Область техники, к которой относится изобретение
Изобретение относится к области химической технологии, химии природных соединений и медицины, а именно к многостадийному способу получения биологически активных соединений, проявляющих антибактериальную активность и применимых в качестве лекарственного препарата в медицинской практике.
Предшествующий уровень техники
В последние годы значительно снижается эффективность существующих антибиотиков при лечении распространенных инфекций. В связи с этим в 2017 году всемирная организация здравоохранения сформировала список смертельно-опасных бактерий, для которых срочно требуется новая терапия [Tacconelli Е, Carrara Е, Savoldi A, et al., Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. The Lancet Infectious Diseases. 2018;18(3):318-327]. К этому списку относится такой патоген, как золотистый стафилококк (Staphylococcus aureus), который является одним из самых распространенных и опасных болезнетворных микроорганизмов в человеческой популяции [Mulani MS, Kamble ЕЕ, Kumkar SN, Tawre MS, Pardesi KR. Emerging Strategies to Combat ESKAPE Pathogens in the Era of Antimicrobial Resistance: A Review. Frontiers in Microbiology. 2019; 10:539]. Природные антибиотики пептидной природы обладают существенным потенциалом применения в клинической практике для борьбы с резистентными патогенными микроорганизмами, яркими примерами коммерчески успешных препаратов являются гликопептидные антибиотики и их производные [Blaskovich MAT, Hansford KA, Butler MS, Jia Z, Mark AE, Cooper MA. Developments in Glycopeptide Antibiotics. ACS Infect Dis. 2018;4(5):715-735] и липопептидный антибиотик даптомицин [Raja A, LaBonte J, Lebbos J, Kirkpatrick P. Daptomycin. Nature Reviews Drug Discovery. 2003;2(12):943-944].
В НИИ по изысканию новых антибиотиков им. Г.Ф. Гаузе обнаружен штамм, продуцирующий антибиотический комплекс ИНА-5812, в котором более 20 химически родственных веществ [Лапчинская O.A., Катруха Г.С., Гладких Е.Г и др. Исследование антибиотического комплекса ИНА-5812. Биоорганическая химия. 2016;42(6):732-740]. Он активен в отношении грамположительных бактерий, в том числе метициллинрезистентного золотистого стафилококка. Был описан штамм продуцент Streptomyces roseoflavus ИНА-Ас-5812, способ получения сырца антибиотика из культуральной жидкости штамма и компонента с молярной массой 1917,8 Да [Патент RU 2572341]. Продуктивность штамма продуцента антибиотического комплекса была существенно повышена и получен новый штамм продуцент Streptomyces tendae ВКПМ Ас-1980, накапливающий большее количество антибиотического комплекса в культуральной жидкости при культивировании [Патент RU 2710733].
Структура, способ определения структуры, способ выделения, брутто-формула, полный аминокислотный состав, биологическая активность заявляемых соединений в патентной и научно-технической литературе не описаны.
Сущность изобретения
Сущность изобретения заключается в разработке технологии выделения индивидуальных компонентов антибиотического комплекса гауземицинов А и В, установлении их структуры, физико-химических свойств и антибактериальной активности.
Способ выделения биологически активных соединений в индивидуальном виде является многостадийным. Можно условно разделить его на три основных этапа.
1. Экстракция культуры штамма-продуцента.
2. Получение концентрата антибиотика из экстракта с помощью комбинации методов (переосаждение, селективная сорбция/десорбция, хроматография).
3. Выделение индивидуальных соединений из концентрата антибиотика с помощью препаративной хроматографии гидрофильных взаимодействий.
Экстракция культуры штамма-продуцента заключается в отделении мицелия с помощью центрифугирования или фильтрования; последующей жидкостной или твердофазной экстракцией водной фазы; жидкостной экстракцией мицелия; обезжиривания полученных экстрактов.
Получение концентрата антибиотика из экстракта может осуществляться двумя способами.
1. Последовательное хроматографическое обогащение экстракта при низком давлении на гель-сорбентах типа Sephadex G15, обращеннофазовом сорбенте (октадецилсиликагель) в нейтральных условиях и завершающем этапе осаждения в кислой среде.
2. Сорбции полученного экстракта твердофазным сорбентом LPS500-H, селективной десорбции, хроматографическом разделении полученного концентрата путем обращеннофазовой хроматографии.
Последующее выделение компонентов в индивидуальном виде осуществляется путем хроматографии гидрофильных взаимодействий в условиях ВЭЖХ. Полученные компоненты характеризуют физико-химическими и биологическими методами.
