Предложенное изобретение относится к новым способам получения кандесартана или защищенной формы кандесартана, соли кандесартана или сложного эфира кандесартана; к соединениям, которые можно использовать в заявленных способах, способам их получения, их применению в заявленных способах; к новой полиморфной форме кандесартана - цилексетилу, способу его получения и применения для получения лекарственного средства.
Биологически активное вещество кандесартан является ангиотензин-II-антагонистом, который сдерживает ангиотензин-II-рецептор типа 1 и допускается для лечения эссенциальной гипертонии. Кандесартан обладает хорошей переносимостью и может применяться перорально в форме кандесартана цилексетила.
Соединение кандесартан (химическое название 2-этокси-1-[[2'-(1H-тетразол-5-ил)бифенил-4-ил]метил]-1H-бензимидазол-7-карбоновая кислота) и его синтез были описаны впервые в ЕР 0459136. Обычно кандесартан реализуют не как свободные карбоновые кислоты, а в виде сложного эфира 1{[(циклогексилокси)карбонил]оксила}, названного также кандесартаном цилексетилом. Согласно ЕР 0459136 при получении кандесартана исходят из уже заранее образованных производных бифенила.
Также и в ЕР 0881212 при синтезе кандесартана исходят из уже предварительно образованных производных бифенила.
Патент CN 1510031 описывает связь С-С, при которой 2-этокси-1-(п-галофенил)метил-1Н-бензимидазол-7-карбоновая кислота-1-[[циклогексилокси)карбонил]окси]этиловый эфир, вступая в реакцию по Гриньяру с 5-(2-галофенил)-2-(1Н)-тетразолом, превращается в кандесартан цилексетил. При этом применяют никелевый катализатор Cl2Ni(PPh3)2.
Н.Matsunuga, Т.Euchi, К.Nishijima, Т.Enomoto, К.Sasaoki, N.Nakamura, Chemical&Pharmaceutical Bulletin 1999, 47(2), 182-186, описывают две полиморфные формы I и II кандесартана цилексетила.
Форму I получают при синтезе, описанном в ЕР 0459136.
В WO 2004/085426 описаны сольват 1,4-диоксана кандесартана цилексетила и другие полиморфные формы III и IV кандесартана цилексетила. В соответствии с этим форма III должна быть получена в результате того, что любую форму кандесартана цилексетила, но только не аморфный кандесартан цилексетил или кандесартан цилексетил полиморфной формы III, перекристаллизовывают из толуола.
Поэтому задачей изобретения является создание новых способов приготовления кандесартана в виде свободных карбоновых кислот, соли, сложного эфира или защищенной формы кандесартана, в частности кандесартана цилексетила, и предоставление новой формы кандесартана цилексетила. При этом, по возможности, необходимо отказаться от ядовитых, аллергенных, карциногенных и/или тератогенных соединений.
Поставленная задача решается при помощи способа получения кандесартана, защищенной формы кандесартана, соли кандесартана или сложного эфира кандесартана, в частности кандесартана цилексетила, который включает следующие стадии:
(а) приготовление и реакция обмена соединения формулы (I)

где R является водородом, незамещенным или замещенным радикалом алкила или арила, (циклогексилоксикарбонилокси)этилом, предпочтительно метилом,
Y1 представляет собой группу, которая при образовании связи С-С должна быть способна вступить в реакцию сочетания, в которую входит, кроме того, группа Y2,
с соединением формулы (II), имеющим группу Y2
 ,
,
где R1 является защитной группой тетразолила или водородом, при образовании защищенной формы кандесартана или кандесартана цилексетила или другого сложного эфира кандесартана, и, при необходимости,
(b) превращения в кандесартан, кандесартан цилексетил или в физиологически совместимую соль.
Выгодным является заявленный способ, в котором в формуле (I) радикал R представляет собой С1-С4-алкил, в частности метил.
Если в формуле (I) R является радикалом С1-С4-алкила, в частности метила, то в отличие от соединения, в котором R является (циклогексилоксикарбонилокси)этилом, имеется сравнительно ограниченно функционализированное соединение, которое менее чувствительно к соответствующим условиям реакции. По причине сравнительно ограниченной функционализации выделенное вещество также меньше подходит для того, чтобы снизить активность реактивов и/или катализаторов, используемых в реакции. Такое нарушение может произойти, например, в результате комплексирования соединений, используемых в реакции и содержащих металлы или металл, при помощи свободных пар электронов атомов кислорода радикала (циклогексилоксикарбонилокси)этила. Таким образом можно сократить число или количество побочных продуктов и повысить выход продукта.
Если в формуле (I) R не является водородом, то стадия (b) заявленного способа включает гидролиз получаемого на стадии (а) сложного эфира предпочтительно при помощи обработки едким натром в EtOH.
Также возможно использование других средств для гидролиза сложных эфиров. Специалист может выбрать такие средства.
Стадия (b) может включать согласно изобретению, кроме того, реакцию обмена кандесартана с соединением формулы (IV)

где Z1 является отходящей группой, при образовании кандесартана цилексетила предпочтительно в присутствии NaI и K2СО3.
В заявленных способах R1 может быть выбран из группы, включающей водород, третичный бутил и трифенилметил. Предпочтительно, если R1 является трифенилметилом.
В заявленных способах Y1 можно выбрать из следующих функциональных групп:
- галоген, предпочтительно бром,
- В(OR4)2, причем каждый из радикалов R4 независимо друг от друга обозначает водород, алкил, арил или алкиларил, предпочтительно водород,
- радикал триалкилолова или
- радикал магний(II)галогенида, причем, если Y2 обозначает галоген, то Y1 обозначает В(OR4), радикал триалкилолова или галогенида магния(II) и наоборот.
В заявленных способах Y2 может обозначать:
- галоген, предпочтительно бром,
- B(OR4)2, причем каждый из радикалов R4 независимо друг от друга обозначает водород, алкил, арил или алкиларил, предпочтительно водород,
- радикал триалкилолова или
радикал магний(II)галогенида, причем, если Y1 обозначает галоген, то Y2 обозначает В(OR4), радикал триалкилолова или галогенида магния(II) и наоборот.
В предпочтительном варианте выполнения изобретения Y1 и Y2 выбраны из одной из следующих комбинаций:
Y1 = галоген, предпочтительно бром, а Y2 = В(OR4)2, причем каждый из радикалов R4 независимо друг от друга обозначает водород, алкил, арил или алкиларил, предпочтительно водород,
Y1 = галоген, предпочтительно бром, а Y2 = радикал триалкилолова,
Y1 = галоген, предпочтительно бром, а Y2 = радикал галогенида магния(II),
Y1=B(OR4)2, причем каждый из радикалов R4 независимо друг от друга обозначает водород, алкил, арил или алкиларил, предпочтительно водород, а Y2 = галоген, предпочтительно бром,
Y1 = радикал триалкилолова и Y2 = галоген, предпочтительно бром,
Y1 = радикал галогенида магния(II), а Y2 = галоген, предпочтительно бром.
Если в формуле (I) R обозначает метил, то появляется сложный метиловый эфир кандесартана, который с помощью реакции обмена с NaOH в EtOH можно перевести в кандесартан или соль кандесартана, которую снова можно перевести в кандесартан цилексетил.
В предпочтительном варианте выполнения изобретения происходит реакция обмена соединения общей формулы (I) с соединением общей формулы (II) с молярным соотношением от 0,2:1 до 2:1, наиболее предпочтительно от 0,3:1 до 0,8:1.
В заявленных способах реакция радикалов Y1 и Y2 приводит к соединению С-С.
Это соединение С-С можно осуществить в присутствии реактивов Гриньяра. Они очень выгодны, так как позволяют реализовать заявленный способ при сравнительно незначительных материальных затратах.
В заявленных способах можно использовать, кроме того, один катализатор или несколько катализаторов, предпочтительно включающих один или несколько переходных металлов, в частности марганец, хром, железо, кобальт, никель или палладий. Эти катализаторы ускоряют течение реакции сочетания.
Применение таких катализаторов делает способ особенно экономичным. Катализатор применяют обычно в количестве от 0,001 до 20 мол.%, предпочтительно от 0,01 до 15 и, в частности, от 0,1 до 10 мол.%, в пересчете на молярное количество соединения формулы (I).
Катализатор или катализаторы можно выбрать из группы, состоящей из MnCl2, CrCl3, FeCl2, Fe(асас)3, FeCl3, Fe(salen)Cl, CoCl2(dppe), CoCl2(dpph), Co(acac)2, CoCl2(dppb), Pd(PPh3)4, NiCl2(PPh3)2.
Особенно предпочтительными являются Pd(PPh3)4, NiCl2(PPh3)2.
В одном из предпочтительных вариантов выполнения изобретения катализаторы можно использовать вместе с активатором. Этот активатор переводит атомы металла катализаторов в нулевую степень окисления.
