Изобретение относится к области получения пестицидов, более конкретно - к способу получения имазапира (2-(4-метил-5-оксо-4-пропан-2-ил-1Н-имидазол-2-ил)пиридин-3-карбоновой кислоты (I), регистрационный номер CAS [81334-34-1], далее по тексту – имазапир (I)). Это соединение является неселективным гербицидом широкого спектра действия из класса имидазолинонов (см. The Imidazolinone Herbicides, Edited by D.L. Shaner, S.L. O’Connor, Boca Raton, FL: CRC Press, Inc., 1991) и применяется в различных областях народного хозяйства для уничтожения однолетних и многолетних трав и широколиственных сорняков, деревянистых кустарников, лиственных деревьев и др. (см., напр., Modern Crop Protection Compounds, Edited by P. Jeschke et al., 3rd Ed., Vol. 1: Herbicides. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA, 2019, p. 95). Имазапир был разработан и представлен на рынке компанией «Американ Цианамид Компани» в середине 1980-х годов.
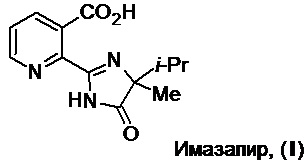
Известно несколько общих методов получения имазапира, которые кратко можно представить в виде Схемы 1:
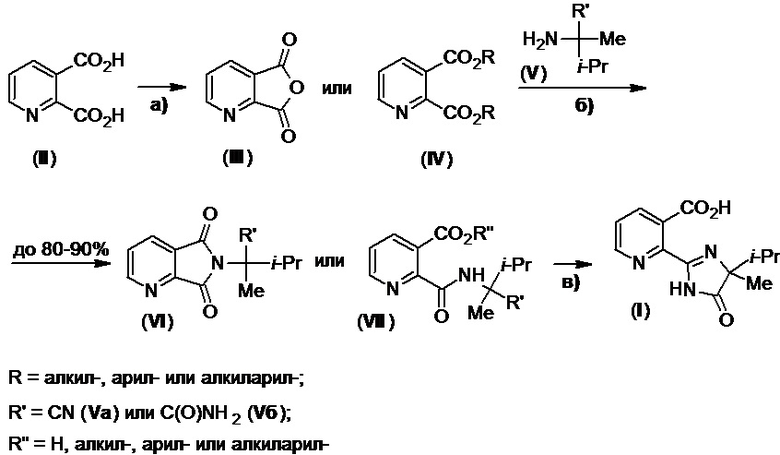
Схема 1.
Все эти методы синтеза реализуются в несколько стадий и, как правило, включают 3 основных этапа: а) трансформацию исходной 2,3-пиридиндикарбоновой кислоты (II) (далее по тексту - кислота (II)) в ангидрид (III) (патент США №4439607) или диэфиры (IV) (см. ниже по тексту); б) реакцию этих соединений с 2-амино-2,3-диметилбутиронитрилом (Vа) или 2-амино-2,3-диметилбутирамидом (Vб) (далее по тексту - амид (Vб)), приводящую, в зависимости от условий, к ключевым циклическим имидам (VI) или моноамидам (VII) (см., напр., патенты США №№4518780, 4562257, 4782157; патенты КНР №№102532102B, 112142729A) и в) дальнейшее превращение последних в целевой продукт под действием оснований, кислот, окислителей и др. (см., напр., патенты США №№4709036, 4719303, 4798619, 4921961, 5334576).
Также известен метод получения имазапира, который отличается от описанных в Схеме 1, и основан на использовании 2-метилпиридина (VIII) в качестве исходного сырья. Его реакция с элементарной серой и амидом (Vб) при нагревании приводит к образованию имидазолинона (IX) (Схема 2), в пиридиновый цикл которого вводят карбоксильную группу с помощью 2 экв. н-бутиллития и твердого диоксида углерода. Подкисление полученной реакционной массы приводит к имазапиру (патент США №4474962).
