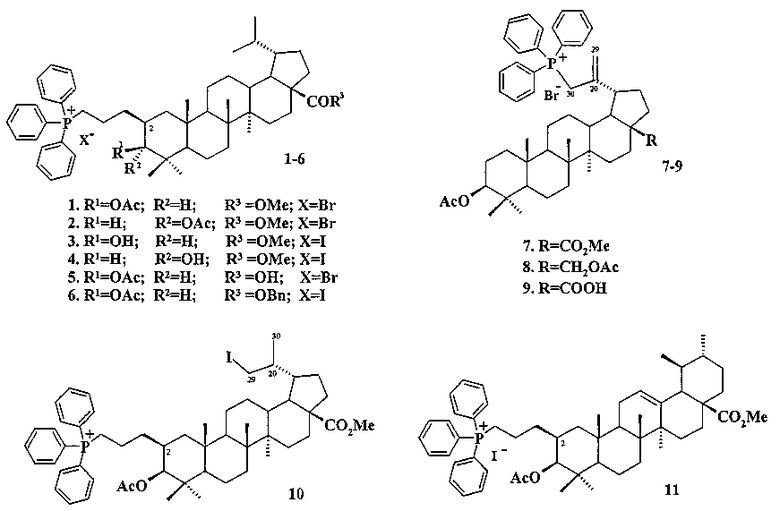
Изобретение относится к области биоорганической и медицинской химии, в частности к созданию новых лекарственных средств для лечения паразитарных (инвазионных) болезней человека и животных, и конкретно касается трифенилфосфониевых производных лупановых и урсановых тритерпеноидов формулы 1-11, которые проявляют шистосомицидную активность против шистосомул (newly transformed schistosoma) и взрослых червей Шистосомы Мансона (Schistosoma mansoni) в низких микромолярных концентрациях
Трифенилфосфониевые соли формулы 1-11 представляют собой производные бетулина формулы 12, бетулиновой кислоты формулы 13, 20,29-дигидробетулиновой кислоты формулы 14, 20,29-дигидро-3-эпи-бетулиновой кислоты формулы 15 и урсоловой кислоты формулы 16.
Использование изобретения позволит расширить ассортимент лекарственных средств, полученных на основе природных продуктов, для лечения шистосомоза - опасного паразитарного заболевания человека и животных.
Шистосомоз - это хроническая паразитарная болезнь, вызываемая кровяными сосальщиками (трематодами) рода Schistosoma. Из существующих паразитарных инфекций шистосомоз находится на втором месте после малярии по негативному влиянию на здравоохранение и экономику общества. На сегодняшний день лечение требуется, по меньшей мере, для 243 миллионов людей и 779 миллионов людей проживает в зоне риска. Передача шистосомоза документально зарегистрирована практически во всех странах, расположенных в зонах субтропического и тропического климата [Информационный бюллетень ВОЗ, 2013, № 215]. Хронический шистосомоз может спровоцировать развитие других серьезных инфекций, таких как ВИЧ-инфекция и малярия. Шистосому S. Haematobium рассматривают как высокую группу риска для развития рака мочевого пузыря. Кишечный шистосомоз (Schistosoma mansoni) может стать причиной рака толстой кишки и гепатоцеллюлярной карциномы. В Африке от шистосомоза ежегодно умирают более 200 тысяч человек. Существуют две основные формы шистосомоза - кишечный и мочеполовой, вызываемые пятью основными видами облигатного паразита. Кишечный шистосомоз вызван кровяными сосальщиками вида Schistosoma msnsoni, Schistosoma japonicum, Schistosoma mekongi, Schistosoma intercalatum, которые живут в венах толстого кишечника и брюшной полости. Мочеполовой шистосомоз вызван трематодами Schistosoma haematobium. Среди перечисленных пяти видов шистосомоза человека S. mansoni является самым распространенным заболеванием. Все виды шистосом, паразитирующих у человека, имеют довольно сложный жизненный цикл. На инвазионной стадии свободно живущие церкарии, после выхода из пресноводного моллюска, проникают через неповрежденную кожу человека или животных и превращаются в шистосомулы. Через 2-3 суток шистосомулы мигрируют по кровеносным сосудам в легкие, а затем в воротную вену, где происходит копуляция зрелых шистосом. Затем, в зависимости от вида шистосом, они мигрируют в венулы брыжейки, мочевого пузыря или мочеточников и начинают откладывать яйца. Для S. mansini сроки миграции и созревания паразитов составляют 4-5 недель.
Список современных лекарственных средств, рекомендованных ВОЗ для лечения шистосомоза, включает два препарата - празиквантел и оксамнихин. Наиболее широко используется препарат празиквантел (2-(циклогексилкарбонил)-1,2,3,6,7,11b-гексагидро-4H-пиразино[2,1-a]изохинолин-4-он) [R. Abdul-Ghani, N. loutfy, A. El Sahn, A. Hassan. Current chemotherapy arsenal for schistosomiasis mansoni: alternatives and challenges. Parasitol Res., 2009, 104, 955-965; D. Ndjonka, L.N. Rapado, A.M. Silber, E. Liebau, С. Wrenger. Natural Products as a source for treating neglected parasitic diseases. Int. Mol Sci, 2013, 14, 3395-3439]
Препарат был получен в середине 1970-х годов. Это безопасное и недорогое лекарство, эффективное для лечения всех форм шистосомоза. Ювенильные паразиты (возраст 7-21 день) менее чувствительны к препарату, чем взрослые особи. Несмотря на длительное использование празиквантела механизм его многофункционального действия на шистосомы до конца не исследован. Предполагается, что празиквантел взаимодействует со специфическими белками паразита, которые модулируют величину трансмембранного потенциала клетки, отвечают за деятельность потенциал-управляемых кальциевых каналов и контролируют величину Ca2+-тока. Под его влиянием происходит поступление в клетку избыточного количества ионов Ca2+, что приводит к нарушениям специфических функций клетки. Кроме того, под воздействием празиквантела нарушается оболочка червя и открывается доступ к антигенам, которые вызывают иммунный ответ организма хозяина [K. Abdul-Ghani, N. Loutfy, A. El Sahn, A. Hassan. Current chemotherapy arsenal for schistosomiasis masnsoni: alternatives and challenges. Parasitol Res., 2009, 104, 955-965].
Второе лекарственное средство, используемое в клинической практике как препарат резерва - оксамнихин, синтетический тетрагидрохинолин (1,2,3,4-тетрагидро-2-{[(1-метилэтил)амино]метил}-7-нитро-6-хинолинометанол)
Это соединение эффективно только при зараженности S. mansoni. В Египте и Южной Америке встречаются устойчивые разновидности возбудителя, что требует высоких доз препарата. Процесс получения оксамнихина включает пять синтетических стадий с применением на заключительной стадии ферментативного катализа, что делает производство препарата сложным и дорогостоящим.
Метрифонат (хлорофос, вермицид-Байер 2349) представляет собой О,О-диметил-2,2,2-трихлор-1-гидроксиэтилфосфат и до применения в медицинской практике использовался как инсектицид для защиты растений от сельскохозяйственных вредителей
Проявляет низкую эффективность при кишечном шистосомозе, пригоден только для лечения мочеполового шистосомоза S. haematobium. Употребление метрифоната приводит к серьезным побочным эффектам в связи с подавлением активности ацетилхолинэстеразы и накоплением ацетилхолина в нервных синапсах. В настоящее время этот препарат проходит клинические испытания в Гане, Замбии и других африканских странах.
Калий тартрат сурьмы и другие трехвалентные производные сурьмы использовались для лечения шистосомоза с 1918 г. на протяжении пятидесяти лет
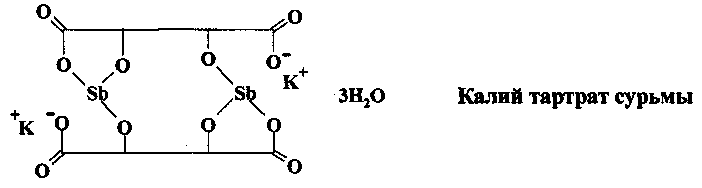
В настоящее время эти чрезвычайно токсичные лекарства, вызывающие серьезные побочные эффекты (тошнота и рвота, диарея, боли в суставах, гепатит), не рекомендованы для использования.
Таким образом, набор современных лекарственных средств, применяемых в клинической практике для лечения шистосомоза, весьма ограничен. Слабый интерес фармацевтических компаний к созданию новых лекарств против шистосомоза и других «игнорируемых» («neglected») тропических болезней объясняется относительно низкой ценой продукции в этой области фармацевтики.
Шистосомоз - серьезное, широко распространенное в мире заболевание, с постоянно расширяющейся географией эндемичных районов по причине развитого туризма и массовой трудовой миграции населения. На сегодняшний день празиквантел является практически единственным эффективным и нетоксичным лекарством, пригодным для массового употребления. Поскольку этот препарат используется в химиотерапии шистосомоза с середины семидесятых годов, существует серьезная проблема резистентности к нему паразитов. В связи с этим крайне важны исследования по разработке и внедрению в практику новых эффективных шистосомицидных лекарственных агентов.
Перспективным представляется поиск новых дешевых препаратов на основе доступных биологически активных веществ растительного происхождения и их направленной химической модификации с учетом специфических особенностей организма паразита [D. Ndjonka, L.N. Rapado, S.M. Silber, E.Liebau, C. Wrenger. Natural products as a source for treating neglected parasitic diseases. Int. J. Mol. Sci., 2013, 14, 3395-3439]. В лабораторных экспериментах in vitro исследовались экстракты эфирных масел различных медицинских растений, овощные масляные экстракты и индивидуальные биологически активные соединения, принадлежащие к классу кумаринов, флаваноидов, производных антрацена и сесквитерпеновых лактонов. В ряду этих природных веществ были выявлены антипаразитарные соединения, однако их антишистосомальная активность была слабо выражена. Так, при действии на взрослые S. mansoni куркумина, гидроксихризофанола, кверцетина и вернодалина высокая смертность паразитов наблюдалась при концентрациях в интервале 40-100 мкг/мл. Более значительное шистосомицидное действие проявили известные противомалярийные препараты, такие как природное соединение артемизинин (трициклический сесквитерпен с пероксидным мостиком), его водорастворимый полусинтетический аналог артесунат и растворимый в масле метиловый эфир артемизинина - препарат артеметер [D. Ndjonka, L.N. Rapado, S.M. Silber, E.Liebau, C. Wrenger. Natural products as a source for treating neglected parasitic diseases. Int. J. Mol. Sci., 2013, 14, 3395-3439; J. Utzinger, X. Shuhua, E.K. N'Goran, R. Bergquist, M. Tanner. The potential of artemethen for the control of schistosomiasis. International Journal for Parasitology 2001, 31, 1549-1562]
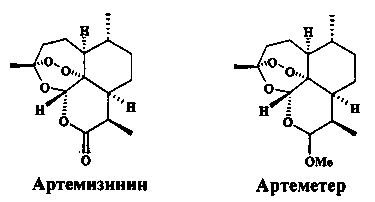
Антишистосомальная активность артемизинина, артесуната и артеметера была открыта китайскими учеными в 1980-х годах при изучении шистосомы S. japonicum. Наиболее химически стабильный и перспективный в этой группе соединений препарат артеметер в испытаниях на животных проявил высокую активность против S. mansoni, S. japonicum и S. haematobium. Ювенильные шистосомы (возраст 1-3 недели) были значительно чувствительней к артеметеру по сравнению с празиквантелом. В комбинированной химиотерапии шистосомоза одновременное использование артеметера и празиквантела показало обнадеживающие результаты в лабораторных исследованиях и клинических испытаниях [J. Utzinger, J. Keiser, X. Shuhua, M. Tanner, B.H. Singer. Combination chemotherapy of schistosomiasis in laboratory studies and clinucal trials. Antimicrobial agents and chemotherapy, 2003, 1487-1495]. Однако использование артемизинина для контроля шистосомоза в эндемичных по малярии районах может привести к необратимому увеличению резистентности малярийных плазмодий к этому лекарству.
