивания с использованием счета, как в спорте, промежутку 10 соответствует выступ 1 и матрицы Хеникофа и Хеникофа "цветение 62", как показано в Таблице 1 (аминокислоты обозначаются стандартными однобуквенными кодами).

Затем следующим образом подсчитывается идентичность, выраженная в процентах:

Фактически гомологические протеины характеризуются тем, что имеют одну или более замен, делиций или добавлений аминокислот. Такие изменения желательно иметь минорными (побочными) по своей природе, то есть быть консервативными заменами аминокислот, что не сильно влияет на укладку или деятельность протеина (см. Таблицу 2); с малыми делициями, обычно от одной до примерно 30 аминокислот; и малая протяженность амино- или карбоксил-терминала, как, например, остаток метионина амино-терминала, малое количество линкерного пептида до 20-25 остатков или малая протяженность, что упрощает процесс очищения, такие как полигистидиновый путь (пространство) антигенный эпитоп или связывающая область. См. в основном Форд и др. Protein Expression and Purification 2:95-107, 1991, что используется в данной заявке в качестве ссылки.

Аминокислоты в ТРО можно обнаружить с помощью методики, известной в данной области науки, например сайт-направленный мутагенез или аланин-сканирующий мутагенез (Каннингэм и Уэллс, Science 244, 1081-1085, 1989). В последнем методе одно-аланинная мутация вводится в каждый остаток молекулы и получаемые молекулы-мутанты испытываются на биолгическую активность (например, связывание рецептора, in vitra или in vivo пролиферативная дейтельность) для обнаружения аминокислотных остатков, которые важны для активности молекул. Сайты взаимодействия лиганда и рецептора также могут быть определены путем анализа кристаллической структуры, которая определяется такими методами, как ядерный магнитный резонанс, кристаллография или фотоаффинное мечение. См., например, де Во и др. Science 255:306-312, 1992; Смит и др. J.Mol.Biol. 224:899-904, 1992; Влодавер и др. FEBS Lett. 309:59-64, 1992.
Обычно предсказывают цитокинам 4-альфа спиральную структуру, в которой первая и четвертая спирали являются наиболее важными во взаимодействии лиганда и рецептора и из всех членов семейства лучше всего сохраняются. Взглянув на человеческую аминокислотную ТРО последовательность, показанную в SEQ ID NO:19, можно сказать, что выравнивание цитокиновых последовательностей предполагает, что эти спирали связаны аминокислотными остатками от 29 до 53, 80 и 99, 108 и 130 и 144 и 168 соответственно (границами являются +4 остатка). Границы спиралей мышиных (SEQ ID NO:2) и других нечеловеческих ТРО можно определить выравниванием с человеческой последовательностью. Другая важная структурная особенность ТРО заключается в том, что ТРО содержит цистеиновые остатки в точках 51, 73, 129 и 195 SEQ ID NO:2 (в соответствии с точками 28, 50, 106 и 172 SEQ ID NO:19).
Кроме того, протеины по настоящему изобретению (или их полипептидные фрагменты) могут быть соединены с другими биоактивными молекулами, особенно с другими цитокинами, для создания многофункциональных молекул. Например, область с-терминала тромбопоэтина может быть соединена с другими цитокинами для усиления их биологических свойств или повышения эффективности производства. Молекула тромбопоэтина оказалась составленной из двух областей. Первая (амино-терминальная) область из примерно 150 аминокислот похожа по размеру эритропоэтину и некоторым другим гематопоэтичным цитокинам и напоминает их по структуре. За этой первой областью идет вторая примерно из 180 аминокислот, структура которой не совсем аналогична какой-либо известной в базе данных структуре протеина. Эта вторая область сильно обогащается в гликосиляционных сайтах с N-связями и в остатках серина, пролила и трионина, которые являются отметкой гликосиляционных сайтов с O-связями. Это очевидно высокое содержание карбогидрата предполагает, что эта область должна участвовать в создании более растворимой первой гидрофобной области. Эксперименты указывают на то, что карбогидрат, связанный со второй областью, входит в нужную межклеточную упорядоченную структуру и секрецию протеина во время биосинтеза. Вторая область также может участвовать в стабилизации первой области против протеолитической деградации и/или продления полупериода жизни молекулы и может сделать возможной передачу биологического сигнала или специфическую активность протеина.
Таким образом, настоящее изобретение предлагает серию новых, гибридных молекул, в которых вторая область ТРО соединена по вторым цитокином. Предпочтительно соединение области С-терминала ТРО с С-концом второго цитокина. Желательно соединение осуществлять путем сплайсинга на уровне ДНК, чтобы допустить экспрессию химеровых молекул в системах рекомбинантного продуцирования. Получаемые при этом молекулы затем анализируются на проверку таких свойств, как улучшенная растворимость, повышенная стабильность, продленный полупериод времени жизни или повышенные уровни экспрессии и секреции и фармакодинамики. В конкретные примеры таких химеровых цитокинов входят те, в которых вторая область ТРО соединяется с С-концом ЕРО, G-CSF, GM-CSF, IL-6, IL-3, IL-11. Как указывалось выше, это обычно делается слиянием ДНК. Слитая сДНК затем сабклонируется в соответствующий экспрессирующий вектор и преобразуется или трансфектируется в клетки-хозяева или организмы обычными способами. Получаемые после слияния протеины очищаются обычными хроматографическими методами очистки (например, способом хроматографии), и их свойства сравниваются со свойствами собственного, неслитого родительского цитокина. Далее такие гибридные молекулы могут содержать дополнительные аминокислотные остатки (например, линкеры полипептида) между компонентами протеинов или полипептидов.
Помимо гематопоэтических протеинов, рассмотренных выше, настоящее изобретение касается фрагментов этих протеинов и изолированных полинуклеотидовых молекул, кодирующих этих фрагменты. Особый интерес представляют фрагменты длиной, по крайней мере, в 10 аминокислот, которые связаны с рецепторами MPL, и полипуклеотидные молекулы длиной, по крайней мере, в 30 нуклеотидов, которые кодируют такие полипептиды. Полипептиды такого типа обнаруживаются известными методами скрининга, например переваривание интактных (целых) протеинов или синтезирование небольших, перекрывающихся полипептидов или полинуклеотидов (и экспрессия последних), по желанию в сочетании с методами структурного анализа, рассмотренного выше. Полученные полипептиды затем испытываются на способность специфически связать рецептор MPL и стимулировать пролиферацию клетки через рецептор MPL. Наличие связки определяется известными способами, например такими, которые описаны у Клотца, Science 217:1247, 1982 ("Scatchard analysis"). Короче говоря, радиомеченный испытуемый полипептид инкубируется (выращивается) вместе с MPL клетками, несущими рецептор, при увеличении концентрации немеченных ТРО. Меченый, с граничными клетками, полипептид отделяется от неграничного меченого полипептида центрифугированием через фталатовое масло. Родство испытуемого полипептида определяется вычерчиванием графика зависимости границы со свободными метками (по оси ординат) от граничной метки (по оси абсцисс). Специфичность связи определяется в сравнении с цитокином, отличным от ТРО. Связывание рецепторов также может быть определено путем осаждения испытуемого состава с помощью лишенного движения рецептора MPL (или связанного с лигандом экстрацеллюлярной области). Короче говоря, рецептор или его протеин лишен подвижности на нерастворимой опоре. Испытуемый состав помечается, например, метаболическим способом мечения клетки-хозяина в случае рекомбинантного испытуемого состава, или обычным in vitro способом мечения (например, радио-иодирование). Затем меченый состав комбинируется с лишенным подвижности рецептором, несвязанное вещество удаляется и обнаруживается связанный, меченый состав. В данной области науки известны методы обнаружения разнообразных меток. Стимулирование пролиферации определяется обычным способом путем калориметрического МТТ анализа клеток с рецептором MPL. Полипептиды анализируются на предмет активности при разных концентрациях, обычно в пределах диапазона от 1 нм до 1 мМ.
Предлагаются также более крупные полипептиды вплоть до 50 и более остатков, желательно 100 или более остатков, еще более предпочтительно 140 и более остатков, размером со зрелый протеин. Например, результаты анализа и моделирования аминокислотной последовательности, показанной в SEQ ID NO:2 от остатка 51 до остатка 195 включительно, или в SEQ ID NO:19 от остатка 28 до остатка 172 включительно, предполагают, что эти части молекул представляют собой области, подобные цитокину, способные самоупорядочиваться. Представляют собой также интерес молекулы, содержащие эту основную цитокино-подобную область плюс один или более дополнительных сегментов или областей продукта первичной трансляции. Таким образом, другие интересующие нас полипептиды включают в себя полипептиды, указанные в Таблице 3.

Специалистам в данной области науки будет очевидно, что промежуточные формы молекул (например, тех, которые имеют С-концы между остатками 196 и 206 в SEQ ID NO:2 или тех, которые имеют N-концы между остатками 22 и 28 в SEQ ID NO:19) так же представляют интерес, также, как и полипептиды с одной или более аминокислотными заменами, делециями, вставками или N- или С-терминальную протяженность, описанную выше. Таким образом, настоящее изобретение предлагает гематопоэтические полипептиды с, по крайней мере, 10 аминокислотными остатками, предпочтительно с, по крайней мере, 50 остатками, более предпочтительно с, по крайней мере, 100 остатками и особенно предпочтительно с, по крайней мере, примерно 140 остатками по длине, в которых указанные полипептиды являются фактически гомологичными относительно таких же по размеру полипептидов из SEQ ID NO:2 или SEQ ID NO:19.
Протеины по данному изобретению могут продуцироваться в клетках-хозяевах, созданных генной инженерией, известными способами. Подходящими клетками-хозяевами являются клетки такого типа, которые можно трансформировать с экзогенной ДНК и выращивать в культуре и которые содержат бактерии, грибковые клетки и культивируются в более высокие эукаритические клетки. Технология манипулирования клонированных молекул ДНК в различные клетки-хозяева рассмотрена в работах Самбрука и др.. Molecular cloning: A Laboratory Manual, 2-е издание, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1989 и Озубел и др., которые использованы в данной заявке в качестве ссылочного материала.
Обычно последовательность ДНК, кодирующая протеин по данному изобретению, в действии связана с промотором транскрипции и терминатором внутри экспрессируюшего вектора. Этот вектор обычно должен содержать один или более селектированных маркеров и одно или более начал репликации, хотя специалисту понятно, что в пределах определенных систем селектированные маркеры можно создать на отдельных векторах, а репликацию экзогенной ДНК можно проводить путем интеграции в геном клетки-хозяина. Селекция промоторов, терминаторов, селективных маркеров, векторов и других элементов - это вопрос обычного расчета для специалистов среднего уровня. Многие из этих элементов описаны в литературе и их можно достать через коммерческих поставщиков.
Для того чтобы направить протеин по данному изобретению на секреторный путь клетки-хозяина, в экспрессирующем векторе предполагается последовательность секреторных сигналов (известных также как лидер последовательность или предварительная последовательность). Последовательность секреторных сигналов соединяется с последовательностью ДНК, кодирующей протеин по данному изобретению в правильной рамке считывания. Последовательность секреторных сигналов обычно располагается под углом 5’ к последовательности ДНК, кодируя нужный протеин, хотя определенная последовательность сигналов может располагаться в любом месте нужной последовательности ДНК (см., например, работу Уэлч и др., Патент США №5,037,743; Холланда и др., Патент США №5,143,830). Последовательность секреторных сигналов может быть из тех, которые обычно связаны с протеином по данному изобретению, или может быть из гена, кодирующего другой выделенный протеин.
Дрожжевые клетки, особенно клетки рода Sacharomyces, являются предпочтительными клетками-хозяевами, применяемыми в данном изобретении. Способы для преобразования дрожжевых клеток с экзогенной ДНК и продуцирования рекомбинантных протеинов из них рассмотрены, например, Кавасаки в Патенте США №4,599,311; Кавасаки и др., Патент США №4,931,373; Брейк, Патент США №4,870,008; Уэлч и др., Патент США №5,037,743 и Мюррей и др., Патент США №4,845,075, которые вошли в данный документ в качестве ссылочного материала. Преобразованные клетки отбираются по фенотипу, определяемому по селектированному маркеру, по стойкости к лекарственным препаратам или по способности расти в отсутствии специального питательного вещества (например, лейцина). Для применения в дрожжах предпочтительной векторной системой является векторная система РОТ1, рассмотренная Кавасаки и др., Патент США №4,931,373, которая позволяет отбирать преобразованные клетки по росту в глюкозо-содержащей среде. Предпочитаемой последовательностью секреторных сигналов для применения в дрожжах является последовательность S.cerevisiae MFal гена (Брейк там же; Куржан и др. Патент США №4,546,082). Подходящими промоторами и терминаторами для дрожжей являются эти элементы из генов глюколитного фермента (энзима) (См., например, Кавасаки, Патент США №4,599,311; Кингсмен и др. Патент США №4,615,974; и Биттер, Патент США №4,977,092, которые включены в данный документ в качестве ссылочного материала) и гены спиртового дегидрагеназа. См. также Патенты США №4,990,446; 5,063,154; 5,139,936 и 4,661,454, которые также включены в настоящий документ в качестве ссылочного материала. Системы преобразования для других дрожжей, включая Hansenula polymorpha, Schizosaccharomyces роmbе, Ustilago maydis, Pichia pastoris, Pichia guillermondi и Candida maltosa, известны в данной области. См., например, работу Глиссон и др., J.Gen.Microbiol. 132:3459-3465, 1986 и Крегг, патент США №4,882,279.
Другие грибковые клетки также подходят в качестве клеток-хозяев. Например, клетки Aspergilus могут быть использованы по методу МакНайта и др., Патент США №4,935,349, который использован в данном документе в качестве ссылочного материала. Способы преобразования Acremonium chrysogenum рассматриваются у Сумино и др., Патент США №5162,228, который используется в данном документе в качесте ссылочного материала. Способы преобразования Neurospora рассматриваются в данном документе в качестве ссылочного материала.
Предпочтительными клетками-хозяевами в рамках данного изобретения также являются культивированные клетки млекопитающих. Способы введения экзогенной ДНК в клетку-хозяина млекопитающего включают в себя трансфекцию посредством фосфата кальция (Винглер и др., Cell 14:725, 1978; Корсаро и Пирсон, Somatic Cell Genetics 7:603, 1981; Грэм и Ван дер Эб, Virology 52: 456, 1973), электропорацию (Ньюман и др., ЕМВО.7.1:841-845, 1982) и трансфекцию посредством DEAE-декстрана (Озубел и др. Current Protocols in Molecular Biology, Джон Уидли и сыновья, Инк7, NY, 1987), которые включены в данный документ в качестве ссылочного материала. Продуцирование рекомбинантных протеинов в культивированных клетках млекопитающих рассмотрено, например, у Левинсона и др. Патент США №4,713,339; Хагена и др. Патент США №4,784,950; Палмитер и др. Патент США №4,579,821 и Рингольд, Патент США №4,656,134, которые включены в данный документ в качестве ссылочного материала. В культивированные клетки млекопитающих входит COS-1 (ATCC No CRL 1650), COS-7 (АТСС No CRL 1651), BHK (ATCC No CRL 1632), BHK 570 (ATCC No CRL 10314), 293 (ATCC No CRL 1573; Грэм и др., J.GEn.Virol. 36:59-72, 1977) и клеточные линии яичника китайского хомяка (например, СНО-К1; АТСС No CCL 61). Известны и другие подходящие клеточные линии, которые можно достать в публичных хранилищах, таких как Американское Собрание Типов Культур, Роквил. Мэридэнд. Обычно предпочтительными являются сильные промоторы транскрипции, такие промоторы, как из SV-40 или цитомегаловирус. См. например. Патент США №4,956,288. Другие подходящие промоторы состоят из промоторов из металлотионеиновых генов (Патенты США №4,579,821 и 4,601,978, которые включены в данный документ в качестве ссылочного материала), и поздние основные аденовирусные промоторы.
Отбор с помощью лекарственных препаратов обычно используется при отборе культивированных клеток млекопитающих, в которые внедряется чужеродная ДНК. Такие клетки обычно называются "трансфектантами". Клетки, которые культивированы в присутствии селективного вещества и способны передать нужный ген своему потомству, называются "Стабильными трансфектантами". Предпочтительным селективным маркером является ген, кодирующий стойкость к неомицину антибиотика.
Отбор проводится в присутствии лекарственного препарата типа неомицина, например G-418 или ему подобный. Системы отбора могут также применяться для повышения уровня экспрессивности нужного гена, такой процесс называется "амплификация". Амплификация проводится культивированием трансфектантов в присутствии небольшого количества селективных веществ с последующим увеличением количества селективных агентов для отбора клеток, которые продуцируют большое количество продуктов введенных генов. Предпочтительным усиливаемым отбираемым маркером является редуктаза лигидрофолата, которая придает стойкость метатрексату. Другие гены, стойкие к воздействию лечебных препаратов (например, стойкость к действию гигромицина, стойкость ко многим лечебным препаратам или пуромициновой ацетилтрансферазы), также могут быть использованы.
Другие эукариотические клетки также могут быть использованы как клетки-хозяева, в том числе клетки насекомых, клетки растений и клетки птиц. Преобразование клеток насекомых и продуцирование из них чужеродных протеинов описано в работе Гуарино и др. Патент США №5,162,222; Бэнга и др. Патент США №4,775,624; и в публикациях WIPO WO 94/06463, которые используются здесь для справки. Применение Agrobacterium rhizogenes в качестве вектора, экспрессирующего гены в клетках растений, рассматривалось в работе Синкер и др. J.Biosci. (Bangalore) 11:47-58, 1987.
Предпочтительные прокариотичные клетки-хозяева для использования по данному изобретению представляют собой штаммы бактерий Escherechia coli, хотя также применимы и Bacillus и другие роды. Технология преобразования этих клеток-хозяев и экспрессии последовательностей чужеродной ДНК, клонированных из них, хорошо известны (см., например, работу Самбрука и др. там же). При экспрессии протеина в бактериях, таких как Е.соli, протеин может сохраняться в цитоплазме, обычно в виде нерастворимых гранул или может направляться в периплазмическую область посредством бактериальной последовательности секреции. В предыдущем случае клетки лизируются, и гранулы восстанавливаются и денатурируются с помощью, например, гуанилинового изотиоцианата. Денатурированный протеин затем опять складывается путем разбавления денатуранта. В последнем случае протеин может быть восстановлен из периплазмической области в растворимую и функционирующую форму путем разрыва клеток (например, обработкой ультразвуком или осмотическим шоком) для того, чтобы выпустить содержимое периплазмаческой области и восстановить протеин.
Преобразованные или трансфектированные клетки-хозяева культивируются в соответствии с известной методикой в культивирующей среде, состоящей из питательных веществ и других компонентов, необходимых для роста выбранных клеток-хозяев. В данной области науки известно большое количество подходящих сред, в том числе среды определенного состава и сложные среды, которые обычно содержат источник углерода, источник азота, основные аминокислоты, витамины и минералы. Среды могут также содержать такие компоненты, как факторы роста или сыворотку, по необходимости. Среда роста обычно выбирается для клеток, содержащих экзогенно добавляемую ДНК, например, выбором лечебного препарата или недостатком основного питания, которые подбираются селективным маркером, находящимся на экспрессирующем векторе, или трансфектируются в клетку-хозяина.
В рамках данного изобретения для продуцирования ТРО можно применять трансгенную технологию. Предпочтительно продуцировать протеины в молочных железах в женском реципиенте млекопитающего. Экспрессия в молочную железу и последующая секреция нужного протеина в молоко позволяет преодолеть многие трудности, которые встречаются при изоляции протеинов от других источников. Молоко легко собирается, может находиться в большом количестве и имеет хорошие биохимические характеристики. Кроме того, в молоке присутствует большая концентрация основных молочных протеинов (от примерно 1 до 15 г/л).
С коммерческой точки зрения очевидно предпочтительным является использование в качестве реципиента образца с большим объемом производства молока. Хотя можно использовать маленьких животных - мышей или крыс (и они предпочтительны на стадии доказательства правильности концепции), в рамках данного изобретения предпочтительно использовать домашний скот, в том числе свиней, коз, овец и крупный рогатый скот, не ограничиваясь перечисленными животными. Особенно предпочтительными являются овцы, благодаря таким факторам, как предыдущая история трансгенезиса в этих образцах, объем производства молока, стоимость и легко доступное оборудование для сбора овечьего молока. См. WIРО публикацию WO 88/00239 для сравнения факторов, воздействующих на выбор образцов реципиентов. Обычно бывает желательно отобрать таких животных-реципиентов, которые выращивались как молочные породы, такие как Восточно-Фризлендские овцы, или ввести молочные породы скота выведением трансгенных линий в более позднее время. В любом случае необходимо использовать животных с хорошим здоровьем.
Для того чтобы получить экспрессию в молочной железе, используется транскрипционный промотор из гена молочного протеина. Гены молочного протеина содержат гены, кодирующие казеины (см. Патент США №5,304,489, включенный в данный документ в качестве ссылочногго материала), бета-лактоглобулин, альфа-лактулбумин и кислый протеин. Здесь предпочтительным является промотор бета-лактоглобулина (BLG). В случае использования гена овечьего бета-лактоглобулина обычно можно использовать часть фланкирующей последовательности гена, по крайней мере, 406 bp 5’, хотя более предпочтительными являются более крупные части 5’ фланкирующей последовательности вплоть до 5 kpb, скажем, -4,25 kpb сегмент ДНК, окружающего 5’ фланкирующий промотор и некодирующую часть гена бета-лактоглобулина. См. работу Уайтлоу и др., Biochem J. 286:31-39, 1992. Подойдут и аналогичные фрагменты промотора ДНК из других образцов.
