Настоящее изобретение относится к способу получения производных хроманола, в частности к способу получения соединений, принадлежащих к семейству витамина Е.
Предшествующий уровень техники настоящего изобретения
Витамин Е является самым важным жирорастворимый антиоксидантом в биологических системах. Термин витамин Е включает все производные токола и токотриенола, обладающие биологической активностью (2R)-2,5,7,8-тетраметил-2-[(4R,8R)-4,8,12-триметилтридецил]-3,4-дигидро-2Н-хромен-6-ола (1.1), который является наиболее важным витамином Е для здоровья человека (см., например, W. Bonrath et al., Angew. Chem. Int. ed., 2012, 51, 12960 - 12990; T. Netscher, Vitamins and Hormons, 2007, Elsevier Inc. volume 76, 155).
Встречающийся в природе витамин Е охватывает соединения токоферола формул I.1 - I.4 (α-, β-, γ- и δ-токоферол), а также соединения токотриенола формул I.5 - I.8 (α-, β-, γ- и δ-токотриенол).



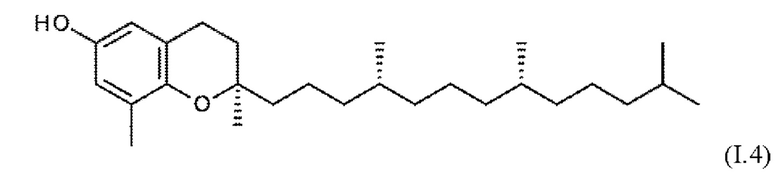

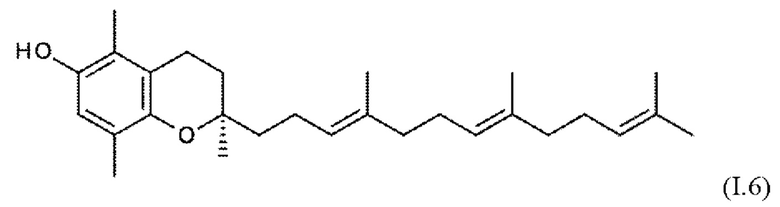
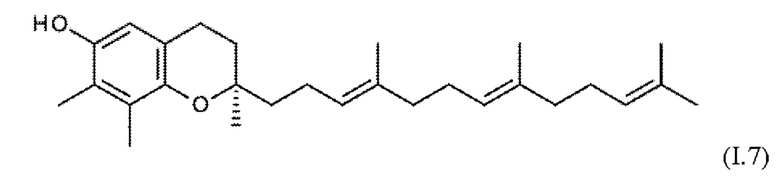

Эти встречающиеся в природе соединения существуют в форме отдельных изомеров, т.е. α-, β-, γ- и δ-токоферолы (I.1 - I.4) имеют конфигурацию 2R, 4R, 8R, и соответствующие α, β-, γ- и δ-токотриенолы (I.5 - I.8) присутствуют в виде изомеров 2R, 3Е, 7Е.
Промышленным образом α-токоферол в основном производят в форме полностью рацемического α-токоферола (all-rac-I.1) и ацетата all-rac-α-токоферола (all-rac I.1a), которые представляют собой эквимолярные смеси всех 8 возможных стереоизомеров. Как правило, полностью рацемический α-токоферол синтезируют путем конденсации триметилгидрохинона (III.1) с полностью рацемическим изофитолом (полностью рацемический изофитол), как показано на схеме 1. Эта реакция конденсации включает алкилирование по Фриделю-Крафтсу триметилгидрохинона (III.1) с последующей реакцией замыкания цикла. Полученный таким образом полностью рацемический α-токоферол (all-rac-1.1) затем превращают в более стабильную ацетатную форму (all-rac-I.1a) путем эстерификации ангидридом уксусной кислоты.
Схема 1: промышленный синтез полностью рацемического α-токоферола (ацетат).

Другие токоферолы, т.е. β-, γ- и δ-токоферол, а также соответствующие токотриенолы, в частности α-токотриенол, обычно производят аналогичным образом.
За последние десятилетия было разработано большое количество способов получения α-токоферола. Ключевой стадией во многих из этих способов является алкилирование по Фриделю-Крафтсу соответствующих предшественников гидрохинона, как показано на схеме 1, которое проводят в присутствии катализатора Фриделя-Крафтса.
Обычно для этой реакции Фриделя-Крафтса в качестве катализаторов используют сильные кислоты Льюиса, такие как хлорид цинка, хлорид алюминия, хлорид олова, хлорид железа, тетрахлорид титана или трифторэтерат бора, а также комбинации сильных кислот Льюиса и сильных кислот Бренстеда, таких как соляная кислота, серная кислота и фосфорная кислота.
В WO 9728151, например, описана реакция 2,3,5-триметилгидрохинона с изофитолом с получением α-токоферола в присутствии циклических карбонатных растворителей посредством применения гомогенных кислот Бренстеда и кислот Льюиса, таких как ортоборная кислота, щавелевая кислота, винная кислота, лимонная кислота или трифторэтерат бора, при повышенных температурах реакции в интервале от 145 до 155°С.
Однако применение этих катализаторов связано с рядом недостатков. Прежде всего, они могут способствовать образованию побочных продуктов (например, использование щавелевой кислоты, винной кислоты или лимонной кислоты обычно увеличивает образование нежелательных фитадиенов). Кроме того, эти катализаторы, как правило, не подлежат восстановлению и повторному использованию из-за их нестабильности по отношению к воде. Кроме того, катализаторы используются в стехиометрических количествах или с высокой каталитической загрузкой. Кроме того, эти катализаторы обычно являются коррозионными, и часто образуются отходы, содержащие тяжелые металлы, такие как цинк или олово, и хлориды.
Чтобы преодолеть эти недостатки, гетерогенные катализаторы были применены в качестве катализатора алкилирования по Фриделю-Крафтсу.
В Odinokov et al., ARKIVOC 2003, (xiii), 101-118 and Scegolev et al., UDK: 547.814.I.07 1982, VINITI 7.09.82, No. 4780-82, например, описано использование цеолитных катализаторов, такие как Tseokar-10 или ASNC-ZP, в реакции гидрохинонов с третичными изопреноидными аллиловыми спиртами. Недостатком использования цеолитов является то, что реакции необходимо проводить при высоких разбавлениях, и что эти цеолиты часто не являются коммерчески доступными.
В Y. Tachibana, Bull. Chem. Soc. Japan, 1977, 50 (9), 2477, описано использование обработанных хлоридом цинка или хлоридом олова сильно кислотных ионообменных смол, таких как амберлист 15, в качестве катализатора в реакции триметилгидрохинона с изофитолом. Однако эти катализаторы обычно имеют низкую каталитическую активность, и образуются отходы, содержащие тяжелые металлы и хлориды.
ЕР 677520 А1 и Matsui et al., Bull. Chem. Soc. Japan, 1996, 69, 137, описывают использование ионообменного бентонита, монтмориллонита или сапонита путем обработки хлоридом скандия или солями других металлов, таких как иттрий, лантан и т.д., в качестве катализатора в реакции триметилгидрохинона с изофитолом, недостаток состоит в том, что требуется большое количество катализатора.
В DE 2404621 описан способ получения α-токоферола путем взаимодействия триметилгидрохинона с фитолом, изофитолом или его производным с использованием твердого кислотного катализатора, имеющего определенную кислотную силу. Среди прочего, в качестве подходящих катализаторов упоминаются встречающиеся в природе минералы, которые проявляют кислотность, такие как кислая глина, бентонит, каолин или морденит. В конкретном примере в качестве катализатора используется бентонит, дающий желаемый α-токоферол с выходом 51,8%. Также здесь требуются большие количества катализатора и получаемые выходы являются умеренными.
2,3,5-Триметилгидрохинон (III. 1), который обычно используется в качестве ароматической структурной единицы в промышленном синтезе полностью рацемического α-токоферола, обычно получают восстановлением 2,3,5-триметилхинона (II. 1), как показано на схеме 2.
Схема 2:

Разработано множество способов восстановления этих хинонов до соответствующих гидрохинонов.
В ЕР 0264823 В2, например, описан способ получения 2,3,5-триметилгидрохинона каталитическим гидрированием 2,3,5-триметилхинона в присутствии катализатора гидрирования, выбранного из металлов платиновой группы, нанесенных на оксид кремния, содержащий щелочные металлы, в качестве носителя. В качестве подходящего растворителя для этих реакций каталитического гидрирования предлагаются алифатические спирты, ароматические спирты, кетоны, органические кислоты, сложные эфиры органических кислот, простые эфиры, циклические простые эфиры, лактоны и их смеси.
Основное внимание в вышеупомянутых разработках уделяется идентификации подходящих условий реакции, которые позволяют избежать вредных для окружающей среды реагентов и катализаторов и упростить процедуру реакции, в частности, упростить выделение продуктов. Таким образом, постоянно ведется поиск новых способов и новых реагентов, альтернативных уже известным.
Сущность изобретения
Таким образом, задача настоящего изобретения состоит в обеспечении способа получения производных витамина Е, исходя из соответствующих бензохиноновых предшественников, который является эффективным и обеспечивает получение желаемых продуктов с высоким выходом и селективностью без необходимости применения дорогих, коррозионных и/или экологически вредных катализаторов и растворителей. Способ должен быть простым, и следует избегать трудоемких процедур очистки. Кроме того, способ должен быть применим в крупномасштабном производстве, а применяемый катализатор должен подлежать вторичной переработке.
Было неожиданно обнаружено, что эти и другие цели достигаются с помощью способа, который включает каталитическое гидрирование бензохиноновых предшественников до соответствующих гидрохинонов в присутствии карбонатного растворителя с последующим алкилированием по Фриделю-Крафтсу полученных таким образом гидрохинонов.
Соответственно настоящее изобретение относится к способу получения соединения общей формулы I

где
R1, R2 и R3 независимо друг от друга выбраны из водорода и метила,
R4 выбран из водорода и C1-С6-алканоила, и
X выбран из C1-С20-алкила и С2-С20-алкенила,
Включающему следующие стадии:
a) обеспечения соединения хинона общей формулы II,

где R1, R2 и R3 имеют значения, как определено выше,
b) каталитического гидрирования соединения хинона формулы II, обеспеченного на стадии а) в присутствии водорода, катализатора гидрирования и в присутствии а карбонатный растворитель с получением соединения гидрохинона общей формулы III,

где R1, R2 и R3 имеют значения, как определено выше,
c) реакции соединения гидрохинона III, обеспеченного на стадии b) с ненасыщенным соединением общей формулы IV.а или IV.b


где
X имеет значение, как определено выше,
Y выбран из ОН, галогена, -O-R11, -S-R12 и -SO2-R12,
R11 выбран из С1-С4-алкила, С1-С4-алканоила и трифторацетила, и
R12 выбран из C1-С6-алкила, трифторметила и фенила, где фенил является незамещенным или замещен 1, 2, 3, 4 или 5 радикалами, выбранными из галогена и метила,
в присутствии а катализатор конденсации, и
d) если R4 выбран из C1-С6-алканоила, реакции продукта конденсации, полученного на стадии с) с С2-С7карбоновой кислотой или с ангидридом С1-С7карбоновой кислоты в присутствии катализатор эстерификации,
или реакции продукта конденсации, полученного на стадии с), с активированной С2-С7карбоновой кислотой в присутствии основания.
Подробное описание настоящего изобретения
В контексте настоящего изобретения термин «алкил», как применяется в настоящем документе, относится к линейному или разветвленному насыщенному углеводородному радикалу, имеющему от 1 до 3 ("C1-С3-алкил"), от 1 до 4 ("C1-С4-алкил"), от 1 до 6 ("C1-С6 -алкил") или от 1 до 20 ("С1-С20-алкил") атомов углерода. C1-С3-Алкил представляет собой метил, этил, пропил и изопропил. C1-С4-Алкил дополнительно представляет собой н-бутил, 1-метилпропил (втор-бутил), 2-метилпропил (изобутил) или 1,1-диметилэтил (трет-бутил). C1-С6-Алкил также дополнительно представляет собой, например, н-пентил, 1-метилбутил, 2-метилбутил, 3-метилбутил, 1,2-диметилпропил, н-гексил, 1-метилпентил, 2-метилпентил, 3-метилпентил, 4-метилпентил или 1,3-диметилбутил. С1-С20-Алкил также дополнительно представляет собой, например, н-гептил, 1-метилгексил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,4-диметилпентил, н-октил, изооктил, 2-этилгексил, н-нонил, изононил, н-децил, изодецил, 2-пропилгептил, н-ундецил, изоундецил, 2,4-диметилнонил, н-додецил, изододецил, н-тридецил, изотридецил, тетрадецил, изотетрадецил, гексадецил, изогексадецил, 4,8,12-триметидтридецил, октадецил, изооктадецил и тому подобное.
В контексте настоящего изобретения термин "С1-С4-алканоил" означает С1-С4-алкильную группу, как определено выше, присоединенную через карбонильную [(С=O)] группу к остальной части молекулы. Термин "C1-С6-алканоил" означает C1-C6-алкильную группу, как определено выше, присоединенную через карбонильную [(С=O)] группу к остальной части молекулы. С1-С4-алканоил представляет собой метилкарбонил, этилкарбонил, н-пропилкарбонил, изопропилкарбонил, н-бутил карбонил, 1-метилпропилкарбонил, 2-метилпропилкарбонил или 1,1-диметилэтилкарбонил. C1-C6-алканоилдополнительно представляет собой, например, н-пентилкарбонил, 1-метилбутилкар бонил, 2-метилбутилкарбонил, 3-метилбутилкарбонил, 1,2-диметилпропилкарбонил, н-гексил-карбонил, 1-метилпентилкарбонил, 2-метилпентилкарбонил, 3-метилпентил карбонил, 4-метилпентил кар бонил или 1,3-диметилбутилкарбонил.
Термин "галоген" означает в каждом случае фтор, бром, хлор или иод, в частности фтор, хлор или бром. Галоген в качестве заместителя при фениле предпочтительно представляет собой Cl или Br.
Соединения, получаемые способом согласно настоящему изобретению, представляют собой соединение общей формулы I

где
R1, R2 и R3 независимо друг от друга выбраны из водорода и метила,
R4 выбран из водорода, C1-С6-алканоила, и
X выбран из C1-С20-алкила и С2-С20-алкенила.
Благодаря своей структуре соединения (I) могут присутствовать в форме чистых энантиомеров или диастереомеров, а также в форме смесей энантиомеров или диастереомеров.
Термин «стереоизомеры» охватывает оптические изомеры, такие как энантиомеры или диастереоизомеры, последние существуют благодаря более чем одному стереогенному центру в молекуле. Соединения формулы (I), где X не является метилом, имеют по меньшей мере один стереогенный центр, а именно атом углерода в положении 2 хроманового кольца. Кроме того, в соединениях (I) радикал X может также иметь по меньшей мере один стереогенный центр, например, если X выбран из 4,8-диметилнонила или 4,8,12-триметилтридецила. Настоящее изобретение относится как к чистым энантиомерам или диастереоизомерам соединений (I), так и к их смесям.
Кроме того, если радикал X выбран из С2-С20-алкенила, соединения (I) также могут иметь по меньшей мере одну двойную связь, которая может иметь Е- или Z-конфигурацию, например, если X представляет собой изопренильные фрагменты 4,8-диметил-3,7-нонадиенил или 4,8,12-триметил-3,7,11-тридекатриенил. Таким образом, настоящее изобретение также относится к соединениям (I), в которых двойная связь (связи), если присутствует, имеет/имеют чистую Е- или Z-конфигурацию и/или присутствует/присутствуют в виде E/Z-смеси (смесей).
Предпочтительно, в соединениях (I) и (III) способа согласно настоящему изобретению, радикалы R1, R2 и R3 имеют следующие значения:
R1 представляет собой метил,
R2 представляет собой метил, и
R3 представляет собой метил,
или
R1 представляет собой метил,
R2 представляет собой водород, и
R3 представляет собой метил,
или
R1 представляет собой водород,
R2 представляет собой метил и
R3 представляет собой метил,
или
R1 представляет собой водород,
R2 представляет собой водород, и
R3 представляет собой метил.
В частности, в соединениях (I) и (III) радикалы R1, R2 и R3 представляют собой метил.
Предпочтительно, в соединениях (I) способа согласно настоящему изобретению, радикал R4 выбран из водорода и С1-С4-алканоила, более предпочтительно из водорода или С1-С2-алканоила, в частности из водорода и этаноила.
Предпочтительно, в соединениях (I), (IV.a) и (IV.b) способа согласно настоящему изобретению, фрагмент X выбран из метила или имеет одно из следующих значений Х-1 - Х-6



где * показывает точку присоединения к хромановому кольцу.
В частности, в соединениях (I), (IV.a) и (IV.b) согласно этому первому варианту осуществления, фрагмент X выбран из (Х-3) или (Х-6)
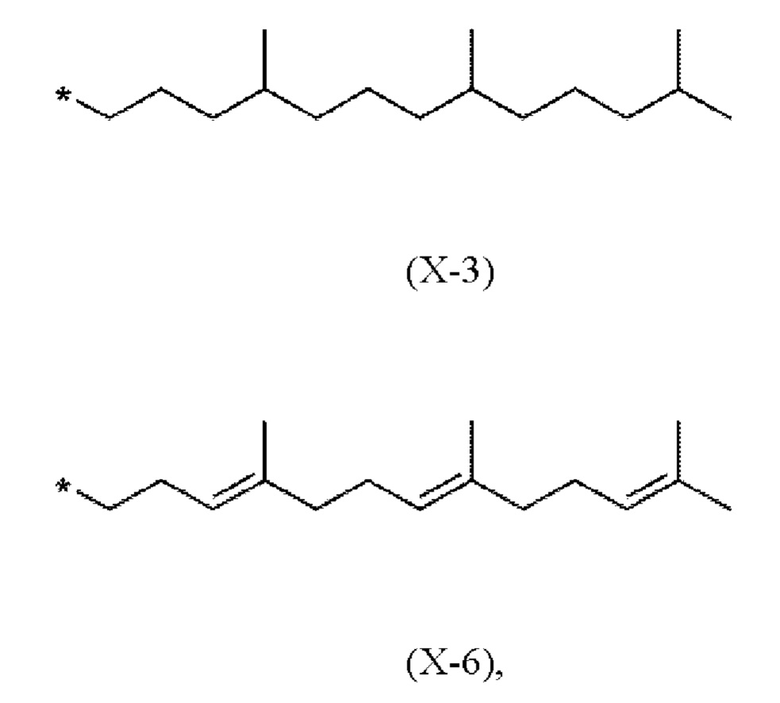
где * показывает точку присоединения к остальной части молекулы.
Согласно предпочтительному варианту осуществления, в соединениях (I)
R1 представляет собой метил,
R представляет собой метил, и
R3 представляет собой метил,
или
R1 представляет собой метил,
R2 представляет собой водород, и
R3 представляет собой метил,
или
R1 представляет собой водород,
R2 представляет собой метил и
R3 представляет собой метил,
или
R1 представляет собой водород,
R представляет собой водород, и
R3 представляет собой метил,
R4 выбран из водорода и С1-С4-алканоила, и
X выбран из метила или радикала формул Х-1 - Х-6.
Согласно более предпочтительному варианту осуществления, в соединениях (I)
R1 представляет собой метил,
R2 представляет собой метил, и
R3 представляет собой метил,
или
R1 представляет собой метил,
R представляет собой водород, и
R представляет собой метил,
или
R1 представляет собой водород,
R представляет собой метил, и
R3 представляет собой метил,
или
R1 представляет собой водород,
R2 представляет собой водород, и
R3 представляет собой метил,
R4 выбран из водорода и С1-С2-алканоила, в частности из водорода и этаноила, и
X представляет собой метил или имеет одно из следующих значений (Х-3) или (Х-6)