Шистосомицидная активность доступных пентациклических тритерпеноидов - бетулина, бетулиновой и урсоловой кислот и их аналогов ранее не исследовалась. Вместе с тем эти растительные метаболиты известны широким спектром биологических свойств: противовоспалительными, противовирусными (анти-ВИЧ), противоопухолевыми, противомолярийными, противолейшманиозными и трипаноцидными [Г.А. Толстиков, О.Б. Флехтер, Э.Э. Шульц, Л.А. Балтина, А.Г. Толстиков. Химия в интересах устойчивого развития, 2005, 13, 1; P. Yogeeswari, D. Sriram, Current Medicinal Chemistry, 2005, 12, 657; G. NS da Silva, N. RG Marial, D.C. Schuch, L.N. Cruz, M. S de Moraes, M. Nakabashi, C. Graebin, G. Gosmann, C. RS Garcia, S. CB Gnoatto. Two series of new semisynthetic triterpene derivatives: differences in anti-malarial activity, cytotoxicity and mechanism of action. Malaria Journal, 2013, 12, 1573-1582; S. Alakurtti, T. Heiska, A. Kiriazis, N. Sacerdoti-Sierra, C.L. Jaffe, J. Yli-Kauhaluoma. Synthesis and anti-leishmanial activity of heterocyclic betulin derivatives. Bioorganic & Medicinal Chemistry. 2010, 18, 1573-1582; D.B. Domínguez-Carmonaa, F. Escalante-Erosaa, K. García-Sosaa, G. Ruiz-Pinellb, D. Gutierrez-Yapub, M.J. Chan-Bacabc, A. Giménez-Turbab, L.M. Pena-Rodrígueza. Antiprotozoal activity of betulinic acid derivatives. Phytomedicine. 2010, 17, 379-382]. В лабораторных экспериментах in vitro бетулиновая и урсоловая кислоты проявили умеренную антималярийную активность в отношении Plasmodium falciparum 3D7 с IC50 13.9 мкМ (для бетулиновой кислоты) и 36 мкМ (для урсоловой кислоты) [G. NS da Silva, N. RG Marial, D.C. Schuch, L.N. Cruz, M. S de Moraes, M. Nakabashi, C. Graebin, G. Gosmann, C. RS Garcia, S. CB Gnoatto. Two series of new semisynthetic triterpene derivatives: differences in anti-malarial activity, cytotoxicity and mechanism of action. Malaria Journal, 2013, 12, 1573-1582]. В результате активных исследований по синтезу и изучению противомалярийной активности большой группы полусинтетических аналогов бетулиновой и урсоловой кислот были выявлены новые потенциальные противомолярийные агенты, проявившие in vitro активность при низких микромолярных или наномолярных концентрациях [H.L. Ziegler, H. Franzyk, M. Sairafianpour, M. Tabatabai, M.D. Tehrani, K. Bagherzadeh, H. Hagerstrand, D. Stark, J.W. Jaroszewski. Erythrocyte membrane modifying agents and the inhibition of Plasmodium falciparum growth: structure-activity relationships for betulinic acid analigues. Bioorganic & Medicinal Chemistry. 2004. 12. 119-127; S.C.B. Gnoatto, S. Susplugas, L.D. Vechia, T.B. Ferreira, A. Dassonville-Klimpt, K.R. Zimmer, C. Demailly, S. Da Nascimento, J. Guillon, P. Grellier, H. Verli, G. Gosmann, P. Sonnet. Pharmacomodulation on the 3-acetylursolic acid skeleton: design, synthesis, and biological evaluation of nivel N-{3-[4-(3-aminopropyl)piperazinyl]propyl}-3-O-acetylursolamide derivatives as antimalarial agents. Bioorganic & Medicinal Chemistry. 2008. 16. 771-782; A.M. Innocente, G.N.S. Silva, L.N. Cruz, M.S. Moraes, M. Nakabashi, P. Sonnet, G. Gosmann, C.R.S. Garcia, S.C.B. Gnoatto. Synthesis and antiplasmodialactivity of betulinic acid and ursolic acid analogues. Molecules. 2012, 17, 12003-12014]. Так, гибридные соединения, содержащие несколько фармакофорных групп (фрагменты урсоловой или бетулиновой кислоты и замещенного пиперазина), показали высокую противомолярийную активность в отношении плазмодия «Plasmodium falciparum 3D7» (IC50=175-220 nM), проявили малую токсичность в отношении эмбриональных клеток почек человека HEK293T (IC50=4 мкМ) и приемлемый терапевтический индекс селективности (SI 18-23) [S.C.B. Gnoatto, S. Susplugas, L.D. Vechia, T.B. Ferreira, A. Dassonville-Klimpt, K.R. Zimmer, C. Demailly, S. Da Nascimento, J. Guillon, P. Grellier, H. Verli, G. Gosmann, P. Sonnet. Pharmacomodulation on the 3-acetylursolic acid skeleton: design, synthesis, and biological evaluation of nivel N-{3-[4-(3-aminopropyl)piperazinyl]propyl}-3-O-acetylursolamide derivatives as antimalarial agents. Bioorganic & Medicinal Chemistry. 2008. 16. 771-782; A.M. Innocente, G.N.S. Silva, L.N. Cruz, M.S. Moraes, M. Nakabashi, P. Sonnet, G. Gosmann, C.R.S. Garcia, S.C.B. Gnoatto. Synthesis and antiplasmodialactivity of betulinic acid and ursolic acid analogues. Molecules. 2012, 17, 12003-12014].
В последние годы усиленное внимание исследователей вызывают липофильные трифенилфосфониевые катионы как молекулы для изучения функций митохондрий и как средство доставки в митохондрии антиоксидантов, противомикробных и противоопухолевых агентов [J.S. Modica-Napolitano, J.R. Aprille, Adv Drug Delivery Rev, 2001, 49, 63; F. Wang, M.A. Ogasawara, P. Huang, Mol Aspect of Medicine, 2010, 31, 75; L. Biasutto, L-F. Dong, M. Zoratti, J. Neuzil, Mitochondrion, 2010, 10, 670; B. Bachowska, J. Kazmierczak-Baranska, M. Cieslak, B. Nawrot, D. Szczesna, J. Skalik, P. Balczewski, Chemistryopen, 2012, 1, 33]. Большая группа моно- и бисфосфониевых солей бензофенонов была исследована in vitro против резистентных линий африканских трипаносом и протозойных паразитов лейшеманий [J.R. Luque-Ortega, P. Reuther, L. Rivas, C. Dardonville. New benzophenone-derived bisphosphonium salt as leishmanicidal leads targeting mitochondria through of respiratory complex II. J. Med. Chem. 2010 53, 1788-1798; A. Taladriz, A. Healy, E.J.F. Pérez, V.H. Garcia, C.R. Martнnez, A.A.M. Alkhaldi, A.A. Eze, M. Kaiser, H.P. de Koning, A. Chana, C. Dardonville. Synthesis and structure - activity analysis of new phosphonium salts with potent activity against african trypanosomes. J. Med. Chem. 2012, 55, 2606-2622]
Фосфониевые производные ингибировали пролиферацию паразитов в низких микромолярных концентрациях при незначительной токсичности по отношению к клеткам человека. Под действием этих соединений, направленных на комплекс II дыхательной цепи паразитов, быстро снижалась цитоплазматическая АТФ и уменьшался электрохимический митохондриальный потенциал.
Фосфониевые липокатионы, полученные на основе фталимидов и 1,4-нафтохинонов, проявили in vitro высокую антипротозойную активность в отношении Plasmodium falciparum и Trypanosoma cruzi [T.E. Long, Xiao Lu, M. Galizzi, R. Docampo, J. Gut, P.J. Rosenthal. Phosphonium lipocations as antiparasitic agents. Bioorganic & Medicinal Chemistry Letters. 2012. 22. 2976-2979].
Недавно нами были получены новые трифенилфосфониевые производные лупановых тритерпеноидов. Новые соединения проявили in vitro значительно более высокое цитотоксическое действие, чем бетулиновая кислота на раковые клеточные линии карциномы Эрлиха и мастоцитомы P-815 [А.Ю. Спивак, Д.А. Недопёкина, Э.Р. Шакурова, Р.Р. Халитова, Р.Р. Губайдуллин, В.Н. Одиноков, У.М. Джемилев, Ю.П. Вельский, Н.В. Вельская, С.А. Станкевич, Е.В. Короткая, В.А. Хазанов. Синтез лупановых тритерпеноидов с трифенилфосфониевыми фрагментами и изучение их противоопухолевой активности. Известия Академии наук. Сер. химическая. 2013, 62, №1, 189-199].
Известно, что наружный покров трематод представляет собой тегумент. Он состоит из слоя клеток, слившихся между собой и образующих общую массу протоплазмы (синцитий) [J.J. Van Hellemond, К. Retra, J. F.H.M. Brouwers, В.W.M. van Balkom, M. Yazdanbakhsh, С.B. Shoemaker, A.G.M. Tielens. Functions of the tegument of schistosomes: Clues from the proteome and lipidome. International Journal for Parasitology. 2006, 36, 691-699]. Наружный слой тегумента - это безъядерная цитоплазма, содержащая большое число митохондрий. Биологической мишенью бетулиновой кислоты являются митохондрии. Бетулиновая кислота способствует накоплению в митохондриях активных кислородсодержащих частиц, что приводит к серьезным дисфункциям в этих органеллах [S. Fulda, G. Kroemer, Drug Discovery Today, 2009, 14, 885]. Мы предположили, что ковалентное связывание липофильного катиона трифенилфосфония с бетулиновой кислотой или с другими производными лупановой группы усилит их направленность к митохондриям, расположенным на поверхности тегумента. Разрушение поверхности тегумента будет способствовать гибели паразитов, поскольку тегумент выполняет жизненноважные для организма шистосом функции, такие как абсорбция питательных веществ и секреции, защита от гуморального и клеточного иммунитета организма хозяина.
В связи с изложенными фактами задачей настоящего изобретения является получение трифенилфосфониевых производных лупановых и урсановых тритерпеноидов, которые могут быть использованы в качестве новых шистосомицидных лекарственных средств. Поставленная задача решается получением трифенилфосфониевых солей 1-11 взаимодействием трифенилфосфина с C(2)- или C(30)-бром(йод)-замещенными C(3)- и С(28)-эфирными производными бетулина 12, бетулиновой кислоты 13, дигидробетулиновой кислоты 14, дигидро-3-эпибетулиновой кислоты 15 и урсоловой кислоты 16, а также выявлением in vitro шистосомицидного действия полученных фосфониевых солей на шистосомулы, и взрослые черви Schistosoma mansoni.
Заявленные соединения 1-6, представляющие собой конъюгаты C(3)- и C(28)-эфирных производных тритерпеновых кислот 13-15 с катионом трифенилфосфония, связанным с молекулой тритерпеноида при C(2) позиции пропильным мостиком, синтезировали следующим образом: дигидробетулонаты 17 и 18 трансформировали в 3β-эпимеры дигидробетулиновой кислоты 19 и 20 путем стереоселективного восстановления 3-кето группы под действием NaBH4, модифицированного CeCl3·7H2O. Дигидробетулонат 17 также подвергали восстановлению в α-эпимер 21 под действием три-втор-бутилборгидрида лития (L-селектрида) (химические реакции I).
