Изобретение относится к физическим методам анализа химического состава вещества, в частности к рентгенофазовому методу, и может быть использовано в различных отраслях промышленности, при исследовании минерального сырья, горных пород и почв, при определении концентраций минералов, составляющих анализируемое вещество. Особенно интересен данный метод в связи с широкими экспериментальными исследованиями по моделированию процессов минералообразования, где данные количественного фазового анализа позволяют судить об изменении условий осадконакопления, помогают оценивать перспективность отложений для накопления и сохранения нефти и газа.
Известны следующие способы определения концентрации при рентгенофазовом анализе.
Метод внутреннего стандарта [1]. Сущность метода состоит в определении концентрации искомой фазы хi по отношению интенсивностей аналитических линий этой фазы и добавляемой в анализируемую пробу в постоянной концентрации фазы внутреннего эталона (хэ=const).
Интенсивность отражения фазы х1 в смеси с внутренним эталоном согласно уравнению:

Для внутреннего эталона
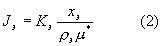
Для отношения интенсивностей получим
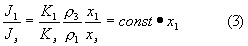
Уравнение (3) означает, что при добавлении в пробу внутреннего эталона в постоянной концентрации содержание искомой фазы x1 пропорционально отношению интенсивностей J1/Jэ. Выявленная здесь прямолинейная зависимость устанавливается по трем искусственным смесям различной концентрации.
Метод внешнего эталона [2]. Попытки избежать необходимости добавления в каждую пробу определенного количества внутреннего эталона и исключение процесса взвешивания и перемешивания пробы и эталона привели к разработке методов, частично или полностью исключающих эту процедуру. В методе используется специальный держатель образца, представляющий собой круглое углубление в плексигласе, заполняемое порошком пробы, в центре которого запрессован металлический стержень. Его торцевая поверхность совпадает с плоскостью образца. При облучении пробы рентгеновским пучком этот стержень дает несколько интенсивных линий и таким образом является эталоном, внешним по отношению к пробе. Предварительная калибровка и получение графических зависимостей проводятся так же, как в методе внутреннего эталона. За счет исключения процедуры взвешивания и перемешивания веществ пробы и эталона сокращается время на анализ каждого образца.
К недостаткам метода относятся ограниченный выбор веществ-эталонов, необходимость переделки стандартного держателя образца, возможность изменения отражающей способности торцевой части эталона при окислении, децентровке или механических повреждениях. Примерное отношение облучаемых поверхностей эталона и анализируемой пробы равно 1:10. Абсолютная погрешность определения концентраций отдельных фаз рассматриваемым методом 2-3%.
В основу изобретения положена задача создания высокоточного и высокоэкспрессного способа определения концентрации фазы в веществах сложного химического состава.
Указанная задача решается тем, что в способе определения концентрации фазы в веществе сложного химического состава, включающем облучение пробы анализируемого вещества монохроматическим гамма- или рентгеновским излучением, регистрацию интенсивности когерентно рассеянного определяемой фазой первичного излучения согласно изобретению одновременно или последовательно с регистрацией указанной интенсивности регистрируют интенсивность некогерентно рассеянного этой же пробой первичного излучения, а затем по отношению указанных интенсивностей устанавливают концентрацию определяемой фазы.
Впервые предложен способ, основанный как на рентгенофазовом, так и на рентгенофлуоресцентном методах анализа.
Определение количественного содержания кристаллических фаз (минералов), входящих в состав исследуемого образца, может быть проведено путем сравнительной оценки интенсивностей дифракционных максимумов на порошковой дифрактограмме. Возможность решения такой задачи основывается на существовании зависимости интенсивности дифракционного максимума от концентрации каждой кристаллической фазы, входящей в состав пробы. В общем случае эта зависимость не является линейной, так как помимо концентрации данной фазы на интенсивность дифракционного максимума влияет коэффициент поглощения образца, зависящий от концентрации всех фаз, то есть матричный эффект.