По сравнению с раннее разработанными изобретениями [Патент RU 2572341, Патент RU 2710733] описываемый способ позволяет получать несколько индивидуальных компонентов из сложной смеси в количестве и с чистотой, достаточной для проведения физико-химических и биологических исследований.
Осуществление изобретения
Пример 1. Экстракция культуры Streptomyces tendae ВКПМ Ас-1980
Культуральную жидкость (271 мл) центрифугируют 20 мин при 5000g. Водную фазу декантируют, подкисляют HCl до рН 4 и экстрагируют н-бутанолом (2×52 мл). Органические вытяжки объединяют и упаривают досуха. Сухой остаток обезжиривают: заливают 19 мл этилацетата и озвучивают на ультразвуковой бане (40 кГц, 60 Вт, 10 мин при температуре 25°С), затем раствор фильтруют. Раствор отбрасывают, а с экстрактом (1,3 г), оставшимся на фильтре, проводят дальнейшую работу.
Мицелий суспендируют в 50 мл этанола, озвучивают в ультразвуковой бане (40 кГц, 60 Вт, 20 мин при температуре 25°С) и фильтруют. Фильтрат упаривают в вакууме. Сухой остаток обезжиривают описанным выше способом. Масса экстракта мицелия 0,8 г. Экстракт мицелия наравне с экстрактом водной фазы может быть использован для получения гауземицинов.
Пример 2. Экстракция культуры Streptomyces roseoflavus ИНА-Ас-5812
Культуральную жидкость (7,0 л) отфильтровывают от мицелия. К водной фазе добавляют сорбент XAD-4 (из расчета 100 г воздушно-сухого сорбента на 1 л КЖ). Перемешивают 4-12 ч при комнатной температуре до насыщения сорбента. Отфильтровывают сорбент, фильтрат отбрасывают и промывают сорбент одним объемом 1%-ной уксусной кислоты. Элюируют активные вещества 3-кратным объемом метанола.
Мицелий суспендируют в 500 мл метанола, озвучивают в ультразвуковой бане (40 кГц, 60 Вт, 20 мин при температуре 25°С) и фильтруют. Метанольные фильтраты объединяют и упаривают. Сухой остаток обезжиривают этилацетатом (2×100 мл) описанным в примере 1 способом.
С полученным экстрактом (17,3 г) проводят дальнейшую работу.
Пример 3. Экстракция культуры Streptomyces tendae ВКПМ Ас-1980
Культуральную жидкость (6,4 л) центрифугируют 15 мин при 5000g. Водную фазу декантируют, подкисляют HCl до рН 4 и экстрагируют изобутанолом (2×900 мл). Органические вытяжки объединяют и упаривают досуха. Сухой остаток обезжиривают этилацетатом (100 мл) описанным в примере 1 способом. Получают 10,7 г экстракта.
Мицелий суспендируют в 500 мл этанола, озвучивают в ультразвуковой бане (40 кГц, 60 Вт, 20 мин при температуре 25°С) и фильтруют. Дополнительно промывают остаток на фильтре 75 мл этанола. Фильтрат упаривают досуха, остаток обезжиривают этилацетатом (2×100 мл) описанным в примере 1 способом. Получают 12,3 г экстракта.
Пример 4. Экстракция культуры Streptomyces roseoflavus ИНА-Ас-5812
Культуральную жидкость (5,0 л) отфильтровывают от мицелия. К водной фазе добавляют сорбент Diaion HP-20 (из расчета 100 г воздушно-сухого сорбента на 1 л КЖ). Перемешивают 4-12 ч при комнатной температуре до насыщения сорбента. Отфильтровывают сорбент, фильтрат отбрасывают и промывают сорбент одним объемом 1%-ной уксусной кислоты. Элюируют активные вещества 3-кратным объемом изопропанола.
Мицелий суспендируют в 450 мл изопропанола, озвучивают в ультразвуковой бане (40 кГц, 60 Вт, 20 мин при температуре 25°С) и фильтруют. Изопропанольные вытяжки объединяют и упаривают. Сухой остаток обезжиривают этилацетатом (3×100 мл) описанным в примере 1 способом. Получают 16.9 г экстракта.
Пример 5. Получение концентрата антибиотика из экстракта
Экстракт (1,3 г) очищают от низкомолекулярных примесей методом гель-фильтрации на колонке (22×70 мм) с сорбентом Sephadex G15 с использованием изократического элюирования (20% метанола в воде, скорость потока 7 мл/мин, детекция по поглощению при 210, 260, 360, 410 нм). Собирают фракцию 1 (0,44 г) с временем удерживания 0-10 мин.
