Изобретение относится к способу получения М-нитрозо-Ы-(бзта-хлорэтил)- карбамоилпептидов или их солей - новых биологически активных соединений, которые могут найти применение в медицине .
Цель изобретения - способ получения новых производных N-нитpoзo-N- -(бэта-хлорэтил)карбамоилпептидов, обладающих более высокой противоопухолевой активностью.
Хроматографию проводят в тонком слое кизельгеля G (фирмы Мерк) в слет дующих системах растворителей: А - хлороформ:метанол 9:1; Б - н-бута- нол:ледяная уксусная кислота:вода 4:1:1; Б -хлороформ:метанол 8:2} Г - этилацетат:пиридин:ледяная уксусная кислота: вод а 60:20:6:11, Д - этилацетат:пиридин:муравьиная кислота:вода 60:20:6-5,5; Е - метанол: этилацетат 1:3; Ж - этилацетат:
сх
158
гииридин:ледяная уксусная кислота: :|вода 240:20:6:11. : Пример. N-HHTpo30-N-(- - хлорэтил)-карбамоил-пролилвалннамид
1).
1,37 г (5 ммоль) Пролил-валинамидацетата суспендируют в 5 мл безводного диметилформамида и добавляют перемешивании и охлаждении сне- ij-OM 1,4 мл (10 ммоль) триэтиламина щ. 1 ,25 г (5 ммоль) сложного N-нитро- O-N-(/ -хлорэтил)-карбаминовая кис- ота-N окси-сукцинимидного эфира, еакционную смесь перемешивают 15 ми Ари 0°С, затем разбавляют,ледяной водой, выпавший в осадок продукт от- :: (зильтровывают, промывают холодной зодой на фильтре. Продукт сушат в эксикаторе над P. - s 0,91 г |(53%), т.пл. (разложение),Rf t 0,8 (В), 5 88 (М-нитрозогруппа, 400 нм)..
Вычислено,%: С 44,85; Н 6,33; С1 10,19; N 20,13,
C,/Hij, (347,8). Найдено,%: С ,83; Н 6,35; |:i 9,86; N 19,76.,
i Пример2. а) Альфа-Ы-нитро- зо-К-(бэта-хлорэтил)-карбамоил-эпси- лон-трет.бутилоксикарбонил-лизил-про пил- в алин ами,г,.
614 мг (1 ммоль) эпсилон-трет.бу- ;токсикарбонил-лизил-пролилвалин-ами.д 1-тозилата растворят в 10 мл безвод
|ного диметилформамида, после чего в приготовленный раствор добавляют |0,14 мл (1 ммоль) триэтиламина, 274 м :(1 ммоль) К-нитрозо-бэта-хлор этил- карбамоил-п-нитрофенола и 136 мл (1 ммоль) 1-оксибензотриазола. Через 2 ч диметилформамид отгоняют в вакууме, остаток растворяют в 30 мл этилацетата и промывают водой. Этил- ацетатный слой отделяют, высушивают, упаривают. Остаток обрабатывают безводным диэтиловым эфиром, эфирный слой отделяют декантацией, масло высушивают.
Полученную при этом твердую пену- чистят в колонке, заполненной сили- кагелем. Фракции собирают, г створи тель отгоняют в вакууме. Остаток растирают в порошок в присутствии безводного петролейного эфир.а, фильтруют .
Получают 250 мл (43%) соединения,
Rf 0,90 (А), Rf 0,80 (Б) 6 90 (N-нитрозогруппа, 397 нм).
5
Q
0
5
0
5
0
55
Вычислено, %: С1 6,15; N 17,02.
. (576,10).
Найдено,%: С1 6,00; N 16,61,
б) Альфа-)l-HHTpo3o-N-(бэта-хлор- этил )-карбамошГ -лизил-пролил-валин- амидгидрохлорид (II).
200 мг альфа- N -нитрозо-Ы-(бэта- -хлорэтил)-карбамоил -эпсилон-трет. бутоксикарбонил-лизил-пролил-валин- амида смешивают с 6 мл 0,17 н. раствора муравьиной кислоты в соляной кислоте и раствору дают постоять в течение 5 мин с последующим его выпариванием. Остаток растворяют в воде и экстрагируют этилацетатом. Водный слой отделяют и лиофилизируют. Получают 140 мг соединения, Rf 0,10 (Б).
Вычислено,%: С 44,49; Н 6,83; С 13,04; N 19,13.
, (512,44).
Найдено,%: С 44,51; Н 6,80; С1 13,72; N 18,81.
П р и м е р 3. Альфа-эпсилон-бис- - К-нитрозо-М-(бэта-хлорэтил)карба- мoилJ-лизил-пpoлил-вaлинaмид (III).
