Изобретение относится к области молекулярной биологии и диагностической медицины и может быть использовано для определения последовательностей экзонов генов BRCA1 и BRCA2.
Мутации генов BRCA1 и BRCA2 приводят к развитию синдрома наследуемого рака молочной железы и яичников (HBOCS, hereditary breast and ovarian cancer syndrome). Диагностика мутаций BRCA1 и BRCA2 имеет существенное значение для оценки индивидуального риска HBOCS и планирования профилактических мер, направленных на снижение риска развития рака. Кроме того, в декабре 2014 г. Компания AstraZeneca получила регистрацию на препарат-ингибитор PARP (olaparib), предназначенного для пациентов с раком молочной железы (РМЖ) и раком яичников (РЯ), несущих дефектный ген BRCA. В связи с этим, молекулярная диагностика BRCA1 и BRCA2 становится необходимым этапом при выборе пациентов для таргетной терапии РМЖ и РЯ.
Золотым стандартом тестирования BRCA1 и BRCA2 является секвенирование по Сэнгеру. Однако большой размер генов (5592 п. н., и 10257 п. н., соответственно) и отсутствие «hot spot» мутаций (см. базу данных Breast Cancer Information Core, http://www.research.nhgri.nih.gov/bic/) делает эту процедуру слишком дорогой. Кроме того, среди всех случаев HBOCS только 10-20% обусловлены мутациями BRCA1 и BRCA2, в остальных же случаях гены BRCA1 и BRCA2 не содержат каких-либо мутаций [1]. Это в купе с высокой стоимостью секвенирования генов BRCA объясняет, почему большинство лабораторий очень тщательно и осторожно подходят к назначению анализа генов BRCA пациентам с HBOCS. На сегодняшний день описаны несколько вариантов анализа мутаций BRCA с использованием NGS-платформ 454 FLX [2, 3] и GS Junior [4, 5] (Roche), Genome Analyzer (Illumina) [6], SOLiD System, Ion PGM/Ion Proton (Invitrogen) [7], HeliScope (Helicos Biosciences) [8]. Во всех перечисленных работах использовались другие наборы праймеров, другие платформы для секвенирования и (или) другое программное обеспечение для анализа полученных данных.
NGS (next-generation sequencing) или высокопроизводительное секвенирование - технология, основанная на массивном параллельном секвенировании пространственно разделенных молекул ДНК; производительность NGS-секвенирования достигает десяти Гб и при этом характеризуется низкой стоимостью в пересчете на один нуклеотид. Таким образом, использование NGS для ресеквенирования BRCA1 и BRCA2 в пулированных образцах нескольких пациентов существенно снижает стоимость анализа мутаций BRCA.
Наиболее близким к заявленному способу - прототипом является способ определения последовательностей экзонов генов BRCA 1/2, предложенный De Leener и соавт. [2], где также был применен подход двухэтапной амплификации целевых участков молекулы ДНК с последующим секвенированием нового поколения (NGS). На первом этапе для каждого образца по отдельности амплифицируются все регионы экзонов генов BRCA1 и BRCA2, после чего смеси объединяют и проводят амплификацию с включением индексирующих последовательностей, определяющих к какому пациенту относится данный ампликон. В работе De Leener и соавт. для секвенирования полученной библиотеки использовали платформу 454 FLX (Roche), что является одним из основных отличий их разработки от заявленного изобретения. Другими отличиями являются использование другого набора праймеров и другого программного обеспечения для анализа данных секвенирования. Помимо этого, данный подход оптимизирован только для ДНК, выделенной из лимфоцитов. Заявленный же способ оптимизирован для ДНК, выделенной и из клеток крови, и из слюны.
Задачей изобретения является получение нуклеотидных последовательностей экзонов генов BRCA 1/2 для выявления мутаций и, следовательно, возможных генетических предпосылок развития HBOCS.
Поставленная задача достигается предлагаемым способом, заключающимся в следующем.