Фракцию 1 растворяют в 4 мл 50%-ного ацетонитрила и хроматографируют на колонке (25×130 мм) с обращеннофазовым сорбентом (октадецилсиликагель), с использованием ступенчатого элюирования (скорость потока 14 мл/мин, ступени по 5 минут: 20%, 30%, 40%, 50%, 60%, 80% ацетонитрил в воде; детекция по поглощению при 210, 260, 360, 410 нм). Емкость колонки - 0,2-0,3 г смеси. Фракция с временем удерживания 16.0-17.0 мин по данным хроматомасс-спектрометрии содержит в основном гауземицины А и В, и примеси. В результате разделения 0,440 г смеси получают 0,062 г данной фракции.
Образец полученной смеси гауземицинов (0,12 г) растворяют в 2,4 мл водного аммиачного раствора с рН 10. Полученный раствор подкисляют водным раствором соляной кислоты до рН 5 и наблюдают образование темно-коричневого осадка. Раствор фильтруют, полученный осадок растворяют в 1,1 мл водного аммиачного раствора с рН 10 и подкисляют водным раствором соляной кислоты до рН 5, вновь наблюдают образование темно-коричневого осадка. В результате получают объединенный раствор после двух осаждений и осадок с массой 0,053 г. Раствор после двух осаждений (3 мл) экстрагируют бутанолом (3×3,85 мл). Органические вытяжки объединяют, промывают подкисленной водой с рН 5 и упаривают досуха. В результате получают порошок (0,036 г) с желтоватым оттенком. Данный концентрат антибиотика содержит в основном гауземицины А и В, его используют для выделения индивидуальных соединений.
Пример 6. Получение концентрата антибиотика из экстракта
Экстракт (1 г) растворяют в 0,25 л воды с добавкой 10 мл концентрированного водного аммиака. Полученный раствор пропускают через колонку с 5 мл воздушно-сухого сорбента LPS500-H. Собирают фракцию «0», содержащую примеси, не связавшиеся с сорбентом. После чего проводят хроматографию с использованием ступенчатого элюирования с возрастающей концентрацией ацетонитрила в воде (ступени 0%, 10%, 20%, 30%, 40%, 50%, 100% по 50 мл). Получают семь фракций. Фракции 20-50% содержат в основном гауземицины А, В. В результате разделения 1 г экстракта получают 0,430 г фракции «0», 0,051 г фракции «0%», 0,044 г фракции «10%», 0,061 г фракции «20%», 0,085 г фракции «30%», 0,043 г фракции «40%», 0,072 г фракции «50%», 0,054 г фракции «100%».
Фракцию «20%» (61 мг) растворяют в 2 мл 50%-ного ацетонитрила и наносят на препаративную колонку ZORBAX SB-C18 (250×21,2 мм, октадецилсиликагель, 7 мкм). Разделение осуществляется путем изократического элюирования раствором В (0,1% трифторуксусная кислота в ацетонитриле, элюент А - 0,1% трифторуксусная кислота в воде) 33% за 30 мин, затем элюированием 50% В за 10 мин, при скорости потока 20 мл/мин. Детектируют по поглощению в УФ при 210, 260, 360, 410 нм или с помощью диодно-матричного детектора. Фракция с временем удерживания 9-13 мин по данным хроматомасс-спектрометрии содержит, в основном, гауземицины А и В. Ее высушивают в вакууме. В результате получают 25 мг концентрата антибиотика в виде белого порошка.
Пример 7. Препаративная жидкостная хроматография гидрофильных взаимодействий
Концентрат антибиотика (2 мг в 500 мкл 50%-ного ацетонитрила) хроматографируют на колонке VDSpher PUR 100 SIL (250×10 мм, 5 мкм) с использованием в качестве элюента смеси растворителей 0,1% трифторуксусной кислоты, 4% воды, 24% пропанола-1 и 71,9% ацетонитрила по объему. Скорость потока 6 мл/мин, детекция по поглощению в УФ при 205 нм. Фракция с временем удерживания 8.0-10.0 мин содержит гауземицин А, фракция с временем удерживания 10.0-13.0 мин содержит гауземицин В. Органические растворители удаляют из полученных фракций при пониженном давлении, получающийся водный раствор замораживают и лиофилизуют. Содержание основных компонентов во фракциях контролируют хроматографически и масс-спектрометрически, стадию хроматографии гидрофильных взаимодействий повторяют до полного удаления примесей.
