Изобретение относится к области синтеза биологически активных соединений и касается Nδ-нитрозо-Nδ-[(2-хлорэтил)карбамоил]-L-орнитина, формулы 1, обладающего противоопухолевой активностью и предназначенного для лечения больных онкологическими заболеваниями [Патент РФ №2503657].

Известен способ получения гомолога соединения 1 - Nε-нитрозо-Nε-[(2-хлорэтил)карбамоил]-L-лизина (2) из смеси Nε-нитрозо-Nε-[(2-хлорэтил)карбамоил]-L-лизина (2) и Nε-[(2-хлорэтил)-N-нитрозокарбамоил]-L-лизина (3) путем выдерживания водного раствора смеси соединений 2-3 в соотношении 75:25 при комнатной температуре в течение 140 ч, в результате чего менее устойчивый изомер 3 разлагается [Левит Г.Л., Радина Л.Б., Краснов В.П., Гопко В.Ф., Никифорова Н.В., Перетолчина Н.М. Nω-Алкилнитрозокарбамоил-α,ω-диаминокарбоновые кислоты. III. Синтез и противоопухолевая активность Nε-нитрозо-Nε-[N′-(2-хлорэтил)карбамоил]-L-лизина и Nε-(N′-(2-хлорэтил)-N′-нитрозокарбамоил]-L-лизина // Хим.-фарм. Ж., 1996. Т. 30 (5), с. 23-25]. После упаривания реакционной массы осадок отделяют и перекристаллизовывают из 50% этанола. Выход соединения 2 53%.

Известен способ получения соединения 1 путем разделения смеси изомеров положения нитрозогруппы - Nδ-нитрозо-Nδ-[(2-хлорэтил)карбамоил]-L-орнитина (1) и Nδ-[(2-хлорэтил)-N-нитрозокарбамоил]-L-орнитина (4) методом высокоэффективной жидкостной хроматографии (ВЭЖХ) в обращенно-фазовом варианте.

Согласно сведениям, приведенным в Патенте РФ №2503657, 2,0 мл раствора смеси изомеров, содержащей изомеры 1 и 4 в соотношении 74,24:25,76, с концентрацией 20,0 мг/мл в 3,0% водном изопропаноле (подвижная фаза) вводят в колонку препаративного жидкостного хроматографа. Используют: насос Gilson 305 25 SC, инжектор Rheodyne 7010 с петлей 2,0 мл, УФ детектор LCD 2070 с фиксированной длиной волны 254 нм, колонка размерами 250×20 мм, заполненная сорбентом марки Reprosil-Pur С18, со средним размером гранул сорбента 10 мкм, удельной поверхностью 300 м2/г и размером пор 100 Å, предварительно уравновешенная подвижной фазой. Температура колонки комнатная (20-24°С). Отбор фракций производят по показаниям детектора и по показаниям самопишущего потенциометра от начала сигнала до выхода показаний на нулевую линию. В качестве подвижной фазы используют раствор изопропилового спирта в воде с концентрацией 3% (неподвижная фаза - октадецил-силикагель) при скорости подачи подвижной фазы 6,0 мл/мин и объеме вводимой пробы не более 2,6% от объема колонки. Отбор фракций производят по показаниям детектора и по показаниям самопишущего потенциометра, от начала сигнала до выхода показаний на нулевую линию, средний объем полученных фракций составляет 20,0 мл. Собирают фракцию с временем удерживания 27,0±1,0 мин и после упаривания получают 28,2 мг изомера 1 (выход 95% от содержания изомера 1 в исходном образце). Таким образом, за один цикл удается получить 28,2 мг целевого продукта 1.
Недостатками данного способа являются большой расход органических растворителей, длительность процесса и его сложное аппаратурное оформление, что делает этот способ малопригодным для наработки значительных количеств соединения 1 для его клинических испытаний и дальнейшего использования в медицине.
С целью создания эффективного способа получения соединения 1 предлагается новый способ получения соединения 1, отличающийся тем, что раствор смеси изомеров 1 и 4 в воде выдерживают в течении 50 ч при +37°С, концентрируют реакционную массу, выдерживают 8 ч при +8°С и после осаждения и промывки осадка водой и спиртом получают соединение 1 с выходом 74,1% от содержания изомера 1 в исходном образце.