Можно использовать и другие области гена бета-лактоглобулина, как геномные области гена, которые следует выразить. Обычно в данной области науки принято, что конструкции, лишенные интронов, например, экспрессируют плохо по сравнению с конструкциями, содержащими последовательности ДНК (см. работу Бринстора и др., Proc.Natl.Acad. Sci. США 85:836-840, 1988; Палмера и др. Proc.Natl.Acad.Sci.США 88:478-482, 1991: Уатлоу и др. Transgenic Res.1:3-13, 1991; WO 89/01343: WO 01/02318). В свете сказанного, обычно предпочтительным является там, где это возможно, использовать геномные последовательности, содержащие все или некоторые собственные интроны гена, кодирующего нужный протеин или полипептид, Таким образом, предпочтительным является дальнейшее введение, по крайней мере, некоторых нитронов, например, предпочтительным является ген бета-лактоглобулина. Одной такой областью сегмента ДНК, которая обеспечивает сплайсинг нитрона и полиадеинляцию, РНК из 3’ некодирующей области гена бета-лактоглобулина овцы. При замене натуральными 3’’ некодирующими последовательностями гена этот бета-лактоглобулиновый сегмент овцы может как повысить, так и стабилизировать уровень экспрессивности нужного протеина или полипептида. В других вариантах изобретения область, окружающая начало ATG последовательности ТРО, заменяется соответствующими последовательностями из специфического гена молочного протеина. Такая замена создает условия для мнимой инициации ткани для усиления экспрессивности. Обычно заменяют целые препро- последовательности ТРО и 5’ некодирующие последовательности с, например, BLG геном, хотя можно заменять и более мелкие области.
Для экспрессии ТРО в трансгенных животных сегмент ДНК, кодирующий ТРО, связан в действиии с добавочными сегментами ДНК, необходимыми для экспрессии, для того чтобы продуцировать единицу экспрессии. Такие добавочные сегменты содержат упомянутый выше промотор, а также последовательности, которые создают условия для окончания транскрипции и для полиадениляции mPHK. Далее эти единицы экспрессии должны содержать сегмент ДНК, кодирующий последовательности секреторных сигналов, в действии соединенный с сегментом, кодирующим ТРО. Последовательность секреторного сигнала может быть собственной последовательностью сигналов ТРО или может быть последовательностью секреторных сигналов другого протеина, такого как, например, протеина молока. См., например, работу вон Хенжа, Nuc.Acids ReS& 14% 4683-4690, 1986; и Мида и др. Патент США №4,873,316, которые включены в данный документ в качестве ссылочного материала.
Построение единиц экспрессии для использования в трансгенных животных обычно выполняется путем вставки последовательности ТРО в плазмидный или в фаговый вектор, содержащие дополнительные сегменты ДНК, хотя единица экспрессии может быть построена фактически с помощью любой последовательности лигатур. В частности, известен метод использования вектора, содержащего сегмент ДНК, кодирующий молочный протеин, и замены кодирующей последовательности для молочного протеина на кодирующую последовательность ТРО полипептида, тем самым создавая генное слияние, в которое входят последовательности регулирования экспрессии гена молочного протеина. В другом случае, клонирование единиц экспрессии в плазмидных или других векторах упрощает амплификацию последовательности ТРО. Амплификация обычно выполняется в бактериальных клетках-хозяевах (например, Е.соli), таким образом, эти вектора обычно содержат начало репликации и селектируемый маркер, действующий в бактериальных клетках-хозяевах.
Затем единица экспрессии вводится в оплодотворенную яйцеклетку (включая эмбрионы на ранней стадии) образца выбранного реципиента. Введение гетерологовой ДНК может производиться одним из нескольких способов, в том числе методом микроинъекции (см. Патент США №4,8736191) ретровирусной инфекции (Джениш, Science 240:1468-1474, 1988) или путем сайт-направленной интеграции с помощью клеток первичного стебелька зародыша (ES) (рассматривается в работе Брэдли и др. Bio/Technology 10:534-539, 1992). Затем эти яйцеклетки имплантируются в яйцепроводы или в матки псевдобеременных женских особей и развиваются там до срока. Потомство, несущее ДНК, введенное в зародышевую линию, может перенести ДНК на свое потомство обычным, способом Менделя, что дает возможность развиваться трансгенному стаду.
В данной области науки известны основные методики продуцирования трансгенных животных. См., например, работу Хогана и др. Manipulating the Mouse Embryo: A laboratoty mаnuаl, Cold Spring Harbor Laboratory, 1986; работу Симонов и др. BIol.REprod.32: 645-651, 1985; работу Бухлера и др. Bio/Technology 8:140-143, 1990; работу Эберта и др. Bio/Technology 9:835-838, 1991; работу Кримпенфорта и др. Bio/Technology 9:844-847, 1991; работу Уолла и др. J.Cell.Bilochem. 49:113-120, 1992; Патенты США №4,873,191 и 4,873,316; WIPO Публикации WO 88/00239, WO 90/05188, WO 92/11757; GB 87/00458, которые включены в данный документ в качестве ссылочного материала. Способы введения чужеродных последовательностей ДНК в млекопитающих и в их зародышевые клетки сначала были отработаны на мышах. См., например, работу Гордона и др. Proc.Natl.Sci.USA 77:7380-7384, 1980; работу Гордона и Ралдла. Science 214% 1244-1246, 1981; работу Палмитера и Бринстера, Cell 41:343-345, 1985; работу Бринстера и др. Proc.Natl.Acad.Sci.USA 82:4438-4442, 1985; и работу Хогана и др. (там же). Эти способы были адаптированы для использования у большого количества животных, в том числе крупного рогатого скота (см., например, WIPO Публикации WO88/00239, WO 90/05188 и WO 92/11757; и работу Симонов и др., Bio/Technology 6:179-183, 1988). Подытоживая, можно сказать, что наиболее эффективным способом из применяемых в настоящее время в генерации трансгенных мышей и крупного рогатого скота является введение нескольких сотен линейных молекул нужной ДНК в одно из ядер оплодотворенной яйцеклетки по стандартному в данной области науки способу. Использовались также инъекции ДНК в цитоплазму зигота.
Можно также использовать продуцирование в трансгенных растениях. Можно обобщить или направить экспрессию на конкретный орган, скажем на клубень. См. работу Хиатта, Nature 344:469-479, 1990; работу Эдельбаума и др. J.Interferon Res.12: 449-453, 1992; работу Сиймонса и др. Bio/Technology 8:217-221, 1990; и Публикации Европейского Патентного ведомства ЕР 255,378.
ТРО, приготовленный по данному изобретению, очищается известными методами, например, аффиннным очищением и отделением по размеру, заряду, растворимости и других свойств протеина. Когда протеин спродуцирован в культивированных клетках млекопитающего, желательно культивировать эти клетки в среде, свободной от сыворотки для того, чтобы ограничить количество примесного (загрязняющего) протеина. Предпочтительный способ фракционирования заключается в аффинной хроматографии на конканавалине А или другом пектине, таким образом используя углевод, имеющийся на протеине. Эти протеины также могут очищаться с помощью лишенных движения MPL рецепторов протеина или его части, связанной с лигандом, или используя аффинную метку (например, полигистидин, вещество Р или другой полипептид или протеин, для которых имеется антитело или специальное связывающее вещество). Между нужным протеином и аффинной меткой можно расположить специальный сайт дробления.
Протеины по данному изобретению терапевтически могут использоваться везде, где требуется увеличение пролиферации клеток в костном мозге, например, при лечении цитопении, которая вызвана альфатической анемией, миелодиспластическими синдромами, химиотерапией или врожденной цитопенией; у пациентов с трансплантацией костного мозга; у пациентов с трансплантацией периферических стволовых клеток крови; и при лечении условий, которые вызывают сбои функционирования костного мозга, например миелодиспластического синдрома. Протеины также пригодны для увеличения продуцирования тромбоцитов, например при лечении тромбоцитопении. Тромбоцитопения связана с большой группой заболеваний и клинических ситуаций, которые могут самостоятельно или в сочетании создавать такие условия. Показания пониженного содержания тромбоцитов могут оказаться следствием, например, дефектов продуцирования тромбоцитов (из-за, скажем, врожденных нарушений, таких как синдром Фанкони, синдром отсутствия ..., аномалия Вискота Олдрича, аномалия Мэя Хеглина, синдромы Бернарда-Сулие, синдром Меннеаполиса, синдром Эпштейна, тромбоцитный синдром Монреаля и синдром Экштейна), аномального распределения тромбоцитов, потери на разведении из-за переливаний крови в больших объемах, аномальные повреждения тромбоцитов или аномальная изоляция тромбоцитов в селезенках у пациентов с увеличенной селезенкой (например, из-за цироза или перенапряжения сердца). Например, лекарственные препараты химиотерапии, применяемые при терапии рака, могут подавлять развитие клеток-предшественников тромбоцитов в костном мозге, в результате чего возникающая тромбоцитопения ограничивает химиотерапию и может настоятельно потребовать переливания крови. Кроме того, ослабить продуцирование и распределение тромбоцитов могут некоторые злокачественные образования. Радиационная терапия, призванная убивать злокачественные клетки, убивает также и клетки предшественников тромбоцитов. Тромбоцитопения может также возникнуть из-за различных автоиммунных нарушений в тромбоцитах, вызванных действием лечебных препаратов, аллоиммунитетом новорожденного, аллоиммунитетом переливания тромбоцитов и вирусной инфекцией (в том числе человеческого вируса Т-клеток). Протеины по данному изобретению могут уменьшить или ликвидировать необходимость в переливании, благодаря чему можно снизить вероятность аллоиммунитета тромбоцитов. Аномальные нарушения тромбоцитов могут возникнуть из-за: (1) увеличенного потребления тромбоцитов трансплантантом сосудистой ткани и травмированной ткани, (2) иммунного механизма, связанного с, например, тромбоцитопенией, вызванной действием лекарственных препаратов, пурпурином идиопатической тромбоцитопении (ITP), автоиммунными заболеваниями, гематологическими нарушениями, такими как лейкемия или лимпома или метастазный рак в костном мозге. Другие показания на применение протеинов по данному изобретению - это альпастиченская анемия и подавление костного мозга, вызванное действием лечебных препаратов, например, из-за химиотерапии или лечения инфекции человеческим вирусом Т-клеток AZT.
Тромбоцитопения проявляется в усилении кровотечения, например кровотечения слизистых тканей из области носоглотки или желудочно-кишечного тракта, а также выделения из ран, язв и мест введения инъекции.
При фармакологическом использовании протеины по данному изобретению рекомендуются для параэнтерального введения, в частности внутривенно или подкожно уже известными способами. Внутривенное введение должно осуществляться инъекцией или вливанием в течение периода от одного до несколько часов. Обычно фармакологические назначения включают в себя гематопоэтический протеин в сочетании с фармацевтически приемлемым вектором, например физиологический раствор, физиологический раствор, содержащий буфер, 5%-ная дикстроза в воде и т.п. В назначениях далее могут быть один или более экципиентов, консерванты, растворители, буферные вещества, альбумин для предотвращения потери протеина на поверхности пробирки. Кроме того, гематопоэтические протеины по данному изобретению могут сочетаться с другими цитокинами, особенно рано-действующими цитокинами, такими как фактор ствольных клеток, IL-3, IL-6, IL-13 или GM-CSF. При использовании такой комбинированной терапии цитокины могут быть совмещены в одну структуру или могут быть введены в отдельные структуры. Способы создания структур хорошо известны в данной области науки и раскрыты, например, в работе Геннаро и др. Reminoton’s Pharmaceutical Science, Mack Publishing Co., Easton PA, 1990, которая включена в данный документ в качестве ссылочного материала. Терапевтические дозы обычно лежат в пределах от 0.1 до 100 мг/кг от веса пациента каждый день, предпочтительно от 0.5 до 20 мг/кг в день, причем доза должна быть точно определена врачом в соответствии с принятыми нормами, принимая во внимание природу и сложность условий, которые необходимо лечить, особенности пациента и т.д. Определение дозы основывается на среднем уровне квалификации специалиста в данной области. Протеины обычно должны вводиться в течение периода до 28 дней после химиотерапии или трансплантации костного мозга или до тех пор, пока количество тромбоцитов не дойдет до >20,000/мм3, предпочтительно >50,000/мм3. Обычно протеины вводятся в течение одной недели или менее часто в течение периода в один-три дня. Как правило, терапевтически эффективным количеством ТРО является количество, достаточное для продуцирования клинически заметных увеличений пролиферации и/или дифференциации предшественников клеток лимпоидов или милоидов, которые будут проявляться в увеличении уровня циркуляции зрелых клеток (например, тромбоцитов или нейтрофилов). Таким образом, лечение тромбоцитного нарушения должно продолжаться до тех пор, пока показатель количества тромбоцитов не дойдет, по крайней мере, до 20,000/мм3, желательно до 50,000/мм3. Протеины по данному изобретению могут также вводиться в сочетании с другими цитокинами, например, с IL-3, -6 и -11; фактора ствольных клеток; эритропоэтина; G-csf и GM-csf. В рамках режимов комбинированной терапии ежедневные дозы других цитокинов, как правило, должны быть: RPO, <150 U/кг; GM-CSF, 5-15 мг/кг; IL-3, 1-5 мг/кг и G-csf, 1-25 мг/кг. Например, комбинированная терапия с ЕРО показана для анемичных больных с низким уровнем ЕРО.
Протеины по данному изобретнию также являются весьма ценным инструментом для изучения in vivo дифференциации и развития гематопоэтических клеток, например для объяснения механизма дифференциации клеток и определения последовательности клеточных поколений клеток зрелых клеток, и могут также найти применение в качестве пролиферативного вещества в культивировании клеток.
Протеины по данному изобретению также могут использоваться ех vivo, например, при автологовом культивировании костного мозга. Короче говоря, костный мозг берется у пациента до химиотерапии и обрабатывается ТРО, по желанию в сочетании с одним или более другими цитокинами. Обработанный костный мозг затем возвращается пациенту после химиотерапии для ускорения выздоровления костного мозга. Кроме того, протеины по данному изобретению также могут использоваться для in vivo увеличения в объеме костного мозга или периферийных кровяных клеток-предшественников (РВРС). До начала химиотерапии костный мозг можно стимулировать фактором ствольных клеток или G-csf для высвобождения ранних клеток предшественников в периферийную циркуляцию. Эти предшественники можно собрать из периферической крови и сконцентрировать, а затем обработать в культуре с ТРО, по желанию, в сочетании с одним или более других цитокинов, в том числе SCF, G-CSF, IL-3, GM-CSF, IL-6 или IL-11, для дифференциации и пролиферации в мегакариоцитные культуры с высокой плотностью, которые затем могут быть возвращены пациенту после проведения высокой дозы химиотерапии.
Предлагаются также антитела, которые связаны с эпитопом на протеине по данному изобретению. Такие антитела могут продуцироваться разнообразными средствами, известными в данной области науки. Продуцирование нечеловеческих, моноклональных антител хорошо известно и может быть выполнено, например, иммунизированием животного высоко-очищенным протеином или полипептидным фрагментом. Желательно также вводить протеин или полипептид в сочетании с адъювантом (стимулятором), таким как адъювант Фронда, для того чтобы усилить иммунную реакцию. Хотя единичной инъекции антигена может быть достаточно для того, чтобы вызвать прдуцирование антитела в животном, обычно предпочтительно введение инъекции большой дозы, за которой следует одна или более повторных инъекций в течение периода времени от нескольких недель до нескольких месяцев. См. работу Харела и др. Monoclonal Hybridoma Antibodies: Techniques and Applications, CRC Press Inc., Roca Raton, FL, 1982, которая включена в данный документ в качестве ссылочного материала. Затем кровь берется у животного и коагулируется (свертывается), и антитела изолируются от сыворотки с помощью известного метода, например осаждение солей, хроматография с ионным обменом, аффинная хроматография или высокоэффективная жидкостная хроматография.
Обычно более предпочтительным является использование моноклонных антител по сравнению с поликлонными антисыворотками. Моноклонные антитела имеют преимущество более простого продуцирования, специфичность и репродуктивность. Способы продуцирования моноклонных антител хорошо известны в данной области науки и рассмотрены, например, в работе Келлера и Милстейна (Nature 256:495, 1975 и в Eur.J.Immunol. 6:531-519, 1976). См. также работу Харелла там же и Харта патент США №5,094,941, которые включены в данный документ в качестве ссылочного материала. Короче говоря, клетки, продуцирующие антитела, полученные из иммунизированных животных, иммортализируются и отцеживаются, или сначала процеживаются для продуцирования антитела, которое связано с ТРО. Положительные клетки затем иммортализируются слиянием с клетками спинного мозга. Нечеловеческие антитела могут быть "очеловечены" известными методами. См., например, Патент США №4,816,397: Публикации Европейского Патентного Ведомства 173,494 и 239,400 и WIPO ПубликацииWO 87/02671 и WO 90/00616, которые включены в данный документ в качестве ссылочного материала. Короче говоря, постоянные человеческие региональные гены соединяются с соответствующими нечеловеческими переменными региональными генами. Например, аминокислотные последовательности, которые представляют сайты связывания антигена (CDR или комплементарно-детерминирующие регионы) родительского (нечеловеческого) моноклонного антитела, трансплантируются на ДНК уровне на человеческие переменные региональные каркасные последовательности. Способы для осуществления этого известны и рассмотрены, например, в работе Джонса и др. (Nature 326:522-525, 1986), в работе Ричмена и др., (Nature 322: 323-327, 1988) и Куина и др. (Proc.Natl.Acad. Sci. USA 86: 10029-10033, 1989). Затем объединенные гены трансфектируются в клетки-хозяева, которые выращиваются в соответствии с известными методами. В другом варианте моноклональное антитело, продуцирующее клетки, может трансфектироваться с клонированными человеческими постоянными региональными генами и с генами химерового антитела, порожденного гомологовой рекомбинацией. Таким образом, возможно сконструировать моноклоновые антитела, значительная часть которых будет человеческой, тем самым создавая антитела, которые больше подходят для множественного введения пациентам-людям.
Антитела с одиночной цепью можно вырастить посредством экспрессии рекомбинантного полипептида, который, как правило, состоит из переменной последовательности легкой цепи, соединенной - обычно через линкерный полипептид - с переменной последовательностью тяжелой цепи. В данной области науки известны способы предуцирования антител одиночной цепи и раскрыты, например, в работе Дэвиса и др. (Bio/Technology 9:165-169, 1991).
Антитела, которые связаны с эпитопом ТРО, полезны, например, при диагностировании заболеваний, характеризующихся пониженным уровнем тромбоцитов, мегакариоцитов или других кровяных клеток или их предшественников, причем эти заболевания относятся к дефициту пролиферации или дифференциации клеток-предшественников. Такой диагноз, как правило, ставится на основания результатов исследования крови и плазмы с помощью известных методов иммунологического анализа, таких как иммуноабсорбционный анализ связанных ферментов или радиоиммунный анализ. Анализы такого рода хорошо известны в данной области науки. См., например, работу Харта и др. Biochem. 29:166-172, 1990; Ma и др. British Journal of Haematology 80:431-436, 1992; и работу Андре и др. Clin.Chem. 38/5:758-763, 1992. Диагностический анализ деятельности ТРО может пригодиться при определении популяции пациента, скорее всего являясь более удобной, чем ТРО терапия. Антитела для ТРО также могут быть использованы при очищении ТРО, например при закреплении антитела на твердой основе, например на основе карпускулярной матрицы, упакованной в столб и проходящей через раствор, содержащий протеин над столбом. Затем связанный протеин элютируется с соответствующим буфером. Обычно протеин связан со столбом при таких физиологических условиях, как низкий уровень ионной силы и почти нейтральный рН. Столб затем промывается для элютирования несвязанных примесей. Элютирование связанного протеина выполняется путем изменения величины ионной силы или рН, например, с помощью 3М KSCN (группа или градиент) или буфера цитрата с низким рН. Как правило, следует избегать величины рН ниже 2,5.
Данное изобретение также предлагает способы продуцирования большого количества мегакариоцитов и тромбоцитов, которые могут быть использованы, например, для создания сДНК библиотек. Поскольку тромбоциты направляются в участки повреждения, они могут считаться носителями заживления ран и при определенных условиях носителями патогенезиса. Следовательно, возможность подробно научить молекулярную биологию тромбоцитов и мегакариоцитов даст понимание как гомеостаза, так и клинических нарушений функций тромбоцитов. Протеины по данному изобретению предлагают улучшенные средства для продуцирования библиотек мегакариоцитов или тромбоцитов ДНК.
Рекомбинантные тромбопоэтины при введении в животных или при наложении на культивированные клетки селезенки или костного мозга вызывают пролиферацию мегакариоцитов из клеток-предшественников. Расширение (распространение) мегакариоцитов и их предшественников и созревание мегакариоцитов после введения ТРО дает возможность изолировать мегакариоциты с высокой степенью чистоты и при достаточном количестве для изоляции структур библиотек mPHK и сДНК. Установив дозы ТРО и режим введения, рано или полностью созревшие мегакариоциты и те из них, которые активно образуют тромбоциты, должны выборочно распространяться из первичных клеток селезенки или костного мозга. Соответственно, репрезентативные сДНК библиотеки могут составляться в соответствии с ранними, промежуточными или поздними стадиями или мегакариопоэзисом.