Примерами таких активаторов являются цинк (предпочтительно в виде цинкового порошка), борогидрид натрия, гидрид лития-алюминия или органические соединения алюминия, магния или лития (предпочтительно бутиллитий или DIBAH (гидрид дизобутилалюминия).
Обычно количественное соотношение активатора и катализатора составляет от 25:1 до 1:1, предпочтительно от 18:1 до 2:1.
В другом предпочтительном варианте выполнения катализаторы можно применять вместе со стабилизатором. Этот стабилизатор стабилизирует атомы металла катализаторов в нулевой степени окисления.
Примерами таких стабилизаторов являются основания Льюиса, предпочтительно фосфаны, особенно предпочтительно триарилфосфаны и триалкилфосфаны, в частности трифенилфосфан.
Обычно количественное соотношение стабилизатора и катализатора составляет от 10:1 до 1:1, предпочтительно от 5:1 до 1,5:1.
В частности предпочтительно, если катализатор, активатор и стабилизатор применяют вместе.
Применение таких катализаторов в реакциях сочетания С-С, в которых присутствуют железо, марганец, хром или кобальт, особенно выгодно, так как содержащиеся здесь металлы сравнительно благоприятны.
В альтернативной форме выполнения изобретения катализатор или катализаторы могут быть выбраны из группы катализаторов, свободных от фосфанов и содержащих предпочтительно железо. Таким образом можно избежать недостатков, связанных с применением фосфансодержащих катализаторов, точнее, в частности, у которых токсичность и тенденция связываться с кислородом воздуха вызывает опасность самовоспламенения.
В заявленных способах, кроме того, можно использовать один или несколько растворителей, представленных в следующем перечне: THF (тетрагидрофуран), THF/NMP (N-метилпирролидон), Et2O (простой диэтиловый эфир), DME (диметоксиэтан), бензол и толуол. Особенно предпочтительным является THF. Растворители можно применять, при необходимости, в смеси с водой.
Задача решается также с помощью способа получения соединения с указанной выше формулой (I), который включает следующие стадии.
Приготовление и реакция обмена соединения формулы (III)
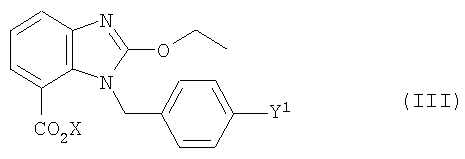
где Y1 имеет значение, указанное выше,
Х обозначает группу, которая при образовании связи O-С должна быть в состоянии вступить в реакцию, в которую входит, кроме того, группа Z1,
с соединением формулы (IV)
где Z1 обозначает отходящую группу,
при образовании соединения формулы (I).
Согласно одному варианту выполнения изобретения Х может обозначать щелочной металл или предпочтительно водород, и/или Z1 может обозначать галоген, предпочтительно йод.
Кроме того, задача решается с помощью способа получения соединения, имеющего приведенную выше формулу (III), который включает следующую стадию:
приготовление и снятие защиты соединения формулы (V)
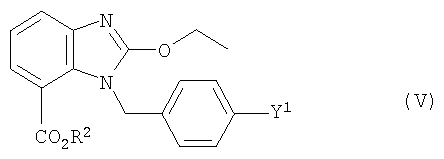
где Y1 имеет значение, указанное выше, а
R2 обозначает группу, заменяемую на Х при образовании соединения формулы (III), причем Х имеет то же самое значение, что и выше в связи с формулой (III).
Согласно изобретению R2 можно выбрать из одной из следующих функциональных групп: замещенный или незамещенный C1-С6-низший алкил, бензил или арил, предпочтительно этил (СН2СН3) и наиболее предпочтительно метил (СН3).
Кроме того, задача решается с помощью способа получения соединения, имеющего приведенную выше формулу (V), который включает следующую стадию:
приготовление и реакция обмена соединения формулы (VI)
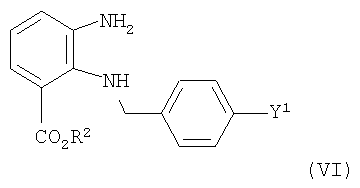
где Y1 и R2 имеют те же значения, что и выше,
с карбонилирующим реактивом или предпочтительно C(OEt)4 при образовании соединения формулы (V).
Кроме того, в таком способе при замещении можно использовать Aс2O.
Также задача решается с помощью способа получения соединения формулы (VI), как определено выше, который включает следующую стадию:
приготовление соединения формулы (VII)
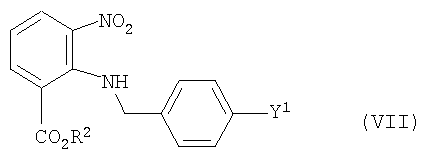 ,
,
где Y1 и R2 имеют те же значения, что и выше, и преобразование имеющейся в ней нитрогруппы в аминогруппу.
Согласно изобретению преобразование нитрогруппы в аминогруппу осуществляется с привлечением неблагородных металлов, каталитического гидрирования, электролитическим методом или предпочтительно с помощью SnCl2.
Кроме того, задача решается с помощью способа получения соединения формулы (VII), как определено выше, который включает следующий этап:
приготовление и снятие защиты соединения формулы (VIII)
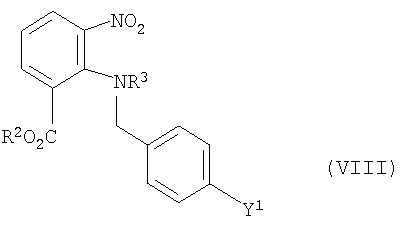
где Y1 и R2 имеют те же значения, что и выше, и R3 обозначает защитную группу, заменяемую на Н,
при образовании соединения формулы (VII), как определено выше.
Согласно изобретению R3 обозначает группу карбоксилалкила, предпочтительно группу карбокси-трет-бутила (-СООС-(СН3)3).
Кроме того, задача решается с помощью способа получения соединения формулы (VIII), как определено выше, который предполагает следующий этап:
приготовление и реакция обмена соединения формулы (IX)
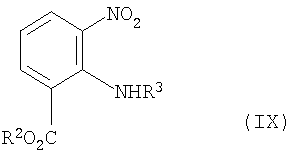
где R2 и R3 имеют те же значения, что и выше, с соединением формулы (X)
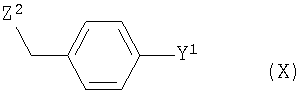
где Y1 имеет то же значение, что и выше, а
Y2 обозначает отходящую группу,
при образовании соединения формулы (VIII).
Согласно изобретению Z2 можно выбрать из одной из следующих функциональных групп: Cl, I и предпочтительно Br.
В одном предпочтительном варианте выполнения изобретения можно вести реакцию обмена соединения формулы (IX) с соединением формулы (X) в присутствии основных соединений, предпочтительно карбонатов щелочных и щелочноземельных металлов, в частности Na2CO3 или K2СО3.
Согласно изобретению можно приготовить по соответствующим заявленным способам получения соединений формул (VII), (VI), (III) или (I) каждый раз необходимое соединение формул (VIII), (VII), (V) или (III) одним или несколькими заявленными способами.
Согласно изобретению можно приготовить, кроме того, по заявленному способу получения кандесартана соли кандесартана, сложного эфира кандесартана или защищенной формы кандесартана соединение формулы (I) с помощью одного или нескольких заявленных способов.
Задача решается, кроме того, с помощью промежуточного продукта, имеющего формулу
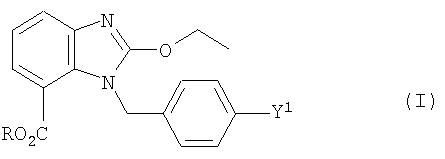
где R обозначает водород, незамещенный или замещенный радикал алкила или арила, предпочтительно (циклогексилоксикарбонилокси)этил, и где Y1 имеет то же значение, что и выше.
В предпочтительном промежуточном продукте формулы (I) Y1 обозначает
- галоген,
- В(OR4)2, причем каждый из радикалов R4 обозначает независимо друг от друга водород, алкил, арил или алкиларил, предпочтительно водород,
- радикал триалкилолова или
- радикал галогенида магния(II).
В особенно предпочтительном промежуточном продукте формулы (I) Y1 обозначает бром.
Кроме того, для заявленного промежуточного продукта формулы (I) действует предпочтительно такое условие, что, если Y1 обозначает Cl, Br или I, то R не является водородом, этилом или {[(циклогексилокси)карбонил]окси}этилом. Это условие, однако, не относится к заявленному способу.
Кроме того, задача решается с помощью промежуточного продукта, имеющего формулу (III)
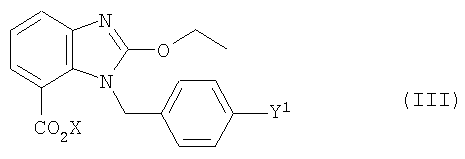
где Y1 и Х имеют значения, указанные выше.
