Настоящее изобретение относится к новым обладающим антагонистической активностью в отношении рецепторов тахикинина 1-[1-(гетеро)арил-1-пергидроксиалкилметил]пиперазиновым соединениям, а также к содержащим эти соединения лекарственным средствам. Изобретение относится далее к способу получения вышеуказанных новых пиперазиновых соединений и к промежуточным продуктам, используемым при осуществлении этого способа.
К тахикининам относятся такие встречающиеся в естественных условиях нейропептиды, как вещество Р, нейрокинин А и нейрокинин В. Тахикинины действуют в качестве агонистов рецепторов, которые присутствуют в организме крупных млекопитающих и человека, таких как рецепторы нейрокинина 1 (NK-1), нейрокинина 2 (NK-2) и нейрокинина 3 (NK-3). Полученные искусственным путем соединения, обладающие антагонистической активностью в отношении рецепторов тахикинина, обычно классифицируют в соответствии с их относительной способностью связываться с одним или несколькими рецепторами из трех их указанных выше подтипов. Тахикинины участвуют в физиологических процессах, например играют важную роль в передаче болевого импульса, при рвоте, нейрогенных воспалениях, воспалениях мочевого пузыря, воспалительных заболеваниях суставов или при астматических осложнениях.
Из ЕР 0474561 А1 уже известны, в частности, пиперазиновые производные, обладающие активностью в качестве антагонистов рецептора NK-2.
В WO 96/10568 описаны другие пиперазиновые производные, которые могут обладать активностью в качестве антагонистов рецепторов тахикинина.
В основу настоящего изобретения была положена задача получить новые действующие вещества, которые обладали бы свойствами антагонистов рецепторов тахикинина и улучшенным профилем действия и которые можно было бы применять прежде всего для лечения периферических нарушений, таких как функциональные и воспалительные нарушения желудочно-кишечного тракта.
При создании изобретения неожиданно было установлено, что группа новых 1-[1-(гетеро)арил-1-пергидроксиалкилметил]пиперазиновых соединений обладает антагонистическими свойствами в отношении рецепторов тахикинина, прежде всего антагонистическими свойствами в отношении рецептора NK-2, a также обладает ярко выраженным действием на периферическую область. В соответствии с этим группа предлагаемых в изобретении соединений наиболее пригодна для лечения периферических нарушений, в которых тахикинины и прежде всего нейрокинин А участвуют в качестве медиаторов, например для лечения и/или профилактики функциональных и воспалительных нарушений желудочно-кишечного тракта. В контексте настоящего описания понятие "(гетеро)арил" может обозначать как арильные, так и гетероарильные радикалы.
Объектом изобретения являются новые 1-[1-(гетеро)арил-1-пергидроксиалкилметил]пиперазиновые соединения общей формулы I
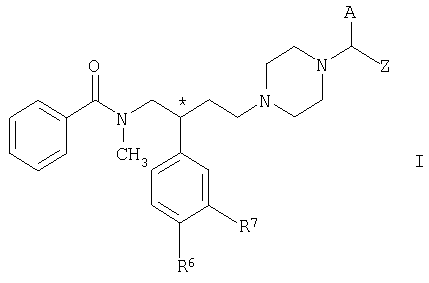
в которой
А обозначает нафтил, фенил, необязательно замещенный гидроксигруппой, моно- или бициклический гетероарил или необязательно замещенный фенилом С3-С6алкенил,
Z обозначает подгруппу общей формулы

где
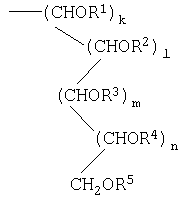
R1 обозначает водород или (низш.)алканоил либо вместе с одним из других заместителей из числа R2, R3, R4 и R5 может образовывать 5- или 6-членное кольцо, связанное мостиком, образованным карбонилом, тиокарбонилом или метиленом, необязательно замещенным (низш.)алкилом или С4-С5алкиленом,
R2 обозначает водород или (низш.)алканоил либо вместе с одним из других заместителей из числа R1, R3, R4 и R5 может образовывать 5- или 6-членное кольцо, связанное мостиком, образованным карбонилом, тиокарбонилом или метиленом, необязательно замещенным (низш.)алкилом или С4-С5алкиленом,
R3 обозначает водород или (низш.)алканоил либо вместе с одним из других заместителей из числа R1, R2, R4 и R5 может образовывать 5- или 6-членное кольцо, связанное мостиком, образованным карбонилом, тиокарбонилом или метиленом, необязательно замещенным (низш.)алкилом или С4-С5алкиленом,
R4 обозначает водород или (низш.)алканоил либо вместе с одним из других заместителей из числа R1, R2, R3 и R5 может образовывать 5- или 6-членное кольцо, связанное мостиком, образованным карбонилом, тиокарбонилом или метиленом, необязательно замещенным (низш.)алкилом или С4-С5алкиленом,
R5 обозначает водород или (низш.)алканоил либо вместе с одним из других заместителей из числа R1, R2, R3 и R5 может образовывать 5- или 6-членное кольцо, связанное мостиком, образованным карбонилом, тиокарбонилом или метиленом, необязательно замещенным (низш.)алкилом или С4-С5алкиленом,
k обозначает 0 или 1,
l обозначает 0 или 1,
m обозначает 0 или 1 и
n обозначает 0 или 1,
R6 обозначает галоген или водород и
R7 обозначает галоген или водород,
а также физиологически приемлемые кислотно-аддитивные соли соединений формулы I. Объектом изобретения являются далее лекарственные средства, содержащие соединения формулы I. Объектом изобретения являются также способ получения соединений формулы I и промежуточные продукты, используемые при осуществлении этого способа.
Если в соединениях формулы I или в других соединениях, подпадающих под объем настоящего изобретения, заместители представляют собой или содержат (низш.)алкил, то он в каждом случае может иметь прямую или разветвленную цепь и содержать от 1 до 4 атомов углерода. Если заместители в соединениях формулы I обозначают галоген, то он может представлять собой фтор, хлор или бром. Предпочтительным является хлор. Если заместители содержат (низш.)алканоил, то он может иметь прямую или разветвленную цепь и содержать от 2 до 4 атомов углерода. Предпочтительным (низш.)алканоилом является ацетил.
Подгруппа А предпочтительно представляет собой моноциклический гетероарил. Таким моноциклическим гетероарилом является прежде всего тиофен, фуран и пиррол. Предпочтительны при этом тиофен и фуран. Если же А представляет собой бициклический гетероарил, то предпочтительны бензотиофен, бензофуран и индол. Если А представляет собой С3-С6алкенил, необязательно замещенный фенилом, то алкенильная цепь может быть прямой или разветвленной и предпочтительно представляет собой 1-алкенил.
Если один из относящихся к подгруппе Z заместителей из числа R1, R2, R3, R4 и R5 совместно с одним из других заместителей этой группы представляет собой 5- или 6-членное кольцо, связанное мостиком, образованным необязательно замещенным (низш.)алкилом или С4-С5алкиленом метиленом, то в этом случае предпочтительны 5- или 6-членные кольца, связанные мостиком, образованным метиленом, 1,1-диметилметиленом, 1,1-спиро-тетраметиленметиленом или 1,1-спиро-пентаметиленметиленом. Соответствующие 5- или 6-членные кольца, соединенные карбонильным мостиком, называют циклическими карбонатами. Соответствующие связанные тиокарбонильным мостиком 5- или 6-членные кольца называют циклическими тиокарбонатами. k предпочтительно равно 1. n предпочтительно равно 0. Предпочтительными значениями Z являются таким образом необязательно замещенный 1,2-диольный остаток, 1,2,3-триольный остаток или 1,2,3,4-тетрольный остаток. Несущие заместители R1, R2, R3 и R4 атомы углерода являются асимметричными и в каждом случае могут находиться в двух различных конфигурациях. Вследствие этого Z может быть представлен в нескольких стереоизомерных формах. В объем настоящего изобретения наряду с соединениями формулы I, содержащими смеси стереоизомерных форм подгруппы Z, включены также соединения формулы I, содержащие чистые изомеры подгруппы Z. Предпочтительными подгруппами Z являются ксило-1,2,3,4-тетрагидроксибутил, ликсо-1,2,3,4-тетрагидроксибутил, арабино-1,2,3,4-тетрагидроксибутил, трео-1,2,3-тригидроксипропил, эритро-1,2,3-тригидроксипропил, а также глицеро-1,2-дигидроксиэтил. Оптимальные результаты достигаются преимущественно при наличии углеводов, выбранных из D-ряда углеводов, входящих в подгруппу Z. Предпочтительными являются чистые диастереоизомеры подгруппы Z.