где * показывает точку присоединения к остальной части молекулы.
Согласно конкретному варианту осуществления в соединениях (I)
R1, R2 и R3 представляют собой метил,
R4 выбран из водорода и этаноила, и
X имеет одно из следующих значений (Х-3) или (Х-6)
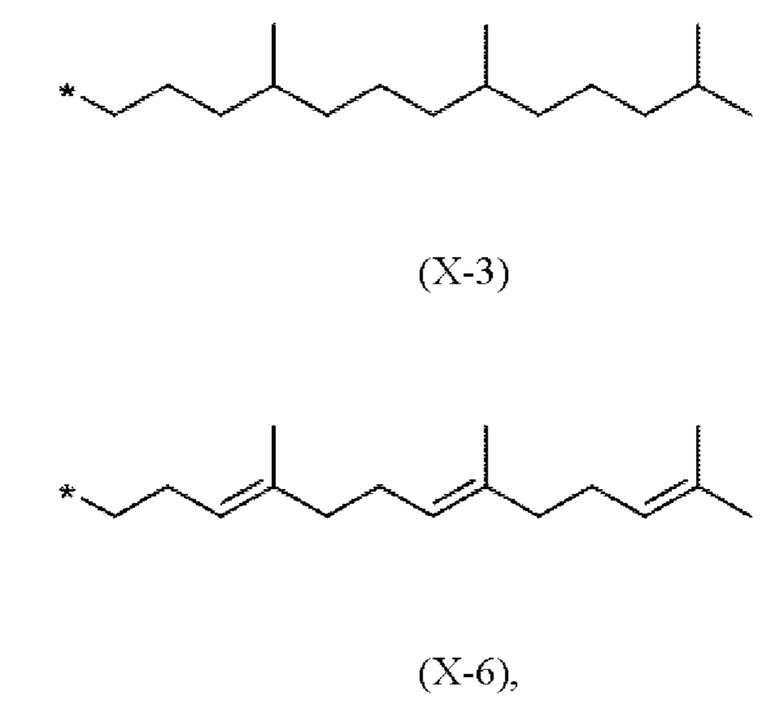
где * показывает точку присоединения к остальной части молекулы.
Предпочтительно, в соединениях (IV.a) и (IV.b), радикал Y выбран из ОН, Cl, Br, I, O-R11, -S-R12 и -SO2-R12, где
R11 выбран из C1-C4-алканоила и трифторацетила, и
R12 выбран из C1-С3-алкила, трифторметила, фенила, 4-метилфенила и пентафторфенила.
Более предпочтительно, в соединениях (IV.a) и (IV.b), радикал Y выбран из ОН, Br, O-R11, -S-R12 и -SO2-R12, где
R11 выбран из ацетила и трифторацетила, и
R12 выбран из метила, трифторметила и 4-метилфенила.
В частности, в соединениях (IV.a) и (IV.b), радикал Y выбран из ОН, OR11 и -SO2-R12, где
R11 выбран из ацетила, и
R12 выбран из метила, трифторметила и 4-метилфенила.
Стадия а):
Стадия а) согласно настоящему изобретению включает обеспечение соединения хинона общей формулы (II).
Эти соединения хинона являются либо химически доступными, либо могут быть получены из легко доступных предшественников способами, известными из уровня техники.
Например, 2,3,5-триметилхинон можно получить каталитическим метилированием м-крезола до 2,3,6-триметилфенола (стадия i) с последующим окислением полученного таким образом 2,3,6-триметилфенола с использованием окисляющего агента (стадия ii)), как показано на схеме 3.
Схема 3:
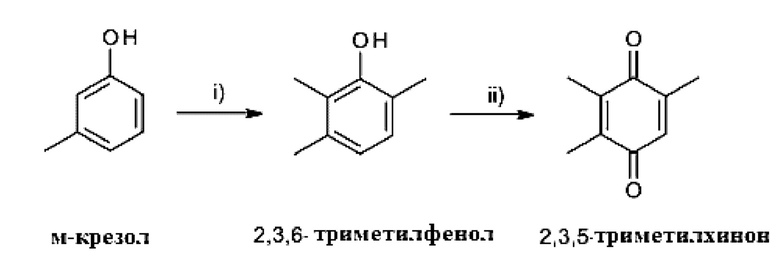
Каталитическое метилирование на стадии i) обычно проводят с метанолом или простым диметиловым эфиром в качестве алкилирующего агента в присутствии катализатора на основе оксида металла, такого как, например, оксид алюминия, оксид кремния, оксид магния, оксид кальция, оксид бария, оксид железа, оксид хрома, оксид цинка, оксид марганца, оксид циркония, оксид тория и т.п., а также катализаторов на основе смешанных оксидов металлов.
Индивидуальные условия реакции каталитического метилирования фенолов, таких как м-крезол, хорошо известны специалисту в данной области техники.
Окисление на стадии ii) можно проводить в соответствии со стандартными процедурами, которые хорошо известны специалисту в данной области техники. Как правило, окисление проводят в присутствии окисляющего агента, необязательно с помощью соли металла или катализатора на основе благородного металла. В принципе, все окисляющие агенты, известные специалисту как способные окислять фенолы до хинонов, могут быть использованы при окислении на стадии ii).
Подходящими окисляющими агентами являются, например:
- пероксид водорода, кислород или кислородсодержащий газ в присутствии каталитических количеств солей металлов, таких как Mg (II) -, Са (II) -, Ва (II) -, Cu (II) -, Fe (II) -, Cr (II) -, Mn (II) -, Со (II) -, Ni (II) -, Zn (II) -, сульфатов или хлоридов, а также смесей этих солей металлов или катализатор на основе благородного металла, такого как, например, рутениевый, родиевый, платиновый или палладиевый катализатор;
- минеральные кислоты, такие как азотная кислота, серная кислота, хлорная кислота, хлорноватистая кислота, перхлорная кислота, йодноватая кислота или периодная кислота; или
- органические пероксикислоты, такие как пербензойная кислота или мета-хлорпербензойная кислота.
Подходящие условия реакции для этой реакции окисления хорошо известны специалисту в данной области техники.
Стадия b):
Стадия b) согласно настоящему изобретению включает каталитическое гидрирование соединения хинона формулы II, обеспеченного на стадии а), в присутствии водорода, катализатора гидрирования и в присутствии карбонатного растворителя с получением соединения гидрохинона общей формулы III,

где R1, R2 и R3 имеют значения, как определено выше.
На стадии b) бензохинон (II) обычно растворяют в карбонатном растворителе и вводят в контакт с водородом и катализатором гидрирования.
Катализаторы гидрирования, которые можно использовать в каталитическом гидрировании на стадии b), представляют собой обычно используемые в данной области техники для катализа гидрирования органических соединений. Некоторые примеры из них включают хлорид палладия на активированном угле, активированный никель, никель-оксид никеля, платину-оксид платины, платиновую чернь, платину на активированном угле, хромит меди, никель Ренея, палладий, палладиевую чернь, палладий на активированном угле, палладиевый сплав, никель, оксид алюминия, пропитанный медью, активированный оксид алюминия, медь Ренея, хром, ванадий, молибден и т.п.
Предпочтительно катализатор гидрирования, применяемый на стадии b), представляет собой гетерогенный катализатор гидрирования, более предпочтительно нанесенный на металлический катализатор.
Подходящими носителями являются, например, углерод, углеродная сажа, активированный уголь, графит, оксид алюминия, оксид кремния, диоксид титана, диоксид циркония, диоксид церия, оксид лантана (III), оксид цинка, силикаты, асбест, карбид кремния, карбонат кальция, карбонат магния, сульфат магния, карбонат бария, сульфат бария, цеолиты, диатомовая земля и их смеси.
Еще более предпочтительно катализатор гидрирования, применяемый на стадии b), представляет собой катализатор на углеродном носителе.
В частности, катализатор гидрирования, применяемый на стадии b), представляет собой катализатор на основе благородного металла на углеродном носителе, то есть катализатор на основе благородного металла, содержащий материал носителя на основе углерода (С), например, активированный уголь.
В принципе, подходящие материалы носителя на основе углерода представляют собой все углеродные материалы, известные специалисту в данной области техники для таких применений. Подходящие материалы-носители на основе углерода включают, например, углерод, сажу, активированный уголь и графит. Материалы носителя могут быть использованы в форме формованных тел, гранул, нитей, пеллет, сколов, таблеток или кусочков. Площадь поверхности по БЭТ материалов носителя (25°С) обычно находится в диапазоне от 1 до 10 000, предпочтительно от 10 до 5000 м2/г, но в большинстве случаев не является критичной для способа согласно настоящему изобретению.
Подходящие благородные металлы выбраны, например, из Pt, Pd, Ru, Rh и Ir.
В частности, катализатор гидрирования, применяемый на стадии b), выбирают из Pd-катализатора, нанесенного на углеродный носитель, а именно из Pd на активированном угле.
Содержание металла в металлическом катализаторе на носителе, используемом на стадии b) согласно настоящему изобретению, обычно составляет от 0,1% до 20 мас. %, предпочтительно в интервале от 1% до 15 мас. %, на основе общей массы катализатора на основе благородного металла, нанесенного на углерод.
Обычно количество катализатора гидрирования, используемого на стадии b) согласно настоящему изобретению, составляет от 0,05% до 20 мас. %, предпочтительно в интервале от 0,1% до 10 мас. %, более предпочтительно в интервале от 0,2% до 5 мас. %, на основе количества хинона, присутствующего в реакционной смеси.
Каталитическое гидрирование можно проводить при атмосферном давлении или при повышенном давлении. Более высокие давления обычно приводят к более высокой скорости гидрирования. Чрезвычайно высокое давление не требуется, поскольку бензохиноны легко восстанавливаются. Каталитическое гидрирование на стадии b) обычно проводят при давлении в диапазоне от 1 до 100 бар, предпочтительно в диапазоне от 1 до 50 бар, особенно в диапазоне от 2 до 30 бар.
Гидрирование проводят при температуре, достаточно высокой, чтобы способствовать восстановлению бензохинона, но не настолько высокой, чтобы вызвать разложение реагентов, реакционной среды или продуктов. Подходящая температура реакции обычно находится в интервале от 20 до 150°С, предпочтительно в диапазоне от 30 до 120°С и особенно в диапазоне от 40 до 100°С.
Согласно настоящему изобретению реакцию каталитического гидрирования на стадии b) проводят в присутствии карбонатного растворителя, выбранного из органических карбонатов.
Предпочтительно, карбонатный растворитель выбран из циклических или линейных карбонатов общей формулы VI.а и VI.b

где
R15, R16 и R17 независимо друг от друга выбраны из водорода, метила и этила, в частности из водорода и метила,
R18 выбран из водорода, фенила и C1-C51-алкила, где C1-C15-алкил является незамещенным или замещен 1, 2, или 3 радикалами, выбранными из С1-С3-алкокси, полиалкиленоксида, фенила и фенокси, в частности из водорода, фенила, C1-С3-алкила и бензила, и
R19 независимо друг от друга выбраны из С1-С4-алкила, в частности из этила и н-пропила.
Более предпочтительно, карбонатный растворитель выбран из циклических или линейных карбонатов общей формулы VI.а и VI.b