460 мг (1 ммоль) лизил-пролил- -валинамиддиацетата растворяют в 6 мл,диметилформамида, затем в полученный раствор -добавляют 0,28 мл (2 ммоль) триэтиламина и 548 мл (2 ммоль)N-нитpoзo-бeтa-xлopэтшlкap- бамоил-п-нитрофенола, а затем 270 мг (2 ммоль) 1-оксибензотриазола, через 2 ч диметилформамид отгоняют в вакууме, остаток растворяют в 30 мл этил- ацетата, промывают водой. Этилацетат- ный слой отделяют, сушат, упаривают. Остаток обрабатывают безводным диэтиловым эфиром, эфир о тделяют декантацией, масло сушат. Полученную твердую пену чистят в колонке, заполненной силикагелем. Фракции собирают, растворитель отгоняют в вакууме. Остаток растирают в порошок в присутствии безводного петролейного эфира, фильтруют. Получают 312 мг (51%) продукта, Rf 0,80 (S), Rf 0,88, 5 185 (N-нитрозогруппа, 397 нм).
Вычислено,%: С 43,17; Н 6,05; С1 11,59; N 20,61.
C iHjyNjOyCU (611,51).
Найдено,%: С 43,21; Н 6,02; С1 11,18; N 20,07
Пример 4. а) 0 - Н-Нитро- зо-Н-/УЗ хлорэтил)-карбамоил- -тре- тичный бутилоксикарбонил-триптофил- -глицил-лизил-пролилвалинамид.
51586520
К смешанному раствору из 1,2 гпаривают в вакууме. Остаток растворя(1,74 ммоль) третичн.бутилоксикарбо- « этилацетате н промывают водой.
Гнил-триптофенилглицил-лизил-пролил-Этилацетатный слой отделяют, сушат
валинамида и 10 мл безводного диметил- с« выпаривают. Получают 1,29 г (80%)
,./,J П QO . n Р z Q 1
фррмамида добавляют при 0,24 мл
(1,74 моль) триэтиламина и 400 мг (1,6 ммоль) Ы-нитрозо-М-(А-хлорэтш1)- -карбамоил-Н-оксисукцинимида. Через 0,5 ч реакционную сме сь упаривают, ос- 10 таток растворяют в этилацетате, раствор промывают водой. Органический слой отделяют, сушат над безводным сульфатом натрия, упаривают. Выход 1,2 г (85%), Rf (А).
Вычислено,%: N 17,09; С1 4,33.
продукта. Rf 0,82 vB), 6 91 (N-нитрозная группа, 397 нм).
Вычислено,%: С1 5,60; N 17,69.
С,Н 5КзОдС1
Найдено,%: С1 5,48; N 17,31.
б) Альфа-К-нитрозо-Ы-(бэта-хлор- зтил)-карбамоил-глицил-лизил-пролил- -валинамидгидрохлорид (V).
633 мг (1 ммоль) альфа-Н-нитроэс- 5 -Н-(бзта-хлорэтил)-карбамоил-эпсилон- -трет.бутилоксикарбонил-ГЛИЦШ1-ЛИСз7«5 ю 1 (819,37).
Найдено,%: N 16,72; С1 4,01.
.6) .Hитpoзo T-(ft -хлорэтил)- -карбамоилД-триптофил-глицил-лизил- -Пролил-валил-амидоацетат (IV).
819 мг (1 ммоль) об-LN-нитрозо- -N-(й-хлорэтил)-карбамокл-третичн. бутилоксикарбонил-триптсфил-глицил- -лизил-пролил-валинамида растворяют в 10 мл 0,12 н. муравьиной кислоты с хлористоводородной кислотой, отстаивают раствор 5 мин. затем раствор упаривают в вакууме. Остаток растворяют в воде, экстрагируют этиЛ- ацетатом. Водную фазу отделяют, лио- филизуют, лиофилизат чиctят хроматографией на колонке, заполненной си- ликагелем, элюируют смё сь этил ацетата, пиридина, ледяной уксусной кислоты и воды 60:20:6:11. Элюат упаривают. Остаток растворяют в воде, лио- филизируют, получают 480 мг (63%) соли заглавия. Rf 0,25 (I); 90 (50 нм).
Вычислено,%: С 52,40; Н 6,59; О 18,47; N 17,97, С1 4,55.
C,4Hj,NioO,Cl (779,27).
Найдено,%: С 52,00; Н 6,29; О 18,01; N 17,00; С1 .
11рймер5. а) Альфа-К-нитро- зо-М-(бета-хлорэтил)-карбамоил-эпси- лон-трет.бутилоксикарбонил глицил- --лизил-пролил-валинамид.
1,30 г (2J6 ммоль) эпсилрн-трет., бутилоксикарбонил-глицил-лизил-про- лил-валинамида растворяют в 13 мл абсолютированного диметилформамида,
зил-пролил-валинамида выдерживают в спокойном состоянии в течение 5 мин в 10 мл 0,14 н. раствора муравьиной 20 кислоты в соляной кислоте, а затем раствор выпаривают.