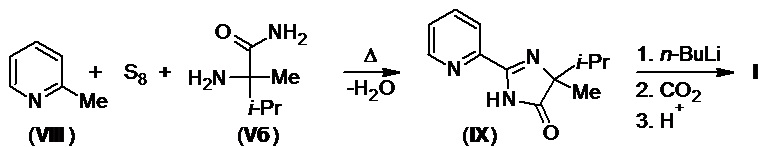
Схема 2.
В целом, представленные выше методы синтеза имазапира (Схемы 1 и 2) позволяют получать его из кислоты (II) с удовлетворительным результатом, однако, их многостадийность, использование нестабильных и чувствительных к влаге и кислороду воздуха промежуточных и исходных соединений (напр., ангидрида (III) или н-бутиллития), а также частое применение высоких температур (>150°С) в ходе проведения реакции оказывают негативное влияние на показатели процесса, усложняют его масштабируемость и накладывают дополнительные требования к его организации и аппаратурному оформлению.
Этих недостатков лишен способ получения имазапира из диэфиров (IV), предложенный в 1988 г. (патент США №4758667). Он включает реакцию последних с амидом (Vб) под действием трет-бутилата калия в среде толуола, с высокой региоселективностью приводящую к образованию калиевой соли имазапира (I-K), подкислением которой минеральными кислотами получают имазапир с выходом 80% (Схема 3). Также известна модификация этого метода, позволяющая получать хиральный R-изомер целевого продукта (патент США №5973154А).
Схема 3.
В свою очередь, диэфиры (IV), необходимые в синтезе имазапира по Схеме 3, доступны несколькими методами. Наиболее широко применяется прямая этерификация исходной кислоты (II) спиртами в присутствии избытка сильной минеральной кислоты при нагревании (Схема 4). Эта реакция проста в исполнении, что облегчает последующее масштабирование процесса (см., напр., международные заявки WO №№2016119706Al, 2013092893Al; патент КНР №105566319A; K. Yoshiizumi et al., Bioorg. Med. Chem., 2003, 11, 433-450; J.B. Sperry et al., Org. Process Res. Dev., 2012, 16, 11, 1854-1860). В зависимости от условий проведения реакции и используемого спирта (ROH), выходы диэфиров (IV) составляют 60-95%, их выделение и очистку после реакции осуществляют стандартными методами (экстракцией, отмывкой, перегонкой в вакууме, колоночной хроматографией и др.). Также известны и другие способы получения диэфиров (IV), включающие, например, использование пропаргиламина (X) и ацетилендикарбоксилатов (XI) в присутствии окислителей (перекиси водорода, персульфата калия и др.) (патент КНР №104447528B; T.A. Nizami, R. Hua, Tetrahedron, 2017, 73, 6080-6084), акролеина (XII) и аминодикарбоксилатов (XIII) (патент США №4948896) или кислоты (II) и спиртов в присутствии SOCl2 или POCl3 вместо минеральной кислоты (международная заявка WO №2008108988А1).
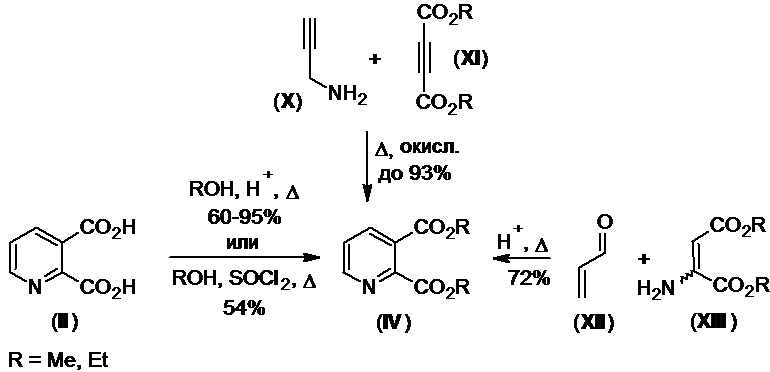
Схема 4.