В предлагаемом способе устранение матричного эффекта достигается тем, что одновременно или последовательно с регистрацией интенсивности когерентно рассеянного определяемой фазой первичного излучения регистрируют интенсивность некогерентно рассеянного этой же пробой первичного излучения, а концентрацию определяемой фазы устанавливают по отношению вышеуказанных интенсивностей.
Теоретические основы метода.
Убыль энергии первичного пучка из-за поглощения в пробе дается выражением
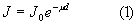
где J0 - интенсивность первичного пучка; J - интенсивность первичного пучка после прохождения слоя вещества толщиной d; μ - линейный коэффициент поглощения, см-1.
Выражение (1) справедливо для строго монохроматического излучения [так как μ=ƒ(λ), где λ - длина волны излучения] и однородного вещества пробы.
В основе всех методов количественного фазового анализа лежит следующее фундаментальное уравнение [3]
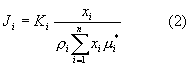
где Ji - интенсивность некоторого выбранного рефлекса фазы i;
Ki - экспериментальная постоянная, зависящая от энергии первичного пучка, от структуры анализируемой фазы, индексов (hkl) и условий съемки; μi * - массовый коэффициент поглощения фазы i.
xi - содержание фазы i в пробе, %; ρi - плотность фазы i.
Массовый коэффициент поглощения не зависит от агрегатного состояния вещества.
При выводе формулы (2) предполагалось, что дифракция происходит от поверхности плоского образца (съемка на отражение), составленного однородной смесью n компонентов, причем толщина образца бесконечна.
В предлагаемом способе показана возможность применения нового, на первый взгляд несколько неожиданного для количественного рентгенофазового анализа способа, сходного со способом стандарта - фона, широко применяемого в рентгенофлуоресцентном анализе [4].
Дифрактограмма представляет собой пики дифракционных максимумов когерентно рассеянного первичного излучения различными фазами вещества анализируемой пробы, наложенные на сплошную линию фона, представляющего в том числе некогерентно рассеянное веществом анализируемой пробы первичное излучение.
Суть данного способа заключается в том, что аналитическим параметром служит отношение Ji - интенсивности измеряемого рефлекса фазы i к интенсивности Js некогерентно рассеянного анализируемым веществом пробы первичного рентгеновского излучения.
Легко увидеть аналогичность выражений (2) для определения Ji интенсивности измеряемого рефлекса фазы i и выражения для определения интенсивности i линии спектра флуоресценции анализируемого элемента, для монохроматического первичного излучения и бесконечно толстой пробы:
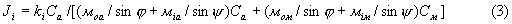
где ki - коэффициент пропорциональности, не зависящий от химического состава пробы;
Са и См - содержание определяемого компонента и элементов наполнителя в пробе (Сa+См=1);
моа и Mia - массовые коэффициенты поглощения первичного и характеристического излучения в определяемом компоненте;
мом и мiм - массовые коэффициенты поглощения первичного и характеристического излучения в наполнителе пробы;
ϕ и ψ - углы падения к поверхности пробы первичного и отбора характеристического излучений.
Так как при снятии дифрактограммы углы падения и отбора равны, а в роли характеристического излучения выступает когерентно рассеянное первичное монохроматическое излучение, то есть моа=мia и мом=мiм, то (3) можно представить как
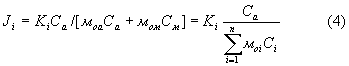
где Ki=кi/sinϕ - коэффициент пропорциональности, зависящий от геометрии съемки, но не зависящий от химического состава пробы.
Полученное выражение полностью коррелирует с выражением (2).
Интенсивность некогерентно рассеянного анализируемой пробой излучения описывается следующим выражением
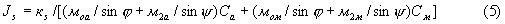
где кs - коэффициент пропорциональности, не зависящий от химического состава пробы;
м2а и м2м - массовые коэффициенты поглощения рассеянного излучения соответственно в определяемой фазе и наполнителе пробы.