Остаток растворяют в воде и экстрагируют этилацетатом. Водную фазу лиофилизуют. Получают 540 мг (95%) . 25 продукта, Rf 0,65 (D), g 84 (N-нитрозная группа, 396 нм).
Вычислено,%: С 42,24.; Н 6,67; С1 12,45; N 19,67.
C -l iaNgOgClg (569,59). 30 Найдено,%: С 42,22; Н 6,69; С1 12,01; N 18,76.
П р и м е р 6. Ы-Нитрозо-М-(бэта--хлорэтил)-карбамоил-триптофил-нор- лейцил-альфа-аспарагил-фенилапанил- амид.
В условиях, описанных в примере 3 проводят взаимодействие 921 мг (1,5 ммоль) триптофил-норлейцил- -альфа-аспарагил-фе нилаланиламид- Q гидрохлорида, 0,21 мл О,5 ммоль) триэтиламина, 420 мг (1,5 ммоль) N-нитрозо-бета-Гхлорзтилкарбамоил - -п-нитрофенола и 200 мг (1,5 ммоль) 1-оксибензтриазола. Получают 800 мг дс соединения, выход 75%, Rf 0,8 (Б),
Вычислено,%: С 55,52; Н 5,75; С1 4,97; N 15,71; N0 4,20. .OgCl (713,20). Найдено,%: С 55,50; Н 5,77; 50 С1 4,58; N 15,32; N0 3,92.
Испытание А. Фармакологическая активность предлагаемых соединений демонстрируется результатами испыта-
35
НИИ с использованием в качестве исГв раствор добавляют 650 мг (2,6ммо ль) 55 пытуемых соединений о6,/}-бис-Гк-нитМ -нитрозо-бэта-хлорэтилкарбамоил-Н- розо-N-(|5-хлорэтил)-карбамоил -лизил-оксисукцинимида и 0,36 мл (2,6 ммоль) -пролил-валинамида (на который далее
триэтиламина. После перемешивания в в тексте ссьшаются, как на соединетечение 15 мин реакционную смесь вы- .ние II) и о(- } -нитрозо-1Ь(/3-хлор« выпаривают. Получают 1,29 г (80%
П QO . n Р z Q 1
продукта. Rf 0,82 vB), 6 91 (N-нитрозная группа, 397 нм).
Вычислено,%: С1 5,60; N 17,69.
С,Н 5КзОдС1
Найдено,%: С1 5,48; N 17,31.
б) Альфа-К-нитрозо-Ы-(бэта-хлор- зтил)-карбамоил-глицил-лизил-пролил- -валинамидгидрохлорид (V).
633 мг (1 ммоль) альфа-Н-нитроэс- -Н-(бзта-хлорэтил)-карбамоил-эпсилон- -трет.бутилоксикарбонил-ГЛИЦШ1-ЛИзил-пролил-валинамида выдерживают в спокойном состоянии в течение 5 мин в 10 мл 0,14 н. раствора муравьиной кислоты в соляной кислоте, а затем раствор выпаривают.
Остаток растворяют в воде и экстрагируют этилацетатом. Водную фазу лиофилизуют. Получают 540 мг (95%) . продукта, Rf 0,65 (D), g 84 (N-нитрозная группа, 396 нм).
Вычислено,%: С 42,24.; Н 6,67; С1 12,45; N 19,67.
C -l iaNgOgClg (569,59). Найдено,%: С 42,22; Н 6,69; С1 12,01; N 18,76.
П р и м е р 6. Ы-Нитрозо-М-(бэта-хлорэтил)-карбамоил-триптофил-нор- лейцил-альфа-аспарагил-фенилапанил- амид.
В условиях, описанных в примере 3 проводят взаимодействие 921 мг (1,5 ммоль) триптофил-норлейцил- -альфа-аспарагил-фе нилаланиламид- гидрохлорида, 0,21 мл О,5 ммоль) триэтиламина, 420 мг (1,5 ммоль) N-нитрозо-бета-Гхлорзтилкарбамоил - -п-нитрофенола и 200 мг (1,5 ммоль) 1-оксибензтриазола. Получают 800 мг соединения, выход 75%, Rf 0,8 (Б),
Вычислено,%: С 55,52; Н 5,75; С1 4,97; N 15,71; N0 4,20. .OgCl (713,20). Найдено,%: С 55,50; Н 5,77; С1 4,58; N 15,32; N0 3,92.
Испытание А. Фармакологическая активность предлагаемых соединений демонстрируется результатами испыта-
этил)-карбамоил -лизил-пролил-валин- амнд гидрохлорида (на которое далее в тексте ссылаются, как на соединение III).