Наиболее предпочтительными соединениями формулы I являются соединения из группы, включающей
N-((2S)-2-(3,4-дихлорфенил)-4-{4-[(2S,3R,4R)-2,3,4,5-тетрагидрокси-1-(тиенил)пентил]-1-пиперазинил}бутил)-N-метилбензамид,
N-((2S)-2-(3,4-дихлорфенил)-4-{4[(2S)-2,3-дигидрокси-1-(2-фурил)пропил]-1-пиперазинил}бутил)-N-метилбензамид,
(2S)-2-(ацетилокси)-3-{4-[(3S)-4-[бензоил(метил)амино]-3-(3,4-дихлорфенил)бутил]-1-пиперазинил}-3-(2-фурил)пропилацетат,
N-[(2S)-2-(3,4-дихлорфенил)-4-(4-{2-фурил[(4S)-2-оксо-1,3-диоксолан-4-ил]метил}-1-пиперазинил)бутил]-N-метилбензамид,
N-((2S)-2-(3,4-дихлорфенил)-4-{4-[(1S,2R)-2,3-дигидрокси-1-(3-тиенил)пропил]-1-пиперазинил}бутил)-N-метилбензамид,
N-((2S)-2-(3,4-дихлорфенил)-4-{4-[(2S)-2,3-дигидрокси-1-(3-фурил)пропил]-1-пиперазинил}бутил)-N-метилбензамид,
N-((2S)-2-(3,4-дихлорфенил)-4-{4-[(2R,3R,4R)-2,3,4,5-тетрагидрокси-1-(3-тиенил)пентил]-1-пиперазинил}бутил)-N-метилбензамид,
N-((2S)-2-(3,4-дихлорфенил)-4-{4-[(2S,3R,4R)-2,3,4,5-тетрагидрокси-1-(2-фурил)пентил]-1-пиперазинил}бутил)-N-метилбензамид,
N-((2S)-2-(3,4-дихлорфенил)-4-{4-[(2R)-2,3-дигидрокси-1-(3-тиенил)пропил]-1-пиперазинил}бутил)-N-метилбензамид,
N-((2S)-2-(3,4-дихлорфенил)-4-{4-[(2R,3S,4S)-2,3,4,5-тетрагидрокси-1-(3-тиенил)пентил]-1-пиперазинил}бутил)-N-метилбензамид,
N-((2S)-2-(3,4-дихлорфенил)-4-{4-[(2S,3R,4R)-2,3,4,5-тетрагидроксиацетил-1-(3-тиенил)пентил]-1-пиперазинил}бутил)-N-метилбензамид,
N-((2S)-2-(3,4-дихлорфенил)-4-{4-[(2S,3R)-2,3,4-тригидрокси-1-(3-тиенил)бутил]-1-пиперазинил}бутил)-N-метилбензамид и
N-((2S)-2-(3,4-дихлорфенил)-4-{2-фурил[(4S)-2-тиоксо-1,3-диоксолан-4-ил]метил}-1-пиперазинил)бутил)-N-метилбензамид.
Соединения формулы I и их кислотно-аддитивные соли можно получать взаимодействием соединения общей формулы II
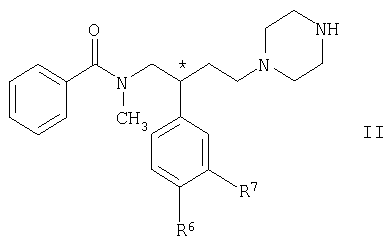
в которой R6 и R7 имеют указанные выше значения, с соединением общей формулы III

в которой А имеет указанное выше значение, и с соединением общей формулы IV

в которой R1, R2, R3, R4, R5, k, l, m и n имеют указанные выше значения, и затем либо в полученном соединении формулы I, в которой по меньшей мере один заместитель из числа R1, R2, R3, R4 и R5 обозначает водород, взаимодействием с соединением общей формулы VIII

в которой R8 обозначает алкил с прямой или разветвленной цепью, содержащий 1-3 атома углерода, при необходимости ацилируют подгруппу Z, либо в полученном соединении формулы I, в которой по меньшей мере два заместителя из числа R1, R2, R3, R4 и R5 обозначают водород, взаимодействием с реакционноспособным эквивалентным с точки зрения синтеза карбонильным или тиокарбонильным соединением при необходимости карбонилируют, соответственно тиокарбонилируют подгруппу Z, либо полученное соединение формулы I, в которой по меньшей мере два заместителя из числа R1, R2, R3, R4, R5 обозначают водород, взаимодействием подгруппы Z с ди(низш.)алкилкетоном или с С5-С6циклоалкилкетоном превращают в производное с 5- или 6-членным кольцом, связанным мостиком, образованным метиленом, необязательно замещенным (низш.)алкилом или C4-C5алкиленом, и полученное соединение формулы I при необходимости переводят в его кислотно-аддитивную соль или кислотно-аддитивную соль переводят в свободное соединение формулы I.
Описанную выше реакцию можно осуществлять известным методом в условиях реакции Манниха с бороновыми кислотами (см., например, N.A.Petasis и др. Journal of the American Chemical Society 120, c.11798-11799 (1998), WO 98/00398 или WO 00/24510). В соответствии с этим соединение формулы II можно подвергать взаимодействию по реакции, проводимой в одном реакционном сосуде, с бороновой кислотой формулы III и углеводом формулы IV, при необходимости несущим приемлемые защитные группы, в инертном в условиях реакции растворителе. Защитные группы, которые можно применять для углеводов, известны и описаны, например, у J.A.W.McOmie, "Protective Groups in Organic Chemistry", изд-во Plenum Press, 1973 или у T.W.Green, P.G.Wuts, "Protective Groups in Organic Synthesis", изд-во Wiley and Sons, 1999. В качестве растворителей можно использовать биполярные протонные органические растворители, такие как (низш.)алканолы, например С1-С4алканолы с прямой или разветвленной цепью, предпочтительно этанол, или смеси указанных выше растворителей с водой или с биполярными апротонными растворителями, такими как (низш.)галоалканы, предпочтительно дихлорметан. Реакцию целесообразно проводить при температуре в интервале от комнатной до температуры кипения растворителя или смеси растворителей. Соединения формул II, III и IV предпочтительно подвергать взаимодействию между собой в указанном порядке. Вместе с тем соединение формулы II можно также сначала подвергать взаимодействию с соединением формулы IV, а затем с соединением формулы III. Вновь образующийся в результате такой реакции сочетания несущий подгруппы А и Z хиральный центр в соединениях формулы I в значительной степени образуется по данным анализа диастереоизомеров преимущественно в виде "антипродукта".