Суммируя Схемы 3 и 4, известный подход к синтезу имазапира из кислоты (II) включает два этапа, первый из которых - синтез диэфиров (IV) [в целях «атомной экономики» обычно используют диметиловый (IVа), диэтиловый (IVб) или дипропиловый (IVв) эфиры (Схема 5)]. Основным недостатком этого подхода является необходимость выделения этих диэфиров (IV) в чистом виде в процессе их получения для дальнейшего использования на втором этапе процесса (трансформации диэфиров (IV) в имазапир), что приводит к использованию в процессе большого количества разных технологических операций, таких как промывка, экстракция, фильтрация, отгонка растворителя, вакуумная перегонка, очистка на сорбенте и др., что снижает общую эффективность и экономические показатели процесса, удлиняет и усложняет его, а также требует использования разнообразного промышленного оборудования.
Задачей предлагаемого технического решения является улучшение технико-экономических показателей процесса производства имазапира за счет упрощения технологии его получения из 2,3-пиридиндикарбоновой кислоты.
Техническим результатом является а) улучшение технико-экономических показателей технологического процесса производства имазапира из 2,3-пиридиндикарбоновой кислоты; б) сокращение количества технологических операций процесса и его продолжительности; в) упрощение его аппаратурного оформления; г) получение целевого продукта высокого качества с выходом 65-78% в зависимости от условий проведения процесса; д) возможность регенерации использованного растворителя с частичным возвратом его в процесс, что сокращает отходы производства.
Технический результат достигается при использовании способа получения имазапира, включающего первоначальную этерификацию 2,3-пиридиндикарбоновой кислоты низшим спиртом в присутствии минеральной кислоты при нагревании с последующей отгонкой избытка низшего спирта, обработкой реакционной массы водным раствором основания и экстракцией продукта реакции ароматическим растворителем с получением раствора соответствующего диэфира 2,3-пиридиндикарбоновой кислоты в этом растворителе, для которого осуществляют корректировку концентрации и обезвоживание путем удаления части растворителя азеотропной отгонкой при атмосферном или пониженном давлении, с последующим введением полученного безводного раствора с необходимой концентрацией диэфира 2,3-пиридиндикарбоновой кислоты, без фактического его выделения в индивидуальном виде, в реакцию с 2-амино-2,3-диметилбутанамидом в присутствии алкоголята щелочного металла в атмосфере инертного газа или на воздухе без доступа влаги с получением целевого продукта, его выделением и очисткой.
В качестве низшего спирта используют метиловый, этиловый или н-пропиловый спирт; в качестве минеральной кислоты используют серную или фосфорную кислоту; в качестве основания используют карбонат натрия или калия, гидрокарбонат натрия или калия или гидроксид натрия, калия или аммония; в качестве ароматического растворителя используют толуол, этилбензол, кумол, ксилол (о-, м- или п-) или их смесь или диэтилбензол (о-, м- или п-) или их смесь; в качестве алкоголята щелочного металла используют метилат натрия или калия, этилат натрия или калия, изопропилат натрия или калия, трет-бутилат натрия или калия, неопентилат натрия или калия; в качестве инертного газа используют азот или аргон.
Изобретение иллюстрируется следующими примерами (Схема 5).
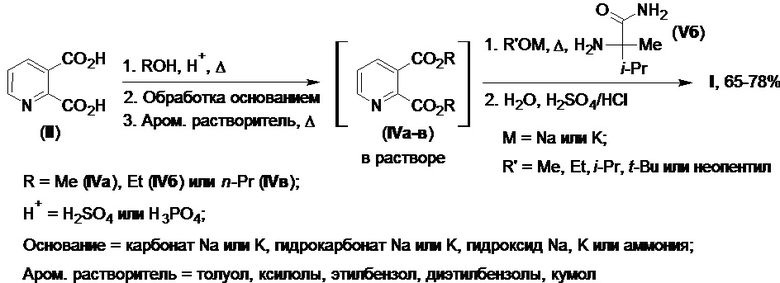
Схема 5.