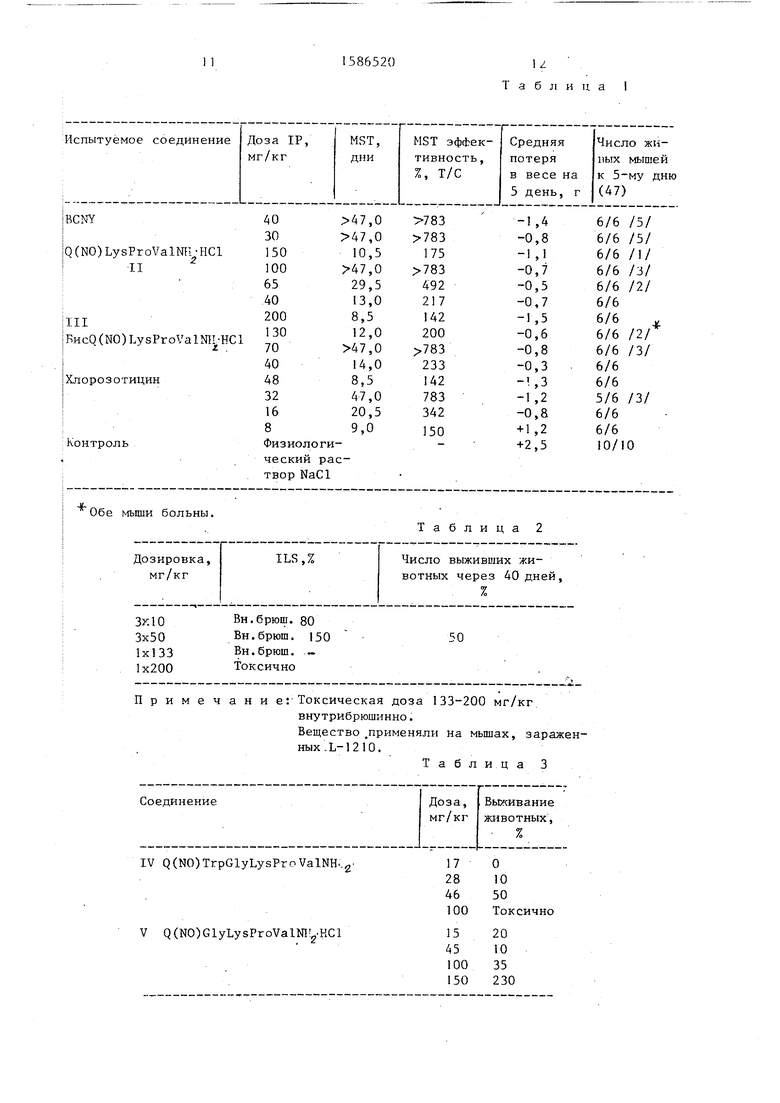
В табл.1 показано влияние соединений II и III на внутрибрюшинно имплантированную мышам лейкемию. Соединения II и III оказались более г1{кти1зными, чем 1 , 3-бис-(/3-хлорэтил)- 4 1--нитрозомочевина (далее обозначен- 1|ую как BCNY) и (2-хЛорэтил)-3- 1 итроуреидо |-2-деокси-0-глюкопирано- ;ia (далее обозначенную как хлорозоти- Иин), используемые в качестве сравни- Цельных веществ (см.табл.1), i Опухолевый прививочный материал - |0 асцитных клеток, внутрибрюшинно; |сивотное-хозяин - самки мышей вида (fDF; лечение - 3 дня; токсичность - пятый день 4/6 остаются в живых; (|)Пенка - MST-средний период-въгашвания;
,, „/„ MST обработанной группы
Эффект - Т/С --:
MST контрольной группы
X 100; критерий - , значительная противоопухлевая активность. ; В табл.2 приведены фармакологичес- ие свойства соединения III. I Фармакологические свойства соеди- )|1ения III i Токсическая доза 133- too мг/кг внутрибрюшинно. Вещество применяли на мьтах, зараженных L-1210 (см.табл.2-7).
В табл.3 приведены результаты исследования связи между эффектом и :|симической структурой соединений, Диалогичных соединению формулы (II), jfia мышах, зараженных лейкемией L-1210 I Результаты влияния соединений фор- улы (II) на выживание мышей, зара- сенных лейкемией L-1210 приведены в табл.4.
Б табл.5 приведены результаты влияния соединений формулы (II) в комби- Нации с фармацевтическими агентами на мышей, зараженных лейкомией L-1 2 IО
Приведенные вьш1е данные показывают, что новые пептидные производные формул II и III значительно более активны и менее токсичны, чем BCNY.H хлорозотицин, используемые в качест- ве контрольных агентов.