В рассматриваемом случае (5) можно представить как:
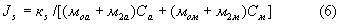
Как следует из выражений (3), (4) и (2), интенсивность определяемой фазы, так же как и интенсивность флуоресценции убывают с ростом поглощающей способности анализируемой пробы. Причем при значительном росте массового коэффициента наполнителя пробы интенсивность может уменьшаться в несколько раз, что может привести к относительной ошибке определения содержания того или иного компонента в несколько сот процентов! Аналогичным образом ведет себя зависимость интенсивности некогерентно рассеянного излучения от поглощающей способности пробы, что следует из выражений (5) и (6). Таким образом, величина отношения этих интенсивностей существенно меньше зависит от поглощающей способности пробы, следовательно, и должна использоваться в качестве аналитического параметра.
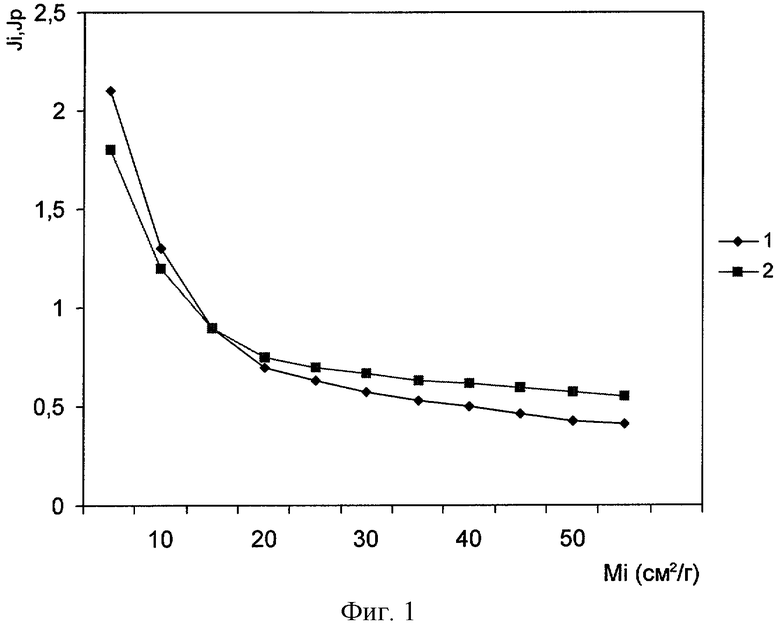
Сущность изобретения поясняется фигурами, где на фигуре 1 представлена зависимость интенсивностей анализируемой фазы определяемой компоненты Ji и некогерентно рассеянного пробой первичного излучения Js от массового коэффициента поглощения наполнителя пробы, при измерении проб с одинаковым содержанием ZrO2 (1%), но различными наполнителями, массовый коэффициент поглощения которых для различных проб изменялся в интервале от 0,5 до 60 см2/г (значения интенсивностей нормированы относительно I0).
Как видно из фигуры, при постоянной концентрации анализируемого элемента, зависимость интенсивности анализируемой фазы определяемой компоненты Ji от значения массового коэффициента поглощения наполнителя пробы не устраняется, что приводит к существенному возрастанию ошибки анализа при значительном изменении вещественного состава проб.
Но из фигуры также видно, что интенсивности рефлекса анализируемой фазы и некогерентно рассеянного излучения незначительно отличаются на всем рассматриваемом интервале изменения поглощающей способности наполнителя. Это подтверждает, что аналитический параметр η=Ji/Js незначительно зависит от матрицы пробы и может использоваться в качестве аналитического параметра.