где
R15, R16 и R17 независимо друг от друга выбраны из водорода и метила,
R18 выбран из водорода, метила, этила, фенила и бензила, и
R19 независимо друг от друга выбраны из этила и н-пропила.
Среди этих карбонатных растворителей предпочтительны те карбонатные растворители, которые имеют точку кипения, равную по меньшей мере 100°С, более предпочтительно по меньшей мере 120°С, в частности по меньшей мере 140°С.
В частности, карбонатный растворитель выбран из этиленкарбоната, пропиленкарбоната, бутиленкарбоната, 2,3-пропиленкарбоната, изобутиленкарбоната, диэтилкарбоната и ди-н-пропилкарбоната.
Эти циклические и ациклические карбонаты не вызывают никаких токсикологических проблем, что очень важно для получения соединений общих формул I и II. Кроме того, эти растворители хорошо биоразлагаемы.
Реакцию на стадии b) можно дополнительно проводить в присутствии аполярного углеводородного растворителя (HS).
Соответственно, согласно альтернативному варианту осуществления настоящего изобретения, стадию b) проводят в смеси растворителей, состоящей из по меньшей мере одного карбонатного растворителя, как определено выше, и по меньшей мере одного аполярного углеводородного растворителя (HS).
Предпочтительно, аполярный углеводородный растворитель (HS), если присутствует, выбран из следующих групп:
HS.1 линейных и разветвленных алканов, имеющих от 5 до 15 атомов углерода, как например пентан, гексаны, гептнаны, октаны, нонаны, деканы, лигроин и петролейный эфир;
HS.2 циклоалканов, имеющих от 5 до 10 атомов углерода, как например циклогексан;
HS.3 ароматических углеводородов, имеющих от 6 до 12 атомов углерода, как например бензол, толуол, ксилолы, этилбензол и тетралин;
и их смесей.
Более предпочтительно, аполярный углеводородный растворитель (HS) выбран из групп HS.1 и HS.2.
В частности, аполярный углеводородный растворитель (HS) выбран из гексана, циклогексана, гептана, октана и нонана, более конкретно из гептана и октана.
Согласно этому альтернативному варианту осуществления содержание карбонатного растворителя в этой смеси растворителей обычно составляет от 35 до 99 мас. %, предпочтительно в интервале от 50 до 99 мас. %, в частности интервале от 50 до 90 мас. %, на основе общей массы смеси растворителей.
Соответственно, массовое соотношение карбонатного растворителя и HS, используемое на стадии b), обычно находится в интервале от 1: 3 до 100: 1, предпочтительно в интервале от 1: 1 до 100: 1, в частности в интервале от 1: 1. до 10: 1.
Согласно этому альтернативному варианту осуществления предпочтительными являются карбонатные растворители, которые не смешиваются или смешиваются лишь в небольшой степени с по меньшей мере одним аполярным углеводородным растворителем (HS), что означает, что стадию b) проводят в двухфазной смеси растворителей, состоящей из фазы карбонатного растворителя и HS-фаза. В этой связи термин «умеренно смешиваемый» означает, что менее 5 мас. %, предпочтительно менее 2 мас. %, более предпочтительно менее 1 мас. %, в частности менее 0,5 мас. % полярного апротонного растворителя (PS) присутствует в HS-фазе.
Согласно этому альтернативному варианту осуществления предпочтительны смеси растворителей, состоящие по меньшей мере из одного карбонатного растворителя и по меньшей мере одного аполярного углеводородного растворителя группы HS. 1.
В частности, согласно этому альтернативному варианту осуществления, смесь растворителей состоит из по меньшей мере одного карбонатного растворителя, выбранного из этиленкарбоната, пропиленкарбоната, бутиленкарбоната, 2,3-пропиленкарбоната, изобутиленкарбоната, диэтилкарбоната и ди-н-пропил карбоната, и по меньшей мере одного аполярного углеводородного растворителя (HS), выбранного из гептана и октана.
Согласно конкретному варианту осуществления настоящего изобретения стадию b) проводят в по меньшей мере одном карбонатном растворителе (PS), как определено выше.
Каталитическое гидрирование можно проводить в различных реакторах, известных для этой цели, таких как последовательный петлевой реактор, как описано в US 5,756,856, но также и в более простых реакторах, как описано, например, в DE 2008128. Предпочтение отдается реакторам с неподвижным слоем, в частности реакторам с орошаемым слоем.
Продукт реакции, то есть соединение формулы (III), полученный на стадии b), обычно перерабатывают, отфильтровывая катализатор гидрирования. Полученную таким образом реакционную смесь можно непосредственно использовать на стадии с) способа согласно настоящему изобретению.
Если желательно, полученная таким образом реакционная смесь может быть подвергнута дальнейшей переработке обычным способом, например, путем смешивания с водой, разделения фаз и, при необходимости, очистки неочищенных продуктов с помощью хроматографических методов, дистилляции или перекристаллизации.
Согласно предпочтительному варианту осуществления согласно настоящему изобретению, реакционную смесь, полученную на стадии b), применяют непосредственно в реакции на стадии с) способа согласно настоящему изобретению, после удаления катализатора гидрирования.
Стадия с):
Стадия с) согласно настоящему изобретению включает реакцию соединения (III), обеспеченного на стадии b), с ненасыщенным соединением общей формулы (IV.a) или (IV.b)
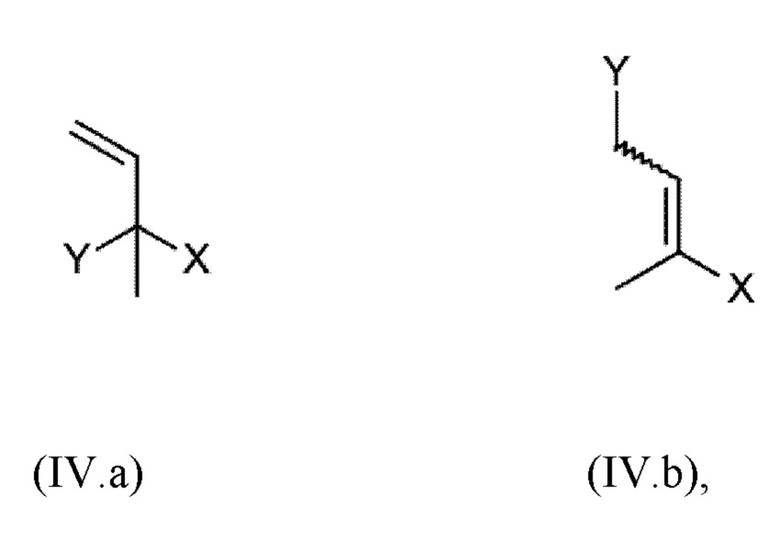
где
X имеет одно из значений, указанных выше,
Y выбран из ОН, галогена, -O-R11, -S-R12 и -SO2-R12,
R11 выбран из С1-С4-алкила, С1-С4-алканоила и трифторацетила, и
R12 выбран из C1-C6-алкила, трифторметила и фенила, где фенил является незамещенным или замещен 1, 2, 3, 4 или 5 радикалами, выбранными из галогена и метила,
в присутствии катализатора конденсации.
В общем, любые катализаторы конденсации, которые способны катализировать реакции алкилирования по Фриделю-Крафтсу и/или (внутримолекулярные) гидроксалкилирования олефинов (реакция внутримолекулярной циклизации), могут применяться на стадии с) способа согласно настоящему изобретению.
Подходящими традиционными катализаторами конденсации являются, например, кислоты Льюиса, такие как хлорид цинка, хлорид алюминия, хлорид олова, хлорид железа, тетрахлорид титана или трифторэтерат бора, кислоты Бренстеда, такие как соляная кислота, серная кислота и фосфорная кислота, и кислотные гетерогенные катализаторы, а также их комбинации.
Согласно предпочтительному варианту осуществления настоящего изобретения, катализатор конденсации, применяемый на стадии с), выбран из кислотного гетерогенного катализатора.
Подходящие кислотный гетерогенный катализатор выбирают, например, из катионообменных смол, например, катионообменные смолы, содержащие группы сульфоновой кислоты, такие как амберлист 15 или Dowex 50W, цеолитов, такие как цеолиты H-BFA, цеолиты H-Y, цеолиты типа пентасил, цеолиты Tseokar-10 или К-10; а также встречающихся в природе минералов, имеющих определенную кислотность, таких как кислая глина, монтмориллонит, бентониты, каолин, сапониты или мордениты.
Предпочтительный катализатор конденсации, применяемый на стадии с) согласно настоящему изобретению, выбирают из силикатных глинистых минералов, содержащих элементы Н, С, О, Si, Al, Mg, Са, Li, Na, K, Fe, Zn, S, F и все их комбинации. Предпочтительные силикатные минералы, которые могут применяться в качестве катализатора конденсации на стадии с) согласно настоящему изобретению, включают, например, пирофиллит, тальк, слюды (например, мусковит, парагонит, флогопит, биотит, лепидолит, циннвальдит, тэниолит, фтор-тетрасиликатную слюду), хрупкие слюды (маргарит, хлоритоид, зейберит, клинтонит), водные слюды, иллиты, хлориты, вермикулиты, смектиты (монтмориллонит, сапонит, нонтронит, бейделлит, сауконит, гекторит, фторгекторит), кандиты, серпентины и/или палыгорскиты (аттапульгит, сепиолит)
Более предпочтительно, катализатор конденсации, применяемый на стадии с) согласно настоящему изобретению, выбран из силикатных глинистых минералов, содержащих монтмориллониты, как описано, например, в Klockmann's textbook of mineralogy, 16th edition, F. Euke Verlag 1978, pages 739-765 и в R.M. Barrer Zeolites and Clay Minerals as Sorbents and Molecular Sieves, Academic Press, и в Y. Izumi, K. Urabe, M. Onaka Zeolite, Clay, and Heteropoly Acid in Organic Reactions, VCH.
В зависимости от их каталитических свойств в их природной или производимой форме, эти гетерогенные катализаторы конденсации могут применяться в необработанной форме или их можно обрабатывать перед использованием, чтобы изменить их каталитические свойства. В этом отношении выражение «обработанный» относится к химической обработке. Обычно химическая обработка гетерогенных катализаторов конденсации включает кислотную обработку, щелочную обработку, обработку солями металлов (катионный обмен) или органическую обработку. Предпочтительно химическая обработка гетерогенного катализатора конденсации включает обработку солями металлов (катионный обмен) или кислотную обработку. Кислотную обработку гетерогенного катализатора конденсации обычно проводят сильными кислотами Бренстеда и/или кислотами Льюиса.
Согласно даже более предпочтительному варианту осуществления настоящего изобретения реакцию на стадии с) проводят в присутствии катализатора на основе обработанного бентонита.
Бентонит образован из высококоллоидной и пластичной глины. Бентонит часто используется в качестве загустителя и наполнителя для красок, в качестве добавки для керамики, а также для товаров для здоровья (например, косметики, пищевых продуктов или фармацевтических препаратов). Бентониты являются хорошими адсорбентами и характеризуются высокой емкостью катионного обмена, сильной способностью к набуханию и низкой проницаемостью.
Обычно основным компонентом бентонита является монтмориллонит, глинистый минерал группы смектита. Монтмориллонит состоит из двух слоев кремниевых тетраэдров с центральным слоем из одного алюминиевого октаэдра между ними. Он имеет гидроксильные группы между слоями, а также на поверхности.
В принципе, все месторождения, содержащие монтмориллонит, как, например, перечисленные в the monograph "The Economics of Bentonite", 8th Edition 1997, Roskill Information Services Ltd, London, можно рассматривать как подходящие источники бентонитов. В зависимости от своего происхождения бентониты могут содержать, помимо монтмориллонита, различные количества различных сопутствующих минералов, как определено выше, и неминеральных компонентов. Такими сопутствующими минералами и неминеральными компонентами являются, в частности, кварц, полевой шпат, каолин, мусковит, цеолиты, оксиды титана, оксиды железа, иллиты, слюдяной кальцит и/или гипс. Предпочтительным сырьем является сырье с высоким содержанием монтмориллонита и, соответственно, низким содержанием вторичных компонентов, поскольку количество чистого монтмориллонита в бентоните определяет его характеристики. Чем выше содержание монтмориллонита в бентоните, тем лучше его характеристики в качестве промышленного сырья. Содержание монтмориллонита можно определить, например, по адсорбции метиленового синего.
Предпочтительные исходные материалы имеют значение метиленового синего по меньшей мере 250 мг/г, предпочтительно по меньшей мере 290 мг/г, в частности по меньшей мере 320 мг/г. Особенно предпочтительным сырьем является такое сырье, в котором обменные катионы состоят из большого процента щелочных металлов, в частности натрия. Что касается эквивалентов заряда, эти исходные материалы содержат по меньшей мере 25%, предпочтительно по меньшей мере 40% одновалентных обменных катионов. Это сырье для натриевых бентонитов встречается в природе, известные источники бентонитов, содержащих натрий, находятся, например, в Вайоминге/США или в Индии, они также известны по своему происхождению как «западные бентониты», «бентониты Вайоминга» или по своим свойствам как «набухающие бентониты». Бентониты с высоким содержанием щелочноземельных катионов, особенно кальция, известны как «суббентониты» или «южные бентониты» и могут быть преобразованы в натрийсодержащие бентониты путем щелочной активации.
Наконец, в принципе также возможно производить подходящие синтетические глинистые минералы, например, путем добавления органических катионов или комплексных катионов металлов (PILC) и использовать их в настоящем изобретении (М.М. Herling et al. Z. Anorg. Allg. Chem. 2014, 640, 3-4. 547-560; G. Poncelet and J. J. Fripiat Handbook of Heterogeneous Catalysis (2nd Edition) 2008, 1, 219-247).
Глинистые минералы природного происхождения могут, помимо минеральных примесей, также содержать неминеральные примеси, особенно соединения углерода. Предпочтительным сырьем являются бентониты с общим содержанием углерода менее 3 мас. %, предпочтительно менее 1 мас. %, особенно предпочтительно менее 0,5 мас. %.
Хорошо известно, что макроскопические свойства и применимость бентонитов тесно связаны с количеством и качеством содержащегося в них монтмориллонита, их значением рН (остаточной кислотностью), размером частиц и их пористой микроструктурой (например, площадью поверхности, пористостью).
Бентониты можно разделить на природные бентониты, то есть необработанные бентониты, и обработанные бентониты (см., например, J. Nones et al., Applied Clay Science, 2015, 105-106, 225-230). Согласно предпочтительному варианту осуществления настоящего изобретения, реакцию на стадии с) проводят в присутствии катализатора на основе обработанного бентонита.
Используемый здесь термин «обработанный бентонит» относится к бентонитам, где структура, текстура и другие свойства бентонита модифицированы химической обработкой и/или термической обработкой. Таким образом, используемый в настоящем документе термин «обработанный бентонит» относится к химически обработанному и/или термически обработанному бентониту. Обычно химическая обработка бентонитов включает кислотную, щелочную или органическую обработку. Бентониты, полученные обработкой кислотой или щелочью, также называются «активированными бентонитами» (бентониты, активированные кислотой, или бентониты, активированные щелочью).
Термин «обработанный кислотой бентонит» или «активированные кислотой бентониты» в контексте настоящего изобретения относится к бентонитам, которые обрабатывают кислотой Бренстеда, например, минеральная кислота, такая как HCl или H2SO4, Н3РО3, HNO3 борная кислота, кремниевая кислота, карбоновой кислоты, например, муравьиная кислота или уксусная кислота, или другими органическими кислотами, такими как трифторуксусная кислота, метансульфоновая кислота, толуолсульфоновая кислота или трифторметансульфоновая кислота. Предпочтение отдается HCl и/или H2SO4 или смесям HCl и/или H2SO4 с другими неорганическими или органическими кислотами. Обычно активированные кислотой бентониты широко используются в качестве отбеливающей земли для обесцвечивания масел.
Также известно, что обработка минеральной кислотой придает поверхностную кислотность глине, что улучшает ее каталитические свойства (P. Komandel, Applied Clay Science, 2016, 131, 84-99; D.A. D'Amico et al. Applied Clay Science, 2014, 99, 254-260). He ограничиваясь конкретной теорией, полагают, что во время кислотной обработки или кислотной активации, соответственно, края силикатных листов глинистых минералов в бентоните открываются, и катионы Al3+ и Mg2+ октаэдрического листа становятся растворимыми. Химия этого процесса активации, где кислотный ион водорода, например кислотный ион водорода из серной кислоты открывает пластинчатую структуру глинистых минералов в бентоните и образует кислотные центры, например, проиллюстрировано в J. Amorim et al. Hydrocarbon Engineering 2016, 21, 11, 83-8. Конечные обработанные кислотой бентониты содержат аморфный, пористый, протонированный и гидратированный диоксид кремния с трехмерной сшитой структурой (Р. Komandel Applied Clay Science, 2016, 131, 84-99).
Способы получения активированных кислотой глинистых минералов, в частности слоистых силикатов, таких как бентониты, хорошо известны в данной области техники; обзор представлен, например, в ЕР0398636 (В1), а подробный способ кислотной активации глинистых минералов, таких как бентониты, можно найти, например, в DE10245198 (А1).
Щелочная обработка бентонитов относится к обработке бентонитов минеральными основаниями, такими как NaOH, КОН или карбонат натрия, или органическими основаниями, такими как аммиак, триметиламин или гидроксиды тетраалкиламмония. Щелочную активацию обычно проводят обработкой карбонатом натрия.
Органическая обработка бентонитов относится к обработке бентонитов органическими соединениями, такими как катионы четвертичного аммония (например, алкиламмоний и α-ω-диалкиламмоний).
Другие органические и неорганические соединения, которые подвергаются обмену с вышеописанными минералами, включают: гидразин, мочевину, формамид, ацетамид, соли Li, Na, K, Rb, Cs и NH4 низших жирных кислот (ацетаты, пропионаты, цианоацетаты), оксалаты, гликолят, аланинат, лизинат, лактат, глицерин, ацетилацетон, α-метоксиацетилацетон, этиловый сложный эфир ацетоуксусной кислоты, нонантрион-2: 5: 8, гександион-2: 5, β: β'-оксидипропионитрил, β-этоксипропионитрил, тетрацианоэтилен, 7, 7, 8, 8-тетрацианохинометан, бис-(2-этоксиэтил)-простой эфир, бис-(2-метоксиэтил)-простой эфир, этиленгликоля диглицидный простой эфир, триэтиленгликоль, диэтиленгликоль, триэтиленгликольдиацетат, диэтиленгликольдиацетат, гександиол-1: 6, пентандиол-1: 5, 2: 4-гексадииндиол-1: 6.
Другими органическими основаниями, которые подвергаются обмену с вышеописанными минералами, являются амины, такие как н-пропиламин, н-бутиламин, н-гексиламин, н-октиламин, бензидин, N,N,N',N'-тетраметилбензидин, диэтиламин, триэтиламин, трифениламин, п-фенилендиамин, N,N'-диметил-п-фенилендиамин, N,N,N',N'-тетраметил-п-фенилендиамин, транс-4,4'-диаминостильбен дигидрохлорид, бензиламин, анилин, отолуидин.
Другими длинноцепочечными солями алкиламмония, которые подвергаются обмену в вышеописанных минералах, являются бромиды 1-н-алкилпиридиния и бромид цетилтриметиламмония.
Кроме того, глицин и его пептиды, множество других аминокислот и лигандов, которые подвергаются обмену с вышеописанными минералами, описаны в R.М. Barrer Zeolites and Clay Minerals as Sorbents and Molecular Sieves, Academic Press, и ссылочных источниках, процитированных в данном документе.
Обычно «катализатор на основе обработанного бентонита» выбирают из обработанных кислотой бентонитов, то есть бентонитов, обработанных кислотами Бренстеда. Предпочтительно «катализатор на основе обработанного бентонита» выбирают из бентонитов, обработанных минеральными кислотами, а также из бентонитов, обработанных сильными органическими кислотами. В частности, «катализатор на основе обработанного бентонита» выбран из бентонитов, обработанных минеральными кислотами.
Эти катализаторы на основе бентонита не вызывают проблем с коррозией для реакционного устройства или с загрязнением сточной воды ионами металлов или неорганическими кислотами и являются достаточно кислотными, чтобы проводить реакцию на стадии с) при скоростях реакции от разумных до высоких.
Обработанные кислотой бентониты либо коммерчески доступны, либо их можно получить с использованием способов, которые хорошо описаны в данной области, как показано выше.
Коммерчески доступные обработанные кислотой бентониты (например, также известные как выщелоченные кислотой бентониты, имеющие номер CAS 70131-50-9), которые можно применять в качестве катализаторов в реакции на стадии с), представляют собой, например:
- монтмориллонит К 10, монтмориллонит К 30, монтмориллонит (алюминиевая сшитая глина) (CAS 139264-88-3), монтмориллонит-KSF (CAS 1318-93-0), приобретаемый, например, у Sigma-Aldrich;
- TONSIL™ катализаторы от компании Clariant Produkte (Deutschland) GmbH.
Как правило, катализатор на основе обработанного бентонита имеет площадь поверхности по БЭТ в интервале от 50 до 800 м2/г, предпочтительно в интервале от 100 до 600 м2/г, более предпочтительно в интервале от 120 до 500 м2/г, в частности в интервале от 150 до 400 м2/г. Выражение «площадь поверхности по БЭТ», как применяется в настоящем документе, относится к хорошо известному способу Брунауэра-Эмметта-Теллера определения площади поверхности. Значения площади поверхности по БЭТ, приведенные в настоящей заявке, определяют посредством адсорбции азота согласно способу БЭТ в основном в соответствии со стандартом DIN 66131 (1973), как подробно описано ниже.
Как правило, катализатор на основе обработанного бентонита имеет остаточную кислотность, измеряемую в мг КОН/г бентонита титрованием с потенциометрической индикацией, в интервале от 3 до 70, предпочтительно в интервале от 5 до 50, более предпочтительно в интервале от 10 до 45, в частности в интервале от 15 до 40. Остаточную кислотность (мг КОН/г бентонита) определяют с помощью следующих стандартных процедур, как описано в экспериментальном разделе ниже.
Вкратце, определение остаточной кислотности катализатора на основе обработанного бентонита проводится таким образом, что сначала готовят водную суспензию с определенным количеством катализатора на основе бентонита. Водный раствор NaOH с определенной концентрацией затем титруют в эту водную суспензию бентонита до тех пор, пока значение рН суспензии бентонита не перейдет в щелочной диапазон (рН>7,0), который соответствует конечной точке титрования. Значение рН определяется потенциометрически с помощью предварительно откалиброванного электрода KCl-рН (потенциометрическая индикация). Затем рассчитывается количество NaOH, необходимое для достижения конечной точки титрования (в миллиграммах) на грамм катализатора, внесенного в водную суспензию. Это расчетное значение соответствует остаточной кислотности в мг КОН/г бентонита.
Обычно количество свободной влаги в катализаторе на основе обработанного бентонита составляет самое большее 30 мас. %, предпочтительно самое большее 25 мас. %, предпочтительно самое большее 20 мас. %.
Количество свободной влаги в обработанном бентоните определяют путем взвешивания отдельного бентонита относительно безводного образца того же бентонита. Безводный образец получают посредством сушки в вакуумной печи при температуре от 100 до 200°С, необязательно при пониженном давлении ниже 200 мбар, предпочтительно при температуре в диапазоне от 100 до 150°С и при пониженном давлении ниже 10 мбар, в частности ниже 1 мбар, до постоянной массы.
Согласно этому более предпочтительному варианту осуществления количество катализатора на основе обработанного бентонита, применяемого на стадии с) способа согласно настоящему изобретению, находится в интервале от 1 до 750 г на моль ненасыщенного алканола общей формулы IV.a или IV.b, применяемого на стадии с). Предпочтительно, количество катализатора на основе обработанного бентонита, применяемого на стадии с), находится в интервале от 5 до 500 г на моль соединения IV.a или IV.b., более предпочтительно в интервале от 10 до 250 г на моль соединения IV.a или IV.b., даже более предпочтительно в интервале от 15 до 200 г на моль соединения IV.a или IV.b, в частности в интервале от 20 до 150 г на моль соединения IV.a или IV.b, применяемый на стадии с).
Как правило, массовое соотношение катализатора на основе обработанного бентонита и соединения (III), применяемый на стадии с), находится в интервале от 0,01:1 до 2,5:1, предпочтительно в интервале от 0,03:1 до 1,3:1, более предпочтительно в интервале от 0,04:1 до 1:1, в частности в интервале от 0,05:1 до 0,7:1.
Пористая структура бентонита может быть дополнительно изменена с помощью способов гидратации и дегидратации, как в случае с термической активацией, например (L.A. Shah et al. Applied Clay Science, 2018, 162, 155-164).
Согласно конкретному варианту осуществления настоящего изобретения катализатор на основе обработанного бентонита подвергают стадии сушки перед его применением на стадии с).
Стадию сушки обычно проводят при температуре в интервале от 50 до 200°С, предпочтительно в интервале от 70 до 170°С, в частности в интервале от 80 до 150°С, особенно в интервале от 100 до 120°С.
Стадия сушки обычно может проводиться при атмосферном давлении или при пониженном давлении. Предпочтительно, чтобы стадия сушки проводилась при пониженном давлении. В частности, стадию сушки проводят при давлении в интервале от 0,1 до 500 мбар, более конкретно в интервале от 1 до 200 мбар.
Время сушки катализатора на основе обработанного бентонита зависит от температуры и давления, применяемых на стадии сушки, и, таким образом, может варьироваться в широком диапазоне. Обычно время сушки катализатора на основе обработанного бентонита составляет от нескольких минут до нескольких дней, но предпочтительно находится в интервале от 30 минут до 2 дней.
Реакция на стадии с) обычно осуществляется при температуре в интервале от 50 до 200°С, предпочтительно в интервале от 70 до 170°С, в частности в диапазоне от 80 до 150°С.
Реакцию на стадии с) в общем можно проводить при атмосферном давлении или при повышенном или пониженном давлении.
Реакция на стадии с) может протекать в отсутствие или в присутствии инертного газа. Выражение «инертный газ», используемое в настоящем документе, в общем означает газ, который в преобладающих условиях реакции не вступает в какие-либо реакции с исходными материалами, реагентами или растворителями, участвующими в реакции, или с образующимися продуктами. Предпочтительно, чтобы реакция на стадии с) протекала в присутствии инертного газа, предпочтительно в присутствии аргона или азота, в частности в присутствии азота.
Реакцию на стадии с) обычно проводят в органическом растворителе. Предпочтительно, чтобы растворитель, применяемый на стадии с) согласно настоящему изобретению, был выбран из по меньшей мере одного полярного апротонного растворителя (PS), а также из смесей растворителей, состоящих по меньшей мере из одного полярного апротонного растворителя (PS) и по меньшей мере одного аполярного углеводородного растворителя (HS), как определено выше.
Подходящими полярными апротонными растворителями (PS), например, выбирают из следующих групп:
PS.1 органические карбонаты, то есть линейные и циклические карбонаты, как например, этиленкарбонат (243°С), пропиленкарбонат, бутиленкарбонат, 2,3-пропиленкарбонат, изобутиленкарбонат, диметилкарбонат (90°С), диэтилкарбонат (128°С) и ди-н-пропилкарбонат;
PS.2 кетоны, как например, диэтилкетон (102°С) или метилизобутилкетон (116°С);
PS.3 лактоны, как например, γ-бутиролактон (204-206°С);
PS.4 лактамы, как например, N-метил-2-пирролидон (NMP, 203°С);
PS.5 нитрилы, как например, ацетонитрил (82°С) и валеронитрил (117°С);
PS.6 нитро со единения, как например, нитрометан (101°С);
PS.7 третичные карбоксамиды, как например, диметилформамид (153°С);
PS.8 производные мочевины, как например, тетраметилмочевина (177°С) и диметилпропиленмочевина (DMPU, 247°С);
PS.9 сульфоксиды, как например, диметилсульфоксид (DMSO, 189°С);
PS. 10 сульфоны, как например, сульфолан (285°С)
PS. 11 алициклические простые эфиры, как например, 1,4-диоксан (101°С);
PS.12 гликолевые простые эфиры, как например, диалкиловый простой эфир алкиленгликоля, диалкиловые простые эфиры диалкиленгликоля и диалкиловые простые эфиры полиалкиленгликоля;
и их смесей.
Среди этих групп предпочтительны те полярные апротонные растворители (PS), которые имеют точку кипения по меньшей мере 100°С, более предпочтительно по меньшей мере 120°С, в частности по меньшей мере 140°С.
Более предпочтительно полярный апротонный растворитель (PS) выбран из групп PS.1, PS.3, PS.4, PS.7, PS.8, PS.9, PS.10 и PS.12, даже более предпочтительно из PS.1, PS.7, PS.8 и PS.12, даже более предпочтительно из PS.1 и PS.12, в частности H3PS.1.
В частности, полярный апротонный растворитель (PS) выбран из циклических или линейных карбонатов общей формулы VIa и VI.b, как определено выше.
Предпочтительно, аполярный углеводородный растворитель (HS) выбран из групп HS.1 и HS.2.
Более предпочтительно, аполярный углеводородный растворитель (HS) выбран из гексана, циклогексана, гептана, октана и нонана, более конкретно из гептана и октана.
Согласно предпочтительному варианту осуществления согласно настоящему изобретению, стадию с) проводят в смеси растворителей, состоящей из по меньшей мере одного полярного апротонного растворителя(PS), как определено выше, и по меньшей мере одного аполярного углеводородного растворителя (HS), как определено выше.
Согласно этому предпочтительному варианту осуществления содержание PS в смеси растворителей, как правило, находится в интервале от 35 од 99 мас. %, предпочтительно в интервале от 50 до 99 мас. %, в частности в интервале от 50 до 90 мас. %, на основе общей массы смеси растворителей.
Соответственно, массовое соотношение PS и HS, применяемых на стадии с), находится, как правило, в интервале от 1:3 до 100:1, предпочтительно в интервале от 1:1 до 100:1, в частности в интервале от 1:1 до 10:1.
Согласно этому предпочтительному варианту осуществления предпочтительны полярные апротонные растворители (PS), которые не смешиваются или смешиваются только умеренно по меньшей мере с одним аполярным углеводородным растворителем (HS), что означает, что стадия с) проводится в двухфазной смеси растворителей, состоящей из PS-фазы и HS- фазы. В этой связи термин «умеренно смешиваемый» означает, что менее 5 мас. %, предпочтительно менее 2 мас. %, более предпочтительно менее 1 мас. %, в частности менее 0,5 мас. % полярного апротонного растворителя (PS) присутствует в HS-фазе.
Согласно этому предпочтительному варианту осуществления предпочтительными являются смеси растворителей, состоящие из по меньшей мере одного полярного апротонного растворителя группы PS.1 и по меньшей мере одного аполярного углеводородного растворителя группы HS.1.
В частности, согласно этому предпочтительному варианту осуществления смесь растворителей состоит из по меньшей мере одного полярного апротонного растворителя (PS), выбранного из этиленкарбоната, пропиленкарбоната, бутиле нкарбоната, 2,3-пропиленкарбоната, изобутиленкарбоната, диэтилкарбоната и ди-н-пропилкарбоната, и по меньшей мере одного аполярного углеводородного растворителя (HS), выбранного из гептана и октана.
Согласно конкретному варианту осуществления настоящего изобретения стадию b) проводят в по меньшей мере одном полярном апротонном растворителе (PS), как определено выше.
Соединения общей формулы (III), применяемые на стадии с), хорошо растворимы в полярном апротонном растворителе (PS-фаза), тогда как аполярный продукт реакции, полученный на стадии с), то есть продукт алкилирования или конденсации, который обычно отделяют от полярного апротонного растворителя после охлаждения реакционной смеси, хорошо растворим в аполярном углеводородном растворителе (HS-фаза). Таким образом, использование вышеупомянутых смесей растворителей имеет то преимущество, что реакционная смесь, полученная на стадии с), может быть легко разделена путем разделения фаз на PS-фазу, содержащую большую часть или практически все непрореагировавшее соединение (III), а также катализатор конденсации, и HS-фазу, содержащую большую часть или практически весь аполярный продукт реакции, полученный на стадии с). При необходимости отделение аполярного продукта реакции, полученного на стадии с), от PS-фазы может быть завершено экстракцией аполярным углеводородным растворителем (HS). Отделенную PS-фазу можно затем вернуть в реакцию на стадии с) или повторно использовать позже в другой реакции на стадии с). Отделенная HS-фаза может быть непосредственно подвергнута следующей стадии способа или стадии очистки. Кроме того, вода, образующаяся во время реакции на стадии с), может быть легко отогнана из PS-фазы, что позволяет легко рециркулировать полярный апротонный растворитель (PS). Кроме того, использование аполярного углеводородного растворителя (HS) позволяет эффективно удалять посредством дистилляции воду, образующуюся во время реакции на стадии с), посредством образования азеотропной смеси.
Согласно особенно предпочтительному варианту осуществления настоящего изобретения стадию с) проводят в том же карбонатном растворителе или в той же смеси карбонатный растворитель/аполярный углеводородный растворитель, которые применяются на стадии b).
Концентрация соединения (III) в полярном органическом растворителе (PS) обычно находится в интервале от 2 до 50 мас. %, предпочтительно в интервале от 3 до 45 мас. %, в частности в интервале от 5 до 40 мас. %.
Молярное соотношение соединения (III) и соединения IV.a или IV.b, применяемых на стадии с), как правило, находится в интервале от 1:1 до 10:1, предпочтительно в интервале от 1,05:1 до 5:1, более предпочтительно в интервале от 1,05:1 до 3:1, в частности в интервале от 1,1:1 до 2:1.
Соединения общей формулы IV.a и IV.b либо коммерчески доступны, либо могут быть получены из легкодоступных предшественников способами, известными из уровня техники, или могут быть получены из природных источников.
Например, соединения IV.a и IV.b, где Y представляет собой гидроксил, легко коммерчески доступны или могут быть получены из природных источников.
Соединения IV.a и IV.b, где Y представляет собой уходящую группу, отличную от гидроксила, как определено выше, могут быть получены из соответствующих предшественников спирта с помощью обычных реакций нуклеофильного замещения. Эти нуклеофильные реакции можно проводить в обычных реакционных условиях, которые хорошо известны специалисту в данной области техники.
Обычно реакция на стадии с) согласно настоящему изобретению сначала протекает с образованием продукта алкилирования по Фриделю-Крафтсу. В случае, если гидроксильные группы, смежные с положением алкилирования, не защищены, реакция алкилирования по Фриделю-Крафтсу обычно сопровождается последующей реакцией замыкания цикла (внутримолекулярное гидроксиалкилирование двойной связи) с образованием конденсированного шестичленного цикла, содержащего атом кислорода. При желании промежуточный продукт алкилирования по Фриделю-Крафтсу также может быть выделен, и реакция замыкания цикла может быть проведена на отдельной стадии. Однако предпочтительно, чтобы алкилирование по Фриделю-Крафтсу и реакция замыкания цикла проводились в одну стадию.
Согласно предпочтительному варианту осуществления настоящего изобретения реакцию на стадии с) проводят с удалением дистилляцией по меньшей мере одной части воды, образовавшейся во время реакции. Как уже упоминалось выше, дистилляционному удалению воды, образовавшейся в ходе реакции, можно способствовать, используя аполярный углеводородный растворитель (HS), такой как циклогексан, гептан, октан или толуол, в дополнение к полярному апротонному растворителю (PS) на стадии с), поскольку аполярные углеводородные растворители (HS) часто образуют с водой азеотропные смеси. Для этого пар удаляют из реакционной системы и конденсируют. В случае, если пар состоит из азеотропной смеси воды с органическим растворителем, применяемым на стадии с), и/или другим компонентом реакционной смеси стадии с), или если вода содержит значительные количества продукта или исходного вещества, образующийся конденсат, как правило, подвергают фазовому разделению с получением водной фазы и органической фазы. Для этого конденсат обычно подают в сепаратор фаз (декантатор), где механическое расслоение приводит к его распаду на две фазы, которые можно извлекать отдельно. Если необходимо, несмешивающийся с водой органический растворитель, предпочтительно органический растворитель, применяемый на стадии с), добавляют к конденсату перед пропусканием конденсата в разделитель фаз. Водную фазу удаляют и выбрасывают, а органическую фазу по меньшей мере до некоторой степени возвращают в реакционную систему. «Возврат в реакционную систему» означает, что органическая фаза проходит в любой желаемый по меньшей мере один реактор реакционной системы.
Любой из подходящих конденсаторов может использоваться для конденсации или частичной конденсации пара. Их можно охлаждать любыми охлаждающими жидкостями. Предпочтение отдается конденсаторам с воздушным и/или водяным охлаждением, при этом особое предпочтение отдается воздушному охлаждению.
Реакция на стадии с) может осуществляться либо в периодическом режиме (прерывистый режим), как описано выше, либо в непрерывном режиме. Предпочтение отдается проведению реакции на стадии с) в непрерывном режиме.
Если реакция на стадии с) проводится в периодическом (прерывистом) режиме, реагенты и катализатор обычно помещают в подходящий реакционный сосуд, например, сосуд с мешалкой или петлевой реактор при температурах, указанных выше, до тех пор, пока не будет достигнуто желаемое превращение. Время реакции может составлять от 0,5 до 30 часов, предпочтительно от 1 до 20 часов, в зависимости от количества добавленного катализатора.
Предпочтительно реакцию на стадии с) проводят таким образом, что сначала органический растворитель и катализатор конденсации, предпочтительно кислотный гетерогенный катализатор, в частности катализатор на основе обработанного бентонита, помещают в подходящий реакционный сосуд, например, сосуд с мешалкой или петлевой реактор, и нагревают до температуры реакции. При необходимости полученную суспензию хранят при температуре реакции в течение нескольких минут, например, в течение 1, 2, 5, 10, 15 или 20 минут до охлаждения суспензии до ниже 80°С. В ходе этих стадий предварительной обработки, инертный газ, предпочтительно аргон или азот, вводят в устройство, чтобы гарантировать, что устройство не содержит кислорода. После этого соединение (III) добавляют одной порцией к предварительно нагретой суспензии растворитель/катализатор, и полученную смесь нагревают до температуры реакции. После этого в реакционную смесь добавляют соединение IV.a или IV.b, необязательно растворенное в аполярном углеводородном растворителе (HS). Обычно соединение IV.a или IV.b добавляют пошагово в реакционную смесь, содержащую органический растворитель, катализатор конденсации и соединение (III), несколькими порциями, например, в виде 2, 3, 4, 5, 10, 15 или 20 порций, или дозируют непрерывно. Предпочтительно, соединение IV.a или IV.b дозируют непрерывно. Скорость добавления соединений IV.a или IV.b в реакционной смеси обычно составляет в интервале от 0,2 до 5 об.%/мин, предпочтительно в интервале от 0,3 до 3% об.%/мин, в частности в интервале от 0,5 до 2об.%/мин, на основе общего объема соединений IV.a или IV.b
В зависимости от того, применяется ли только полярный апротонный растворитель (PS) или смесь, состоящая из полярного апротонного растворителя (PS) и аполярного углеводородного растворителя (HS), в качестве органического растворителя на стадии с), желаемый продукт реакции отделяют от полученной реакционной смеси разделением фаз и/или экстракцией аполярным углеводородным растворителем (HS). Таким образом, получают две фазы, то есть PS-фазу, содержащую в основном катализатор конденсации и в конечном итоге непрореагировавшее соединение (III), и HS-фазу, содержащую в основном желаемый продукт реакции и в конечном итоге непрореагировавшее соединение IV.a или IV.b. После разделения фаз и/или экстракции, продукт реакции может быть очищен хроматографическими методами, дистилляцией и/или кристаллизацией, предпочтительно дистилляцией, или продукт реакции может быть непосредственно подвергнут следующей стадии реакции d).
В случае использования гомогенного катализатора конденсации на стадии с) способа согласно настоящему изобретению реакционную смесь также можно нейтрализовать водным щелочным раствором и/или водой, чтобы удалить гомогенный катализатор конденсации из реакционной смеси путем экстракции.
В случае, если кислотный гетерогенный катализатор, в частности катализатор на основе обработанного бентонита, используется на стадии с) способа согласно настоящему изобретению, кислотный гетерогенный катализатор, в частности катализатор на основе обработанного бентонита, предпочтительно отделяют от реакционной смеси или PS- фазы после завершения реакции и повторно используют в следующей реакции на стадии с).
Для отделения кислотного гетерогенного катализатора, в частности катализатора на основе обработанного бентонита, от реакционной смеси или PS-фазы, в частности могут использоваться все способы, известные специалисту в данной области техники, которые подходят для отделения твердых веществ от жидких смесей. Предпочтительно катализатор удаляют из полученной реакционной смеси фильтрованием. После разделения кислотный гетерогенный катализатор, в частности катализатор на основе обработанного бентонита, сушат в потоке инертного газа, лучше в потоке азота. Время сушки кислотного гетерогенного катализатора, в частности катализатора на основе обработанного бентонита, в потоке инертного газа может варьироваться в широком диапазоне, в зависимости от природы растворителя, применяемого в реакции на стадии с). Время сушки кислотного гетерогенного катализатора в потоке инертного газа обычно составляет в интервале от нескольких минут до нескольких дней, то есть от 5 минут до 5 дней. Время сушки кислотного гетерогенного катализатора в потоке инертного газа может составлять, например, 10 минут, 30 минут, 1 час, 5 часов, 12 часов, 1 день, 3 дня или 5 дней.
Согласно другому предпочтительному варианту осуществления настоящего изобретения PS-фазу, содержащую в основном катализатор конденсации и в конечном итоге непрореагировавшее соединение (III), которое получают после разделения и/или экстракции реакционной смеси, полученной на стадии с), с аполярным углеводородным растворителем HS), непосредственно повторно используется в дальнейшей реакции на стадии с).
Непрерывную реакцию обычно проводят по меньшей мере в одном реакторе, например, 1, 2, 3, 4 или 5 реакторах, предпочтительно в одном реакторе, содержащем катализатор на основе обработанного бентонита в виде неподвижного слоя или движущегося слоя, предпочтительно в виде неподвижного слоя, в который, например, подают смесь органического растворителя с соединением III и соединением IV.a или IV.b. В предпочтительном режиме работы с неподвижным слоем реактор может работать в режиме работы отстойника, т.е. реакционная смесь направляется снизу вверх, или в режиме работы струйным потоком, т.е. реакционная смесь будет проходить через реактор сверху вниз. Воду, образующуюся во время реакции, удаляют путем отвода пара из верхней части реактора, который конденсируют и разделяют на органическую фазу, в конечном итоге содержащую аполярный углеводородный растворитель (HS) и незначительные количества непрореагировавшего соединения III и/или продукта реакции, и водную фазу, как описано выше. Органическую фазу необязательно возвращают по меньшей мере в один реактор. Поток реакционной смеси, содержащей полярный апротонный растворитель (PS), аполярный углеводородный растворитель (HS), если присутствует, продукт реакции и в конечном итоге непрореагировавшее соединение III, отводят из нижней части реактора. В зависимости от того, используется ли только полярный апротонный растворитель (PS) или смесь, состоящая из полярного апротонного растворителя (PS) и аполярного углеводородного растворителя (HS) в качестве органического растворителя на стадии с), желаемый продукт реакции отделяют от полученной реакционной смеси разделением фаз и/или экстракцией аполярным углеводородным растворителем (HS). Продукт реакции может быть очищен или продукт реакции может быть непосредственно подвергнут следующей стадии реакции.
Часовая объемная скорость катализатора в реакции на стадии с) находится в диапазоне от 0,1 до 50 кг соединения IV.a или IV.b на кг катализатора в час, в частности в интервале от 0,2 до 30 кг соединения IV.a или IV.b на кг катализатора в час.
По меньшей мере один реактор может быть выбран из любых желаемых реакторов, которые подходят для проведения гетерогенно катализируемых химических реакций в жидкой фазе.
Подходящими реакторами являются реакторы без обратного перемешивания, такие как трубчатые реакторы или контейнеры с выдержкой времени при остановке, снабженные внутренними устройствами, но предпочтительно реакторы с обратным перемешиванием, такие как реакторы с мешалкой или петлевые реакторы. Однако также возможно использовать комбинации последовательных реакторов с обратным перемешиванием и реакторов без обратного перемешивания.
При необходимости несколько реакторов также могут быть объединены в многоступенчатое устройство. Такими реакторами являются, например, петлевые реакторы со встроенными ситчатыми тарелками, каскадные контейнеры, трубчатые реакторы с промежуточной точкой подачи или колонки с мешалкой.
Стадия d):
Если R4 в соединении I выбран из C1-C6-алканоила, продукт конденсации, полученный на стадии с), реагирует с С2-С7-карбоновой кислотой или с ангидридом С2-С7-карбоновой кислоты в присутствии катализатора эстерификации.
Подходящие катализаторы эстерификации, которые могут применяться на стадии d) способа согласно настоящему изобретению, хорошо известны специалистам. Подходящими катализаторами эстерификации являются, например, катализаторы на основе металлов, например, катализаторы на основе железа, кадмия, кобальта, свинца, цинка, сурьмы, магния, титана и олова, в форме металлов, оксидов металлов или солей металлов, таких как алкоксилаты металлов; минеральные кислоты, такие как серная кислота, соляная кислота или фосфорная кислота; или органические сульфоновые кислоты, такие как метансульфоновая кислота или пара-толуолсульфоновая кислота.
В качестве альтернативы продукт конденсации, полученный на стадии d), реагирует с активированной С2-С7карбоновой кислотой в присутствии основания.
Подходящими активированными С2-С7-карбоновыми кислотами, которые могут применяться на стадии d) способа согласно настоящему изобретению, являются, например, галогениды С2-С7-карбоновой кислоты, такие как хлориды, бромиды или иодиды С2-С7-карбоновой кислоты.
Подходящие основания, которые можно применять на стадии ii), представляют собой, например, органические основания, такие как, например, третичные амины, например, триметиламин, триэтиламин, трипропиламин, этилдиизопропиламин и т.п. или основные N-гетероциклы, такие как морфолин, пиридин, лутидин, DMAP, DABCO, DBU или DBN.
Индивидуальные реакционные условия для этих реакций этерификации хорошо известны специалисту.
Предпочтительно, на стадии d) способа согласно настоящему изобретению продукт конденсации, полученный на стадии b), реагирует с С2-С7-карбоновой кислотой или с ангидридом С2-С7-карбоновой кислоты в присутствии катализатора эстерификации.
Согласно предпочтительному варианту осуществления настоящего изобретения катализатор эстерификации, применяемый на стадии d), выбран из кислотных гетерогенных катализаторов, как определено выше.
Согласно более предпочтительному варианту осуществления настоящего изобретения стадию d) проводят в присутствии того же кислотного гетерогенного катализатора, как применяется на стадии с).
Предпочтительно, согласно этому более предпочтительному варианту осуществления кислотный гетерогенный катализатор, применяемый на стадии с) и d), отделяют от реакционной смеси после завершения реакции на стадии d) и повторно используют в дальнейшей реакции на стадии с). Рецикл гетерогенных катализаторов конденсации проводят, как описано выше, на стадии с) для катализатора на основе обработанного бентонита.
Согласно даже более предпочтительному варианту осуществления настоящего изобретения реакцию эстерификации на стадии d) проводят в присутствии а катализатора на основе обработанного бентонита, как определено выше, в частности в присутствии кислотного катализатора на основе обработанного бентонита.
Согласно конкретному варианту осуществления настоящего изобретения реакцию на стадии с) и реакцию эстерификации на стадии d) проводят в присутствии одного и того же катализатора на основе обработанного бентонита, в частности в присутствии кислотного катализатора на основе обработанного бентонита.
Предпочтительно, также согласно этому конкретному варианту осуществления, катализатор на основе обработанного бентонита, применяемый на стадии с) и d), отделяют от реакционной смеси после завершения реакции на стадии d) и повторно используют в дальнейшей реакции на стадии с), как описано выше для стадии с).
Согласно специфическому варианту осуществления настоящего изобретения реакции на стадиях с) и d) проводятся в присутствии по меньшей мере одного полярного апротонного растворителя (PS), как определено выше, или в смеси растворителей, состоящей из по меньшей мере одного полярного апротонного растворителя (PS), как определено выше, и по меньшей мере одного аполярного углеводородного растворителя (HS), как определено выше.
Согласно более конкретному варианту осуществления настоящего изобретения стадии с) и d) проводят в присутствии карбонатного растворителя.
Согласно другому специфическому варианту осуществления настоящего изобретения реакцию на стадиях с) и d) проводят в смеси карбонатный растворитель/аполярный углеводородный растворитель, как описано выше.
Подходящие и предпочтительные карбонатные растворители, а также подходящие и предпочтительные аполярные углеводородные растворители (HS), если они присутствуют, имеют значения, как определено выше.
Еще более предпочтительно, в этом специфическом варианте осуществления стадию d) проводят в том же карбонатном растворителе и, если он присутствует, в том же аполярном углеводородном растворителе (HS), которые применяются на стадии с). В частности, реакционную смесь, полученную на стадии с), применяют непосредственно в реакции на стадии d), то есть стадию с) и стадию d) проводят как реакцию в одном реакторе.
Продукт реакции, то есть соединение формулы (I), полученное на стадии d), можно переработать обычным способом, например, путем отфильтровывания любого твердого катализатора, если он присутствует, при необходимости добавив аполярный углеводородный растворитель (HS), разделения фаз и, при необходимости, очистки неочищенных продуктов с помощью хроматографических методов, дистилляции или перекристаллизации.
Согласно особому варианту осуществления настоящего изобретения стадии b), с) и d) проводят в одном и том же карбонатном растворителе и, если он присутствует, в одном и том же аполярном углеводородном растворителе (HS). Предпочтительно, согласно этому особому варианту осуществления, реакционная смесь, полученная на стадии b), непосредственно используется в реакции на стадии с) после удаления катализатора гидрирования, а реакционная смесь, полученная на следующей стадии с), непосредственно используется в реакции на стадии d).
Способ согласно настоящему изобретению обеспечивает соединения (I) с высокими выходами и селективностью. Обычно соединения (I) дополнительно очищают перекристаллизацией, дистилляцией, если применимо, или с помощью хроматографических методов.
В общем, получают только незначительные количества побочных продуктов.
Обычными побочными продуктами, которые получают с использованием способов, описанных в предшествующем уровне техники для получения соединений общей формулы (I), являются, например, диеновые соединения общей формулы X.1 - Х.3, которые образуются из соединений IV. а или IV.b в результате нежелательных реакций элиминирования, как показано на схеме 4.
Схема 4:  где
где
X имеет значение, как определено выше, и
Х-2 предпочтительно выбирают из фрагментов формул Х-2.а и Х-2.b
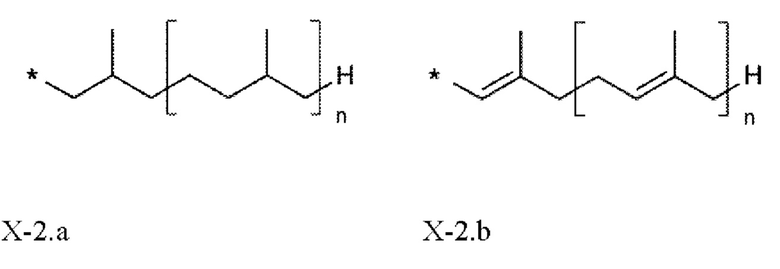
где
n представляет собой целое число от 0 до 2 и
* показывает точку присоединения к остальной части молекулы.
Образование этих диеновых продуктами обычно увеличивается, когда карбоновые кислоты, такие как щавелевая кислота, винная кислота или лимонная кислота, применяют в качестве катализатора конденсации. Эти кислоты способны образовывать сложноэфирные промежуточные соединения с соединениями IV.a или IV.b., которые могут легко подвергаться элиминации до соединений общей формулы X. 1 - Х.3.
Диеновые соединения общей формулы X. 1 - Х.3 также могут реагировать с соединениями (III) на стадии с). Однако реакция протекает очень медленно по сравнению с реакцией с соединениями IV.a или IV.b. Таким образом, следует избегать образования диеновых соединений X. 1 - Х.3.
Кроме того, изомеры бензофурана формулы XI-1 могут быть образованы в результате реакции соединений гидрохинона (III) с соединениями IV.a, как показано на схеме 5, которые трудно отделить от желаемых соединений (I).
Схема 5:

Используя способ согласно настоящему изобретению можно успешно подавить образование этих общих побочных продуктов.
Приведенные ниже примеры обеспечивают дальнейшее пояснение настоящего изобретения. Эти примеры не следует понимать, как ограничивающие настоящее изобретение.
Примеры
Аббревиатуры:
GC означает газовую хроматографию,
ВЭЖХ высокоэффективная жидкостная хроматография,
ТМН означает триметилгидрохинон (2,3,5-триметилгидрохинон),
TMQ означает триметилхинон (2,3,5-триметилхинон),
PC пропиленкарбонат
ЕС этиленкарбонат
1. Аналитические исследования:
1.1 Определение чистоты продукта:
Чистоту продуктов определяли посредством определения % площади методом газовой хроматографии. Выход соединений I и II определяли посредством определения % массы методом GC, используя докозан в качестве внутреннего стандарта и н-гептан в качестве растворителя.
GC-система: Agilent 6980N;
GC колонка: Agilent DB-1: 30 м (длина), 0,25 мм (внутренний диаметр), 0,25 микрометра (толщина пленки);
Температурная программа: 80°С - 350°С при 10°/мин, 350°С в течение 10 минут, общее время цикла: 37 минут.
Количество соединений (III) в (конечной) реакционной смеси определяли посредством определения % массы методом ВЭЖХ:
ВЭЖХ система: Agilent Series 1200
ВЭЖХ колонка: Zorbax Eclipse РАН, 1,8 мкм, 50*4,6 мм от AgilentÒ
Элюент:
А: вода с 0,1 об.% H3PO4;
В: ацетонитрил