Применений у получающейся при этом сДНК библиотеки много. Такие библиотеки можно, например, использовать для идентифицирования и клонирования протеинов в небольшом количестве, что имеет определенное значение при различных дисфункциях тромбоцитов. Простота, с которой мегакариоциты пациента могут распространяться, а его mРНК изолироваться для проведения анализа, в значительной степени способствует молекулярной десекции заболевания. Библиотеки также являются источниками клонирования новых факторов роста и других протеинов при потенциальном применении в терапии. Полезные протеины уже клонированных тромбоцитов содержат производный от тромбоцитов фактор роста (Росс и др. Cell 26:155-169, 1986); трансформирующий фактор роста (Милетич и др. Blood 54:1015-1023, 1979); Робертс и Спрон, Growth Factors 8: 1-9, 199-3); фактор роста эндотельных клеток, производных от тромбоцитов (Милетич и др. Blood 54:1015-1023, 1979) и PF-4 (Дуа и др. Mol.Cell.Biol. 7:898-904, 1987); Понц и др. Blood 69:219-223, 1987). Новые факторы роста могут быть идентифицированы путем подробного функционального анализа библиотеки экспрессии сДНК или гидрадизированным анализом при пониженной струновидности известных зондов факторов роста. Изолирование новых факторов роста можно также провести на реакции полимеризованной цепи, используя праймеры вырождения в сохраненных регионах известных факторов роста. Кроме того, систематическое и полное секвенирование ДНК из библиотеки создает базу данных мегакариацитной сДНК последовательности. Такая база данных может быть использована в последовательностях при применении разнообразных компьютерных поисковых алгоритмов.
Мегакариоциты, приготовленные, как указывалось выше, могут также применяться для создания протеиновой библиотеки. Эта протеиновая библиотека комплементарна библиотеке сДНК. Информация об аминокислотной последовательности, получаемая из протеиновой библиотеки, позволяет быстро проводить изоляцию сДНК, кодирующую нужные протеины. Использование сведений о протеиновой последовательности при конструировании праймеров для изолирования ДНК, ликвидируют проблемы, возникающие при использовании известных способов создания библиотек, благодаря относительно большому количеству mPHK. Объединение библиотек протеинов и сДНК также упрощает меченое клонирование последовательностей, которые представляют особый интерес.
Протеиновая библиотека создается выделением протеинов (целых протеинов или их фрагментов) из мегакариоцитов в соответствии с известными методами, затем в отделении протеинов с помощью двухмерного гель-электрофореза. Изолированные протеины затем подвергаются триптическому сжиганию in situ, после чего следует сепарация с помощью жидкостной хроматографии высокого разрешения (ЖХВР). Сепарированные фрагменты затем исследуются методом масс-спектрометрии. Полученный масс-профиль изучается на основе базы данных протеиновой последовательности для заключения об идентичности протеинов. Неидентифицированные пептиды могут быть секвенсированы по деградации Эдмана.
Библиотеки сДНК и протеинов являются ценными источниками новых протеинов последовательностей, их кодирующих. Тромбоциты считаются важными веществами для заживления ран и при некоторых условиях патогенезисом. Были идентифицированы и охарактеризованы многие важные тромбоцитные протеины, в том числе факторы роста, производные от тромбоцитов, преобразующий фактор роста-β, фактор роста эндотельных клеток, производных от тромбоцитов, и тромбоцитный фактор 4. Идентификация и характеристика других тромбоцитных протеинов могли бы быть особенно полезны при разъяснении процессов, лежащих в основе заживления ран и патогенеза, и предполагается, что даст много терапевтических веществ и стратегических методов.
Как будет более подробно описано ниже, гены человеческого ТРО локализованы в хромосоме 3q26. Эта информация, соединенная с последовательностью гена человеческого ТРО (SEQ ID NO:28), позволяет делать прямую диагностику путем генетического анализа нарушений в гене ТРО или регулировать их экспрессию. К таким нарушениям относятся изменения последовательностей промоторов, ведущие к увеличению или уменьшению уровня экспрессии, хромосомным транслокациям в кодирующих или некодирующих регионах, и наложение прямой мутации и супрессора в новых регулирующих последовательностях в локусе ТРО. Методы диагностики, которые могут здесь применяться, хорошо известны в данной области науки. Например, праймеры или зонды гибридизации из, по крайней мере, 5 нуклеотидов, а предпочтительно из 15-20 нуклеотидов, в длину могут быть построены из геномной последовательности и использоваться при обнаружении хромосомных аномалий или при измерении уровней mPHK. В данной области науки известно огромное количество подходящих методов обнаружения и измерения, к которым относятся "Южный" блоттинг, цепная реакция полимеризации (Мюллис, Патент США №4,683,202), цепная реакция лигазации (Бэрани, PCR Methods and Application 1:5-16, Cold Spring Harbor Laboratory Press, 1991). Например, ДНК пациента может быть поглощена одним или более ферментами и преобразована в нитроцеллюлозу для продуцирования Южного блота. Затем этот блот зондируется для обнаружения серьезных изменений в размерах фрагмента, возникающих из-за мутации в последовательности распознавания сайта ограничения. По другой методике анализ аномальных генных последовательностей и сравнение нормальных и ненормальных последовательностей дают возможность создания праймеров, которые можно использовать для идентификации аномального гена (например, разорванного или транслокированного). ДНК пациента амплифицируется цепной реакцией полимеризации для обнаружения характеристик амплифицированных продуктов нормального гена или, в частности, генной реорганизации.
Далее настоящее изобретение иллюстрируется следующими неограничивающими примерами.
Пример 1. Изолирование рецептора ДНК человеческого MPL
Изоформа человеческого MPL-P MPL-K рецептора, кодирующего сДНК, была изолирована от человеческих эритроидных лейкемических клеток (Мартин и Папаянопуло, Science 216:1233-1235, 1982) путем цепной реакции обратной транскриптазы полимеразы, в которой применяются праймеры, известной последовательности, кодирующей аминные и карбоксиловые окончания рецепторов (Вигон и др. Рrос.Natl.Acad.USA 89:5640-5644, 1992). Матричная сДНК человеческой эритроидной лейкемической клетки была синтезирована из поли-d(Т)-выбранной поли(А)+РНК с использованием праймера ZC5499 (SEQ ID N0:3). Тиртин м1 поли(А)+РНК человеческой эритроидной лейкемической клетки при концентрации 1 мг/мл смешивался с 3 мл 20 рмоль/мл первого ствольного праймера ZC5499 (SEQ ID NО:3). Эта смесь нагревалась при температуре 65°С в течение 4 минут и охлаждалась на льду.
Синтез первой ствольной ДНК начинался с добавления 8 мл первого ствольного буфера (250 мМ Трис-HCl, рН 8.3, 375 mM RCl, 15 mM MgCl2) (5x SUPERSCRIPT TM буфер; GIBCO BRL, Gaithersbm’g, MD), 4 мл из 100 мМ дитиотреитола и 3 мл раствора трифосфата деоксинуклеотида, содержащего по 10 мл dATP, dGTP, dTTP и 5-метилdСТР (Phatmacia LKB Biotechnology Inc., Piscataway, NJ). Реактивная смесь была инкубирована при 45°С в течение 4 минут, затем добавлялось 10 мл из 200 U/мл обратной транскриптазы RNase H- (обратная транскриптаза SUPERSCRIPT TM; GTBCO BRL) в смесь РНК-праймер. Эта реакция была инкубирована при 45°С в течение 1 часа, а затем при 50°С в течение 15 минут. К этой реакции добавлялось 60 мл из ТЕ (10 мМ Трис:НСl, рН 80, 1 vV EDTA), а затем следовала хроматография через гелиевую фильтрационную колонну с 400 отверстиями (CHROMA SPTN+ ТЕ-400 tm); Clontech Laboratories Inc., Palo Alto, CA) для удаления лишнего праймера.
Первая ствольная ДНК человеческой эритроидной лейкемической клетки использовалась, как тромбоцит при амплификации ДНК человеческого MPL-рецептора с помощью праймеров, соответствующих регилину, кодирующему амино и карбоксиловые окончания протеина рецептора (Вигон и др., там же). Также каждый праймер содержит сайт рестрикционного ферментативного гидролиза, который способствует направленному клонированию амплифицированного продукта (ZC5746, SEQ ID NO:4, содержащий Eco RI сайт: SEQ ID NO:5, содержащий XhoI сайт).
Была проведена реакция при 100 мл, содержащая 10 нг тромбоцитной сДНК, по 50 рмолей от каждого праймера; 200 мл каждого трифосфата деоксинуклеотида (Pharmacia LKB Biotechnology Inc.); 1 мл из 1-х PCR буфера (Promega Corp., Madison, WI) и 10 единиц Taq полимеразы (Roche Molecular Systems, Inc., Branchburg, NJ). Цепная реакция полимеразы была проведена в течение 35 циклов (1 минута при 95°С, 1 минута при 60°С и 2 минуты при 72°С при добавлении 1 лишней секунды к каждому последующшему циклу), а затем следовал инкубационный период в течение 10 минут при 72°С.
сДНК человеческого MPL-рецептора изолировалась путем амплификации цепной реакции полимеразы из сДНК человеческой эритроидной лейкемической клетки способом, идентичным для сДНК рецептора MPL-P, описанного выше, за исключением того, что праймер ZC5762 (SEQ ID NO:5) был заменен на ZC5742 (SEQ ID NO:6). PCR праймер ZC5742 специфичен для 3’ окончаний человеческой сДНК MPL-K и входит в состав рестрикционного сайта Xho I для упрощения процесса клонирования.
Продукты реакции были экстрагированы дважды с помощью фенола/хлороформа (1:1), затем один раз с помощью хлороформа и осаждены с помощью этанола. После поглощения Eco RI и Xho I эти продукты были Фракционированы на агарозном геле с низкой точкой плавления 0.8% (SEA PLAQUE GTG tm агароза с низкой точкой плавления; FMC Corp., Rockland, ME). Из срезов возбужденного геля путем поглощения гелиевой матрицы с агаразой I был восстановлен 1.9 тысяч пар нуклеотидов (т.п.н.) амплифицированный продукт в соответствии с сДНК человеческим MPL-P рецептором и 1.7 т.п.н. в соответствии с сДНК человеческим MPL-K рецептором (New England Biolabs, Inc., Beverly, MA), затем следовало этаноловое осаждение. Кислоты сДНК были субклонированы в вектор pBluescript @ SK+(Stratagene Cloning Systems, La Jolla, CA) для валидации путем секвенсирования.
Пример II. Изолирование сДНК мышинового MPL-рецептора
У мышей C57BL/KsJ-db/db были удалены селезенки и немедленно помещены в жидкий азот. РНК полностью была приготовлена из ткани селезенки с помощью изотиоцианата гуанидина (Ширгвин и др. Biochemistry 18:52-94, 1979), затем следовала стадия центрифугирования с CsCl. PHК поли+(А) селезенки была изолирована с помощью олиго d(T) целлюлозной хроматографии (Авив и Лендер, Рrос. Natl.Acad.Aci.USA 69:1408-1412, 1972).
Семь с половиной мл поли d(Т)-отобранной из поли-(А)+ РНК мышиной селезенки при концентрации 1.7 мг/мл были смешаны с 3 мл из 20 рмоль/мл праймера первого ствола ZC66091 (SEQ ID NO:7), содержащего рестрикционный сайт Not I. Эта смесь нагревалась при 65°С в течение 4 минут и охлаждалась на льду. Синтез сДНК первого ствола был начат добавлением 81 мл из 250 MM Трис-HCl, рН 8.3, 375 mM KCl, 15 mМ MgCl2 (5хSUPERSCRIPT tm буфер; GIBCO ВRL), 4 мл из 100 мМ литиотреитола и 3 мл раствора трифосфата деоксинуклеотида, содержащего по 10 мМ dATP, dGTP. dTTP и 5-метил-dCTP (Pharmacia LCB Biotechnology Inс.) в смесь РНК-праймер. Эта реактивная смесь инкубировалась при 45°С в течение 4 минут, а затем добавлялось 10 мл из 200 U/мл RNase H- обратной транскриптазы (GIBCO BKL). Эффективность синтеза первого ствола анализировался проведением параллельной реакции при добавлении 10 mCi32p-adCTP в объем реактивной смеси, кратный 10 мл для мечения реакции для проведения анализа. Эти реакции инкубировались при 45°С в течение 1 часа, а затем инкубировались при 50°С в течение 15 минут. 32p-adCTP, не вошедшая в меченную реакцию, удалялась путем хроматографирования на гелиевой фильтрационной колонне размером в 400 отверстий (CHROMA SPIN + ТЕ-400 tm); Clontech Laboratories Inc.). Нуклеотиды, не вошедшие в немеченую реакцию первого ствола, удалялись двойным осаждением сДНК в присутствии 8 мг носителя гликогена, 2.5 ацетата аммония и 2.5 объема этанола. Немеченая сДНК переводилась во взвешенное состояние в 50 мл воды для использования синтеза второго ствола. Длина меченной сДНК первого ствола определялась электрофорезом агарозного геля.
Синтез второго ствола проводился на сДНК первого ствола при условии, что стимулированное примирование по первому стволу второго ствола приведет к образованию шпильки (петли) ДНК. Реактивная смесь создавалась при комнатной температуре и состояла из 50 мл немеченой сДНК первого ствола, 16.5 мл воды, 20 мл буфера 5х полимераза I (100 мМ Трис:НСl, рН 7&4, 500 мМ КСl, 25 мМ MgCl2, 50 мМ (NH4)2SO4), 1 мл из 100 мМ дитиотриэтола, 2 мл раствора, содержащего по 10 мМ трифосфата леоксинуклеотида, 3 мл из 5 мМ -NAD, 15 мЛ из 3U/мл E.coli лигазы ДНК (New England Biolabs Inc., Beverly, MA) и 5 мл 10/мл Е.coli полимеразы I ДНК (Amersham Corp., Arlington Heights. IL). Эта реакция инкубировалась при комнатной температуре в течение 5 минут, а затем добавлялось 1.5 мл 2 U/мл RNase H (GIBCO BRL). Параллельная реакция, в которой объем, кратный 10 мл, синтезирующей смеси второго ствола помечался добавлением 10 мСi 32р-СТР, использовалась для управления эффективностью синтеза второго ствола. Эти реакции были инкубированы при 15°С в течение 2 часов, а затем инкубировались в течение 14’5 минут при комнатной температуре. Не вошедший в меченую реакцию 32p-adCTP удалялся методом хроматографии через гелиевую фильтрационную колонну размером в 400 отверстий (Clontech Laboratories, Inc.) перед проведением анализа электрофорезом агарозного геля. Эта немеченая реакция заканчивалась двумя экстрагированиями с экстрагированием фенол/хлороформа и хлороформа путем этанолового осаждения в присутствии 2.5 М ацетата аммония.
Одноствольная ДНК со шпилечной структурой расщеплялась с помощью нуклеазы фасоли золотистой. Реактивная смесь содержала 100 мл сДНК второго ствола, 20 мл 10× буфер нуклеазы фасоли золотистой (Stratagene Cloning Systems, La Jolla, CA), 16 мл из 100 мМ дитиотреитола, 51.5 мл воды и 12.5 мл 1:10 раствора нуклеазы фасоли золотистой (Promega Corp.; окончательная концентрация 10.5 U/мл) в буфере нуклеазы фасоли золотистой. Эта реакция инкубировалась при температуре 37°С в течение 15 минут. Реакция заканчивалась добавлением 20 мл из 1 М Трис:НСl, рН 8.0, затем следовали последовательные экстрагирования фенола/хлороформа и хлороформа, как описывалось выше. После экстрагирования ДНК осаждалась в этаноле и переводилась во взвешенное состояние в воде.
сДНК, вновь переведенная во взвешенное состояние, затуплялась с концов полимеразой Т4 ДНК. сДНК, которая была вновь введена во взвешенное состояние, в 190 мл воды, смешивалась с 50 мл буфером полимеразы 5× Т4 ДНК (250 мМ Трис:НСl, рН 8.0, мМ КСl, 25 mM MgCl2), 3 мл 0.1 М дитиотреитола в 3 мл раствора, содержащего по 10 мМ трифоофата деоксинуклеотида и 4 мл 1U/мл полимеразы Т4 ДНК (Boehringer Mannheim Соrр,, Indianapolos, IN). После инкубирования в течение 1 часа при 10°С реакция прекращалась добавлением 10 мл 0.5 М EDTA, после чего следовала серия экстрагировании фенола/хлороформа и хлороформа, как описывалось выше. ДНК хроматографировалась через гельную фильтрационную колонну размером в 400 отверстий (Clontech Laboratories Inc., Palo Alto, CA) для удаления небольшого количества протеина и удаления коротких сДНК, длина которых менее -400 bр(пара нуклеотидов). ДНК осаждалась в этаноле в присутствии 12 мг носителя гликогена и 2.5 М ацетата аммония и была переведена вновь во взвешенное состояние в 10 мл воды. Поскольку 32p-adCTP была использована, выход сДНК прогнозировался порядка -2 мг от начальной величины 12.5 мг тромбоцитов.
Eco RI адаптеры лигировались на 5’ концах сДНК, давая возможность провести клонирование в ламбда-фаг вектор. Объем сДНК, кратный 10 мл (-2 мг) и 10 мл 65 рмоль/мл Eco RI адаптера (Phannacia LKB Biotechnology Inc.) смешивался с 2.5 мл 10× буфер лигазы (Promega Corp.), 1 мл 10 мМ АТР и 2 мл из 15 U/мл Т4 лигазы ДНК (Promega Соrр.). Эта реакция инкубировалась с вечера и всю ночь (примерно 18 часов) при температурном градиенте от 0°С до 18°С. Затем эта реакция инкубировалась с вечера и всю ночь при 12°С. Заканчивалась реакция добавлением 75 мл воды и 10 мл из 3 М ацетата Na, после чего проводилась инкубация при 65°С в течение 30 минут. После инкубации сДНК экстрагировалась с фенолом/хлороформом и хлороформом, как описывалось выше, и осаждалась в присутствии 2.5 М ацетата аммония и 1.2 объема изопропанола. После центрифугирования осадок сДНК промывался 70%-ным этанолом, просушивался на воздухе и вновь возвращался во взвешенное состяние в 89 мл воды.
Для упрощения направленного клонирования сДНК в ламбда-фаг вектор, сДНК поглощалась Not I, в результате чего у сДНК появлялись 5’ Eco RI и 3’’ Not I липкие концы. Ристрикционный сайт Not Iy 3’ конца сДНК вводился через праймер ZG6091 (SEQ ID NO:7). Поглощение ристрикционного фермента проходило во время реакции, содержащей 89 мл сДНК, описанной выше, 1 мМ ДТТ (дитиотреитол) (10× D буфер; Promega Corp., Madison, WI) и 1 мкл из 12 U/мкл NOt I (Promega Corp). Поглощение проходило при 37°С в течение 1 часа. Реакция заканчивалась серией экстрагирований фенола/хлороформа и хлороформа. сДНК осаждалась в этаноле, промывалась 70%-ным этанолом, просушивалась на воздухе и возвращалась во взвешенное состояние в 20 мл из 1х загруженный гелем буфер (10 мМ Трис:НСl, рН 8.0, 1 mМ EDTA, 5%-ный глицерол и 0.125% бромфенолового синего).
Возвращенная во взвешенное состояние сДНK нагревалась при 65°C в течение 5 минут, остужалась на льду и подвергалась электрофорезу на 0.8% агарозном геле с низкой точкой плавления (SEA PLAQUE GTG ТМ агароза с низкой точкой плавления); FMC соrр.). Неиспользованные адаптеры и сДНК менее 1.6 Kb (тысяча пар нуклеотидов) в длину выщипливались из геля. Электродам меняли знаки, и сДНК подвергалась электрофорезу до тех пор, пока она не начнет концентрироваться вблизи начала пути (полосы). Область геля, содержащую сконцентрированную сДНК, вырезали и помещали в трубку микроцентрифуги, и определяли примерный объем среза геля. Объем воды (300 мл) примерно в три раза больше объема среза геля добавлялся в трубку и агароза плавилась при нагревании до 65°С в течение 15 минут. После уравновешивания этого образца добавлялся образец при 42°С, 10 мл из 1U/мл -агарозы I (New England Biolabs, Inc.) и смесь инкубировалась в течение 90 минут для проведения поглощения агарозы. После инкубации 40 мл из 3М ацетата Na были добавлены в образец, и смесь инкубировалась на льду в течение 15 минут. Этот образец центрифугировался при 14,000×g в течение 15 минут при комнатной температуре, что позволяло убрать непоглощенную агарозу. сДНК, находящуюся в надосадочной жидкости, осаждали в этаноле, промывали в 70%-ном этаноле, сушили воздухом и возвращали во взвешенное состояние в 37 мл воды для проведения реакции киназы для фосфориляции лигированных Eco RT адаптеров.
В раствор 37 мл сДНК, описанного выше, добавлялся 10 мл× буфер лигазы (Stratagene Cloning Systems) и смесь нагревалась до 65°С в течение 5 минут. Смесь охлаждалась на льду и в нее добавлялись 5 мл 10 мМ АТР и 3 мл из 10 U/мл Т4 киназа полинуклеотидов (Stratagene Cloning Systems). Реакция инкубировалась при 37°С в течение 45 минут и заканчивалась нагревом до 65°С в течение 10 минут, вслед за чем шла серия экстрагирования с фенолом/хлороформом и хлороформом. Фосфорилированная сДНК осаждалась в этаноле в присутствии 2.5 М ацетата аммония, промывалась 70%-ным этанолом, просушивалась воздухом и вновь возвращалась во взвешенное состояние в 12.5 мл воды. Концентрация фосфорилированной сДНК оценена как примерно -40 моль/мм.
Полученная сДНК клонировалась в ламбда-фаг вектор ЕxСеll tm (Pharmacia LKB Biotechnology Inc.), подготовленный с Eco RI и Not I и дефосфорилирована. Лигирование сДНК с вектором выполнялось путем реакции, содержащей 2 мл из 20 fмоль/мл фаговых отростков.. ExCell tm, 4 мл воды, 1 мл 10× буфер лигазы (Promega соrр.), 2 мл из 40 fмоль/мл сДНК и 1 мл из 15 U/мл Т4 лигазы ДНК (Promega Corp.). Лигирование выполнялось при 4°С в течение 48 часов. Примерно 50% лигированной смеси помещалось в фаг с помощью уплотняющего экстракта GIGAPACK II Gold (Stratagene Cloning Systems) в соответствии с направлениями вектора. Полученная при этом библиотека содержала свыше 1,5×107 независимых рекомбинантов с уровнями фона безвставочного фага менее 1.5%.
