Область техники
Перекрестные ссылки на родственные заявки
В настоящей заявке испрашивается приоритет по Корейской патентной заявке No. 10-2020-0023899, поданной 26 февраля 2020, и Корейской патентной заявке No. 10-2021-0025655, поданной 25 февраля 2021, в Ведомство по интеллектуальной собственности Республики Корея, раскрытие которых во всей своей полноте включены в данную заявку посредством ссылки.
Настоящее изобретение относится к способу получения производного гетероциклического амина.
Уровень техники
ITK (Интерлейкин-2 тирозинкиназа) и ВТК (тирозинкиназа Брутона) представляют собой тип тирозинкиназы, вместе с Тес (тирозинкиназа, экспрессируемая при гепатоцеллюлярной карциноме), RLK (киназа покоящихся лимфоцитов) и ВМХ (ген тирозинкиназы костного мозга на хромосоме X), который не имеет рецептора семейства ТЕС и воздействует на различные иммунные ответы.
ITK экспрессируется не только в Т-клетках, но также в NK-клетках и тучных клетках и играет важную роль в пролиферации Т-клеток и в производстве важных цитокинов, таких как IL-2, IL-4, IL-5, IL-10, IL-13 и IL-17 (Schaeffer et al. Nat. Immune 2001, 2, 1183; Fowell et al. Immunity, 1999, 11, 399). T-клетки активируются путем передачи сигналов TCR, и активированные Т-клетки продуцируют воспалительные цитокины и активируют В-клетки и макрофаги, вызывая аутоиммунные заболевания, такие как ревматоидный артрит (РА) (Sahu N. et al. Curr Top Med Chem. 2009, 9, 690). Ранее было известно, что клетки Th1 активируются Т-клетками, чтобы вызвать заболевания РА, но недавно появились сообщения о том, что не только клетки Th17/Treg, но и Th1 клетки действуют как патогенез PA (J Leipe J. et al. Arthritis Rheum. 2010, 62, 2876). Кроме того, ITK ранее разрабатывалась в качестве мишени иммунотерапевтического лекарственного средства для такого заболевания, как астма, но ITK никогда не разрабатывалась в качестве терапевтического лекарственного средства для лечения PA (Lo Н. Y Expert Opin Ther Pat. 2010, 20, 459). Вместе с тем, недавно сообщалось, что она регулирует размножение клеток Th17 и Treg через ITK -/- мышей и обладает широким потенциалом в качестве терапевтической мишени для лечения PA (Gomez-Rodriguez J. et al. J. Exp.Med. 2014, 211, 529).
В исследовании с использованием ITK ингибитора PRN694, исследовании по снижению TNF-a, который является типичным воспалительным цитокином заболеваний РА, сообщалось, что возможность разработки в качестве терапевтического агента для РА подтверждается путем регуляции экспрессии Th17 посредством ингибирования ITK (Zhong Y. et al. THE JOURNAL OF BIOLOGICAL CHEMISTRY 2015, 290, 5960).
ВТК действует как регулятор раннего развития В-клеток, а также активации, передачи сигналов и выживания зрелых В-клеток. В-клетка получает сигнал от В-клеточного рецептора (BCR), который распознает антиген, прикрепленный к поверхности антиген-резонентной клетки, и активируется в зрелой клетке, продуцирующей антитела. Однако аберрантная передача сигналов через BCR приводит к аномальной пролиферации В-клеток и образованию патологических аутоантител и, таким образом, может вызывать рак, аутоиммунные и/или воспалительные заболевания. Таким образом, при аномальной пролиферации В-клеток передача сигналов через BCR может быть заблокирована при дефиците ВТК. Следовательно, ингибирование ВТК может блокировать процессы заболевания, опосредованные В-клетками, и, таким образом, использование ВТК ингибиторов может представлять полезный подход к лечению заболеваний, опосредованных В-клетками.
