Настоящее изобретение относится к органической химии и медицине, в частности к дигидроксидиаминосоединениям, а именно к способу получения этамбутола (ЭБ) формулы
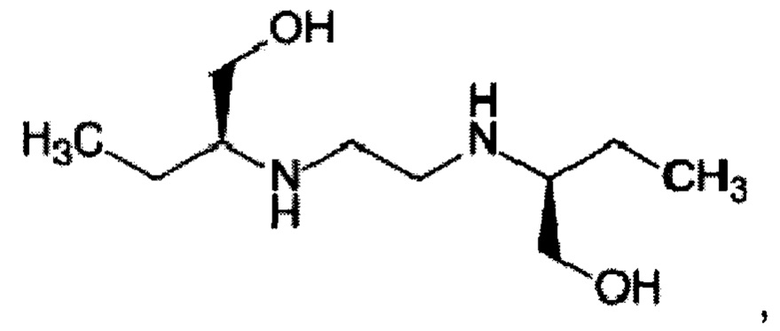
обладающего противотуберкулезной активностью. Изобретение наиболее успешно может быть использовано в фармацевтической промышленности для получения известного препарата, применяемого для лечения туберкулеза.
В последние годы во всем мире наблюдается постоянный рост заболеваемости туберкулезом, ежегодно от этой болезни умирает около 2 млн человек [О. Danilchanka, М. Pavlenok, and М. Niederweis, Antimicrob. Agents Chemother., 2018, 52(9), 3127-3134]. Успешное лечение туберкулеза требует постоянного применения сильных лекарственных препаратов, одним из таких препаратов нового поколения является этамбутол (Ethambutol, (S,S)-2,2'-(1,2-этандиилдиимино)бис(1-бутанол)), который применяют в лечении впервые выявленного туберкулеза легких и его рецидивов в комбинации с другими препаратами. Несмотря на получение многочисленных аналогов, ЭБ остается одним из наиболее эффективных противотуберкулезных средств [R. Yendapally, R.E. Lee., Bioorg. & Med. Chem. Lett., 2008, 18, No. 5, 1607; М.Д. Машковский, «Лекарственные средства», РИА, Новая волна, Москва, 2008, С 861-862; М.В. Мавров, Н.И. Симирская, Хим.-фарм. Ж., 2012, 46, №12, 34; D. Lednicer, L.A. Mitscher, "Organic Chemistry of Drug Synthesis", J. Wiley & Sons, 1977, v. 1, p. 222.].
Этамбутол был впервые получен Томасом и Вилкинсоном [R.G. Wilkinson, R.G. Shepherd, J.P. Thomas, С. Baughn, J. Am. Chem. Soc., 1961, 83, 2212]. ЭБ содержит оба хиральных центра в (+)-форме, т.е. только (S,S)-диастереомер обладает сильной противотуберкулезной активностью [D. Lednicer, L.A. Mitscher, "Organic Chemistry of Drug Synthesis", J. Wiley & Sons, 1977, v. 1, p. 222; D. Kulig, Z. Bugielska, K. Rokicka, Diss. Pharm. Pharmacol, 1971, 22, 46]. Мезо-(S,R)-диастереомер в 16 раз менее активен, в то время как (R,R)-диастереомер практически неактивен и, даже опасен для теплокровных, т.к. вызывает слепоту [R.G. Wilkinson, М.В. Cantrall, R.G. Shepherd, J. Med. Pharm. Chem., 1962, 5, 835]. Таким образом, целевым является (S,S)-диастереомер (CAS Registry Number 74-55-5).
Известны способы получения ЭБ, в которых сначала осуществляют многостадийный синтез важного интермедиата, например синтез рацемического 2-амино-3-бутан-1-ола, затем этот интермедиат разделяют на стереомеры через образование диастереомеров с оптически активным разделяющим реагентом, выделяют требуемый (S)-стереомер, который далее переводят в целевой (S,S)-диастереомер. [R.G. Wilkinson, R.G. Shepherd, J.P. Thomas, C. Baughn, J. Am. Chem. Soc., 1961, 83, 2212; B. Singh, Патент США US, 1976; Б. Луиджи, Т. Алдемио, Ф. Маурицио, Патент СССР №498902, опубл. 05.01.1976, Бюл. №1].