Зонд сДНК человеческого MPL-K рецептора, помеченного 32р, использовался при изолировании сДНК мышиного MPL рецептора от библиотеки фагов сДНК селезенки. Библиотека сДНК была посеяна на SURE штамме клеток E.coli. (Stratagene Cloning Systems) при плотности в 40,000-50,000 PFU/на чашу диаметром 150 мм. Чумные фаги из 33 чашек были перенесены на нейлоновые мембраны (Hybond ntm; Amersham Corp., Arlington Heights, IL) и обрабатывались в соответствии с указаниями изготовителя. Обработанные фильтры обжигались в течение 2 часов при 80°С в вакуумной печи, после чего следовало несколько промывок при 70°С в промывочном буфере (0.25×SSC, 0.25% SDS, 1mМ EDTA) и подвергались предварительной гибридизации в течение ночи при 65°С в гибридизирующем растворе (5×SSC, 5 [раствор Денхардта, 0.1% SDS, 1 mМ EDTA и 100 мг/мл подогретой ДНК денатурированной спермы лосося) в гибридизационной печи (модель НВ-2; Techne Inc., Princeton, NJ). После прегибридизации гибридизирующий раствор выбрасывается и заменяется свежим гибридизирующим раствором, содержащим приблизительно 2×106 cpl/мл из человеческой MPL-K ДНК, меченной 32р, приготовленной с помощью набора мечения, имеющегося в продаже (набор MEGAPRIME tm; Amersham Heights, ID). Зонд денатурировался при 98°С в течение 5 минут перед тем, как его добавили в гибридизирующий раствор. Гибридизация проводилась при 65°С в течение ночи. Фильтры промывались при 55°С в промывочном буфере (0.25-SSC, 0.25∧ SDS, 1 mM EDTA) и радиоавтографированы с помощью интенсифицирующих экранов в течение 4 дней при 70°C на пленке ХАR-5 (Kodak Inc., Rochester, NJ). При использовании радиоавтографии в качестве матрицы масса агара восстанавливалась в регионах, где находились чаши, в соответствии с первичными сигналами и отсасывалась в SM (0.1 М NaCl; 50 mM Трис:НСl, рН 7.5, 0.02% желатин) для элютирования фага для очистки от вируса чумы. Было изолировано семь очищенных от бляшек фагов, у которых были вставки, гибридизирующие с зондом человеческого MPL-Крецептора. Фагемиды, которые находились в фаге ExCell tm, были восстановлены с помощью in vivo рекомбинационной системы в соответствии с направлениями вектора. Идентичность вставок сДНK подтверждалась секвенсированием ДНК.
Изолированные клоны кодировали протеин, выказывающий большую степень секвенированной идентичности человеческому MPL-P рецептору и недавно объявленному мышиному MPL рецептору (работа Скоды и др., EMBO J.12:2645-2653, 1993). Эти клоны распадаются на два класса, отличающиеся друг от друга тремя клонами, имеющими делецию в последовательностях, кодирующих участок их 60 аминокислотных остатков вблизи N-окончаний. сДНК, кодирующая протеин без делеций, была названа мышиная сДНК Типа I MPL рецептора. сДНК по Типу II рецептора не имела последовательностей, кодирующих остатки рецепторов по Типу I, с 131 по 190 из SEQ ID NO:17. Кроме того, рецепторы Типа I и Типа II отличались от последовательности мышиных MPL рецепторов (Скода и др., там же) присутствием последовательности, кодирующей аминокислотные остатки Val-Arg-Thu-Ser-Pro-Ala-Gly-Glu (SEQ ID NO:9), вставленные после аминокислотного остатка 222, и заменой остатка глицина на сыворотку в пункте 241 (пункты соответствуют мышиному рецептору Типа I).
Кислоты сДНК мышиных MPL рецепторов по Типу I и Типу II были субклонированы в плазмидный вектор pHZ-1 для экспрессирования в клетки млекопитающих. Плазмид pHZ-1 является экспрессирующим вектором, который можно использовать для экспрессирования протеина в клетках млекопитающих или системы оцит-трансляции лягушки из mPHK, которая была транскрибирована in vitro. Экспрессирующая единица pHZ-1 содержит мышиный промотор металлотионеина-1, бактериофаговый Т7 промотор, фланкированный банками множественного клонирования, содержащими уникальные ристрикционные сайты для вставки, кодирующей последовательности, терминатор человеческого гормона роста и терминатор Т7 бактериофага. Кроме того, pHZ-1 содержит E.Coli начало репликации; генбактериальной бета-лактомазы; экспрессирующую единицу селективного маркера млекопитающего, содержащего SV40 промотор начало, ген сопротивления неомицину и терминатор SV40 транскрипции. Для упрощения направленного клонирования в pHZ-1 использовалась цепная реакция полимеразы с соответствующими праймерами для создания Eco RI сайта и Xho I сайта выше по сравнению с началом трансляции кодона и ниже по сравнению с окончанием трансляции кодона соответственно. Проводилась цепная реакция полимеразы в смеси, содержащей 10 мл 10х ULTMA tm буфер полимеразы ДНК (Roche Molecular Systems, Inc., Branchburg, NJ), 6 Мл из 25 mМ MgCl2, 0.2 мл из раствора трифосфата деоксинуклеотида, содержащего по 10 мМ dATP, dGTP, dTTP и dCTP (Pharmacia LKB Biotechnology Inc.), 2.5 мл из 20 рмоль/мл ZC6603 праймеpa (SEQ ID NO:8), 2.5 мл из 20 рмоль/мл ZG5762 праймера (SEQ ID NO:5), 32.8 мл воды, 1 мл из лог-фазы бактериальной культуры, содержащей либо плазмид мышиного MPL рецептора по Типу I или Типу II и 1 мл из 6 U/мл ДНК полимеразы (ULTMA tm полимераза; Roche Molecular Systems, Inc., Branchburg, NJ). В реакции в соответствии с направлениями вектора использовалась AmpliWax tm (Roche Molecular Systems, Inc.). Цепная реакция полимеразы проводилась в течение 25 циклов (в течение одной минуты при 95°C, 1 минуты при 55°С и 3-х минут при 75°С), после чего следовало инкубирование в течение 10-ти минут при 72°С. Амплифицированные продукты серийно экстрагировались с фенолом/хлороформом и хлороформом, затем осаждались в этаноле в присутствии 6-ти мг носителя гликогена и 2.5 М ацетата аммония. Осадки вновь переводились во взвешенное состояние в 87 мл воды, к которой добавлялось 10 мл из 10*Н буфер (Boehringer Manneheim Corp.), 2 мл из 10 U/ml Xho I Eco RI (Boehringer Manneheim) и 1 мл из 40 U/ml Xho I (Boehringer Manneheim Corp.). Поглощение происходило при 37°С в течение одного часа. Оканчивалась реакция нагреванием до 65°С в течение 15 минут и хроматографированием через гелевую фильтрационную колонну размером в 400 отверстий (CHROMA SPIN + TE-400tm; Clontech Laboratories Inc.).
Вставки изолированного рецептора, описанные выше, были легированы в поглощенный и дефосфорилированый Eco RI и Xho I pHZ-1 вектор. Реакция легирования содержала 1 мл из 50-ти ug/ml pHZ-1 вектора, 5 мл из 5-ти ng/ml вставки сДНК, 2 мл из 10*буфер легазы (Promega Corp.), 11.75 мл воды и 0.25 мл из 4U/MA T4 лигазы ДНК (Stratagene Cloning Systems). Лигирование проводилось при 10°С в течение ночи. Лигированные кислоты ДНК трансфектировались в E.coli (MAX EFFICIENCY DH10B tm компетентные клетки, GIBCO BRL). Валидность вставок мышиного MPL рецептора и человеческого MPL-P рецептора по Типу I и Типу II подтверждала последовательность ДНК. Получаемые плазмиды pSLmpl-8 и pSLmpl-9 были носителями мышиных рецепторов MPL по Типу I и Типу II соответственно. Плазмид pSLmpl-44 был носителем вставки человеческой MPL-P сДНК.
ПРИМЕР III. СОЗДАНИЕ ЛИНИЙ КЛЕТОК ВаF3, ЭКСПРЕССИРУЮЩИХ MPL РЕЦЕПТОРЫ
BaF3 - линия пре-лимпоидных клеток, зависимых от интерлейкина-3, отведенная от мышиного костного мозга (работа Паластоса и Стейнмеца, Cell 41:727-734, 1985; Мати-Прево и др., Mol.Cell.Biol.6:4133-4135, 1986), сохранялась в полной среде (среда RPMI 1640 (JRH Bioscience Inc., Lenexa, KS), дополненной 10% фетальной телячьей сывороткой, инактивированной при нагреве, 4% кондиционированной средой из культивированных WEHI-3 клеток (Becton Dikinson, Bedford, NA), 2 mM L-глютамин, 2-меркаптоэтанол (окончательная концентрация 1:280,000) и PSN антибиотики (GIBCO BRL). Плазмиды pSLmpl-8, pSLmpl-9 и pSLmpl-44, очищенные хлоридом цезия, линеаризировались в сайте Nde I перед электропорацией в клетки BaF3. Для электропорации клетки ВаF3 один раз промывались в среде RPMI 1640 и вновь переводились во взвешенное состояние в среде BMPI 1640 при плотности клеток 107 клеток/мл. Один мл вновь переведенных во взвешенное состояние клеток ВаF3 смешивался с 30 мг ДНК линеаризированного плазмида и переносился в отдельно расположенные камеры электропорации (GIBCO BRL). После 15 минут инкубации при комнатной температуре клетки подвергались нескольким ударам (800 mFAD/300 В; 1180 mFAD/300 В), поступающим от аппаратуры электропорации (CELL-PORATOR tm; GIBCO BRL). По прошествии 5 минут времени восстановления электропорированные клетки переносились в 10 мл полной среды и помещались в инкубатор на 15-24 часа (37°С, 5% СO2). Затем эти клетки скручивались и возвращались во взвешенное состояние в 10 мл полной среды, содержащей 1600 мг/мл G418 и высевались при ограниченном разбавлении в чашах для культивирования ткани с 96 лунками для того, чтобы изолировать клоны, стойкие к действию G-418. Выражение MPL рецепторов в клоны BaF3, стойкие к действию G-418, было выполнено путем анализа методом блоттинга Нортерна mРНК BaF3 для присутствия транскрипта MPL рецептора. Оказалось, что линия клеток, обозначенная BaF3/MPLR, I, выражает высокие уровни mРНК MPL мышиных рецепторов по Типу I и Типу II и использовались при последующем изучении деятельности MPL лиганда в кондиционированной среде трансфектированных клеток ВНК 570. Линия клеток BaF3, выражающая mPHK рецепторы Типа II, обозначалась BaF3/MPLR2.
ПРИМЕР IV. ПРОДУЦИРОВАНИЕ РАСТВОРИМЫХ МЫШИНЫХ MPL РЕЦЕПТОРОВ
Экспрессирующий плазмид млекопитающего, кодирующий растворимый MPL мышиный рецептор Типа I (pLEtapl-53), продуцировался комбинированием сегментов ДНК из pSLmpl-9, экспрессирующего плазмида млекопитающего, содержащего сДНК, кодирующую мышиный MPL рецептор полной длины, описанный выше, с сегментом ДНК из pSLmpl-26, экспрессирующим плазмидом, созданным для продуцирования растворимого мышиного MPL рецептора Типа I в бактерии. Сегмент сДНК, кодирующий растворимый мышиный NPL рецептор Типа I, был изолирован с помощью PCR, используя праймеры ZC6704 (SEQ ID NO:10) и ZC6703 (SEQ ID NО:11), где в качестве матрицы использовался плазмид pSLmpl-9 рецептора полной длины. Для упрощения направленного клонирования в праймеры ZC6704 и ZС6703 входили Eco RI и Xho I ристрикционные сайты соответвенно на их концах 5’. Также праймер ZC6703 кодировал последовательность консенсусной мишени для киназы протеина, что дало возможность in vitro метить очищенный растворимый рецептор с 32р...-АТР (Ли и др. Proc.Natl.Acad. Sci. USA. 86:558-562, 1989). PCR проводился в смеси, содержащей 10 мл 10× буфер ДНК ULTMA tm (Roche Molecular Systems, Inc.), 6 мл из 25 мМ MgCl2, 0.2 мл из раствора трифосфата деоксинуклеотида, содержащего по 10 мМ dATP, dGTP, dTTP и dCTP (Pharmacia LКB Biotechnology Inc.), 11 мл из 4.55 рмоль/мл праймера ZC6704 (SEQ ID NO:10), 21 мл из 2.43 рмоль/мл праймера ZC6703 (SEQ ID NO:11), 50.3 мл воды, 1 мл 50 нг/мл pSLmpl-9 поглощенного Hind III и Xba и 1 мл из 6U/мл полимеразы ДHKULTNA tm (Roche Molecular Systems, Inc.). В реакции в соответствии с указаниями продавца использовался AmpliWaz tm (Roche Molecular Systems Inc.). Цепная реакция полимеразы проводилась в 3 цикла (1 минута при 95°С, 1 минута при 50°С и 2 минуты при 72°С), после чего шло 11 циклов при повышенных требованиях к гибридизации (1 минута при 95°С, 30 секунд при 55°С и 2 минуты при 72°С), затем следовала инкубация в течение 10 минут при 72°С. Амплифицированный продукт затем последовательно экстрагировался с фенолом/хлороформом и хлороформом, после чего проводилась хроматография через гелевую фильтрационную колонну размером в 400 отверстий (Clontech Laboratories, Inc.). PRC продукт осаждался в этаноле в присутствии 20 мг носителя гликогена и 2.5 М ацетата аммония. Осадок вновь переводился во взвешенное состояние в 32 мл воды. К 16 мл возвращенного во взвешенное состояние продукта PCR добавлялось 2 мл 10× буфер Р (Boehringer Mannheim Corp.), 1 мл из 10U/мл Eco RI (Boehringer Mannheim Corp.) и 1 мл из 40 U/мл Xho I (Boehring Mannheim Corp.). Поглощение происходило при 37°С в течение 1 часа. Это поглощение заканчивалось нагреванием до 65°С в течение 15 минут, затем проводилась очистка на 0.7% геле агоразы с низкой температурой плавления. Восстановление фрагментов из агарозы с низкой температурой плавления проводилось посредством поглощения гелевой матрицы с бета-агарозой I (New England Biolabs).
Полученный таким образом PCR продукт кодировал внеклеточную область N-терминала мышиного MPL рецептора Типа I (остатки 27-480 из SEQ ID NO:17). При отсутствии транс-мембранной области предполагаемого рецептора (остатки 483-504 из SEQ ID NO:17) предполагается, что экспрессированный протеин выделялся в присутствии сигнального пептида. Мышиный MPL рецептор Типа II, кодирующий сДНК, получался при помощи PCR условий, описанных выше, за исключением того, что в качестве матрицы использовался pSLmpl-8. Валидность обоих фрагментов рецептора подтверждается секвенсированием ДНК.
Растворимые мышиные MPL рецепторы Типа I и Типа II клонировались в вектор рОmрА2-5, поглощенный Eco RI и Xho I, для получения pSLmpl-26 и pSLmpl-27 соответственно. рОmрА2-5 плазмид представляет собой модификацию рОmрА2 (работа Грэйаб и др. ЕМВО J.3:2437-2442, 1984), бактериального экспрессирующего вектора, поглощенного для размещения рекомбинантного протеина в периплазменный промежуток. pOmpA2-5 конструировался путем замены последовательности из 13 пар нуклеотидов между сайтами Eco RI и Ваm Н рОmрА2 на синтетическую последовательность из 42 пар нуклеотидов. Эта последовательность создавалась путем отжига двух комплементарных олигонуклеотидов из 42 нуклеотидов (ZC6707, SEQ ID NO:12; ZC 6706, SEQ ID NO:13), которая образовывала липкие концы ДНК, упрощая направленное клонирование в рОmоА2, поглощенном Eco RI и Ваm HI. Внутри вставленной последовательности находится сайт Xho I, встроенный с учетом бактериальной лидерной последовательности и мышиного MPL растворимого рецептора, кодирующего сДНК, описанную выше, а также встроенный тракт (участок) из 6 кодонов гистидина, расположенных в 3’ от сайта Xho I, что позволяет очистить рекомбинантный протеин путем аффинной хроматографии хелата металлов (работа Хоучули и др. Bio/Technol.6:1321-1325 1988). За последовательностью, кодирующей тракт гистидина, следовал встроенный конечный кодон. Валидность рОmрА2-5, pSLmpl-26 и pSLmpl-27 подтверждалась сенквенсированием ДНК.
pLDmpl-53, экспрессирующий плазмид млекопитающего, продуцирующий растворимый мышиный MPL рецептор Типа I, конструировался комбинированием сегментов ДНК из pSLmpl-9 и pSLmpl-26 в экспрессирующий вектор pHZ-200 (pHZ-1, в котором последовательность редуктазы дигидрофолата замещалась геном, стойким к воздействию неомицина). Фрагмент сДНК Eco RI/Bam HI из 1164 пар нуклеотидов из рSImрl-9 заменял сигнальную последовательность млекопитающего, делецированную во время конструирования бактериального экспрессирующего плазмида pSLmpl-26. Фрагмент Bam HI из 416 пар нуклеотидов из pSLmpl-26 обеспечил кодирующей последовательностью конечной карбоксидовой части растворимого MPL рецептора, областью мечения киназы, трактом полигистидина и терминатором транслирования. Два фрагмента прошли очистку гелем и были клонированы в сайты Еcо RI/Bam HI из pBluescrilt R KS+ (Stratagene Cloning System) для получения плазмида pBS8.76LD-5. Правильная ориентация фрагмента Bam HI, отведенного от pSLmpl-26 из 416 пар нуклеотидов, относительно фрагмента Eco RI/Bam HI, отведенного от pSLmpl-9 из 1164 пар нуклеотидов в pBS8.76LD-5, определялась с помощью PCR, использующего праймер ZC6603 (SEQ ID NO:8) и ZC 6703 (SEQ ID NO:11). Сайт Xba I внутри поли-линкерной последовательности pBS8.76LD-5 дал возможность вырезать воссозданную сДНК рецептора, как фрагмент Eco RI/Xba в 1,5 тысяч пар нуклеотидов для клонирования в pHZ-200, после поглощения вектора Eco RI и Xba I. Получаемый в результате экспрессирующий плазмид млекопитающего, pLDmpl-53, приготовлялся в больших количествах для трансфекции в ВНК клетки.
20 микрограмм очищенного pLDmpl-53 плазмида было трансфектировано в 570 клеток ВНК, используя метод осаждения фосфата натрия. Спустя 5 часов на клетки действовали 15% глицерола в течение 3 минут для упрощения поглощения ДНК. В течение ночи добавлялась свежая среда роста. На следующий день клетки расщеплялись при разных концентрациях раствора и была добавлена избирательская среда, содержащая 3 мМ метотрексата. Примерно спустя 2 недели стали видны дискретные, стойкие к действию метотрексата, колонии. Эти колонии либо группировались, либо сохранялись в определенных клонах.
Израсходованные среды немедленно проверялись на наличие растворимого протеина MPL рецептора. Протеин растворимого MPL рецептора изолировался через взаимодействие тракта поли-гистидина, присутствовавшего на карбоксидном окончании протеина со смолой металлического хелата, содержащей лишенный движения Ni2+(HISBIND tm; Novagen, Madison WI).Среда культуры, свободной от сыворотки, из группы pLDmpl-53 проходила поверх смолы и граничный протеин был элютирован 1М имидазола. Анализ SDS-PAGE выявил единичную полоску при -67 килодальтонах (кДА). Этот протеин подвергался анализу с помощью аминокислоты с N-окончанием и было подтверждено, что это мышиный MPL рецептор.
Растворимый мышиный MPL рецептор очищался от трансфектантов ВНК, которые были трансфектированы с растворимым мышиным MPL рецептором Типа I, экспрессирующим плазмид pLDmpl-53. Очищенный растворимый рецептор был лишен движения на CNBr-активированной матрице SEPHAROSE tm 4B (Pharmacia LKB Biotechnology, Inc.), особенно, если на это есть указания изготовителя, и использовался для аффинной очистки ативности MPL в кондиционированных средах клеток 24-11-5. Аффинная матрица упаковывалась в колонку ХК16 (Pharmacia LKB Biotechnology Inc.). Кондиционированные среды из 24-11-5 клеток концентрировались на срезанной полой волокнистой мембране из 10 Kd (A/G Technology Corp., Needham, NA) и погружались на дно аффинной колонки MPL рецептора при скорости течения 1 мл/минута. Эта колонка промывалась в фосфатном физиологическом растворе, содержащем 0.5 М NaCl и 0.01% азид натрия. Деятельность MPL элютировалась из колонки с 3М тиоцианата калия (Sigma Chemical Company, St.Louis, МО) при скорости течения 0.5 мл/минута. Тиоцианат калия удалялся диализом против фосфатного физиологического раствора. Активные фракции обнаруживались с помощью МТТ пролиферационного анализа (рассмотрен в Примере VII).
ПРИМЕР V. ИЗОЛИРОВАНИЕ И ОПРЕДЕЛЕНИЕ ХАРАКТЕРИСТИК ЛИНИИ КЛЕТОК, ЭКСПРЕССИРУЮЩИХ ЛИГАНД MPL РЕЦЕПТОРА
Клетки BaF3/MPLR1.1 являются IL-3 зависимыми клетками, экспрессирующими устойчиво трансфектированный мышиный MPL рецептор по Типу I. Были придуманы мутагенезис и схема селекции для изолирования клеточных линий, экспрессирующих лиганд MPL рецептора с помощью клеток мутагенизирующего BaF3/MPLR.1 и выбирающие рост аутокрина в отсутствии экзогенного IL-3.