Химические реакции I
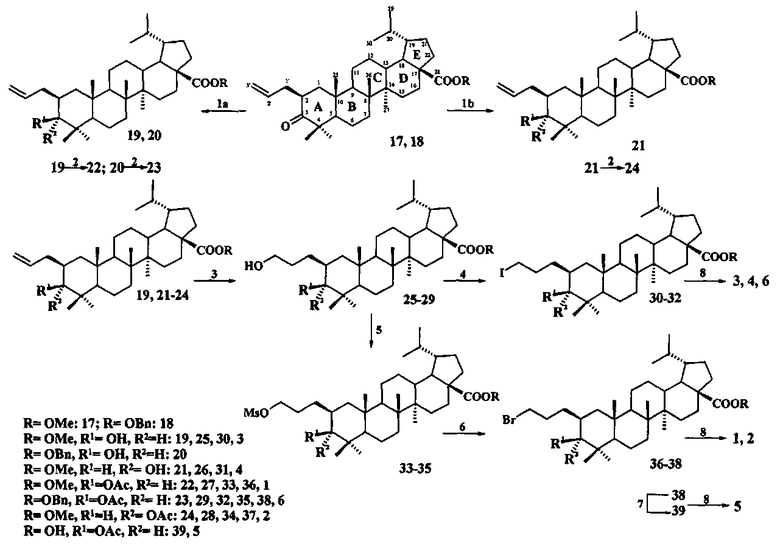
Реагенты и условия: (1a) NaBH4, CeCl3·7H2O, MeOH-ТГФ, -30°C→20°C, Ar; (1b) L-селектрид, ТГФ, -78°C→20°C, Ar; (2) Ac2O или AcCl; (3) BH3·ТГФ или BH3Me2S, ТГФ 20°C, Ar; (4) I2, PPh3, имидазол, ТГФ, 0°C; (5) MsCl, Py, CH2Cl2, DMAP, 20°C; (6) LiBr, (CH3)2CO, кипячение, Ar; (7) Pd/C, -Et2O; (8) PPh3, CH3C6H5 или CH3CN, кипячение, Ar.
При получении солей 1, 2, 5 и 6 спирты 19-21 трансформировали в 3-O-ацетилированные дигидробетулонаты 22-24 по типовой методике под действием Ac2O в пиридине или в AcCl в ТГФ в присутствии каталитических количеств пиридина и 4-диметиламинопиридина (DMAP). Затем соединения 19, 21-24 подвергали последовательным трансформациям, включающим гидроборирование двойной связи в аллильном заместителе с использованием комплекса ВН3·ТГФ или BH3·Me2S с последующим окислением борорганического соединения в спирты 25-29 под действием 30% H2O2 и 10% водного раствора NaOH. Затем следовало получение йодидов 30-32 путем нуклеофильного замещения первичной гидроксильной группы в спиртах 25, 26, 29 с использованием кристаллического йода в присутствии имидазола и трифенилфосфина. Бромиды 36-38 получали из спиртов 27-29 в две стадии через промежуточные мезилаты 33-35 путем взаимодействия спиртов с метансульфохлоридом (MsCl) в присутствии пиридина и DMAP, а затем нуклеофильного замещения мезильной группы под действием LiBr в ацетоне.
При получении соли 5 бензилдигидробетулинат 38 трансформировали в соответственную кислоту 39 путем гидрогенолиза эфирной связи с использованием в качестве катализатора 10% Pd/C. Заключительную стадию получения целевых трифенилфосфониевых солей 1-6 проводили путем взаимодействия галогенидов 30-32, 36, 37, 39 с избытком трифенилфосфина при кипячении в толуоле или в ацетонитриле в течение 16-48 часов в атмосфере аргона.
При получении аллильной соли 8 бетулин 12 ацетилировали в эфир 40 по типовой методике под действием Ac2O в пиридине или AcCl в ТГФ в присутствии каталитических количеств пиридина и 4-диметиламинопиридина (DMAP) или эфир 40 получали непосредственно из бересты березы путем ее кипячения в уксусной кислоте в течение 12 ч по методу [Кузнецова С.А., Кузнецов Б.Н., Редькина Е.С., Соколенко В.А., Скворцова Г.П. Патент РФ №2324700]. Затем соединение 40 трансформировали в аллильный бромид 41 под действием N-бромсукцинимида (NBS) в CCl4 по методу [N.V. Uzenkova, N.I. Petrenko, M.M. Shakirov, E.E.S hul`ts, G.A. Tolstikov. Chemistry of Natural Compounds. 2005, 41, 692-700]. Бромид 41 кипятили с избытком трифенилфосфина в CH3CN в атмосфере аргона (химические реакции II). При замене CH3CN на толуол реакция протекала со скелетной перегруппировкой лупанового остова с получением смеси соединении.
Химические реакции II
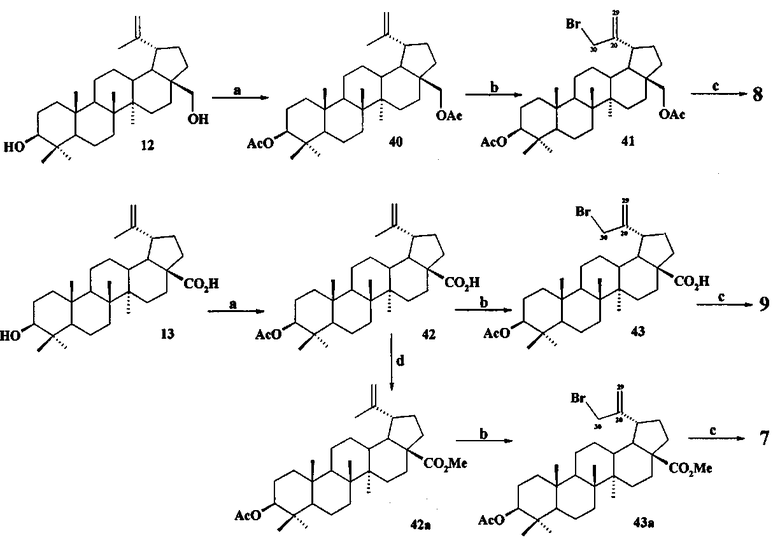
Реагенты и условия: a. Ac2O, Py, DMAP или AcCl, ТГФ, Py, DMAP, 20°C; b. NBS, CCl4; c. PPh3, CH3CN, кипячение, Ar; d. CH2N2, Et2O.
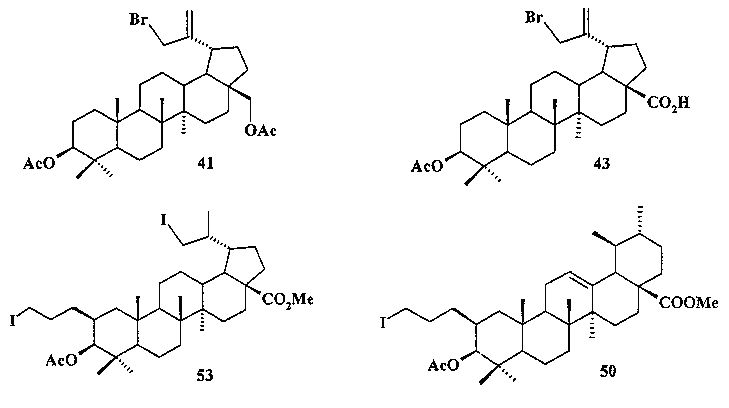
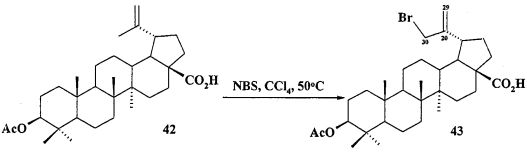
Аллильные соли 7 и 9 получали из бетулиновой кислоты 13 через промежуточную 3β-OAc-бетулиновую кислоту 42, ее метиловый эфир 42a и промежуточные бромиды 43 и 43a, как описано для получения соли 8.
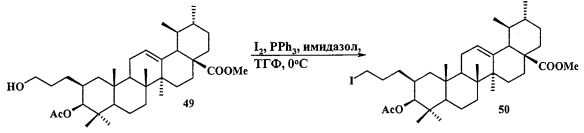
Синтез соединения 11 осуществляли по описанному нами способу получения солей 1-6. Урсоловую кислоту 16 трансформировали в метилурсоноат 44 под действием CH2N2 в Et2O, окисляли в метиловый эфир урсоновой кислоты 45 под действием PCC (пиридиния хлорхромат) в CH2Cl2 и трансформировали в 2β-аллилзамещенный эфир 46 взаимодействием с KN(SiMe3)2, Et3B и аллилбромидом в диметоксиэтане. Соединение 46 трансформировали в 3β-OH-эпимер 47 путем стереоселективного восстановления 3-кето группы под действием NaBH4-CeCl3·7H2O. Соединение 47 ацетилировали в эфир 48, как описано для эфиров 40 и 42. Затем эфир 48 подвергали последовательным трансформациям, включающим гидроборирование двойной связи в аллильном заместителе с использованием комплекса BH3·ТГФ или BH3·Me2S с последующим окислением борорганического соединения в спирт 49 под действием 30% H2O2 и 10% водного раствора NaOH. Затем следовало получение йодида 50 путем нуклеофильного замещения первичной гидроксильной группы в спирте 49 с использованием кристаллического йода в присутствии имидазола и трифенилфосфина и получение целевой соли 11 путем взаимодействия йодида 50 с избытком трифенилфосфина при кипячении в толуоле или ацетонитриле в атмосфере аргона (химические реакции III).
Химические реакции III
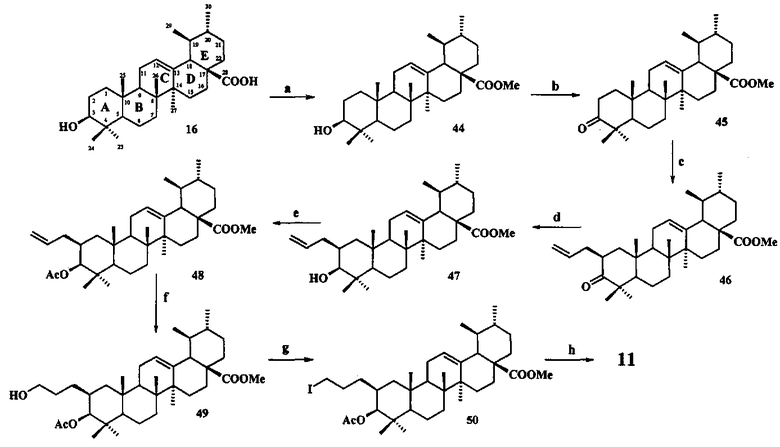
Реагенты и условия: a. CH2N2, Et2O, 20°C; b. PCC, CH2Cl2, 20°C; c. KN(SiMe3)2, BEt3, C3H5Br, диметоксиэтан, 20°C, Ar; d. NaBH4, CeCl3·7H2O, MeOH-ТГФ, 30°C→20°C, Ar; е. Ac2O, Py, DMAP или AcCl, ТГФ, Py, DMAP, 20°C; f. BH3·ТГФ или BH3·Me2S, ТГФ 20°C, Ar; g. I2, PPh3, имидазол, ТГФ, 0°C; h. PPh3, CH3C6H5, кипячение, Ar.