Кроме того, для заявленного промежуточного продукта действует предпочтительно такое соответствие, что, если Y1 обозначает Cl, Br или I, то R не является водородом. Это условие не относится, однако, к заявленному способу.
Кроме того, задача решается с помощью промежуточного продукта, имеющего формулу (V)
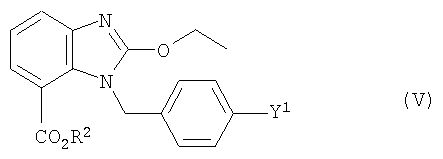
где Y1 и R2 имеют значения, приведенные выше.
В предпочтительном промежуточном продукте формулы (V) Y1 обозначает бром, а R2 обозначает группу метила или низшего С3-С6-алкила.
Кроме того, для заявленного промежуточного продукта формулы (V) действует предпочтительно такое соответствие, что, если Y1 обозначает Cl, Br или I, то R2 не является этилом. Это условие, однако, не относится к заявленному способу.
Кроме того, задача решается с помощью промежуточного продукта, имеющего формулу (VI)
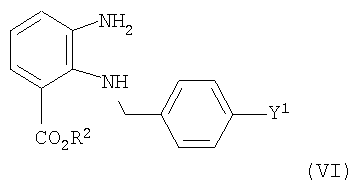
где Y1 и R2 имеют значения, приведенные выше.
В предпочтительном промежуточном продукте формулы (VI) Y1 обозначает бром, а R2 обозначает группу метила или низшего С3-С6-алкила.
Кроме этого, для заявленного промежуточного продукта формулы (VI) действует предпочтительно такое соответствие, что, если Y1 обозначает Cl, Br или I, то R2 не является этилом. Это условие, однако, не относится к заявленному способу.
Кроме того, задача решается с помощью промежуточного продукта, имеющего формулу
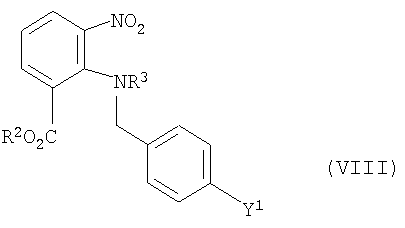
где Y1, R2 и R3 имеют значения, приведенные выше.
В предпочтительном промежуточном продукте формулы (VIII) Y1 обозначает бром, R2 обозначает группу метила или низшего С3-С6-алкила, a R3 обозначает группу карбоксиалкила, предпочтительно группу карбокси-трет-бутила (СООС(СН3)3).
Кроме того, для заявленного промежуточного продукта формулы (VIII) действует предпочтительно такое соответствие, что, если Y1 обозначает Cl, Br или I, то R2 не является этилом, а R3 не является карбокси-трет-бутилом. Эта условие, однако, не относится к заявленному способу.
Согласно изобретению задача решается, кроме того, благодаря применению заявленных промежуточных продуктов и/или соединения (VII) в способах получения кандесартана, сложных эфиров кандесартана или кандесартана цилексетила, причем предпочтительно R2 = метил, а Y1=Br.
Задача решается, кроме того, с помощью способа представления полиморфной формы кандесартана цилексетила, включающей
- обработку кандесартана цилексетила дихлорметаном и простым диэтиловым эфиром с получением прозрачного раствора,
- сгущение прозрачного раствора и выпадение в осадок кандесантана цилексетила.
Задача решается также с помощью полиморфной формы кандесартана цилексетила, которую получают с помощью заявленного способа.
Задача решается с помощью полиморфной формы кандесартана цилексетила, которую можно описать с помощью одного или нескольких следующих физических параметров:
- сигналы на дифрактограмме рентгеноструктурного анализа с применением Cu-Кα-излучения, выраженного в 2 θ при 7,32, 8,20, 9,10, 14,68, 18,88, 24,18°, причем предпочтительно все значения включают стандартное отклонение, составляющее ±0,2°;
- полученные с помощью дифракции рентгеновских лучей (XRD) расстояния между плоскостями решетки d=12,065, 10,773, 9,711, 6,029, 4,696, 3,678 Ǻ (ангстрем), причем предпочтительно все значения включают стандартное отклонение, составляющее ±0,1 Ǻ;
- полученная при помощи дифференциальной сканирующей калориметрии (DSC) точка плавления примерно при 130,7°С и/или
- характерная полоса спектра поглощения в инфракрасном спектре примерно при 1733 см-1.
В заявленной полиморфной форме кандесартана цилексетила можно наблюдать на дифрактограмме рентгеноструктурного анализа всякий раз относительную интенсивность сигналов, равную примерно 100, 29,6, 20,2, 45,2, 20,5, 11,4.
Задача решается, наконец, путем приготовления заявленной полиморфной формы кандесартана цилексетила с помощью заявленного способа.
Согласно изобретению заявленную полиморфную форму кандесартана цилексетила можно использовать для получения лекарственного препарата.
Изобретение поясняется далее более подробно со ссылками на фиг.1-3 и при помощи примеров выполнения.
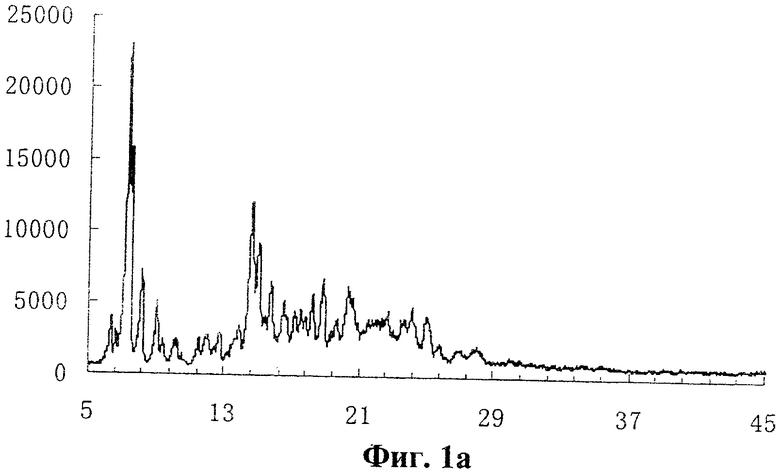
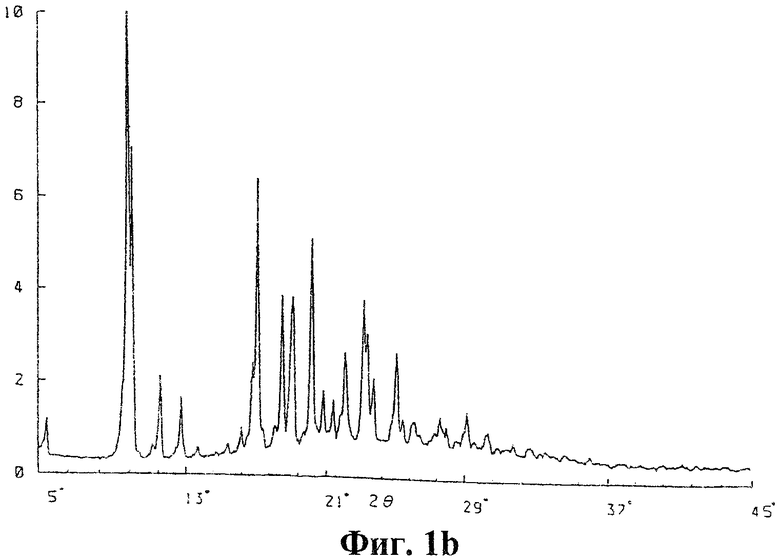
фиг.1a дифрактограмма рентгеноструктурного анализа заявленной полиморфной формы кандесартана цилексетила;
фиг.1b дифрактограмма рентгеноструктурного анализа кандесартана цилексетила полиморфной формы I согласно уровню техники;
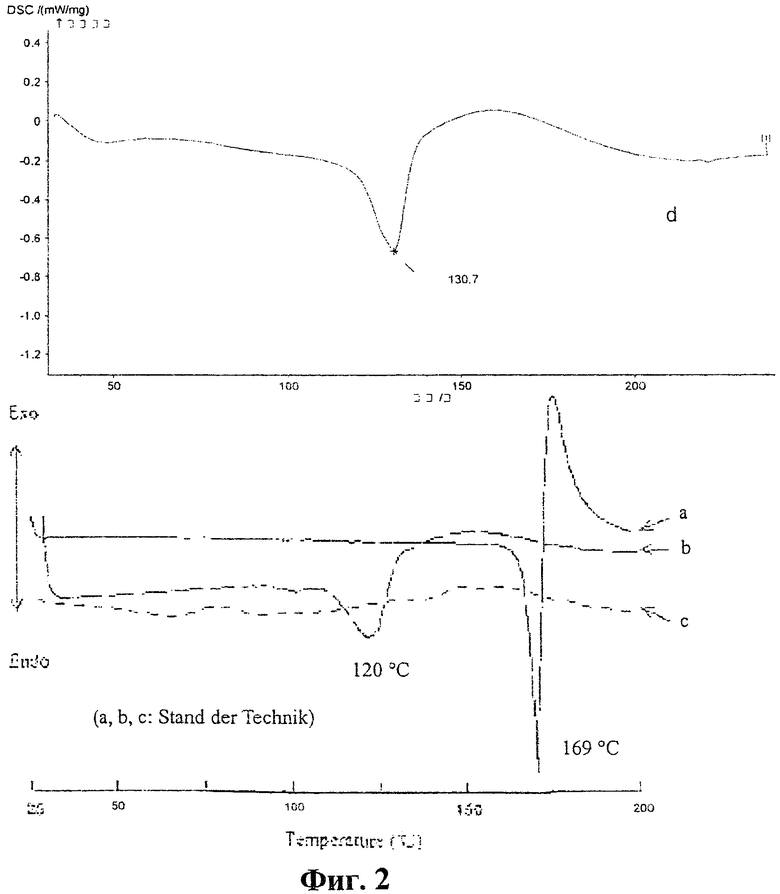
фиг.2 кривая "d" дифференциальной сканирующей калориметрии (DSC)заявленной полиморфной формы, а также кривые DSC полиморфных форм I и II («а» и «b»), а также аморфного кандесартана цилексетила «с» согласно уровню техники;
фиг.3 инфракрасный спектр заявленной полиморфной формы.