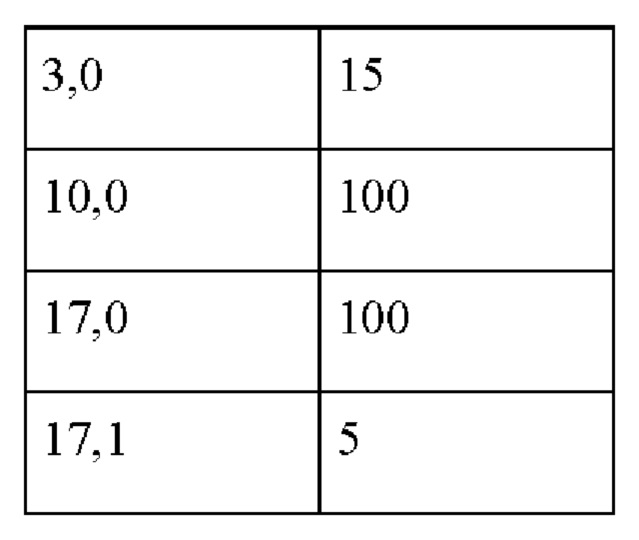
Детектор: УФ-детектор λ=210 нм, BW=5 нм, скорость потока: 1,2 мл/мин, впрыск: 2 мкл, температура: 60°С, время работы: 20 мин, давление: около 130 бар.
1.2 Определение площади поверхности по БЭТ:
Система: Quantachrome Autosorb Automated Gas Sorption System 6B, serial-#: 10896010901;
Программное обеспечение: Autosorb для Windows® для AS-3 и AS-6 версии 1.22;
Масса образца: 0,28-0,43 г твердого катализатора (например, катализатор на основе обработанного бентонита);
Температура ванны: 77,4 К;
Продолжительность: 64 - 106,7 мин;
Газ для измерения: азот; чистота газа: азот 5,0;
Сушка перед измерением: центробежный лопастной насос и, наконец, турбомолекулярный насос в течение 16 часов при 120°С, <1 мбар;
Параметры системы: площадь поперечного сечения 16,2 Å/molec;
Многоточечная БЭТ: 5 точек р/р0; 0,05≤р/р0≤0,30.
1.3 Определение остаточной кислотности твердого катализатора (мг КОН/г твердого катализатора):
Определение остаточной кислотности твердого катализатора (например, катализатора на основе обработанного бентонита) проводили таким образом, что сначала готовили водную суспензию с определенным количеством твердого катализатора следующим образом: от 1,0 г до 1,5 г твердого катализатора суспендировали в 50 мл деионизированной воды и перемешивали в течение 1 ч. В эту суспензию помещали предварительно откалиброванный КС1-рН-электрод. Водный раствор NaOH с определенной концентрацией 0,1 моль/л затем титровали в эту водную суспензию до тех пор, пока значение рН суспензии твердого катализатора не перешло в щелочной диапазон (точка перегиба), что представляет собой конечную точку титрования. Регистрировали объем VI в мл раствора NaOH, использованный для достижения точки перегиба.
Кроме того, холостое определение проводили таким же образом с использованием 50 мл деионизированной воды. Регистрировали объем использованного раствора NaOH V2 в мл.
Остаточная кислотность образца твердого катализатора (в мг КОН/г твердого катализатора), которая затем определяется как общее кислотное число, рассчитывается по следующей формуле:

56,1 г/моль представляет собой константу (молярная масса КОН в г/моль);
m1 представляет собой массу в граммах пробы для испытания, т.е. образца твердого катализатора;
V1 представляет собой объем в миллилитрах раствора NaOH, использованного для нейтрализации суспензии катализатора (объем до точки перегиба);
V2 представляет собой объем в миллилитрах раствора NaOH, использованный при холостом определении (объем до точки перегиба - обычно объем не израсходован/бланк обычно равен нулю);
С представляет собой концентрацию раствора NaOH в молях на литр;
t представляет собой титр раствора NaOH.
Определение остаточной кислотности повторяют один раз и, таким образом, определяют дважды.
1.4 Определение плотности катализатора:
Устройство: Pycnometer серии AccuPyc II 1340
Фирма: Micromeritics
Инертный газ: гелий
Масса образца: 2,1-4,8 г
Камера для образца: 10 мл
Использовалась программа «Условия анализа», включающая 99 циклов и каждый цикл с 5 повторениями.
Перед измерениями образцы обрабатывали в течение 16 ч при 120°С в вакууме <1 мбар.
Плотность [г/см3]=масса образца [г] / объем образца [см3]
1. Примеры получения
Происхождение и спецификация применяемого кислотного катализатора на основе обработанного бентонита:
Обработанные кислотой бентониты, которые используются в качестве катализаторов в следующих примерах получения, представляют собой либо обработанные кислотой бентониты от компании BASF SE, которые были разработаны собственными силами (внутренний материал BASF SE), либо обработанные кислотой бентониты, доступные от Sigma Aldrich или от компании Clariant Produkte GmbH.
Применяли катализаторы на основе бентонита со следующими спецификациями:
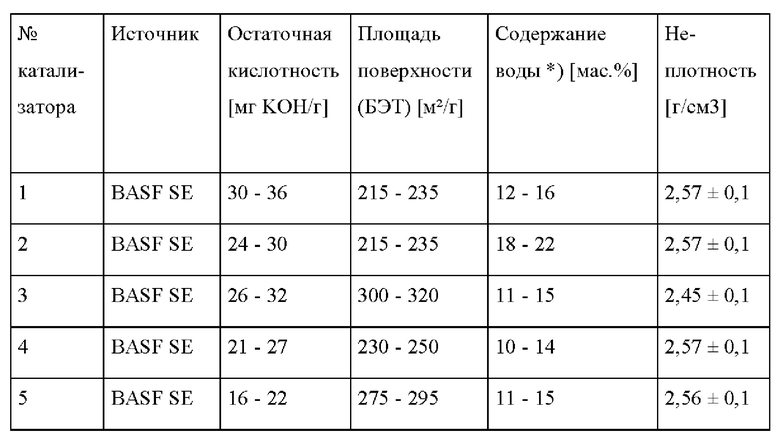

*) определяется титрованием по Карлу-Фишеру и/или по потере массы при сушке (16 ч при 120°С и при давлении <1 мбар).
Для получения катализатора на основе обработанного кислотой бентонита от компании BASF SE (катализаторы 1-5) в качестве исходного природного бентонита использовалась абердинская глина, известная своим высоким качеством. Эти бентониты активируются серной кислотой с последующим превращением в конечные гранулированные минеральные катализаторы.
Общая процедура реакции и примечания:
За ходом реакции следят с помощью тонкослойной хроматографии и GC.
Стадия 1: восстановление TMQ до ТМН
Стадия 2: Алкилирование по Фриделю-Крафтсу и конденсация
Если не указано иное, все реакции проводят в стеклянной колбе с использованием лопастной мешалки и ловушки Дина-Старка, которая
- если в качестве растворителя используется смесь HS и PS, заполняется применяемым HS,
- в случае использования PS в качестве растворителя, который не образует азеотропа с водой, остается пустой, и
- в случае использования растворителя, который образует азеотроп с водой, заполняется используемым растворителем (в отличие от воды), который азеотропно удаляется с водой.
Стадия 3: Эстерификация
2.1 Получение 2,3,5-триметилгидрохинона (стадия 1)
4 г 2,3,5-триметилхинона (99,6%, 26,5 ммоль) растворяли в 76 г (63,1 мл) пропиленкарбоната при комнатной температуре. 0,4 г палладия на активированном угле (10%, 0,38 ммоль, 0,01 экв.) добавляли, и полученную реакционную смесь гидрировали в течение 23 ч при давлении водорода 8 бар и при 64°С. После времени реакции 6 ч отфильтрованная реакционная смесь представляет собой лишь слегка желтоватый раствор, после времени реакции 23 ч
- бесцветный раствор. В результате анализа GC получили:

2.2 Получение 2,5,7,8-тетраметил-2-[4,8,12-триметилтридецил]-3,4-дигидро-2Н-хромен-6-ола (полностью рацемический альфа-токоферол) (Стадия 1 и 2):
Получение2,3,5-триметилгидрохинона
14 г 2,3,5-триметилхинон (99,6%, 92,85 ммоль) растворяли в 79,3 г (65,9 мл) пропиленкарбоната при комнатной температуре. 2,8 г палладия на активированном угле (10%, 2,6 ммоль, 0,03 экв.) добавляли, и полученную реакционную смесь гидрировали в течение 23 ч при давлении водорода 8 бар и 88°С. Затем, реакционную смесь немедленно фильтровали и 83,3 г почти бесцветного элюата (97,6 GC-площадь-% ТМН и 0,05 GC-площадь-% TMQ) получали.
Получение полностью рацемического альфа-токоферола (катализатор 2):
К неочищенному ТМН в пропиленкарбонате (81,4 г после аналитического анализа, принимают: 100% выход: 92,85 ммоль, 1,7 экв.) 0,19 г катализатора 2 (высушенный в течение ночи при 120°С / 50 мбар в вакуумной сушильной печи) / г ТМН (2,7 г катализатора 2) добавляли. Реакционную смесь доводили до 120-125°С и перемешивали в течение 15 мин. Затем, 16,45 г изофитола (97,4%, 54 ммоль) непрерывно добавляли в реакционную смесь в течение периода времени, равного 2 ч (температура реакционной смеси: 120-125°С) при удалении воды, образующейся в ходе реакции, посредством дистилляции. После последующего времени реакции 4 ч при 125°С, при комнатной температуре всю ночь, и еще 4 ч при 125°С реакционную смесь доводили до комнатной температуры, 20 мл гептана добавляли, далее перемешивали в течение 15 мин и затем фильтровали через целит для удаления катализатора 2. Фильтровальную лепешку промывали с помощью 6×20 мл гептана и 3×20 мл пропиленкарбоната. После разделения фаз, пропиленкарбонатную фазу экстрагировали с помощью 4×25 мл гептана. Объединенные фазы гептана сушили над сульфатом натрия. Растворитель удаляли при пониженном давлении 50°С / 5 мбар плюс 15 мин масляный вакуум-насос. 24,09 г неочищенного альфа-токоферола (91,6% площади согласно GC и 85,66% массы согласно GC) получали в виде прозрачного вязкого остатка темно-красного цвета с выходом 89% (на основе % массы согласно GC за две стадии). Каждая стадия имела средний выход 94,5%.
2.3 Получение 2,5,7,8-тетраметил-2-[4,8,12-триметилтридецил]-3,4-дигидро-2Н-хромен-6-ола (полностью рацемический альфа-токоферол) (Стадия 2; катализатор 1):
233,57 г (193,99 мл) пропиленкарбоната и 0,22 г катализатора 1 (высушенного при 120°С всю ночь, 50 мбар) / г ТМН (21,75 г катализатора 1) нагревали под потоком газообразного азота до 120 125°С и перемешивали в течение 15 мин. Суспензию затем охлаждали до <90°С и 100,11 г (652,5 ммоль) триметилгидрохинона (1,5 экв.) добавляли. Смесь затем снова нагревали до 120-125°С. Затем, 136,35 г (162,13 мл) изофитола (447,9 ммоль, 97,4% чистота, 1 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 2 ч (температура реакционной смеси: 120-125°С) при удалении воды, образующейся в ходе реакции, посредством дистилляции. После последующего времени реакции 6 ч при 120-125°С, реакционную смесь охлаждали до комнатной температуры. Смесь фильтровали через стеклянный вакуум-фильтр D4, загруженный цеолитом, для удаления катализатора 1. Фильтровальную лепешку промывали с помощью 6×130 мл гептана и 3×130 мл пропиленкарбоната. Все маточные растворы и промывочные растворы собирали и объединяли. Фазы элюата отделяли. Пропиленкарбонатную фазу экстрагировали с помощью 4×190 мл гептана. Объединенные фазы гептана сушили над сульфатом натрия, и летучие соединения удаляли при пониженном давлении при 50-55°С / 5 мбар плюс 15 мин масляный вакуум-насос: 200 г неочищенного альфа-токоферола (94,6% площади согласно GC и 94,53% массы согласно GC) получали в виде прозрачного вязкого остатка темно-красного цвета. Это соответствует выходу 98%.
2.4 Получение 2,5,7,8-тетраметил-2-[4,8,12-триметилтридецил]-3,4-дигидро-2Н-хромен-6-ола (полностью рацемический альфа-токоферол) (Стадия 2; катализатор 1):
138,08 г (141,91 мл) диэтилкарбоната и 0,22 г катализатора 1 (высушенного при 120°С всю ночь, 50 мбар) / г ТМН (7,5 г катализатора 1) нагревали под потоком газообразного азота до 120-123°С и перемешивали в течение 15 мин. Суспензию затем охлаждали до <90°С и 34,52 г (225 ммоль) триметилгидрохинона (1,5 экв.) добавляли. Смесь затем снова нагревали до 120-125°С. Затем, 45,76 г (54,41 мл) изофитола (150 ммоль, 97,2% чистота, 1 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 2 ч (температура реакционной смеси: 124-119°С) при удалении воды, образующейся в ходе реакции, посредством дистилляции. После последующего времени реакции 6 ч при 120-125°С, реакционную смесь охлаждали до комнатной температуры. 45 мл гептана добавляли в реакционную смесь, которую перемешивали в течение 15 мин при комнатной температуре. Затем, смесь фильтровали через стеклянный вакуум-фильтр D4, загруженный цеолитом, для удаления катализатора 1. Фильтровальную лепешку промывали с помощью 6×45 мл гептана. Все маточные растворы и промывочные растворы собирали и объединяли. Элюат, из которого наблюдалось выпадение осадка, далее фильтровали, и растворитель удаляли при пониженном давлении при 50-55°С / 5 мбар плюс 15 мин масляный вакуум-насос: 68,84 г неочищенного альфа-токоферола (84,8% площади согласно GC и 78,79% массы согласно GC) получали в виде вязкого мутного остатка красновато-коричневого цвета. Это соответствует выходу 84%.
2.5 Получение 2,5,7,8-тетраметил-2-[4,8,12-триметилтридецил]-3,4-дигидро-2Н-хромен-6-ола (полностью рацемический альфа-токоферол) (Стадия 2; катализатор 1):
80,55 г (71,92 мл) гамма-бутиролактона и 0,22 г катализатора 1 (высушенного при 120°С всю ночь, 50 мбар) / г ТМН (7,5 г катализатора 1) нагревали под потоком газообразного азота до 120-125°С и перемешивали в течение 15 мин. Суспензию затем охлаждали до <90°С и 34,52 г (225 ммоль) триметилгидрохинона (1,5 экв.) добавляли. Смесь затем снова нагревали до 120-125°С. Затем, 45,76 г (54,3 мл) изофитола (150 ммоль, 97,4% чистота, 1 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 2 ч (температура реакционной смеси: 120-125°С) при удалении воды, образующейся в ходе реакции, посредством дистилляции. После последующего времени реакции 6 ч при 120-125°С, реакционную смесь охлаждали до комнатной температуры. 45 мл гептана добавляли в реакционную смесь, которую перемешивали в течение 15 мин при комнатной температуре. Затем, смесь фильтровали через стеклянный вакуум-фильтр D4, загруженный цеолитом, для удаления катализатора 1. Фильтровальную лепешку промывали с помощью 6×45 мл гептана и 3×45 мл гамма-бутиролактона. Все маточные растворы и промывочные растворы собирали и объединяли. Фазы элюата отделяли. Фазу гамма-бутиролактона экстрагировали с помощью 4×65 мл гептана. Объединенные фазы гептана сушили над сульфатом натрия, и летучие соединения удаляли при пониженном давлении при 50-55°С / 5 мбар плюс 15 мин масляный вакуум-насос: 68,71 г неочищенного альфа-токоферола (89,7% площади согласно GC и 78,38% массы согласно GC) получали в виде прозрачного вязкого остатка темно-красного цвета. Это соответствует выходу 83%.
2.6 Получение 2,5,7,8-тетраметил-2-[4,8,12-триметилтридецил]-3,4-дигидро-2Н-хромен-6-ола (полностью рацемический альфа-токоферол) (Стадия 2; катализатор 1):
Эту реакцию проводили без удаления воды и, таким образом, без ловушки Дина-Старка. Используемое устройство состоит из колбы, лопастной мешалки и обратного конденсатора.
80,55 г (66,9 мл) пропиленкарбоната и 0,22 г катализатора 1 (высушенного при 120°С всю ночь, 50 мбар) / г ТМН (7,5 г катализатора 1) нагревали под потоком газообразного азота до 120-125°С и перемешивали в течение 15 мин. Суспензию затем охлаждали до <90°С и 34,52 г (225 ммоль) триметилгидрохинона (1,5 экв.) добавляли. Смесь затем снова нагревали до 120-125°С. Затем, 45,86 г (54,5 мл) изофитола (150 ммоль, 97% чистота, 1 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 2 ч (температура реакционной смеси: 120-125°С). После последующего времени реакции 4 ч при 123-124°С, при комнатной температуре всю ночь, и еще 2 ч при 124°С, реакционную смесь охлаждали до комнатной температуры. 45 мл гептана добавляли в реакционную смесь, которую перемешивали в течение 15 мин при комнатной температуре. Затем, смесь фильтровали через стеклянный вакуум-фильтр D4, загруженный цеолитом, для удаления катализатора 1. Фильтровальную лепешку промывали с помощью 6×45 мл гептана и 3×45 мл пропиленкарбоната. Все маточные растворы и промывочные растворы собирали и объединяли. Фазы элюата отделяли. Пропиленкарбонатную фазу экстрагировали с помощью 4×65 мл гептана. Объединенные фазы гептана сушили над сульфатом натрия, и летучие соединения удаляли при пониженном давлении при 50-55°С / 5 мбар плюс 15 мин масляный вакуум-насос: 66,04 г неочищенного альфа-токоферола (92,7% площади согласно GC и 84,55% массы согласно GC) получали в виде прозрачного вязкого остатка темно-красного цвета. Это соответствует выходу 86%.
2.7 Получение 2,5,7,8-тетраметил-2-[4,8,12-триметилтридецил]-3,4-дигидро-2Н-хромен-6-ола (полностью рацемический альфа-токоферол) (Стадия 2; катализатор 2):
103,14 г (78,08 мл) этиленкарбоната, 47,73 г (67,9 мл) н-октана (плюс 60 мл октана в ловушке Дина-Старка) и 0,65 г катализатора 2 (высушенного при 120°С всю ночь, 50 мбар) / г ТМН (22,5 г катализатора 2) нагревали под потоком газообразного азота с небольшим возвратом флегмы н-октана (температура реакционной смеси: 120-125°С) и перемешивали в течение 15 мин с возвратом флегмы. Суспензию затем охлаждали до <80°С и 34,38 г (225 ммоль) триметилгидрохинона (1,5 экв.) добавляли. Смесь затем снова нагревали до 120°С. Затем, 45,39 г (53,97 мл) изофитола (150 ммоль, 98% чистота, 1 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 2 ч (температура реакционной смеси: 120-125°С) при удалении воды, образующейся в ходе реакции, посредством дистилляции. После последующего времени реакции 6 ч при 123-126°С, реакционную смесь охлаждали до <60°С и 50 г пропиленкарбоната добавляли. Затем, реакционную смесь далее охлаждали до комнатной температуры и фильтровали через стеклянный вакуум-фильтр D4, загруженный цеолитом, для удаления катализатора 2. Фильтровальную лепешку промывали с помощью 3×45 мл октана и 3×45 мл пропиленкарбоната. Все маточные растворы и промывочные растворы собирали и объединяли. Фазы элюата отделяли. Фазу этилена/пропиленкарбоната экстрагировали с помощью 4×65 мл октана. Объединенные фазы октана сушили над сульфатом натрия, и летучие соединения удаляли при пониженном давлении при 50-55°С / 5 мбар плюс 15 мин масляный вакуум-насос: 63,66 г неочищенного альфа-токоферола (95,5% площади согласно GC и 91,44% массы согласно GC) получали в виде прозрачного вязкого остатка темно-красного цвета. Это соответствует выходу 90%.
2.8 Получение 2,5,7,8-тетраметил-2-[4,8,12-триметилтридецил]-3,4-дигидро-2Н-хромен-6-ола (полностью рацемический альфа-токоферол) (Стадия 2; катализатора 2):
232,63 г (193,21 мл) пропиленкарбоната, 118,06 г (167,9 мл) н-октана (плюс 60 мл октана в ловушке Дина-Старка) и 0,22 г катализатора 2 (высушенного при 120°С всю ночь, 50 мбар) / г ТМН (21,75 г катализатора 2) нагревали под потоком газообразного азота с небольшим возвратом флегмы н-октана (температура реакционной смеси: 120-125°С) и перемешивали в течение 15 мин с возвратом флегмы. Суспензию затем охлаждали до <80°С и 99,7 г (625,5 ммоль) триметилгидрохинона (1,5 экв.) добавляли. Смесь затем снова нагревали до 123°С. Затем, 137,22 г (163,17 мл) изофитола (435 ммоль, 94% чистота, 1 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 2 ч (температура реакционной смеси: 125-123°С) при удалении воды, образующейся в ходе реакции, посредством дистилляции. После последующего времени реакции 4 ч при 123 - 125°С, при комнатной температуре всю ночь, и еще 2 ч при 125°С, реакционную смесь охлаждали до комнатной температуры и фильтровали через стеклянный вакуум-фильтр D4, загруженный цеолитом, для удаления катализатора 2. Фильтровальную лепешку промывали с помощью 3×130 мл гептана и 3×130 мл пропиленкарбоната. Все маточные растворы и промывочные растворы собирали и объединяли. Фазы элюата отделяли. Пропиленкарбонатную фазу экстрагировали с помощью 4×190 мл гептана. Объединенные фазы гептана / н-октана сушили над сульфатом натрия, и летучие соединения удаляли при пониженном давлении при 50-55°С / 5 мбар плюс 15 мин масляный вакуум-насос: 199,16 г неочищенного альфа-токоферола (92,6% площади согласно GC и 87,57% массы согласно GC) получали в виде прозрачного вязкого остатка темно-красного цвета. Это соответствует выходу 93%.
2.9 Получение 2,5,7,8-тетраметил-2-[4,8,12-триметилтридецил]-3,4-дигидро-2Н-хромен-6-ола (полностью рацемический альфа-токоферол) (Стадия 2; катализатор 2):
80,55 г (66,9 мл) пропиленкарбоната и 0,22 г катализатора 2 (высушенного при 120°С всю ночь, 50 мбар) / г ТМН (7,5 г катализатора 2) нагревали до 50 - 90°С и 34,52 г (225 ммоль) триметилгидрохинона (1,5 экв.) добавляли. Смесь затем нагревали до 150°С и 45,67 г (54,3 мл) изофитола (150 ммоль, 97,4% чистота, 1 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 2 ч. После последующего времени реакции 6 ч реакционную смесь охлаждали до комнатной температуры. 45 мл гептана добавляли в реакционную смесь, которую перемешивали в течение 15 мин при комнатной температуре. Затем, смесь фильтровали через стеклянный вакуум-фильтр D4, загруженный цеолитом, для удаления катализатора 2. Фильтровальную лепешку промывали с помощью 6×45 мл гептана и 3×45 мл пропиленкарбоната. Все маточные растворы и промывочные растворы собирали и объединяли. Фазы элюата отделяли. Пропиленкарбонатную фазу экстрагировали с помощью 4×65 мл гептана. Объединенные фазы гептана сушили над сульфатом натрия, и летучие соединения удаляли при пониженном давлении при 50°С/5 мбар плюс 15 мин масляный вакуум-насос: 65,61 г неочищенного альфа-токоферола (92,5% площади согласно GC и 84,73% массы согласно GC) получали в виде прозрачного вязкого остатка темно-красного цвета. Это соответствует выходу 86%.
2.10 Получение 2,5,7,8-тетраметил-2-[4,8,12-триметилтридецил]-3,4-дигидро-2Н-хромен-6-ола (полностью рацемический альфа-токоферол) (Стадия 2; катализатор 2):
80,55 г (66,9 мл) пропиленкарбоната и 0,33 г катализатора 2 (высушенного при 120°С всю ночь, 50 мбар) / г ТМН (11,25 г катализатора 2) нагревали под потоком газообразного азота до 120-125°С и перемешивали в течение 15 мин. Суспензию затем охлаждали до <90°С и 34,52 г (225 ммоль) триметилгидрохинона (3 экв.) добавляли. Смесь затем снова нагревали до 120-125°С. Затем, 22,88 г (27,21 мл) изофитола (75 ммоль, 97,2% чистота, 1 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 1 ч (температура реакционной смеси: 121-125°С). После последующего времени реакции 6 ч при 120-125°С, реакционную смесь охлаждали до комнатной температуры. 45 мл гептана добавляли в реакционную смесь, которую перемешивали в течение 15 мин при комнатной температуре. Затем, смесь фильтровали через стеклянный вакуум-фильтр D4, загруженный цеолитом, для удаления катализатора 2. Фильтровальную лепешку промывали с помощью 6×45 мл гептана и 3×45 мл пропиленкарбоната. Все маточные растворы и промывочные растворы собирали и объединяли. Фазы элюата отделяли. Пропиленкарбонатную фазу экстрагировали с помощью 4×65 мл гептана. Объединенные фазы гептана сушили над сульфатом натрия и летучие соединения удаляли при пониженном давлении при 50°С / 5 мбар плюс 15 мин масляный вакуум-насос: 33.99 г неочищенного альфа-токоферола (95,2% площади согласно GC и 88,45% массы согласно GC) получали в виде прозрачного вязкого остатка темно-красного цвета. Это соответствует выходу 93%.
2.11 Получение 2,5,7,8-тетраметил-2-[4,8,12-триметилтридецил]-3,4-дигидро-2Н-хромен-6-ола (полностью рацемический альфа-токоферол) (Стадия 2; катализатор 2):
80,55 г (66,9 мл) пропиленкарбоната, 39,79 г (58,18 мл) н-гептана (плюс 60 мл гептана в ловушке Дина-Старка) и 0,22 г катализатора 2 (высушенного при 120°С всю ночь, 50 мбар) / г ТМН (7,5 г катализатора 2) нагревали под потоком газообразного азота до небольшого возврата флегмы н-гептана (температура реакционной смеси: 100°С) и перемешивали в течение 15 мин с возвратом флегмы. Суспензию затем охлаждали до <80°С и 34,52 г (225 ммоль) триметилгидрохинона (1,5 экв.) добавляли. Смесь затем снова нагревали до 100°С. Затем, 45,86 г (54,52 мл) изофитола (150 ммоль, 97% чистота, 1 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 2 ч (температура реакционной смеси: 100-102°С) при удалении воды, образующейся в ходе реакции, посредством дистилляции. После последующего времени реакции 6 ч при 103-104°С, при комнатной температуре всю ночь, и еще 2 ч при 102°С, реакционную смесь охлаждали до комнатной температуры и фильтровали через стеклянный вакуум-фильтр D4, загруженный цеолитом, для удаления катализатора 2. Фильтровальную лепешку промывали с помощью 3×45 мл гептана и 3×45 мл пропиленкарбоната. Все маточные растворы и промывочные растворы собирали и объединяли. Фазы элюата отделяли. Пропиленкарбонатную фазу экстрагировали с помощью 4×65 мл гептана. Объединенные фазы гептана / н-октана сушили над сульфатом натрия и летучие соединения удаляли при пониженном давлении при 50°С / 5 мбар плюс 15 мин масляный вакуум-насос: 63,99 г неочищенного альфа-токоферола (88,9% площади согласно GC и 85,26% массы согласно GC) получали в виде прозрачного вязкого остатка темно-красного цвета. Это соответствует выходу 84%.
2.12 Получение 2,5,7,8-тетраметил-2-[4,8,12-триметилтридецил]-3,4-дигидро-2Н-хромен-6-ола (полностью рацемический альфа-токоферол) (Стадия 2; катализатор 3):
80,55 г (66,9 мл) пропиленкарбоната и 0,22 г катализатора 3 (высушенного при 120°С всю ночь, 50 мбар) / г ТМН (7,5 г катализатора 3) нагревали под потоком газообразного азота до 120-125°С и перемешивали в течение 15 мин. Суспензию затем охлаждали до <90°С и 34,52 г (225 ммоль) триметилгидрохинона (1,5 экв.) добавляли. Смесь затем снова нагревали до 120-125°С. Затем, 45,86 г (54,52 мл) изофитола (150 ммоль, 97% чистота, 1 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 2 ч (температура реакционной смеси: 122-125°С). После последующего времени реакции 4 ч при 120-125°С, при комнатной температуре в течение выходных, и еще 2 ч при 120-125°С, реакционную смесь охлаждали до комнатной температуры. 45 мл гептана добавляли в реакционную смесь, которую перемешивали в течение 15 мин при комнатной температуре. Затем, смесь фильтровали через стеклянный вакуум-фильтр D4, загруженный цеолитом, для удаления катализатора 3. Фильтровальную лепешку промывали с помощью 6×45 мл гептана и 3×45 мл пропиленкарбоната. Все маточные растворы и промывочные растворы собирали и объединяли. Фазы элюата отделяли. Пропиленкарбонатную фазу экстрагировали с помощью 4×65 мл гептана. Объединенные фазы гептана сушили над сульфатом натрия и летучие соединения удаляли при пониженном давлении при 50°С / 5 мбар плюс 15 мин масляный вакуум-насос: 65,43 г неочищенного альфа-токоферола (94,1% площади согласно GC и 87,05% массы согласно GC) получали в виде прозрачного вязкого остатка темно-красного цвета. Это соответствует выходу 88%.
2.13 Получение 2,5,7,8-тетраметил-2-[4,8,12-триметилтридецил]-3,4-дигидро-2Н-хромен-6-ола (полностью рацемический альфа-токоферол) (Стадия 2; катализатор 4):
80,22 г (66,63 мл) пропиленкарбоната, 40,71 г (57,91 мл) н-октана (плюс 60 мл октана в ловушке Дина-Старка) и 0,65 г катализатора 4 (высушенного при 120°С всю ночь, 50 мбар) / г ТМН (22,5 г катализатора 4) нагревали под потоком газообразного азота с небольшим возвратом флегмы н-октана (температура реакционной смеси: 120 122°С) и перемешивали в течение 15 мин с возвратом флегмы. Суспензию затем охлаждали до <80°С и 34,38 г (225 ммоль) триметилгидрохинона (1,5 экв.) добавляли. Смесь затем снова нагревали до 120°С. Затем, 45,39 г (53,97 мл) изофитола (150 ммоль, 98% чистота, 1 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 2 ч (температура реакционной смеси: 118 120°С) при удалении воды, образующейся в ходе реакции, посредством дистилляции. После последующего времени реакции 6 ч при 121-124°С, реакционную смесь охлаждали до комнатной температуры и фильтровали через стеклянный вакуум-фильтр D4, загруженный цеолитом, для удаления катализатора 4. Фильтровальную лепешку промывали с помощью 3×45 мл н-октана и 3×45 мл пропиленкарбоната. Все маточные растворы и промывочные растворы собирали и объединяли. Фазы элюата отделяли. Пропиленкарбонатную фазу экстрагировали с помощью 4×65 мл н-октана. Объединенные фазы н-октана сушили над сульфатом натрия и летучие соединения удаляли при пониженном давлении при 55°С / 5 мбар плюс 15 мин масляный вакуум-насос: 64,06 г неочищенного альфа-токоферола (90,5% площади согласно GC и 86,86% массы согласно GC) получали в виде прозрачного вязкого остатка темно-красного цвета. Это соответствует выходу 86%.
2.14 Получение 2,5,7,8-тетраметил-2-[4,8,12-триметилтридецил]-3,4-дигидро-2Н-хромен-6-ола (полностью рацемический альфа-токоферол) (Стадия 2; катализатор 5):
80,22 г (66,63 мл) пропиленкарбоната, 40,71 г (57,91 мл) н-октана (плюс 60 мл октана в ловушке Дина-Старка) и 0,65 г катализатора 5 (высушенного при 120°С всю ночь, 50 мбар) / г ТМН (22,5 г катализатора 5) нагревали под потоком газообразного азота с небольшим возвратом флегмы н-октана (температура реакционной смеси: 120-121°С) и перемешивали в течение 15 мин с возвратом флегмы. Суспензию затем охлаждали до <80°С и 34,38 г (225 ммоль) триметилгидрохинона (1,5 экв.) добавляли. Смесь затем снова нагревали до 120°С. Затем, 45,39 г (53,97 мл) изофитола (150 ммоль, 98% чистота, 1 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 2 ч (температура реакционной смеси: 115-120°С) при удалении воды, образующейся в ходе реакции, посредством дистилляции. После последующего времени реакции 4 ч при 119-123°С, при комнатной температуре в течение выходных, и еще 2 ч при 122°С, реакционную смесь охлаждали до комнатной температуры и фильтровали через стеклянный вакуум-фильтр D4, загруженный цеолитом, для удаления катализатора 5. Фильтровальную лепешку промывали с помощью 3×45 мл н-октана и 3×45 мл пропиленкарбоната. Все маточные растворы и промывочные растворы собирали и объединяли. Фазы элюата отделяли. Пропиленкарбонатную фазу экстрагировали с помощью 4×65 мл н-октана. Объединенные фазы н-октана сушили над сульфатом натрия и летучие соединения удаляли при пониженном давлении при 55°С / 5 мбар плюс 15 мин масляный вакуум-насос: 65,08 г неочищенного альфа-токоферола (87,3% площади согласно GC и 85,15% массы согласно GC) получали в виде прозрачного вязкого остатка темно-красного цвета. Это соответствует выходу 86%.
2.15 Получение 2,5,7,8-тетраметил-2-[4,8,12-триметилтридецил]-3,4-дигидро-2Н-хромен-6-ола (полностью рацемический альфа-токоферол) (Стадия 2; катализатор 6):
80,55 г (66,9 мл) пропиленкарбоната и 0,33 г катализатора 6 (невысушенный) / г ТМН (11,25 г катализатора 6) нагревали под потоком газообразного азота до 120-125°С и перемешивали в течение 15 мин. Суспензию затем охлаждали до <90°С и 34,52 г (225 ммоль) триметилгидрохинона (1,5 экв.) добавляли. Смесь затем снова нагревали до 120-125°С. Затем, 45,67 г (54,3 мл) изофитола (150 ммоль, 97,4% чистота, 1 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 1 ч (температура реакционной смеси: 122-125°С). После последующего времени реакции 6 ч при 122-125°С, реакционную смесь охлаждали до комнатной температуры. 45 мл гептана и целит добавляли в реакционную смесь, которую перемешивали в течение 15 мин при комнатной температуре. Затем, смесь фильтровали через стеклянный вакуум-фильтр D4 с удалением целита и катализатора 6. Фильтровальную лепешку промывали с помощью 6×45 мл гептана и 3×45 мл пропиленкарбоната. Все маточные растворы и промывочные растворы собирали и объединяли. Фазы элюата отделяли. Пропиленкарбонатную фазу экстрагировали с помощью 4×65 мл гептана. Объединенные фазы гептана сушили над сульфатом натрия и летучие соединения удаляли при пониженном давлении при 50°С / 5 мбар плюс 15 мин масляный вакуум-насос: 64,75 г неочищенного альфа-токоферола (86,7% площади согласно GC и 81,19% массы согласно GC) получали в виде прозрачного вязкого остатка темно-красного цвета. Это соответствует выходу 81%.
2.16 Получение 2,5,7,8-тетраметил-2-[4,8,12-триметилтридецил]-3,4-дигидро-2Н-хромен-6-ола (полностью рацемический альфа-токоферол) (Стадия 2; катализатор 7):
80,55 г (66,9 мл) пропиленкарбоната и 0,33 г катализатора 7 (невысушенный) / г ТМН (11,25 г катализатора 7) нагревали под потоком газообразного азота до 120-122°С и перемешивали в течение 15 мин. Суспензию затем охлаждали до <90°С и 34,52 г (225 ммоль) триметилгидрохинона (1,5 экв.) добавляли. Смесь затем снова нагревали до 120-125°С. Затем, 45,67 г (54,3 мл) изофитола (150 ммоль, 97,4% чистота, 1 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 1 ч (температура реакционной смеси: 120-124°С). После последующего времени реакции 6 ч при 120-125°С, реакционную смесь охлаждали до комнатной температуры. 45 мл гептана добавляли в реакционную смесь, которую перемешивали в течение 15 мин при комнатной температуре. Затем, смесь фильтровали через стеклянный вакуум-фильтр D4, загруженный цеолитом, для удаления катализатора 7. Фильтровальную лепешку промывали с помощью 6×45 мл гептана и 3×45 мл пропиленкарбоната. Все маточные растворы и промывочные растворы собирали и объединяли. Фазы элюата отделяли. Пропиленкарбонатную фазу экстрагировали с помощью 4×65 мл гептана. Объединенные фазы гептана сушили над сульфатом натрия и летучие соединения удаляли при пониженном давлении при 50°С / 5 мбар плюс 15 мин масляный вакуум-насос: 64,76 г неочищенного альфа-токоферола (91,8% площади согласно GC и 86,01% массы согласно GC) получали в виде прозрачного вязкого остатка темно-красного цвета. Это соответствует выходу 86%.
2.17 Получение 2,5,7,8-тетраметил-2-[4,8,12-триметилтридецил]-3,4-дигидро-2Н-хромен-6-ола (полностью рацемический альфа-токоферол) (Стадия 2; катализатор 8):
80,55 г (66,9 мл) пропиленкарбоната и 0,44 г катализатора 8 (невысушенный) / г ТМН (15 г катализатора 8) нагревали под потоком газообразного азота до 120°С и перемешивали в течение 15 мин. Суспензию затем охлаждали до <90°С и 34,52 г (225 ммоль) триметилгидрохинона (1,5 экв.) добавляли. Смесь затем снова нагревали до 120-125°С. Затем, 45,67 г (54,3 мл) изофитола (150 ммоль, 97,4% чистота, 1 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 2 ч (температура реакционной смеси: 121-125°С). После последующего времени реакции 4 ч при 121-125°С, при комнатной температуре всю ночь, и еще 2 ч при 120-125°С, реакционную смесь охлаждали до комнатной температуры. 45 мл гептана добавляли в реакционную смесь, которую перемешивали в течение 15 мин при комнатной температуре. Затем, смесь фильтровали через стеклянный вакуум-фильтр D4, загруженный цеолитом, для удаления катализатора 8. Фильтровальную лепешку промывали с помощью 6×45 мл гептана и 4×45 мл пропиленкарбоната. Все маточные растворы и промывочные растворы собирали и объединяли. Фазы элюата отделяли. Пропиленкарбонатную фазу экстрагировали с помощью 4×65 мл гептана. Объединенные фазы гептана сушили над сульфатом натрия и летучие соединения удаляли при пониженном давлении при 50°С / 5 мбар плюс 15 мин масляный вакуум-насос: 64,27 г неочищенного альфа-токоферола (75,8% площади согласно GC и 87,13% массы согласно GC) получали в виде прозрачного вязкого остатка темно-красного цвета. Это соответствует выходу 87%.
2.18 Получение 2,5,7,8-тетраметил-2-[4,8,12-триметилтридецил]-3,4-дигидро-2Н-хромен-6-ола (полностью рацемический альфа-токоферол) (Стадия 2; катализатор 9):
80,55 г (66,9 мл) пропиленкарбоната и 0,33 г катализатора 9 (невысушенный) / г ТМН (11,25 г катализатора 9) нагревали под потоком газообразного азота до 120°С и перемешивали в течение 15 мин. Суспензию затем охлаждали до <90°С и 34,52 г (225 ммоль) триметилгидрохинона (1,5 экв.) добавляли. Смесь затем снова нагревали до 120-125°С. Затем, 45,67 г (54,3 мл) изофитола (150 ммоль, 97,4% чистота, 1 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 1 ч (температура реакционной смеси: 123-125°С). После последующего времени реакции 6 ч при 125°С реакционную смесь анализировали: 30% площади согласно GC ТМН, 5% площади согласно GC фитадиенонов, 19% площади согласно GC фитил-ТМН, без альфа-токоферола. Реакционную смесь удаляли.
2.19 Непрерывное получение 2,5,7,8-тетраметил-2-[4,8,12-триметилтридецил]-3,4-дигидро-2Н-хромен-6-ола (полностью рацемический альфа-токоферол) (Стадия 2; катализатор 9)
Применяемый катализатор:
Монтмориллонит К10 (рН 3-4, площадь поверхности) «разбавленный» целитом в массовом соотношении 1: 1 от компании Thales Nano, упакованный в картридж (так называемый CatCart). Используемый картридж CatCart имел размер 70 × 4 мм и массу заполнения 459 мг.
Непрерывная процедура получения:
8 мас. % раствора ТМН в бис(2-метоксиэтиловом) простом эфире (=диглим) и 10,4 мас. % раствора изофитола в бис(2-метоксиэтиловом) просом эфире (=диглим) перекачивали с объемным расходом 5 мл/ч. на описанный выше CatCart, заполненный К10 и целитом, и нагревали до 200°С. Катализатор CatCart находится в вертикальном положении.
За ходом реакции следили с помощью GC: 61,3% площади согласно GC 2,5,7,8-тетраметил-2-[4,8,12-триметилтридецил]-3,4-дигидро-2Н-хромен-6-ола (полностью рацемический альфа-токоферол), получено 19,4% площади согласно GC ТМН и 13,5% площади согласно GC фитадиенов.
2.20 Получение полностью рацемического альфа-токоферол ацетата (Стадии 1, 2 и 3):
Получение 2,3,5-триметилгидрохинона
14 г 2,3,5-триметилхинона (99,6%, 92,85 ммоль) растворяли в 79,7 г (66,2 мл) пропиленкарбоната при комнатной температуре. 2,8 г палладия на активированном угле (10%, 2,63 ммоль, 0,03 экв.) добавляли, и полученную реакционную смесь гидрировали в течение 22,5 ч при давлении водорода 8 бар и при 87-93°С. Реакционную смесь фильтровали при температуре и в атмосфере азота с применением теплого стеклянного вакуум-фильтра D4 с бумажным фильтром и получали 84,31 г элюата (95,3% площади согласно GC триметилгидрохинона и 0,28% площади согласно GC триметилхинона).
Получение полностью рацемического альфа-токоферола (катализатор 2)
К неочищенному триметилгидрохинону в пропиленкарбонате, полученному на первой стадии, (82,83 г, 1,73 экв., принимают выход 100%, 92,85 ммоль) 0,19 г катализатора 2 (высушенного при 120°С всю ночь, 50 мбар)/г ТМН (2,69 г катализатора 2) добавляли и нагревали до 120°С и перемешивали в течение 15 мин при 120-124°С. Затем, 16,38 г (19,47 мл) изофитола (53,8 ммоль, 97,4% чистота, 1 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 2 ч (температура реакционной смеси: 124-125°С) при удалении воды, образующейся в ходе реакции, посредством дистилляции. После последующего времени реакции 5 ч при 125°С реакционную смесь перемешивали всю ночь при комнатной температуре.
Получение полностью рацемического альфа-токоферол ацетата (катализатор 2)
Суспензию коричневого цвета, полученную на второй стадии, нагревали до 50°С и 22,19 г (20,55 мл) уксусного ангидрида (220 ммоль, 4 экв.) непрерывно добавляли в течение периода времени, равного 15 мин. Затем, реакционную смесь нагревали до 100°С и перемешивали в течение 4 ч. После охлаждения до комнатной температуры 20 мл гептана добавляли в реакционную смесь, которую перемешивали в течение 15 мин. Затем, реакционную смесь фильтровали через стеклянный вакуум-фильтр D4, загруженный цеолитом, для удаления катализатора 2. Фильтровальную лепешку промывали с помощью 6×20 мл гептана и 3×20 мл пропиленкарбоната. Все маточные растворы и промывочные растворы собирали и объединяли. Фазы элюата отделяли. Пропиленкарбонатную фазу экстрагировали с помощью 4×25 мл гептана. Объединенные фазы гептана сушили над сульфатом натрия и летучие соединения удаляли при пониженном давлении при 50°С / 5 мбар плюс 15 мин масляный вакуум-насос: 25,71 г неочищенного полностью рацемического альфа-токоферол ацетата (89,28% площади согласно GC) получали в виде прозрачного вязкого остатка коричневого цвета. Это соответствует выходу 90% (на основе % площади согласно GC за 3 стадии).
2.21 Получение полностью рацемического альфа-токоферол ацетата (Стадии 2 и 3):
Получение полностью рацемического альфа-токоферола (катализатор 2)
100,29 г (83,3 мл) пропиленкарбоната и 35,15 г катализатора 2 добавляли и нагревали до 120-125°С и перемешивали в течение 15 мин. Суспензию затем охлаждали до <90°С и 22,92 г (150 ммоль) триметилгидрохинона (1,0 экв.) добавляли. Смесь затем снова нагревали до 120°С. Затем, 46,75 г (55,59 мл) изофитола (154,5 ммоль, 98% чистота, 1,03 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 2 ч (температура реакционной смеси: 120-123°С) при удалении воды, образующейся в ходе реакции, посредством дистилляции. После последующего времени реакции 4 ч при 123-124°С реакционную смесь перемешивали в течение выходных при комнатной температуре.
Получение полностью рацемического альфа-токоферол ацетата (катализатор 2)
К суспензии коричневого цвета, полученной на предшествующей стадии, 30,94 г (28,64 мл) уксусного ангидрида (300 ммоль, 2 экв.) непрерывно добавляли в течение периода времени, равного 10 мин. Затем, реакционную смесь подвергали реактивной дистилляции в течение 2 ч (500 мбар, внутренняя температура 87 92°С, температура перехода 28-33°С, температура масляной бани 100°С). Затем, реакционную смесь доводили до комнатной температуры и 45 мл гептана добавляли, и реакционную смесь перемешивали в течение 15 мин. Затем, фильтровали через стеклянный вакуум-фильтр D4 для удаления катализатора 2. Фильтровальную лепешку промывали с помощью 3×45 мл гептана и 3×45 мл пропиленкарбоната. Все маточные растворы и промывочные растворы собирали и объединяли. Фазы элюата отделяли. Пропиленкарбонатную фазу экстрагировали с помощью 4×65 мл гептана. Объединенные фазы гептана сушили над сульфатом натрия, и летучие соединения удаляли при пониженном давлении при 50°С / 5 мбар плюс 15 мин масляный вакуум-насос: 71,79 г неочищенного полностью рацемический альфа-токоферол ацетата (88,08% площади согласно GC) получали в виде коричневого прозрачного вязкого остатка, содержащего 0,18% площади согласно GC альфа-токоферол. Это соответствует выходу 89% (на основе % площади согласно GC за 2 стадии).
2.22 Влияние температуры сушки катализатора на основе бентонита:
Был оценен эффект сушки катализатора на основе бентонита перед его использованием в реакции конденсации ТМН с IP. Две реакции проводили по аналогии с примером 2,8, за исключением того, что здесь применяли 225 ммоль ТМН вместо 625,5 ммоль ТМН (количество других реагентов было адаптировано соответственно). Результаты представлены в таблице 1.