Пример 1.
В реактор синтеза диэфиров (IV) помещают кислоту (II) (100,0 г, 0,60 моль) и безводный метанол, после чего при перемешивании при комнатной температуре дозируют к образовавшейся смеси концентрированную серную кислоту (1,5-3,0 экв.). Содержимое реактора нагревают до кипения и выдерживают при этой температуре без доступа влаги до полного срабатывания исходной кислоты (II) (контроль ТСХ), после чего полученный раствор охлаждают и избыток метанола отгоняют при атмосферном или пониженном давлении для последующей его регенерации и возврата в процесс. К маслянистому остатку при эффективном перемешивании медленно добавляют 10-15%-ный водный раствор карбоната натрия до значения pH раствора равного 7-8, следя за тем, чтобы выделение углекислого газа было не слишком интенсивным. Полученную массу дважды экстрагируют толуолом, органические вытяжки отделяют и объединяют, после чего объединенный толуольный экстракт закачивают в реактор синтеза имазапира, где его промывают водой, водную и органическую фазу разделяют и водную фазу отделяют. Оставшийся толуольный раствор концентрируют при атмосферном или пониженном давлении до необходимой для следующей стадии синтеза концентрации диэфира (IVa) с сопутствующим удалением оставшейся влаги в виде азеотропной смеси. Полученный после отгонки раствор представляет собой 10-15%-ный раствор диметилового эфира (IVa) в безводном толуоле [выход диэфира (IVa) - ~111,8 г (0,57 моль, 96% в расчете на кислоту (II)) по данным ГЖХ], к которому при комнатной температуре добавляют амид (Vб) (76,8 г, 0,59 моль) и включают подачу азота. Полученную суспензию перемешивают при комнатной температуре до полного растворения амида (Vб), после чего прибавляют трет-бутилат натрия (115,4 г, 1,20 моль) двумя равными порциями с интервалом в 5 минут. Реакционную массу нагревают при перемешивании при непрерывной подаче азота до полного срабатывания диэфира (IVa) (контроль ТСХ), после чего ее охлаждают до комнатной температуры и прибавляют воду для растворения натриевой соли имазапира. Перемешивание продолжают до полного растворения твердой фазы в воде, после чего его останавливают и полученной двухфазной системе дают разделиться. Представляющую собой раствор натриевой соли имазапира водную фазу отделяют и направляют в реактор очистки, полученную толуольную фазу отправляют на регенерацию растворителя для возврата его в процесс. К щелочной водной фазе в реакторе очистки прибавляют 5-10 масс.% активированного угля (в расчете на диэфир (IVa)), перемешивают 1-2 часа и отфильтровывают от отработанного угля в реактор подкисления. К очищенному раствору натриевой соли имазапира при перемешивании прибавляют концентрированную серную или соляную кислоту до значения рН раствора равного 2-4. Полученную суспензию фильтруют, осадок промывают водой и высушивают на воздухе. Получают 122,3 г (78,2% в расчете на исходную кислоту (II)) имазапира в виде белого кристаллического порошка с т.пл. 170-172°С и чистотой не менее 98,0% (по данным ВЭЖХ).
Пример 2.