Кроме того, ВТК может экспрессироваться другими клетками, которые могут быть связаны с заболеванием помимо В-клеток. В одном примере ВТК является важными компонентами для передачи сигналов Fc-gamma в клетках костного мозга и экспрессируются тучными клетками. В частности, тучные клетки, индуцированные костным мозгом с дефицитом ВТК, демонстрируют нарушение антиген-индуцированной дегрануляции, и известно, что ингибирование активности ВТК полезно для лечения патологических реакций тучных клеток, таких как аллергия и астма (Iwaki et al. J. Biol Chem. 2005 280: 40261). Кроме того, известно, что моноциты пациентов с XLA, в которых активность ВТК отсутствует, снижают выработку TNF альфа после стимуляции, и, таким образом, воспаление, опосредованное TNF альфа, может быть ингибировано ингибиторами ВТК (см. Horwood et al., J. Exp.Med. 197: 1603, 2003).
В настоящее время не было случая, чтобы было разработано вещество, которое ингибирует и ВТК, и ITK. Однако в качестве БТК ингибитора в WO 2008/039218 описаны производные 4-аминопиразоло[3,4-d]пиримидинилпиперидина, в WO 2015/061247 описаны гетеросоединения, такие как пиридин, пиримидин, пиразин и пиридазин, а в WO 2014/055934 описаны производные пиримидинилфенилакриламида. В качестве ITK ингибитора в WO 2005/066335 описаны аминобензимидазолы, в WO 2005/056785 описаны пиридоны, в WO 2002/050071 описаны производные аминотиазола, а недавно в WO 2014/036016 описаны производные бензимидазола.
Учитывая эти обстоятельства, авторы настоящего изобретения обнаружили, что производное гетероциклического амина, имеющее химическую структуру, отличную от ВТК и ITK ингибиторов, о которых до сих пор сообщалось, проявляют превосходный ингибирующий эффект двойной активности в отношении ВТК и ITK. Таким образом, авторы настоящего изобретения провели интенсивные исследования способа получения, обеспечивающего получение нового производного гетероциклического амина, и в результате обнаружили, что при использовании описанного ниже способа получения возможно промышленное массовое производство, общий выход улучшается, а содержание примесей снижается, тем самым завершив настоящее изобретение.
Подробное описание изобретения
Техническая задача
Одна из целей настоящего изобретения состоит в разработке способа получения производного гетероциклического амина.
Техническое решение
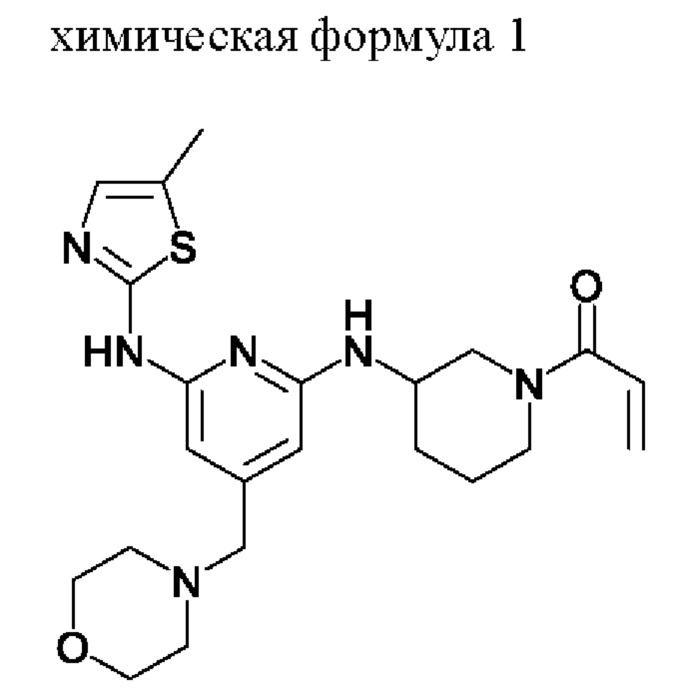
Для достижения вышеуказанных целей в настоящем изобретении предложен способ получения, показанный на схеме реакции 1, и, более конкретно, предложен способ получения, включающий следующие стадии:
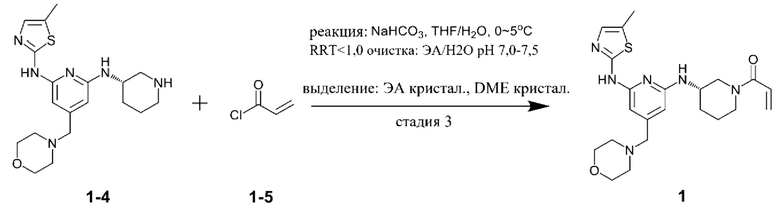
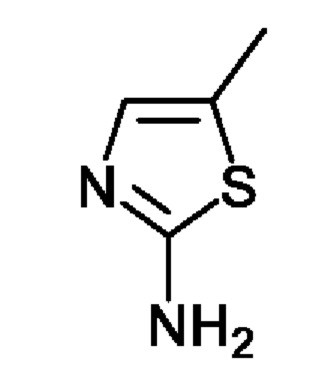
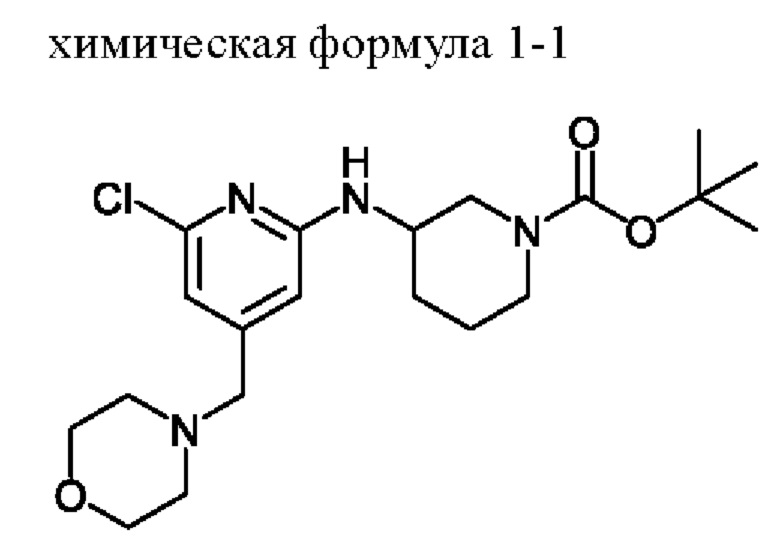
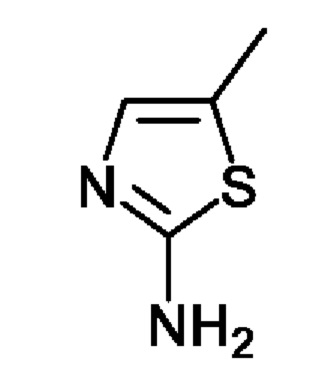
1) получение соединения, представленного химической формулой 1-3, путем взаимодействия соединения, представленного химической формулой 1-1, с соединением, представленным химической формулой 1-2, в присутствии палладиевого катализатора и основания;
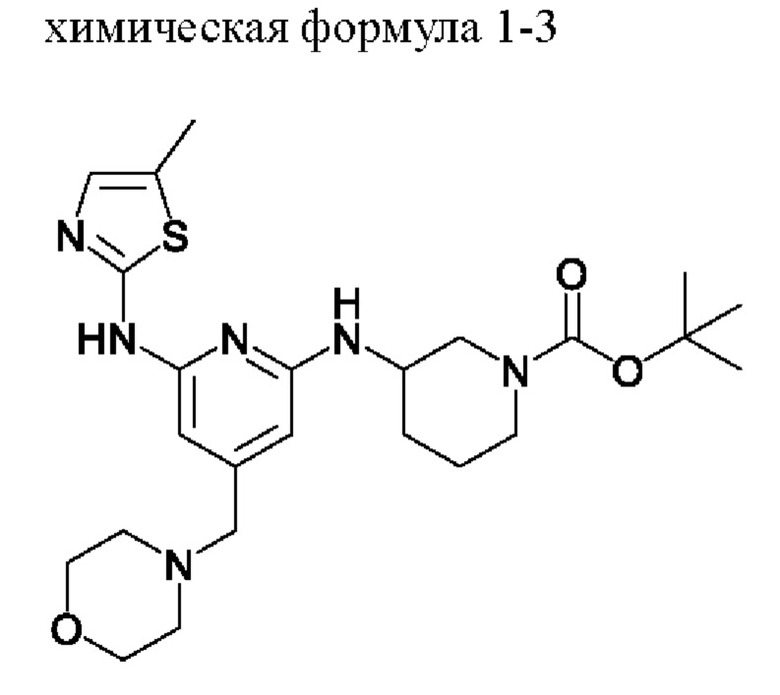
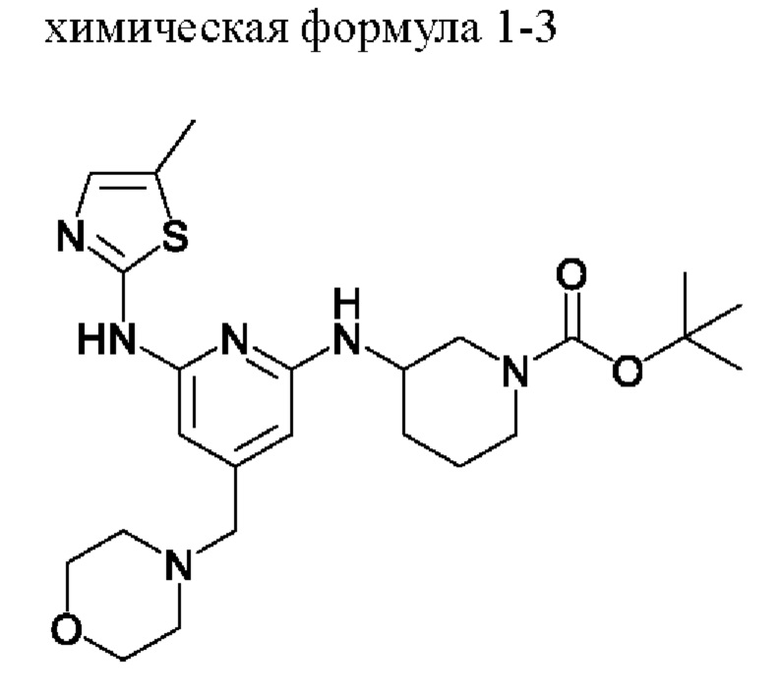
2) получение соединения, представленного химической формулой 1-4, путем взаимодействия соединения, представленного химической формулой 1-3, в присутствии кислоты; и
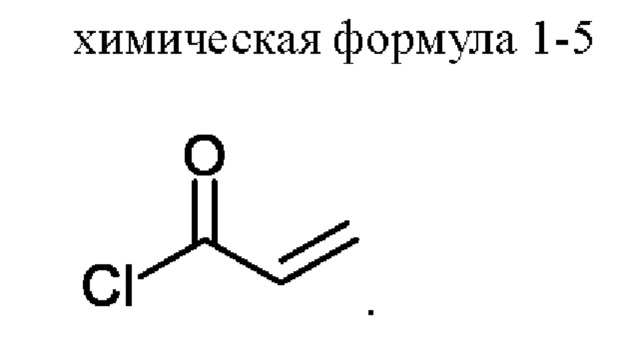
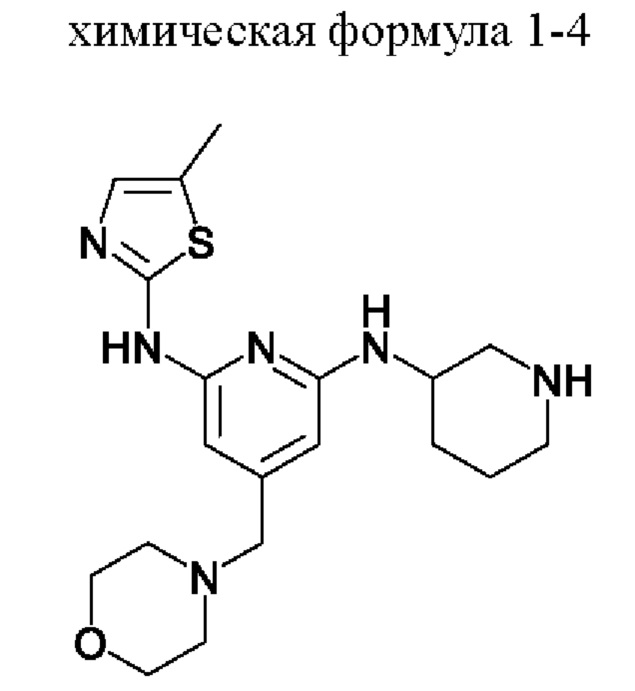
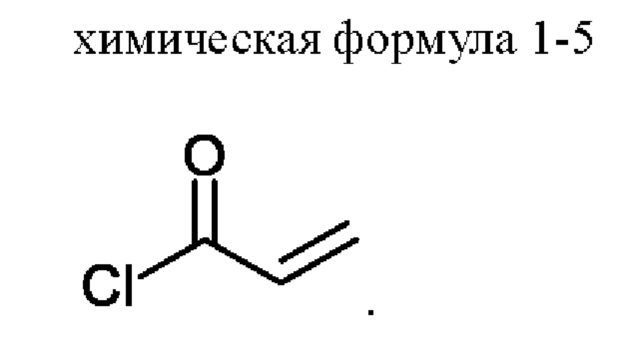
3) получение соединения, представленного химической формулой 1, путем взаимодействия соединения, представленного химической формулой 1-4, с соединением, представленным химической формулой 1-5, в присутствии основания.