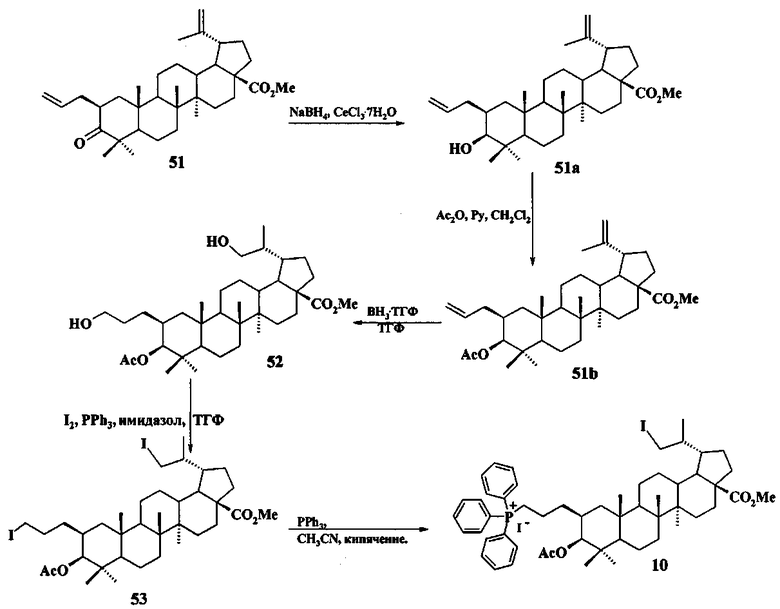
Трифенилфосфониевую соль 10 получали из 2β-аллилбетулоната 51, который в две стадии трансформировали в эфир 51b. Соединение 51b гидроборировали под действием комплекса BH3·ТГФ в ТГФ с получением диола 52. Диол 52 йодировали кристаллическим йодом в присутствии имидазола и трифенилфосфина. Полученный дийодид 53 вовлекали во взаимодействие с избытком трифенилфосфина при кипячении в толуоле или ацетонитриле в атмосфере аргона с получением соединения 10 (химические реакции IV).
Химические реакции IV
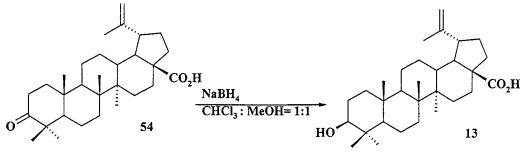
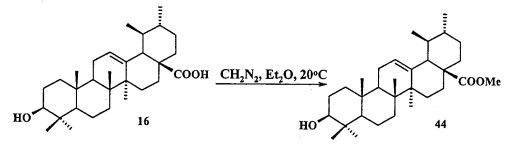
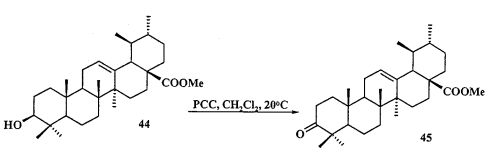
Исходные соединения в синтезе фосфониевых солей 1-11 получали следующим образом. Урсоловую кислоту получали из черноплодной рябины по методу [Л.П. Козлова, Е.В. Малыхин, СМ. Обут, С.П. Понов, О.П. Шеремет, патент РФ 2329048]. Бетулин выделяли из коры березы по методу [Г.А. Толстиков, М.И., Горяев, Х.О. Ким, Р.А. Хегай, Журн. Прикл. Химии, 1967, 40, 920]. Бетулоновую кислоту 54 получали окислением бетулина 12 под действием реагента Джонса в ацетоне. Бетулиновую кислоту 13 в виде смеси 3α- и 3β-эпимеров (3α:3β=5:95) получали восстановлением бетулоновой кислоты NaBH4 в смеси растворителей CHCl3:MeOH=1:1. Кристаллизация смеси продуктов из MeOH дала индивидуальный 3β-эпимер [D.S.H.L. Kim, Z. Chen, T. Nguyen, J.M. Pezzuto, S. Qiu, Z.-Z.Lu. Synthetic Communications. 1997, 27, 1607-1612] (химические реакции V). Гидрирование бетулононовой кислоты 54 в дигидробетулоновую кислоту 55 проводили по методу [L. Pohjala, S. Alakurtti, T. Ahola, J. Yli-Kauhaluoma, P. Tammela. Journal of Natural Products. 2009, 72, 1917-1926].
Химические реакции V
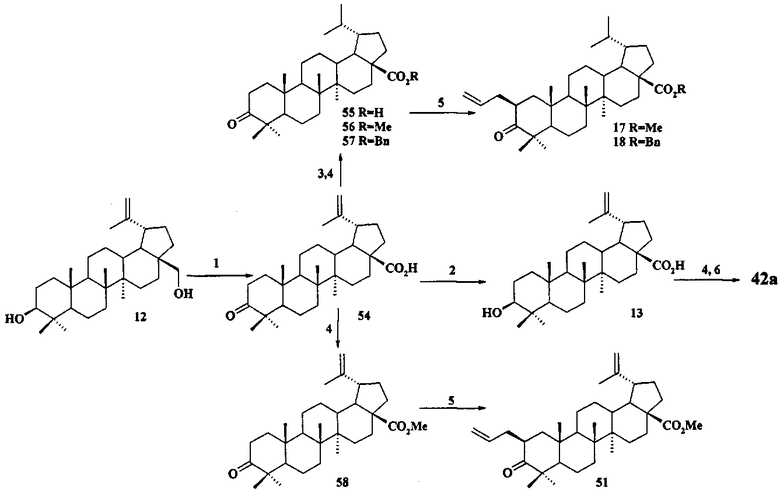
Реагенты и условия: (1) CrO3, H2SO4, CH3COCH3, 0°C; (2) NaBH4, CHCl3-MeOH (50/50), 20°C; (3) H2, 10% Pd/C, MeOH-ТГФ (50/50), 20°C; (4) CH2N2, Et2O, 20°C; BnCl, ДМФА, K2CO3, 55°C; (5) KN(SiMe3)2, BEt3, C3H5 Br, диметоксиэтан, 20°C, Ar; (6) Ac2O или AcCl, 20°C.
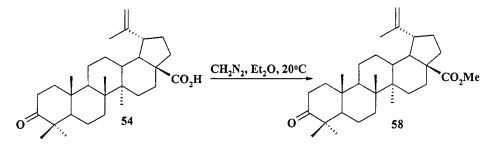
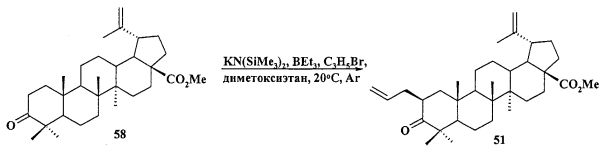
Защиту 28-COOH и 3-OH групп в бетулиновой, бетулоновой и дигидробетулоновой кислотах осуществляли по типовым методикам. Метил бетулонат 51 и дигидробетулонаты 17 и 18 получали взаимодействием эфиров бетулоновой и дигидробетулоновой кислот 56-58 с KN(SiMe3)2, Et3B и аллилбромидом в диметоксиэтане по опубликованному нами ранее способу [А.Ю. Спивак, Э.Р. Шакурова, Д.А. Недопекина, Р.Р. Халитова, Л.М. Халилов, В.Н. Одиноков, Ю.П. Бельский, А.Н. Иванова, Н.В. Бельская, М.Г. Данилец, А.А. Лигачева. Известия Академии Наук. - Сер. химическая. 2011, №4, 681-688].
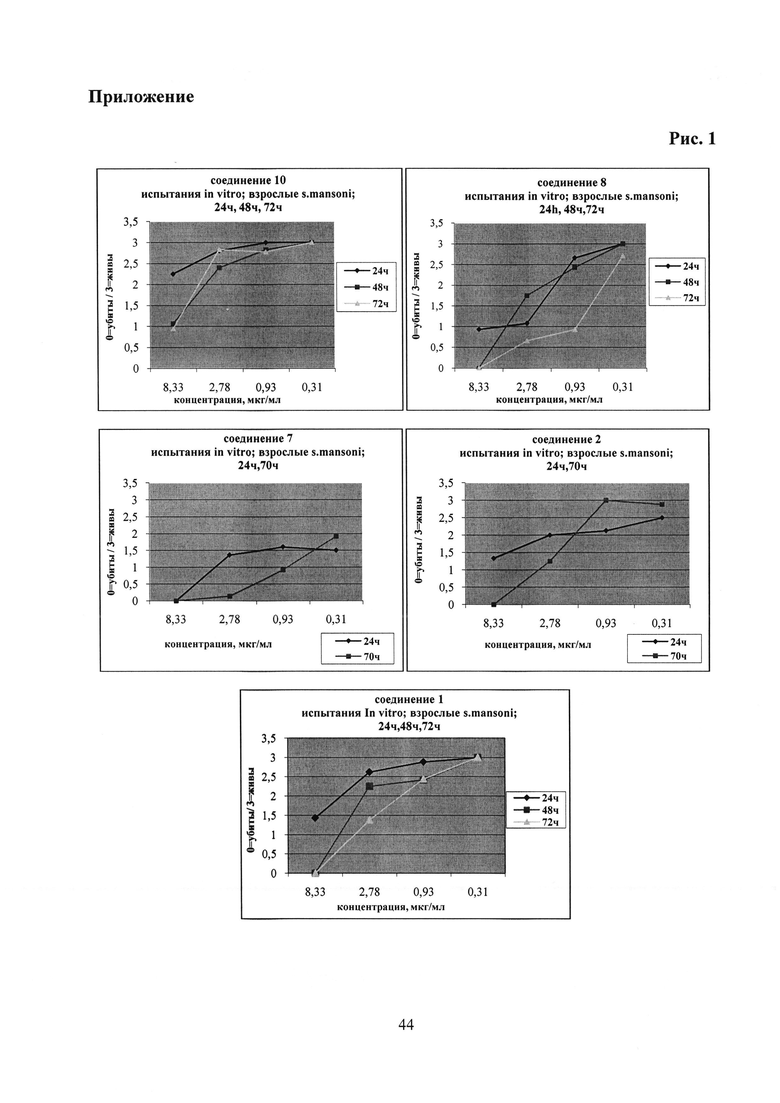
Шистосомицидный эффект синтезированных солей 1-11 (11 соединений) исследовался in vitro в отношении шистосомул и взрослых S. mansoni и классифицировался в следующих категориях относительно контроля: 3 - «все живы»; 0 - «все убиты». Для сравнения тестировались бетулин 12, бетулиновая кислота 13, урсоловая кислота 16 и коммерчески доступная трифенилфосфониевая соль бромбутановой кислоты 59. Первоначально соединения были испытаны против S. mansoni schistosomula в концентрациях 12.5 мкг/мл и 6.25 мкг/мл. Действие всех фосфониевых солей в этих концентрациях привело к полной гибели шистосомул через 72 часа. Бетулиновая и урсоловая кислоты проявили в этих условиях умеренную активность, бетулин и синтетическая соль 59 не проявили активности. Трифенилфосфониевые соли 1-6 и 11 были дополнительно испытаны против шистосомул в следующих шести концентрациях: 0.39 мкг/мл, 0.78 мкг/мл, 1.56 мкг/мл, 3.13 мкг/мл, 6.25 мкг/мл, 12.5 мкг/мл. Наибольшую шистосомицидную активность против шистосомул в ряду соединений 1-6 и 11 проявили фосфониевые соли 1 (IC50 0.56 мкг/мл) и 2 (IC50 0.52 мкг/мл) (Табл. 1).
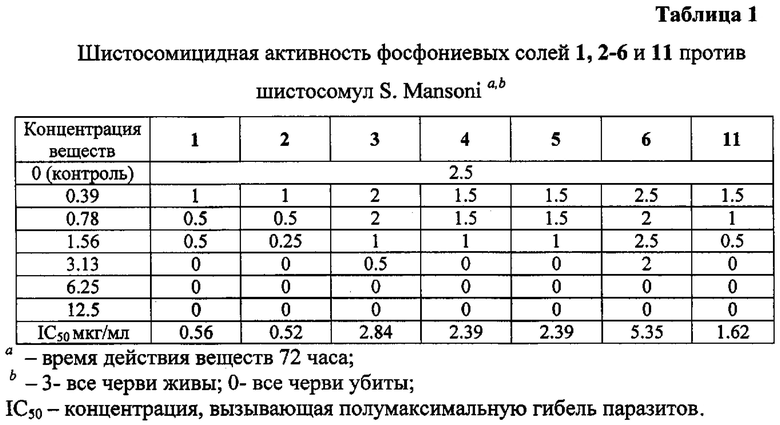
Выбранные пять фосфониевых солей 1, 2, 7, 8 и 10, а также бетулиновая и урсоловая кислоты, были испытаны против взрослых S. mansoni при концентрации 25 мкг/мл. Действие всех фосфониевых солей привело к полной гибели паразитов через 24 часа и 72 часа. Бетулиновая и урсоловая кислоты в этих условиях не проявили активности. Далее пять фосфониевых солей были испытаны при следующих концентрациях: 0.31 мкг/мл, 0.93 мкг/мл, 2.78 мкг/мл, 8.33 мкг/мл. Антипаразитарная активность всех солей начинала проявляться при концентрации 2.78 мкг/мл, далее дозозависимо возрастала и при концентрации 8.33 мкг/мл через 24 ч или 72 ч наблюдалась 100% гибель взрослых шистосом (Табл. 2; Приложение. Рис. 1).