Соединения формулы I, несущие в подгруппе Z по меньшей мере одну свободную гидроксигруппу, можно затем при необходимости подвергать взаимодействию с соединениями формулы VIII, в результате чего происходит ацилирование свободных гидроксигрупп в подгруппе Z. Как правило, в этих условиях происходит перацилирование свободных гидроксигрупп в подгруппе Z. В качестве ацилирующего агента при этом можно использовать кислоты формулы VIII или их реакционноспособные производные. В качестве таких реакционноспособных производных можно применять прежде всего ангидриды кислот и галогенангидриды кислот. Ацилирование можно проводить с применением инертного в реакционных условиях органического растворителя предпочтительно при температуре от -20°С до комнатной температуры. В качестве растворителей при этом можно использовать прежде всего ароматические углеводороды, такие как бензол или толуол, циклические или имеющие разомкнутую цепь ди(низш.)алкиловые эфиры, такие как диэтиловый эфир, тетрагидрофуран (ТГФ) или диоксан, частично галогенированные (низш.)углеводороды, такие как дихлорметан, либо смесь этих растворителей. Если в качестве ацилирующего агента используют ангидрид или галогенангидрид кислоты формулы VIII, то ацилирование целесообразно проводить в присутствии связывающего кислоту реагента. В качестве подобного связывающего кислоту реагента можно использовать растворимые в реакционной смеси ненуклеофильные органические основания, такие как пиридин, триэтиламин или 4-диметиламинопиридин. Применяемые в избытке органические основания одновременно могут служить в качестве растворителей.
Соединения формулы I, которые несут в подгруппе Z по меньшей мере две свободные гидроксильные группы, можно после их получения описанным выше способом при необходимости подвергать взаимодействию не с соединениями формулы VIII, а с реакционноспособными, эквивалентными, с точки зрения синтеза, карбонильными или тиокарбонильными соединениями, что позволяет карбонилировать, соответственно тиокарбонилировать подгруппу Z. Такое взаимодействие можно осуществлять известным методом. Так, например, соединение формулы I можно подвергать реакции в инертном в реакционных условиях органическом растворителе. В качестве примера реакционноспособных, эквивалентных, с точки зрения синтеза, карбонильных соединений можно назвать фосген или реагирующие по типу фосгена соединения, такие как бис(трихлорметил)карбонат (трифосген), трихлорметиловый эфир хлормуравьиной кислоты (дифосген) или прежде всего карбонилдиимидазол. Для применения в качестве реакционноспособного, эквивалентного, с точки зрения синтеза, тиокарбонильного соединения пригоден прежде всего N,N'-тиокарбонилдиимидазол. В реакционную смесь целесообразно добавлять связывающий кислоту реагент. В качестве таких связывающих кислоту реагентов можно применять связывающие кислоту реагенты, указанные выше при описании взаимодействия соединений формулы I с соединениями формулы VIII. Реакцию преимущественно проводят при температуре от -20°С до комнатной температуры.
В соединениях формулы I, которые несут в подгруппе Z по меньшей мере две свободные гидроксигруппы, после их получения вышеописанным способом подгруппу Z можно при необходимости подвергать взаимодействию не с соединениями формулы VIII или с реакционноспособными, эквивалентными, с точки зрения синтеза, карбонильными или тиокарбонильными соединениями, а с ди(низш.)алкилкетоном или С3-С6циклоалкилкетоном с получением производного с 5- или 6-членным кольцом, связанным мостиком, образованным метиленом, необязательно замещенным (низш.)алкилом или C4-C5алкиленом. Предпочтительным ди(низш.)алкилкетоном для применения в вышеуказанных целях является ацетон. Предпочтительными С3-С6циклоалкилкетонами являются циклопентанон и циклогексанон.
При необходимости получить соединения формулы I, в которой входящие в подгруппу Z заместители R1, R2, R3, R4 и/или R5 имеют отличные от водорода значения, следует исходить предпочтительно из углеводных соединений формулы IV, которые по меньшей мере в α-положении относительно альдегидной функциональной группы содержат свободные гидроксигруппы. При этом целесообразно использовать соединения формулы IV, в которой R1, R2, R3, R4, R5 обозначают водород. После этого свободные гидроксигруппы можно при необходимости подвергать описанным выше путем ацилированию, карбонилированию, тиокарбонилированию или взаимодействию с пригодным для этой цели кетоном.
Соединения формулы II являются новыми соединениями, которые пригодны преимущественно для применения в качестве промежуточных продуктов для получения новых действующих веществ, например для получения соединений формулы I, обладающих антагонистической активностью в отношении рецепторов тахикинина.
Соединения формулы II можно получать взаимодействием соединения общей формулы V
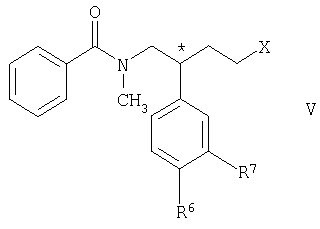
в которой R6 и R7 имеют указанные выше значения, а Х обозначает галоген, прежде всего йод, с защищенным пиперазиновым производным общей формулы VI
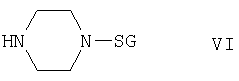
в которой SG обозначает отщепляемую защитную группу, прежде всего трет-бутоксикарбонил, с последующим отщеплением защитной группы SG известным методом. Вышеуказанную реакцию можно проводить в инертном в реакционных условиях органическом растворителе, таком как ароматический углеводород, прежде всего толуол, или в циклическом или имеющим разомкнутую цепь ди(низш.)алкиловом эфире, прежде всего ТГФ, либо предпочтительно в смеси указанных растворителей и в присутствии основания. В качестве оснований при этом можно использовать ненуклеофильные органические азотсодержащие основания, такие как третичные (низш.)алиламины, например триэтиламин. Реакцию целесообразно проводить при температуре от 50 до 100°С, предпочтительно от приблизительно 70 до 90°С.
Соединения формулы V можно получать осуществляемым известным методом взаимодействием соединений общей формулы VII

в которой R6 и R7 имеют указанные выше значения, с галогенидом щелочного металла общей формулы MX, в которой М обозначает щелочной металл, прежде всего натрий, а Х имеет указанное выше значение и прежде всего обозначает йод. Соединения формулы VII и их стереоизомерные формы известны, например, из заявки ЕР 0474561 А1 и их можно получать описанным в указанной заявке способом или аналогичным ему способом.
Соединения формул III, IV и VI являются известными или их можно получать из известных соединений по методам, известным специалистам в данной области. Предпочтительными для применения соединениями формулы IV являются D-ксилоза, D-ликсоза, D-арабиноза, D-треоза, D-эритроза, а также D- и L-глицеринальдегид.
Соединения формулы I можно выделять из реакционной смеси и очищать известными методами. Кислотно-аддитивные соли можно обычным путем переводить в свободные основания, а последние при необходимости можно по известным методам переводить в физиологически приемлемые кислотно-аддитивные соли. Физиологически приемлемыми солями соединений формулы I являются их обычные соли с неорганическими кислотами, такими, например, как серная кислота, фосфорные кислоты или галогенводородные кислоты, предпочтительно хлористоводородная кислота, с органическими кислотами, такими, например, как низшие алифатические моно-, ди- или трикарбоновые кислоты, в частности малеиновая кислота, фумаровая кислота, молочная кислота, винная кислота, лимонная кислота, с сульфоновыми кислотами, такими, например, как (низш.)алкансульфоновые кислоты, в частности метансульфоновая кислота или трифторметансульфоновая кислота, или с бензолсульфоновыми кислотами, необязательно замещенными в бензольном кольце галогеном или (низш.)алкилом, такими, например, как пара-толуолсульфоновая кислота.