Примеры выполнения
Следующие примеры выполнения показывают соединения (е), (g), (h), (i) и (j). Соответствующие соединения формул (е), (g), (h), (i) и (j) соответствуют заявленным промежуточным продуктам, имеющим формулы (VIII), (VI), (V), (I) и (III), где Y1=Br, R2 = метил, R3 = CO2tBu, Х=H, R={[(циклогексилокси)карбонил]окси}этил.
Целевое соединение кандесартан или кандесартан цилексетил можно представить, исходя из соответствующих промежуточных соединений; специалисту нетрудно это узнать на основании примеров выполнения.
Общие условия реакций
Все сухие растворители (CH2Cl2, тетрагидрофуран (THF), Et2O, бензол, толуол, DMF, MeCN) высушивали стандартными способами, т.е. путем удаления воды и кислорода и дистилляции перед использованием. Реакции, описанные ниже, проводили, если необходимо, в атмосфере инертного газа (N2 или Ar) и проверяли с помощью тонкослойной хроматографии (Thin Layer Chromatography, TLC). При экстракциях использовали простой диэтиловый эфир, этилацетат или хлороформ. Экстракты высушивали обычным способом с помощью безводных MgSO4 или Na2SO4, если не дано ничего другого. Продукты реакции, если необходимо, очищали методом колоночной хроматографии с применением, например, петролейного эфира (60-90°С)/этилацетата или петролейного эфира (30-60°С)/этилацетата в качестве элюентов. Если использовали пластинки типа GF254, то в качестве индикаторов применяли йод или этанольный раствор фосфорномолибденовой кислоты. Силикагель для хроматографии (зернистость 200-300) и TLC (GF254) изготавливали по Qingdao Sea Chemical Factory и Yantai Chemical Factory. Все растворители и реактивы имели аналитическую или химическую чистоту.
Точку кипения определяли с помощью ХТ4-100х micro-melting point tester. Поглощение инфракрасных спектров происходило при помощи прессованных изделий из KBr или пленок из ПЭ на спектрометрах Nicolet AVATAR 360 FT-IR и Nicolet NEXUS 670 FT-IR. Измерения ядерно-магнитного резонанса проводили с помощью спектрометров для измерения ядерно-магнитного резонанса фирм Varian (Mercury-300) и Bruker (AM-400) с SiMe4 в качестве внутреннего стандарта в COCl3, если не дано ничего другого. LRMS определяли с помощью масс-спектрометра HP-5988 с применением ЕI при 70 электрон-вольт, если не дано ничего другого. HRMS измеряли с помощью масс-спектрометра Bruker Daltonics APEX П 47е FT-ICR.
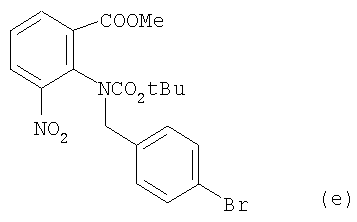
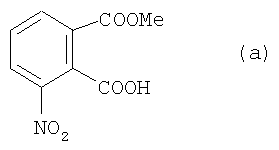
1. Получение (е)
а) 3-Нитрофталевую кислоту (35 г) растворяли в 215 мл метанола, содержащего 20 мл концентрированного H2SO4, в 500-миллилитровой круглодонной колбе. После 24-часового кипячения с обратным потоком реакционную смесь сгущали в вакууме. С помощью водного, насыщенного раствора К2СО3 устанавливали водородный показатель рН остатка 11-12. После этого реакционную смесь экстрагировали с этилацетатом, а показатель рН составлял 2-3 после добавления концентрированной соляной кислоты. Остаток экстрагировали (2×1000 мл CP2Cl2). Органические фазы соединяли и промывали водой и водным раствором NaCl, высушивали с MgSO4 и сгущали.
Полученное голубое твердое вещество можно сразу использовать дальше без дополнительной очистки; продукт имеет формулу
b) 30 г соединения (а) со стадии а) и 16 мл SOCl2 растворяли в 200 мл сухого бензола в 500-миллилитровой круглодонной колбе. Смесь кипятили с обратным потоком в течение 3 часов, а затем сгущали, причем получали белый порошок; продукт имеет формулу

c) 30 г соединения (b) со стадии b) растворяли в 120 мл ацетона в 500-миллилитровой круглодонной колбе. В реакционную смесь добавляли медленно, по каплям, водный раствор 120 мл NaN3 (184 ммоля, 12 г). После этого реакционную смесь мешали в течение часа. Смесь фильтровали, промывали ледяной водой и высушивали в вакууме. Полученный продукт имеет формулу
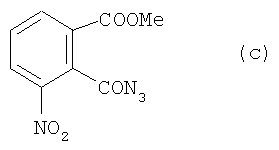
d) 28 г соединения (с) со стадии с) и 112 мл третичного бутанола медленно нагревали в 250-миллилитровой круглодонной колбе и кипятили с обратным потоком в течение 2 часов. Реакционную смесь сгущали в вакууме и очищали при помощи колоночной хроматографии (РЕ: AcOEt, от 16:1 до 4:1). Полученный таким образом сырой продукт перекристаллизовался в метаноле в желтые кристаллы и имеет формулу

e) 10,8 г соединения (d) со стадии d) (36 ммолей) и бромистый 4-бромбензил (40 ммолей, 10,0 г) растворили в CH3CN (200 мл) в 500 -миллилитровой круглодонной колбе. Добавили порошок К2СО3 (36 ммоля, 5,0 г). Реакционную смесь кипятили с обратным потоком в течение 10 часов, сгущали в вакууме и экстрагировали этилацетатом (2×500 мл). Экстракт промывали водой и водным раствором NaCl, высушивали безводным Na2SO4, сгущали в вакууме и перекристаллизовывали из AcOEt/РЕ, что дало целевое соединение (е) (15,4 г, точка плавления 107-108°С) в виде бесцветных кристаллов. Выход составил 92%. Данные анализа целевого соединения (е): 1H ЯМР (CDCl3, 300 МГц): δ 1,30 (9Н, s, t-Bu), 3,67 (3Н, s, OMe), 4,57 (2Н, dd, J=14,7 Гц, CH2), 7,01 (2Н, d, J=8,1 Гц, ArH), 7,32 (2Н, d, J=8,1 Гц ArH), 7,46 (1H, t, J=8,1 Гц, ArH), 7,89 (1H, d, J=8,1, Гц, ArH), 8,00 (1H, d, J=8,1 Гц, ArH); 13С ЯМР (CDCl3, 75 МГц): δ 27,8, 52,7, 53,1, 81,2, 121,9, 127,8, 128,1, 128,3, 131,0, 131,3, 131,6, 132,2, 134,8, 134,9, 135,3, 148,6, 153,5, 164,7; масс-спектрометрия (EI) m/z (%): 464 (M+, 0,1) 365(9), 348(10), 316(3), 302(3), 235(4), 185(27), 169(31), 57(100).
Инфракрасн. (пленка, см-1) λмакс = 3087, 2978, 2952, 1711, 1601, 1536, 1484, 1453, 1384, 1367, 1293, 1164, 1128, 1014, 984, 864, 766, 704.