В реактор синтеза диэфиров (IV) помещают кислоту (II) (130,0 г, 0,78 моль) и безводный этанол, после чего при перемешивании при комнатной температуре дозируют к образовавшейся смеси 100%-ную фосфорную кислоту (1,5-3,0 экв.). Содержимое реактора нагревают до кипения и выдерживают при этой температуре без доступа влаги до полного срабатывания исходной кислоты (II) (контроль ТСХ), после чего полученный раствор охлаждают и избыток этанола отгоняют при атмосферном или пониженном давлении для последующей его регенерации и возврата в процесс. К маслянистому остатку при эффективном перемешивании медленно добавляют 5-10%-ный водный раствор гидрокарбоната натрия до значения pH раствора равного 7-8, следя за тем, чтобы выделение углекислого газа было не слишком интенсивным. Полученную массу дважды экстрагируют этилбензолом, органические вытяжки отделяют и объединяют, после чего объединенный этилбензольный экстракт закачивают в реактор синтеза имазапира (I), где его промывают водой, водную и органическую фазу разделяют и водную фазу отделяют. Оставшийся этилбензольный раствор концентрируют при атмосферном или пониженном давлении до необходимой для следующей стадии синтеза концентрации диэфира (IVб) с сопутствующим удалением оставшейся влаги в виде азеотропной смеси. Полученный после отгонки раствор представляет собой 10-15%-ный раствор диэтилового эфира (IVб) в безводном этилбензоле [выход диэфира (IVб) - ~163,0 г (0,73 моль, 94% в расчете на кислоту (II)) по данным ГЖХ], к которому при комнатной температуре добавляют амид (Vб) (97,9 г, 0,75 моль) и включают подачу аргона. Полученную суспензию перемешивают при комнатной температуре до полного растворения амида (Vб), после чего прибавляют трет-бутилат калия (172,0 г, 1,54 моль) двумя равными порциями с интервалом в 5 минут. Реакционную массу нагревают при перемешивании при непрерывной подаче аргона до полного срабатывания диэфира (IVб) (контроль ТСХ), после чего ее охлаждают до комнатной температуры и прибавляют воду для растворения калиевой соли имазапира. Перемешивание продолжают до полного растворения твердой фазы в воде, после чего его останавливают и полученной двухфазной системе дают разделиться. Представляющую собой раствор калиевой соли имазапира (I-K) водную фазу отделяют и направляют в реактор очистки, полученную этилбензольную фазу отправляют на регенерацию растворителя для возврата его в процесс. К щелочной водной фазе в реакторе очистки прибавляют 5-10 масс.% активированного угля (в расчете на диэфир (IVб)), перемешивают 1-2 часа и отфильтровывают от отработанного угля в реактор подкисления. К очищенному раствору калиевой соли имазапира (I-K) при перемешивании прибавляют концентрированную серную или соляную кислоту до значения рН раствора равного 2-4. Полученную суспензию фильтруют, осадок промывают водой и высушивают на воздухе. Получают 155,5 г (76,5% в расчете на исходную кислоту (II)) имазапира (I) в виде белого кристаллического порошка с т.пл. 171-172°С и чистотой не менее 98,0% (по данным ВЭЖХ).
Пример 3.
Способ получения имазапира осуществлялся аналогично примеру 1 с загрузкой 150,0 г (0,90 моль) кислоты (II). Отличие состояло в использовании н-пропанола вместо метанола; 25%-ного водного раствора гидроксида аммония вместо водного раствора карбоната натрия; о-, м- или п-ксилола или их смеси вместо толуола; неопентилата натрия (197,2 г, 1,80 моль) вместо трет-бутилата натрия. Промежуточно в синтезе получают 10-15%-ный раствор дипропилового эфира (IVв) (выход ~95% в расчете на кислоту (II) по данным ГЖХ) в безводном о-, м- или п-ксилоле или их смеси. Получают 176,0 г (75,0%) имазапира (I) в виде белого кристаллического порошка.
Пример 4.
Способ получения имазапира осуществлялся аналогично примеру 1 с загрузкой 150,0 г (0,90 моль) кислоты (II). Отличие состояло в использовании 100%-ной фосфорной кислоты вместо концентрированной серной кислоты; 20-25%-ного водного раствора карбоната калия вместо водного раствора карбоната натрия; о-, м- или п-диэтилбензола или их смеси вместо толуола; метилата натрия (97,2 г, 1,80 моль) или этилата натрия (122,5 г, 1,80 моль) вместо трет-бутилата натрия. Получают 153,1 г (65,3%) имазапира (I) в виде белого кристаллического порошка.