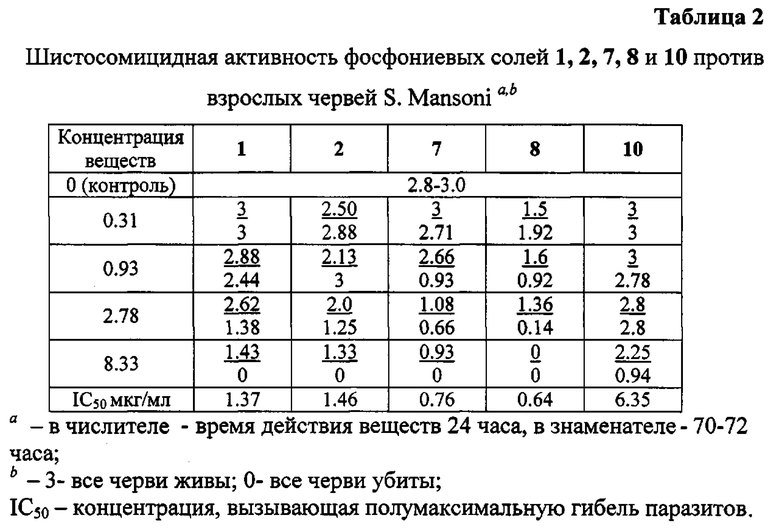
Наибольшую антипаразитарную активность против взрослых особей S. mansoni проявили аллильные фосфониевые соли 7 (IC50 0.76 мкг/мл) и 8 (IC50 0.64 мкг/мл).
Преимущества предлагаемого способа:
1. Впервые исследованы в качестве потенциальных шистосомицидных лекарственных агентов новые ионные производные тритерпеноидов лупановой и урсановой группы - трифенилфосфониевые соли бетулина бетулиновой и урсоловой кислот. В испытаниях in vitro трифенилфосфониевые производные тритерпеноидов проявили высокую шистосомицидную активность против шистосомул и взрослых червей шистосомы мансона.
2. В ряду испытанных трифенилфосфониевых производных лупановых и урсановых тритерпеноидов наибольшую антишистосомную активность проявили аллильные фосфониевые соли 7 и 8 с величиной IC50 - 0.76 и 0.64 мкг/мл соответственно. Эти соединения получены в 2-4 стадии из доступного растительного метаболита бетулина с использованием простых и дешевых реагентов. Простой в препаративном выполнении синтез позволяет получать соединения 7 и 8 в любых количествах, необходимых для научных исследований и медицинских испытаний.
3. Высокая антишистосомальная активность трифенилфосфониевых солей 1-11 в сравнении с активностью бетулина, бетулиновой, урсоловой кислот и коммерчески доступной соли 59 свидетельствует о взаимном синергетическом влиянии катиона трифенилфосфония и фрагментов бетулиновой или урсоловой кислот на антишистосомальную активность гибридных молекул 1-11.
Специфическое строение кожно-мускульной оболочки трематод с высоким содержанием на поверхности тегумента митохондрий делает наружный покров паразитов уязвимым местом для потенциальных митохондриально-направленных лекарственных средств, к которым относятся трифенилфосфониевые производные бетулиновой и урсоловой кислот. Бетулиновая кислота и катион трифенилфосфония выполняют роль прооксидантов [J.S. Modica-Napolitano, J.R. Aprille, Adv Drug Delivery Rev, 2001, 49, 63; S. Fulda, G. Kroemer, Drug Discovery Today, 2009, 14, 885]. Присутствие этих соединений в митохондриях индуцирует образование агрессивных кислородсодержащих радикалов, разрушающих мембраны митохондрий. Шистосомы существуют в аэробной среде и в условиях окислительного стресса, спровоцированного присутствием в организме фосфониевых солей бетулиновой и урсоловой кислот, шистосомы должны дать адекватный антиоксидантный ответ. Вместе с тем, резерв окислительно-восстановительной системы шистосом значительно слабее потенциала биологической редокс-системы человека [Hsin-Hung Huang, С. Rigouin, D.L. Williams. The redox biology of schistosome parasites and applications for drug development. Curr Pharm Des. 2012. 18. 3595-3611]. Так, у шистосом отсутствует эндогенный антиоксидант каталаза, а белки шистосомальной глутатионпероксидазы проявляют низкую активность в отношении пероксида водорода. У шистосомул наблюдается низкая экспрессия цитозольной и пептид-содержащей супероксиддисмутазы.
В связи с изложенными фактами трифенилфосфониевые производные тритерпеноидов лупановой и урсановой группы представляют интерес в качестве новых потенциальных антишистосомальных лекарственных агентов, которые, по-видимому, выполняют роль прооксидантов, разрушающих митохондрии тегумента паразитов.
Изобретение поясняется примерами.
Пример 1.
Бромид метил-3β-ацетокси-2β-(3-трифенилфосфониопропил)-20,29-дигидробетулинат (1): К раствору соединения 56 (0,42 ммоль) в DME (2,50 мл) при комнатной температуре в атмосфере аргона при перемешивании прибавили KN(SiMe3)2 (1М раствор в ТГФ) (0,55 мл, 0,55 ммоль). Через 15 мин, к раствору добавили Et3B (1М раствор в ТГФ) (0,55 мл, 0,55 ммоль) и перемешивали в течение 1 ч. Затем прибавили раствор аллилбромида в 1 мл DME (0,55-0,84 ммоль, преимущественно 0,84). Реакционную смесь перемешивали в течение контрольного времени (3-4 ч, преимущественно 3 ч, контроль ТСХ), нейтрализовали 3М раствором HCl, разбавляли водой (1,5 мл) и экстрагировали EtOAc. Объединенные экстракты сушили MgSO4. Остаток упарили и хроматографировали на колонке SiO2 (элюент - гексан: EtOAc, 30:1→1:1), получили метил 2β-аллил-3-оксолуп-20,29-дигидробетулонат (17). Выход 77%, белые кристаллы, т.пл.=84-86°C (EtOH), 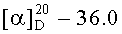
Соединение 17 (0,3 ммоль) растворили в смеси растворителей ТГФ-метанол (1:2, 7,5 мл) в атмосфере Ar и охладили до -30°C. К этому раствору прибавили по каплям раствор CeCl3·7H2O (0,15 г, 0,4 ммоль) в смеси растворителей ТГФ-метанол (1:1, 1 мл). Затем прибавили NaBH4 (0,023 г, 0,6 ммоль) небольшими порциями в течение 5 мин, подняли температуру до комнатной и перемешивали при этой температуре в течение 2 ч (ход реакции контролировали с помощью ТСХ). После завершения прибавили 5% раствор HCl и смесь экстрагировали этилацетатом (25 мл). Экстракт промывали насыщенным водным раствором NaHCO3, водой, сушили (MgSO4) и упарили на вакууме. Остаток очистили с помощью колоночной хроматографии на SiO2 (элюент - гексан: EtOAc, 30:1→1:1), получили метил 2β-аллил-3β-гидрокси-20,29-дигидробетулинат (19). Выход 74%. Физические характеристики, ИК- и УФ-спектры, спектры ЯМР 1H и 13C соединения 19, а также описанных ниже солей 1-6 и промежуточных соединений 20-39 (схема 1) приведены в работе [А.Ю. Спивак, Д.А. Недопекина, Э.Р. Шакурова, P.P. Халитова, P.P. Губайдуллин, В.Н. Одиноков, У.М. Джемилев, Ю.П. Вельский, Н.В. Вельская, С.А. Станкевич, Е.В. Короткая, В.А. Хазанов. Известия АН, Сер. химическая, 2013, №1, 189-199].
К раствору соединения 19 (0.47 ммоль) в 4 мл сухого пиридина, охлажденному до 0°C при перемешивании, прибавили уксусный ангидрид (0.07 мл, 0.69 ммоль), 4-диметиламинопиридин (DMAP) (0.029 г, 0.24 ммоль) и перемешивали при комнатной температуре 10-16 ч преимущественно 16 ч (контроль ТСХ). Затем прибавили 10% раствор HCl и смесь экстрагировали этилацетатом (4×15 мл). Экстракт промывали насыщенным водным раствором соли, сушили (MgSO4) и упарили в вакууме. Остаток очистили с помощью колоночной хроматографии на SiO2 (элюент - гексан: EtOAc, 15:1). Получили метил 2β-аллил-3β-ацетокси-20,29-дигидробетулинат (22). Выход 95%.
Комплекс BH3·ТГФ (1М раствор в ТГФ) (0.86 мл, 0.86 ммоль) добавили в атмосфере аргона при комнатной температуре к перемешиваемому раствору соединения 22 (0,24 г, 0,43 ммоль) в сухом ТГФ (5 мл). Через 3 ч реакционную смесь охладили до 0°C и осторожно по каплям добавили 10% NaOH (1 мл), а затем 30% H2O2 (1 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, нейтрализовали 3М HCl и экстрагировали этилацетатом (3×20 мл). Объединенные органические фазы промывали рассолом, сушили (MgSO4) и удаляли растворитель в вакууме. Остаток очищали с помощью колоночной хроматографии на SiO2 (элюент - гексан: EtOAc, 30:1→1:1), получили метил 3β-гидрокси-2β-(3-гидроксипропил)-20,29-дигидробетулинат (27). Выход: 76%.
Соединение 27 (0,44 ммоль), пиридин (0.06 г, 0.77 ммоль) и DMAP (0.03 г, 0.25 ммоль) растворили в CH2Cl2 (2 мл) и охладили до 0°C. К этому раствору добавили по каплям раствор метансульфонилхлорида (0,07 г, 0,65 ммоль) в CH2Cl2 (1 мл) и перемешивали при комнатной температуре в течение 20-24 ч, преимущественно 24 ч (контроль ТСХ). После окончания реакции добавили холодный 5% раствор HCl и смесь экстрагировали этилацетатом (25 мл). Экстракт промывали насыщенным раствором NaHSO3, водой, сушили (MgSO4) и упарили в вакууме. Остаток очищали с помощью колоночной хроматографии на SiO2 (элюент гексан - EtOAc, 10:1→1:1), получили метил 3β-ацетокси-2β-(3-мезилоксипропил)-20,29-дигидробетулинат (33). Выход 93%.
Мезилат 33 (0.42 ммоль) растворили в сухом ацетоне (12 мл) и добавили LiBr (0.06-0.07 г, 0.7-0.8 ммоль, преимущественно 0.8 ммоль). Смесь кипятили в течение 3-4 ч, преимущественно 3 ч и затем охладили, осадок отфильтровали и промыли ацетоном (2 мл). Фильтрат и промывки объединяли и упарили на вакууме. Остаток очищали с помощью колоночной хроматографии на SiO2 (элюент - гексан: EtOAc, 30:1→1:1). Получили метил 3β-ацетокси-2β-(3-бромпропил)-20,29-дигидробетулинат (36). Выход 79%.