Соединения формулы I содержат в γ-положении относительно атома азота, находящегося в 4-м положении пиперазинового кольца, асимметричный атом углерода, а именно атом углерода *С, несущий фенильное кольцо, замещенное остатками R6 и R7. Соединения формулы I благодаря наличию этого асимметричного атома углерода и наличию асимметричных атомов углерода, несущих подгруппы А и Z, а также благодаря наличию при определенных условиях асимметричных атомов углерода, входящих в подгруппу Z, могут быть представлены в различных стереоизомерных формах. В объем настоящего изобретения включены и смеси оптических изомеров, и чистые изомеры соединений формулы I. Предпочтительными являются соединения формулы I, в которой атом углерода *С, несущий фенильное кольцо, замещенное остатками R6 и R7, имеет S-конфигурацию. В том случае, если при синтезе соединений формулы I используют смеси оптических изомеров исходных соединений, например соединений формулы II или соединений формулы IV, то соединения формулы I также получают в виде смесей оптических изомеров. Использование имеющих однородное стереохимическое строение исходных соединений позволяет получать соединения формулы I, которые также имеют однородное стереохимическое строение. Имеющие однородное стереохимическое строение соединения формулы I можно получать известными методами из смесей оптических изомеров, например хроматографическим разделением на хиральных разделяющих материалах либо взаимодействием с пригодными для этой цели оптически активными кислотами, например с винной кислотой или камфор-10-сульфоновой кислотой, с последующим разделением образующихся диастереоизомерных солей на оптически активные антиподы с помощью фракционированной кристаллизации.
Соединения формулы I и их кислотно-аддитивные соли обладают свойствами антагонистов рецепторов тахикинина и поэтому пригодны для лечения у крупных млекопитающих, прежде всего у человека, болезненных состояний, в которых тахикинины участвуют в качестве медиаторов. Предлагаемая в изобретении группа соединений отличается наиболее благоприятным профилем действия, который характеризуется высокоизбирательной аффинностью к рецепторам NK-2. Помимо этого, предлагаемая в изобретении группа соединений характеризуется хорошей переносимостью даже в течение продолжительного периода их применения, а также сравнительно хорошей доступностью при пероральном введении. Соединения формулы I благодаря их профилю действия пригодны прежде всего для ингибирования процессов, в которых участвуют связывающиеся с рецепторами NK-2 тахикинины, такие как нейрокинин А. Соединения формулы I благодаря тому, что их действие направлено преимущественно на периферическую область, пригодны прежде всего для лечения и/или профилактики у крупных млекопитающих и прежде всего у людей обоего пола функциональных или воспалительных нарушений желудочно-кишечного тракта, которые сопровождаются повышенной болевой чувствительностью и/или нарушением прохождения стула в области ободочной кишки. К функциональным нарушениям желудочно-кишечного тракта, которые можно лечить с помощью предлагаемых в изобретении соединений, относятся прежде всего нарушения в нижней части кишечника, известные под названием "Irritable Bowel Syndrome" (IBS) или синдром раздраженного кишечника. Типичные симптомы, позволяющие диагностировать IBS, описаны, например, у W.G.Thompson и др. Gastroenterology International, 2, c.92-95 (1989), или у W.G.Thompson и др. GUT 45/II, c.1143-1147 (1999), и хорошо известны среди специалистов под названием "римские критерии". В соответствии с этим наиболее существенными симптомами IBS являются боли в подчревной области, которые, как полагают, обусловлены гиперсенсибилизацией висцеральной афферентной нервной системы, и аномальные проявления процесса испражнения, такие как запор, диарея или чередующиеся запор и диарея. К другим обусловленным воспалительными процессами нарушениям желудочно-кишечного тракта, на которые предлагаемая в изобретении группа соединений позволяет оказывать положительное влияние, относятся, например, воспалительные нарушения в области тонкой и толстой кишки, которые в большинстве случае собирательно называют воспалительным заболеванием кишечника или "Inflammatory Bowel Disease" (IBD), например язвенный колит и болезнь Крона. Помимо этого, предлагаемые в изобретении соединения благодаря их механизму действия можно применять для лечения других нарушений, в которых тахикинины и прежде всего нейрокинин А участвуют в качестве медиаторов. К таким нарушениям относятся, например, нейрогенные воспаления, воспалительные заболевания суставов, такие как ревматоидный артрит, астматические осложнения, аллергические нарушения, нарушения иммунорегуляции, воспаления мочевого пузыря, а также функциональная диспепсия.
Описание фармакологических методов исследования
Номера примеров, указанные ниже в фармакологических исследованиях для соединений формулы I, использованных в качестве тестируемых соединений, соответствуют приведенным далее примерам получения.
1. Оценка способности тестируемых соединений связываться с рецепторами NK-2 в опытах in vitro
Аффинность тестируемых соединений к человеческим рецепторам NK-2 оценивали в опытах in vitro. При этом выявляли способность тестируемых соединений вытеснять используемое в качестве контрольного лиганда и являющееся антагонистом рецептора NK-2 соединение SR 48969 (саредутант) из его соответствующего сайта связывания.
Связывание с рецепторами исследовали с использованием радиоактивно-меченного соединения [3H]-SR 48968 (фирма Amersham) в качестве лиганда. В эксперименте по оценке связывания различные образцы препарата мембран клеток СНО (клетки яичника китайского хомячка), экспрессирующие человеческий рецептор NK-2 (полученные по методу, описанному у Gerard и др., Journal of Biological Chemistry, 265/33, c.20455-20462 (1990)), инкубировали в течение 90 мин с раствором меченого лиганда, при этом для инкубации использовали образцы без тестируемого соединения и образцы, содержащие тестируемое соединение в различных концентрациях. После этого в образцах в каждом случае путем фильтрации связанные с мембраной лиганды отделяли от свободных лигандов. Оставшуюся на фильтре фракцию несколько раз промывали буферным раствором, после чего с помощью жидкостного сцинтилляционного счетчика измеряли ее радиоактивность. За значение IC50 принимали такую концентрацию каждого из тестируемых соединений, которая приводила к половинному от максимального уровню вытеснения связанного контрольного лиганда. На основании конкретного значения IC50 рассчитывали константу ингибирования (значение Кi) тестируемого соединения, которую для дальнейших целей выражали в виде величины ее отрицательного логарифма (pKi).
Аффинность соединений из примеров 1-39 к человеческим рецепторам NK-2 в каждом случае оценивали проведением по меньшей мере 3-кратных измерений с использованием тестируемых соединений в концентрациях от 10-6 до 10-10 молей/л. Поскольку проводили несколько измерений, каждый раз определяли среднее значение. Для всех вышеуказанных тестируемых соединений при использовании данной модели исследования значение pKi составило по меньшей мере 7,0. Для соединений из примеров 1-27 и 39 значение рКi составило по меньшей мере 8,0. Для соединений из примеров 1-6 и 39 значение pKi составило по меньшей мере 9,0.
2. Оценка функционального антагонизма тестируемых соединений в опытах in vitro на выделенной ткани морских свинок
Действие тестируемых соединений в качестве антагонистов рецептора NK-2 оценивали с использованием выделенных, содержащихся в насыщенном кислородом питательном растворе препаратов желчного пузыря морских свинок породы Pirbright-White. С этой целью одну сторону препарата закрепляли с помощью специальных зажимов для органов в питательном растворе, а другую сторону прикрепляли с помощью нити к динамометру.
В этом опыте присутствующие в препаратах желчного пузыря рецепторы NK-2 стимулировали нейрокинином A (NKA, 0,1 мкмоля/л), который является природным агонистом рецептора NK-2, и измеряли обусловленную такой стимуляцией силу сокращения препаратов в мН (начальное значение). После этого NKA вымывали из препаратов с помощью не содержащего NKA раствора и добавляли тестируемые соединения в концентрации 10-7 молей/л. После двухчасовой инкубации препаратов с тестируемыми соединениями вновь измеряли сокращение препаратов, которое еще наблюдалось после повторного добавления NKA, и результаты выражали в процентах по отношению к измеренной в начале эксперимента силе сокращения, обусловленной введением только одного NKA. В последующих экспериментах концентрацию тестируемых соединений в зависимости от результата итеративно повышали на целый, соответственно половинный, логарифмический шаг до тех пор, пока по меньшей мере при одной концентрации тестируемого соединения ингибирование сокращения не достигло уровня выше, соответственно ниже 50%-ного ингибирования (вплоть до максимальной концентрации, составляющей 10-5 молей/л). Для каждой концентрации рассчитывали среднее значение ингибирования сокращения с использованием 2-4 препаратов. В качестве критерия для каждого из тестируемых соединений рассчитывали концентрацию, обеспечивающую 50%-ное ингибирование (IC50). Данные для каждого тестируемого соединения выражали в виде значения pIC50 [в молях/л], представляющего собой логарифм величины IC50. При использовании подобной модели исследования для тестируемых соединений были получены приведенные в таблице 1 значения pIC50.