Предлагаемый способ отличается от способа-прототипа тем, что не требует сложного аппаратурного оформления, использует минимальный объем органического растворителя (этилового спирта), позволяет за один цикл получать значительные количества целевого продукта.
По сравнению со способом получения гомолога соединения 1, соединения 2, предлагаемый способ также имеет ряд существенных особенностей, связанных с отличиями в химических свойствах исходных соединений, продукта реакции и побочных продуктов.
Так, смесь соединений 1 и 4 значительно менее растворима в воде по сравнению со смесью соединений 2 и 3 (1 г/л по сравнению с 2 г/л), что определяет исходную концентрацию раствора. В результате сравнительного изучения кинетики разложения соединений 1, 4, 2 и 3 показано, что гидролитическая устойчивость соединения 1 (период полураспада 124 мин) значительно меньше таковой для гомолога 2 (период полураспада 140 мин). А гидролитическая устойчивость соединения 4 значительно меньше таковой для гомолога 3 (Табл., рис.).
Сравнительное изучение кинетики гидролитического разложения изомеров положения нитрозогруппы 1 и 4, 2 и 3 проводили при +37°С, рН 7,2. Изменение концентрации определяли спектрофотометрически в максимуме поглощения N-нитрозогруппы при ~230 нм. На основании зависимости ln(С/С0) от времени t (Рис.) рассчитывали значения констант скорости гидролитического разложения (k) и периодов полураспада (τ0,5) по методу наименьших квадратов (Таблица).
На основании полученных результатов выбраны оптимальные условия проведения заявляемого процесса, в частности, сокращено время процесса по сравнению с методом [Левит Г.Л., Радина Л.Б., Краснов В.П., Гопко В.Ф., Никифорова Н.В., Перетолчина Н.М. Nω-Алкилнитрозокарбамоил-α,ω-диаминокарбоновые кислоты. III. Синтез и противоопухолевая активность Nε-нитрозо-Nε-[N′-(2-хлорэтил)карбамоил]-L-лизина и Nε-[N′-(2-хлорэтил)-N′-нитрозокарбамоил]-L-лизина // Хим.-фарм. Ж., 1996. Т. 30 (5), с. 23-25].
Изобретение иллюстрируется следующим примером.
Пример 1. 6 г смеси изомеров 1-4 с содержанием изомера 1 76% растворяют в 600 мл воды. Раствор выдерживают при температуре +37°С 50 ч. Отгоняют воду в вакууме на роторном испарителе при температуре 35-40°С до объема 100 мл. Реакционную массу выдерживают в холодильнике при +8°С 8 ч, осадок отфильтровывают, промывают водой и этанолом, сушат в вакууме над Р2О5 и получают 2,86 г соединения 1. Фильтрат и промывные растворы объединяют, упаривают до объема 40 мл, выдерживают в холодильнике при +8°С 8 ч, осадок отфильтровывают, промывают водой и этанолом, сушат в вакууме над Р2О5 и получают дополнительно 0,52 г соединения 1. Общий выход соединения 1 3,38 г, 74,1% от содержания изомера 1 в исходном образце.
Nε-Нитрозо-Nε-[(2-хлорэтил)карбамоил]-L-орнитин (1) имеет следующие физико-химические характеристики: Т. пл. 157-160°С (разл.); [α]D +11,2 (С 2, 1 н. HCl); 1Н ЯМР спектр (D2O, δ, м.д.): 3,88 (м, 2Н, CH2-N(NO)); 3,78 (с, 4Н, ClCH2-CH2-NH); 3,69 (дд, 1Н, СН); 1,83-1,69 (м, 2Н, СН2-СН); 1,61-1,45 (м, 2Н, СН2-СН2-СН2). Что соответствует литературным данным.
Найдено, %: С 35,94; Н 5,77; N 20,85; Cl 13,34. C8H15ClN4O4. Вычислено, %: С 36,03; Н 5,67; N 21,01; Cl 13,31.
Предлагаемый способ отличается от способа-прототипа тем, что не требует сложного аппаратурного оформления, использует минимальный объем органического растворителя (этилового спирта), позволяет за один цикл получать значительные (более чем в сто раз по сравнению с прототипом) количества целевого продукта.
| название | год | авторы | номер документа |
|---|---|---|---|
| N-НИТРОЗО-N-[(2-ХЛОРЭТИЛ)КАРБАМОИЛ]-L-ОРНИТИН | 2012 |
|
RU2503657C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИНДИВИДУАЛЬНЫХ N-НИТРОЗО- N-[(2-ХЛОРЭТИЛ)КАРБАМОИЛ]-L-ЛИЗИНА И N-[(2-ХЛОРЭТИЛ)-N-НИТРОЗОКАРБАМОИЛ]-L-ЛИЗИНА | 2008 |
|
RU2408576C2 |
| ПРОТИВООПУХОЛЕВОЕ СРЕДСТВО | 2014 |
|
RU2569729C1 |
| СПОСОБ ПОЛУЧЕНИЯ N (2-ХЛОРЭТИЛ)- N НИТРОЗО-L-ГОМОЦИТРУЛЛИНА ИЛИ N (2-ХЛОРЭТИЛ)- N НИТРОЗО-2-ГОМОЦИТРУЛЛИНА ДЛЯ ИНЪЕКЦИЙ | 1995 |
|
RU2111742C1 |
| Способ получения N-нитрозо-N-(бета-хлорэтил)карбамоилпептидов или их солей | 1984 |
|
SU1586520A3 |
| Способ получения N-нитрозо-N-(бэта-хлорэтил)-карбамоилпептидов или их кислотно-аддитивных солей | 1982 |
|
SU1424739A3 |
| N-Нитрозо-N-(бэта-хлорэтил)-карбамоилпептиды или их хлоргидраты,обладающие противоопухолевой активностью | 1983 |
|
SU1414851A1 |
| СПОСОБ СЕЛЕКЦИИ КЛЕТОК IN VIVO С ПОМОЩЬЮ ЛИЗОМУСТИНА | 2010 |
|
RU2425149C1 |
| ПРОТИВООПУХОЛЕВОЕ СРЕДСТВО | 1988 |
|
SU1834006A1 |
| ГЛИКОКОНЪЮГАТЫ 20(S)-КАМПТОТЕЦИНА | 1998 |
|
RU2184122C2 |