Пример 5.
Способ получения имазапира осуществлялся аналогично примеру 2 с загрузкой 100,0 г (0,60 моль) кислоты (II). Отличие состояло в использовании концентрированной серной кислоты вместо 100%-ной фосфорной кислоты, 30-40%-ного водного раствора гидроксида натрия или гидроксида калия вместо водного раствора гидрокарбоната натрия, кумола вместо этилбензола, метилата калия (84,8 г, 1,21 моль) или этилата калия (101,8 г, 1,21 моль) вместо трет-бутилата калия. Получают 108,0 г (69,1%) имазапира (I) в виде белого кристаллического порошка.
Пример 6.
Способ получения имазапира осуществлялся аналогично примеру 1 с загрузкой 130,0 г (0,78 моль) кислоты (II). Отличие состояло в использовании 20-25%-ного водного раствора гидрокарбоната калия вместо водного раствора карбоната натрия, изопропилата натрия (127,8 г, 1,56 моль) вместо трет-бутилата натрия. Получают 147,8 г (72,7%) имазапира (I) в виде белого кристаллического порошка.
Пример 7.
Способ получения имазапира осуществлялся аналогично примеру 2 с загрузкой 170,0 г (1,02 моль) кислоты (II). Отличие состояло в использовании н-пропанола вместо этанола, о-ксилола вместо этилбензола, изопропилата калия (197,4 г, 2,01 моль) или неопентилата калия (254,4 г, 2,01 моль) вместо трет-бутилата калия. Промежуточно в синтезе получают 10-15%-ный раствор дипропилового эфира (IVв) (выход ~95% в расчете на кислоту (II) по данным ГЖХ) в безводном о-ксилоле. Получают 202,0 г (76,0%) имазапира (I) в виде белого кристаллического порошка.
В результате использования предлагаемого способа получения имазапира (I) удается улучшить технико-экономические показатели технологического процесса его производства из 2,3-пиридиндикарбоновой кислоты (II) за счет использования в синтезе промежуточных диэфиров 2,3-пиридиндикарбоновой кислоты (IV) без выделения, позволяющего а) сократить количество технологических операций процесса и его продолжительность; б) упростить его аппаратурное оформление; в) получать целевой продукт высокого качества с выходом 65-78% в зависимости от условий проведения процесса; г) регенерировать использованные растворители с частичным их возвратом в процесс, что сокращает отходы производства.
Изобретение относится к области получения пестицидов, более конкретно - к способу получения имазапира, являющегося неселективным гербицидом широкого спектра действия из класса имидазолинонов и имеющего химическую структуру (2-(4-метил-5-оксо-4-пропан-2-ил-1Н-имидазол-2-ил)пиридин-3-карбоновой кислоты
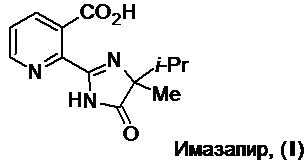
Способ получения имазапира включает первоначальную этерификацию 2,3-пиридиндикарбоновой кислоты низшим спиртом в присутствии минеральной кислоты при нагревании с последующей отгонкой избытка низшего спирта. Далее реакционную массу отрабатывают водным раствором основания и осуществляют экстракцию продукта реакции ароматическим растворителем с получением раствора соответствующего диэфира 2,3-пиридиндикарбоновой кислоты в этом растворителе. Затем осуществляют корректировку концентрации диэфира 2,3-пиридиндикарбоновой кислоты и обезвоживание путем удаления части растворителя азеотропной отгонкой при атмосферном или пониженном давлении. Далее осуществляют введение полученного безводного раствора с необходимой концентрацией диэфира 2,3-пиридиндикарбоновой кислоты, без фактического его выделения в индивидуальном виде, в реакцию с 2-амино-2,3-диметилбутанамидом в присутствии алкоголята щелочного металла в атмосфере инертного газа или на воздухе без доступа влаги с получением целевого