В реакции ТМН с IP, катализируемой бентонитом, сушка катализатора на основе бентонита перед его использованием может улучшить оборот катализатора в условиях реакции, применяемых в данном случае (например, применяемой смеси растворителей РС/октан).
Выход альфа-токоферола увеличивается с увеличением температуры сушки катализатора 2 на основе бентонита от 79% (невысушенный катализатор 2) до 93% (катализатор 2 высушен при 120°С, 50 мбар, вакуумная сушильная печь, всю ночь).
Пример 2.22.1:
Получение полностью рацемического альфа-токоферола
80,22 г (66,6 мл) пропиленкарбоната, 40,71 г (57,91 мл) н-октана (плюс 60 мл октана в ловушке Дина-Старка) и 0,74 г катализатора 2 (невысушенный, как есть) / г ТМН (25,43 г катализатора 2) нагревали под потоком газообразного азота до небольшого возврата флегмы (температура реакционной смеси: 109°С) и перемешивали в течение 15 мин с возвратом флегмы. Суспензию затем охлаждали до <80°С и 34,38 г (225 ммоль) триметилгидрохинона (1,5 экв.) добавляли. Смесь затем снова нагревали до небольшого возврата флегмы при 113°С. Затем, 45,39 г (53,97 мл) изофитола (150 ммоль, 98% чистота, 1 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 2 ч (температура реакционной смеси: 113-112°С) при удалении воды, образующейся в ходе реакции, посредством дистилляции. После последующего времени реакции 6 ч при 118-123°С, реакционную смесь охлаждали до комнатной температуры и фильтровали через стеклянный вакуум-фильтр D4, загруженный цеолитом, для удаления катализатора 2. Фильтровальную лепешку промывали с помощью 3×45 мл н-октана и 3×45 мл пропиленкарбоната. Все маточные растворы и промывочные растворы собирали и объединяли. Фазы элюата отделяли. Пропиленкарбонатную фазу экстрагировали с помощью 4×65 мл н-октана. Объединенные фазы н-октана сушили над сульфатом натрия и летучие соединения удаляли при пониженном давлении при 55°С / 5 мбар плюс 15 мин масляный вакуум-насос: 59,31 г неочищенного альфа-токоферола (89,7% площади согласно GC и 85,47% массы согласно GC) получали в виде прозрачного вязкого остатка темно-красного цвета. Это соответствует выходу 79%.
Пример 2.22.2:
Получение полностью рацемического альфа-токоферола
80,22 г (66,6 мл) пропиленкарбоната, 40,71 г (57,91 мл) н-октана (плюс 60 мл октана в ловушке Дина-Старка) и 0,65 г катализатора 2 (высушенного при 60°С, всю ночь, 50 мбар) / г ТМН (22,5 г катализатора 2) нагревали под потоком газообразного азота од небольшого возврата флегмы (температура реакционной смеси: 115-117°С) и перемешивали в течение 15 мин с возвратом флегмы. Суспензию затем охлаждали до <80°С и 34,38 г (225 ммоль) триметилгидрохинона (1,5 экв.) добавляли. Смесь затем снова нагревали до небольшого возврата флегмы при 118°С. Затем, 45,39 г (53,97 мл) изофитола (150 ммоль, 98% чистота, 1 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 2 ч (температура реакционной смеси: 119-117°С) при удалении воды, образующейся в ходе реакции, посредством дистилляции. После последующего времени реакции 6 ч при 120 124°С, реакционную смесь охлаждали до комнатной температуры и фильтровали через стеклянный вакуум-фильтр D4, загруженный цеолитом, для удаления катализатора 2. Фильтровальную лепешку промывали с помощью 3×45 мл н-октана и 3×45 мл пропиленкарбоната. Все маточные растворы и промывочные растворы собирали и объединяли. Фазы элюата отделяли. Пропиленкарбонатную фазу экстрагировали с помощью 4×65 мл н-октана. Объединенные фазы н-октана сушили над сульфатом натрия и летучие соединения удаляли при пониженном давлении при 55°С / 5 мбар плюс 15 мин масляный вакуум-насос: 61,10 г неочищенного альфа-токоферола (91,1% площади согласно GC и 86,31% массы согласно GC) получали в виде прозрачного вязкого остатка темно-красного цвета. Это соответствует выходу 82%.
Пример 2.22.3:
Получение полностью рацемического альфа-токоферола
80,22 г (66,6 мл) пропиленкарбоната, 40,71 г (57,91 мл) н-октана (плюс 60 мл октана в ловушке Дина-Старка) и 0,65 г катализатора 2 (высушенного при 100°С, всю ночь, 50 мбар) / г ТМН (22,5 г катализатора 2) нагревали под потоком газообразного азота до небольшого возврата флегмы (температура реакционной смеси: 120-121°С) и перемешивали в течение 15 мин с возвратом флегмы. Суспензию затем охлаждали до <80°С и 34,38 г (225 ммоль) триметилгидрохинона (1,5 экв.) добавляли. Смесь затем снова нагревали до небольшого возврата флегмы при 122°С. Затем, 45,39 г (53,97 мл) изофитола (150 ммоль, 98% чистота, 1 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 2 ч (температура реакционной смеси: 122-121°С) при удалении воды, образующейся в ходе реакции, посредством дистилляции. После последующего времени реакции 6 ч при 123-125°С, реакционную смесь охлаждали до комнатной температуры и фильтровали через стеклянный вакуум-фильтр D4, загруженный цеолитом, для удаления катализатора 2. Фильтровальную лепешку промывали с помощью 3×45 мл н-октана и 3×45 мл пропиленкарбоната. Все маточные растворы и промывочные растворы собирали и объединяли. Фазы элюата отделяли. Пропиленкарбонатную фазу экстрагировали с помощью 4×65 мл н-октана. Объединенные фазы н-октана сушили над сульфатом натрия и летучие соединения удаляли при пониженном давлении при 55°С / 5 мбар плюс 15 мин масляный вакуум-насос: 65,91 г неочищенного альфа-токоферола (92,1% площади согласно GC и 86,47% массы согласно GC) получали в виде прозрачного вязкого остатка темно-красного цвета. Это соответствует выходу 88%.
Пример 2.22.4:
Получение полностью рацемического альфа-токоферола
80,22 г (66,6 мл) пропиленкарбоната, 40,71 г (57,91 мл) н-октана (плюс 60 мл октана в ловушке Дина-Старка) и 0,65 г катализатора 2 (высушенного при 120°С, всю ночь, 50 мбар) / г ТМН (22,5 г катализатора 2) нагревали под потоком газообразного азота до небольшого возврата флегмы (температура реакционной смеси: 120-121°С) и перемешивали в течение 15 мин с возвратом флегмы. Суспензию затем охлаждали до <80°С и 34,38 г (225 ммоль) триметилгидрохинона (1,5 экв.) добавляли. Смесь затем снова нагревали до небольшого возврата флегмы при 121°С. Затем, 45,39 г (53,97 мл) изофитола (150 ммоль, 98% чистота, 1 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 2 ч (температура реакционной смеси: 115-121°С) при удалении воды, образующейся в ходе реакции, посредством дистилляции. После последующего времени реакции 6 ч при 118-123°С, реакционную смесь охлаждали до комнатной температуры и фильтровали через стеклянный вакуум-фильтр D4, загруженный цеолитом, для удаления катализатора 2. Фильтровальную лепешку промывали с помощью 3×45 мл н-октана и 3×45 мл пропиленкарбоната. Все маточные растворы и промывочные растворы собирали и объединяли. Фазы элюата отделяли. Пропиленкарбонатную фазу экстрагировали с помощью 4×65 мл н-октана. Объединенные фазы н-октана сушили над сульфатом натрия и летучие соединения удаляли при пониженном давлении при 55°С / 5 мбар плюс 15 мин масляный вакуум-насос: 68,47 г неочищенного альфа-токоферола (91,2% площади согласно GC и 87,93% массы согласно GC) получали в виде прозрачного вязкого остатка темно-красного цвета. Это соответствует выходу 93%.
2.23 Рециклизация катализатора на основе бентонита на стадии 2:
Была оценена возможность рециркуляции катализатора на основе бентонита, применяемого в реакции алкилирования по Фриделю-Крафтсу и конденсации. Было проведено несколько реакций, как описано ниже (пример 2.23.1), в которых катализатор на основе бентонита 2 извлекали в каждой реакции и повторно использовали в следующей идентичной реакции.
Рециркуляция катализатора на основе бентонита 2 была пятикратной для реакции алкилирования по Фриделю-Крафтсу и конденсации в идентичных условиях (см. Примеры 2.23.1 - 2.23.6 в таблице 2). Выход может быть воспроизведен в пределах ошибки.
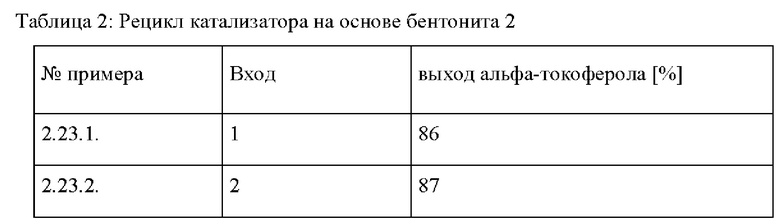
Пример 2.23.1:
Получение полностью рацемического альфа-токоферола
80,22 г (66,63 мл) пропиленкарбоната, 40,71 г (57,91 мл) н-октана (плюс 60 мл октана в ловушке Дина-Старка) и 0,65 г катализатора 2 (высушенного при 120°С, всю ночь, 50 мбар) / г ТМН (22,5 г катализатора 2) нагревали под потоком газообразного азота с небольшим возвратом флегмы н-октана (температура реакционной смеси: 120-121°С) и перемешивали в течение 15 мин с возвратом флегмы. Суспензию затем охлаждали до <80°С и 34,38 г (225 ммоль) триметилгидрохинона (1,5 экв.) добавляли. Смесь затем снова нагревали до 121°С. Затем, 45,39 г (53,97 мл) изофитола (150 ммоль, 98% чистота, 1 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 2 ч (температура реакционной смеси: 116-121°С) при удалении воды, образующейся в ходе реакции, посредством дистилляции. После последующего времени реакции 4 ч при 120-122°С, при комнатной температуре всю ночь, и еще 2 ч при 122°С, реакционную смесь охлаждали до комнатной температуры и фильтровали через стеклянный вакуум-фильтр D4 для удаления катализатора 2. Фильтровальную лепешку промывали с помощью 3×45 мл н-октана и 3×45 мл пропиленкарбоната. Остаток отсасывали до сухости и далее сушили в потоке азота всю ночь (влажная масса: 40,20 г, сухая масса: 38,11 г). Рециклизованный таким образом катализатор на основе бентонита 2 снова применяли в примере 2.23.2.
Все маточные растворы и промывочные растворы собирали и объединяли. Фазы элюата отделяли. Пропиленкарбонатную фазу экстрагировали с помощью 4×65 мл н-октана. Объединенные фазы н-октана сушили над сульфатом натрия и летучие соединения удаляли при пониженном давлении при 55°С / 5 мбар плюс 15 мин масляный вакуум-насос: 64,90 г неочищенного альфа-токоферола (92,0% площади согласно GC и 86,02% массы согласно GC) получали в виде красного прозрачного вязкого остатка. Это соответствует выходу 86%.
Пример 2.23.2:
Получение полностью рацемического альфа-токоферола
80,22 г (66,63 мл) пропиленкарбоната, 40,71 г (57,91 мл) н-октана (плюс 60 мл октана в ловушке Дина-Старка) и 38,11 г катализатора 2 из примера 2.23.1. нагревали под потоком газообразного азота с небольшим возвратом флегмы н-октана (температура реакционной смеси: 120-124°С) и перемешивали в течение 15 мин с возвратом флегмы. Суспензию затем охлаждали до <80°С и 34,38 г (225 ммоль) триметилгидрохинона (1,5 экв.) добавляли. Смесь затем снова нагревали до 122°С. Затем, 45,39 г (53,97 мл) изофитола (150 ммоль, 98% чистота, 1 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 2 ч (температура реакционной смеси: 119-122°С) при удалении воды, образующейся в ходе реакции, посредством дистилляции. После последующего времени реакции 2 ч при 122°С, при комнатной температуре всю ночь, и еще 4 ч при 123-124°С, реакционную смесь охлаждали до комнатной температуры и фильтровали через стеклянный вакуум-фильтр D4 для удаления катализатора 2. Фильтровальную лепешку промывали с помощью 3×45 мл н-октана и 3×45 мл пропиленкарбоната. Остаток отсасывали до сухости и далее сушили в потоке азота в течение выходных (сухая масса: 33,33 г). Рециклизованный таким образом катализатор на основе бентонита 2 снова применяли в примере 2.23.3.
Все маточные растворы и промывочные растворы собирали и объединяли. Фазы элюата отделяли. Пропиленкарбонатную фазу экстрагировали с помощью 4×65 мл гептана. Объединенные фазы гептана/н-октана сушили над сульфатом натрия и летучие соединения удаляли при пониженном давлении при 55°С / 5 мбар плюс 15 мин масляный вакуум-насос: 65,62 г неочищенного альфа-токоферола (92,6% площади согласно GC и 86,04% массы согласно GC) получали в виде красного, прозрачного вязкого остатка. Это соответствует выходу 87%.
Примеры 2.23.3 - 2.23.6 проводят как описано в Примере 2.23.2.
2.24 Рецикл катализатора на основе бентонита на стадии 2 и 3:
Пример 2.24.1:
Получение полностью рацемического альфа-токоферола
100,29 г (83,3 мл) пропиленкарбоната и 1,01 г катализатора 2 (высушенного при 120°С, всю ночь, 50 мбар) / г ТМН (23,17 г катализатора 2) добавляли и нагревали до 123-124°С и перемешивали в течение 15 мин. Суспензию затем охлаждали до <90°С и 22,92 г (150 ммоль) триметилгидрохинона (1,0 экв.) добавляли. Смесь затем снова нагревали до 120°С. Затем, 46,75 г (55,59 мл) изофитола (154,5 ммоль, 98% чистота, 1,03 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 2 ч (температура реакционной смеси: 120-121°С) при удалении воды, образующейся в ходе реакции, посредством дистилляции. После последующего времени реакции 4 ч при 120-125°С реакционную смесь перемешивали всю ночь при комнатной температуре.
Получение полностью рацемического альфа-токоферол ацетата
К суспензии коричневого цвета, полученной на предшествующей стадии, 30,94 г (28,64 мл) уксусного ангидрида (300 ммоль, 2 экв.) непрерывно добавляли в течение периода времени, равного 10 мин. Затем, реакционную смесь подвергали реактивной дистилляции в течение 1 ч (310-335 мбар, внутренняя температура 74-88°С, температура перехода 28-45°С, температура масляной бани 100°С). Реакционную смесь перемешивали при комнатной температуре при нормальном давлении в течение выходных. Затем, реакционную смесь далее подвергали реактивной дистилляции в течение 1 ч (310-335 мбар, внутренняя температура 64-67°С, температура перехода 32-34°С, температура масляной бани 75°С). Затем, реакционную смесь доводили до комнатной температуры и 45 мл гептана добавляли, и реакционную смесь перемешивали в течение 15 мин. Затем, фильтровали через стеклянный вакуум-фильтр D4 для удаления катализатора 2. Фильтровальную лепешку промывали с помощью 3×45 мл гептана и 3×45 мл пропиленкарбоната. Остаток отсасывали до сухости и далее сушили в потоке азота в течение 3 дней (влажная масса: 42,76 г, сухая масса: 35,15 г). Рециклизованный таким образом катализатор на основе бентонита 2 снова применяли в примере 2.24.2.
Все маточные растворы и промывочные растворы собирали и объединяли. Фазы элюата отделяли. Пропиленкарбонатную фазу экстрагировали с помощью 4×65 мл гептана. Объединенные фазы гептана сушили над сульфатом натрия, и летучие соединения удаляли при пониженном давлении при 50°С / 5 мбар плюс 15 мин масляный вакуум-насос: 68,80 г неочищенного полностью рацемического альфа-токоферол ацетата (79,90% площади согласно GC) получали в виде охристо-желтого прозрачного вязкого остатка, содержащего 8,34% площади согласно GC альфа-токоферол. Это соответствует выходу 78% (на основе % площади согласно GC за 2 стадии).
Пример 2.24.2:
Получение полностью рацемического альфа-токоферола
100,29 г (83,3 мл) пропиленкарбоната и 35,15 г катализатора 2 из примера 2.24.1 добавляли и нагревали до 120-125°С и перемешивали в течение 15 мин. Суспензию затем охлаждали до <90°С и 22,92 г (150 ммоль) триметилгидрохинона (1,0 экв.) добавляли. Смесь затем снова нагревали до 120°С. Затем, 46,75 г (55,59 мл) изофитола (154,5 ммоль, 98% чистота, 1,03 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 2 ч (температура реакционной смеси: 120-123°С) при удалении воды, образующейся в ходе реакции, посредством дистилляции. После последующего времени реакции 4 ч при 123-124°С реакционную смесь перемешивали в течение выходных при комнатной температуре.
Получение полностью рацемического альфа-токоферол ацетата
К суспензии коричневого цвета, полученной на предшествующей стадии, 30,94 г (28,64 мл) уксусного ангидрида (300 ммоль, 2 экв.) непрерывно добавляли в течение периода времени, равного 10 мин. Затем, реакционную смесь подвергали реактивной дистилляции в течение 2 ч (500 мбар, внутренняя температура 87-92°С, температура перехода 28-33°С, температура масляной бани 100°С). Затем, реакционную смесь доводили до комнатной температуры и 45 мл гептана добавляли, и реакционную смесь перемешивали в течение 15 мин. Затем, фильтровали через стеклянный вакуум-фильтр D4 для удаления катализатора 2. Фильтровальную лепешку промывали с помощью 3×45 мл гептана и 3×45 мл пропиленкарбоната. Все маточные растворы и промывочные растворы собирали и объединяли. Фазы элюата отделяли. Пропиленкарбонатную фазу экстрагировали с помощью 4×65 мл гептана. Объединенные фазы гептана сушили над сульфатом натрия, и летучие соединения удаляли при пониженном давлении при 50°С / 5 мбар плюс 15 мин масляный вакуум-насос: 71,79 г неочищенного полностью рацемического альфа-токоферол ацетата (88,08% площади согласно GC) получали в виде коричневого прозрачного вязкого остатка, содержащего 0,18% площади согласно GC альфа-токоферола. Это соответствует выходу 89% (на основе % площади согласно GC за 2 стадии).
2.25 Рециклизация ТМН, применяемого в избытке, в карбонатном растворителе:
Была оценена возможность рециркуляции непрореагировавшего избытка ТМН вместе с карбонатным растворителем, применяемым в реакции алкилирования по Фриделю-Крафтсу и конденсации. Две реакции были проведены по аналогии с примером 2,8, за исключением того, что здесь применяли 225 ммоль ТМН вместо 625,5 ммоль ТМН (количества других реагентов были адаптированы соответственно) и что непрореагировавший избыток ТМН извлекали в карбонатном растворителе после завершения реакции и повторно использовали в следующей идентичной реакции. В обеих реакциях полученные выходы были идентичными (примеры 2.25.1 и 2.25.2 ниже, выход 87% в каждом случае). Таким образом, непрореагировавший избыток ТМН может быть успешно рециклизован вместе с карбонатным растворителем.
Пример 2.25.1.
Получение полностью рацемического альфа-токоферол
80,55 г (66,9 мл) пропиленкарбоната, 40,9 г (58,18 мл) н-октана (плюс 60 мл октана в ловушке Дина-Старка) и 0,22 г катализатора 2 (высушенного при 120°С, всю ночь, 50 мбар) / г ТМН (7,5 г катализатора 2) нагревали под потоком газообразного азота с небольшим возвратом флегмы н-октана (температура реакционной смеси: 120-125°С) и перемешивали в течение 15 мин с возвратом флегмы. Суспензию затем охлаждали до <80°С и 34,52 г (225 ммоль) триметилгидрохинона (1,5 экв.) добавляли. Смесь затем снова нагревали до 121°С. Затем, 45,86 г (54,52 мл) изофитола (150 ммоль, 97% чистота, 1 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 2 ч (температура реакционной смеси: 120-125°С) при удалении воды, образующейся в ходе реакции, посредством дистилляции. После последующего времени реакции 4 ч при 124-125°С, при комнатной температуре всю ночь, и еще 2 ч при 125°С, реакционную смесь охлаждали до комнатной температуры и фильтровали через стеклянный вакуум-фильтр D4, загруженный цеолитом, для удаления катализатора 2. Фильтровальную лепешку промывали с помощью 3×45 мл н-октана и 3×45 мл пропиленкарбоната. Все маточные растворы и промывочные растворы собирали и объединяли. Фазы элюата отделяли. Пропиленкарбонатную фазу экстрагировали с помощью 4×65 мл н-октана. Объединенные фазы н-октана сушили над сульфатом натрия и летучие соединения удаляли при пониженном давлении при 55°С / 5 мбар плюс 15 мин масляный вакуум-насос: 64,58 г неочищенного альфа-токоферола (93,1% площади согласно GC и 87,24% массы согласно GC) получали в виде прозрачного вязкого остатка темно-красного цвета. Это соответствует выходу 87%. Кроме того, 172,31 г красной прозрачной пропиленкарбонатной фазы, содержащей 93,6% площади согласно GC, 6,2% массы согласно ВЭЖХ непрореагировавшего ТМН получали. Это соответствует извлечению 93% ТМН (70 ммоль из 75 ммоль применяемого ТМН в избытке). 85,87 г этой пропиленкарбонатной фазы рециклизовали в Пример 2.25.2.