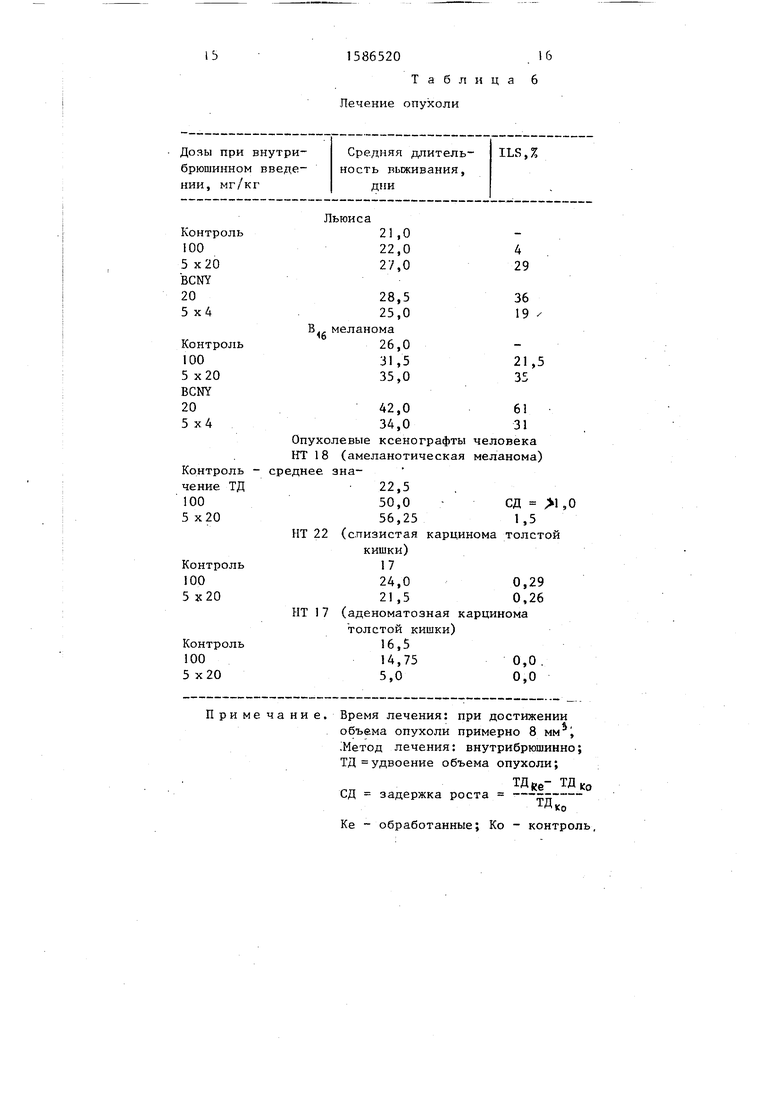
Испытание В. Исследовано влияние соединения II на твердую опухоль.
А) S180 опухоль (подкожно).
Дозировка 100 или 5x20 мг/ кг внутрибрюшинно (лечение начиналось на следующий день после трансплантации) оказалось неэффективной.
В) Легочная меланома Льюиса и ме- ланома В , - опухоли, дающие на мышах метастазы (i.m.)
Результаты приведены в табл.6.
Испытание С.
Фармакология.
Влияние пептидо-нитрозомочевиновы производных на рост экспериментальных твердых опухолей и опухолевых ксенографтов человека.
Объекты:
а) экспериментальная твердая опухоль мыши S 180, легочная опухоль Льюиса и меланома мыши В, ;
в) человеческие опухолевые ксе- нотрансплантаты; беспигментная ме- ланола НТ 18, колоректальная адено- карцинома НТ 17, колоректальна аде- нокарцинома НТ 22 (муциноз кожи), карцинома поджелудочной железы НТ 27.
Опыты на экспериментальных твердых опухолях: S ал .
Опухоль разрезают на маленькие кусочки и кусочки пересаживают под- кржно под кожу спины мышей CLFP. Лечение начинают на следующий день после трансплантации. Размер опухолей измеряют при помощи кронциркуля и их объем рассчитывают от их диаметра.
Соединение SV370 вводят внутрибрюшинно при дозах 100 или 5x20 мг/к (одна доза на пять дней). Влияния на рост опухоли не обнаружено.
Опухоль легкого по Льюису (ОЛЛ).
Эту опухоль пересаживают в виде суспензии клеток в бедренную мьшщу ин бредной мыши С57В1 с количеством клеток 10 . Растуща.я опухоль дает регулярные метастазы легкого. Лечение ; начинают на следующий день после трансплантации. Наблюдают выживаемость обработанных и контрольных животных.
Меланома .
Опухоль пересаживают под кожу спины инбредной мьш1и С57В1 в виде кусочков опухоли. Растущая опухоль дает метастазы в легком. Лечение и оценку проводят аналогичным способом, как описано в слз чае опухоли легкого по Льюису.