Схема реакции 1
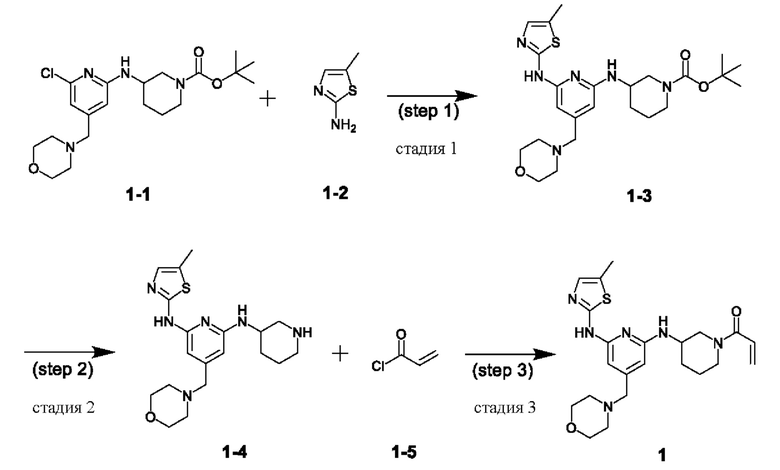
При этом соединение, представленное химической формулой 1, следует понимать как понятие, охватывающее следующие три типа соединений в зависимости от хиральности:
(1) (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинометил)пиридин-2-ил)амино)пиперидин-1 -ил)проп-2-ен- 1-он
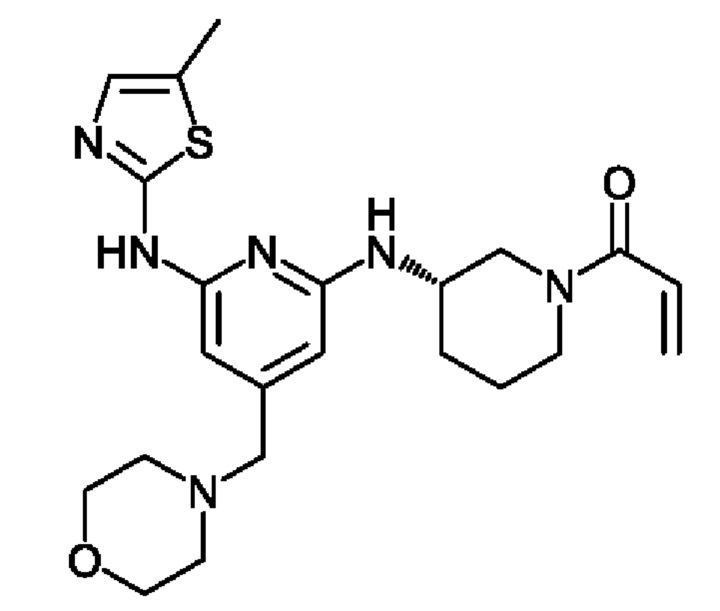
(2) (R)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинометил)пиридин-2-ил)амино)пиперидин-1 -ил)проп-2-ен- 1-он
 5
5
(3) 1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинометил)пиридин-2-ил)амино)пиперидин-1 -ил)проп-2-ен- 1-он
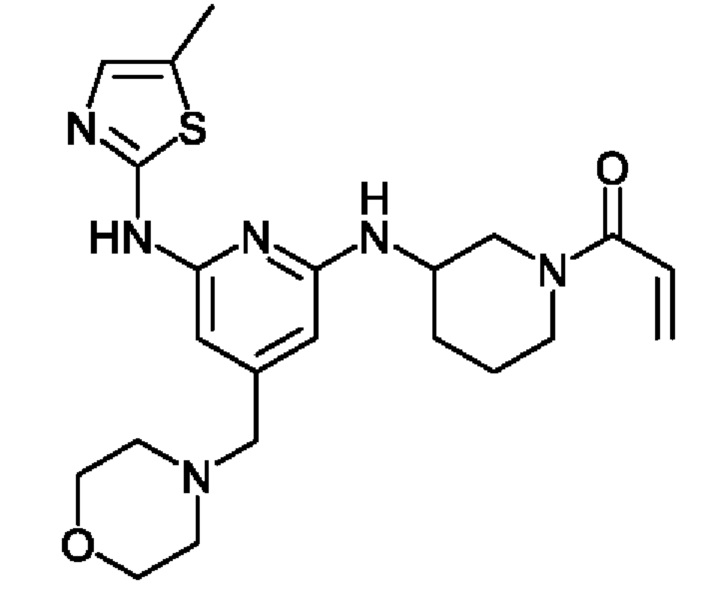
Далее настоящее изобретение будет подробно описано в отношении каждой стадии.
Стадия 1
Стадия 1 представляет собой стадию получения соединения, представленного химической формулой 1-3, путем взаимодействия соединения, представленного химической формулой 1-1, с соединением, представленным химической формулой 1-2, причем эта реакция представляет собой реакцию замещения с амином, которую проводят в присутствии палладиевого катализатора и основания.
При этом соединение, представленное химической формулой 1-1, и соединение, представленное химической формулой 1-2, может быть использовано в молярном отношении от 1:0,1 до 1:2. В частности, соединение, представленное химической формулой 1-1, и соединение, представленное химической формулой 1-2, может быть использовано в молярном отношении от 1:0,5 до 1:1,7, от 1:0,7 до 1:1,5 или от 1:0,9 до 1:1,5.
Кроме того, палладиевый катализатор может представлять собой катализатор палладия (0) с нулевой валентностью палладия (Pd) в соединении или катализатор палладия (II) с валентностью +2. Например, в качестве палладиевого катализатора можно использовать по меньшей мере один, выбранный из группы, состоящей из трис(дибензилиденацетон)дипалладия (0), тетракис(трифенилфосфин) палладия (0), бис[трис(2-метилфенил)фосфин]палладия и ацетата палладия (II).
Кроме того, основание, используемое в реакции на стадии 1, может представлять собой по меньшей мере одно, выбранное из группы, состоящей из карбоната цезия, карбоната калия, карбоната натрия, трет-бутоксида натрия и трет-бутоксида калия. Из них с точки зрения скорости реакции и выхода предпочтительно использовать карбонат цезия.
Кроме того, реакцию можно проводить в присутствии соединения на основе фосфина вместе с палладиевым катализатором и основанием. В качестве соединения на основе фосфина можно использовать по меньшей мере одно, выбранное из группы, состоящей из 2,2'-бис(дифенилфосфино)-1,1'-бинафтила, 4,5-бис(дифенилфосфино)-9,9-диметилксантена, 2-(дициклогексилфосфино)-3,6-диметокси-2,4',6'-триизопропил-1,1'-бифенила и дициклогексилфосфино-2',6'-диизопропоксибифенила.
Кроме того, в качестве растворителя для реакции можно использовать растворитель, неактивный в реакции замещения с амином. Например, можно использовать такой растворитель, как толуол, диоксан, диметилформамид (ДМФА), бутиловый спирт или диметилацетамид (ДМАА), но с точки зрения скорости реакции и выхода предпочтительно использовать толуол и диметилацетамид (ДМАА).
При этом растворитель можно использовать в количестве (мл/г) от 10 до 30 раз (по объему), более конкретно в количестве (мл/г) от 15 до 25 раз (по объему), больше относительно массы соединения, представленного химической формулой 1-1.
Кроме того, реакцию можно проводить при температуре от 80 до 150°С в течение от 3 ч до 15 ч. Когда реакцию проводят в условиях более низкой температуры и/или в течение более короткого времени реакции, чем в указанном выше диапазоне, реакция проходит недостаточно, и выход продукта может быть снижен. Кроме того, даже если реакцию проводят при более высоких температурах и/или в течение более длительного времени реакции, чем в указанном выше диапазоне, выход продукта не обязательно увеличивается, что нежелательно с точки зрения производственных затрат. Более конкретно, реакцию можно проводить при температуре от 90 до 130°С в течение от 4 ч до 12 ч, предпочтительно от 4 ч до 10 ч, более предпочтительно от 6 ч до 10 ч.
Кроме того, при использовании сочетания указанного выше палладиевого катализатора, основания и соединения на основе фосфина реакцию можно проводить в обычном известном реакторе без использования микроволнового реактора. При этом стадия (1) может применяться в качестве способа промышленного производства соединения, представленного химической формулой 1.
С другой стороны, после завершения реакции, без стадии очистки или выделения полученного соединения, представленного химической формулой 1-3, способ вплоть до стадий 1 и 2, описанных ниже, может проводиться in-situ в одном и том же реакционном сосуде. Как описано выше, поскольку стадия очистки или выделения соединения на стадии 1 не требуется, способ получения соединения, представленного химической формулой 1, включающий стадию 1, может иметь преимущество с точки зрения промышленного массового производства соединения.
Стадия 2
Стадия 2 представляет собой стадию получения соединения, представленного химической формулой 1-4, путем взаимодействия соединения, представленного химической формулой 1-3, в присутствии кислоты, которая представляет собой стадию удаления трет-бутоксикарбонильной группы, которая представляет собой защитную группу, замещенную на пиперидиновом кольце соединения, представленного химической формулой 1-3, в присутствии кислоты.
В качестве кислоты, используемой в реакции на стадии (2), можно использовать соляную кислоту, уксусную кислоту, трихлоруксусную кислоту, трифторуксусную кислоту или тому подобное. Из них соляная кислота является предпочтительной с точки зрения выхода и затрат.
В качестве растворителя для реакции можно использовать такой растворитель, как этилацетат, метиловый спирт, диоксан или толуол.