Примерно 1.2×106 клеток BaF3/MPLR1.1 осаждалось и промывалось в GM (среда RPMI 1640, дополненная 2-меркаптоэталоном (1:240,000 - окончательная концентрация), 2 мМ L-глитамина, 110 мг/мл пирувата натрия, 50 мг/мл G418 и 10% неактивированная фетальная бычья сыворотка). Эти клетки были вновь переведены во взвешенное состояние в 2 мл GM, содержащего 0-15% (v/v) мутагенного 2-этилметанэзулфанат (EMS), и инкубировались в течение 2 часов при 37°С. После инкубирования эти клетки промывались один раз в PBS и один раз в GM и высевались на чаши в 10 см с плотность примерно 40,000 клеток/мл в GM, дополненным 5% WEHI-3 кондиционированной средой (Becton Dickinson Labware, MA) в качестве источника IL-3. Этим клеткам был предоставлен период восстановления в семь дней и икубирование проходило при 37°С в 5% CO2 перед отбором для независимого роста IL-3. После периода восстановления культура уплотнялась жизнеспособными клетками. Клетки промывались с помощью GM и культивировались в GM при отсутствии кондиционированных сред WEHI-3. После 11 дней селекции стали наблюдаться в небольшом количестве жизнеспособные клетки. Плотность жизнеспособных клеток в культуре, независимой от IL-3, прогнозировалась равной 250 клеток/мл. Один мл независимой от IL-3 культуры высевался на каждую из 19 лунок в 24-лунковой чаше для дальнейшего определения характеристик.
Кондиционированные среды из упомянутых выше независимых от IL-3 клеток ВаF3/MPLR1.1 исследовались на пролиферативную деятельность относительно клеток BaF3/MPLR. Оказалось, что кондиционированные среды из 19 независимых от IL-3 групп роста имеют активность в испытуемом образце (рассмотрен в Примере VII). Положительные среды были проанализированы на предмет пролиферативной активности в присутствии 2 мг/мл крысиного антимышиного IL-3, антимышиного IL-4 или в присутствии обоих нейтрализующих тел (Pharmingen, San Diego, CA) для идентификации мутантов, независящих от роста IL-3, экспрессирующих эти цитокинезы. (В предыдущих экспериментах было обнаружено, что клетки BaF3 также реагировали на IL-4). Только у кондиционированной среды клеток из чаши №11 (клетка с обозначением "24-11") была обнаружена активность, которая не нейтрализовалась антителами IL-3 и IL-4.
Схема мутагенеза и селекции, описанная выше, применялась к пяти другим BaF3/MPLR1 клонам (BaF3/MPLR1 клоны №4696126 15 и 18, обозначенные как BaF3/MPLR1-4, -9, -12, -15 и -18 соответственно). Оказалось, что у семнадцати изолятов (штаммов) имелась кондиционированная среда, которая стимулировала пролиферацию клеток BaF3/MPLR1. Активность всех этих сред оказалась нейтрализованной антителами анти-IL-3 или IL-4 по одному или в сочетании. Характеристики этих клонов далее не определялись.
Пролиферативная активность кондиционированных сред из группы 24-11 была охарактеризована подробно. Группа 24-11 была разделена на 19 подгрупп, и кондиционированные среды были повторно проверены на активность. Все 19 подгрупп (то есть 24-11-1 по 24-11-19 включительно) стимулировали пролиферацию клеток BaF3/MPLR1, независимых от роста IL-3, в отсутствии экзогенного IL-3. Эта активность не подавлялась нейтрализующими антителами IL-3 или IL-4 или комбинацией этих антител.
Проводилось два эксперимента для определения особенности активности 24-11. Кондиционированные среды исследовались на пролиферативную активность на контрольных клетках BaF3, которые не экспрессируют рецептор MPL. В отсутствии экзогенного IL-3 пролиферации контрольных клеток BaF3 не наблюдалось в кондиционированных средах ни в одной из девятнадцати 24-11 подгрупп. Во втором эксперименте пролиферативная активность исследовалась на подавление очищенным растворимым MPL рецептором. Клетки BaF3/MPLR1 культивировались в среды GM, в которые добавлялись 5-% кондиционированных сред 24-11. К каждому образцу добавлялся растворимый мышиный MPL рецептор по Типу I до окончательной концентрации 0.0, 0.625, 1.25, 2.5 или 5.0 мг/мл. Результаты сказались через четыре дня при анализе пролиферации МТТ клеток. Пролиферативная активность кондиционированной среды 24-11 была полностью блокирована при концентрации 0.625-1.25 мг/мл растворимого MPL рецептора. Концентрации растворимого рецептора, которые полностью останавливают активность, не действовали на стимулирование клеток BaF3/MPLR1 с помощью IL-3 или IL-4. Результаты показали, что растворимый MPL рецептор может конкурировать с гипотезой о том, что 24-11 клетки экспрессировали лиганд MPL рецептора.
Клоны, отводимые от клеток 24-11, изолировались высеванием при ограничивающих концентрациях растворов. Один клон, обозначенный 24-11-5 №3, показал высокий уровень пролиферативной активности в его кондиционированных средах относительно группы 24-11. Пролиферативная активность оказалась равной 1:2000 разбавлению кондиционированных сред из клеток WEHI-3 (Becton Dickinson Labware).
ПРИМЕР VI. КОНСТРУИРОВАНИЕ БИБЛИОТЕКИ сДНК 24-11-5 №3
Общая РНК была приготовлена из -2.7×108 клеток 24-11-5 №3 с помощью изотиоционата гуанидида, после чего проводилось центрифугирование в CsCl (работа Чиргвина и др., там же). Поли(А)+ был изолирован с помощью набора для изолирования OLIGOTEX-dT-mPHK (Qiagen Inc., Chatsworth, CA) в соответствии с указаниями изготовителя.
Кислота сДНК первого ствола из клеток 24-11-5 №3 была синтезирована в 4 параллельные реакции. Каждая ракция содержала 7 мл полиd(Т)-отобранную поли(А)+24-11-5 №3 РНК при концентрации 1.6 мг/мл и 2.5 мл из 20 рмоль/мл праймера ZC6172 первого ствола (SEQ ID NO:14), содержащего ристрикционный сайт Xho I. Эта смесь нагревалась при 65°С в течение 4 минут и охлаждалась на льду. Синтез сДНК первого ствола инициировался добавлением 8 мл буфера первого ствола (5х буфер SUPERSCRIPT tm; GIBCO BRL), 4 мл из 100 мМ дитиотреитола и 2 мл раствора деоксинуклеотидного трифосфата, содержащего по 10 мМ dATP, dGTP, dTTP и 5-метил-dСТР (Pharmacia LKB Biotechnology Inc.) в смесь РНК-праймер. Реактивная смесь инкубировалась при 45°С в течение 4 минут, после чего следовало добавление 10 мл из 200 U/мл обратной транскриптазы RNase H- (GIBCO BRL). Эффективность синтеза первого ствола анализировалась в параллельной реакции путем добавления 10 мСi из 32p-adCTP в объем, кратный 10 мл, из одной из реактивных смесей для мечения реакции для проведения анализа. Эти реакции инкубировались при 45°С в течение 1 часа, после чего проводилась инкубация при 50°С в течение 15 минут. Неиспользуемый 32p-adCTP в отмеченной реакции удалялся методом хроматографии на гелевой фильтрационной колонне размером в 400 отверстий (Clontech Laboratories). Немеченые реакции первого ствола были сгруппированы, а неиспользуемые нуклеотиды удалялись двойным осаждением сДНК в присутствии 32 мг носителя гликогена, 2.5 М ацетата аммония и 2.5 объема этанола. Немеченая сДНК вновь переводилась во взвешенное состояние в 144 мл воды для использования при синтезе второго ствола. Длина меченой сДНК первого ствола определялась гель-электрофорезом агарозы.
Синтез второго ствола проводился на сДНК первого ствола при условии, что синтез второго ствола, стимулированный примированием первого ствола, приведет к образованию петли ДНК. Проводились три самостоятельных параллельных реакции второго ствола. Каждая реакция второго ствола содержала 48 мл немеченой сДНК первого ствола, 16.5 мл воды, 20 мл из 5хбуфер полимеразы I (100 мл Трис:НСl, рН 7.4, 500 mМ КСl, 25 mМ MgCl2, 50 mM (NH4)2SO4), 1мл из 100 мМ дитиотреитола, 1 мл раствора, содержащего 10 мМ деоксинуклеотидного трифосфата, 3 мл из 5 мМ бета-NAD, 1 мл из 3U/мл Е.coli лигазы ДНК (New England Biolabs Inc.) и 5 мл из 10 U/мл E.coli полимеразы I ДНК (Amersham Corp.). Реакция проводилась при комнатной температуре и инкубировалась при комнатной температуре в течение 5 минут, после чего добавлялось 1.5 мл из 2 U/мл RNase Н (GIBCO BRL). 10 мл из одной из реакций синтеза второго ствола помечались добавлением 1- мСi 32-p-adCTP для регулирования эффективности синтеза второго ствола. Эти реакции инкубировались при 15°С в течение 2 часов, после чего следовало инкубирование в течение 15 минут при комнатной температуре. Неиспользованный в меченой реакции 32p-adCTP удалялся методом хроматографии через гель-фильтрационную колонну размером 400 отверстий (Clontech Laboratories) перед проведением анализа гель-электрофорезом агарозы. Немеченные реакции группировались и экстрагировались с фенолом/хлороформом и хлороформом, после чего проводилось осаждение в этаноле в присутствии 2.5 М ацетата аммония.
Одноствольная ДНК с петлевой структурой расщеплялась с помощью нуклеазы фасоли золотистой. Реактивная смесь содержала 100 мл сДНК второго ствола, 20 мл 10х буфер нуклеазы фасоли золотистой (Stratagene Cloning Systems), 16 мл из 100 мМ дитиотреитола, 48 мл воды, 10 мл диллютивного буфера нуклеазы фасоли золотистой (Stratagene Cloning Systems) и 6 мл из 50 U/мл нуклеазы фасоли золотистой (Promega Соrр.). Реакция инкубировалась при 37°С в течение 30 минут. Реакция заканчивалась добавлением 20 мл из 1 М Трис:НСl, рН 8.0, после этого шло последовательное экстрагирование фенола/хлороформа и хлороформа, как описывалось выше. После экстрагирования ДНК осаждалась в этаноле и повторно переводилась во взвешенное состояние.
Вновь переведенная во взвешенное состояние сДНК имела затупленные Т4 ДНК полимеразой концы. сДНК, которая была переведена во взвешенное состояние в 188 мл воды, смешивалась с 50 мл 5х буфер полимеразы Т4 ДНК (250 мМ Трис:НС1, рН 8.0, 250 mМ RCl, 25 mM MgCl2), 3 мМ деоксинуклеотидного трифосфата и 5 мл из 1U/мл полимераэы Т4 ДНК (Boehringer Mannheim Corp.). После инкубации в течение 30 минут при 15°С реакция заканчивалась добавлением 10 мл из 0.5 EDTA, после этого следовало последовательное экстрагирование фенола/хлороформа и хлороформа, как описывалось выше. ДНК хроматографировалась через гель-фильтрационную колонну размером 400 отверстий (Clontech Laboratories Inc.) для удаления небольшого количества протеина и удаления коротких ДНК, длина которых менее -400 пар нуклеотидов. ДНК осаждалась в этаноле в присутствии 10 мг носителя гликогена и 2.5 М ацетата аммония и была вновь переведена во взвешенное состояние в 15 мл воды. На основании использования 32p-adCTP размер получаемой ДНК прогнозируется равным -8 мг из начального количества mРНК матриц, равного 40 мг.
Адаптеры Eco RI были лигированы на 5’ концах сДНК, описанной выше, для того, чтобы дать возможность провести клонирование в экспрессирующий вектор. 10 мл сДНК (-5 мг) и 21 мл 65рмоль/мл адаптера Eco RI (Pharmacia LKB Biotechnology Inc.) смешивались с 4 мл 10× буфер лигазы (Proinega Corp.), 3 мл из 10 мМ АТР и 3 мл из 15 U/мл Т4 лигазы ДНК (Promega Corp.). Реакция инкубировалась в течение ночи (-48 часов) при 9°С. Реакция заканчивалась добавлением 140 мл воды, 20 мл 10×Н буфер (Boehringer Mannheim Corp.) и инкубировалась при 65°С в течение 40 минут. После инкубирования сДНК экстрагировалась с фенолом/хлороформом и хлороформом, как описывалось выше, и осаждалась в присутствии 2.5 М ацетата аммония и 1.2 объема изопропанола. После центрифугирования осадок сДНК промывался 70% этанолом, просушивался воздухом и возвращался во взвзешенное состояние в 89 мг воды.
Для упрощения направленного клонирования сДНК в экспрессирующий вектор сДНК поглощалась Xho I, что привело к тому, что у сДНК образовался 5’ Есо RI липкий конец и 3’ Xho I липкий конец. Предварительно был внедрен ристрикционный сайт Xho I с 3’конца сДНК с помощью праймера ZC6172 (SEQ ID NO:14). Поглощение ристрикционного фермента проводилось в реакционной смеси, содержащей 89 мл сДНК, описанной выше, 10 мл из 10× Н буфер (Promega Corp.) и 1.5 мл из 40U/мл Хсо I (Boehringer Mannheim Corp.). Поглощение проводилось при 37°С в течение 1 часа. Реакция заканчивалась последовательными экстрагированиями фенола/хлороформа и хлороформа и хроматографией через гель-фильтрационную колонну размером 400 отверстий (Clontech Laboratories Inc.).
сДНК осаждалась в этаноле, промывалась в 70% этаноле, просушивалась на воздухе и вновь возвращалась во взвешенное состояние в 20 мл 1 × груженный гелем буфер (10 мМ трис:НСl, рН 8.0, 1 mМ EDTA, 5% глицерола и 0.125% бромфенола синего. Возвращенная во взвешенное состояние сДНК нагревалась до 65°С в течение 5 минут, охлаждалась на льду и подвергалась электрофорезу на 0.8% геле агарозы с низкой температурой плавления (SEA PLAQUE GTG tm агароза с низкой температурой плавления; FMC Corp.). Примесные адаптеры и сДНК длиной менее 0.5 тыс. пар нуклеотидов были вырезаны из геля. У электродов меняли знаки и сДНК подвергалась электрофорезу до тех пор, пока она не сконцентрируется вблизи начала промежутка. Область геля, содержащая концентрированную сДНК, вырезалась и помещалась в трубку микроцентрифуги, и определялся примерный объем среза геля. Объем воды, примерно в три раза больше объема среза геля (300 мл), добавлялся в трубку и агароза плавилась при нагревании 65°С в течение 15 минут. После достижения равновесного состояния образца при 45°С добавлялось 5 мл из 1U/мл бета-агарозы I (New England Biolabs, Inc.) и смесь инкубировалась в течение 90 минут при 45°С для поглошения агарозы. После инкубирования 40 мл из 3 М ацетата Na добавлялось к образцу и смесь инкубировалась на льду в течение 15 минут. Этот образец центрифугировался при 14,000×g в течение 15 минут при комнатной температуре для удаления непоглощенной агарозы, после чего проводилась хроматография через гель-фильтрационную колонну размером 400 отверстий (Clontech Laboratories). сДНК осаждалась в этаноле, промывалась в 70% этаноле, просушивалась на воздухе и возвращалась во взвешенное состояние в 70 мл воды для проведения реакции киназы для фосфорилирования лигированных Eco RI адаптеров.
К 70 мл раствора сДНК добавлялось 10 мл 10× буфер лигазы (Stratagene Cloning System), и смесь нагревалась до 65°С в течение 5 минут. Смесь охлаждалась на льду, и 16 мл из 10 мМ АТР и 4 мл из 10 U/мл Т4 полинуклеотидной киназы (Stratagene Cloning Systems) были добавлены. Реактивная смесь инкубировалась при 37°С в течение 1 часа и реакция заканчивалась нагреванием 65°С в течение 10 минут, после чего следовало последовательное экстрагирование с фенолом/хлороформом и хлороформом. Фосфорилированная сДНК осаждалась в этаноле в присутствии 2.5 М ацетата аммония, промывалась 70% этанолом, просушивалась на воздухе и возвращалась во взвешенное состояние в 10 мл воды. Концентрация фосфорилированной сДНК оценивалась как -40 fмоль/мл.
Экспрессирующий pDX вектор млекопитающего (рассмотрен в Патенте США №4,959,318, Фигура) была смодифицирована для восприятия 24-11-5 №3 сДНК, которая синтезировалась с концами Eco RI-Xho I. Эндогенный Sal сайт на pDX был ликвидирован поглощением плазмида с Sal I и рециркуляризацией плазмида, которая следовала после затупления липких концов Sal I полимеразами Т4 ДНК. Рециркулированный плазмид поглощался Есо RI и к нему лигировалась короткая полилинкерная последовательность двух комплементарных олигонуклеотидов, ZC6936 (SEQ ID NO:15) и ZC6937 (SEQ ID NO:16), для получения плазмида pDX.ES. Внедренная полилинкерная последовательность на pDX.ES состояла из сайтов Есо RI и Sal I, что позволяло упростить направленное клонирование сДНК 24-11-5, синтезированную с концами Есо RI-Xho I.
Библиотека сДНК плазмида создавалась лигированием сДНК Есо RI-Xho I 24-11-5 в pDX.ES, поглощенный Есо RI/Sal I. Смесь лигирования была электропорирована в E.coli (компетентные клетки ELECTROMAX DH10B tm; GIBCO BRL, Gaithersburg, MD) с помощью импульсного контроллера и 0.2 см кюветы (Bio-Rad Laboratories, Hercules, CA), применяя 0.2 KB, 400 Ом и 25 mFAD. Эти клетки разбавлялись в бульоне Луриа и инкубировались при 37°С в течение 45 минут, после чего добавляли 0.75 мл 50%-ного глицерола. Транфектированные клетки были аликвотированы и хранились при -70°С до использования. 80 fмолей сДНК породили более 700,000 независимых рекомбинантных плазмидов.
ПРИМЕР VII. ЭКСПРЕССИВНЫЕ ИССЛЕДОВАНИЯ БИБЛИОТЕКИ сДНК 24-11-5 НА MPL АКТИВНОСТЬ
Библиотека сДНК 24-11-5 №3 высевалась на примерно 2 тысячи агаровых чашек диаметром 10 см с бульоном Луриа, в который добавлялось 100 мг/мл ампицилина. Плотность высевания равнялась от 200 до 250 бактериальных колоний на чашку. ДНК плазмида для трансфекции в ВНК 570 клеток готовилась из каждой бактериальной чаши с помощью очищающей ДНК смолы MAGIC MINIPREPS tm (Promega Corp.), в соответствии с указаниями изготовителя. ДНК плазмиды хранились при -20°С до начала трансфекции в ВНК 570 клеток.
Группы плазмидов сДНК 24-11-5 №3, каждая из которых содержала примерно 200-250 клонов сДНК, трансфектировались в ВНК 570 клетки, используя рецептуру 3:1 липосомы 2,3-диолеилокси-N-{2(сперминекарбоксиамидо)этил}-N,N-диметил-1-пропанаминиумтрифлюороацетат и диолеолифосфатидилетаноламин в воде (LIPOFECTAMINE tm; GIBCO BRL). 20 мл из 30 нг/мл ДНК добавлялся к 20 мл из раствора LIPOFECTAMINE tm, разбавленный в пропорции 1:10, и проводили инкубирование при комнатной температуре в течение 30 минут. После инкубирования 160 мл сред, свободных от сыворотки (Hams F12: Dulbeccos MEM (1:1) с добавлением 2 мМ L-глютамина, 0.11 мг/мл пирувата натрия, 5 мг/мл инсулина, 5 мг/мл фетуина, 10 мг/мл трансферрина, 2 нг/мл окиси IV селениума и 25 мМ буфера HEPES) в смесь ДНК/LIPOFECTAMINE tm. Затем это переносилось на 42-лунковый титрационный микропланшет, содержащий -100,000 ВНК 570 клеток. Эти клетки были инкубированы при 37°С в 5% CO2 в течение 4 часов, после чего добавлялось 200 мл ВНК Сред Роста (Среды Иглеса, модифицированные Дулбекко, к которым добавлены 2 мМ L-глитамина, 0.11 мг/мл пирувата натрия, 5%-ной телячьей фетальной инактивированной сыворотки и 100x PSN антибиотики (GIBCO BRL). Эти клетки были инкубированы в течение 16 часов. Среды удалялись и заменялись 0.5 мл свежей Среды Роста ВНК, которая кондиционировалась в течение 48 часов до исследования на наличие MPL активности.
Для определения присутствия активности MPL в кондиционированной среде библиотеки, трансфектировавшей ВНК 570 клетки, использовали анализ пролиферации клеток. 100 мл кондиционированных сред добавлялось к 100 мл из 10 6/мл промытых клеток BaF3/MPLR1.1 в средах RMPI 1640 (JRH Вioscience Inc., Lenexa, KS) дополненных 2 мМ L-глютамина, PSN антибиотиками (GIBCO BRL), 0.00036% 2-меркаптоэтанола и 10% нагретой фетальной телячьей инактивированной сыворотки. Испытуемые клетки инкубировались в течение 3 суток при 37°С в 5% CO2 до исследования на пролиферацию.