2. Получение (g)

а) соединение (е) из примера 1 (3,2 ммоля, 15,4 г) растворяли в смеси из CF3COOH (32 мл) и CH2Cl2 (20 мл) в 100-миллилитровой круглодонной колбе и мешали в течение часа при комнатной температуре. Реакционную смесь сгущали в вакууме. После этого добавляли метанол (30 мл) и добавлением концентрированного водного раствора NaHCO3 устанавливали показатель рН 10. Желтый осадок отфильтровывали и перекристаллизовывали из этанола, что давало соединение формулы
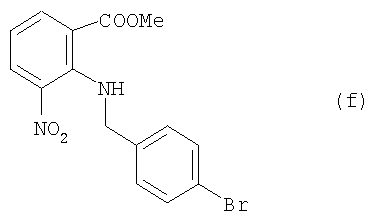
(9,45 г, точка плавления 111-112°С) в виде желтых игольчатых кристаллов. Выход составил 84%. Данные анализа (f): 1H ЯМР (CDCl3, 300 МГц): δ 3,87 (3Н, s, ОМе), 4,09 (2Н, d, J=5,1 Гц, CH2), 6,71 (1H, t, J=7,5 Гц, ArH), 7,16 (2Н, d, J=8,4 Гц ArH), 7,45 (2Н, d, J=8,4 Гц, ArH), 7,96 (1H, d, J=7,5 Гц, ArH), 8,10 (1H, d, J=7,5 Гц, ArH), 8,77 (1H, br, ArH); 13С ЯМР (CDCl3, 75 МГц): δ 50,2, 52,4, 115,0, 116,6, 121,8, 129,6, 131,6, 131,9, 136,6, 136,9, 137,4, 145,1, 167,7; мacc-спектрометрия (EI) m/z (%): 364 (M+, 2) 346 (16), 302 (15), 235 (13), 207 (8), 183 (100), 169 (80), 89 (64). Инфракрасн. (пленка, см-1) λмакс = 3306, 3094, 3011, 2953, 1937, 1715, 1692, 1601, 1575, 1527, 1486, 1441, 1402, 1339, 1260, 1198, 1116, 107, 1010, 971, 893, 833, 808, 766, 734, 717, 670, 644, 589.
b) Соединение (f) стадии а) (7,4 г, 20 ммоля) и сухой этанол (40 мл) помещали в 100-миллилитровую круглодонную колбу. Порциями добавляли SnCl2·2Н2О (102 миллимоля, 23,0 г). Реакционную смесь нагревали в течение 2 часов до температуры 80°С и удаляли этанол. Остаток растворяли в этилацетате (60 мл), а водородный показатель рН устанавливали 4 N NaOH на 11-12. Органическую фазу отделяли, а водную фазу экстрагировали с этилацетатом. Объединенные органические фазы промывали водой и водным раствором NaCl, высушивали безводным Na2SO4 и сгущали. Полученный продукт
можно использовать в дальнейшем сразу без дополнительной очистки. Данные анализа соединения (g): 1Н ЯМР (диметилсульфоксид, 300 Мгц) δ 3,72 (3Н, s), 4,36 (2Н, s), 7,08 (1Н, m), 7,31 (2Н, brd, J=8,1 Гц), 7,44-7,51 (4H, m), 8,69 (3Н, brs); 13С ЯМР (диметилсульфоксид, 75 Гц) δ 51,1, 53,2, 121,9, 123,0, 124,4, 126,4, 127,2, 131,8, 131,9, 132,0, 132,1, 132,2, 136,2, 167,8; масс-спектрометрия (ESI) [M+H]+ 335,0088 (в пересчете 335,0089).
3. Получение (h)
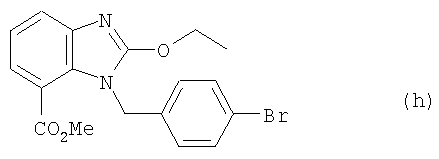
Соединение (g) (31 ммоль) из примера 2, C(OEt)4 (46,5 ммоль, 9,8 мл) и уксусную кислоту (31 ммоль, 1,8 мл) смешивали в 50-миллилитровой круглодонной колбе и перемешивали в течение 6 часов при температуре 80°С. После этого реакционную смесь сгущали, добавляли насыщенный раствор NaHCO3 и устанавливали водородный показатель рН 10, смесь экстрагировали этилацетатом (2×500 мл), высушивали безводным Na2SO4 и сгущали при пониженном давлении. Полученное твердое вещество перекристаллизовывали из этилацетата, что давало целевое соединение (h) (7,9 г, точка плавления 122-123°С). Исходя из использованного количества исходного соединения (f) (см. выше, пример 2), выход составил 65%. Данные анализа соединения (h): 1H ЯМР (CDCl3, 300 МГц) δ 1,45 (3Н, t, J=7,2 Гц, Me), 3,74 (3H, s, OMe), 4,63 (2Н, q, J=7,2 Гц, СН2), 4,63 (2Н, q, J=7,2 Гц СН2), 5,56 (2Н, s, СН2), 6,85 (2Н, d, J=9 Гц), 7,16 (1H, t, J=8,4 Гц), 7,34 (2H, d, J=9 Гц), 7,57 (1H, d, J=8,4 Гц), 7,72 (1H, d, J=8,4 Гц); 13С ЯМР (CDCl3, 75 МГц) δ 14,6, 46,7, 52,2, 66,7, 115,5, 120,9, 122,1, 123,7, 128,2, 131,5, 136,4, 141,9, 158,6, 166,7; масс-спектрометрия (EI) m/z (%): 388 (M+, 14) 341 (2), 327 (3), 299 (2), 249 (5), 221 (5), 192 (3), 169 (100), 89 (28). Инфракрасн. (пленка, см-1) λмакс = 3407, 3058, 2991, 2952, 2852, 1903, 1709, 1615, 1549, 1479, 1430, 1382, 1248, 1128, 1036, 927, 869, 800, 743, 687.
4. Получение (i)
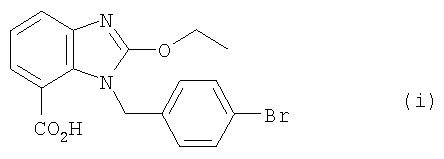
Соединение (h) (10,3 ммоля, 4,0 г), 1 М NaOH (30 мл) и этанол (30 мл) смешивали в 100-миллилитровой круглодонной колбе и мешали в течение часа при температуре 80°С. После этого реакционную смесь для удаления растворителя сгущали при пониженном давлении. Затем к остатку добавляли воду (50 мл) и этилацетат (50 мл), а водную фазу отделяли. При добавлении концентрированной соляной кислоты устанавливали водородный показатель рН=2-3 и получали соединение (i) в виде белого твердого вещества, которое высушивали в вакууме. Выход составлял 3,6 г, 95%. Полученное таким образом соединение (i) можно сразу перерабатывать дальше без дополнительной очистки. Данные анализа соединения (i): 1Н ЯМР (d-диметилсульфоксид, 300 МГц): δ 1,36 (3Н, t, J=6,9 Гц, СН3), 4,56 (2Н, q, J=6,9 Гц, CH2), 5,55 (2Н, s, CH2), 6,89 (2Н, d, J=8,1 Гц), 7,15 (1Н, t, J=7,8 Гц), 7,43-7,51 Гц (3Н, m), 7,64 (1Н, d, J=8,1 Гц); 13С ЯМР (d-диметилсульфоксид, 75 МГц) δ 15,0, 46,8, 67,2, 117,3, 120,9, 121,5, 122,2, 124,1, 129,2, 131,8, 132,1, 132,2, 137,7, 142,3, 158,9, 168,1; масс-спектрометрия (ESI) [М+Н]+ 375,0193 (в пересчете 375,0339).