Кроме того, реакцию можно проводить при комнатной температуре в течение от 30 мин до 20 ч, предпочтительно от 30 мин до 4 ч. Реакция удаления трет-бутоксикарбонильной группы, которая представляет собой защитную группу, представляет собой реакцию, которая легко протекает без дополнительной теплообработки, и, таким образом, может проводиться при комнатной температуре. Однако, если время реакции составляет менее 30 мин, реакция может протекать недостаточно, и, если время реакции превышает 20 ч, выход продукта существенно не повышается. Таким образом, указанное выше время реакции является предпочтительным.
С другой стороны, после завершения реакции, для очистки соединения, представленного химической формулой 1-4, если необходимо, также можно включить стадию кристаллизации продукта реакции. На стадии кристаллизации кристаллизованное соединение, представленное химической формулой 1-4, получают путем использования растворителя кристаллизации в продукте реакции. Другими словами, стадию (2) можно проводить через стадии взаимодействия соединения, представленного химической формулой 1-3, в присутствии кислоты, и затем кристаллизации продукта реакции с получением соединения, представленного химической формулой 1-4.
В частности, стадию кристаллизации можно проводить путем первичной кристаллизации с водой и вторичной кристаллизации с метанолом. Как описано выше, когда продукт реакции, образованный после завершения стадии (2) реакции, последовательно кристаллизуют с водой и метанолом, можно эффективно удалять палладиевый катализатор, лиганд, остаток аминотиазола и т.д., которые являются побочными продуктами, оставшимися после реакции. При этом соединение высокой степени чистоты может быть получено с высоким выходом по сравнению со стадией очистки в виде суспензии с использованием растворителя, такого как этилацетат.
Первичную стадию кристаллизации с водой можно проводить путем добавления воды в продукт реакции и его перемешивания при температуре от примерно 0 до 5°С в течение от 10 мин до 2 ч. Кроме того, кристаллизацию предпочтительно проводят в присутствии основания, такого как гидроксид натрия, при поддержании рН продукта реакции на уровне 11 или более или 12 или более. Когда кристаллы образуются при указанных выше условиях, после завершения реакции реакционный раствор можно отфильтровать при пониженном давлении для получения кристаллов.
При этом воду можно использовать в количестве (мл/г) от 5 до 30 раз (по объему), более конкретно в количестве (мл/г) от 5 до 25 раз (по объему), больше относительно массы соединения, представленного химической формулой 1-1.
Далее, после высушивания полученных кристаллов можно проводить вторичную стадию кристаллизации с метанолом. Вторичную стадию кристаллизации с метанолом можно проводить путем добавления метанола к полученным кристаллам и их перемешивания при температуре о примерно 50 до 80°С в течение от 10 мин до 2 ч.
При этом метанол можно использовать в количестве (мл/г) от 5 до 40 раз (по объему), более конкретно в количестве (мл/г) от 10 до 35 раз (по объему), больше относительно массы соединения, представленного химической формулой 1-1.
Стадия 3
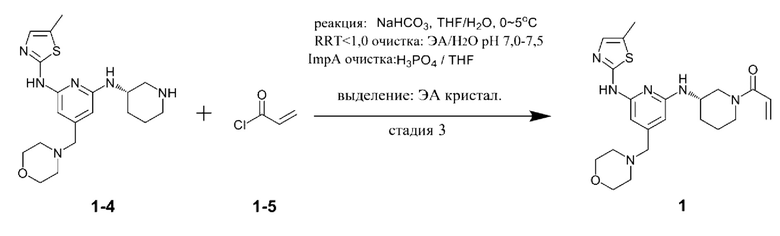
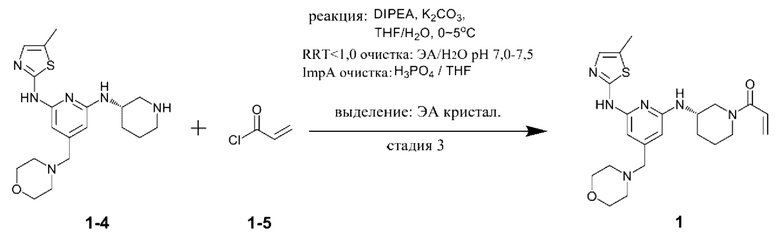
Стадия 3 представляет собой стадию получения соединения, представленного химической формулой 1, путем взаимодействия соединения, представленного химической формулой 1-4, с соединением, представленным химической формулой 1-5. Реакция представляет собой реакцию амидирования, которую предпочтительно проводят в присутствии основания.
При этом соединение, представленное химической формулой 1-4, и соединение, представленное химической формулой 1-5, можно использовать в молярном отношении от 1:0,5 до 1:2,0. В частности, соединение, представленное химической формулой 1-4, и соединение, представленное химической формулой 1-5, можно использовать в молярном отношении от 1:0,5 до 1:1,5, от 1:0,7 до 1:1,3 или от 1:0,9 до 1:1,1.
В качестве основания, используемого в реакции на стадии (3), можно использовать по меньшей мере одно, выбранное из группы, состоящей из карбоната калия, гидроксида натрия, гидроксида лития, гидроксида калия, триэтиламина, диизопропиламина, диизопропилэтиламина, гидрокарбоната натрия, гидрокарбоната калия, карбоната цезия, карбоната натрия, метилата натрия и бутирата калия. Из них карбонат калия, карбонат натрия, гидрокарбонат натрия или гидрокарбонат калия предпочтительно использовать с точки зрения завершения реакции и образования побочных продуктов.
В качестве растворителя этой реакции можно использовать смешанный растворитель тетрагидрофурана (ТГФ) и воды. При этом тетрагидрофуран можно использовать в количестве (мл/г) от 10 до 40 раз (по объему) больше относительно массы соединения, представленного химической формулой 1-4, а воду можно использовать в количестве (мл/г) от 2 до 10 раз (по объему) больше относительно массы соединения, представленного химической формулой 1-4.
С другой стороны, соединение, представленное химической формулой 1-5, можно добавлять в виде смеси с N,N-диизопропилэтиламином или триэтиламином. Акрилоилхлорид, который представляет собой соединение, представленное химической формулой 1-5, имеет небольшое количество соляной кислоты, оставшейся в соединении. При этом соляная кислота участвует в реакции и генерирует, в качестве побочного продукта, примеси (здесь и далее обозначена как примесь С (ImpC)), такие как соединение, в котором HCl присоединяется по двойной связи соединения, представленного химической формулой 1, и соединение, в котором HCl присоединяется по двойной связи акриламида, что вызывает проблему, заключающуюся в снижении чистоты конечного соединения. Однако, когда соединение, представленное химической формулой 1-5, используют в виде раствора, в котором он растворен в растворителе, таком как тетрагидрофуран вместе с N,N-диизопропилэтиламином (DIPEA), небольшое количество соляной кислоты, оставшейся в соединении, представленном химической формулой 1-5, может быть заранее удалено благодаря присутствию N,N-диизопропилэтиламина или триэтиламина, и таким образом, примесь С может не образовываться.
При этом в растворе, в котором соединение, представленное химической формулой 1-5, смешивают с N,N-диизопропилэтиламином, N,N-диизопропилэтиламин может содержаться в количестве от 0,01 до 0,2 моль относительно 1 моль соединения, представленного химической формулой 1-5. В указанном выше диапазоне, соляная кислота в соединении, представленном химической формулой 1-5, может быть эффективно удалено.
Кроме того, реакцию можно проводить при температуре от -10°С до 10°С, предпочтительно при температуре 0°С или ниже и более предпочтительно при температуре -10°С или выше и ниже 0°С. Димер соединения, представленного химической формулой 1, (здесь и далее обозначен как примесь В (ImpB)) может быть получен путем указанной выше реакции. Когда реакция протекает при низкой температуре, можно минимизировать образование такой примеси, как примесь В. В частности, температура реакции от -10 до 0°С, более конкретно, температура реакции от -8 до -3°С, предпочтительна с точки зрения уменьшения примеси и повышения выхода. Следовательно, предпочтительно, чтобы все компоненты реакции, такие как реагенты и органические растворители, которые используют для снижения роста температуры реакции во время реакции, используют после охлаждения до температуры 0°С или ниже.
С другой стороны, после завершения реакции, если необходимо, можно включить по меньшей мере одну из стадий экстракции продукта реакции, очистки путем концентрирования при пониженном давлении и кристаллизации, при этом выделяя и очищая соединение, представленное химической формулой 1. Чтобы получить соединение высокой чистоты, предпочтительно последовательно проводить все стадии экстракции, очистки и кристаллизации продукта реакции.
В частности, после завершения реакции на стадии (3) может быть также включена стадия экстракции продукта реакции с использованием этилацетата. В частности, продукт реакции может быть экстрагирован с использованием этилацетата и воды. То есть соединение, представленное химической формулой 1, может быть экстрагировано с использованием этилацетата и воды после завершения реакции. При экстракции таким способом примеси, имеющие относительное время удерживания менее 1,0, которые выделяются с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ), могут эффективно удаляться водным слоем по сравнению с экстракцией с использованием смешанного растворителя метиленхлорид/вода.
Стадию экстракции этилацетатом можно также проводить при рН от 6,5 до 7,5. Если рН ниже 6,5, существует опасение, что выход снижается, а если рН выше 7,5, удаление примесей может быть затруднено. Более конкретно, стадию экстракции этилацетатом можно проводить в диапазоне рН от 7,0 до 7,5.