Пролиферация клеток в присутствии MPL определялась количественно с помощью калориметрического анализа, основанного на метаболическом распаде 3-(4,5-диметилтиазол-2-ил)-2.5-дифенил тетразолийного бромида (МТТ) (Мосман, J.Immunol.Meth.65:55-63, 1983). 20 мл из 10 мг/мл раствора МТТ (Polyscience, Inc., Warrington, PA) добавлялись к 100 мл испытуемых клеток BaF3/MPLR1.1, и эти клетки инкубировались при 37°С. Спустя 4 часа добавляли 200 мл из 0.04 N НСl в изопропаноле, этот раствор смешивался, и адсорбенты образца рассматривались при 570 нм на ридер-планшете модели EL320 ELISA (Bio-Tek Instruments Inc., Highland Park, VT).
Одна из плазмидных групп оказалась позитивной, обозначенная Т1081, трансфектировалась в клетки ВНК 570. Надосадочная жидкость из трансфектантов давала позитивный сигнал при изучении пролиферации МТТ. Эксперименты по нейтрализации PСR и антитела показали, что активность происходила не благодаря IL-3 и IL-4.
Плазмиды из позитивных групп использовались для преобразования Е coli DH10B, и клетки высевались (42 чаши с примерно 15-20 колоний на чашу, 10 чаш с примерно 90 колоний на чашу и 8 чаш с примерно 250 колоний на чашу). С каждой чаши был сделан отпечаток (реплика) и сохранялся при 4°С. Колонии счищались с оригинальных чаш и свободно проростали в жидкую культуру в течение еще нескольких часов, затем приготавливалась ДНК.
Плазмидная ДНК из подгрупп трансфектировалась в ВНК 570 клетки, и клеточная надосадочная жидкость собиралась и исследовалась, как описывалось выше. Спустя примерно два часа одна подгруппа (№22) оказалась позитивной в результате микроскопических исследований (клетка удлиненной формы). Несколькими часами позже еще две подгруппы (№19 и №28) оказались также позитивными. Оставшаяся надосадочная жидкость из каждой позитивной группы анализировалась в сравнении с контрольными клетками BaF3 и оказалось, что у нее нет активности. Кроме того, оказалось, что активность этих трех позитивных подгрупп подавлялась растворимым MPL рецептором Типа I.
Отпечатки чаш из трех позитивных подгрупп росли еще в течение нескольких часов. Затем индивидуальные (отдельные) колонии собирались и использовались для инокуляции (прививки) 3-мл культур. Культуры выращивались примерно в течение 8 часов при 37°С, затем приготавливалась ДНК методом, описанным выше. Плазмидная ДНК трансфектировалась в ВНК 570 клетки и надосадочная жидкость выращивалась примерно 10 часами позже и исследовалась на активность. Спустя 1 час один клон (обозначенный Т1081-19-215, соответствующий подгруппе №19) оказался позитивным. Этот клон высевался полоской вновь для единичных колоний. ДНК приготавливалась из 12 колоний и трансфектировалась в ВНК 570 клетки. Все 12 трансфектантов позже оказались позитивными при исследовании. ДНК из одной из 12 позитивных колоний преобразовывалась в Е-соli DH5a. Плазмид обозначался pZGmp1-1081. Этот трансформант был депозитирован 14 февраля 1994 года в Американском Собрании Типов Культур, 12301 Parklawn Drive, Rockville, MD под каталожным номером 69566.
Нуклеотидная последовательность сДНК, кодирующая гематопоэтический протеин (тромбопоэтин), была определена (SEQ ID NO:1). Анализ закодированной аминокислотной последовательности (SЕQ ID NO:2) показал, что амино окончание зрелого протеина представляет собой аминокислотный остаток 45. Два метиониновых кодона, в пунктах 105 и 174 SEQ ID NO:1 оказались кодонами инициации, причем основной сайт инициации предполагается в пункте 174.
ПРИМЕР VIII. ГЕМАТОПОЭТИЧЕСКАЯ АКТИВНОСТЬ РЕКОМБИНАНТНОГО ТРОМБОПОЭТИНА
Костное вещество из бедренной кости и голени женских особей мышей CD-1 в послеродовом периоде культивировали в 25 мл буфера CATCH (99 мг теофилина, 0.75 г цитрата натрия, 75 мг аденозина, 20 мл 10× сбалансированного физиологического раствора, свободного от Ca++Mg++, на 200 мл dH2O; рН 7.4). Клетки были взвешены в единой клеточной суспензии путем раскапывания из 25-миллилитровой пипетки. Этот объем выращивался до 50 мл с помощью буфера CATCH, и клетки осаждались центрифугированием со скоростью 1000 об/мин в течение 7 минут. Этот осадок вновь переводился во взвешенное состояние в 20 мл буфера CATCH и инкубировался в колбе для культивирования ткани Т75 в течение первого цикла пластической адгезии при 37°С в течение 2 часов. Неадгезированные клетки собирались центрифугированием при скорости вращения 1000 об/мин в течение 7 минут, позволяющей осаждать клетки. Этот осадок вновь переводился во взвешенное состояние в 15 мл альфа-МЕМ+ 10% FBS (+L-глютамин NаПируват и PSN антибиотики) и инкубировался в колбе Т75 для второго цикла пластической адгезии, как описано выше, для первого цикла. После окончательного центрифугирования и переведения во взвешенное состояние клетки подсчитывались. Половина мл всех клеток при 576,000 клеток/мл были посеяны в 24-лунковые чаши для культивирования ткани вместе с пробными средами из контрольных ВНК клеток или с кондиционированными средами из ВНК клеток, трансфектированных с pZGmpl-1081. После трех дней инкубирования при 37°С клетки собирались и окрашивались, как описано выше.
150 мл клеток было собрано с контрольной лунки при обработке в стандартной кондиционированной среде. 50 мл клеток были собраны из лунки, обрабатываемой кондиционированной средой из ВНК клеток, трансфектированных pZGmpl-1081. Эти образцы сплетались и изготавливались стандартные микроскопичекие слайды.
Эти слайды сохранялись в 100% метаноле, затем заливались раствором Райта в пропорции 1:1 (0.5 г краски Райта в 300 мл метанола)/Н2O на 6 минут, промывались водой и просушивались. Затем слайды заливались краской Гайемса (Sigma Chemical Corp.) в буфере Соренсена (2.28 г КН2РО4/2&38 г NaРО4 в 250 мл Н2O), промытом в воде и высушенном.
После отрегулирования используемых объемов образец среды BHK/pZGmpl-1081 состоял из 120 мегакариоцитов на 150 мл по сравнению с 9 мегакариоцитами на 150 мл контрольной среды. Кроме того, мегакариоциты в обрабатываемом экспериментальном образце, наблюдаемые в микроскоп, оказались значительно больше по размеру, чем контрольные клетки, и имели гораздо более окрашенные полинуклеотиды.
Кондиционированные среды мутантной BaF3/MPLR1.1 линии 24-11-5 №3 собирались в отсутствии сыворотки и концентрированной 20-складки 10 кД срезе фильтрующего прибора Amicon Inc. (Beverly, MA). Из мышиной бедренной кости собиралось вещество костного мозга и взвешивалось в Средах Дулбекко, модифицированных Исков (GIBCO BRL)+ 15% фетальной телячьей сыворотки (FCS). После перевода во взвешенное состояние подсчитывались нуклеотидные клетки и высевались при 75,000 клеток/мл и 0,9 мл/чаша в среду, содержащую 50% метилцеллюлозу, 15% FSC, 10% BSA и 0.6%PSN (полутвердая среда) в чашах для культивирования ткани объемом 1 мл. Добавлялись разнообразные кондиционированные среды и контрольные образцы для получения объема в 1 мл. Чаши инкубировались при 37°С/5% CO2 в течение 6 суток, а затем исследовались под микроскопом для подсчета колоний гранулоцитных/ макрофаговых колоний. Оказалось, что чаши, инкубированные в присутствии кондиционированной среды 24-11-5 №3, обладали слабой GMCSF-подобной активностью, выражающейся в количестве 25 по сравнению с нулевым значением в случае негативного контрольного образца и в количестве 130 для чаш, стимулируемых положительным контролем (среда, приготовленная обработкой селезенки фитолаккой американской (PWMSCM) методом инкубирования измельченной мышиной селезенки в течение 1 недели в присутствии митогена фитолакки американской (полученной из Boehringer Mannheim, Indianapolis, IN) + 2 единицы/мл эритропоэтина).
Из мышиной бедренной кости бралось вещество костного мозга и взвешивалось в Средах Дулбекко, модифицированных Исков (GIBCO-BRL), содержащее 15% FCS, и нуклеированные клетки подсчитывались и высевались в полутвердую среду, как описывалось выше. Эти клетки использовались для испытания колонии мегакариоцитов, создающей активность протеина, закодированного вставкой pZGmpl-1081.
Группа ВНК 570 клеток, стабильно трансфектируемых с pZGmpl-1081, культивировалась в отсутствии сыворотки, и собиралась кондиционированная среда. Эта кондиционированная среда испытывалась индивидуально и в сочетании со средой селезенки, обработанной фитолаккой американской, с рекомбинантными мышиными IL-3, IL-4 (Genzyme Corp., Cambridge, МА), IL-11 (Genzyme Corp.) с разными комбинациями этих факторов. PWMSCM применялся в качестве позитивного контроля. В качестве негативного контроля использовалась некондиционированная среда культивирования.
Испытуемые или контрольные образцы добавлялись к культурам вещества костного мозга для добавления до объема в 1 мл. Чаши инкубировались в течение 6 суток при 37°С в 5% CO2, затем исследовались под микроскопом для подсчета мегакариоцитных колоний. Результаты приведены в Таблице 4. Подытоживая, можно сказать, что кондиционированная среда BHK/pZGmpl-1081 проявила наличие мегакариоцитной колонии, формирующей активность, которая усиливалась в присутствии рано действующих факторов до уровней, значительно выше, чем любой из рано действующих факторов отдельно.

In vivo активность кондиционированной среды BHK/pZGmpl-1081 исследовалась на мышах. Была собрана безсывороточная среда и сконцентрирована в 5 складок с помощью 10 Kd с помощью прибора для фильтрации среза (Amicon Inc., Beverly, NA). Контрольная (не кондиционированная) среда концентрировалась таким же образом. 6 мышей BALB/c (Simonsen Laboratories Inc., Gilroy, CA) обрабатывались семью ежедневными внутрибрюшными инъекциями из 0.5 мл либо контрольной, либо кондиционированной среды. Пробы крови забирались в следующие дни: 0. 3 и 7 и было подсчитано содержание тромбоцитов. Результаты, приведенные в Таблице 5, показали, что эта кондиционированная среда из клеток BHK/pZGmpl-1081 имела тромбопоэтическую активность.
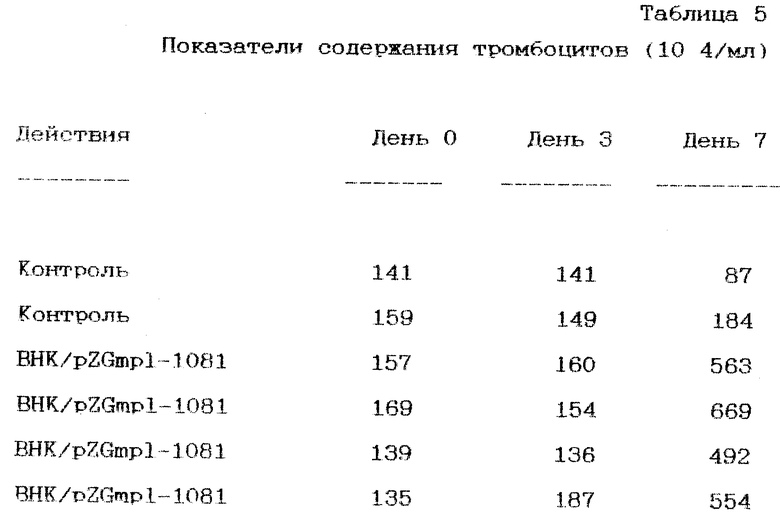
ПРИМЕР IX. ИЗОЛИРОВАНИЕ ЧЕЛОВЕЧЕСКОГО ТРОМБОПОЭТИЧЕСКОГО ГЕНА
Амплифицированная генотека человеческого легкого LAMBDA FIX R (Stratagene Cloning Systems) была подробно исследована на предмет гена человеческого тромбопоэтина с помощью сДНК лиганда мышиного mpl рецептора в качестве зонда. Эта генотека была титрована, и 30 чашек размером 150 мм, инокулированными с Е-соli ствольными LE-392 клетками (Stratagene Cloning Systems), были заражены 4×10 4 бляшкообразующими единицами (PFU). Чаши инкубировались в течение ночи при 37°С. Были изготовлены фильтрационные подъемники этих бляшек с помощью найлоновых мембран HYBOND - N tm (Amersham) в соответствии с инструкцией, рекомендванной изготовителем. Эти фильтры обрабатывались денатуратом в растворе, содержащем 1.5 М NaCl и 0.5 М NaOH в течение 7 минут при комнатной температуре. Эти фильтры были быстро блоттированы на фильтрационной бумаге для удаления лишнего раствора денатурата, после чего проводилась нейтрализация в течение 5 минут в 1 М Трис-НСl (рН 7.5) и 1.5 М NaCl. Фаговая ДНК фиксировалась на фильтрах устройства для поперечного сшивания (Stratagene Cloning Systems) STATALINKER R с УФ энергией в 1,200 мДжоулей. После фиксирования фильтры промывались три раза в 0.25×SSC, 0.25% SDS и 1 мМ EDTA при 65°С. После промывки эти фильтры предварительно гибридизировались в гибридизирующем растворе (5×SSC, 5× раствор Денхарта, 0.2% SDS и 1 мМ EDTA), который фильтровался через 0.45 мМ фильтр. Непосредственно перед употреблением добавляли обработанную подогретым денатуратом, фрагментированную ДНК спермы лосося (окончательная концентрация 100 мг/мл). Фильтры предварительно гибридизировались при 65°С в течение ночи.
Мышиная сДНК ТРО полной длины из pZGmpl-1081 была помечена 32р произвольным приммированием с помощью Системы Мечения ДНК MEGAPRIME tm (Amersham) методом, рекомендованным изготовителем. Прегибридизационный раствор был заменен свежим гибридизационным раствором, содержащим зонд с примерно 1×106 импульсов в минуту, и гибридизированным в течение ночи при 65°С. После гибридизации этот гибридизационный раствор удалялся, а фильтры прополаскивались 4 или 5 раз в промывочном растворе, содержащем 0.25× SSC, 0.25% SDS и 1 мМ EDTA. После прополаскивания эти фильтры промывались последовательно 8 раз при 50°С в промывочном растворе. После последней промывки фильтры экспонировались перед авторадиографической пленкой (XAR-5; Eastman Kodak Co.; Rochester, NY) в течение 4 дней при -70°С с помощью интенсифицирующего экрана.
Изучение автофотографий выявило несколько сотен регионов, которые гибридизировали с меченым зондом. Агаровые пробки брались в 100 регионах для очистки. Каждая агаровая пробка в течение ночи пропитывалась 1 мл SM, содержащем 1% хлороформ (Маниатис и др. Molecular Cloning: A Laboratory Manual, Cold Spring Harbor, NY, 1982). После инкубирования в течение ночи фаг с каждой пробки растворялся в пропорции 1:1,000 в SN. Объемы, кратные 5 мл, высевались на клетки штамма Е-соli LE392. Чаши инкубировалсь в течение ночи при 37°С и были подготовлены подъемники для фильтров, прегибридизированные, гибридизированные, промытые и авторадиографированные, как описывалось выше.
Изучение полученных авторадиофотографий выявило наличие сильных позитивных сигналов из двух основных изолянтов и слабые сигналы из других 18. Агаровые пробки брались из позитивных зон для каждого из 20 сигналов. Агаровые пробки обрабатывались, как описывалось выше. Фаг, элютированный из каждой агаровой пробки, разбавлялся в пропорции 1:100 в SM и объемы, кратные 1 мл, высевались с клетками штамма E.coli LE392. Чаши инкубировались и подготавливались и гибридизировались подъемники фаговых фильтров, как описано выше. Фильтры промывались при 55°С в промывочном буфере. Авторадиографии выявили области гибридизации, соответствующие сигналу, дискретные фаговые бляшки из трех первоначальных изолянтов, 8-3-26 10-1-1 и 29-2-1.
Фаговые изолянты 8-3-2, 10-1-1 и 29-2-1 были обозначены лямбдаZGmрl-Н10 и лямбда ZGmpl-H29 соответственно. ДНК из изолянтов лямбда ZGmpl-H8, лямбдаZGmрl-Н10 и лямбдаZGmр1-Н29 очищалась с помощью фагового абсорбента LAMBDASORB tm (Promega Corp. Madison, WI) в соответствии с указаниями изготовителя. Вставки человеческой геномной ДНК из фага были отделены от фагового вектора ДНК поглощением Хbа I и очищены гель-электрофорезом агарозы. Все три фаговые изолянта содержали последовательности, которые гибридизировались с зондом сДНК лиганда мышиного mpl рецептора, как показано в Южном блот-анализе (Маниатис и др., там же). Фаговый лямбдаZGmрl-Н8 анализировался и оказалось, что гибридизирующие регионы лямбдаZGmрl-Н8 осаждаются на фрагменты ДНК Xba I длиной 9-5 тыс. пар нуклеотидов, 2,5 т.п.н. и 1 т.п.н. Фрагмент в 2.5 т.п.н. был субклонирован в фагемит BLUESCRIPT II SК+, поглощаемый Xba I (Stratagene Cloning Systems) для того, чтобы получить плазмидовый pZGmpl-H82.5.
Последовательность человеческого ТРО гена и закодированная аминокислотная последовательность показаны в SEQ ID NO:28 и SEQ ID NO:29.
ПРИМЕР X. ИЗОЛИРОВАНИЕ ТРОМБОПОЭТИЧЕСКОЙ ЧЕЛОВЕЧЕСКОЙ сДНК ПОЛНОЙ ДЛИНЫ
Человеческая ДНК, кодирующая ТРО, полной длины изолировалась посредством цепной реакции полимеразы из тромбоцитов сДНК человеческой печени и почек, с помощью специальных праймеров, отведенных от эксонных последовательностей, идентифицированных на pZGmpl-H82&5 и из сохраненной 5’ нетранслированной последовательности мышиной ТРО сДНК.
Поли d(T) человеческих почек, печени и легких, селектированный поли (А)+ РНК (Clontech, Palo Alto, CA) применялись для синтезирования сДНК первого ствола. Каждая реакция подготавливалась с помощью 4 микрограммов поли(А)+РНК, смешанных с 1 мг олиго d(T)18 (без 5’ фосфата) праймера mPHK (New England Biolab, Beverly, MA) с окончательным объемом 19 мл. Смеси нагревались до 65°С в течение 5 минут и охлаждались на льду. Синтез сДНК был инициирован добавлением 8 мл 5× буфер SUPERSCRIPT tm (GIBCO BRL), 2 мл из 100 мМ дитиотреитола, 2 мл раствора трифосфата деоксинуклеотида, содержащего по 10 мМ dATP, dGTP, dTTP и dCTP (Pharmacia LKB Biotechnology Inc., Piscataway, NJ), 2мл из 1 мСi/мл 32р-a-dCTP (Аmеrshаm, Arlington Heights, IL) и 8 мл из 200 U/мл обратной транскриптазы SUPERSCRIPT tm (GIBCO BRL) для каждой смеси РНК-праймер. Реакции инкубировались при 45°С в течение 1 часа и были разбавлены до 120 мл с помощью ТЕ (10 мМ Трис:НС1, рН 8.0, 1 vV EDTA). сДНК осаждались два раза добавлением 50 мл 8 М ацетата аммония и 160 мл изопропанола. Полученный осадок сДНК был переведен вновь во взвешенное состояние в 10 мл ТЕ. Количество получаемой сДНК первого ствола для каждой реакции прогнозировалось равным уровням внедрения 32p-dCTP.
сДНК первого ствола из mPHK печени, легких и почек использовались при выработке двух сегментов сДНК, N-окончание, равное 1/3, и С-окончание, равное 2/3 последовательности, с помощью цепной реакции полимеразы. Ристрикционный сайт Крn I внедрялся в сегменты сДНК путем единичного изменения основания геномной последовательности с помощью мутагенеза PCR, с использованием праймеров ZC7422 (SEQ ID NO:20) и ZC7423 (SEQ ID NO:21). Результирующее изменение нуклеотидов, создаваемое обычным ристрикционным сайтом Kpnl без изменения в заданном кодировании аминокислот.
Сегмент с N-окончанием был амплифицирован в 50 мл реактива, содержащего 5 нг сДНК тромбоцитов (в отдельных реакциях для сДНК почек, печени и легких), по 80 рмолей олигонуклеотидов ZC7424 (SEQ ID NO:22) и ZC7422 (SEQ ID NO:20), 5 мл из 2-5 мМ раствора трифосфата деоксинуклеотида (Cetus Corp., Emerville, CA), 5 мл из 10× буфер PCR (Promega Cotp., Madison, WI) и 2.5 единиц Taq полимеразы (Boehringer, Mannheim). Цепная реакция полимеразы проводилась в 35 циклов (1 минута при 94°С, 1 минута при 58°С и 1.5 минуты при 72°С), после чего проводилось инкубирование в течение 7 минут при 72°С. Праймер Чувства ZC7424 (SEQ ID NO:22) перекрывал нетранслированный регион 5’ лиганда мышиного mрlрецептора и включал в себя ATG кодон инициации. Праймер античувства ZC7422 (SEQ ID NO:20) включал в себя последовательность из региона, соответствующего эксонам 4 и 5 человеческой геномной ДНК ТРО.