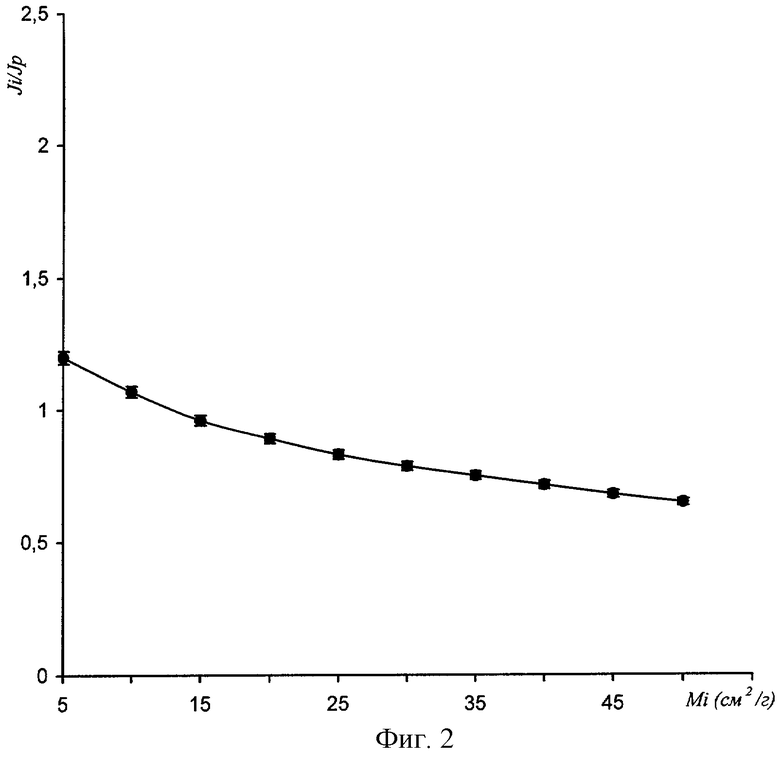
На фиг.2 приведена зависимость аналитического параметра η от массового коэффициента поглощения наполнителя пробы при фиксированных прочих параметрах. Как видно из фигуры, зависимость аналитического параметра η от массового коэффициента поглощения наполнителя пробы существенно меньше, чем зависимости интенсивностей Ji и Js.
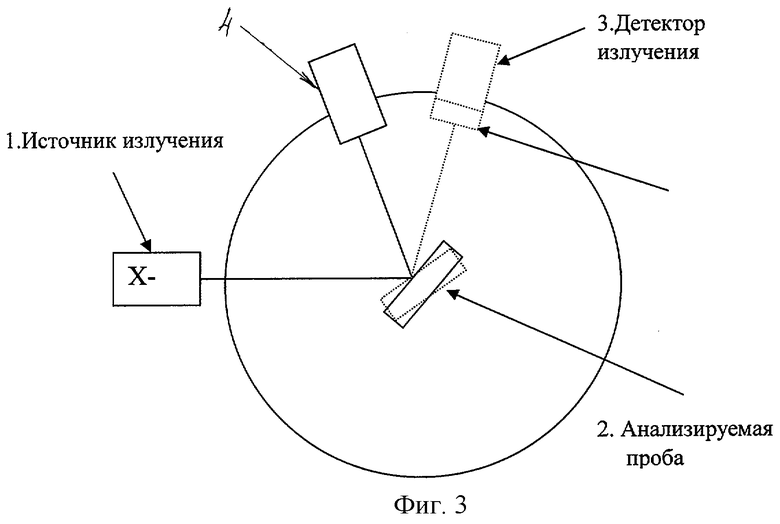
На фиг.3 представлена схема измерения по предлагаемому способу: 1 - источник изучения; 2 - образец; 3 - детектор излучения; 4 - детектор излучения.
Техническая реализация способа в силу своей простоты легко осуществима на серийных отечественных дифрактометрах типа ДРОН. При параллельном измерении одновременно регистрируются интенсивности измеряемой фазы и с помощью дополнительного детектора рассеянного излучения. При последовательном измерении регистрируется интенсивность измеряемой фазы, затем детектор перемещается на малый угол (рядом с измеряемой фазой), причем угловое положение пробы остается неизменным и регистрируется интенсивность некогерентно рассеянного излучения. А концентрация измеряемой фазы определяется по отношению вышеуказанных интенсивностей по градуировочному графику, построенному по эталонам с гарантированными концентрациями определяемой фазы.
Предложенный способ позволяет существенно снизить влияние химического состава пробы на ошибку измерения, понизить систематическую составляющую ошибки на порядок и ниже случайной составляющей. Созданные на основе данного способа методики анализа вольфрамовой и молибденовой продукции УзКТЖМ по метрологическим характеристикам соответствуют требованиям ГОСТов и отличаются высокой экспрессностью, точностью и простотой реализации.