Существуют способы получения ЭБ, в которых используют оптически чистые исходные соединения. Известен способ, в котором в качестве исходного соединения используют неприродный (S)-норвалин, полученный сложными специальными методами (например, через хиральные комплексы никеля(II) [Y.N. Belokon, K.A. Kochetkov, Т.D. Churkina, N.S. Ikonnikov, S. Viscochil, H.В. Kagan, Tetrahedron: Asymm., 1999, 10, 1723]. Восстановлением (S)-норвалина действием LiAlH4 получают ключевое соединение (S)-2-амино-3-бутан-1-ол и по стандартной схеме переводят его в этамбутол [R.G. Wilkinson, R.G. Shepherd, J.P. Thomas, С. Baughn, J. Am. Chem. Soc., 1961, 83, 2212].
Известен способ получения ЭБ из оптически активного (L)-метионина, осуществляемый без выделения промежуточного (S)-стереомера аминобутанола, но с двукратной стадией восстановления и использованием предварительной защиты сложноэфирной группы обработкой оксалилхлоридом [С.S. Stauffer, A. Datta, Tetrahedron, 2002, 58, No. 49, 9765-9767; R.S.В. Goncalves, E.T. da Silva, M.V.N. de Souza, Lett. Org. Chem., 2015, 12, No. 7, 478].
Известен способ получения ЭБ из малодоступного оптически чистого соединения - (R)-бутан-1,2-диола стереонаправленным синтезом в 6 стадий [R.S.В. Goncalves,; Е.Т. da Silva, М.V.N. de Souza, Lett. Org. Chem., 2015, 12(7), 478].
Известны каталитические способы получения этамбутола, в которых используют каталитическое асимметрическое аллилирование на дорогих хиральных палладиевых катализаторах или применяют дефицитные оптически активные вещества и азосоединения [В.М. Trost, R.C. Bunt, R.С. Lemoine, T.L. Calkins, J. Am. Chem. Soc., 2000, 122, No. 25, 5968; S.P. Kotkar, A. Sudalai, Tetrahedron: Asymm., 2006, 17, 1738].
Известен близкий к заявляемому способ, выбранный за прототип, согласно которому на ключевой стадии расщепляют рацемическое производное 2-амино-3-бутен-1-ола, с помощью оптически активных веществ, образующих смесь разделяемых в дальнейшем диастереомеров, из одного из которых выделяют (S)-2-амино-3-бутен-1-ол [Б. Луиджи, Т. Алдемио, Ф. Маурицио, Патент СССР №498902, опубл. 05.01.1976, Бюл. №1]. Для получения промежуточного рацемического производного - 2-бензиламино-3-бутен-1-ола, исходный 1,2-эпокси-3-бутен подвергают взаимодействию с 1-2 эквивалентами фосгена в присутствии пиридина с выделением хлорформиата 2-хлор-3-бутен-1-ола. Полученный хлорформиат взаимодействует с бензиламином при пониженной температуре с образованием 2-хлор-3-бутен-1-ол-N-бензилуретана, его обрабатывают щелочью в водно-спиртовом растворе и выделяют 4-винил-3-бензилоксазолидин, который в присутствии избытка щелочи дает (±)-2-бензиламино-3-бутен-1-ол. Рацемический (±)-2-бензиламино-3-бутен-1-ол разделяют на его оптические антиподы с помощью оптически активных кислот (миндальной кислоты или производных дибензоилвинной кислоты) с образованием солей. При этом выпадает в осадок соль - (S)-(+)-2-бензиламино-3-бутен-1-ола, а в растворе остается его стереоизомер. При обработке осадка водным раствором основания, освобождается (S)-(+)-2-бензиламино-3-бутен-1-ол и переходит в органический слой, из которого его выделяют известными приемами и по известному способу переводят в целевой продукт.
Недостатками способа-прототипа являются: 1) использование токсичных фосгена и пиридина в качестве реагентов и малодоступного исходного соединения 1,2-эпокси-3-бутена на первой стадии, 2) трудоемкость стадий получения рацемического производного и его расщепления на оптические антиподы с использованием дорогих оптически активных кислот с применением большого количества воды и основания; 3) большое число стадий, необходимых для получения целевого этамбутола.