Сегмент с С-окончанием был амплифицирован в 50 мл реактив, содержащий 5 нг сДНК тромбоцитов (человеческие почки, печень и легкие, как описано выше), по 80 рмолей из олигонуклеотидов ZC7423 (SEQ ID NO:21) и ZC7421 (SEQ ID NO:23), 5 мл из 2.5 мМ раствора трифосфата деокси нуклеотида (Cetus Corp.), 5 мл из 10× буфер PCR (Promega Corp.) и 2.5 единиц Taq полимеразы (Boehringer Mannheim). Цепная реакция полимеразы проводилась в 35 циклов (1 минута при 94°С, 1 минута при 65°C и 1.5 минут при 72°С), после чего проводилось инкубирование в течение 7 минут при 72°С. Праймер чувства ZC7423 (SEQ ID NO:21) включал в себя последовательность из регионов, соответствующих эксонам 4 и 5 человеческой ТРО ДНК. Праймер античувства ZC7421 (SEQ ID NO:23) включал в себя последовательность из региона, соответствующего некодирующей 3’ последовательности человеческого гена, и включал в себя кодон окончания трансляции.
Амплифицированные продукты PRC анализировались прямым секвенсированием ДНК и были субклонированы в pGEM-T (Promega Corp.) для проведения дальнейшего анализа путем сравнения с последовательностью мышиной сДНК и с человеческой геномной последовательностью. Последовательность ДНК, кодирующая человеческий ТРО, показана на SEQ ID NO:18, а кодированная аминокислотная последовательность показана на SEQ ID NO:19. Анализ последовательности показывает, что расщепление сигнального пептида встречается в аминокислоте 22 (SEQ ID NO:19), а зрелый протеин начинается в аминокислоте 22 (SEQ ID NO:39).
Фрагменты человеческих N-окончаний и С-окончаний вырезались из pGEM-T, как фрагменты EcoRI-Kpnl и лигировались в сайт EcoRI экспрессирующего вектора Zem229R. Этот плазмид трансфектировался в ВНК 570 клетки с помощью Lipofectamine tm (GIBCO BRL). Спустя 24 часа после трансфекции среда культивирования была заменена на свежую среду и клетки инкубировались в течение 48 часов в отсутствии селективных агентов. Кондиционированная среда исследовалась на предмет пролиферативной активности с помощью линии клеток BaF3/MPLR1.1, как описывалось ранее. Результаты ясно показали, что человеческий ТРО в среде культивирования стимулирует пролиферацию клеток BaF3, экспрессирующих мышиные MPL рецепторы.
сДНК делали из mРНК человеческой печени и почек (полученных из Clontech Laboratories, Inc.) с помощью обратной транскриптазы SUPERSCRIPT tm (GIBCO BRL) в соответствии со спецификацией изготовителя. Затем получали клоны ДНК человеческих ТРО, отведенных от печени и почек, с помощью 2 PCR реакций (условия указаны в Таблице 6). Реакции проводились в 35 циклов при 94°С в течение 1 минуты, 58°С в течение 1 минуты. 72°С в течение 1.5 минуты; затем следовало инкубирование в течение 7 минут при 72°С.

Продукты PCR обрабатывались фенолом/хлороформом/изоамилом алкоголя и осаждались с 95Ж ЕТОН, высушенным и переведенным во взвешенное состояние в 20 мл H2O. Затем каждый продукт разрезался ристрикционными ферментами Аsр718 и EcoRI и подвергался электрофорезу на 1% геле агарозы. Фрагменты длиной 410 т.п.н. (печень или почки) из реакции №1 и фрагмент в 699 т.п.н. (печень и почки) из Реакции №2 вырезались из геля и элютировались центрифугированием пластинок геля через нейлоновую шерсть. PCR продукты Реакции №l и Реакции №2 лигировались вместе с вектором Zem229R (депозитирован в Американском Собрании Культур Типа, 12301 Parklawn Drive, Rockvilie, MD 28 сентября 1993 года с каталожным номером 69447), который был отрезан EcoRI, благодаря чему соединил эти два продукта в созданном сайте Asp718. Получаемые плазмиды обозначались №10 (в соответствии с сДНК, отводимой от почек) и №28 (содержащий сДНК, отведенную от печени).
После секвенсирования ДНК были обнаружены единичные вызванные PCR ошибки на 5’ и 3’ от уникального сайта AvrII в плазмидах №28 и №10 соответственно. Для создания ДНК ТРО фрагмент 5’ от EcoRI-AvrII из 826 т.п.н. изолировался от №10 и фрагмент 3’ от AvrII-EcoRI был изолирован от №28. Эти два фрагмента лигировались вместе с вектором Zem 229R, который был отрезан EcoRI. Получаемый плазмид обозначался pZGmpl-124. Этот плазмид был депозитирован в Американском Собрании Культуры Типа, 12301 Раrklawn Drive, Rockville, MD 4 мая1994 года в качестве трансформанта с каталожным номером 69615.
ПРИМЕР XI. БИБЛИОТЕКА сДНК МЕГАКАРИОЦИТА
Для амплификации предшественников мегакариоцитов in vivo 20 мышам вводили в брюшную полость 40,000 единиц активности (единиц, определяемых, как 500/мл, для получения половины максимальной скорости пролиферации клеток ВаF3/MPLR1.1 при анализе МТТ (Пример VII) рекомбинантного мышиного тромбопоэтина ежедневно (концентрированные безсывороточные кондиционированные среды из ВНК 570 клеток, стабильно транфектировали с сДНК мышиного тромбопоэтина). На пятый день инъекций удалялись селезенки и помещались в CATCH буфер+HEPES (сбалансированный соляной раствор Хенка (HBSS), свободный от кальция и магния, 10 мМ Hepes (GIBCO BRL), 1.4 мМ аденозина, 2.74 мМ теофилина (Sigma Chemical Со., St.Louis, МО) и 0.38% цитрата натрия (J.Т.Baker Inc., Philipsburg, NJ) с рН, отрегулированным до 7.40 с гидроокисью натрия). Пять селезенок обрабатывались одновременно, на них делались надрезы, извлекая клетки между двух сит из нержавеющей стали в смесь буфера GATCH+Hepes. После разбивания скопления клеток 25 мл-й пипеткой объем увеличился до 50 мл, и клетки сплетались в течение 7 минут в центрифуге Sorval TJ-6 при 208×g. Каждый клеточный осадок переводился вновь во взвешенное состояние в 10 мл смеси CATCH буфера+Hepes и фильтровался через 130 мм-е найлоновое сито для получения единой клеточной суспензии. Объемы были увеличены до 50 мл благодаря CATCH буфер+Hepes, и клетки сплетались в течение 15 минут при 33×g. Эти клетки промывались в дополнительных 50 мл CATCH+Hepes и сплетались в течение 10 минут при 33×g. Клеточные осадки вновь переводились во взвешенное состояние в 10 мл CATCH буфера+Hepes и разделялись на слои при трехступенчатом Percoll (Pharmacia LKB Biotechnology AB, Uppsala, Швеция) градиенте (65, 40 и 27% в 1 x CATCH буфер+Hepeы, по 12 мл каждого в 50 мл трубки центрифуги) и центрифугировались в течение 45 минут при 833×g. Клетки между 40 и 63% слоями Percoll собирались, и объемы увеличивались до 50 мл за счет CATCH буфера+Hepes. Клетки сплетались в течение 7 минут при 208×G и вновь переводились во взвешенное состояние в 50 мл сред роста мегакариоцитов (минимальная существенная альфа-модификация среды), свободная рибонуклеозида и деокси рибонуклеозида с 15% инактивированной фетальной телячьей сыворотки, 2 мМ L-глютамина (компоненты сред получены от JRH Biosciences, Lenexa, КБ), 1 мМ пирувата натрия (Irvine Acience, Santa Ana, CA), 1 × PSN смесь антибиотиков (GIBCO BRL) и добавлялось 1,000 единиц активности рекомбинантного мышиного тромбопоэтина/мл (безсывороточные кондиционированные среды из ВНК 570 клеток стабильно трансфектировались с сДНК мышиным тромбопоэтином). Затем клетки высевались на 150 мм тарелки для культивирования ткани при 106 мононуклеотидных клеток/мл и выращивались в полностью увлажненном инкубаторе с содержанием 6.0% СО2 в воздухе при 37°С. После трех суток роста неприлипшие клетки собирались в 50 мл трубку центрифуги и охлаждались на льду. Большие клетки высевались центрифугированием при 33-g в течение 15 минут при 4°С. Клеточный осадок вновь переводился во взвешенное состояние в 50 мл CATCH буфера+Hepes при комнатной температуре и сплетались в течение 10 минут при 33×g. (Все дальнейшие стадии выполнялись при комнатной температуре). Эта промывка повторялась вновь для достижения более высокой степени чистоты созревших мегакариоцитов. Оставшиеся клетки переводились вновь во взвешенное состояние в 15 мл САТСН буфера+Hepes (объем группировки) и раскладывались на слои по трем ступенчатым градиентам фетальной бычьей сывротки (JRH Biosciences) (65% и 40% САТСН буфера+Hepes) седиментации (отстаивания) при 1×g в течение 30 минут. Нижние 5 мл 64% фракций группировались, разбавлялись до 50 мл смесью CATCH буфер+Hepes и переплетались в течение 10 минут при 33×g. Осадок содержал более, чем 107 клеток. Клетки исследовались на ацетилхолинестеразу методом Бурштейна и др. (J.Cell.Physiol.122:159-165, 1985), и оказалось, что это зрелые мегакариоцитные клетки с чистотой более чем 00%. Затем осажденные клетки лизированы (разрушены) в растворе тиоцината гуанида/2-меркаптоэтанола для изолирования РНК с помощью центрифугирования градиента плотности хлорида цезия.
сДНК приготовляется из мегакариоцитной РНК, как описано в выше Примере VI.
ПРИМЕР XII. ФЛЮОРЕСЦЕНЦИЯ in situ ГИБРИДИЗАЦИОННОГО КАРТИРОВАНИЯ ЧЕЛОВЕЧЕСКОГО ТРОМБОПОЭТИЧЕСКОГО ГЕНА
К 1.5 мл трубкам микроцентрифуги на льду было добавлено следующее: 1 мг лямбдаZGmрl-Н8, лямбдаZGmрl-Н10 или лямбдаZGmрl-Н29,содержащие человеческий тромбопоэтический ген, 5 мл 10×буфер ник-трансляции (0.5 М Трис/HCl, 50 мМ MgCl2, 0.5 мг/мл BSA (без нуклеазы), 5 мл раствора dNTP, содержащих 0.5 мМ dATP, 0.5 dGTP и 0.5 мМ dCTP, 5 мл 5мМ Bio-11-dUTP (5-(N-{N-биотинил-е-аминокаприл-3-аминоалил)-2’-деоксиуридин 5’-трифосфат, Sigma Chemical Co.), 5 мл 10 мМ DTT, 5 мл DNase I (1000× разбавление из 10U/мл запаса, Boehringer Mannheim, без RNase), 2.5 мл полимеразы I ДНК (5 U/мл, Boehringer Mannheim), Н2О до окончательного объема 50 мл. После смешивания реакции инкубировались при 15°С в течение 2 часов в микроохладителе Бокела. Реакции прекращались добавлением 5 мл 0.5 М ЕDTA, рН 7.4. Зонды очищались с помощью колонок для очистки ДНК Sephadex G-50 (Worthington Biochemical Corporation, Freehold, NJ) в соответствии с указаниями изготовителя. Для контроля размера меченных зондов по 5-10 мл каждого очищающего зонда смешивались с 5 мл загружающего гель буфера (12.5% фиколла, 0.2% бромфенола синего, 0.2 М Трис-ацетата, 0.1 М ацетата натрия, 1 мМ EDTA) и затем обрабатывались 0.7% мингеля агарозы при напряжении 80 В, Фрагменты лямбда-Hind III (GIBCO BRL) и фрагменты ФХ-Нае III (GTBCO BRL) использовались в качестве маркеров размеров основных пар (т.п.н.). Центромерный зонд, меченый дигоксигенином, характерный для хромосомы 3 (D3Z1),получался из Oncor (Gaithersburg, MD).
Метафазные хромосомы были получены из клеточной культуры Hell. 100 мл Colcemid (GIRCO BRL, 10 мг/мл штамм) добавлялось к средам в 100×15 мм чаше Петри, используемым для культивирования клеток, и инкубировались при 37°С. Спустя 275-3 часа эти среды удалялись и переносились в 15 мл полипропиленовую коническую трубку (Blue Max tm, Becton Dickinson). 2 мл 1× PBS (140 мМ NaCl, 8 мМ КСl, 8 мМ Na2HPO4, 1.5 мМ КН2РO4, рН 7.2) было добавлено в чашу Петри для промывки с помощью 5 мл стерильной пластиковой пипетки, а затем все это было перенесено в коническую трубку. 2 мл трипсина (GIBCO BRL, раствор штамма) добавлялось в чашу Петри с помощью стерильной 5 мл пластиковой пипетки, затем эту чашу Петри слегка потрясли и поместили в инкубатор при 37°С на 3-5 минут. Затем клетки смывались с чаши Петри с помощью 5 мл стерильной пипетки и добавлялись в трубку со средами. Трубка для культивирования центрифугировалась при 250×g в течение 8 минут и все, кроме 0.5 мл надосадочной жидкости, удалялось. Осадок вновь переводился во взвешенное состояние путем постукивания, затем осторожно и медленно туда доливали 8 мл 0.075 М КCl (предварительно нагретого до 37°С). Эта суспензия осторожно перемешивалась и помещалась в водяную ванну при 37°С на 10 минут. Раствор центрифугировался при 250×g в течение 5 минут и все, за исключением 0.5 мл надосадчной жидкости, удалялось. Осадок переводился во взвешенное состояние постукиванием по трубке. Два мл холодной метанол:ацетатной кислоты (3:1) добавлялось по капле при потряхивании для фиксирования (затвердевания) клеток. Таким образом, в целом было добавлено 8 мл. Трубку помещали в рефрижераторе на 20 минут, затем следовали 5 минут центрифугирования при 250×g. Надосадочная жидкость отсасывалась и процесс фиксирования повторялся еще два раза. Для нанесения капель метафазы на 25×75 мм предварительно очищенные, замороженные стеклянные пластины (VWR Scientific, Media, PA) на каждую пластину наносили 5 мл 50% уксусную кислоту с помощью 20 миллилитровой Pipetman tm (Gilson Medical Electonics, Inc., Middleton, WI), после чего переводили 5 мл клеток в суспензию. Пластины оставлялись высыхать на воздухе, а затем старели в течение ночи при 42°C в печи (Вoekel Industries, Inc., Philadelphia, PA) перед употребелением. Пластины проверялись на соответствующее разрастание метафаз с помощью микроскопа, снабженного фазовым контрастным конденсером. Некоторые препараты метафазных хромосом были исчерчены G-полосой краской Гимеза R66 (BDH Ltd., Dorset, Англия), сфотографированы и очищены от краски перед употреблением в экспериментах по гибридизации. Препараты на пластинах с разросшимися человеческими метафазными хромосомами инкубировались в течение 2 часов в 2×SSC (0.3 М NaCl, 0.03 М цитрат натрия, рН 7.0), быстро прополаскивались в H2O и окрашивались краской Гурра-Гиемза, которая была разбавлена в пропорции 1:4 в растворе буфера Гиемза, рН 6.5 (BDH Ltd) и фильтровалась перед употреблением через фильтр Вотмана №1. Некоторые препараты инкубировались сначала в течение 45 минут - 1 часа в печи при 90°С и оставлялись остывать перед инкубированием в SSC. Эти препараты затем дифференцировались в растворе буфера Гиемза, прополаскивались в H2O и просушивались на воздухе. Соответствующие разрастания метафазного хромосома, исчерченного G-полосой, фотографировались на микроскопе OlympuS с помощью пленки на 400 слайдов kodak Ektachrome tm и пронумеровывались и сохранялись на цветной видео-системе Optronics (Goleta, CA) ZVS-47E CCD RGB и аппаратуры Optimus (BioScan Inс., Edmonds, WA). Препараты очищались от краски в течение примерно 20 минут в 100% EtOh и просушивались на воздухе перед дальнейшим употреблением. Неиспользованные препараты на пластинах метафазного хромосома хранились при -70°С.
Гибридизационные смеси готовились в 1.5 мл стерильных трубках микроцентрифуги, комбинируя 2.5 мг конкурентной ДНК (Cot-1 ДНК, GIВСО BRL), 40-60 нг меченных биотином фагов лямбдаZGmрl-Н8, лямбдаZGmрl-Н10 или лямбдаZGmрl-Н29, (содержащих человеческий тромбопоэтический ген), 7 мг ДНК носителя (ДНК денатурированного лосося, Sigma Chemical Co.), 1 мл 3 M NaAc и 2 объема этанола просушивались в вакууме в вакуумном концентраторе. Осадок разбавлялся в 10 мл гибридизационного раствора, содержащего 10% сульфата декстрана, 2×SSC и 50% формамида (ЕМ Science, Gibbstown, NJ). Этот зонд и конкурентная дНК денатурировались при 70-80°С в течение 5 минут, охлаждались на льду и проходили предварительный отжиг при 37°С в течение 1-2 часов. Хромосомы денатурировались погружением каждой пластины в 70% формамид, 2×SSC при 70-80°С в течение 5 минут, после чего следовало немедленное охлаждение в остуженном льдом 70% этаноле, затем в 100% этаноле в течение 5-10 минут каждая. Затем эти пластины просушивались на воздухе и нагревались до 42°С непосредственно перед нанесением на них с помощью 20 мл Gilson Pipetman пипетки гибридизирующих смесей. Эти гибридизирующие смеси и хромосомы затем покрывались покровными стеклами размером 18×18 мм, №1 (VWR Scientific). Гибридизация проводилась во влажной камере в течение ночи при 37°С. В некоторых случаях после примерно 6 часов гибридизации 5-10 нг денатурированного, меченного дигоксигенином D3Z2 центромерного зонда (в гибридизационном растворе 10% сульфата декстрана, 2×SSC и 65% формамида) добавлялось в препараты.
После удаления покровных стекол пластины промывались 3×5 минут в 50% формамиде, 2×SSC при 42°С, 3×5 минут в 2×SSC при 42°С и 1-3 минуты в 4×SSC, 0.05% полиоксиэтиленсорбитан монолаурата (Tween-20, Sigma Chemical Co.). После этого шло предварительное инкубирование в течение 20 минут с 4×SSC, содержащим 5% сухого обезжиренного молока во влажной камере (100 мл под покровным стеклом размером 24×50 мм). Для препаратов, в которые входил центрометрический хромосом 3D3Z1, проводилось 45 минутное инкубирование при разбавлении в пропорции 1:100 меченным биотином мышиным антидиоксином (Sigma Chemical Со.) в 4×SSX/5% BSA, после шли 3 промывания по три минуты в 4xSSC, 0.05% Tween-20. Затем проводились стадии послегибридизации для всех препаратов при 20 минутном инкубировании савидином, меченным флюорисценцией (Fluorescein Avidin DCR, Vector Laboratories, Burlingame, CA) (100 мл, 5 мг/мл в 4×SSC, 5% обезжиренного сухого молока) под покровным стеклом размером 24×50 мм. Затем пластины промывались 3×3 минуты в 4×SSC, 0.05% Tween-20, затем шло 20-минутное инкубирование сбиотинилированным козьим антиавилином D (аффинная очистка,vector Laboratorues) (5 мг/мл в 4×SSC, 5% сухого обезжиренного молока) под покровным стеклом размером 24×50 мм. Эти пластины промывались опять 3×3 минуты в 4×SSC, 0.05% Tween 20, после чего шло еще одно инкубирование с авидином, меченным флуорисцином (100 мг/мл в 4×SSC, 5% сухого обезжиренного молока) под покровным стеклом размером 24×50 мм. В некторых случаях процедура сигнальной амплификации повторялась еще один раз. Окончательные промывки продолжались 2×3 минуты в 4×SSC, 0.05% Tween-20 и 1×3 минут в 1×PBS. Пластины устанавливались в противообесцвечивающей среде, состоящей из 9 частей глицерола, содержащего 2% 1,4-диазобицикло(2,2,2)-октан (DABCO, растворяемый при 70°С) и одну часть 0.2 М Трис/HCl, рН 7.5 и 0.25-0.5 мг/мл иодида пропида. Пластины просматривались в микроскоп Olympus BH2, снабженный приставкой для отраженного флюоресцентного света BH2-RFC, автоматической фотомикрографической системой, системой цветной видео-камеры ZVS-47E CCD RGB и красным фильтром FITC/Texas (Brattlebow, VT) производства СРНrоmа Technology Corp. для FITC визуализации. Изображения разрастаний метафазного хромосома были пронумерованы и сохранялись в системе камеры видео изображения и в аппаратуре Optimus.
Предварительные результаты процедуры физического картирования показали, что локус человеческого тромбопоэтического гена является дистальным на q плече хромосома 3 в регионе 3q26.
ПРИМЕР XIII. ЭКСПРЕССИЯ ЦИТАКИННОЙ ОБЛАСТИ МЫШИНОГО ТРО В Sacharomyces cerevisiae
Плазмид pBJ3-5 содержит промотор S. cerevisiae ТРII, секреторный лидер а-фактора, мышиный ТРО, кодирующий последовательность (SEQ ID NО:1) от 237 т.п.н 7 до 692, транскрипционный терминатор ТРII<2 м последовательности для репликации в дрожжах и ген изомеразы фосфата триозы Schizosaccharomyces pombe (POTI ген) для селекции в дрожжах. Этот плазмид сконструирован для прямой секреции протеина мышиного ТРО, содержащего аминокислоты 45-196 из SEQ ID NO:2.
Для построения рBJ3-5, pMVRl (Фигура 2) поглощался вместе с SphI и XbaI, и каркас вектора, содержащий 5’ часть промотора ТРI1 и терминатора ТРI1, был восстановлен. Затем в этот каркас вектора были вставлены следующие фрагменты:
1) Фрагмент SphI/Hind III, отводимый от рВS114, который содержит 3’ часть промотора ТРI1 и лидер а-фактора. Плазмид pBS114 представляет собой дрожжевой челночный вектор, который содержит промотор ТРI1 и лидер а-фактора, после которого полилинкерная последовательность, которая включает сайт Hind III.