Пример 2.25.2:
Получение полностью рацемического альфа-токоферола
85,87 г пропиленкарбонатной фазы из примера 2.25.1 (содержащей 5,32 г, 34,96 ммоль, 0,23 экв ТМН), 40,9 г (58,18 мл) н-октана (плюс 60 мл октана в ловушке Дина-Старка) и 0,22 г катализатора 2 (высушенного при 120°С, всю ночь, 50 мбар) / г ТМН (7,5 г катализатора 2) нагревали под потоком газообразного азота с небольшим возвратом флегмы н-октана (температура реакционной смеси: 120-123°С) и перемешивали в течение 15 мин с возвратом флегмы. Суспензию затем охлаждали до <80°С и 29,16 г (190,04 ммоль) триметилгидрохинона (1,23 экв., в общем: 1,46 экв.) добавляли. Смесь затем снова нагревали до 120°С. Затем, 47,2 г (56,12 мл) изофитола (154,4 ммоль, 97% чистота, 1 экв.) непрерывно добавляли в реакционную смесь в течение периода времени, равного 2 ч (температура реакционной смеси: 120-125°С) при удалении воды, образующейся в ходе реакции, посредством дистилляции. После последующего времени реакции 4 ч при 124-125°С, при комнатной температуре всю ночь, и еще 2 ч при 125°С, реакционную смесь охлаждали до комнатной температуры и фильтровали через стеклянный вакуум-фильтр D4, загруженный цеолитом, для удаления катализатора 2. Фильтровальную лепешку промывали с помощью 3×45 мл н-октана и 3×45 мл пропиленкарбоната. Все маточные растворы и промывочные растворы собирали и объединяли. Фазы элюата отделяли. Пропиленкарбонатную фазу экстрагировали с помощью 4×65 мл н-октана. Объединенные фазы н-октана сушили над сульфатом натрия, и летучие соединения удаляли при пониженном давлении при 55°С / 5 мбар плюс 15 мин масляный вакуум-насос: 65,70 г неочищенного альфа-токоферола (92,9% площади согласно GC и 87,71% массы согласно GC) получали в виде прозрачного вязкого остатка темно-красного цвета. Это соответствует выходу 87%. Кроме того, 196,85 г красной прозрачной пропиленкарбонатной фазы, содержащей 91,5% площади согласно GC, 5,5% массы согласно ВЭЖХ непрореагировавшего ТМН получали. Это соответствует извлечению >99% ТМН (71 ммоль из 71 моля применяемого ТМН в избытке).
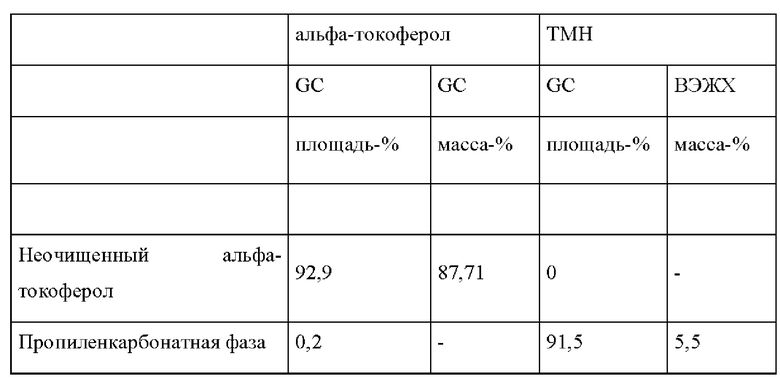
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ DL-АЛЬФА-ТОКОФЕРОЛА ИЛИ ЕГО АЦЕТАТА | 1994 |
|
RU2098416C1 |
| СПОСОБ ПОЛУЧЕНИЯ DL-α-ТОКОФЕРОЛА ИЛИ DL-α-ТОКОФЕРИЛАЦЕТАТА | 1997 |
|
RU2160258C2 |
| СПОСОБ ПОЛУЧЕНИЯ α-ТОКОФЕРОЛА ИЛИ α-ТОКОФЕРИЛАЦЕТАТА ПУТЕМ ВЗАИМОДЕЙСТВИЯ ТРИМЕТИЛГИДРОХИНОНА И ФИТОЛА ИЛИ ИЗОФИТОЛА С ВОЗВРАТОМ В ПРОЦЕСС ЦИНКГАЛОГЕНИДНОГО КАТАЛИЗАТОРА КОНДЕНСАЦИИ | 1997 |
|
RU2188823C2 |
| СПОСОБ ПОЛУЧЕНИЯ α--ТОКОФЕРОЛА (ВИТАМИНА Е) | 2000 |
|
RU2186064C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНОИДНЫХ ПРОИЗВОДНЫХ 2,3,5-ТРИМЕТИЛ-1,4-БЕНЗОХИНОНА | 2000 |
|
RU2197469C2 |
| Способ очистки @ -токоферола | 1982 |
|
SU1154279A1 |
| ЭФФЕКТИВНЫЙ ПРОЦЕСС ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 6-КАРБОКСИБЕНЗОКСАЗОЛА | 2020 |
|
RU2810493C1 |
| Способ получения 6-ацетокси-2,5,7,8-тетраметил-2-(4",8",12"-триметилтридецил)-хромана | 1973 |
|
SU460723A1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ БЕНЗОПИРАН-2-ОЛА | 2007 |
|
RU2397163C2 |
| Способ получения 6-ацетокси-2,5,7,8тетраметил-2-(4"8"12-триметилтридецил) -хромана | 1977 |
|
SU696020A1 |
Настоящее изобретение относится к способу получения производных хроманола, более конкретно к способу получения соединения общей формулы I
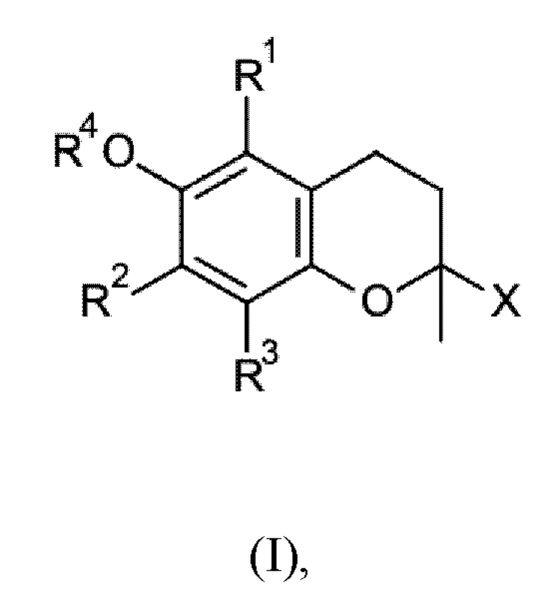
где R1, R2 и R3 независимо друг от друга выбраны из водорода и метила, R4 выбран из водорода и C1-С6-алканоила и X выбран из C1-С20-алкила и С2-С20-алкенила. Предлагаемый способ включает стадии а)-d). На стадии а) обеспечивают соединение хинона общей формулы II

и на стадии b) осуществляют каталитическое гидрирование соединения хинона формулы II в присутствии водорода, катализатора гидрирования и карбонатного растворителя с получением соединения гидрохинона общей формулы III
 .
.
Далее на стадии c) проводят реакцию соединения гидрохинона III с ненасыщенным соединением общей формулы IV.а или IV.b
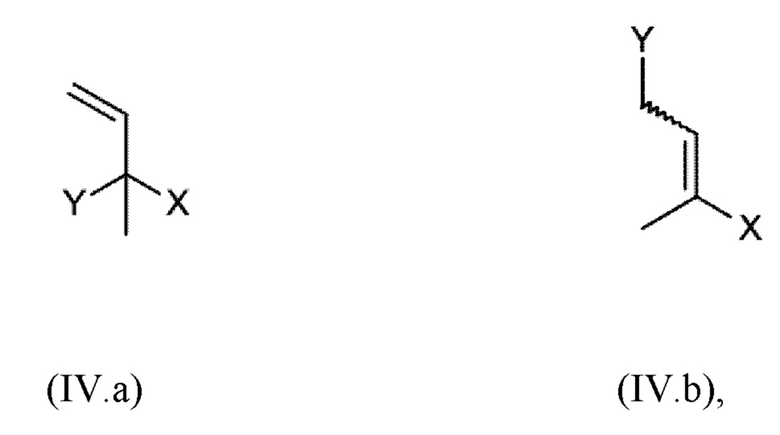
где X имеет значение, как определено выше, Y выбран из ОН, галогена, -O-R11, -S-R12 и -SO2-R12, R11 выбран из С1-С4-алкила, С1-С4-алканоила и трифторацетила и R12 выбран из C1-С6-алкила, трифторметила и фенила, где фенил является незамещенным или замещен 1, 2, 3, 4 или 5 радикалами, выбранными из галогена и метила, в присутствии катализатора конденсации, и на стадии d), если R4 выбран из C1-C6-алканоила, осуществляют реакцию продукта конденсации, полученного на стадии с), с С2-С7карбоновой кислотой или с ангидридом С2-С7-карбоновой кислоты в присутствии катализатора эстерификации или реакцию продукта конденсации, полученного на стадии с), с активированной С2-С7карбоновой кислотой в присутствии основания. Технический результат - обеспечение способа получения производных витамина Е, который является эффективным и обеспечивает получение желаемых продуктов с высоким выходом и селективностью без необходимости применения дорогих, коррозионных и/или экологически вредных катализаторов и растворителей. 17 з.п. ф-лы, 2 табл., 37 пр.
1. Способ получения соединения общей формулы I

где R1, R2 и R3 независимо друг от друга выбраны из водорода и метила,
R4 выбран из водорода и C1-С6-алканоила и
X выбран из C1-С20-алкила и С2-С20-алкенила, включающий следующие стадии:
а) обеспечение соединения хинона общей формулы II

где R1, R2 и R3 имеют значения, как определено выше,
b) каталитическое гидрирование соединения хинона формулы II, обеспеченного на стадии а), в присутствии водорода, катализатора гидрирования и в присутствии карбонатного растворителя с получением соединения гидрохинона общей формулы III

где R1, R2 и R3 имеют значения, как определено выше,
c) реакция соединения гидрохинона III, обеспеченного на стадии b), с ненасыщенным соединением общей формулы IV.а или IV.b

где X имеет значение, как определено выше,
Y выбран из ОН, галогена, -O-R11, -S-R12 и -SO2-R12,
R11 выбран из С1-С4-алкила, С1-С4-алканоила и трифторацетила и
R12 выбран из C1-С6-алкила, трифторметила и фенила, где фенил является незамещенным или замещен 1, 2, 3, 4 или 5 радикалами, выбранными из галогена и метила,
в присутствии катализатора конденсации, и
d) если R4 выбран из C1-C6-алканоила, реакция продукта конденсации, полученного на стадии с), с С2-С7карбоновой кислотой или с ангидридом С2-С7-карбоновой кислоты в присутствии катализатора эстерификации или реакция продукта конденсации, полученного на стадии с), с активированной С2-С7карбоновой кислотой в присутствии основания.
2. Способ по п. 1, где карбонатный растворитель выбран из по меньшей мере одного органического карбоната и из смесей, состоящих из по меньшей мере одного органического карбоната и по меньшей мере одного аполярного углеводородного растворителя.
3. Способ по п. 2, где содержание органического карбоната в смесях, состоящих из по меньшей мере одного органического карбоната и по меньшей мере одного аполярного углеводородного растворителя, находится в интервале от 50 до 99 мас.%, на основе общей массы смесей.
4. Способ по п. 2, где аполярный углеводородный растворитель выбран из группы, состоящей из линейных и разветвленных алканов, имеющих от 5 до 15 атомов углерода, циклоалканов, имеющих от 5 до 10 атомов углерода, и ароматических углеводородов, имеющих от 6 до 12 атомов углерода.
5. Способ по п. 2, где органический карбонат выбран из циклических или линейных карбонатов общей формулы V.a и V.b

где R5, R6 и R7 независимо друг от друга выбраны из водорода, метила и этила,
R8 выбран из водорода, фенила и C1-C15-алкила, где C1-C15-алкил является незамещенным или замещен 1, 2 или 3 радикалами, выбранными из С1-С3-алкокси, полиалкиленоксида, фенила и фенокси, и
R9 независимо друг от друга выбраны из С1-С4алкила.
6. Способ по любому из пп. 2-5, где органический карбонат выбран из этиленкарбоната, пропиленкарбоната, бутиленкарбоната, 2,3-пропиленкарбоната, изобутиленкарбоната, диметилкарбоната, диэтилкарбоната и ди-н-пропилкарбоната.
7. Способ по п. 1, где в соединениях общей формулы I, II и III
R1, R2 и R3 представляют собой метил и
R4, если присутствует, выбран из водорода или этаноила.
8. Способ по п. 1, где X представляет собой метил или имеет одно из следующих значений Х-1 - Х-6
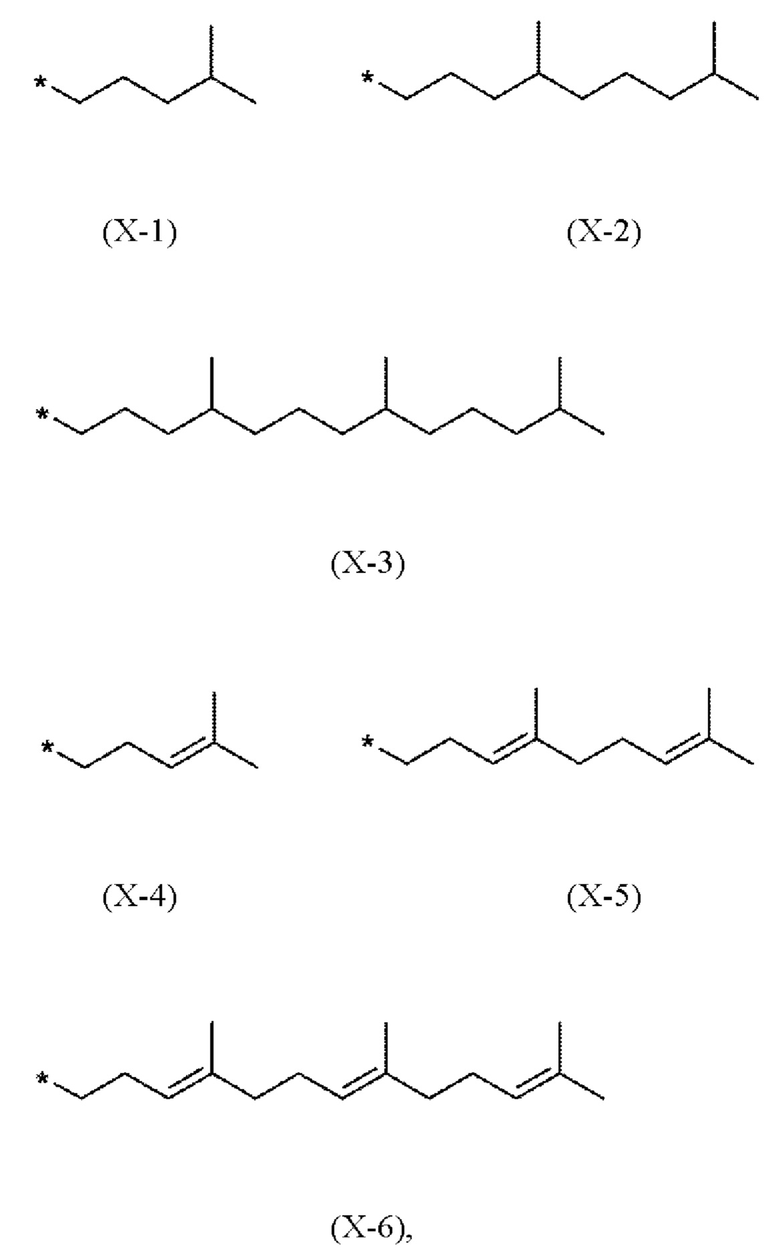
где * показывает точку присоединения к хромановому кольцу.
9. Способ по п. 1, где стадию с) проводят в карбонатном растворителе.
10. Способ по п. 1, где один и тот же карбонатный растворитель применяют на каждой из стадий b) и с).
11. Способ по п. 1, где стадию с) проводят в карбонатном растворителе, применяемом на стадии b).
12. Способ по п. 1, где реакционную смесь, полученную на стадии b), применяют непосредственно в реакции на стадии с) после удаления катализатора гидрирования.
13. Способ по п. 1, где катализатор конденсации, применяемый на стадии с), выбран из кислотного гетерогенного катализатора и, в частности, из катализатора на основе обработанного бентонита.
14. Способ по любому из пп. 1-13, где стадию d) проводят в карбонатном растворителе.
15. Способ по п. 14, где стадии b), с) и d) проводят в одном и том же карбонатном растворителе.
16. Способ по п. 14, где катализатор эстерификации, применяемый на стадии d), выбран из кислотного гетерогенного катализатора и, в частности, из катализатора на основе обработанного бентонита.
17. Способ по п. 16, где стадии с) и d) проводят в присутствии одного и того же катализатора на основе обработанного бентонита.
18. Способ по п. 17, где реакционную смесь, полученную на стадии с), применяют непосредственно в реакции на стадии d).
| EP 0677520 A1, 18.10.1995 | |||
| ДИФФЕРЕНЦИАЛЬНЫЙ ДАТЧИК ДАВЛЕНИЯ | 1981 |
|
SU970953A1 |
| УСТРОЙСТВО для АВТОМАТИЧЕСКОГО СЧЕТА ШТУЧНЫХ ГРУЗОВ НА КОНВЕЙЕРЕ | 0 |
|
SU264823A1 |
| WO 2012025587 A1, 01.03.2012 | |||
| Способ получения производных хромана | 1973 |
|
SU518135A3 |
| СПОСОБ ПОЛУЧЕНИЯ РАЦЕМИЧЕСКОГО НЕБИВОЛОЛА | 2006 |
|
RU2392277C2 |