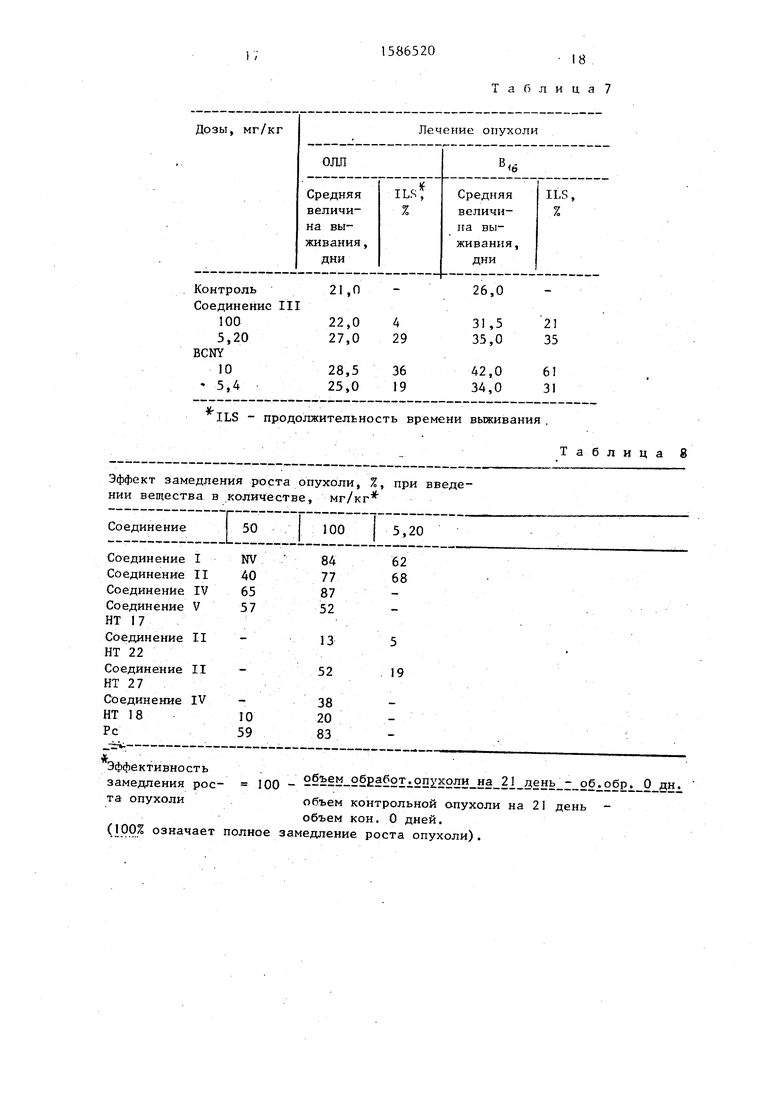
В соответствии с результатами сое- .динение SV370 умеренно удлиняет продолжительность жизни животных включая случаи ОЛЛ и В меланомы. Порядок эффективности замедления роста опухоли в основном равен эффекту BCNY,применяемому как контрольное соединение (см.табл.7).
Проверка ксенотрансплантатов человеческих опухолей.
Кусочки серийно пересаживаемых опухолей (зрелая тимeктo a я, полная облучение 8,5 GY, восстановление косного мозга за счет 5«10 синтетических клеток костного мозга) пересаживают под кожу спины мьши СВА/СА с искусственно подавленным иммунитетом. Лечение внутрибрюшинным методом начинают, когда опухоли достигают диаметра 0,7-1,0 см и можно наблюдать их рост в прогрессии. Диаметры- опухолей измеряют при помощи кронциркуля, и и объем рассчитывают на основании их диаметра (V ц /б xD , D-средний диа
метр). Порядок эффективности замедления роста опухоли определяют на ос- н овании разницы объемов опухолей, измеренных на 21 день после пересадки в контрольной и обработанной группах (каждая экспериментальная группа включала по крайней мере 6 опухолей), см,табл. 8.
1 время выживания мыше
ILS % - --5 ВЬН; 5Э5™2 15251
- время вьокивания конт
Критерий: ILS.% - значительный эффек замедления роста опухоли.
Полученные результаты -показывают, что соединения, полученные в условия предлагаемого способа, являются эффективными цитостатическими агентами причем более активными, чем хлорозо- тицин и BCNY.
Формула изобретения
Способ получения М-нитрозо-М-(бэ- та-хлорэтил)карбамоилпептидов или их солей формулы I
N0
clCll..Nн,iU
U2;, .
-Pro-ValMl, -Lys-Pro-Vaim:
Gly-Lys-Pro-ValNH, -Trp-Gly-Lys-Pro-ValllH, -Trp-Leu-Asp-PheNK,
где
1 R
Результаты экспериментов показывают, что производные пептиднитрозо - мочевины значительно замедляют рост беспигментной мелаьомы в то время, как их эффект на рост опухолей толстой кишки и поджелудочной железы ниже.