В результате разделения 36 мг концентрата антибиотика (полученного по методу, описанному в примере 3) получают 0,8 мг гауземицина А и 1,1 мг гауземицина В в виде белых порошков.
В результате разделения 25 мг концентрата антибиотика (полученного по методу, описанному в примере 4) получают 1,9 мг гауземицина А и 5,3 мг гауземицина В в виде белых порошков.
Пример 8. Аналитическая хроматография для контроля содержания гауземицинов А и В
Для анализа содержания минорных компонентов антибиотического комплекса и низкомолекулярных примесей проводят аналитическую хроматографию. Для этого готовят раствор анализируемой пробы с концентрацией 4 мг/мл в метаноле, наносят на колонку Waters Sunfire (С 18; 5 мкм, 4,6×250 мм) 20 мкл анализируемого раствора и проводят градиентное элюирование (20-80% раствора В за 25 мин, где раствор А - 0,1% трифторуксусной кислоты в воде, раствор В - 0,1% трифторуксусной кислоты в ацетонитриле) со скоростью потока 1 мл/мин. Время удерживания группы соединений, включающий гауземицины А, В и компонент с массой 1930 мг составляет 10,2 мин.
Хроматографический контроль содержания гауземицинов А, В и компонента с массой 1930 Да проводят в условиях нормальнофазовой аналитической ВЭЖХ. Готовят раствор анализируемой пробы с концентрацией 4 мг/мл в метаноле, наносят на колонку Waters Spherisorb Silica (Силикагель, 5 мкм, 4,6×250 мм) 10 мкл раствора. Проводят градиентное элюирование (5-17% раствора В за 20 мин, где раствор А - 0,1% TFA, 25% PrOH, 74,9% CH3CN, раствор В - 0,1% TFA в HrO, также на протяжении всей хроматографии была добавка) со скоростью потока 1 мл/мин. Время удерживания гауземицина А составляет 13,8 мин, гауземицина В 14,7 мин, компонента с массой 1930 Да - 15,5 мин. Для обеспечения воспроизводимости времени удерживания поддерживают постоянное насыщение нормальнофазовой колонки водой и периодически калибруют время удерживания по стандартным смесям.
Масс-спектрометрический контроль получаемых образов проводят с помощью детектора Agilent 6340 Ion Trap с источником ионизации электрораспылением, соединенного с ВЭЖХ-системой Agilent 1100 HPLC system. Разделение пробы проводилось на колонке YMC-Triart С18 (YMC) (50×2.1 mm, 1.9 μ.м) путем элюирования линейным градиентом (2-98% ацетонитрила в воде с добавкой 0.1% муравьиной кислоты в оба элюента (при проведении анализа в хроматомасс-спектрометрическом режиме). Отслеживают содержание в образцах гауземицинов А и В по общему ионному току двухзарядных ионов с массами 924 Да и 959 Да соответственно, содержание компонента с массой 1930 отслеживают по ионному току двухзарядного иона с массой 967 Да. Содержание сторонних примесей также подтверждают масс-спектрометрически для хроматографически чистых образцов в связи с заметной ассоциацией гауземицинов с низкомолекулярными примесями.
Пример 9. Определение основных физико-химических и спектральных характеристик гауземицинов А, В
Полученные образцы гауземицинов А и В представляют собой аморфные твердые вещества.
Растворы веществ обладают оптической активностью. Измеренное удельное вращение: гауземицин А [α]22D -6.9 (с 0.3, метанол); гауземицин В [α]25D=-9.7 (с 0.4, метанол).
Гауземицины обладают характерным спектром поглощения в УФ-диапазоне:
гауземицин А (метанол) λmax, нм (log ε) 203 (4.7), 229 (4.5), 262 (4.4), 363 (3.7);
гауземицин В (метанол) λmax, нм (log ε) 204 (4.7), 229 (4.5), 262 (4.4), 363 (3.7).
При облучении в УФ-диапазоне для обоих веществ наблюдается флуоресценция с максимумом эмиссии при 448 нм (96% этанол, С = 1 мкМ, λвозб 350 нм).
Спектры поглощения в ИК-диапазоне имеют характерные максимумы:
гауземицин А (в KBr) νmax, см-1: 3330, 3070, 2960, 2930, 1660, 1540, 1450, 1200, 1180, 1070;
гауземицин В (в KBr) νmax, см-1: 3330, 3070, 2960, 2930, 1660, 1540, 1450, 1230, 1200, 1180, 1070.