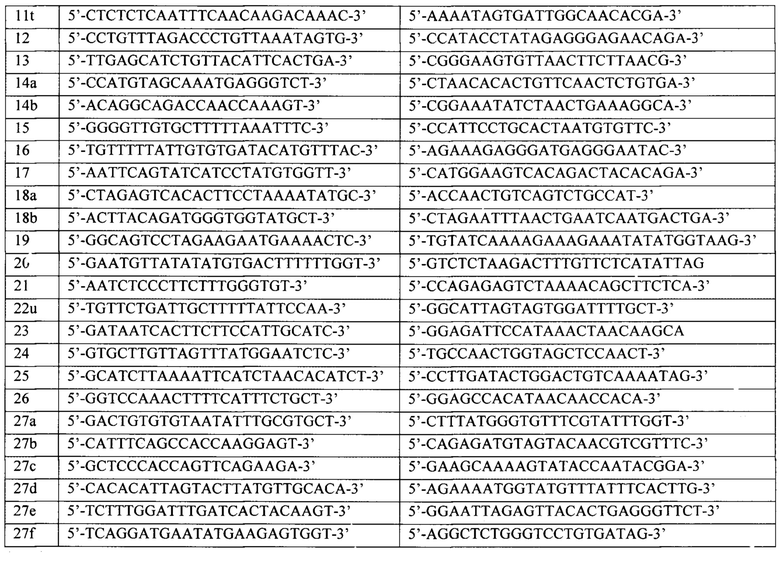
Для анализа используется ДНК, выделенная из крови или слюны стандартными методами молекулярной биологии. Очищенную ДНК каждого пациента подвергают первому раунду амплификации. Каждый праймер для первого раунда содержит уникальную часть, комплементарную последовательности ДНК генов BRCA1 и BRCA2 и универсальную последовательность из 20 нуклеотидов на 5'-конце (прямой праймер - acacgacgctcttccgatct, обратный праймер - gacgtgtgctcttccgatct). Структуры праймеров приведены в таблицах 1 и 2.
Для каждого образца ДНК выполняют 86 моноплексных ПЦР с использованием ампликон-специфичных праймеров. Амплификация образцов ДНК выполняют в 16 мкл реакционной смеси, содержащей 10 мМ TrisHCl (рН 8.9), 2.5 мМ MgCl2, 55 мМ KCl, dNTP (каждый в концентрации 0.2 мМ), 1.25 нМ Syto13, 0.5 U AmpliTaq Gold (Life Technologies), прямой и обратный праймеры (каждый в концентрации 300 нМ) и ДНК (10-25 нг). Протокол амплификации включает этапы: инкубация при 94°С в течение 12 минут; 38 циклов, состоящих из денатурации при 94°С (6 с), отжига праймеров при 58°С (10 с), элонгации при 72°С (50 с); заключительная элонгация при 72°С (2 мин).
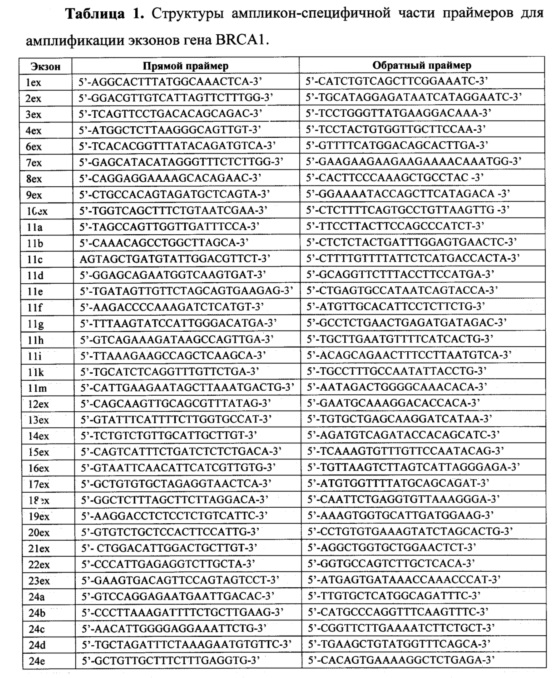

Все продукты ПЦР, полученные при амплификации одного образца геномной ДНК, объединяют в эквимолярных количествах и очищают на магнитных частицах AmpPURE Beads (Agilent Technologies) согласно инструкции фирмы-производителя. Нормализацию количества продуктов ПЦР для получения эквимолярных пулов осуществляют на основании значений итоговой флуоресценции амплификационной смеси.
Далее от каждого пулированного образца отбирают по аликвоте и разбавляют 1:1000; 2 мкл разбавленного пулированного образца используют в качестве матрицы для второго раунда ПЦР.
В процессе второго раунда ПЦР происходит амплификация индивидуальных пациент-специфичных библиотек и включение в состав ампликонов библиотек индексирующих (баркодов) и адаптерных нуклеотидных последовательностей. Второй раунд ПЦР выполняют с праймерами-адаптерами, содержащими на 3'-конце 20-нуклеотидную последовательность, аналогичную таковой на 5'-конце ампликон-специфичных праймеров; уникальную 8-нуклеотидную последовательность для индексирования индивидуальных библиотек и адаптерные последовательности на 5'-конце (таблицы 3 и 4). Все последовательности праймеров-адаптеров были взяты из протоколов рекомендуемых компанией Illumina (9). Амплификация пациент-специфичных библиотек осуществляют в тех же условиях, что и первый раунд ПЦР. Протокол амплификации включает этапы: инкубация при 94°С в течение 12 минут; 20 циклов, состоящих из денатурации при 94°С (6 с), отжига праймеров при 58°С (10 с), элонгации при 72°С (50 с); заключительная элонгация при 72°С (2 мин).