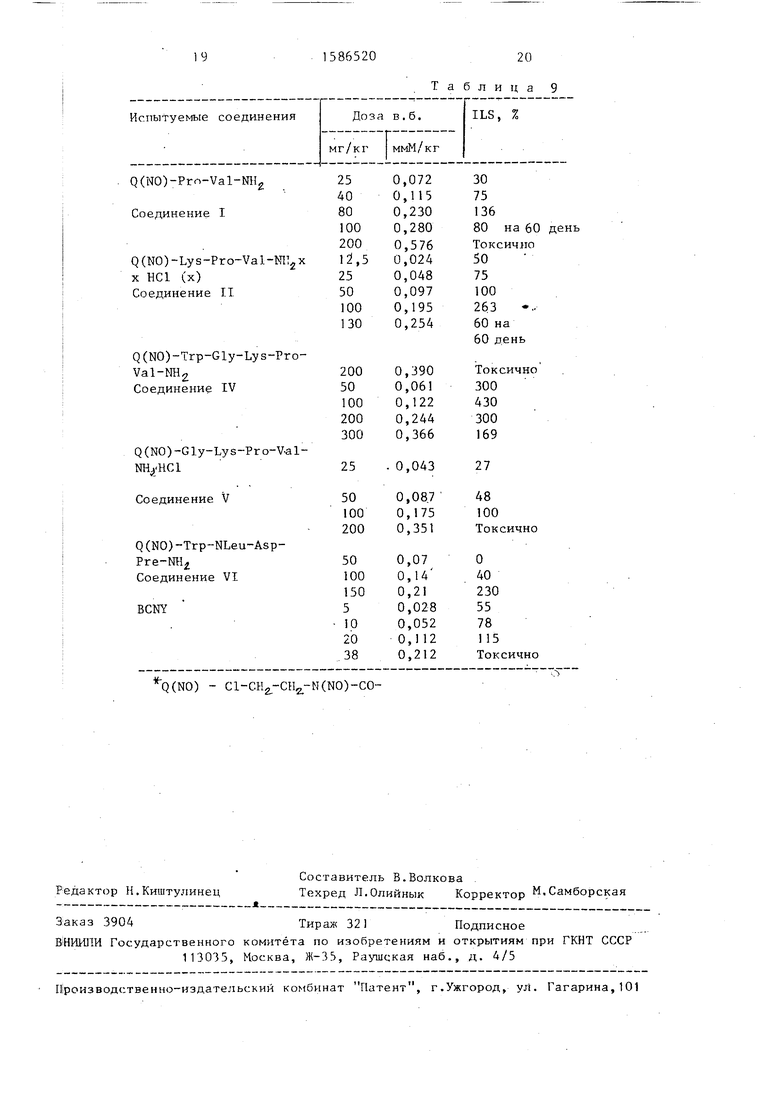
Эффект, оказываемый N-нитрозо- -(А-хлорэтил)-карбамоилпептидными фрагментами, на замедление роста опухоли мышей L-1210, имеющих лейкемию, приведены в табл.9.
15
Измерение эффекта замедления роста опухоли у L-1210 осуществляют следующим образом, 10 L-1210 лейке- миновых клеток пересаживают внутри- брюшинно DBA мьшгам. Лечение начинают на следующий день после пересадки; Испытуемые соединения добавляют в физиологический раствор хлорида натрия или в 2%ный буффер Твина 80, содержащий. 5% диметилсульфоксида.
, прошедших лечение
или их солей, о тличающий - с я тем, что вводят во взаимодействие пептиды общей формулы II
XHN-R, где X - водород или Вое;
R - имеет указанные значения, с активированным сложным эфиром общей формулы III
NU
Cl-CH -ClI-N-CO-Q ,
Д Q -п - нитрофенил, галогенфенил, оксисукцинимид или фталимидо- оксигруппа.
причем при получении соединений 1-- где п , используют эквимолярные
количества исходных, если п 2, исодные II и III берут в молярном соот- нош-гнии 1:2, с последующим снятием в случае необходимости защитных групп и выделением целевого продукта
в свободном виде или в виде соли.