Функциональный антагонизм тестируемых соединений в отношении рецепторов NK-2 в опытах на выделенной ткани морских свинок
3. Оценка эффективности тестируемых соединений в качестве антагонистов рецептора NK-2 в опытах in vivo
Активность тестируемых соединений в качестве антагонистов рецепторов NK-2 и NK-1 после их внутривенного (в.в.) и перорального (п.о.) введения исследовали в опытах in vivo на наркотизированных морских свинках. Рассматриваемая модель позволяет на одном животном одновременно исследовать как антагонистическое действие в отношении рецептора NK-2 в трех различных системах организма (дыхательные пути, ободочная кишка и система кровообращения), так и антагонистическое действие в отношении рецептора NK-1 (быстрое падение кровяного давления).
Морских свинок породы Pirbright-White весом 500-700 г наркотизировали с помощью кетамина/ксилазина (начальная доза 67/13 мг/кг, подкожно, затем при необходимости вводили дополнительные дозы). Животным вводили внутривенный катетер для введения соединений и внутриартериальный катетер для измерения кровяного давления. Животных переводили на искусственную вентиляцию легких через введенную в трахею канюлю и с помощью датчика давления регистрировали давление дыхания. Для манометрической регистрации моторики ободочной кишки с помощью датчика давления животным в дистальную область ободочной кишки вводили баллонный катетер. У каждого животного непрерывно измеряли кровяное давление, частоту сердечных сокращений, давление дыхания и давление в ободочной кишке и данные регистрировали с помощью самописца, а также с помощью цифровой системы сбора данных. В качестве опытного раздражителя для стимуляции рецепторов NK-1 и NK-2 внутривенно вводили в виде болюса нейрокинин A (NKA, 200 пмолей/животное). Такое введение NKA приводило к значительному повышению давления дыхания (в результате сокращения бронхов) и давления в ободочной кишке, а также к двухфазному снижению кровяного давления. Первая фаза гипотензии (фаза максимальной гипотензии в течение первой минуты после введения NKA) опосредуется рецепторами NK-1, поскольку ее можно полностью блокировать с помощью антагонистов, специфических в отношении рецепторов NK-1. В отличие от этого вторая фаза замедленной гипотензии (фаза максимальной гипотензии через 2-5 мин) опосредуется рецепторами NK-2, поскольку ее можно блокировать с помощью антагонистов, специфических в отношении рецепторов NK-2. За характерные величины для каждого из измеряемых параметров, а именно сокращения бронхов, давления в ободочной кишке, а также опосредуемого рецептором NK-1, соответственно NK-2, изменения кровяного давления, принимали значения ED50, т.е. те дозы тестируемых соединений, которые приводили к 50%-ному от исходного значения уменьшению реакции на введение опытного стимула NKA.
Сначала антагонистическое действие тестируемых соединений исследовали по кумулятивной схеме, при этом опытный стимул NKA вводили через 1 мин после окончания введения каждой из доз тестируемых соединений. Значения ED50, полученные на основании зависимости действия от кумулятивной дозы, приведены ниже в таблице 2 (строка 1). С целью дополнительно выявить зависимость антагонистического действия тестируемых соединений от времени оценивали действие опытного стимула NKA в различные моменты времени (через 1, 30, 60, 90, 120, 150 и 180 мин) после введения тестируемых соединений. Антагонистическое действие тестируемых соединений оценивали затем по площади под кривой (AUC от англ. "area under curve") за период времени после введения тестируемых соединений, в течение которого проводили исследование (при в.в. введении: 120 мин после введения, при п.о. введении: 180 мин после введения), при этом полученные в результате значения ED50 приведены ниже в таблице 2 (строки 2 и 3).
Антагонистическое действие тестируемых соединений формулы I в отношении рецептора NK-2 в опытах in vivo на морских свинках
(кумулятивное действие)
Представленные в приведенной выше таблице 2 результаты измерений свидетельствуют среди прочего о том, что соединения из примеров 1, 5, 13, 14 и 15 после их в.в. введения в кумулятивных дозах (антагонистическое действие анализировали через 1 мин по окончании введения тестируемого соединения) оказывают ярко выраженное антагонистическое в отношении рецептора NK-2 действие на моторику ободочной кишки, на запаздывание снижения кровяного давления и на сопротивление дыханию.
Представленные в приведенной выше таблице 2 результаты измерений свидетельствуют также о том, что указанные соединения и прежде всего соединение из примера 1 более эффективно ингибируют механизм действия рецептора NK-2 в ободочной кишке (преимущественное действие в отношении ободочной кишки) по сравнению с ингибирующим действием на сокращающую бронхи, соответственно гипотензивную, активность рецептора NK-2. Предлагаемые в изобретении соединения и прежде всего соединение из примера 1 отличаются также медленно наступающим и продолжительным действием.
Ни для одного из исследованных соединений при их применении в соответствующих дозах не было выявлено антагонистическое действие в отношении рецептора NK-1.
Соединения формулы I можно применять в виде обычных фармацевтических композиций. Назначаемые дозы при этом могут быть различными в каждом конкретном случае и, как очевидно, варьируются в зависимости от состояния, подлежащего лечению, и конкретно применяемого соединения. В целом для введения человеку и крупным млекопитающим можно использовать лекарственные формы с содержанием действующего вещества от 0,2 до 200 мг, предпочтительно от 1 до 50 мг, из расчета на один прием. Согласно изобретению предлагаемые в нем соединения можно перерабатывать с обычными фармацевтическими вспомогательными веществами и/или носителями в твердые или жидкие фармацевтические композиции. В качестве примера твердых лекарственных препаратов можно назвать препараты для перорального введения, такие как таблетки, драже, капсулы, порошки или гранулы, а также суппозитории. В состав таких препаратов наряду с обычными фармацевтическими вспомогательными веществами, например смазывающими веществами или разрыхлителями для таблеток, могут входить обычные фармацевтические неорганические и/или органические носители, такие как тальк, лактоза или крахмал. Жидкие препараты, такие как суспензии или эмульсии действующих веществ, могут содержать обычные разбавители, такие как вода, масла и/или суспендирующие агенты, такие как полиэтиленгликоль и т.п. Помимо этого, дополнительно можно добавлять другие вспомогательные вещества, например консерванты, вкусовые добавки и т.п.
Действующие вещества можно по известной технологии смешивать и включать в состав фармацевтической композиции в сочетании с фармацевтическими вспомогательными веществами и/или носителями. При изготовлении твердых лекарственных форм действующее вещество можно, например, обычным методом смешивать со вспомогательными веществами и/или носителями и подвергать влажной или сухой грануляции. Гранулят или порошок можно непосредственно расфасовывать в капсулы или обычным методом спрессовывать в таблетки. При необходимости последние можно подвергать дражированию известным методом.
Ниже изобретение более подробно поясняется на примерах, которые не ограничивают его объем.