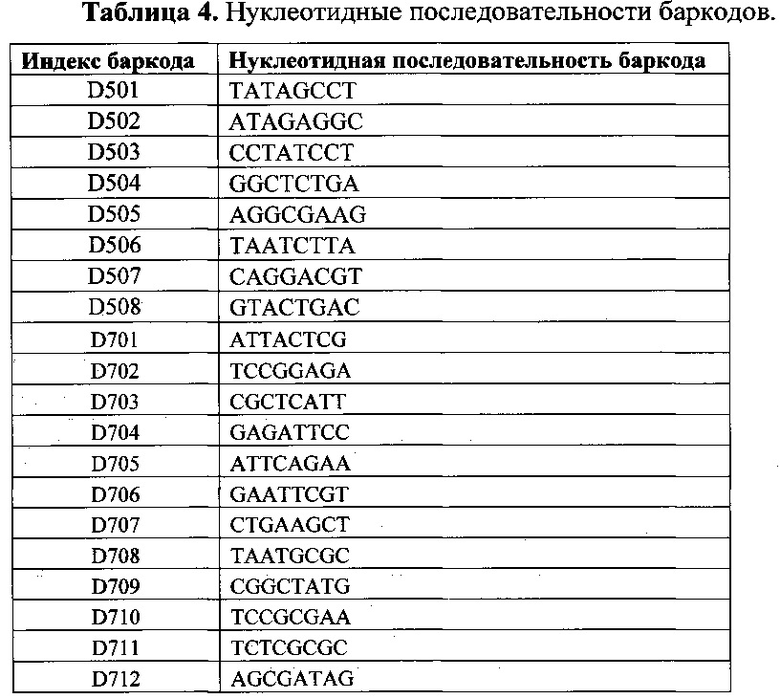
Все полученные в результате второго раунда индивидуальные библиотеки ампликонов нормализуют и объединяют в эквимолярном количестве. Нормализацию количества продуктов ПЦР в индивидуальных библиотеках осуществляют на основании значений итоговой флуоресценции амплификационной смеси. Пулированный образец очищают на магнитных частицах AmpPURE Beads (Agilent Technologies). Концентрацию ДНК в образце оценивают при помощи количественной ПЦР с праймерами 5'-AATGATACGGCGACCACCGA-3', 5'-CAAGCAGAAGACGGCATACGA-3' и TaqMan зондом 5'-FAM-TCCCTACACGACGCTCTTCCG-FQ-3'. Для построения калибровочной кривой используют образцы, приготовленные при помощи серийного разведения стандартной библиотеки PhiX. На основании полученных результатов концентрацию пулированного образца доводят до 10 пМ.
Секвенирование библиотеки (10 пМ) выполняют при помощи набора реагентов MiSeq Reagent Kit v2 (500cycle) (MS-102-2003) с помощью платформы MiSeq (Illumina) согласно инструкциям фирмы производителя.
Анализ полученных данных производят, используя разработанный нами алгоритм. Все полученные в результате секвенирования последовательности разделяют по пациентам на основании баркодов, нуклеотидные последовательности которых содержались в описании каждого рида. Разделение проводят разработанным нами скриптом fastqSep.py с допущением одной замены в баркоде. Риды, имеющие больше замен, в дальнейшем не используют.
Далее проводят картирование прочтений каждого пациента на последовательности 13й и 17й хромосом (версия сборки человеческого генома GRCh37.pl3). Перед картированием каждую референсную последовательность индексируют программой Samtools (http://samtools.sourceforge.net) (10). Картирование осуществляют функцией mem программы BWA (http://bio-bwa.sourceforge.net) (11). Далее полученный SAM-файл конвертируют в ВАМ-файл функцией view программы Samtools, а последний сортируют и индексируют функциями sort и index Samtools, соответственно. Выявление SNP (single nucleotide polymorphism, однонуклеотидная замена) и инсерций-делеций проводят программой GenomeAnalysisToolKit (GATK) (https://www.broadinstitute.org/gatky) (12) со следующими значениями параметров: "-glm BOTH". Затем все найденные SNP и инсерции-делеции аннотируют с помощью ANNOVAR (http://www.openbioinformatics.org/annovar/) (13). Дальнейшие фильтры накладывают в программе Microsoft Office или Open Office. Отбор проводят только тех вариаций, которые удовлетворяют следующим требованиям: покрытие не ниже 20 прочтений, не менее 14% всех прочтений данного локуса должны быть с альтернативным аллелем. Оставшиеся вариации считаются выявленными и подтверждаются секвенированием по Сэнгеру.