Задачей настоящего изобретения является разработка нового способа получения этамбутола, который позволил бы уменьшить трудоемкость стадий расщепления рацемического N-производного и выделения S-энантиомера и исключить применение токсичных реагентов.
Технический результат - новый технологичный способ получения этамбутола, позволяющий упростить сложное и трудоемкое выделение нужного (S)-энантиомера из рацемата и исключить использование токсичных веществ.
Поставленная задача решается заявляемым способом получения этамбутола из рацемического 2-аминобутан-1-ола, включающим защиту его аминогруппы под действием карбобензоксихлорида (КБЗ-Cl) в присутствии NaOH в с образованием рацемического N-карбобензокси-2-аминобутан-1-ола; стереоселективное ацилирование полученного рацемата этилацетатом, катализируемое липазой PPL, выделение (S)-N-карбобензокси-2-аминобутан-1-ола хроматографией на силикагеле; последующее снятие защитной группы с образованием (S)-2-аминобутан-1-ола и алкилирование последнего 1,2-дихлорэтаном с образованием целевого этамбутола, причем рацемический 2-аминобутан-1-ол получают конденсацией параформа с 1-нитропропаном с образованием 2-нитробутан-1-ола и последующим его восстановлением водородом над Pd/C в метаноле (схема 1).
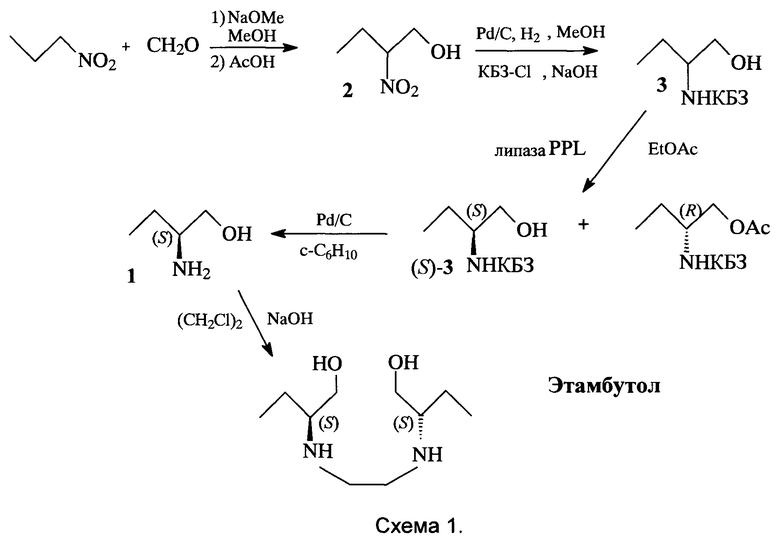
Ключевое промежуточное соединение - стереоизомер ((S)-(+)-2-амино-3-бутан-1-ол) 1 получают следующим образом. Сначала получают нитроспирт 2 конденсацией параформа с нитропропаном с высоким выходом. Нитроспирт 2 восстанавливают до амина и переводят его в рацемическое карбобензоксипроизводное 3 реакцией с карбобензоксихлоридом (КБЗ-Cl). На ключевой стадии расщепления рацемического N-карбобензокси-2-аминобутан-1-ола 3 проводят его стереоселективное ацилирование этилацетатом, катализируемое липазой PPL. Для расщепления производного 3 используется доступная липаза свиньи (PPL - липаза), полученную после ацилирования реакционную смесь без очистки хроматографируют на силигагеле, выделяя требуемый непрореагировавший энантиомер (S)-3 с высоким выходом. После снятия защиты полученный (S)-аминоспирт 1 алкилируют 1,2-дихлорэтаном в присутствии NaOH по известному способу в стандартных условиях Шоттен-Баумана [R.G. Wilkinson, R.G. Shepherd, J.P. Thomas, С. Baughn, J. Am. Chem. Soc., 1961, 83, 2212; B. Singh, Патент США №3944618, 1976].