Кроме того, стадия 3 может включать стадию растворения продукта реакции в тетрагидрофуране; стадию смешивания раствора, содержащего фосфорную кислоту, с полученным раствором; и стадию очистки полученной смеси путем фильтрации с последующим концентрированием фильтрата при пониженном давлении. Такую стадию концентрирования при пониженном давлении предпочтительно проводят после стадии экстракции этилацетатом.
Другими словами, стадия очистки может включать стадию растворения продукта, экстрагированного этилацетатом, в тетрагидрофуране с получением раствора; стадию смешивания раствора, содержащего фосфорную кислоту, с этим раствором с получением смеси, в которой образуется фосфатная соль; и стадию фильтрации смеси, в которой образуется фосфатная соль, с последующим концентрированием фильтрата при пониженном давлении.
Раствор, содержащий фосфорную кислоту, представляет собой раствор, в котором фосфорную кислоту растворяют в растворителе тетрагидрофуране, при этом фосфорная кислота может быть растворена в количестве от 0,05 до 0,5 моль на 1 моль соединения, представленного химической формулой 1-4. При добавлении к продукту раствора, содержащего фосфорную кислоту в указанном выше диапазоне, соединение, в котором связаны соединение, представленное химической формулой 1, и соединение, представленное химической формулой 1-4, полученное во время реакции (здесь и далее обозначено как примесь A (ImpA)) реагирует с фосфат-анионом (РО4-3) с образованием фосфатной соли. Однако полученная фосфатная соль может быть легко удалена путем фильтрации, и, таким образом, содержание примеси А в конечном полученном соединении, представленном химической формулой 1, может быть снижено.
Кроме того, стадия 3 может включать стадию кристаллизации продукта реакции этилацетатом. Эту стадию кристаллизации предпочтительно проводят после стадии очистки.
Другими словами, стадию кристаллизации можно проводить после стадии очистки путем добавления этилацетата в продукт, концентрированный при пониженном давлении, с последующим перемешиванием при температуре от примерно 20 до 40°С в течение от 30 минут до 4 ч. Когда кристаллы получают в вышеуказанных условиях, реакционный раствор может быть высушен при пониженном давлении после завершения реакции, с получением, таким образом, соединения, представленного химической формулой 1-1, которое представляет собой конечный продукт.
Таким образом, когда все стадии экстракции, очистки и кристаллизации продукта реакции на стадии 3 последовательно проводят для получения соединения, представленного химической формулой 1, стадию 3 можно проводить посредством следующих стадий:
стадия взаимодействия соединения, представленного химической формулой 1-4, с соединением, представленным химической формулой 1-5, в присутствии основания;
стадия экстракции продукта реакции с использованием этилацетата;
стадия растворения продукта, экстрагированного этилацетатом, в тетрагидрофуране для получения раствора; стадия смешивания раствора, содержащего фосфорную кислоту, с этим раствором с получением смеси, в которой образуется фосфатная соль; и стадия фильтрации смеси, в которой образуется фосфатная соль, и последующего концентрирования фильтрата при пониженном давлении; и
стадия кристаллизации продукта, концентрированного при пониженном давлении этилацетатом.
Когда стадии дополнительной экстракции, очистки и кристаллизации проводят после реакции так, что можно получить соединение, представленное химической формулой 1, высокой чистоты, в котором содержание примеси, имеющей RRT менее 1,0 (RRT<1,0), примеси А (ImpA), примеси В (ImpB) и примеси С (ImpC) существенно низкое, это может быть подтверждено путем сравнения примеров и сравнительных примеров, описанных далее.
Преимущества
Как описано выше, способ получения согласно настоящему изобретению имеет преимущество, состоящее в том, что с высоким выходом можно получить производное гетероциклического амина, имеющее пониженное содержание примесей.
Подробное описание воплощений
Далее настоящее изобретение будет описано более подробно со ссылкой на следующие примеры. Однако эти примеры представлены только в иллюстративных целях и не ограничивают объем настоящего изобретения. Кроме того, под эквивалентом (экв.) в дальнейшем следует понимать количество моль относительно исходного материала в реакции, протекающей на каждой стадии.
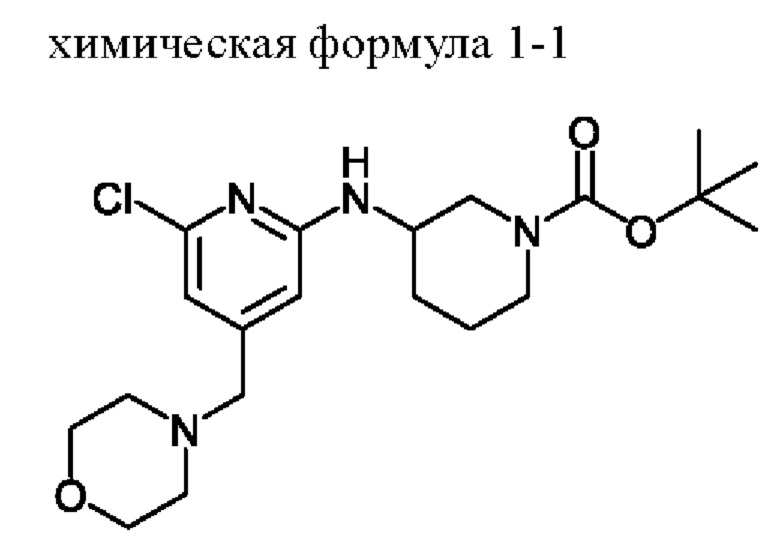
Пример получения: Получение соединения, представленного химической формулой 1-1, ((S)-трет-бутил-3-((6-хлор-4-(морфолинометил)пиридин-2-ил)амино)пиперидин-1-карбоксилата)
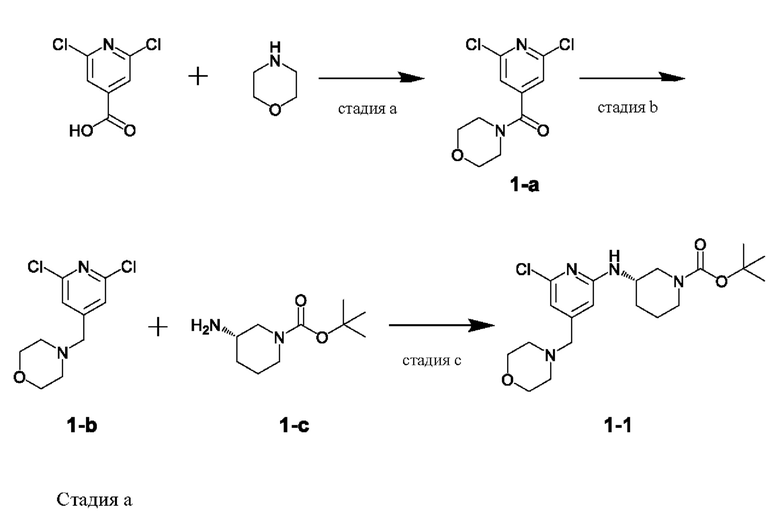
2,6-дихлоризоникотиновую кислоту (10,0 г, 1,0 экв.) растворяли в диметилформамиде (100,0 мл), и затем добавляли туда 1,1-карбонилдиимидазол (1,0 г, 1,2 экв.). Смесь перемешивали при комнатной температуре (25-30°С) под газообразным азотом в течение 1 ч, затем добавляли морфолин (5,4 мл, 1,2 экв.) и перемешивали при той же температуре в течение 2 ч для завершения реакции. Этилацетат (200,0 мл) и воду (200,0 мл) добавляли и экстрагировали, и водный слой повторно экстрагировали трижды с использованием этилацетата (200,0 мл). Слой этилацетата сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии (этилацетат:гексан=1:5) с получением соединения, представленного химической формулой 1-а, (2,6-дихлорпиридин-4-ил)(морфолино)метанона (13,0 г, 93,0%).
1H-ЯМР (500 МГц, CDCl3): 7,27 (с, 2Н), 3,77 (м, 4Н) 3,65 (м, 2Н), 3,37 (м, 2Н).
Стадия b
Соединение, представленное химической формулой 1-а (10,0 г, 1,0 экв.), полученное на стадии (а), растворяли в дихлорметане (100,0 мл) и затем охлаждали до от 0 до 10°С под газообразным азотом. К нему медленно по каплям добавляли 1М комплекса боран-тетрагидрофуран (115,0 мл, 3,0 экв.) и затем перемешивали при комнатной температуре в течение 12 ч для завершения реакции. Реакционный раствор охлаждали до 0-10°С, затем медленно по каплям добавляли водный раствор 6н соляной кислоты (256,0 мл, 20,0 экв.) и затем перемешивали при той же температуре в течение 1 ч. Уровень рН доводили до от 9 до 12, используя 10н водный раствор гидроксида натрия, и затем дважды экстрагировали дихлорметаном. Слой дихлорметана отделяли, сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии (этилацетат: гексан=1:1) с получением соединения, представленного химической формулой 1-b, 4-((2,6-дихлорпиридин-4-ил)метил)морфолина (8,1 г, выход: 90,0%).