Источники информации, принятые во внимание:
1. Герасимов В.Н. и др. Руководство по рентгеновскому исследованию минералов. Ленинград: Недра, 1975, с.100.
2. Герасимов В.Н. и др. Руководство по рентгеновскому исследованию минералов. Ленинград: Недра, 1975. с.101.
3. Герасимов В.Н. и др. Руководство по рентгеновскому исследованию минералов. Ленинград: Недра, 1975, с.100.
4. Мамиконян С.В. Аппаратура и методы флуоресцентного рентгенорадиометрического анализа. М.: Атомиздат, 1976, с.29.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОПРЕДЕЛЕНИЯ КОНЦЕНТРАЦИЙ ЭЛЕМЕНТА И ФАЗЫ, ВКЛЮЧАЮЩЕЙ ДАННЫЙ ЭЛЕМЕНТ, В ВЕЩЕСТВЕ СЛОЖНОГО ХИМИЧЕСКОГО СОСТАВА | 2008 |
|
RU2362149C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ КОНЦЕНТРАЦИИ ЭЛЕМЕНТА В ВЕЩЕСТВЕ СЛОЖНОГО ХИМИЧЕСКОГО СОСТАВА | 2013 |
|
RU2524454C1 |
| Способ количественного рентгенофлуоресцентного анализа трехкомпонентных сред | 1971 |
|
SU444970A1 |
| СПОСОБ РЕНТГЕНОФЛУОРЕСЦЕНТНОГО АНАЛИЗА КОНЦЕНТРАЦИИ ЭЛЕМЕНТНОГО СОСТАВА ВЕЩЕСТВА | 2020 |
|
RU2753164C1 |
| Способ определения рассеивающей способности вещества | 1982 |
|
SU1087856A1 |
| Способ рентгенофлуоресцентного анализа состава вещества | 1987 |
|
SU1580232A1 |
| Способ рентгенофлуоресцентного анализа | 1975 |
|
SU648890A1 |
| СПОСОБ РЕНТГЕНОРАДИОМЕТРИЧЕСКОГО АНАЛИЗА СОСТАВА ВЕЩЕСТВА | 2010 |
|
RU2442147C2 |
| СПОСОБ СЕПАРАЦИИ АЛМАЗОВ И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2021 |
|
RU2772789C1 |
| СПОСОБ РЕНТГЕНОФЛУОРЕСЦЕНТНОГО АНАЛИЗА ЭЛЕМЕНТНОГО СОСТАВА ВЕЩЕСТВА | 2002 |
|
RU2240543C2 |
Использование: для определения концентрации фазы в веществе сложного химического состава. Сущность: заключается в том, что проводят облучение пробы анализируемого вещества насыщенной толщины монохроматическим гамма- или рентгеновским излучением, регистрацию интенсивностей когерентно рассеянного определяемой фазой некогерентно рассеянного веществом пробы первичного излучения. Концентрацию определяемого элемента в анализируемой пробе рассчитывают по аналитическому сигналу, представляющему собой отношение вышеуказанных интенсивностей, регистрируемых одновременно или последовательно. Технический результат: повышение точности измерений. 3 ил.
Способ определения концентрации фазы в веществе сложного химического состава, включающий облучение пробы анализируемого вещества монохроматическим гамма- или рентгеновским излучением, регистрацию интенсивностей когерентно рассеянного определяемой фазой первичного излучения, отличающийся тем, что одновременно или последовательно с регистрацией указанной интенсивности регистрируют интенсивность некогерентно рассеянного этой же пробой первичного излучения, а затем по отношению указанных интенсивностей устанавливают концентрацию определяемой фазы.
| Способ количественного рентгеноструктурного фазового анализа | 1986 |
|
SU1376015A1 |
| Способ рентгенострукторного количественного фазового анализа твердых веществ | 1972 |
|
SU488122A1 |
| WO 0225257 A1, 28.03.2002 | |||
| WO 03062805 A2, 31.07.2003 | |||
| US 5105454 A, 14.04.1992. | |||