Пример 1
N-((2S)-2-(3,4-дихлорфенил)-4-{4-[(2S,3R,4R)-2,3,4,5-тетрагидрокси-1-(тиенил)пентил]-1-пиперазинил}бутил)-N-метилбензамид
А) 45,0 г N-[(2S)-(3,4-дихлорфенил)-4-метансульфонилокси]-N-метилбензамида растворяли в атмосфере защитного газа в 550 мл ацетона. В этот раствор добавляли 84,6 г NaI и образовавшуюся суспензию перемешивали при комнатной температуре в течение 20 ч. Далее растворитель выпаривали в вакууме и образовавшийся остаток растворяли в 650 мл метил-трет-бутилового эфира (МТБЭ) и 500 мл воды. После добавления 120 г Na2S2O4 водную фазу отделяли, а оставшуюся органическую фазу четырежды промывали насыщенным водным соляным раствором порциями по 100 мл. Органическую фазу сушили над сульфатом натрия и выпаривали растворитель в вакууме. После сушки образовавшегося остатка в вакууме получили 45,9 г N-[(2S)-2-(3,4-дихлорфенил)-4-йодбутил]-N-метилбензамида в виде стекловидного соединения, которое непосредственно использовали в последующей реакции без дополнительной очистки.
Б) 15,38 г N-трет-бутоксикарбонилпиперазина растворяли при комнатной температуре в атмосфере защитного газа в 200 мл толуола и смешивали с 32 мл триэтиламина. Полученный раствор нагревали до 84°С. Затем к этому раствору медленно по каплям добавляли 45,9 г полученного на предыдущей стадии йодида, растворенного в смеси 100 мл ТГФ и 200 мл толуола. Полученную в результате реакционную смесь нагревали до 80-85°С и выдерживали при этой температуре в течение 5 ч, а затем перемешивали еще в течение 8 ч при комнатной температуре. Смесь растворителей практически полностью выпаривали в вакууме и полученный остаток растворяли в 600 мл этилового эфира уксусной кислоты (ЭУ). После отделения выпавшего осадка органическую фазу последовательно промывали дважды водой порциями по 100 мл 15%-ного водного раствора винной кислоты. Затем к органической фазе добавляли 8,0 г NaOH и снова дважды промывали водой порциями по 200 мл воды. После сушки органической фазы над сульфатом натрия и выпаривания растворителя в вакууме получили 44,9 г трет-бутил-4-[(3S)-4-[бензоил(метил)амино]-3-(3,4-дихлорфенил)бутил]-1-пиперазинкарбоксилата в виде маслянистого соединения, которое непосредственно использовали в последующей реакции без дополнительной очистки.
В) 44,5 г полученного на предыдущей стадии пиперазинкарбоксилатного соединения растворяли при комнатной температуре в 600 мл метанола, добавляли 150 мл 6 н. HCl и перемешивали в течение 60 ч. Затем добавляли 500 мл воды и метанольную фазу практически полностью выпаривали в вакууме. Оставшуюся водную фазу четырежды экстрагировали ЭУ порциями по 100 мл и четырежды МТБЭ порциями по 100 мл. После этого к водной фазе добавляли раствор 36,0 г NaOH в 200 мл воды и образовавшуюся щелочную водную фазу еще дважды экстрагировали ЭУ порциями по 350 мл. Объединенные органические фазы промывали 100 мл воды, сушили над сульфатом натрия и упаривали в вакууме. После сушки остатка получили 25,8 г N-[(2S)-2-(3,4-дихлорфенил)-4-(пиперазинил)бутил]-N-метилбензамида в виде желтоватого твердого масла, которое непосредственно использовали в последующей реакции без дополнительной очистки.
Г) 25,0 г полученного на предыдущей стадии пиперазинового соединения с удаленной защитной группой растворяли при 30°С в атмосфере защитного газа в 250 мл этанола. После нагревания этого раствора до 50-60°С к нему добавляли сначала 10,0 г тиофен-3-бороновой кислоты, а затем 8,93 г D-ксилозы. Реакционную смесь кипятили с обратным холодильником в течение 15 ч, после чего перемешивали при комнатной температуре еще в течение 8 ч. Далее добавляли 500 мл воды и смесь растворителей практически полностью выпаривали в вакууме. К образовавшемуся остатку добавляли 20 мл 6 н. HCl и последовательно промывали ЭУ сначала однократно порцией 200 мл, а затем шестикратно порциями по 100 мл. Добавлением соответствующего количества 4 н. NaOH значение pH водной фазы устанавливали на 9-10 и затем экстрагировали 600 мл дихлорметана. Органическую фазу отделяли, сушили над сульфатом натрия и в завершение упаривали в вакууме. В результате получили 32,0 г указанного в заголовке соединения в виде аморфного твердого вещества.
Угол оптического вращения [α]D 20=-14,8° (с=1 в метаноле).
1H-ЯМР (d6-ДМСО, 90°С): 3,81 (d, 1Н); 4,15 (dd, 1H); 3,79 (dd, 1H); 3,66 (ddd, 1H); 3,48 (m, 2H); 7,12 (dd, 1H); 7,12 (d, 1H); 7,38 (dd, 1H).
Пример 2
N-((2S)-2-(3,4-дихлорфенил)-4-{4[(2S)-2,3-дигидрокси-1-(2-фурил)пропил]-1-пиперазинил)бутил)-N-метилбензамид
2,08 г N-[(2S)-2-(3,4-дихлорфенил)-4-(пиперазинил)бутил]-N-метилбензамида (полученного по описанной выше методике согласно примеру 1В) растворяли в 100 мл этанола и нагревали приблизительно до 50°С. К этому раствору добавляли 740 мг 2-фуранбороновой кислоты и 550 мг 80%-ного раствора D-глицеринальдегида в воде. Образовавшийся раствор в течение 10 ч кипятили с обратным холодильником. Затем избыток растворителя выпаривали в вакууме. После очистки остатка с помощью колоночной хроматографии на силикагеле (растворитель: дихлорметан/EtOH/NH4OH в соотношении 87:11:2) получили 2,1 г указанного в заголовке соединения в виде пены бежевого цвета.
Угол оптического вращения [α]D 20=-6,1° (с=1 в метаноле).
1H-ЯМР (CDCl3, комнатная температура): 3.65 (d, 1H); 4,23 (ddd, 1H); 3,72 (dd, 1H); 3,78 (dd, 1H); 6,27 (d, 1H); 6,37 (1H); 7,42 (1H).
Пример 3
(2S)-2-(ацетилокси)-3-{4-[(3S)-4-[бензоил(метил)амино]-3-(3,4-дихлорфенил)бутил]-1-пиперазинил}-3-(2-фурил)пропилацетат
К раствору 400 мг N-((2S)-2-(3,4-дихлорфенил)-4-{4[(2S)-2,3-дигидрокси-1-(2-фурил)пропил]-1-пиперазинил}бутил)-N-метилбензамида (полученного по описанной выше методике согласно примеру 2) в 10 мл пиридина при комнатной температуре добавляли 1,5 г ангидрида уксусной кислоты. Реакционную смесь перемешивали в течение 72 ч и сливали в раствор 2,2 г Na2СО3 в 40 мл воды. Органическую фазу экстрагировали 60 мл толуола и толуольную фазу промывали трижды водой порциями по 30 мл и дважды насыщенным водным соляным раствором. Объединенные органические фазы сушили над сульфатом натрия и выпаривали растворитель в вакууме. В результате получили 463 мг указанного в заголовке соединения в виде пены желтоватого цвета.
[α]D 20=+9,6° (с=1 в метаноле).
1H-ЯМР (CDCl3, КТ): 3,81 (d, 1H); 5,62 (ddd, 1H); 4,58 (dd, 1H); 4,21 (dd, 1H); 1,87 (s, 3H); 2,05 (s, 3H); 6,16 (d, 1H); 6,31 (1H); 7,35 (1H).