Пример 10. Установление состава и структуры гауземицинов А, В
Брутто-формулы гауземицинов А и В устанавливают на основании масс-спектров высокого разрешения. В масс-спектре высокого разрешения гауземицина А наблюдается ион с m/z 1845.788, что согласуется с брутто-формулой C84H116ClN17O28 (расчетное значение m/z=1845.786 для [М+Н]+), масс-спектр высокого разрешения гауземицина В содержит двухзарядный ион с m/z 959.4187, что согласуется с брутто-формулой C87H121ClN18O29 (расчетное значение m/z=959.4191 для [М+2Н]2+).
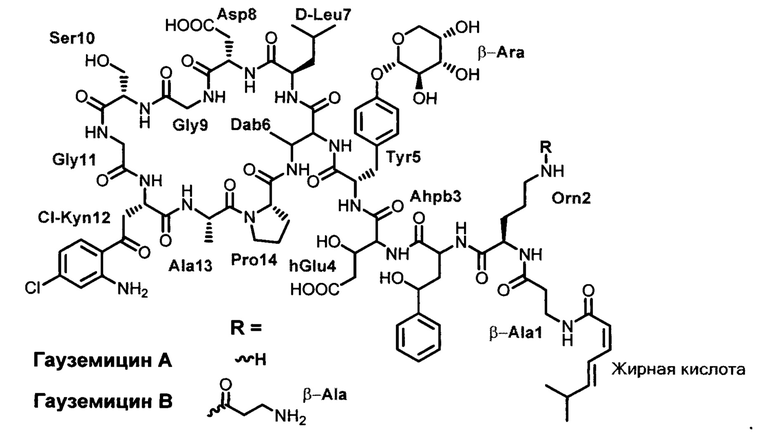
Структура гауземицинов А и В, представленная на Рисунке 1, была установлена на основании данных ЯМР (Таблица 1). Отнесение сигналов в спектрах ЯМР 1Н, 14N и13С проведено на оснований двумерных спектров, включающих как гомоядерные, так и гетероядерные корреляции. Спектры зарегистрированы для растворов индивидуальных веществ в пердейтеродиметилсульфоксиде при повышенной температуре (45°С) для упрощения наблюдаемой при более низких температурах сложной конформационной динамики. При более высоких температурах наблюдается частичная деструкция образца.
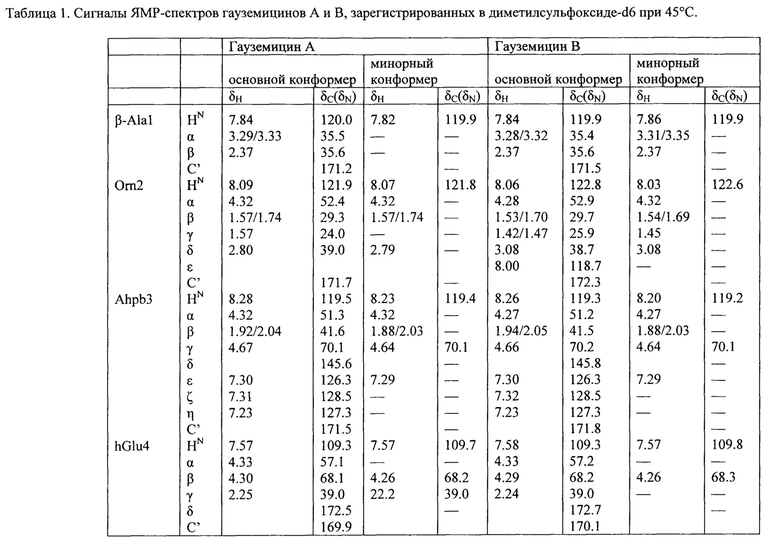
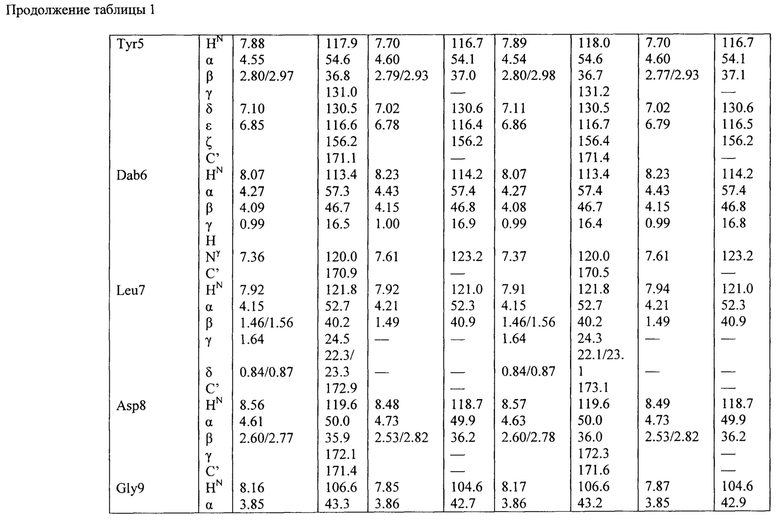
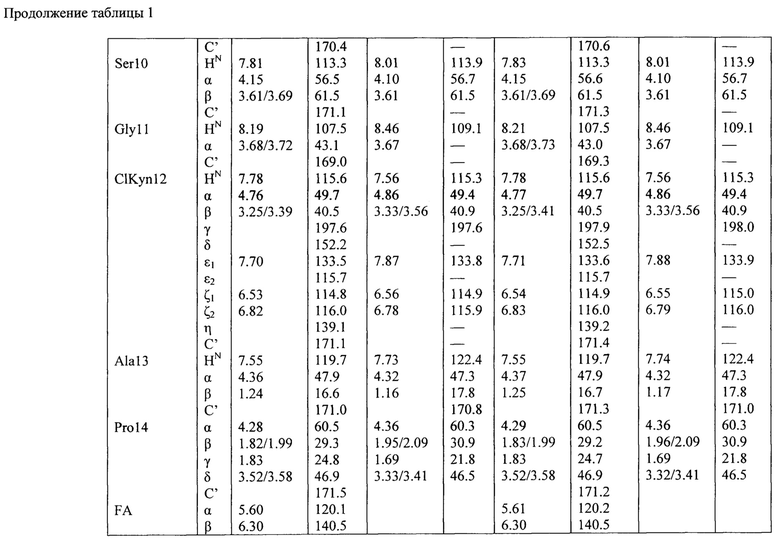
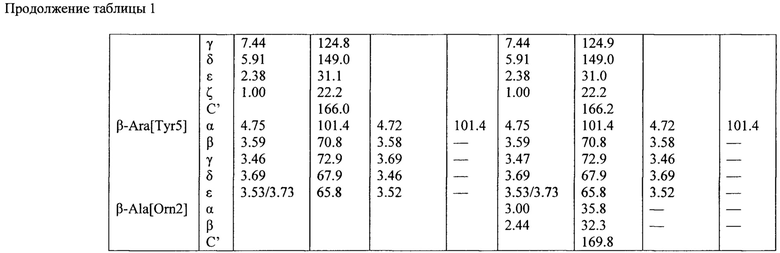
Пример 11. Оценка антибактериальных свойств гауземицинов А, В