5. Получение (j)

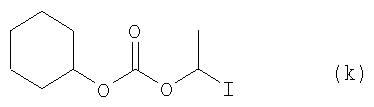
Соединение (i) из примера 4 (9,7 ммоля, 3,64 г), 1-{[(циклогексилокси)карбонил]окси}-1-йодэтан (к) из следующего примера 6 (19,5 ммоля, 4,05 г), безводный К2СО3 (9,7 ммоля, 1,34 г), NaI (42,7 ммоля, 6,4 г) и сухой DMF (40 мл) смешивали в 100-миллилитровой круглодонной колбе и перемешивали при температуре 60°С в течение 13 часов. Затем сгущали при пониженном давлении, реакционную смесь экстрагировали этилацетатом (2×200 мл). Органические фазы отделяли и промывали водой и водным раствором NaCl, высушивали Na2SO4, сгущали и очищали с помощью колоночной хроматографии (РЕ: AcOEt, от 16:1 до 4:1), получая целевое соединение (j) (2,8 г). Добавив концентрированную соляную кислоту, устанавливали водородный показатель рН=2-3. Затем реакционную смесь экстрагировали с этилацетатом (100 мл). Органическую фазу отделяли, высушивали безводным Na2SO4 и сгущали для получения следующих 1,2 г соединения (j). Выход составил 79%. Данные анализа соединения (j): 1H ЯМР (CDCl3, 300 МГц) δ 0,84-1,94 (16Н, m), 4,65 (3H, m, СН2, СН), 5,24 (2Н, s, СН2), 6,88 (3H, m), 7,16 (1H, t, J=8,4 Гц), 7,34 (2H, d, J=8,l Гц), 7,60 (1H, d, J=8,4 Гц), 7,73 (1Н, d, J=8,4 Гц); 13С ЯМР (CDCl3, 75 МГц) δ 14,6, 19,5, 23,6, 25,1, 31,3, 46,7, 66,7, 77,5, 91,6, 114,4, 120,9, 121,1, 122,7, 124,1, 128,6, 131,6, 131,9, 136,3, 141,9, 152,5, 158,6, 164,0; масс-спектрометрия (FAB): найдено 567,5 (М++Na), 545,5 (М++1); Инфракрасн. (пленка, см-1) λмакс = 3413, 2938, 2860, 1754, 1722, 1618, 1551, 1485, 1458, 1428, 1280, 1243, 1077, 1038, 1008, 989, 911, 871, 802, 747, 608.
6. Получение 1-{[(циклогексилокси)карбонил]окси}-1-йодэтан (k)
а) В 100-миллилитровую круглодонную колбу при температуре -40°С к суспензии уксусного альдегида (360 ммолей, 20 мл) и PhCH2 +Et3Cl- (18 ммолей, 4,1 г) добавили трифосген (10 ммолей, 39,0 г). Реакционную смесь мешали в течение 5 часов. Избыточный трифосген удаляли при пониженном давлении. Остаток перегоняли при пониженном давлении, а дистиллят собрали при 41-42°С/4,2 мм Hg, в результате чего получили 1-хлоркарбонилокси-1-хлорэтан (соединение (m) (21,2 г, выход 41,2%). Данные анализа соединения (m): 1H ЯМР (CDCl3, 300 МГц) δ 1,85 (3Н, d, J=5,7 Гц, СН3), 6,42 (1Н, q, J=5,7 Гц, СН).
b) В 250-миллилитровую круглодонную колбу при охлаждении на ледяной бане соединение (m), полученное на стадии а), по каплям добавляли в раствор циклогексанола (91,5 ммоля, 9,15 г) и пиридина (91,8 ммоля, 7,38 мл) в CH2Cl2 (150 мл). Реакционную смесь перемешивали в течение 16 часов, промывали насыщенным водным раствором NaCl, высушивали безводным Na2SO4, а затем отгоняли растворитель. Остаток перегоняли при пониженном давлении и собирали дистиллят при 130-132°С/5 мм Hg, в результате чего получили 1-{[(циклогексилокси)карбонил]окси}-1-хлорэтан (соединение (n)) (16,7 г). Данные анализа соединения (n): 1H ЯМР (CDCl3, 300 МГц) δ 1,20-1,53 (6Н, m, CH2), 1,70 (2Н, m, CH2), 1,78 (3Н, d, J=6 Гц, СН3), 1,89 (2Н, m, CH2), 4,64 (1Н, m, СН), 6,39 (1Н, q, J=6 ГЦ, СН).
c) Соединение (n), полученное на стадии b) (6,7 ммоля, 1,4 г) растворили в 100-миллилитровой круглодонной колбе в 50 мл MeCN. Добавили NaI (26,8 ммоля, 4,4 г) и реакционную смесь перемешивали в течение 90 мин при температуре 60°С. Затем при пониженном давлении сгущали, а остаток экстрагировали простым эфиром. Органическую фазу отделяли, высушивали с безводным Na2SO4 и очищали с помощью колоночной хроматографии для получения целевого соединения (к) (810 мг, выход 40%). Данные анализа соединения (к): 1H ЯМР (CDCl3, 300 МГц) δ 1,23-1,93 (10Н, m, CH2), 2,23 (3Н, d, J=6 Гц, СН3), 4,68 (1Н, m, CH), 6,75 (1H, q, J=6 Гц, CH).
7. Получение соединения (о)

а) Бензонитрил (10,3 г, 100 ммолей), NH4Cl (6,9 г, 1,3 экв.), NaN3 (8,5 г, 1,3 экв.) и LiCl (300 мг) растворили в 100 мл N,N-диметилформамида и реакционную смесь перемешивали в течение 12 часов при температуре 100°С. Затем большую часть растворителя удалили при пониженном давлении. Остаток сделали щелочным с помощью 10%-ного водного раствора NaOH до водородного показателя рН 12. После экстракции с этилацетатом водную фазу отделили и подкислили концентрированной соляной кислотой до рН 2. Осадок отфильтровали при помощи воронки Трихтера, промыли водой и высушили, получив соединение (р) (13,5 г, точка плавления 208-209°С)
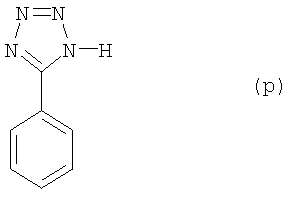
Выход составил 96%. Данные анализа соединения (р): 1H ЯМР (d-диметилсульфоксид, 300 МГц) δ 7,55-7,57 (3Н, m), 8,01-8,03 (2Н, m); 13C ЯМР (d-диметилсульфоксид, 75 МГц) δ 129,5, 132,4, 134,8, 136,7, 160,7; масс-спектрометрия (EI) m/z (%): 146 (M+, 42), 118 (100), 103 (17), 91 (46), 77 (32), 63 (48); инфракрасн. (пленка, см-1) λмакс = 3055, 2982, 2837, 2607, 2545, 1607, 1562, 1485, 11463, 1409, 1163, 1056, 1013, 725, 703, 686.
b) Соединение (р) стадии а) (6,6 г, 45 ммолей) растворили в 20 мл CH2Cl2 и прибавили Net3 (8 мл, 1,3 экв.). Реакционную смесь охлаждали до 0°С в водно-ледяной ванне и добавляли в течение 10 минут 3 порции Ph3CCl (13,2 г, 1,0 экв.). Затем нагревали до комнатной температуры и 3 часа перемешивали. Реакционную смесь фильтровали, промывали водой и высушивали для получения соединения (q) (16,5 г, точка плавления 163-164°С). Выход составлял 94%.
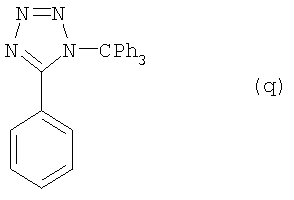
Данные анализа соединения (q): 1H ЯМР (CDCl3,300 МГц) δ 7,21-7,24 (6Н, m), 7,37-7,39 (9Н, m) 7,47-4,49 (3Н, m), 8,19-8,20 (2Н, m); 13С ЯМР (CDCl3, 75 Мгц) δ 83,0, 127,0, 127,5, 127,7, 128,3, 128,7, 130,3, 141,3, 164,0; инфракрасн. (пленка, см-1) λмакс = 3058, 1490, 1465, 1445, 1186, 1028, 874, 763, 748, 697, 635.
с) Для раствора соединения (q) стадии b) (10 г, 25,8 ммолей) в тетрагидрофуране (30 мл) в атмосфере аргона (атмосфере защитного газа) устанавливали температурный режим до -20°С. Затем добавляли BuLi (1 M, 27 мл, 1,05 экв.). Температуру повышали до -5°С и мешали в течение часа. За это время в осадок выпало большое количество твердого вещества. Снова охлаждали до -25°С и через шприц медленно добавляли В(ОМе)3 (4,3 мл, 1,5 экв.). После этого реакционную смесь нагревали до 20°С и мешали полчаса. Количество растворителя сократили при пониженном давлении до 1/3 от первоначального, причем образовалось белое твердое вещество. Твердую фазу отфильтровывали, промывали 20%-ным раствором тетрагидрофурана (THF) в Н2О (40 мл) и водой (40 мл) и высушивали, получая целевое соединение (о) (10,4 г). Выход составил 94%. Соединение (о) можно перерабатывать дальше без дополнительной очистки.
8. Получение кандесартана цилексетила (сочетание С-С)
Примеры 8-a1) - 8-а4) показывают 4 возможных условия реакций, благодаря которым может произойти сочетание С-С соединения (j) из примера 5 с соединением (о) из примера 7.
Пример 8-b) описывает удаление защитной группы с последующей обработкой.