Таким образом, предложенный способ обладает высокой специфичностью и чувствительностью при анализе ДНК, выделенной из крови или слюны пациентов. Скрининг больных HBOCS или раком молочных желез с использованием предложенного нами подхода, учитывая его быстроту и низкую себестоимость, может быть эффективен для установления наследственной формы заболевания. Данное утверждение основывается на предположении, что мутации генов BRCA1 и BRCA2 приводят к развитию HBOCS.
Разработанный способ определения нуклеотидной последовательности генов BRCA 1/2 и, соответственно, выявления мутаций в данных генах, обладает высокой чувствительностью и специфичностью (100%), а также наиболее низкой стоимостью среди описанных к настоящему времени альтернативных подходов, имеет высокий уровень надежности и воспроизводимости. Способ апробирован на представительной выборке в 96 образцов ДНК пациентов клинической лаборатории больных семейной формой рака молочных желез. Разработанный подход может быть использован клиническими лабораториями как для выявления уже известных мутаций (однонуклеотидных замен - SNP, многонуклеотидных замен - MNP, инсерций и делеций) в генах BRCA1 и BRCA2, так и для еще ранее не выявленных. Данный метод хорошо адаптируем к инструментальным возможностям современных клинико-диагностических лабораторий и, таким образом, уже в настоящее время может быть использован в онкологических диспансерах и лабораториях.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ выявления вариаций и изменений числа копий в генах BRCA1 и BRCA2 по данным таргетного массового параллельного секвенирования генома | 2020 |
|
RU2759953C2 |
| Способ анализа терминальных мутаций в генах BRCA1, BRCA2, ATM и PALB2 с использованием мультиплексной ПЦР и последующей гибридизацией с олигонуклеотидным биологическим микрочипом (биочипом) | 2020 |
|
RU2729360C1 |
| Способ секвенирования экзонов гена HLA-DPA1 и набор синтетических олигонуклеотидов для его реализации | 2024 |
|
RU2837865C1 |
| Способ секвенирования экзонов гена HLA-DQA1 и набор синтетических олигонуклеотидов для его реализации | 2024 |
|
RU2833819C1 |
| Способ пробоподготовки образцов изолятов коронавируса SARS-CoV-2 и олигонуклеотидные праймеры для его реализации | 2021 |
|
RU2762759C1 |
| Способ молекулярно-генетической диагностики наследственных форм рака молочной железы | 2019 |
|
RU2702755C1 |
| Способ пробоподготовки образцов для типирования генов главного комплекса гистосовместимости HLA-A, HLA-B, HLA-C, HLA-DPB1, HLA-DQB1, HLA-DRB1 и олигонуклеотидные праймеры для его реализации | 2023 |
|
RU2829344C1 |
| Способ диагностики предрасположенности к раку молочной железы в русской популяции на основе ПЦР-ПДРФ | 2018 |
|
RU2723585C2 |
| Способ получения полноразмерной последовательности митохондриальной ДНК человека с использованием набора олигонуклеотидов методом мультиплексной амплификации для работы с образцами деградированной ДНК | 2021 |
|
RU2818323C2 |
| СПОСОБ ПРОГНОЗИРОВАНИЯ РИСКА ЗЛОКАЧЕСТВЕННЫХ ЗАБОЛЕВАНИЙ МОЛОЧНОЙ ЖЕЛЕЗЫ И/ИЛИ ЯИЧНИКОВ У ПАЦИЕНТОВ ПОСЛЕ ТРАНСПЛАНТАЦИИ ПОЧКИ | 2023 |
|
RU2821583C1 |
Изобретение относится к области молекулярной биологии и диагностической медицины. Описан способ определения последовательностей экзонов генов BRCA1 и BRCA2. Способ включает в себя два раунда амплификации экзонов с сайтами сплайсинга генов BRCA1 и BRCA2, секвенирование пулированной ДНК после амплификации с помощью MiSeq Illumina и анализ данных. В первом раунде амплификации для каждого образца ДНК проводят 86 моноплексных полимеразных цепных реакций (ПЦР), после чего продукты амплификации объединяют в эквимолярных количествах и очищают на магнитных частицах. Во второй раунд амплификации используют объединенную ДНК с первого раунда, разведенную 1:1000. На данном этапе происходит включение индексирующих и адаптерных нуклеотидных последовательностей. После этого продукты амплификации снова нормализуют и объединяют в эквимолярных количествах. Секвенирование осуществляют на MiSeq Illumina согласно инструкциям производителя. Изобретение обеспечивает снижение времени и стоимости анализа генов BRCA1/2, а также необходимость малого количества ДНК для выполнения данного анализа. 1 з.п. ф-лы, 3 табл.