Предварительные эксперименты показали, что гауземицины обладают заметной антибактериальной активностью в отношении грамположительных бактерий, прежде всего стафилококков. Антибактериальные свойства выделенных гауземицинов А и В оценивали методом серийных микроразведений в жидкой питательной среде согласно клиническим рекомендациям «Определение чувствительности микроорганизмов к антимикробным препаратам», версия 2018-03, утверждены 18.10.2017 г. экспертным совещанием профильной комиссии по специальности «Клиническая микробиология и антимикробная резистентность» при МЗ РФ. В качестве контроля использовали стандартные образцы пептидных антибиотиков (даптомицин, ванкомицин). Полученные значения минимальной подавляющей концентрации представлены в табл. 2.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ выделения и очистки нафтохиноновых противогрибковых антибиотиков астолидов А и В | 2019 |
|
RU2725187C1 |
| СПОСОБ ПОЛУЧЕНИЯ АНТИБИОТИКА АКТИНОПЛАЦИНА, ШТАММ STREPTОMYCES (KITASATOA) SPECIES 834 ВНИИСХМ Д-484-ПРОДУЦЕНТ АНТИБИОТИКА АКТИНОПЛАЦИНА | 1997 |
|
RU2151793C1 |
| ШТАММ STREPTOMYCES GRISEOCARNEUS SUBSP. BLEOMYCINI ВКПМ-S1086 - ПРОДУЦЕНТ БЛЕОМИЦЕТИНА И СПОСОБ ПОЛУЧЕНИЯ АНТИБИОТИКА БЛЕОМИЦЕТИНА | 2007 |
|
RU2358008C2 |
| СПОСОБ ПОЛУЧЕНИЯ АНТИБИОТИКА | 1990 |
|
RU2029783C1 |
| СПОСОБ ПОЛУЧЕНИЯ АНТИБИОТИЧЕСКОГО КОМПЛЕКСА | 1972 |
|
SU352469A1 |