а1) Соединение (j) из примера 5 (2,5 г, 4,6 ммоля), соединение (о) из примера 7 (3,4 г, 1,2 экв.) и Na2CO3 1,46 г, 3 экв.) растворяли в 20 мл раствора из толуола и воды (7: 3) и систему трижды орошали аргоном. Затем добавляли Pd(PPh3)4 (266 мг, 0,05 экв.) и реакционную смесь нагревали в течение 13 часов до температуры 80°С. После этого реакционную смесь экстрагировали с этилацетатом и очищали методом колоночной хроматографии (ПЭ: простой эфир, 3: 2), получая соединение (r) (3,2 г, выход 82%).
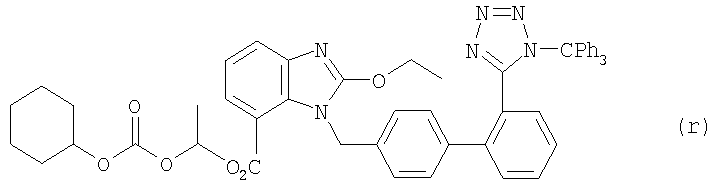
Данные анализа оединения (r): 1H ЯМР (CDCl3, 400 МГц) δ 1,19-1,51 (11Н, m), 1,67-1,71 (3H, m), 1,91 (2H, m), 4,59-4,65 (3H, m, CH2 и СН), 5,56 (2Н, q, J=16 Гц, CH2), 6,78-7,47 (24H, m), 7,56 (1H, d, J=8 Гц), 7,76 (1H, d, J=8,4 Гц), 7,87 (1Н, d, J=6,8 Гц); 13С ЯМР (CDCl3, 100 МГц) δ 14,6, 19,5, 23,6, 25,1, 31,4, 47,0, 66,7, 77,5, 82,2, 91,7, 114,8, 120,8, 122,5, 124,0, 126,2, 126,3, 127,4, 127,6, 128,2, 129,4, 129,8, 130,2, 130,3, 130,6, 135,7, 140,0, 141,2, 141,8, 142,0, 152,5, 158,7, 163,9, 164,0; масс-спектрометрия (FAB): найдено 875 (М++Na), 853 (M++1); инфракрасн. (пленка, см-1) λмакс = 2939, 2860, 1753, 1723, 1550, 1447, 1429, 1279, 1242, 1078, 1036, 909, 733, 699.
a2) NiCl2(PPh3)2 (33 мг, 0,05 ммоля), PPh3 (26 мг, 0,1 ммоля) растворяли в 3 мл DME (диметоксиэтана) или бензола в атмосфере аргона (атмосфере инертного газа). После этого добавили по каплям BuLi (0,13 мл, 0,2 ммоля, 1,6 М в гексане) и перемешивали 10 минут. Добавляли соединение (j) из примера 5 (0,5 ммоля), K3PO4 (1,5 ммоля), соединение (о) из примера 7 (1,1 ммоля) и реакционную смесь нагревали в течение 12 часов до температуры 80°С. Реакционную смесь дважды экстрагировали с этилацетатом, а органические фазы промывали водой и насыщенным водным раствором NaCl. Органическую фазу отделяли, высушивали безводным Na2SO4 и очищали методом колоночной хроматографии.
а3) Реакцию проводят так же, как описано в примере 8-а2), причем вместо BuLi альтернативно использовали DIBAH (гидрид дизобутилалюминия) (0,045 мл, 0,2 ммоля).
а4) NiCl2(PPh3)2 (33 мг, 0,05 ммоля), PPh3 (26 мг, 0,1 ммоля) и цинковый порошок (55 мг, 0,85 ммоля) растворяли в 1 мл THF в атмосфере аргона (атмосфере защитного газа) и нагревали в течение часа до 50°С. Затем добавляли соединение (j) из примера 5 (0,5 ммоля), K3PO4 (1,5 ммоля) и соединение (о) из примера 7 (1,5 ммоля), а также 2 мл THF. Реакционную смесь кипятили 48 часов с обратным потоком и обрабатывали так же, как это было описано в а2).
b) Соединение (r) стадии а) (3 г, 3,5 ммоля) растворяли в 51 мл CH2Cl2: МеОН: 1 N HCl (10: 36: 5,5) и реакционную смесь перемешивали в течение 3,5 часов при комнатной температуре. После этого добавляли насыщенный водный раствор NaHCO3 до получения водородного показателя примерно рН=3, а преобладающую часть растворителя удаляли при пониженном давлении. Остаток экстрагировали с этилацетатом и очищали методом колоночной хроматографии (РЕ: AcOEt 1:1), получая соединение кандесартан цилексетил (s) (2,05 г, точка плавления 128-129°С). Выход составил 95%.
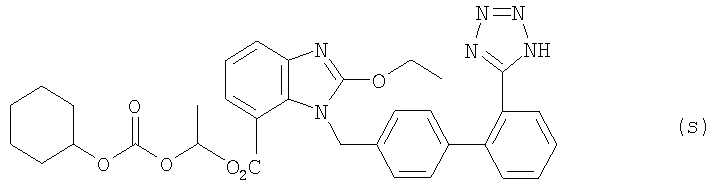
Данные анализа соединения (s): 1H ЯМР (CDCl3, 300 МГц) δ 0,97-1,39 (11Н, m), 1,46-1,48 (1H, m), 1,63 (2H, s, br), 1,78-1,82 (m, 2H), 3,92-4,00 4,00 (m, 1H), 4,28-4,36 (m, 1H), 4,46-4,52 (m, 1H), 5,55 (2H, dd, J=18, 20 Гц, СH2), 6,55-6,60 (4Н, m), 6,69-6,75 (2H, m), 6,81 (1H, t, J=7,8 Гц), 7,24 (1H, d, J=7,8 Гц), 7,39 (1H, d, J=7,5 Гц), 7,52-7,61 (2H, m), 7,93 (1H, d, J=8,1 Гц); 13С ЯМР (CDCl3, 75 МГц) δ 14,4, 19,0, 23,4, 24,9, 31,2, 46,7, 67,7, 77,6, 91,7, 115,3, 120,6, 121,2, 123,3, 124,1, 124,9, 128,2, 129,4, 130,2, 130,4, 131,1, 136,1, 138,0, 139,5, 140/8, 152,2, 154,7, 157,7, 163,1; масс-спектрометрия (FAB): найдено 633 (M++Na), 611 (M++1); инфракрасн. пленка, см-1) λмакс = 3060, 2939, 2860, 1753, 1725, 1613, 1550, 1473, 1434, 1281, 1245, 1078, 1038, 992, 911, 730.
9. Получение новой полиморфной формы кандесартана цилексетила
3 г кандесартана цилексетила растворяли в 3 мл дихлорметана. Затем при кипячении с обратным потоком медленно добавляли 50 мл простого диэтилового эфира. В том случае, если осаждалось твердое вещество, снова добавляли дихлорметан, пока твердое вещество не растворится. Прозрачный раствор сгущали при нормальном давлении примерно до 5 мл, а затем постепенно охлаждали до комнатной температуры, причем получалась новая полиморфная форма кандесартана цилексетила в виде белого твердого вещества.
Новую полиморфную форму можно получить описанным способом.
Новую полиморфную форму можно описать при помощи одного или нескольких следующих физических параметров:
- сигналы на дифрактограмме рентгеноструктурного анализа, выраженные в 2 θ при 7,32; 8,20; 9,10; 14,68; 18,88; 24,18°, причем относительные интенсивности сигналов могут составлять 100; 29,6; 20,2; 45,2; 20,5; 11,4 (cp. фиг.1a, 2 и таблицу);
- полученные с помощью рентгенографии расстояния между плоскостями решетки d=12,065; 10,773; 9,711; 6,029; 4,696; 3,678 Ǻ (ангстрем); ср. таблицу);
- точка плавления, определенная при помощи дифференциальной сканирующей калориметрии (DSC), составляет примерно 130,7°С (ср. фиг.2, кривую d);
- характерная полоса спектра поглощения в инфракрасном спектре при √=1733 см-1 (cp. фиг.3).
С помощью описанных примеров было продемонстрировано, как можно получить кандесартан цилексетил заявленным способом, исходя из любого из заявленных промежуточных продуктов.
Кроме того, впервые описана новая форма кандесартана цилексетила, а также способ ее получения. Примеры служат лишь для наглядности изобретения без ограничения его объема.