продукта, его выделением и очисткой. В качестве низшего спирта используют метиловый, этиловый или н-пропиловый спирт. В качестве минеральной кислоты используют серную или фосфорную кислоту. В качестве основания используют карбонат натрия или калия, гидрокарбонат натрия или калия или гидроксид натрия, калия или аммония. В качестве ароматического растворителя используют толуол, этилбензол, кумол, о-, м- или п-ксилол или их смесь или о-, м- или п-диэтилбензол или их смесь. В качестве алкоголята щелочного металла используют метилат натрия или калия, этилат натрия или калия, изопропилат натрия или калия, трет-бутилат натрия или калия, неопентилат натрия или калия. В качестве инертного газа используют азот или аргон. Предлагаемый способ получения имазапира обеспечивает улучшение технико-экономических показателей технологического процесса производства имазапира из 2,3-пиридиндикарбоновой кислоты, сокращение количества технологических операций процесса и его продолжительности, упрощение его аппаратурного оформления, получение целевого продукта высокого качества с выходом 65-78%, возможность регенерации использованного растворителя с частичным возвратом его в процесс, что сокращает отходы производства. 6 з.п. ф-лы, 7 пр.
1. Способ получения имазапира, заключающийся в первоначальной этерификации 2,3-пиридиндикарбоновой кислоты низшим спиртом в присутствии минеральной кислоты при нагревании с последующей отгонкой избытка низшего спирта, обработкой реакционной массы водным раствором основания и экстракцией продукта реакции ароматическим растворителем с получением раствора соответствующего диэфира 2,3-пиридиндикарбоновой кислоты в этом растворителе, для которого осуществляют корректировку концентрации и обезвоживание путем удаления части растворителя азеотропной отгонкой при атмосферном или пониженном давлении, с последующим введением полученного безводного раствора с необходимой концентрацией диэфира 2,3-пиридиндикарбоновой кислоты, без фактического его выделения в индивидуальном виде, в реакцию с 2-амино-2,3-диметилбутанамидом в присутствии алкоголята щелочного металла в атмосфере инертного газа или на воздухе без доступа влаги с получением целевого продукта, его выделением и очисткой.
2. Способ по п. 1, отличающийся тем, что в качестве низшего спирта используют метиловый, этиловый или н-пропиловый спирт.
3. Способ по п. 1, отличающийся тем, что в качестве минеральной кислоты используют серную или фосфорную кислоту.
4. Способ по п. 1, отличающийся тем, что в качестве основания используют карбонат натрия или калия, гидрокарбонат натрия или калия или гидроксид натрия, калия или аммония.
5. Способ по п. 1, отличающийся тем, что в качестве ароматического растворителя используют толуол; этилбензол; кумол; о-, м- или п-ксилол или их смесь или о-, м- или п-диэтилбензол или их смесь.
6. Способ по п. 1, отличающийся тем, что в качестве алкоголята щелочного металла используют метилат натрия или калия, этилат натрия или калия, изопропилат натрия или калия, трет-бутилат натрия или калия, неопентилат натрия или калия.
7. Способ по п. 1, отличающийся тем, что в качестве инертного газа используют азот или аргон.
| US 4518780 A1, 21.05.1985 | |||
| US 4474962 A1, 02.10.1984 | |||
| US 4758667 A1, 19.07.1988 | |||
| US 5973154 A1, 26.10.1999 | |||
| ЛИНИЯ ПОЛУЧЕНИЯ ИМИДАЗОЛА | 1996 |
|
RU2127262C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФЕНИЛИМИДАЗОЛИДИНОНОВ | 1972 |
|
SU453839A3 |
| Способ получения производных имидазола | 1972 |
|
SU450805A1 |