1H-ЯМР (500 МГц, CDCl3): 7,27 (с, 2Н), 3,72 (м, 4Н) 3,46 (с, 2Н), 2,45 (м, 4Н).
Стадия с
Соединение, представленное химической формулой 1-b (1,0 г, 1,0 экв.), полученное на стадии (b), растворяли в 1,4-диоксане (10,0 мл) и затем добавляли туда трис(дибензилиденацетон)дипалладия (0) (465,8 мг, 0,2 экв.) и Xantphos (1,5 г, 0,4 экв.). Добавляли (S)-трет-бутил-3-аминопиперидин-1-карбоксилат (780,0 мкл, 1,0 экв.), который представляет собой соединение, представленное химической формулой 1-c, и затем добавляли карбонат натрия (1,3 г, 3,0 экв.), и затем смесь кипятили с обратным холодильником в течение 12 ч для завершения реакции. После охлаждения до 30°С или ниже добавляли туда воду (20,0 мл) и этилацетат (20,0 мл) и затем слои разделяли. Слой этилацетата сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии (этилацетат: гексан=1:1) с получением соединения, представленного химической формулой 1-1, (S)-трет-бутил-3-((6-хлор-4-(морфолинометил)пиридин-2-ил)амино)пиперидин-1-карбоксилата (900,0 мг, выход: 54,1%).
1Н-ЯМР (500 МГц, CDCl3): 6,63 (с, 1H), 6,31 (с, 1H), 4,66-4,65 (м, 1H), 3,84-3,82 (м, 1H), 3,75-3,70 (м, 5Н), 3,59-3,54 (м, 1H), 3,37 (с, 2Н), 3,27-3,23 (м, 2Н), 2,46 (т, 4Н), 1,98-1,94 (м, 1H), 1,73 (с, 1H), 1,63-1,56 (м, 2Н), 1,50 (с, 9Н).
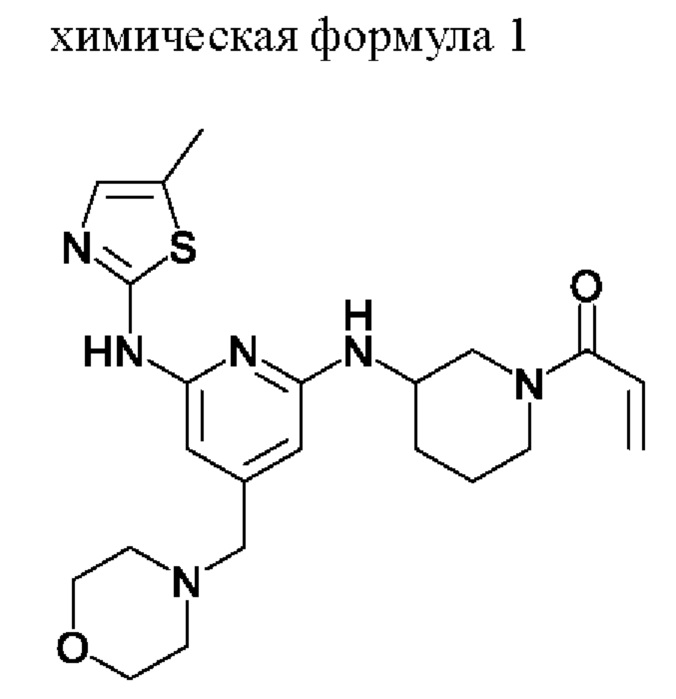
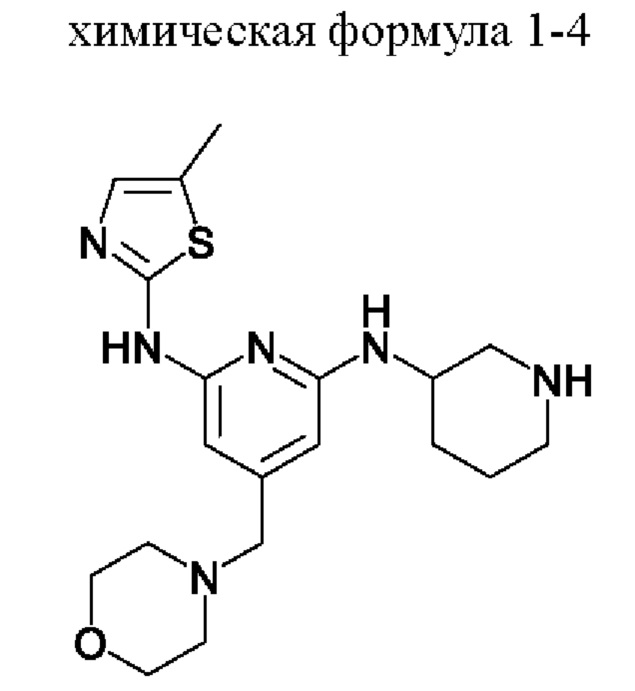
Пример: Получение (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинометил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
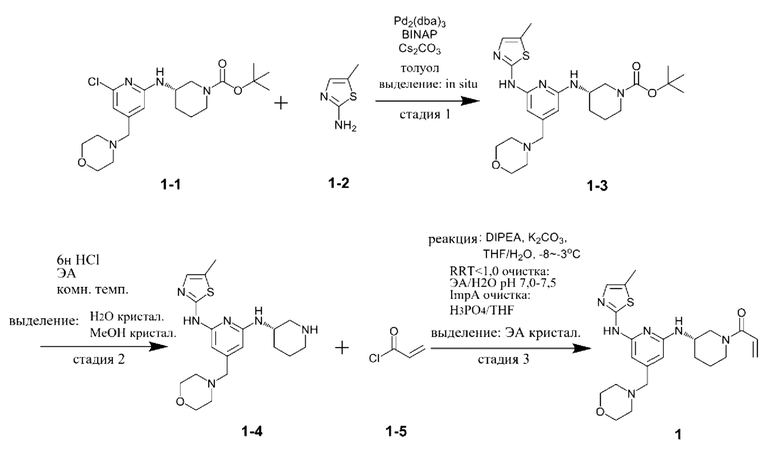
Стадия 1
Соединение, представленное химической формулой 1-1, (50,0 г, 1,0 экв.), полученное в примере получения, соединение, представленное химической формулой 1-2, (16,7 г, 1,2 экв.), трис(дибензилиденацетон)дипалладия (0) (11,1 г, 0,1 экв.), 2,2'-бис(дифенилфосфино)-1,1'-бинафтил (BINAP) (30,3 г, 0,4 экв.), карбонат цезия (118,9 г, 3,0 экв.) и 1000 мл толуола объемом в 20 раз больше относительно массы соединения, представленного химической формулой 1-1, добавляли в колбу и перемешивали смесь при комнатной температуре в течение 10 мин. После повышения температуры в колбе до 110°С, реакционную смесь перемешивали в диапазоне температур 105-111°С в течение 8 ч. Полученный остаток отфильтровывали через Celite для удаления катализатора и полученной неорганической соли. Затем органический слой концентрировали при пониженном давлении и добавляли туда 250 мл очищенной воды объемом в 5 раз больше относительно массы соединения, представленного химической формулой 1-1, и 1000 мл этилуксусной кислоты объемом в 20 раз больше относительно массы соединения, представленного химической формулой 1-1, и органический слой экстрагировали. Водный слой, образованный на данном этапе, отбрасывали. Затем органический слой концентрировали при пониженном давлении при 40-45°С с получением соединения, представленного химической формулой 1-3, которое далее использовали на стадии 2 ниже без дальнейшей очистки.
1H-ЯМР (500 МГц, ДМСО): 10,51 (с, 1H), 6,93 (с, 1H) 6,40 (с, 1H), 6,06 (с, 1H), 5,98 (с, 1H), 4,01 (с, 2Н), 3,55 (с, 5Н), 3,21 (с, 4Н), 2,32 (с, 4Н), 2,24 (с, 3Н), 1,97 (с, 1H), 1,72 (с, 1H), 1,32 (шир. с, 2Н),1,15 (с, 9Н).
Стадия 2
Этилацетат (ЭА) (500 мл) и 6н HCl (250 мл, 5,0 экв.) добавляли в соединение, представленное химической формулой 1-3, полученное на стадии 1, и затем смесь перемешивали при комнатной температуре в течение 2 ч для завершения реакции.
Стадия кристаллизации с водой и метанолом
Затем водный слой экстрагировали и добавляли в органический слой 1000 мл очищенной воды (количество (мл/г) в 20 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-1) и подвергали вторичной экстракции. Водные слои, полученные в 1-ой и 2-ой экстракциях, объединяли, температуру внутри поддерживали 0-5°С с помощью ледяной бани, 700 мл 4н NaOH (количество (мл/г) в 14 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-1) добавляли в течение примерно 30 мин и перемешивали смесь в течение 30 минут при поддержании рН 12 или выше. Полученные кристаллы отфильтровывали при пониженном давлении и промывали фильтрат 500 мл очищенной воды (количество (мл/г) в 10 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-1) и 500 мл гексана (количество (мл/г) в 10 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-1). При этом промытый фильтрат помещали в сушилку и затем сушили под вакуумом при температуре 40-45°С в течение 12 ч или более, затем туда добавляли 1,65 л метанола (количество (мл/г) в 33 раза (по объему) большее относительно массы соединения, представленного химической формулой 1-1) и смесь перемешивали при температуре внутри 65°С в течение примерно 30 мин и затем охлаждали до комнатной температуры. Полученные кристаллы отфильтровывали при пониженном давлении, полученный фильтрат помещали в сушилку и затем сушили под вакуумом при температуре 40-45°С в течение 12 ч или более с получением 28,5 г соединения, представленного химической формулой 1-4 (выход: 60,3% (включая стадии 1 и 2)).