Смесь бромида 36 (0.24 ммоль) и трифенилфосфина (0.72 ммоль-1,2 ммоль, преимущественно 0.72 ммоль) кипятили в толуоле (8 мл) в течение 28-32 ч, преимущественно 32 ч (контроль ТСХ). После окончания реакции раствор охладили и упарили в вакууме, полученный твердый продукт промыли горячим гексаном (7 мл × 2), растворили в минимальном объеме EtOAc (2 мл) и осадили гексаном (8 мл). Осадок отфильтровали, получили бромид метил-3β-ацетокси-2β-(3-трифенилфосфониопропил)-20,29-дигидробетулинат (1). Выход 94%.
Пример 2.
Бромид метил-3α-ацетокси-2β-(3-трифенилфосфониопропил)-20,29-дигидробетулинат (2): Соединение 17 (0,8 ммоль) растворили в сухом ТГФ (15 мл) в атмосфере Ar, охладили до -78°C и добавили 1 М раствор L-селектрида в ТГФ (2,4 мл, 2,4 ммоль). Затем раствор перемешивали при комнатной температуре в течение 2 ч и прибавили 2М раствор NaOH (18 мл) и H2O2 (30%, 4 мл), перемешивали в течение 1 ч. Смесь упарили до небольшого объема и экстрагировали этилацетатом. Органическую фазу промыли водой, сушили (MgSO4) и упарили на вакууме. Остаток очистили с помощью колоночной хроматографии на SiO2 (элюент - гексан: EtOAc, 30:1→1:1). Получили метил 2β-аллил-3α-гидрокси-20,29-дигидробетулинат (21). Выход 68%.
Затем соединение 21 трансформировали в целевую соль 2 (выход 89%) через промежуточные ацетат 24 (выход 96%), первичный спирт 28 (выход 74%), мезилат 34 (выход 87%), бромид 37 (выход 78%) по способу, описанному в примере 1.
Пример 3.
Йодид метил-3β-гидрокси-2β-(3-трифенилфосфониопропил)-20,29-дигидробетулинат (3): Получили соединение 25 восстановлением соединения 17 реагентом NaBH4-CeCl3·7H2O и гидроборированием аллильного фрагмента в соединении 19 по способу, описанному в примере 1.
К раствору соединения 25 (0.35 ммоль) в 6 мл сухого ТГФ при 0°C в атмосфере аргона при перемешивании прибавили Ph3P (трифенилфосфин) (0.220 г, 0.84 ммоль), имидазол (0.117 г, 1.17 ммоль), кристаллический I2 (0.191 г, 0.75 ммоль) и перемешивали при 0°C 0.5-1 ч, преимущественно 1 ч. После завершения реакции, раствор разбавили этилацетатом (10-15 мл) и упарили на вакууме. Остаток очистили с помощью колоночной хроматографии на SiO2 (элюент - гексан: EtOAc, 30:1→1:1). Получили метил 3β-гидрокси-2β-(3-йодпропил)-20,29-дигидробетулинат (30). Выход 57%.
Йодид 30 трансформировали в соль 3 (выход 21%) по способу, описанному в примере 1, но отличающемуся тем, что смесь йодида и трифенилфосфина кипятили в толуоле в течение 16-20 ч, преимущественно 16 ч.
Пример 4.
Йодид метил-3α-гидрокси-2β-(3-трифенилфосфониопропил)-20,29-дигидробетулинат (4): Получили соединение 21 восстановлением соединения 17 с использованием L-селектрида по способу, описанному в примере 2.
Соединение 21 трансформировали в первичный спирт 26 реакцией гидроборирования по способу, описанному в примере 1.
Соединение 26 трансформировали в йодид 31 (выход 73%) взаимодействием с кристаллическим йодом и имидазолом по способу, описанному в примере 3, йодид 31 трансформировали в соль 4 (выход 23%) по способу, описанному в примере 3.
Пример 5.
Бромид 3β-ацетокси-2β-(3-трифенилфосфониопропил)-20,29-дигидробетулиновой кислоты (5): Бензилдигидробетулонат 18 восстановили в спирт 20 (выход 72%) с последующим ацетилированием в соединение 23 (выход 87%). Ацетат 23 трансформировали в первичный спирт 29 (выход 79%), мезилат 35 (выход 90%) и бромид 38 (выход 81%) по способу, описанному в примере 1. Бензильную защиту снимали гидрогенолизом в присутствии Pd/C в Et2O с получением соединения 39 (выход 98%), которое трансформировали в соль 5 (выход 81%) по способу, описанному в примере 1.
Пример 6.
Йодид бензил-3β-ацетокси-2β-(3-трифенилфосфониопропил)-20,29-дигидробетулинат (6): Получен по способу, описанному в примере 5, но отличающемуся тем, что первичный спирт 29 транформировали в первичный йодид 32 (выход 94%) и затем в трифенилфосфониевую соль 6 (выход 78%) по способу, описанному в примере 3.
Пример 7.
Бромид метилового эфира 3β-ацетокси-30-трифенилфосфониолуп-20(29)-ен-28-оиковой кислоты (7). Смесь соединения 42a (2.09 г, 4.08 ммоль) и NBS (1.45 г, 8.16 ммоль) в CCl4 (83 мл) перемешивали при 50°C 4 дня. Осадок отфильтровали, фильтрат упарили и хроматографировали на колонке SiO2 (элюент - гексан: EtOAc, 30:1→10:1). Перекристаллизация продукта из гексана дала соединение 43a (1.55 г, 64%). Метиловый эфир 3β-ацетокси-30-бромолуп-20(29)-ен-28-оиковой кислоты (43a): Выход 64%. Белые кристаллы Т.пл.=216-218°C, 218-220°C лит. [N.V. Uzenkova, N.I. Petrenko, М.М. Shakirov, Е.Е. Shul`ts, and G.A. Tolstikov. Chemistry of Natural Compounds, V 41, №6, 2005, 692-700]. ЯМР 1H (400 МГц, CDCl3, δ, м.д., J/Гц,): 0.72 (м, 1H, H(5)); 0.83 (с, 3H, H(24)); 0.84 (с, 6H, H(23), H(25)); 0.91 (с, 3H, H(26)); 0.97 (c, 3H, H(27)); 1.00-2.30 (м, 23H, CH, CH2, в пентациклическом скелете); 2.05 (с, 3H, Me (OAc)); 3.04 (м, 1H, Н(19)); 3.68 (с, 3H, OMe); 3.99 (уш.с, 2H, Н(30)); 4.46 (д.д, 1H, Н(3), J=10.5, J=5.8); 5.04, 5.14 (уш.с, 2H, Н(29)).
Смесь бромида 43a (0.24 г, 0.4 ммоля) и трифенилфосфина (2 ммоля) кипятили в ацетонитриле (20 мл) в течение 4-5 часов (контроль ТСХ). После окончания реакции раствор охладили и упарили в вакууме, полученный твердый продукт промыли горячим гексаном (10 мл × 2), растворили в минимальном объеме EtOAc (2-4 мл) и осадили гексаном (12 мл). Осадок отфильтровали, получили соединение 7 (0.31 г, 90%). Белые кристаллы. Т.пл.=174-175°C, 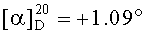
Пример 8.
Бромид 3β,28-диацетокси-30-трифенилфосфониолуп-20(29)-ена (8). Смесь соединения 40 (2.15 г, 4.08 ммоль) и NBS (1.45 г, 8.16 ммоль) в CCl4 (83 мл) перемешивали при 50°C 4 дня. Осадок отфильтровали, фильтрат упарили и хроматографировали на колонке SiO2 (элюент - гексан: EtOAc, 30:1→10:1). Перекристаллизация продукта из гексана дала соединение 41 (1.48 г, 60%). 3β,28-Диацетокси-30-бромолуп-20(29)-ен (41): Белые кристаллы. Т.пл.= 183-185°C, лит., 185°C, [I-Ch. Sun, H-K. Wang, Y. Kashiwada, J-K. Shen. L.M. Cosentino, Ch-HChen, L-M. Yang, and K-H. Lee. J. Med. Chem. 1998, 41, 4648-4657]; лит., 195-196°C. [N.V. Uzenkova, N.I. Petrenko, M.M. Shakirov, E.E. Shul`ts, and G.A. Tolstikov. Chemistry of Natural Compounds, V 41, №6, 2005, 692-700]. ЯМР 1H (400 МГц, CDCl3, 5,м.д., J/Гц,): 0.78 (м, 1H, H(5)); 0.84, 0.85, 0.87 (с, по 3H, Н(23), Н(24), Н(25)); 0.99 (с, 3H, Н(27)); 1.08(с, 3H, Н(26)); 0.95-2.25 (м, 23H, CH, CH2, в пентациклическом скелете); 2.04, 2.08 (оба с по 3H, Me (OAc)); 2.44 (д.т, 1H, Н(19), J=5.5, J=10.8); 3.85, 4.28 (д, 2H, Н(28), J=11.2); 3.98 (с, 2H, Н(30)); 4.47 (д.д, 1H, Н(3), J=5.5, J=10.5); 5.04, 5.14 (с, 2H, Н(29)).
Смесь бромида 41 (0,24 г, 0.4 ммоля) и трифенилфосфина (0.52 г, 2 ммоля) кипятили в ацетонитриле (18 мл) в течение 4-5 ч (контроль ТСХ). После окончания реакции раствор охладили и упарили в вакууме, полученный твердый продукт промыли горячим гексаном (10 мл × 2), растворили в минимальном объеме EtOAc (2-4 мл) и осадили гексаном (12 мл). Осадок отфильтровали, получили соединение 8 (0.32 г, 92%). Белые кристаллы Т.пл.=177-179°C, 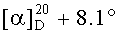
Пример 9.
3β-Ацетокси-30-трифенилфосфониолуп-20(29)-ен-28-оиковая кислота (9). Смесь соединения 42 (2.04 г, 4.08 ммоль) и NBS (1.45 г, 8.16 ммоль) в CCl4 (83 мл) перемешивали при 50°C 4 дня. Осадок отфильтровали, фильтрат упарили и хроматографировали на колонке SiO2 (элюент - гексан: EtOAc, 30:1→10:1). Перекристаллизация продукта из гексана дала соединение 43 (1.32 г, 56%). 3β-Ацетокси-30-бромолуп-20(29)-ен-28-оиковая кислота (43): Белые кристаллы. ЯМР 1H (400 МГц, CDCl3, δ, м.д., J/Гц,): 0.72 (м, 1H, H(5)); 0.84 (с, 3H, H(24)); 0.85, 0.86 (оба с по 3H, H(23), H(25)); 0.94 (с, 3H, H(26)); 0.99 (с, 3H, H(27)); 1.08-2.40 (м, 23H, CH, CH2, в пентациклическом скелете); 2.05 (с 3H, Me (OAc)); 3.05 (т.д, 1H, H(19), J=10.8, J=4.5); 4.04 (уш.с, 2H, H(30)); 4.49 (д.д, 1H, H(3), J=10.4, J=6); 5.06, 5.16 (оба с, 2H, H(29)) 5.68 (м, 1H, CO2H). Соединение было использовано в дальнейшей трансформации в трифенилфосфониевую соль 9 без дополнительной очистки.
Смесь бромида 43 (0,23 г, 0.4 ммоль) и трифенилфосфина (0.52 г, 2 ммоль) кипятили в ацетонитриле (20 мл) в течение 4-5 ч (контроль ТСХ). После окончания реакции раствор охладили и упарили в вакууме, полученный твердый продукт промыли горячим гексаном (10 мл × 2), растворили в минимальном объеме EtOAc (2-4 мл) и осадили гексаном (12 мл). Осадок отфильтровали, получили соединение 9 (0.29 г, 87%). Белые кристаллы. Т.пл.=221-222°C, 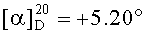
Пример 10.