Изобретение относится к способу получения кандесартана или защищенной формы кандесартана, соли кандесартана или сложного эфира кандесартана с использованием катализатора или нескольких катализаторов, включающих предпочтительно один или несколько переходных металлов, и одного или нескольких следующих растворителей: тетрагидрофуран (THF), THF/NMP (N-метилпирролидон), Et2O, DME (диметоксиэтан), бензол, толуол, включающему следующие стадии: (а) предоставление и взаимодействие соединения формулы (I), где R означает водород, незамещенный или замещенный радикал алкила или арила, предпочтительно метил или (циклогексилоксикарбонилокси)этил, Y1 означает группу, способную к реакции сочетания, в которую вступает группа Y2, с образованием связи С-С, с соединением формулы (II), имеющим группу Y2, где R1 выбран из группы, включающей водород, трет-бутил и трифенилметил, предпочтительно трифенилметил, с образованием кандесартана, защитной формы кандесартана или сложного эфира кандесартана, или кандесартана цилексетила, или другого сложного эфира кандесартана, причем (i) Y1 означает B(OR4)2, причем каждый из радикалов R4 независимо друг от друга означает водород, алкил, арил или алкиларил, предпочтительно водород, а Y2 означает галоген, предпочтительно бром, или (ii) Y1 означает галоген, предпочтительно бром, а Y2 означает В(OR4)2, причем каждый из радикалов R4 независимо друг от друга означает водород, алкил, арил или алкиларил, предпочтительно водород, и, при необходимости, (b) превращение в кандесартан, кандесартана цилексетил или соль.
Технический результат - разработка нового способа получения кандесартана с высоким выходом. 23 з.п. ф-лы, 3 ил., 1 табл.
1. Способ получения кандесартана, соли кандесартана или сложного эфира кандесартана, или защитной формы кандесартана, соли кандесартана или сложного эфира кандесартана, в частности кандесартана цилексетила,
причем применяют катализатор или несколько катализаторов, включающих предпочтительно один или несколько переходных металлов, и
используют один или несколько следующих растворителей: тетрагидрофуран (THF), THF/NMP (N-метилпирролидон), Et2O, DME (диметоксиэтан), бензол, толуол,
включающий следующие стадии:
(а) предоставление и взаимодействие соединения формулы (I)
,
где R означает водород, незамещенный или замещенный радикал алкила или арила, предпочтительно метил или (циклогексилоксикарбонилокси)этил,
Y1 означает группу, способную к реакции сочетания, в которую поступает группа Y2, с образованием связи С-С,
с соединением формулы (II), имеющим группу Y2,
,
где R1 выбран из группы, включающей водород, трет.бутил и трифенилметил,
предпочтительно трифенилметил,
с образованием кандесартана, защитной формы кандесартана или сложного эфира кандесартана или кандесартана цилексетила или другого сложного эфира кандесартана,
причем
(i) Y1 означает B(OR4)2, причем каждый из радикалов R4 независимо друг от друга означает водород, алкил, арил или алкиларил, предпочтительно водород, a Y2 означает галоген, предпочтительно бром,
или
(ii) Y1 означает галоген, предпочтительно бром, а Y2 означает B(OR4)2, причем каждый из радикалов R4 независимо друг от друга означает водород, алкил, арил или алкиларил, предпочтительно водород,
и, при необходимости,
(b) превращение в кандесартан, кандесартан цилексетил или соль.
2. Способ по п.1, где R означает метил.
3. Способ по п.1, причем R означает радикал алкила, предпочтительно метил, а стадия b) включает переэтерификацию сложного эфира, полученного на стадии а).
4. Способ по п.3, где стадия b) включает гидролиз сложного эфира, полученного на стадии а), с помощью обработки гидроокисью натрия (NaOH) в EtOH.
5. Способ по п.4, где стадия b) дополнительно включает взаимодействие кандесартана в виде свободной карбоновой кислоты с соединением формулы IV
,
где Z1 обозначает уходящую группу, предпочтительно галоген, предпочтительно йод,
с образованием кандесартана цилексетила, предпочтительно в присутствии NaI и K2CO3.
6. Способ по п.1, где Y1=Br.
7. Способ по п.1, где катализатор или катализаторы выбраны из группы, состоящей из MnCl2, CrCl3, FeCl2, Fe(асас)3, FeCl3, Fe(salen)Cl, CoCl2(dppe), CoCl2(dpph), Co(acac)2, CoCl2(dppb), Pd(PPh3)3 или NiCl2(PPh3)2.
8. Способ по п.1, где катализатор применяют вместе активатором и/или стабилизатором.
9. Способ по п.1, где катализатор или катализаторы выбран или выбраны из группы свободных от фосфана катализаторов, содержащих предпочтительно железо.
10. Способ по п.1, дополнительно включающий получение соединения формулы (I), определенной в п.1, включающий следующие стадии:
предоставление и взаимодействие соединения формулы (III)
,
где Y1 имеет указанное в п.1 значение,
Х обозначает группу, способную к реакции, в которую поступает группа Z1, с образованием связи O-С,
с соединением формулы (IV)
,
где Z1 означает уходящую группу,
с образованием соединения формулы (I).
11. Способ по п.10, где Х означает щелочной металл или предпочтительно водород, и/или Z1 обозначает галоген, предпочтительно йод.
12. Способ по п.10, где Y1=Br.
13. Способ по п.10, дополнительно включающий получение соединения, имеющего приведенную в п.10 формулу (III), включающий следующую стадию:
предоставление соединения формулы (V) и снятие защитной(ых) групп(ы) с него
,
где Y1 имеет указанное в п.1 значение, а
R2 означает группу, способную к замене на Х с образованием соединения формулы (III), причем Х имеет указанное в п.10 или 11 значение.
14. Способ по п.13, где R2 выбран из одной из следующих функциональных групп: замещенный или незамещенный низший C1-С6-алкил, бензил или арил, предпочтительно этил (СН2СН3), и наиболее предпочтительно метил (СН3).
15. Способ по п.13, где Y1=Br, а R2 означает группу метила или низшего С3-С6-алкила.
16. Способ по п.13, дополнительно включающий получение соединения, имеющего приведенную в п.13 формулу (V), включающий следующую стадию:
предоставление и взаимодействие соединения формулы (VI)
,
где Y1 имеет значение, указанное в п.1, а
R2 имеет значение, указанное в п.13 или 14,
с карбонилирующим реагентом или предпочтительно с C(OEt)4,
с образованием соединения формулы (V).
17. Способ по п.16, где при взаимодействии дополнительно используют Ас2О.
18. Способ по п.16, где Y1=Br, a R2 означает группу метила или низшего С3-С6-алкила.
19. Способ по п.16, дополнительно включающий получение соединения формулы (VI), как оно определено в п.16, включающий следующие стадии: предоставление соединения формулы (VII)
,
где Y1 имеет значение, указанное в п.1, а
R2 имеет значение, указанное в п.13 или 14,
и преобразование имеющейся в нем нитрогруппы в аминогруппу.
20. Способ по п.19, где преобразование нитрогруппы в аминогруппу проводят с использованием неблагородных металлов, каталитического гидрирования, электролитическим способом или предпочтительно с помощью SnCl2.
21. Способ по п.19, дополнительно включающий получение соединения формулы (VII), как оно определено в п.19, включающий следующий этап: предоставление соединения формулы (VIII) и снятие защитной(ых) групп(ы) с него
,
где Y1 имеет значение, указанное в п.1, а
R2 имеет значение, указанное в п.13 или 14,
и R3 означает защитную группу, способную к замене на Н,
с образованием соединения формулы (VII), как определено в п.19.
22. Способ по п.21, где
Y1 означает Br,
R2 означает группу метила или низшего С3-С6-алкила, и
R3 означает группу карбоксилалкила, предпочтительно группу карбокси-трет.-бутила (-СООС-(СН3)3).
23. Способ по п.21 или 22, дополнительно включающий получение соединения формулы (VIII), как определено в п.21, включающий следующий этап:
предоставление и взаимодействие соединения формулы (IX)
,
где R2 имеет указанное в п.13 или 14 значение,
и R3 имеет указанное в п.21 или 22 значение,
с соединением формулы (X)
,
где Y1 имеет указанное в п.1 значение, а
Z2 означает уходящую группу,
с образованием соединения формулы (VIII).
24. Способ по п.23, где Z2 выбран из одной из следующих функциональных групп: Cl, I и предпочтительно Br.
| Штепсельная розетка | 1987 |
|
SU1510031A1 |
| WO 2004065383 A2, 05.08.2004 | |||
| WO 9310106 A1, 27.05.1993 | |||
| LARSENR.D | |||
| et al, J | |||
| Org | |||
| Chem | |||
| Устройство для охлаждения водою паров жидкостей, кипящих выше воды, в применении к разделению смесей жидкостей при перегонке с дефлегматором | 1915 |
|
SU59A1 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА, ИХ ТАУТОМЕРЫ ИЛИ ИХ СОЛИ И ЛЕКАРСТВЕННОЕ СРЕДСТВО С АНТАГОНИСТИЧЕСКИМ В ОТНОШЕНИИ АНГИОТЕНЗИНА II ДЕЙСТВИЕМ | 1993 |
|
RU2126401C1 |
| Производные бензимидазола, их изомеры, смеси изомеров, гидраты или их физиологически переносимые соли, обладающие антагонистическими в отношении ангиотензина свойствами | 1991 |
|
SU1836357A3 |