| СПОСОБ ПОЛУЧЕНИЯ НИБОМИЦИНА | 2018 |
|
RU2718802C1 |
| Способ получения компонентов А @ ,А @ ,А @ ,А @ ,В @ или В @ антибиотического комплекса ВВМ-1675 , обладающих антимикробным и противоопухолевым действием, штамм актиномицета АстINомаDURа VeRRUcoSoSpoRa АТСС 39334 и штамм актиномицета АстINомаDURа VeRRUcoSoSpoRa АТСС 39638, используемый для получения компонентов А @ ,А @ ,А @ ,А @ ,В @ или В @ антибиотического комплекса ВВМ-1675 , обладающих антимикробным и противоопухолевым действием | 1984 |
|
SU1344249A3 |
| Способ получения антибиотического комплекса | 1978 |
|
SU1039446A3 |
| МАКРОЦИКЛИЧЕСКИЙ ЛАКТОН, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТИБИОТИЧЕСКОЙ АКТИВНОСТЬЮ, И ИНСЕКТОАКАРИЦИДНАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2100354C1 |
| Штамм Amycolatopsis orientalis - продуцент антибиотика диметилванкомицина и способ получения антибиотика | 2016 |
|
RU2633511C1 |
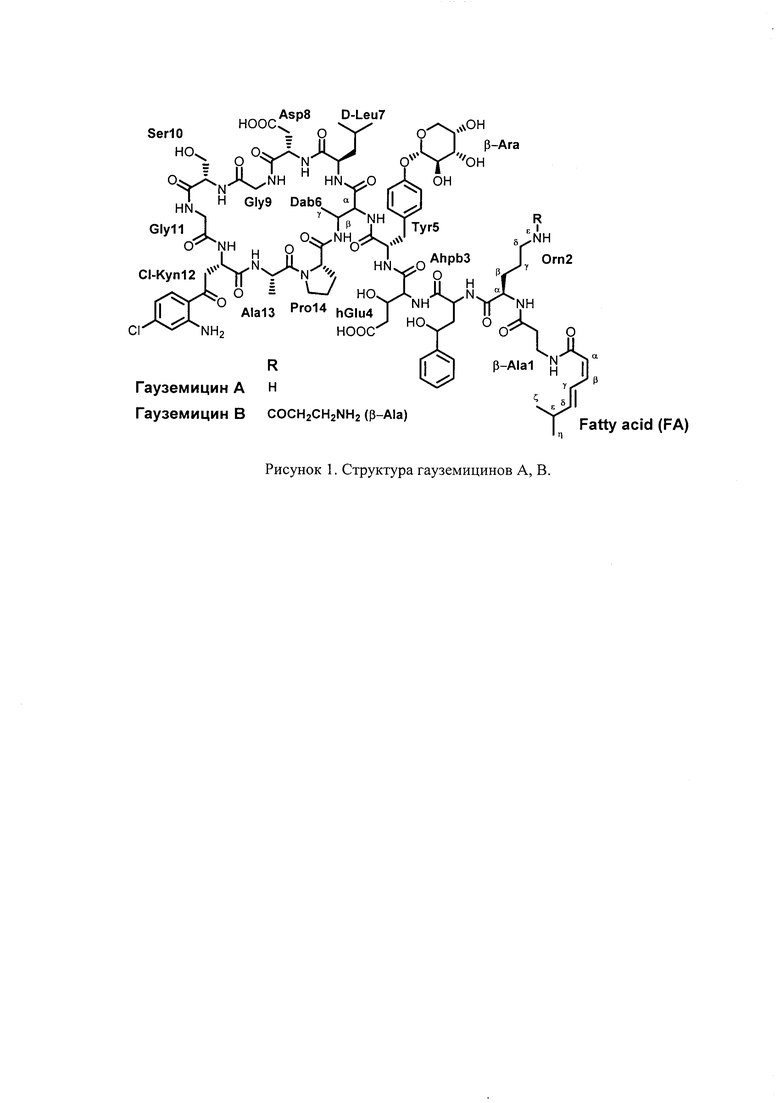
Изобретение относится к соединению, а именно к гауземицинам А и В, формулы:
Изобретение также относится к способу выделения указанных гауземицинов А и В. Технический результат – получены новые соединения, обладающие антибактериальной активностью в отношении грамположительных бактерий, включая метициллинрезистеный золотистый стафилококк, которые могут найти применение в медицине. 2 н. и 4 з.п. ф-лы, 1 ил., 2 табл., 11 пр.