Йодид метил-3β-ацетокси-2β(3-трифенилфосфониопропил)-29-йодметил-20(29)-дигидробетулината (10): Комплекс BH3·ТГФ (1М раствор в ТГФ) (1.8 мл, 1.8 ммоль) добавили в атмосфере аргона при комнатной температуре к перемешиваемому раствору соединения 51 (0.24 г, 0.43 ммоль) в сухом ТГФ (5 мл). Через 3 часа реакционную смесь охладили до 0°C и осторожно по каплям добавили 10% NaOH (4 мл), а затем 30% H2O2 (4 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, нейтрализовали 3М HCl и экстрагировали этилацетатом. Объединенные органические фазы промывали рассолом, сушили MgSO4 и удаляли растворитель в вакууме. Остаток очищали с помощью колоночной хроматографии на SiO2 (элюент - гексан: EtOAc, 30:1→1), получили соединение 52 (0.14 г, 54%) в виде белых кристаллов. Метил-3β-ацетокси-2α(3-гидроксипропил)-29-гидроксиметил-20(29)-дигидробетулинат (52): ЯМР 1H (400 МГц, CDCl3, δ, м.д., J/Гц,): 0.68 (т, 1H, H(1), J=12); 0.80 (д, 3H, H(30), J=6.8); 0.87, 0.91, 0.95, 0.96, 0.98 (все с по 3H, H(23)-H(27)); 1.05-2.40 (м, 23H, CH, CH2, в пентациклическом скелете, 1H, H(20), 2H, H(1′), 2H, H(2′)); 2.08 (с, 3H, Me (OAc)); 3.42 (м, 1H, H(29)); 3.77 (д.д. 1H, H(29), J=10, J=4); 3.58 (м, 2H, H(3′)); 3.60 (с, 3H, CO2Me); 4.45 (д, 1H, H(3), J=11,2). Соединение было использовано в дальнейшей трансформации в йодид 53 без дополнительной очистки.
К раствору соединения 52 (0,12 г, 0,20 ммоль) в сухом ТГФ (6 мл) при 0°C в атмосфере аргона при перемешивании прибавили Ph3P (трифенилфосфин) (0,13 г, 1.96 ммоль), имидазол (0.13 г, 1.96 ммоль), кристаллический 12 (0.16 г, 0.86 ммоль) и перемешивали при 0°C 1 ч. После завершения реакции раствор разбавили этилацетатом (10 мл) и упарили на вакууме. Остаток очистили с помощью колоночной хроматографии на SiO2 (элюент - гексан: EtOAc, 30:1→1:1), получили соединение 53 (0.11 г, 68%) в виде белых кристаллов. Метил-3β-ацетокси-2β(3-йодпропил)-29-йодметил-20(29)-дигидробетулинат (53): ЯМР 1H (400 МГц, CDCl3, δ, м.д., J/Гц,): 0.69 (т, 1H, H(1), J=12.8); 0.82, 0.83, 0.87, 0.91, 0.97 (все с по 3H, H(23)-H(27)); 1.07 (д, 3H, H(30), J=6.4); 1.10-2.45 (м, 23H, CH, CH2, в пентациклическом скелете, 1H, H(20), 2H, H(1′), 2H, H(2′)); 2.12 (с, 3H, Me (OAc)); 2.85 (м, 1H, H(29)); 3.12 (м, 2H, H(3′)); 3.41 (д, 1H, H(29), J=7.2); 3.66 (с, 3H, CO2Me); 4.46 (д, 1H, H(3), J=10,8). Соединение было использовано в дальнейшей трансформации в трифенилфосфониевую соль 10 без дополнительной очистки.
Смесь йодида 53 (0.11 г, 0.14 ммоль) и трифенилфосфина (0.36 г, 1.36 ммоль) кипятили в ацетонитриле или толуоле (5 мл) в течение 18 ч (контроль ТСХ). После окончания реакции раствор охладили и упарили в вакууме, полученный твердый продукт промыли горячим гексаном (10 мл × 2), растворили в EtOAc (4 мл) и осадили гексаном (12 мл). Осадок отфильтровали, получили соединение 10 (0.10 г, 69%). Белые кристаллы. Т.пл.=154-156°C, 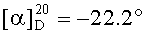
Пример 11.
Йодид метилового эфира 3β-ацетокси-2β-(3-трифенилфосфониопропил)урс-12ен-28-оиковой кислоты (11): К раствору соединения 44 (0.50 г, 1.06 ммоль) в CH2Cl2 (13 мл) при 0°C и интенсивном перемешивании прибавили PCC (пиридиния хлорхромат) (0.684 г, 3.18 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение контрольного времени (1-2 ч, контроль ТСХ). Затем прибавили силикагель (3 г) (ООО «Имид» Силикагель, фракция 50±160 мкм), EtOAc (15 мл), перемешивали 5 минут и отделились от осадка на фильтре Шотта, фильтрат промыли насыщенным водным раствором соли, водой и сушили MgSO4. Остаток упарили и хроматографировали на колонке с SiO2 (элюент - гексан - EtOAc), получили соединение 45 (0.40 г, 94%). Метиловый эфир урсоновой кислоты (45): Белые кристаллы Т.пл.=192-194°C, лит., 192-193°C, [S.M. Jainand С.К. Atal. IndianJ. Chem., Sec. В., 1986, 25B(4), 427]. Лит, 191-193°C (EtOH), [α]24+87.8 (с 0.5, CHCl3) [A.V. Korovin and A.V Tkachev. Russian Chemical Bulletin, International Edition, 2001, V 50, №2, 304-310]. Спектр ЯМР 1H (5, м.д., J/Гц): 0.80, 0.95, 1.05, 1.09, 1.10 (с, 3H, H(23)-H(27)); 0.84, 0.94 (оба д, 3H, H(29), H(30), J=6); 1.25-2.05 (м, 22 H, CH, CH2 в скелете); 2.25 (д, 1H, H(18), J=11.6); 3.62 (с, 3H, OMe); 5.27 (т, 1H, H(12), J=3.6).
К раствору соединения 45 (1 г, 2.13 ммоля) в DME (10 мл) и ТГФ (8 мл) при комнатной температуре в атмосфере аргона при перемешивании прибавили KN(SiMe3)2 (1М раствор в ТГФ), (2.77 мл, 2.77 ммоль). Через 15 мин к раствору добавили Et3B (1 М раствор в ТГФ), (2.77 мл, 2.77 ммоль) и перемешивали в течение 1 ч. Затем прибавили раствор аллилбромида в 5 мл DME (4.69 ммоль). Реакционную смесь перемешивали в течение контрольного времени (4 ч, контроль ТСХ), нейтрализовали 3М раствором HCl, разбавили водой (17 мл) и экстрагировали EtOAc (10 мл × 3). Объединенные экстракты сушили MgSO4. Остаток упарили и хроматографировали на колонке с SiO2 (элюент - гексан-EtOAc, 30:1→1:1), получили соединение 46 (0.87 г, 94%). Метиловый эфир 2β-аллил-3-оксоурс-12ен-28-оиковой кислоты (46): Белые кристаллы Т.пл.=109-112°C (EtOH), 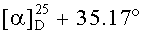
Соединение 46 (0,50 г, 0.98 ммоль) растворили в смеси растворителей ТГФ - метанол (1:2, 22,5 мл) в атмосфере Ar и охладили до -30°C. К этому раствору прибавили по каплям раствор CeCl3 ·7H2О (0.48 г, 1.27 ммоль) в смеси растворителей ТГФ - метанол (1:1, 1 мл). Затем прибавляли NaBH4 (0.075 г, 1.96 ммоль) небольшими порциями в течение 5 мин, подняли температуру до комнатной и перемешивали при этой температуре в течение 2 ч (ход реакции контролировали с помощью ТСХ). После завершения прибавили 5% раствор HCl и смесь экстрагировали EtOAc (10 мл × 3). Экстракт промывали насыщенным водным раствором NaHCO3, водой, сушили MgSO4 и удаляли растворитель в вакууме. Остаток очищали с помощью колоночной хроматографии на SiO2 (элюент - гексан: EtOAc 30:1→1:1), получили соединение 47 (0.40 г, 80%) в виде белых кристаллов. Метиловый эфир 2β-аллил-3β-гидроксиурс-12ен-28-оиковой кислоты (47): Белые кристаллы. Т.пл.=203-206°C (EtOH), 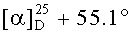
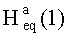
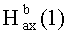
К раствору соединения 47 (0.39 г, 0.77 ммоль) в 17 мл сухого ТГФ, охлажденного до 0°C, при перемешивании прибавили пиридин (0.16 мл, 1.92 ммоль), ацетил хлорид (0.13 мл, 1.54 ммоль), 4-диметиламинопиридин (DMAP) (0.047 г, 0.39 ммоль) и перемешивали при комнатной температуре 24 ч (контроль ТСХ). Затем прибавили 10% раствор HC1 и смесь экстрагировали этилацетатом (15 мл × 4). Экстракт промывали насыщенным водным раствором соли, сушили MgSO4 и растворитель упарили в вакууме. Остаток очистили с помощью колоночной хроматографии на SiO2 (элюент - гексан: EtOAc, 30:1→15:1). Получили соединение 48 (0.35 г, 83%). Метиловый эфир 2β-аллил-3β-ацетоксиурс-12ен-28-оиковой кислоты (48): Выход 95%. Белые кристаллы Т.пл.=186-188°C (EtOH), 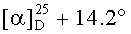
Комплекс BH3·ТГФ (1M раствор в ТГФ) (1.24 мл, 1.24 ммоль) добавили в атмосфере аргона при комнатной температуре к перемешиваемому раствору соединения 48 (0.34 г, 0.62 ммоль) в сухом ТГФ (7 мл). Через 3 ч реакционную смесь охладили до 0°C и осторожно по каплям добавили 10% раствор NaOH (1.6 мл), а затем 30% H2O2 (1.6 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, нейтрализовали 3М HCl и экстрагировали этилацетатом (20 мл × 3). Объединенные органические фазы промывали рассолом, сушили MgSO4 и удаляли растворитель в вакууме.
Остаток очищали с помощью колоночной хроматографии на SiO2 (элюент-гексан:EtOAc, 30:1→1:1), получили соединение 49 (0.21 г, 60%).