Таблица 1





| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения N-нитрозо-N-(бэта-хлорэтил)-карбамоилпептидов или их кислотно-аддитивных солей | 1982 |
|
SU1424739A3 |
| N-Нитрозо-N-(бэта-хлорэтил)-карбамоилпептиды или их хлоргидраты,обладающие противоопухолевой активностью | 1983 |
|
SU1414851A1 |
| Способ получения производных дульцита или их солей | 1981 |
|
SU1205769A3 |
| Способ получения производных пиридо/1,2-а/пиримидина или их солей с щелочными металлами или их оптических изомеров | 1980 |
|
SU1024007A3 |
| Способ получения 1,2-5,6-диангидро-3,4-бис-( @ -карбоксипропионил)-дульцита или его динатриевой соли | 1982 |
|
SU1225487A3 |
| Способ получения конденсированных производных пиримидина или их солей | 1980 |
|
SU1082324A3 |
| Производные дульцита,обладающие ингибирующим действием в отношении злокачественных опухолей | 1982 |
|
SU1194863A1 |
| Способ получения 3-замещенных тетрагидропирроло/1,2- @ / пиримидинов,их кислотно-аддитивных или четвертичных солей | 1980 |
|
SU1048986A3 |
| Способ получения полиоксиметиленовой композиции | 1978 |
|
SU784758A3 |
| Способ получения геминальных дигалоидных производных конденсированных пиримидин-4-онов,рацематов или оптически активных антиподов | 1980 |
|
SU1151210A3 |
Изобретение относится к дипептидам, в частности к получению производных N-нитрозо-N-(бэта-хлорэтил) карбамоилпептидов ф-лы I [CLCH 2CH 2N (NO)CONH] NR где N = 1,2, R--PRO-VALNH 2, -LYS-PRO-VALNH 2, -GLY-LYS-PRO-VALNH 2, - TRP - GLY - LYS - PRO- VALNH 2, - TRP-LEU-ASP - PHENH 2, или их солей, которые обладают противоопухолевой активностью. Цель - разработка способа получения более активных соединений указанного класса. Получение ведут реакцией пептидов ф-лы XHN - R, где X - H или BOC
R имеет указанные значения с активированным сложным эфиром ф-лы CL - CH 2 - CH 2 - N(NO)-CO - Q, где Q - N-нитрофенил, галогенфенил, оксисукцинимид или фталимидооксигруппа, причем при получении соединений ф-лы I, где N = 1, используют эквимолярные количества исходных, если N = 2, исходные продукты берут в молярном соотношении 1:2 с последующим снятием в случае необходимости защитных групп и выделением целевого продукта в свободном виде или в виде соли. 8 табл.
sProValNfl -HCl -II
)LysProValNlI-HCl
ТИЦИН
ь
40 30 150 100 65 40 200 130 70 40 48 32 16 8
Физиологический раствор NaCl
47,0
47,0 10,5
47,0 29,5 13,0 8,5 12,0
47,0 14,0 8,5 47,0 20,5 9,0
J/
Обе ыши больны.
римечани е: Токсическая доза 133-200 мг/кг
внутрибрюшинно.
Вещество .применяли на мьшах, зараженных.L-12 10.
Т а б л и.ц а 3
IV Q(NO)TrpGlyLysProValNH..2
V Q(NO)GlyLysProValNll,,.KCl
783
783 175
783 492 217 142 200
783 233 142 783 342
150
-1,4 -0,8
-1,1 -0,7 -0,5 -0,7 -1,5 -0,6 -0,8 -0,3 - . ,3 -1,2 -0,8 + 1,2 +2,5
6/6 /5/
6/6 /5/
6/6 /1/
6/6 /3/
6/6 /2/
6/6
6/6 ,
6/6 /2/
6/6 /3/
6/6
6/6
5/6 /3/
6/6
6/6
10/10
Таблица 2
О
10
50
Токсично
20 10 35 230
13
Внутрибрюшинно,применение в отмеченный день после трансплантации;
выживание обработанных мышей 7 -2---9-25-21-йУ- 2тСтНг
время выживания необработанных
X 100;
Выживание на 60-й день после трансплантации.
325
325
1x25 2 44
1x50 2 44
1x50 2 О
1x10 2 77
77
44
1x50 3 33
1x20 3 70
1586520
Таблица 4
14
Таблица 5
70
60
15
Таблица 6 Лечение опухоли
Средняя длительность выживания,
дни
ль
ль
ль ТД
ль
ль
Льюиса
21,0 22,0 27,0
28,5 25,0 В меланома
26,0 31,5 35,0
4 29
36
19
21,5 35
42,061
34,031
Опухолевые ксенографты человека НТ 18 (амеланотическая меланома) среднее зна22,5
50,0 - СД 1,0
56,251,5
НТ 22 (слизистая карцинома толстой кишки)
17
24,00,29
21,50,26
НТ 1 7 (аденоматозная карцинома толстой кишки)
16,5
14,750,0.
5,00,0
мечание,
Время лечения: при достижении объема опухоли примерно 8 Метод лечения: внутрибрюшинно; ТД удвоение объема опухоли;
ТДке: ТДко
тд;г
Ке - обработанные; Ко - контроль,
СД задержка роста
ILS,%
4 29
36
19
21,5 35
Контроль 21 ,П Соединение III
ILS - продолжительность времени выживания ,
Эффект замедления роста опухоли, %, при введении вещества в .количестве, мг/кг
Эффективность
замедления рос- 100 - --S5 - бEaP т oпxxoли нa 2J дeнь ; oб o6p 0 дн.
та опухолиобъем контрольной опухоли на 21 день объем кон. О дней. (IP.Q. означает полное замедление роста опухоли) .
Таблица
26,0
Таблица 8
Q(NO) - C1-C..-N(NO)-COТаблиц
| Шредер Э., Любке К | |||
| Пептиды | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