1. Гауземицины А и В, обладающие антибактериальной активностью в отношении грамположительных бактерий, включая метициллинрезистеный золотистый стафилококк, и соответствующие формуле:
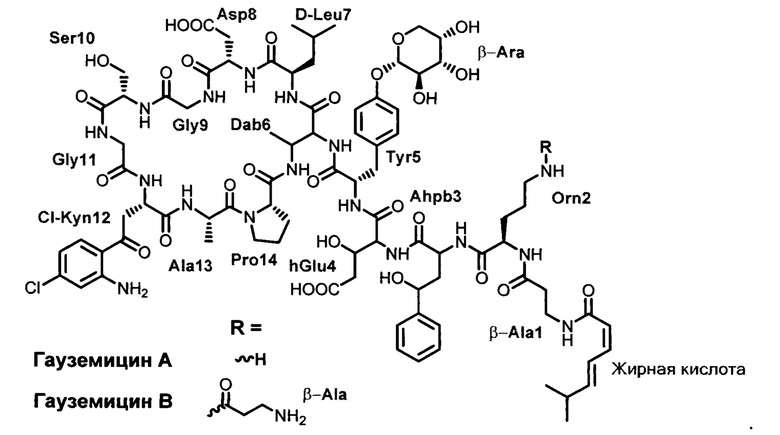
2. Способ выделения пептидных антибиотиков гауземицинов А и В из п. 1, заключающийся в выделении экстракта путем экстракции культуральной жидкости штамма-продуцента; получении концентрата антибиотика с помощью комбинации методов: переосаждение, селективная сорбция/десорбция, хроматография, препаративном разделении полученного концентрата путем хроматографии гидрофильных взаимодействий в условиях ВЭЖХ и физико-химической и биологической характеристикой полученных образцов.
3. Способ выделения пептидных антибиотиков гауземицинов А и В из п. 1 в виде экстракта по п. 2, в котором проводят жидкостно-жидкостную экстракцию культуральной жидкости штамма-продуцента органическим растворителем, выбранным из н-бутанола или изобутанола; получение концентрата антибиотика проводят с помощью последовательных хроматографических разделений при низком давлении: гель-фильтрация на сорбенте Sephadex G15, обращеннофазовом сорбенте в нейтральных условиях; и последующем осаждении при подкислении.
4. Способ выделения пептидных антибиотиков гауземицинов А и В из п. 1 в виде экстракта по п. 2, в котором проводят экстракцию культуральной жидкости твердофазным сорбентом на основе полистирола.
5. Способ выделения пептидных антибиотиков гауземицинов А и В из п. 1 в виде экстракта по п. 2, в котором экстракцию мицелия штамма-продуцента проводят органическим растворителем, выбранным из метанола, этанола, пропанола-2 или их смесями.
6. Способ выделения пептидных антибиотиков гауземицинов А и В из п. 1 в виде концентрата по п. 2, в котором используется сорбция экстракта твердофазным сорбентом LPS500-H, последующая селективная десорбция и хроматографическое разделение путем обращеннофазовой хроматографии в условиях ВЭЖХ.
| Штамм Streptomyces tendae - продуцент противобактериального антибиотика ИНА 5812 | 2019 |
|
RU2710733C1 |
| АНТИБИОТИК ИНА 5812, ШТАММ-ПРОДУЦЕНТ Streptomyces roseoflavus ИНА-Ас-5812 И СПОСОБ ПОЛУЧЕНИЯ АНТИБИОТИКА | 2013 |
|
RU2572341C2 |
| СПОСОБ ОЧИСТКИ ЛИПОПЕПТИДОВ | 2010 |
|
RU2526391C2 |
| НОВЫЕ ПРОТИВОБАКТЕРИАЛЬНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ ГРАМПОЛОЖИТЕЛЬНЫХ ИНФЕКЦИЙ | 2009 |
|
RU2512396C2 |
| RU 2005108995 A, 10.11.2005 | |||
| Катушка переменной самоиндукции | 1928 |
|
SU10294A1 |