В заявляемом способе получения ЭБ не применяют дорогих оптически активных кислот. На стадии расщепления рацемического N-производного 2-аминобутанола 3 используется доступная липаза свиньи (PPL - липаза). Оказалось, что ацилирование производного 3 происходит стереоселективно, и хроматография реакционной смеси без предварительной очистки сразу позволяет выделить требуемый энантиомер (S)-3. Тогда как при осуществлении способа-прототипа выделение после расщепления рацемического производного приводит в итоге к смеси солей, из которой соответствующее производное требуемого стереомера (S)-(+)-2-амино-3-бутен-1-ола выделяется только после разделения образовавшихся солей и дополнительной обработки одной из них щелочью.
Заявляемый способ имеет ряд преимуществ перед прототипом: способ включает меньше стадий, для его осуществления используются доступные исходные соединения, исключается применение токсичного фосгена, уменьшается количество используемой воды, устраняются трудоемкие стадии выделения целевого S-энантиомера из рацемата. Способ может быть легко масштабирован для получения целевого продукта. В качестве рацемического производного 2-амино-3-бутен-1-ола могут быть использованы и другие производные, такие как (±)-N-трет.-бутилоксикарбонил-2-аминобутан-1-ол.
Изобретение иллюстрируется приведенными ниже примерами.
Пример 1. Получение 2-нитробутан-1-ола (2) конденсацией параформа с нитропропаном
К 3.12 г (0.136 г-атома) натрия медленно добавляют 30 мл МеОН при 5°С, затем еще 10 мл и осторожно нагревают до 40°С. Раствор охлаждают и медленно при перемешивании добавляют к смеси 3.60 г (0.12 моля) параформа и 10.68 г (0.12 моля) 1-нитропропана в 30 мл МеОН при 0°С в течение 2 часов. Реакционную смесь перемешивают при 0°С еще в течение 2 часов до выпадения Na-соли и оставляют в холодильнике на ночь. Смесь фильтруют, промывают МеОН и сушат при 20°С в вакууме в течение 2 часов. Получают белый кристаллический порошок Na-соли аци-формы 2-нитробутан-1-ола. Вес 13.3 г (79%). Найдено: С 34.19; 34.31, Н 5.61; 5.50, N 9.99; 9.93%. Вычислено для C4H8NO3Na: С 34.05, Н 5.71, N 9.93%. Спектр ПМР (CDCl3, δ, м.д.): 1.0 (т, 3Н, Me), 2.25 (м, 2Н, СН2), 4.25 (с, 2Н, СН2ОН), 4.6 (т, 1Н, ОН). К взвеси полученной соли в 80 мл эфира при интенсивном перемешивании при 20°С сразу добавляют раствор 7.8 мл АсОН в 40 мл эфира. Осадок соли исчезает и появляется мелкозернистый осадок ацетата натрия. После часа перемешивания его отфильтровывают, промывают эфиром, фильтрат упаривают и получают 7.76 г (68%) 2-нитробутан-1-ола 2, nD20=1.4380. Найдено: С 40.26; 40.21, Н 7.64; 7.65, N 12.03; 11.91%. Вычислено для C4H9NO3: С 40.33, H 7.62, N 11.76%. Спектр ПМР (CDCl3, δ, м.д.): 1.0 (т, 3Н, Me), 1.9-2.0 (м, 2Н, СН2), 3.25 (ш.с., 1Н, ОН), 3.9-4.1 (м, 3Н, СН, СН2ОН).