1. Способ определения нуклеотидных последовательностей генов BRCA1 и BRCA2 с использованием двухэтапной амплификации целевых участков молекулы ДНК, когда на первом этапе для каждого образца по отдельности амплифицируются все регионы экзонов генов BRCA1 и BRCA2, после чего смеси объединяют и проводят амплификацию с включением индексирующих последовательностей, определяющих к какому пациенту относится данный ампликон, с последующим секвенированием нового поколения (NGS), отличающийся тем, что используется технология MiSeq (Illumina) и протокол обработки данных секвенирования, в котором для анализа используется последовательность программ из fastqSep.py, Samtools, BWA, Samtools, Samtools, GenomeAnalysisToolKit, ANNOVAR и оставляют вариации, имеющие покрытие не ниже 20 прочтений и не менее 14% прочтений с альтернативным аллелем, и способ подходит для ДНК, выделенной и из клеток крови, и из слюны.
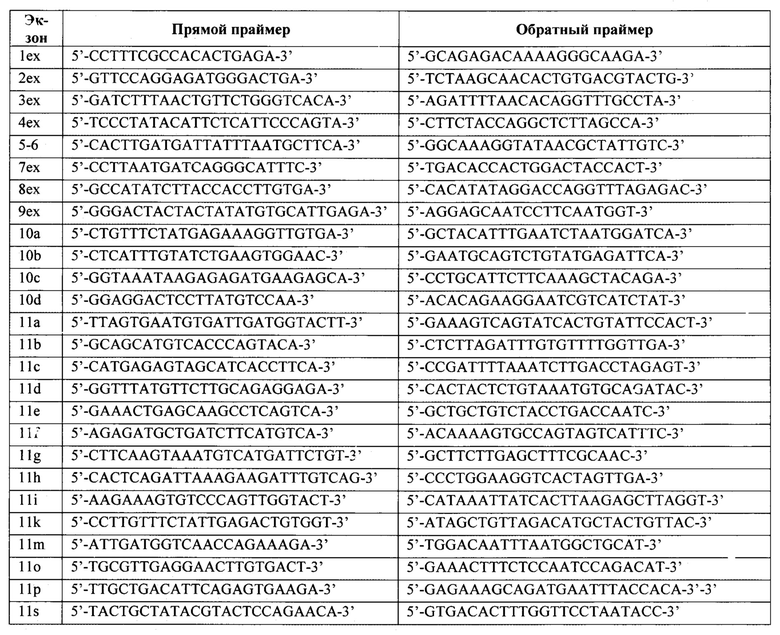
2. Способ определения по п. 1, отличающийся тем, что для амплификации ДНК используются следующие праймеры для генов BRCA1 и BRCA2, соответственно:


| ELLINGSON MS и др., Exome sequencing reveals frequent deleterious germline variants in cancer susceptibility genes in women with invasive breast cancer undergoing neoadjuvant chemotherapy, Breast Cancer Res Treat | |||
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| SIMARD J и др., Evaluation of BRCA1 and BRCA2 mutation prevalence, risk prediction models and a multistep testing approach in French-Canadian families with high risk of breast and ovarian cancer, J Med Genet | |||
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| СПОСОБ ГЕНОТИПИРОВАНИЯ ШТАММОВ Mycobacterium tuberculosis | 2013 |
|
RU2547598C1 |
| ДВИГАТЕЛЬ ДИЗЕЛЯ ДЛЯ ТЕПЛОВОЗА | 1926 |
|
SU6512A1 |
| НАБОР СИНТЕТИЧЕСКИХ ОЛИГОНУКЛЕОТИДОВ ДЛЯ ОПРЕДЕЛЕНИЯ НУКЛЕОТИДНОЙ ПОСЛЕДОВАТЕЛЬНОСТИ КОДИРУЮЩЕЙ ЧАСТИ ГЕНА BRCA1 И ВЫЯВЛЕНИЯ МУТАЦИЙ, АССОЦИИРОВАННЫХ С НАСЛЕДСТВЕННЫМИ ФОРМАМИ РАКА МОЛОЧНОЙ ЖЕЛЕЗЫ | 2010 |
|
RU2440415C1 |