Метиловый эфир 3β-ацетокси-2β-(3-гидроксипропил)урс-12ен-28-оиковой кислоты (49): Белые кристаллы, т.пл. 123-125°C (EtOH), 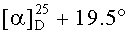
К раствору соединения 49 (0.20 г, 0.35 ммоль) в 6 мл сухого ТГФ при 0°C в атмосфере аргона при перемешивании прибавили Ph3P (трифенилфосфин) (0.22 г, 0.84 ммоль), имидазол (0.12 г, 1.72 ммоль), кристаллический I2 (0.19 г, 0.75 ммоль) и перемешивали при 0°C 1 ч. После завершения реакции раствор разбавили этилацетатом (10-15 мл), упарили на вакууме. Остаток очистили с помощью колоночной хроматографии на SiO2 (элюент - гексан: EtOAc, 30:1→1:1). Получили соединение 50 (0.21 г, 88%). Метиловый эфир 3β-ацетокси-2β-(3-йодпропил)урс-12ен-28-оиковой кислоты (50): Белые кристаллы Т. пл.=116-118°C (EtOH), 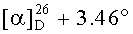
Смесь йодида 50 (0.21 г, 0.28 ммоль) и трифенилфосфина (0.37 г, 1.40 ммоль) кипятили в толуоле (8 мл) в течение 16 ч (контроль ТСХ). После окончания реакции раствор охладили и упарили в вакууме, полученный твердый продукт промыли горячим гексаном (7 мл × 2), растворили в EtOAc (2 мл) и осадили гексаном (8 мл). Осадок отфильтровали, получили соединение 11 (0.24 г, 95%). Т.пл.=151-153°C (EtOH), 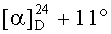
| название | год | авторы | номер документа |
|---|---|---|---|
| ТРИФЕНИЛФОСФОНИЕВЫЕ СОЛИ ЛУПАНОВЫХ ТРИТЕРПЕНОИДОВ, СПОСОБ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОТИВООПУХОЛЕВЫХ ВЕЩЕСТВ | 2012 |
|
RU2551647C2 |
| Фосфониевые соли на основе гликозидов бетулиновой кислоты, обладающие противоопухолевой активностью | 2022 |
|
RU2803739C1 |
| СПОСОБ ПОЛУЧЕНИЯ МЕТИЛОВОГО ЭФИРА 3-ОКСО-3'-(НИТРОМЕТИЛ)-4'-(ХЛОРМЕТИЛ)-СПИРО[ЛУПАН-2,1'-ЦИКЛОПЕНТАН]-28-ОВОЙ КИСЛОТЫ | 2010 |
|
RU2448975C1 |
| 7-ТРИАЗОЛИЛ-ЗАМЕЩЕННЫЕ ГИДРОКСИСПИРОСТАНЫ, ОБЛАДАЮЩИЕ ЦИТОТОКСИЧЕСКОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ ОПУХОЛЕВЫХ КЛЕТОК ЧЕЛОВЕКА | 2023 |
|
RU2798105C1 |
| СПОСОБ ПОЛУЧЕНИЯ СУЛЬФОБЕТАИНОВ НА ОСНОВЕ БЕТУЛИНОВОЙ КИСЛОТЫ | 2015 |
|
RU2588138C1 |
| АНТАГОНИСТЫ ЭНДОТЕЛИНОВЫХ РЕЦЕПТОРОВ, ФАРМКОМПОЗИЦИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СПОСОБ ПОДАВЛЕНИЯ ЭНДОТЕЛИНОВЫХ РЕЦЕПТОРОВ | 1992 |
|
RU2125980C1 |
| БИОКОНЪЮГАТЫ ТРИТЕРПЕНОВЫХ КИСЛОТ ЛУПАНОВОГО РЯДА С ГИДРАЗИДОМ КИСЛОТЫ "ТРОЛОКС", СПОСОБ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИММУНОТРОПНЫХ И ПРОТИВОВОСПАЛИТЕЛЬНЫХ ВЕЩЕСТВ | 2010 |
|
RU2464273C2 |
| ДИАРИЛ-5,6-КОНДЕНСИРОВАННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ КИСЛОТЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЗАЩИТЫ ОТ ВОЗДЕЙСТВИЯ ЛЕЙКОТРИЕНОВ | 1993 |
|
RU2154065C2 |
| ПРОИЗВОДНЫЕ 3-Н-1,2,3-ТРИАЗОЛО-[4,5-D]ПИРИМИДИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1996 |
|
RU2174518C2 |
| Фосфониевые соли на основе бетулиновой кислоты, обладающие цитотоксической активностью в отношении аденокарциномы предстательной железы | 2018 |
|
RU2665922C1 |
Изобретение относится к применению трифенилфосфониевых солей лупановых и урсановых тритерпеноидов формулы 1-11 в качестве средств с шистосомицидной активностью, новым соединениям 8-11, а также способу их получения. Использование изобретения позволит расширить ассортимент лекарственных средств, полученных на основе природных продуктов, для лечения шистосомоза - опасного паразитарного заболевания человека и животных. 3 н. и 4 з.п. ф-лы, 1 ил., 3 табл., 11 пр.
1. Применение трифенилфосфониевых солей лупановых и урсановых тритерпеноидов формулы 1-11 в качестве средств с шистосомицидной активностью:
2. Трифенилфосфониевые соли лупановых и урсановых тритерпеноидов формулы 8-11.
3. Способ получения трифенилфосфониевых солей лупановых и урсановых тритерпеноидов формулы 8-11, отличающийся тем, что соли синтезируют взаимодействием трифенилфосфина с бромидом диацетата бетулина 41, с бромидом бетулиновой кислоты 43, с дийодидом метилбетулоната 53 и с йодидом метилурсоноата 50:
4. Способ получения трифенилфосфониевой соли 8 по п. 3, отличающийся тем, что включает следующие стадии:
a) получают эфир 40 ацетилированием бетулина 12 под действием Ac2O в пиридине или AcCl в ТГФ в присутствии пиридина и 4-диметиламинопиридин (DMAP) или непосредственно из бересты березы путем ее кипячения в уксусной кислоте в течение 12 ч:

b) эфир 40 трансформируют в аллильный бромид 41 под действием N-бромсукцинимида (NBS) в CCl4 при нагревании при 50°С в течение 4 дней:
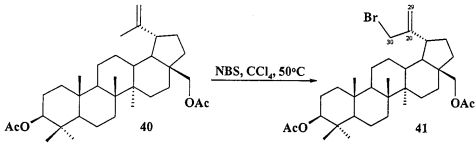
c) полученный на стадии 4b галогенид 41 подвергают взаимодействию с избытком трифенилфосфина при кипячении в ацетонитриле в атмосфере аргона в течение 4-5 ч с получением соли 8, охарактеризованной в п. 1.
5. Способ получения трифенилфосфониевой соли 9 по п. 3, отличающийся тем, что включает следующие стадии:
a) получают бетулоновую кислоту 54 окислением бетулина 12 под действием реагента Джонса в ацетоне:
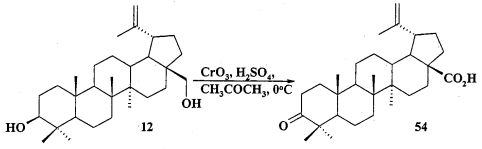
b) получают бетулиновую кислоту 13 восстановлением бетулоновой кислоты 54 NaBH4 в смеси растворителей CHCl3:МеОН=1:1 с последующей очисткой продукта перекристаллизацией из МеОН:
c) бетулиновую кислоту 13 превращают в 3β-ОАс бетулиновую кислоту 42 с помощью Ac2O в пиридине или AcCl в ТГФ в присутствии пиридина и DMAP:

d) ацетилированную кислоту 42 превращают в бромпроизводное 43 под действием N-бромсукцинимида (NBS) в CCl4 при нагревании при 50°C в течение 4 дней:
e) полученный на стадии 5d галогенид 43 подвергают взаимодействию с избытком трифенилфосфина при кипячении в ацетонитриле в атмосфере аргона в течение 5 ч с получением соли 9, охарактеризованной в п. 1.
6. Способ получения трифенилфосфониевой соли 10 по п. 3, отличающийся тем, что включает следующие стадии:
а) получают метиловый эфир бетулоновой кислоты 58 из бетулоновой кислоты 54 под действием диазометана в эфире при комнатной температуре:
b) получают 2β-аллилбетулонат 51 из метилбетулоната 58 путем аллильного алкилирования под действием KN(SiMe3)2, Et3B и аллилбромида в диметоксиэтане:
c) стереоселективно восстанавливают метилбетулонат 51 под действием NaBH4 модифицированного CeCl3·7H2O с получением 3β-ОН-эпимера 51а:
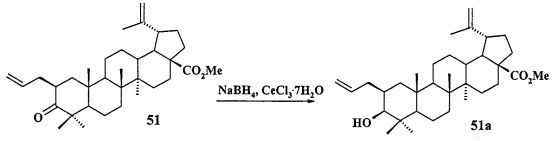
d) осуществляют трансформацию спирта 51а в ацетат 51b с помощью Ac2O в пиридине или AcCl в ТГФ в присутствии пиридина и DMAP:

e) гидроборируют двойные связи в соединении 51b под действием ВН3·ТГФ или BH3·Me2S с последующим окислением борорганического соединения в диол 52 под действием 30% H2O2 и 10% водного раствора NaOH:

f) диол 52 йодируют кристаллическим йодом в присутствии имидазола и трифенилфосфина в атмосфере аргона в растворе сухого ТГФ при 0°C с получением дийодида 53:
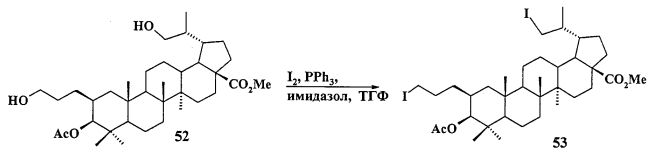
g) полученный на стадии 6f галогенид 53 подвергают взаимодействию с избытком трифенилфосфина при кипячении в толуоле или ацетонитриле в атмосфере аргона в течение 18 ч с получением соли 10, охарактеризованной в п. 1.
7. Способ получения трифенилфосфониевой соли 11 по п. 3, отличающийся тем, что включает следующие стадии:
a) урсоловую кислоту 16 превращают в метилурсоноат 44 под действием диазометана в эфире:
b) метилурсоноат 44 окисляют в метиловый эфир урсоновой кислоты 45 под действием пиридиния хлорхромата (РСС) в CH2Cl2 при 0°C:
c) эфир 45 трансформируют в 2β-аллилзамещенный эфир 46 под действием KN(SiMe3)2, Et3B и аллилбромида в диметоксиэтане при 20°C в атмосфере аргона:
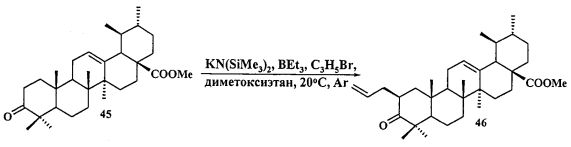 B
B
d) эфир 46 подвергают стереоселективному восстановлению под действием NaBH4, модифицированного CeCl3·7H2O с получением 3β-ОН-эпимера 47:
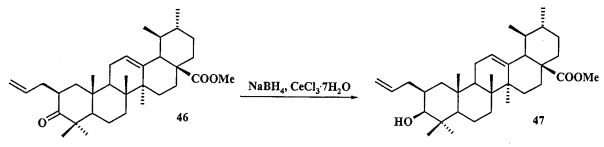
e) осуществляют трансформацию спирта 47 в ацетат 48 с помощью Ac2O в пиридине или AcCl в ТГФ в присутствии пиридина и DMAP:

f) гидроборируют двойную связь аллильного заместителя в соединении 48 под действием ВН3·ТГФ или BH3·Me2S с последующим окислением борорганического соединения в спирт 49 под действием 30% H2O2 и 10% водного раствора NaOH:

g) спирт 49 йодируют кристаллическим йодом в присутствии имидазола и трифенилфосфина с получением йодида 50:
h) полученный на стадии 7g галогенид 50 подвергают взаимодействию с избытком трифенилфосфина при кипячении в толуоле в атмосфере аргона в течение 16 ч с получением соли 11, охарактеризованной в п. 1.
| А | |||
| Ю | |||
| Спивак, Д | |||
| А | |||
| Недопекина, Э | |||
| Р | |||
| Шакурова и др., "Синтез лупановых тритерпеноидов с трифенилфосфониевыми фрагментами и изучение их противоопухолевой активности", Известия академии наук, Серия химическая, 2013 (1), стр | |||
| Питательный кран для вагонных резервуаров воздушных тормозов | 1921 |
|
SU189A1 |
| ТРИФЕНИЛФОСФОНИЕВЫЕ СОЛИ ЛУПАНОВЫХ ТРИТЕРПЕНОИДОВ, СПОСОБ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОТИВООПУХОЛЕВЫХ ВЕЩЕСТВ | 2012 |
|
RU2551647C2 |
| N'-{N-[3-ОКСО-20(29)-ЛУПЕН-28-ОИЛ]-9-АМИНОНОНАНОИЛ}-3-АМИНО-3-ФЕНИЛПРОПИО НОВАЯ КИСЛОТА, ОБЛАДАЮЩАЯ ИММУНОСТИМУЛИРУЮЩЕЙ И ПРОТИВОВИРУСНОЙ АКТИВНОСТЬЮ | 2002 |
|
RU2211843C1 |
| US 20060063749 A1, 23.03.2006 | |||
| Abdul-Ghani R, Loutfy N, el-Sahn | |||