Пример 4
N-[(2S)-2-(3,4)-дихлорфенил)-4-(4-{2-фурил[(4S)-2-оксо-1,3-диоксолан-4-ил]метил}-1-пиперазинил)бутил]-N-метилбензамид
К раствору 576 мг N-((2S)-2-(3,4-дихлорфенил)-4-{4[(2S)-2,3-дигидрокси-1-(2-фурил)пропил]-1-пиперазинил}бутил)-N-метилбензамида (полученного по описанной выше методике согласно примеру 2) в 30 мл безводного дихлорметана при комнатной температуре добавляли 124 мг 4-диметиламинопиридина (ДМАП) и 410 мг N-карбонилдиимидазола. Реакционную смесь перемешивали в течение 15 ч при комнатной температуре и затем добавляли 3,7 г силикагеля. Образовавшуюся суспензию перемешивали еще в течение 1 ч, жидкую фазу отделяли вакуум-фильтрацией и фильтрат концентрировали в вакууме. Образовавшийся остаток растворяли в 50 мл ЭУ и органическую фазу пятикратно промывали смесью 50:50 (об./об.) 5%-ного водного раствора КН2PO4 и 1%-ного водного раствора К2HPO4 порциями по 10 мл. Органическую фазу сушили над сульфатом натрия и в завершение удаляли растворитель в вакууме. В результате получили 460 мг указанного в заголовке соединения в виде пены белого цвета.
1H-ЯМР (CDCl3, КТ): 3,74 (d, 1H); 5,07 (ddd, 1H); 4,55 (dd, 1H); 4,48 (dd, 1H); 6,3 (d, 1H); 6,378 (1H); 7,41 (1H).
Полученное указанное в заголовке соединение растворяли в 4 мл метанола и добавляли 0,31 мл 1,6-молярного раствора HCl в изопропаноле. В результате получили дигидрохлорид указанного в заголовке соединения, [α]D 20=-28° (с=1 в метаноле).
По описанным в приведенных выше примерах методам или аналогичным им методам можно получать также соединения формулы I, представленные в приведенной ниже таблице 3.
Соединения из примеров 5-38, указанные в приведенной ниже таблице 3, получали с помощью автоматизированного метода синтеза. С этой целью для каждой партии в микрореакционный сосуд дозировали по 200 мкл 0,25 н. водного расходного раствора соответствующего углеводного соединения формулы IV и воду практически полностью выпаривали в вакууме. Остаток растворяли в 200 мкл этанола. К этому раствору добавляли по 200 мкл расходного этанольного раствора рацемического, соответственно энантиомерно чистого (см. соответствующие данные в таблице 3) N-[2-(3,4-дихлорфенил)-4-(1-пиперазинил)бутил]-N-метилбензамида формулы I с концентрацией 0,25 моля/л, а также 200 мкл 0,25 н. расходного этанольного раствора соответствующей бороновой кислоты (дигидроксиборановое соединение) формулы III. Реакционную смесь сначала нагревали до 80°С и выдерживали при этой температуре в течение 2 ч, а затем охлаждали до комнатной температуры и смешивали с 1 мл этанола. После этого добавляли 100 мг основной ионообменной смолы Amberjet® и реакционный сосуд встряхивали в течение 2 ч. Ионообменник отфильтровывали, остаток дважды промывали этанолом порциями по 500 мкл и растворитель досуха выпаривали в вакууме. С целью определить чистоту полученного продукта, соответственно структуру полученного соединения из полученного остатка без дальнейшей очистки, отбирали образец для жидкостной хроматографии высокого разрешения (ЖХВР) и образец для автоматической масс-спектроскопии.
Другие соединения формулы I
Другие соединения формулы I (продолжение)
Пример 39
N-((2S)-2-(3,4-дихлорфенил)-4-{2-фурил[(4S)-2-тиоксо-1,3-диоксолан-4-ил]метил}-1-пиперазинил)бутил)-N-метилбензамид
К раствору 303 мг N-((2S)-2-(3,4-дихлорфенил)-4-{4[(2S)-2,3-дигидрокси-1-(2-фурил)пропил]-1-пиперазинил}бутил)-N-метилбензамида (полученного по описанной выше методике согласно примеру 2) в 10 мл безводного дихлорметана при комнатной температуре добавляли 240 мг N,N'-тиокарбонилдиимидазола. Реакционную смесь перемешивали в течение 20 ч при комнатной температуре (КТ) и затем концентрировали в вакууме, создаваемом водоструйным насосом. Образовавшийся остаток растворяли в 50 мл ЭУ и органическую фазу пятикратно промывали водой. Органическую фазу сушили над сульфатом натрия и затем растворитель выпаривали в вакууме (создаваемом сначала с помощью водоструйного насоса, а затем с помощью масляного насоса). После колоночной хроматографии образовавшейся пены желтоватого цвета (стационарная фаза: силикагель; подвижная фаза: н-гексан/ацетон в соотношении 1:1) получили 118 мг указанного в заголовке соединения в виде аморфного вещества.
1Н-ЯМР (CDCl3, КТ): 3,79 (d, 1Н); 5,10 (ddd, 1Н); 6,27 (1Н); 6,36 (1Н).
Пример 1
Капсулы, содержащие N-((2S)-2-(3,4-дихлорфенил)-4-{4-[(2S,3R,4R)-2,3,4,5-тетрагидрокси-1-(тиенил)пентил]-1-пиперазинил}бутил)-N-метилбензамид
Изготавливали капсулы со следующим составом из расчета на одну капсулу:
Действующее вещество, кукурузный крахмал и лактозу с использованием ЭУ перерабатывали в гомогенную пастообразную смесь. Эту пасту измельчали и полученный гранулят помещали на поддон и сушили при 45°С для удаления растворителя. Высушенный гранулят пропускали через измельчитель и смешивали в смесителе со следующими дополнительными вспомогательными веществами:
после чего смесь расфасовывали в капсулы вместимостью 400 мг (размер капсул 0).
Настоящее изобретение относится к 1-[1-(гетеро)арил-1-пергидроксиалкилметил]пиперазиновым соединениям общей формулы I
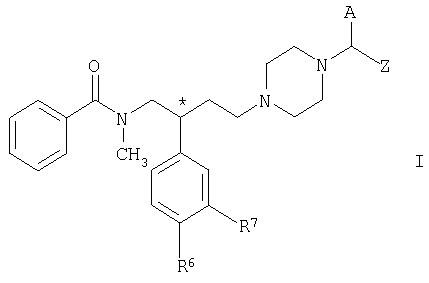
А обозначает нафтил, фенил, необязательно замещенный метокси, гетероарил, выбранный из группы, включающей тиенил, фурил, индолил или необязательно замещенный фенилом С3-С6алкенил,
Z обозначает подгруппу общей формулы
k, l, m и n обозначают 0 или 1; R6 и R7 обозначают галоген, и их физиологически приемлемым кислотно-аддитивным солям. Соединения обладают антагонистической активностью в отношении рецепторов тахикинина и могут быть использованы при лечении функциональных и воспалительных нарушений желудочно-кишечного тракта. Описан способ получения соединений и промежуточные продукты, используемые при осуществлении этого способа, а также содержащие указанные соединения лекарственные средства. 4 н. и 5 з.п. ф-лы, 3 табл.