Пример 2. Восстановление 2-нитробутан-1-ола (2) и получение (±)-N-карбобензокси-2-амино-бутан-1-ола (3)
В автоклав из нержавеющей стали, снабженный магнитной мешалкой, загружают 0.51 г 5% Pd/C и 2.55 г 2-нитробутан-1-ола 2 в 30 мл МеОН. Начальное давление водорода - 50 атм. Перемешивание проводят в течение 20 часов. Конечное давление водорода 30 атм. Реакционную смесь фильтруют, промывают МеОН и упаривают. Получают 1.80 г (94%) 2-аминобутан-1-ола, т. кип. 84°С (3 мм); nD20=1,4225. К 1.8 г (20 ммол) полученного 2-аминобутан-1-ола при 0°С добавляют 25 мл 1N NaOH. К реакционной смеси медленно прибавляют 7.5 мл 50% раствора (3.75 г, 22 ммол) КБЗ-Cl в толуоле, рН-раствора поддерживают не менее 9, периодически добавляя 1N NaOH. Перемешивание продолжают 2 часа, реакционную смесь нейтрализуют и экстрагируют эфиром. После упаривания получают 4.25 г (95%) (±)-N-карбобензокси-2-аминобутан-1-ола (3), Rf=0.5 (эфир/петр. эфир = 3/1). Продукт 3 очищают на колонке с SiO2 (Rf=0.25, эфир/петр. эфир = 3/1), т.пл. 37-39°С, Спектр ПМР (CDCl3, δ, м.д.): 0.95 (т, 3Н, Me), 1.4-1.7 (м, 2Н, СН2), 2.25 (ш.с., 1Н, ОН), 3.5-3.8 (м, 3Н, СН, СН2ОН), 4.95 (ш.с., 1Н, NH), 5.10 (с, 2Н, PhCH2), 7.35 (м, 5Н, Ph).
Пример 3. Ацилирование (±)-N-карбобензокси-2-аминобутан-1-ола (3). Получение (S)-(+)-N-карбобензокси-2-аминобутан-1-ола ((S)-3)
К раствору 0.236 г (1 ммоль) (±)-N-карбобензокси-2-аминобутан-1-ола 3 в 20 мл EtOAc прибавляют 0.240 г липазы PPL (Fluka, N-62300). Суспензию перемешивают при 30°С до 50%-ной конверсии по исходному соединению (около 16 ч). После завершения реакции липазу отфильтровывают и промывают EtOAc и CH2Cl2. Объединенные органические фазы фильтруют и упаривают. Остаток хроматографируют на колонке (SiO2, эфир/петр. эфир = 3/1), разделяя (S)-(+)-аминоспирт и (R)-(-)-аминоацетат. Выход (S)-(+)-N-карбобензокси-2-аминобутан-1-ола, (S)-3: 0.108 г (92%), [α]D20 +23.4 (с 0.71, CHCl3), спектр ПМР идентичен предыдущему. Выход (R)-(-)-(N-карбобензокси-2-амино)бутилацетата, (R)-3: 0.128 г (96%), Rf=0.5, эфир/петр. эфир = 3/1, т.пл. 66-67°С, [α]D20 -25.2 (с 0.52, CHCl3), Спектр ПМР (CDCl3, δ, м.д.): 0.95 (т, 3Н, Me), 1.55 (м, 2Н, СН2), 2.0 (с., 3Н, Me), 3.8 (м, 3Н, СН), 4,10 (м, 2Н, СН2ОАс), 4.80 (ш.с., 1Н, NH), 5.10 (с, 2Н, PhCH2), 7.35 (м, 5Н, Ph).
Пример 4. Получение (S)-(+)-2-аминобутан-1-ола 1
Снятие КБЗ-защитной группы проводят, используя 5% Pd/C, который прибавляют к 1 ммоль (S)-(+)-N-карбобензокси-2-аминобутан-1-ола (1:1 по весу) в 4 мл абс. этанола и 2 мл циклогексена. Смесь кипятят 2 часа, отфильтровывают, растворитель упаривают и получают (S)-(+)-2-аминобутан-1-ол 1 с выходом 95%. [α]D20 +10.5 (с 2.05, EtOH), (лит. [В. Singh, Патент США US 3944618, 1976]: [α]D20 +10.0 (с 2.0, EtOH) 98% ее).
Пример 5. Получение этамбутола (ЭБ)
Получение этамбутола из (S)-(+)-2-аминобутан-1-ола проводят в стандартных условиях согласно [В. Singh, Патент США №3944618, 1976].