Изобретение относится к способу получения Nδ-нитрозо-Nδ-[(2-хлорэтил)карбамоил]-L-орнитина формулы 1, обладающего противоопухолевым действием. Согласно предлагаемому способу Nδ-нитрозо-Nδ-[(2-хлорэтил)карбамоил]-L-орнитин получают из смеси изомеров Nδ-нитрозо-Nδ-[(2-хлорэтил)карбамоил]-L-орнитина и Nδ-[(2-хлорэтил)-N-нитрозокарбамоил]-L-орнитина, при этом раствор смеси изомеров в воде выдерживают в течение 50 ч при +37°С, концентрируют реакционную массу и выдерживают 8 ч при +8°С. Предлагаемый способ не требует сложного аппаратурного оформления и позволяет за один цикл получать значительные количества целевого продукта. 1 ил., 1 табл., 1 пр.
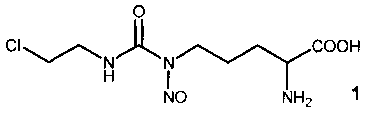
Способ получения Nδ-нитрозо-Nδ-[(2-хлорэтил)карбамоил]-L-орнитина формулы 1
из смеси изомеров Nδ-нитрозо-Nδ-[(2-хлорэтил)карбамоил]-L-орнитина и Nδ-[(2-хлорэтил)-N-нитрозокарбамоил]-L-орнитина, отличающийся тем, что раствор смеси изомеров в воде выдерживают в течение 50 ч при +37°С, концентрируют реакционную массу и выдерживают 8 ч при +8°С.
| N-НИТРОЗО-N-[(2-ХЛОРЭТИЛ)КАРБАМОИЛ]-L-ОРНИТИН | 2012 |
|
RU2503657C1 |
| ЛЕВИТ Г.Л | |||
| и др., N-Алкилнитрозокарбамоил-α,ω-диаминокарбоновые кислоты | |||
| II | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| ЖУРН., 1996, 30(4), с.15-17 | |||
| СПОСОБ ПОЛУЧЕНИЯ ИНДИВИДУАЛЬНЫХ N-НИТРОЗО- N-[(2-ХЛОРЭТИЛ)КАРБАМОИЛ]-L-ЛИЗИНА И N-[(2-ХЛОРЭТИЛ)-N-НИТРОЗОКАРБАМОИЛ]-L-ЛИЗИНА | 2008 |
|
RU2408576C2 |