2) HindIII/Sall фрагмент, созданный PCR, содержащий сайт Hind III, сконструированный так, что он соответствует сайту Hind III в лидере а-фактора, сайт протеолитического расщепления Кех2 и последовательность мышиного ТРО от 237 т.п.н. до 335 т.п.н. из SEQ ID NО:1.
3) Фрагмент Sall/EcoRI, содержащий основные пары мышиного ТРО от 336 до 692 из SЕQ ID NO:1, который отводился от плазмида pZGmpl 1-1 081 с помощью праймеров ZC319 (SEQ ID NO: 27) и ZC7318 (SEQ ID NO:26), поглощающий с Есо RI и клонирующий фрагмент, содержащий последовательность цитокинной области ТРО и 5’ некодирующая последовательность в сайт Есо RI из Zem229R {ATCC 69447}. Этот фрагмент изменялся в фрагмент Sall/Xbal клонированием его в рIС19Н, который сначала поглощался Sall и EcoRI.
Получаемый плазмид, обозначенный pBJ3 (Фигура 2), затем поглощался BgIII и XhoI для выделения в свободном состоянии полной экспрессионной кассеты, содержащей промотор, лидер, последовательность, кодирующую ТРО и терминатор. Этот фрагмент BgIII/Xhol вставлялся в pRPOT (раскрыто в Патенте США №5,128,321, который включен в данный документ в качестве ссылки), который поглощался BamHI и Xhol. Результирующий плазмид обозначался pBJ3-5.
S.cerevisiae штамм JG134 (МАТа urа3-52 lеu2-А2 рер4-А1 atpil:URA3 {fcir о} преобразовывался с помощью pBJ3-5 и pRPOT с помощью обработки ацетатом лития (как в общих чертах раскрыто у Ито и др., J.Bacteriol. 153:163-168, 1983). Трансформанты отбирались по росту на глюкозосодержащей среде. JG134/pBJ3-5 и JG134/pRPOT выращивались в жидкой YEPO среде в течение трех суток. Среда культивирования отделялась от клеток центрифугированием и анализировалась путем исследования пролиферации клеток в ВаF3 клетках, содержащих MPL рецептор. Среда из штамма JG134/pBJ3-5 содержала 5000-7000 единиц/мл ТРО активности, в то время как негативный контроль JG134/pRPOT не имел активности. Этот результат указывает на то, что дрожжи могут выделять биологически активную форму ТРО.
ПРИМЕР XIV. АКТИВНОСТЬ РЕКОМБИНАНТНОГО ЧЕЛОВЕЧЕСКОГО ТРО
ДНК плазмида из двух 5 мл бактериальных культур, культивированных в течение ночи, трансформированных с pZGmpl-124, приготаливалась с помощью лизиса клеток алкалина, после чего ДНК связывалась со смолой при высоком содержании соли (с помощью набора пробоотборника Magic Minipreps tm от Propega Corp.). ДНК элютировалась с 75 мл 10 мМ Трис, 1 мМ EDTA, рН 8,0.
Культуры клеток ВНК 570 при 50,000 клетка/лунку были трансфектированы с ДНК pZGmpl-124. 20 мл при разбавлении LIPOFECTAMINE tm (GIBCO BRL) в пропорции 1:10 добавлялись к 20 мл ДНК плазмида и 160 мл безсывороточных сред (среды F/DV {смесь в пропорции 1:1 DMEM и F12 Наm} при добавлении 10 мг/мл фетуина, 2 нг/мл селениума, 5 мг/мл инсулина, 10 мг/мл трансферина, 2 мМ L-глютамина, 110 мг/мл пирувата натрия, 25 мМ HEPES и 0.1 мМ раствора несущественной аминокислоты (GIBCO BRL) в течение 30 минут при комнатной температуре перед добавлением к клеткам ВНК 570 и инкубированием в течение 4 часов при 37°С. 200 мл Сред Роста (DMEM (Biowhittaker), дополненные 2 мМ L-глютамина, 110 мг/мл пирувата натрия, 0.05 мг/мл пеницилина, 0.05 мг/мл стрептомицина, 0.01 мг/мл неомицина, 25 мМ HEPES, 10% фетальная телячья сыворотка) и эти клетки инкубировались при 37°С в течение ночи. Среды культивирования затем заменялись Средой Роста, содержащей 5% фетальной телячьей сыворотки, и инкубировались при 37°С в течение 4 часов.
Кондиционированные среды из трансфектантов ВНК 570 затем исследовались на способность вызывать пролиферацию клеток в клетках BaF3, экспрессирующих мышиный MPL рецептор. Эти клетки выращивались в средах BaF3 (RPMI 1640 среды (JRH Biosciences), дополненных 10% фетальной телячьей сыворотки, 2 мМ L-глютамина, 1 мМ пирувата натрия, 10 мМ HEPES, 57 мМ бета-Меркаптоэтанола, 0,5 мг/мл пеницилина, 0,5 мг/мл стрептомицина, 0,1 мг/мл неомицина и 4% V/V кондиционированные среды из культур клеток WEHI-3 (мышиный интерлейкин-3, дополнение к культуре. Совместно изготовленные Биомедицинские Продукты)). До исследований клетки ВаF3 разбавлялись и вновь переводились во взвешенное состояние в среде ВаF3, свободной от IL-3, до 10.000 клеток/100 мл кондиционированной среды из клеток ВНК 570, трансфектированных pZGmpl-124, и эти культуры инкубировались при 37°С. Затем эти клетки исследовались низуально на удлинение клеток после 30 минут и после 24 часов. Негативный контроль, состоящий из среды ВаF3 без IL-3, и позитивный контроль кондиционированной среды из клеток ВНК 570, трансфектированные с ДНК мышиного ТРО, также исследовались. Результаты исследований показали, что не было ни одного удлинения клеток в клетках ВаF3 в негативном контроле, несколько удлинений клеток было в позитивном контроле и значительное удлинение клеток наблюдалось в клетках, трансфектированных pZGmpl-124.
ПРИМЕР XV. АФФИННОЕ ОСАЖДЕНИЕ РЕЦЕПТОРОВ
150-мм чаши для культивирования ткани, содержащие клетки, продуцирующие ТРО или нормальные ВНК клетки были помечены в течение 18 часов 10 мл MEM Дулбеко без метоинина, содержащего 2 мМ L-глютамин, антибиотики и 200 мСi из 35S-Express (Amersham, Arlington Heghts, IL).
После инкубирования в течение ночи использованные среды собирались и концентрировались 15 раз с помощью концентратора Centriprep-10tm (Amicon, Inc.). Полученную 0.7 мл концентрированную надосадочную жидкость смешивали с 40 мл растворимого MPL рецептора с окончанием в виде поли-гистидина, который был связан с CNBr-Sepharose 4В (Pharmacia), что указывалось поставщиком. Смесь инкубировалась в течение 2 часов на льду при потряхивании.
Клетки промывались один раз PBS, затем разрушались 1 мл буфера RIP А (10 мМ Трис, рН 7.4, 1% деоксихолата, 1% Triton Х-100, 0.1% SDS, 5 мМ EDTA, 0.15 M NaCk). Лизаты центрифугировались для удаления нерастворимых веществ, и добавлялось 10 мл MPL-Sepharose, как указывалось ранее.
Затем MPL-Sepharose осаждалась при малой скорости центрифуги, и использованные среды и надосадочная жидкость клеточного лизата удалялись. Осадок промывался четыре раза в PBS, содержащем 0.5 M NaCl. После последней промывки PBS удалялся и добавлялся 40 мл 2× пробный буфер (10% глицерола, 4% SDS, 50 мМ Трис, рН 7.0, 1 мМ EDTA, 0.05% бромфенола синего), содержащий 4% бета-меркаптоэтанола.
Пробы кипятились в течение 5 минут, и по 18 мл загружалось на ини-гель с 10-20% градиентом (Интегральная Сепарационная Система), затем подвергались электрофорезу при 100 В примерно в течение 2 часов. Гель фиксировался в течение 30 минут (в 40% метанола, 16% глациала уксусной кислоты в дистиллированной воде), затем отсасывался в AMPLIFY tm (Amersham) в течение 20 минут. После просушки гель экспонировался перед пленкой в течение ночи. Полоса -70 KD была очень заметна в промежутке, соответствующем использованным средам из клеток, трансфектированных сДНК ТРО. Эта полоса не присутствует в использованных средах из клеток ВНК или является лизатами клеток из каждой линии клеток.
Из всего сказанного можно понять, что хотя конкретные примеры использования изобретения описаны здесь в целях иллюстрирования, различные модификации можно проводить, не отклоняясь от сути и духа данного изобретения. Соответственно, данное изобретение не ограничивается ничем, кроме прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЙ ЦИТОКИН ZALPHA11-ЛИГАНД | 2000 |
|
RU2258710C2 |
| НОВЫЙ ЛИГАНД РЕЦЕПТОРА ЦИТОКИНА ZCYTOR17 | 2003 |
|
RU2490276C2 |
| ТРОМБОПОЭТИН | 1994 |
|
RU2245365C2 |
| ДЕГРАДИРОВАННОЕ АНТИТЕЛО, ЯВЛЯЮЩЕЕСЯ АГОНИСТОМ TPO | 2001 |
|
RU2287534C2 |
| СПОСОБЫ И ПРЕПАРАТЫ ДЛЯ СТИМУЛИРОВАНИЯ РОСТА И ДИФФЕРЕНЦИРОВКИ МЕГАКАРИОЦИТОВ | 1995 |
|
RU2158603C2 |
| МОДИФИЦИРОВАННОЕ АГОНИСТИЧЕСКОЕ АНТИТЕЛО | 2001 |
|
RU2295537C2 |
| ГЕМОПОЭТИЧЕСКИЙ БЕЛОК (ВАРИАНТЫ), ПОСЛЕДОВАТЕЛЬНОСТЬ ДНК, СПОСОБ ПОЛУЧЕНИЯ ГЕМОПОЭТИЧЕСКОГО БЕЛКА, СПОСОБ СЕЛЕКТИВНОЙ ЭКСПАНСИИ КРОВЕТВОРНЫХ КЛЕТОК, СПОСОБ ПРОДУЦИРОВАНИЯ ДЕНДРИТНЫХ КЛЕТОК, СПОСОБ СТИМУЛЯЦИИ ПРОДУЦИРОВАНИЯ КРОВЕТВОРНЫХ КЛЕТОК У ПАЦИЕНТА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ | 1997 |
|
RU2245887C2 |
| ВЫДЕЛЕННЫЙ РАСТВОРИМЫЙ IL-20 РЕЦЕПТОР (ВАРИАНТЫ) | 2000 |
|
RU2279441C2 |
| АНТИТЕЛА К ЧЕЛОВЕЧЕСКОМУ IL-1БЕТА | 2001 |
|
RU2286351C2 |
| ПОЛИПЕПТИД ТРОМБОПОЭТИН (ТРО), ДНК, КОДИРУЮЩАЯ ПОЛИПЕПТИД ТРО (ВАРИАНТЫ), СПОСОБ ПОЛУЧЕНИЯ ПОЛИПЕПТИДА (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ (ВАРИАНТЫ), АНТИТЕЛО К ПОЛИПЕПТИДУ ТРО | 1995 |
|
RU2194074C2 |
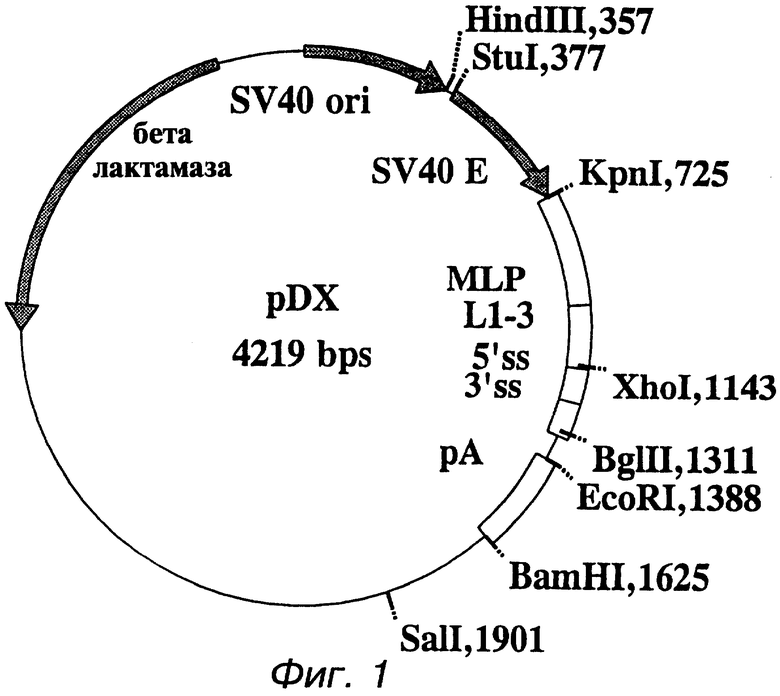















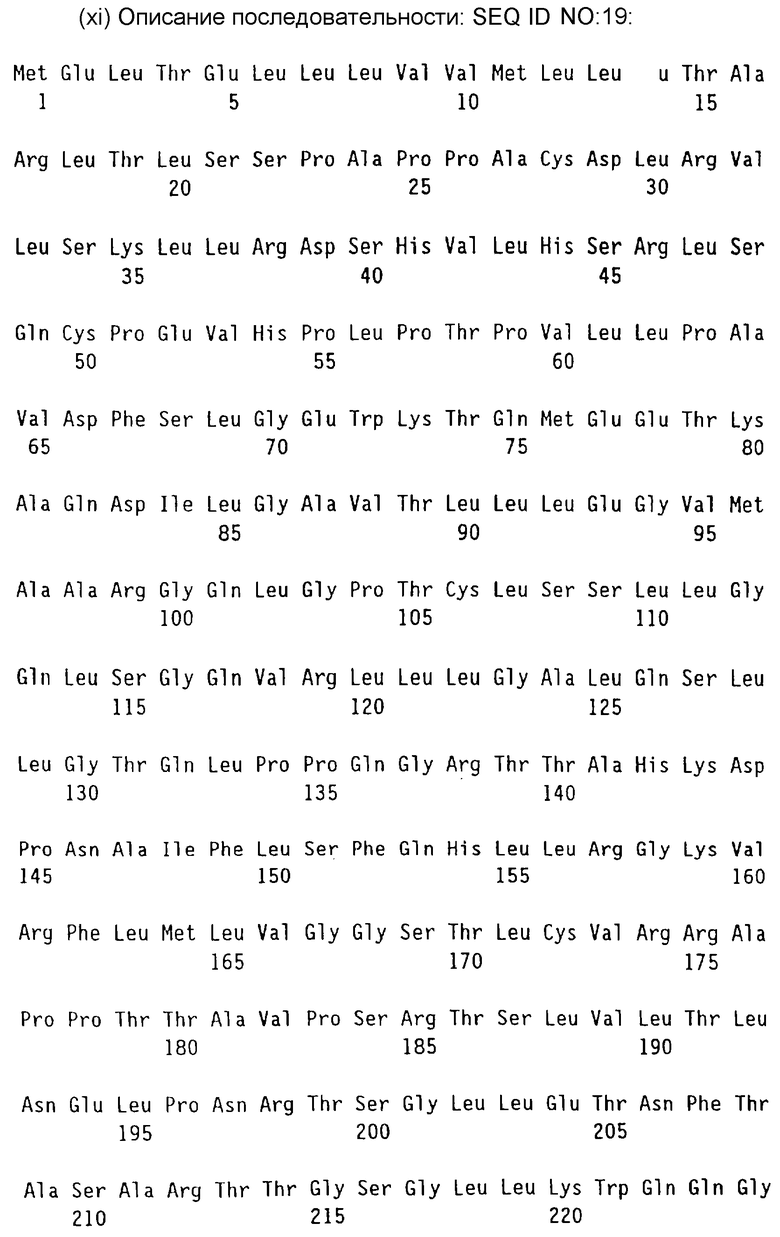











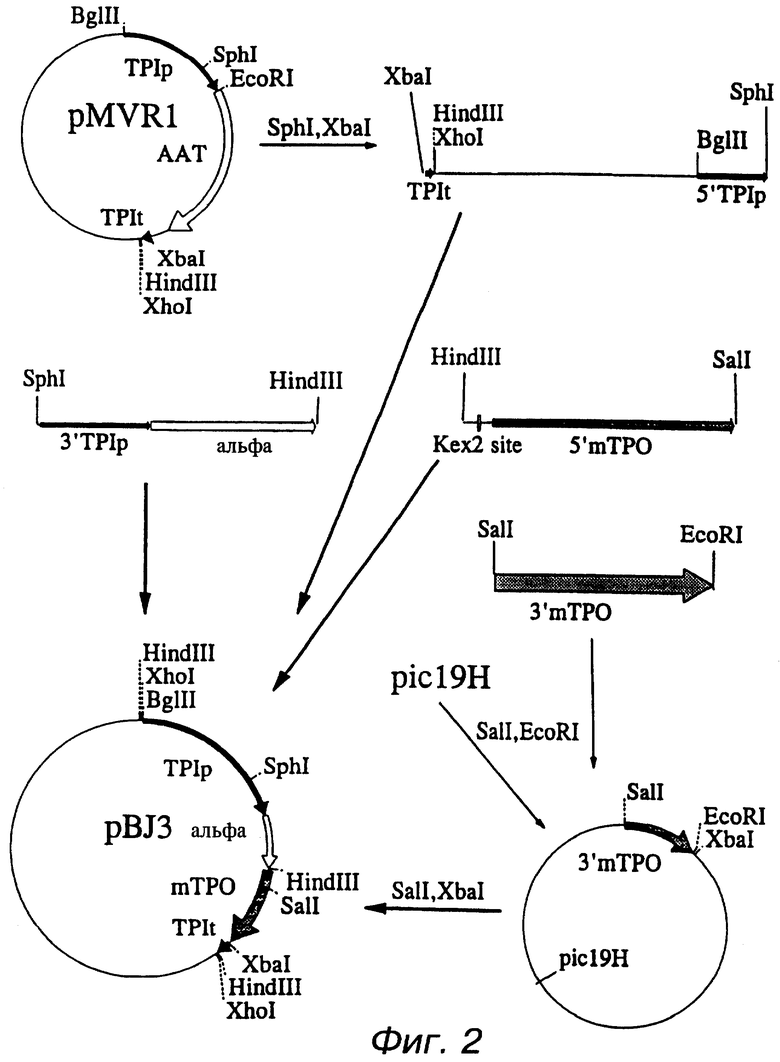
Изобретение относится к области генной инженерии и может быть использовано в медико-биологической промышленности. Путем скрининга кДНК-библиотеки мыши изолирована нуклеотидная последовательность, кодирующая белок(тромбопоэтин), обладающий способностью стимулировать MPL-зависимую пролиферацию или дифференцировку предшественников клеток миелоидного или лимфоидного ряда. Сконструированы векторы для экспрессии полученной кодирующей последовательности в клетках дрожжей и млекопитающих. Культивирование эукариотических клеток хозяев, трансформированных предложенными экспрессирующими векторами, позволяет получить значительные количества рекомбинантного тромбопоэтина. 8 с. и 5 з.п. ф-лы, 2 ил., 6 табл.
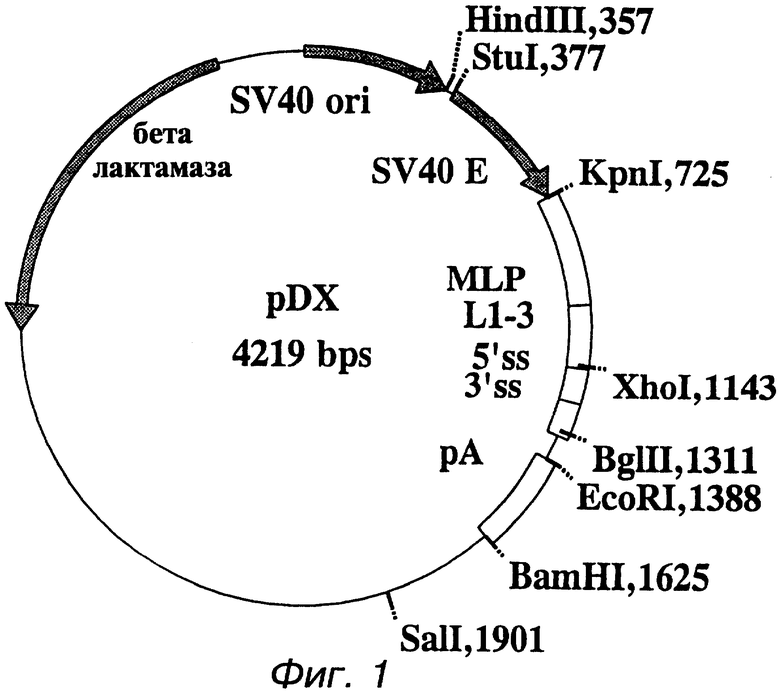
Приоритет по пунктам
| МС DONALD Т.Р | |||
| et al | |||
| Experimental Hematology, v.17, 8, р.865-871, 1989 | |||
| MC DONALD T.P | |||
| et al | |||
| Experimental Hematology, v.16, 3, р.201-205, 1988 | |||
| CHEMICAL ABSTRACTS, v.113, no 19, abst.no 170137u, 1990 | |||
| CHEMICAL ABSTRACTS, v.91, no 21, abst | |||
| БУНКЕРНОЕ УСТРОЙСТВО | 0 |
|
SU186949A1 |