1Н-ЯМР (500 МГц, ДМСО): 10,47 (с, 1H), 6,93(с, 1H) 6,28 (д, 1H), 6,02 (с, 1H), 5,94 (с, 1H), 3,91 (с, 1H), 3,56 (с, 4Н), 3,20 (с, 2Н), 3,06 (д, 1H), 2,79 (д, 1H), 2,44-2,42 (м, 1H), 2,37-2,34 (м, 1H), 2,32 (с, 3H), 2,28 (с, 4Н), 2,03-2,01 (м, 2Н), 1,63-1,60 (м, 1H), 1,44-1,42 (м, 1H), 1,30-1,28 (м, 1H).
Стадия 3
В соединение, представленное химической формулой 1-4 (5,0 г, 1,0 экв.), полученное на стадии 2, добавляли 90,0 мл ТГФ (количество (мл/г) в 18 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) и 20,0 мл H2O (количество (мл/г) в 4 раза (по объему) большее относительно массы соединения, представленного химической формулой 1-4) для получения раствора при комнатной температуре. Затем температуру внутри понижали до -8 - -3°С с помощью внешней бани с температурой -8 - -5°С (смесь льда, H2O и МеОН), затем добавляли К2СО3 (2,6 г, 1,5 экв.) и растворяли. Теперь в 5 мл ТГФ, охлажденного до -8 - -3°С, по каплям добавляли смешанный раствор N,N-диизопропилэтиламина (DIPEA) (111,5 мкл, 0,05 экв.) и соединения, представленного химической формулой 1-5, (1,1 мл, 1,05 экв.) при температуре внутри -8 - -3°С. Полученную смесь перемешивали при температуре -8 - -3°С в течение 0,5 ч для завершения реакции.
Стадия экстракции этилацетатом
В реакционный сосуд, в котором были завершены реакции, добавляли 100,0 мл этилацетата при температуре -8 - -3°С (количество (мл/г) в 20 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) и 50,0 мл насыщенного водного раствора NH4Cl (количество (мл/г) в 10 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4), слои разделяли и только органический слой отделяли. Затем 50,0 мл этилацетата (количество (мл/г) в 10 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) добавляли в водный слой при температуре -8 - -3°С и повторно экстрагировали. Затем только органический слой собирали и концентрировали при пониженном давлении при температуре снаружи 35°С. Добавляли туда 100,0 мл этилацетата (количество (мл/г) в 20 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) при температуре -8 - -3°С и 50,0 мл Н2О (количество (мл/г) в 10 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) при температуре 0 - 5°С, рН доводили до 7,0-7,5 с помощью 1н раствора HCl и слои разделяли дважды. Выделенный органический слой собирали, сушили над Na2SO4 и затем концентрировали при пониженном давлении при температуре снаружи 35°С.
Стадия очистки фосфорной кислотой (Н3РО4)
Добавляли туда 90,0 мл ТГФ (количество (мл/г) в 18 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) при температуре -8 - -3°С и доводили до растворения. Затем раствор H3PO4 (9,3 г, 0,095 экв.), растворенный в 5,0 мл ТГФ при температуре -8 - -3°С, по каплям добавляли при температуре -8 - -3°С и затем смесь перемешивали в течение 30 минут. Смесь отфильтровывали через Celite для удаления солей, а раствор H3PO4 (8,3 г, 0,085 экв.), растворенный в 5,0 мл ТГФ, при температуре -8 - -3°С по каплям добавляли в фильтрат, и раствор перемешивали в течение 30 минут при температуре -8 - -3°С. Полученный остаток отфильтровывали через Celite для удаления солей, а фильтрат концентрировали при пониженном давлении при температуре снаружи 35°С. Добавляли туда 25,0 мл метиленхлорида (MX) (количество (мл/г) в 5 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) при температуре -8 - -3°С и 10,0 мл H2O (количество (мл/г) в 2 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) при температуре от 0 до 5°С для доведения рН до 9,0-9,5 и затем органический слой отделяли. Выделенный органический слой сушили с помощью Na2SO4 и затем концентрировали при пониженном давлении при температуре снаружи 35°С.
Стадия кристаллизации с этилацетатом
Добавляли туда 75,0 мл этилацетата (количество (мл/г) в 15 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) и затем, когда образовались кристаллы, их перемешивали при температуре 20-30°С в течение 2 ч и отфильтровывали. Фильтрат сушили при пониженном давлении при комнатной температуре в течение 12 ч с получением 3,4 г соединения, представленного химической формулой 1, (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинометил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она (выход: 60,0%).
1Н ЯМР (500 МГц, ДМСО): 10,57 (м, 1H), 6,91-6,90 (м, 1H), 6,80-6,85 (м, 0,5Н), 6,70-6,40 (м, 1,5Н), 6,10-5,96 (м, 3Н), 5,65-5,63 (д, 0,5Н), 5,42-5,40 (д, 0,5Н), 4,42-4,40 (м, 0,5Н), 4,10-4,0 (м, 1H), 3,90-3,87 (м, 1,5Н), 3,56 (м, 4Н), 3,20 (с, 2Н), 3,14-3,10 (м, 1H), 2,68-2,63 (м, 0,5Н), 2,32 (м, 4Н), 2,19 (с, 3H), 1,90-2,0 (м, 1H), 1,80 (м, 1H), 1,50-1,40 (м, 2,5Н).
Сравнительный пример 1
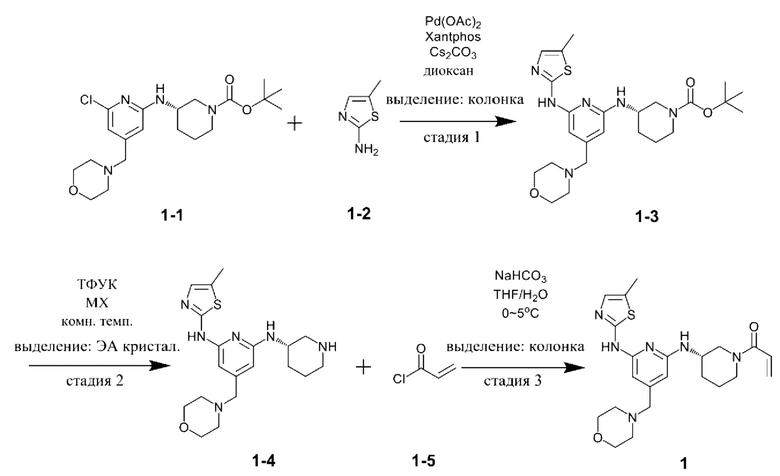
Стадия 1
Соединение, представленное химической формулой 1-1, (730,0 мг, 1,0 экв.), полученное в примере получения, растворяли в 1,4-диоксане (14,0 мл). Последовательно добавляли ацетат палладия (40,0 мг, 0,1 экв.), Xantphos (204,7 мг, 0,2 экв.), соединение, представленное химической формулой 1-2, (203,6 мг, 1,0 экв.) и карбонат цезия (1,7 г, 3,0 экв.). Реакцию проводили в микроволновом реакторе при температуре 150°С в течение 30 минут. Реакционную смесь охлаждали до 30°С или менее, затем добавляли туда воду (15,0 мл) и этилацетат (15,0 мл) и затем слои разделяли. Слой этилацетата сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии (ЭА 100%) с получением 564,0 мг соединения, представленного химической формулой 1-3 (выход: 65,0%).
1Н-ЯМР (500 МГц, ДМСО): 10,51 (с, 1H), 6,93 (с, 1H) 6,40 (с, 1H), 6,06 (с, 1Н), 5,98 (с, 1H), 4,01 (с, 2Н), 3,55 (с, 5Н), 3,21 (с, 4Н), 2,32 (с, 4Н), 2,24 (с, 3H), 1,97 (с, 1H), 1,72 (с, 1H), 1,32 (шир. с, 2Н),1,15 (с, 9Н).
Стадия 2
Соединение, представленное химической формулой 1-3, (500,0 мг, 1,0 экв.), полученное на стадии 1, растворяли в дихлорметане (10,0 мл) и затем охлаждали до 0-10°С. Добавляли туда медленно по каплям трихлоруксусную кислоту (1,6 мл, 20,0 экв.) и затем перемешивали смесь в течение 1 ч. Затем рН доводили до 9-12 с помощью 12н водного раствора гидроксида натрия, затем выделенный слой дихлорметана сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Добавляли в полученный остаток этилацетат (10,0 мл) с образованием кристаллов в течение 30 минут. Полученные кристаллы отфильтровывали и затем сушили с получением 357,5 мг соединения, представленного химической формулой 1-4 (выход: 90,0%).