Смесь 44 ммоль (S)-(+)-2-аминобутан-1-ола 1 и 3.2 ммоль 1,2-дихлорэтана нагревают при перемешивании до 80°С, при этом температура экзотермически повышается до 120°С. Затем при 95°С медленно добавляют 6.4 ммоль NaOH, при этом температура опять повышается в течение часа до 110°С. После охлаждения смеси до 70°С избыток аминоспирта 1 (который можно использовать повторно в реакции) удаляют в вакууме выделением целевого соединения (ЭБ) с т пл. 88-89°С с выходом 96%, [α]D25 +13.5 (с 2.03, H2O), (лит. т.пл. 87.5-88.8°С, [α]D25 +13.7 (с 2.0, H2O) (The Merck - Encyclopedia Chemicals, Drugs, and Biologicals. 2006, р. 638]. Продукт стабилизировали переводом в дигидрохлорид с т пл. 199-201°С, [α]D20 +7.4 (с 2.0, H2O) (лит. т.пл. 198.5-200.3°С, [α]D20 +7.6 (с 2.0, H2O) [The Merck - Encyclopedia Chemicals, Drugs, and Biologicals. 2006, р. 638] известным путем с использованием избытка хлористого водорода [В. Singh, Патент США №3944618, 1976].
Изобретение относится к усовершенствованному способу получения этамбутола формулы
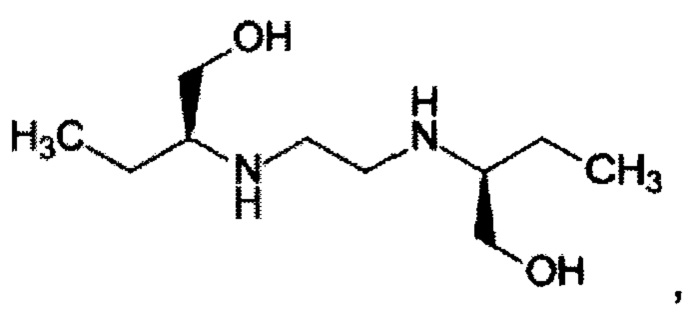
применяемого в медицине в качестве противотуберкулезного препарата. Согласно изобретению этамбутол получают из рацемического 2-аминобутан-1-ола, способ включает защиту его аминогруппы действием карбоксибензилхлорида в присутствии NaОН с получением N-карбобензоксипроизводного 2-аминобутан-1-ола, который стереоселективно ацилируют по гидроксигруппе этилацетатом, при катализе липазой PPL, хроматографией на силикагеле. Затем выделяют (S)-энантиомер его восстановлением с получением (S)-2-аминобутан-1-ола, который алкилируют 1,2-дихлорэтаном с образованием целевого продукта. Исходный 2-аминобутан-1-ол получают конденсацией параформа с 1-нитропропаном и последующим восстановлением образовавшегося 2-нитробутан-1-ола. 1 з.п. ф-лы, 5 пр.
1. Способ получения этамбутола из рацемического 2-аминобутан-1-ола, включающий защиту его аминогруппы действием карбобензоксихлорида в присутствии NaOH с образованием рацемического N-карбобензокси-2-аминобутан-1-ола; стереоселективное ацилирование последнего этилацетатом, катализируемое липазой PPL; выделение (S)-N-карбобензокси-2-аминобутан-1-ола хроматографией на силикагеле, последующее снятие защитной группы, приводящее к образованию (S)-2-аминобутан-1-ола, алкилирование которого 1,2-дихлорэтаном в присутствии NaOH дает целевой этамбутол.
2. Способ по п. 1, отличающийся тем, что рацемический 2-аминобутан-1-ол получают конденсацией параформа с 1-нитропропаном и последующим восстановлением образовавшегося 2-нитробутан-1-ола водородом над Pd/C в метаноле.
| US 3944618 A, 16.03.1976 | |||
| US 3847991 A, 11.11.1974 | |||
| Способ получения этамбутола | 1972 |
|
SU498902A3 |
| Способ получения -аминоспирта или его солей | 1974 |
|
SU556723A3 |
| Trost, Barry M.; et al., Dynamic kinetic asymmetric transformation of diene monoepoxides: a practical asymmetric synthesis of vinylglycinol, vigabatrin, and ethambutol, Journal of the American Chemical Society, 2000, 122(25), 5968-5976 | |||
| Wang, | |||