в которой А обозначает нафтил, фенил, необязательно замещенный метокси, гетероарил, выбранный из группы, включающей тиенил, фурил, индолил или необязательно замещенный фенилом С3-С6алкенил,
Z обозначает подгруппу общей формулы
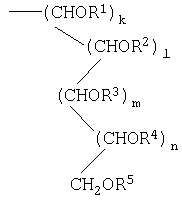
где R1 обозначает водород или (низш.)алканоил, либо вместе с одним из других заместителей из числа R2, R3, R4 и R5 может образовывать 5- или 6-членное кольцо, связанное мостиком, образованным карбонилом или тиокарбонилом,
R2 обозначает водород или (низш.)алканоил, либо вместе с одним из других заместителей из числа R1, R3, R4 и R5 может образовывать 5- или 6-членное кольцо, связанное мостиком, образованным карбонилом или тиокарбонилом,
R3 обозначает водород или (низш.)алканоил, либо вместе с одним из других заместителей из числа R1, R2, R4 и R5 может образовывать 5- или 6-членное кольцо, связанное мостиком, образованным карбонилом или тиокарбонилом,
R4 обозначает водород или (низш.)алканоил, либо вместе с одним из других заместителей из числа R1, R2, R3 и R5 может образовывать 5- или 6-членное кольцо, связанное мостиком, образованным карбонилом или тиокарбонилом,
R5 обозначает водород или (низш.)алканоил, либо вместе с одним из других заместителей из числа R1, R2, R3 и R4 может образовывать 5- или 6-членное кольцо, связанное мостиком, образованным карбонилом или тиокарбонилом,
k обозначает 0 или 1,
l обозначает 0 или 1,
m обозначает 0 или 1, и
n обозначает 0 или 1,
R6 обозначает галоген, и
R7 обозначает галоген,
а также физиологически приемлемые кислотно-аддитивные соли соединений формулы I.
N-((2S)-2-(3,4-дихлорфенил)-4-{4-[(2S,3R,4R)-2,3,4,5-тетрагидрокси-1-(3-тиенил)пентил]-1-пиперазинил}бутил)-N-метилбензамид;
N-((2S)-2-(3,4-дихлорфенил)-4-{4[(2S)-2,3-дигидрокси-1-(2-фурил)пропил]-1-пиперазинил}бутил)-N-метилбензамид;
(2S)-2-(ацетилокси)-3-{4-[(3S)-4-[бензоил(метил)амино]-3-(3,4-дихлорфенил)бутил]-1-пиперазинил}-3-(2-фурил)пропилацетат;
N-[(2S)-2-(3,4-дихлорфенил)-4-(4-{2-фурил[(4S)-2-оксо-1,3-диоксолан-4-ил]метил}-1-пиперазинил)бутил]-N-метилбензамид;
N-((2S)-2-(3,4-дихлорфенил)-4-{4-[(1S,2R)-2,3-дигидрокси-1-(3-тиенил)пропил]-1-пиперазинил}бутил)-N-метилбензамид;
N-((2S)-2-(3,4-дихлорфенил)-4-{4-[(2S)-2,3-дигидрокси-1-(3-фурил)пропил]-1-пиперазинил}бутил)-N-метилбензамид;
N-((2S)-2-(3,4-дихлорфенил)-4-{4-[(2R,3R,4R)-2,3,4,5-тетрагидрокси-1-(3-тиенил)пентил]-1-пиперазинил}бутил)-N-метилбензамид;
N-(2S)-2-(3,4-дихлорфенил)-4-{4-[(2S,3R,4R)-2,3,4,5-тетрагидрокси-1-(2-фурил)пентил]-1-пиперазинил}бутил)-N-метилбензамид;
N-((2S)-2-(3,4-дихлорфенил)-4-{4-[(2R)-2,3-дигидрокси-1-(3-тиенил)пропил]-1-пиперазинил}бутил)-N-метилбензамид;
N-((2S)-2-(3,4-дихлорфенил)-4-{4-[(2R,3S,4S)-2,3,4,5-тетрагидрокси-1-(3-тиенил)пентил]-1-пиперазинил}бутил)-N-метилбензамид;
N-((2S)-2-(3,4-дихлорфенил)-4-{4-[(2S,3R,4R)-2,3,4,5-тетрагидроксиацетил-1-(3-тиенил)пентил]-1-пиперазинил}бутил)-N-метилбензамид;
N-((2S)-2-(3,4-дихлорфенил)-4-{4-[(2S,3R)-2,3,4-тригидрокси-1-(3-тиенил)бутил]-1-пиперазинил}бутил)-N-метилбензамид и
N-((2S)-2-(3,4-дихлорфенил)-4-{2-фурил[(4S)-2-тиоксо-1,3-диоксолан-4-ил]метил}-1-пиперазинил)бутил)-N-метилбензамид.
в которой А обозначает нафтил, фенил, необязательно замещенный метокси, гетероарил, выбранный из группы, включающей тиенил, фурил, индолил или необязательно замещенный фенилом С3-С6алкенил,
Z обозначает подгруппу общей формулы
где R1 обозначает водород или (низш.)алканоил, либо вместе с одним из других заместителей из числа R2, R3, R4 и R5 может образовывать 5- или 6-членное кольцо, связанное мостиком, образованным карбонилом или тиокарбонилом,
R2 обозначает водород или (низш.)алканоил, либо вместе с одним из других заместителей из числа R1, R3, R4 и R5 может образовывать 5- или 6-членное кольцо, связанное мостиком, образованным карбонилом или тиокарбонилом,
R3 обозначает водород или (низш.)алканоил, либо вместе с одним из других заместителей из числа R1, R2, R4 и R5 может образовывать 5- или 6-членное кольцо, связанное мостиком, образованным карбонилом или тиокарбонилом,
R4 обозначает водород или (низш.)алканоил, либо вместе с одним из других заместителей из числа R1, R2, R3 и R5 может образовывать 5- или 6-членное кольцо, связанное мостиком, образованным карбонилом или тиокарбонилом,
R5 обозначает водород или (низш.)алканоил, либо вместе с одним из других заместителей из числа R1, R2, R3 и R4 может образовывать 5- или 6-членное кольцо, связанное мостиком, образованным карбонилом или тиокарбонилом,
k обозначает 0 или 1,
l обозначает 0 или 1,
m обозначает 0 или 1, и
n обозначает 0 или 1,
R6 обозначает галоген, и
R7 обозначает галоген,
а также физиологически приемлемых кислотно-аддитивных солей, отличающийся тем, что соединение общей формулы II
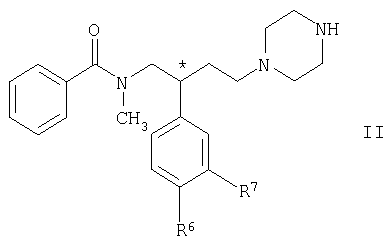
в которой R6 и R7 имеют указанные выше значения, подвергают взаимодействию с соединением общей формулы III
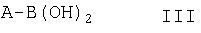
в которой А имеет указанное выше значение, и с соединением общей формулы IV

в которой R1, R2, R3, R4, R5, k, l, m и n имеют указанные выше значения, и затем либо в полученном соединении формулы I, в которой по меньшей мере один заместитель из числа R1, R2, R3, R4 и R5 обозначает водород, взаимодействием с соединением общей формулы VIII

в которой R8 обозначает алкил с прямой или разветвленной цепью, содержащий 1-3 атома углерода, при необходимости ацилируют подгруппу Z, либо в полученном соединении формулы I, в которой по меньшей мере два заместителя из числа R1, R2, R3, R4 и R5 обозначают водород, взаимодействием с реакционноспособным эквивалентным с точки зрения синтеза карбонильным или тиокарбонильным соединением при необходимости карбонилируют, соответственно тиокарбонилируют подгруппу Z, и полученное соединение формулы I при необходимости переводят в его кислотно-аддитивную соль или кислотно-аддитивную соль переводят в свободное соединение формулы I.
в которой R6 обозначает галоген и R7 обозначает галоген.
| Способ получения производных фенилпиперазина или их солей | 1977 |
|
SU727146A3 |
| Сырьевая смесь для изготовления керамзита | 1980 |
|
SU903349A1 |
| Способ очистки жидкости от окисляемыхОРгАНичЕСКиХ пРиМЕСЕй | 1978 |
|
SU802196A1 |