1Н-ЯМР (500 МГц, ДМСО): 10,47 (с, 1H), 6,93 (с, 1H) 6,28 (д, 1H), 6,02 (с, 1H), 5,94 (с, 1H), 3,91 (с, 1H), 3,56 (с, 4Н), 3,20 (с, 2Н), 3,06 (д, 1H), 2,79 (д, 1H), 2,44-2,42 (м, 1H), 2,37-2,34 (м, 1H), 2,32 (с, 3H), 2,28 (с, 4Н), 2,03-2,01 (м, 2Н), 1,63-1,60 (м, 1H), 1,44-1,42 (м, 1H), 1,30-1,28 (м, 1H).
Стадия 3
Соединение, представленное химической формулой 1-4 (350,0 мг, 1,0 экв.), полученное на стадии 2, растворяли в тетрагидрофуране (7,0 мл), в который добавляли воду (7,0 мл), бикарбонат натрия (226,8 мг, 3,0 экв.) и затем охлаждали до 0-10°С. Соединение, представленное химической формулой 1-5, (73,1 мкл, 1,0 экв.) медленно по каплям добавляли туда и затем смесь перемешивали в течение 30 минут для завершения реакции. Проводили разделение слоев с помощью дихлорметана, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток очищали методом колоночной хроматографии (дихлорметан:метанол=15:1) с получением 318,0 мг соединения, представленного химической формулой 1 (выход: 60,0%).
1Н ЯМР (500 МГц, ДМСО): 10,57 (м, 1H), 6,91-6,90 (м, 1H), 6,80-6,85 (м, 0,5Н), 6,70-6,40 (м, 1,5Н), 6,10-5,96 (м, 3H), 5,65-5,63 (д, 0,5Н), 5,42-5,40 (д, 0,5Н), 4,42-4,40 (м, 0,5Н), 4,10-4,0 (м, 1H), 3,90-3,87 (м, 1,5Н), 3,56 (м, 4Н), 3,20 (с, 2Н), 3,14-3,10 (м, 1H), 2,68-2,63 (м, 0,5Н), 2,32 (м, 4Н), 2,19 (с, 3H), 1,90-2,0 (м, 1H), 1,80 (м, 1H), 1,50-1,40 (м, 2,5Н).
Сравнительный пример 2
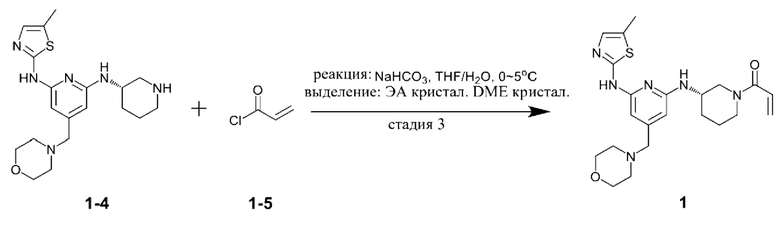
Стадии 1 и 2
Соединение, представленное формулой 1-4, получали теми же способами, что и стадии 1 и 2 получения в примере.
Стадия 3
Соединение, представленное химической формулой 1-4 (350,0 мг, 1,0 экв.), полученное на стадии 2, растворяли в тетрагидрофуране (7,0 мл), в который добавляли воду (7,0 мл), бикарбонат натрия (226,8 мг, 3,0 экв.) и затем охлаждали до 0-10°С. Соединение, представленное химической формулой 1-5, (73,1 мкл, 1,0 экв.) медленно по каплям добавляли туда и смесь перемешивали в течение 30 минут для завершения реакции. Проводили разделение слоев с помощью дихлорметана, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Этилацетат добавляли в полученный остаток в количестве (мл/г), в 20 раз больше (по объему) относительно массы остатка и затем перемешивали при комнатной температуре в течение 3 ч с образованием кристаллов. Полученные кристаллы отфильтровывали, сушили при пониженном давлении при комнатной температуре и добавляли туда диметоксиэтан в количестве (мл/г), в 15 раз (по объему) больше относительно массы кристаллов, затем растворяли при кипении с обратным холодильником и медленно охлаждали до комнатной температуры, затем перемешивали в течение 2 ч для образования кристаллов. Их отфильтровывали и сушили при пониженном давлении при комнатной температуре с получением 0,12 г соединения, представленного химической формулой 1, в качестве 6-го материала (выход: 30,0%).
1Н ЯМР (500 МГц, ДМСО): 10,57 (м, 1H), 6,91-6,90 (м, 1H), 6,80-6,85 (м, 0,5Н), 6,70-6,40 (м, 1,5Н), 6,10-5,96 (м, 3H), 5,65-5,63 (д, 0,5Н), 5,42-5,40 (д, 0,5Н), 4,42-4,40 (м, 0,5Н), 4,10-4,0 (м, 1H), 3,90-3,87 (м, 1,5Н), 3,56 (м, 4Н), 3,20 (с, 2Н), 3,14-3,10 (м, 1H), 2,68-2,63 (м, 0,5Н), 2,32 (м, 4Н), 2,19 (с, 3H), 1,90-2,0 (м, 1H), 1,80 (м, 1H), 1,50-1,40 (м, 2,5Н).
Сравнительный пример 3
Стадии 1 и 2
Соединение, представленное химической формулой 1-4, получали теми же способами, что и стадии 1 и 2 получения в примере.
Стадия 3
В соединение, представленное химической формулой 1-4, (2,34 г, 1,0 экв.), полученное на стадии 2, добавляли 46,8 мл ТГФ (количество (мл/г) в 20 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) и 9,4 мл H2O (количество (мл/г) в 4 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) для получения раствора при комнатной температуре. Затем температуру внутри снижали до 0-5°С с помощью бани с температурой 0-5°С (смесь льда и H2O) и добавляли затем NaHCO3 (1,51 г, 3,0 экв.) и растворяли. Раствор, в котором было смешано соединение, представленное химической формулой 1-5, (0,51 мл, 1,05 экв.), по каплям добавляли туда при температуре внутри 0-5°С. Смесь перемешивали при температуре 0-5°С в течение 0,5 ч для завершения реакции.
Стадия экстракции этилацетатом
В реакционный сосуд, в котором были завершены реакции, добавляли 46,8 мл этилацетата (количество (мл/г) в 20 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) и 23,4 мл H2O (количество (мл/г) в 10 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4), и затем проводили разделение слоев для выделения только органического слоя. Затем 23,4 мл этилацетата (количество (мл/г) в 10 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) добавляли в водный слой и повторно экстрагировали, и затем только органический слой собирали и концентрировали при пониженном давлении при температуре снаружи 40°С. Добавляли 46,8 мл этилацетата (количество (мл/г) в 20 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) и 23,4 мл H2O (количество (мл/г) в 10 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4), рН доводили до 7,0-7,5 1н раствором HCl и затем слои разделяли дважды. Выделенный органический слой собирали, сушили над Na2SO4 и затем концентрировали при пониженном давлении при температуре снаружи 40°С.
Стадия кристаллизации с этилацетатом
Добавляли туда 46,8 мл этилацетата (количество (мл/г) в 20 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) и затем, когда образовались кристаллы, их перемешивали при температуре 20-30°С в течение 2 ч и отфильтровывали. Фильтрат сушили при пониженном давлении при комнатной температуре в течение 12 ч с получением 1,75 г соединения, представленного химической формулой 1, (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинометил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она (выход: 68,9%).
Стадия кристаллизации с диметоксиэтаном
7,5 мл диметоксиэтана (количество (мл/г) в 15 раз (по объему) большее относительно массы крситаллов с этилацетатом) добавляли в 0,5 г кристаллов с этилацетатом и затем растворяли при температуре 50-60°С, и затем естественным путем охлаждали до 20-30°С с образованием кристаллов. После образования кристаллов их перемешивали при температуре 20-30°С в течение 2 ч и отфильтровывали. Фильтрат сушили при пониженном давлении при комнатной температуре в течение 12 ч с получением 0,25 г соединения, представленного химической формулой 1, (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинометил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она (выход: 35%).
1Н ЯМР (500 МГц, ДМСО): 10,57 (м, 1H), 6,91-6,90 (м, 1H), 6,80-6,85 (м, 0,5Н), 6,70-6,40 (м, 1,5Н), 6,10-5,96 (м, 3H), 5,65-5,63 (д, 0,5Н), 5,42-5,40 (д, 0,5Н), 4,42-4,40 (м, 0,5Н), 4,10-4,0 (м, 1H), 3,90-3,87 (м, 1,5Н), 3,56 (м, 4Н), 3,20 (с, 2Н), 3,14-3,10 (м, 1H), 2,68-2,63 (м, 0,5Н), 2,32 (м, 4Н), 2,19 (с, 3H), 1,90-2,0 (м, 1H), 1,80 (м, 1H), 1,50-1,40 (м, 2,5Н).
Сравнительный пример 4
Стадии 1 и 2
Соединение, представленное формулой 1-4, получали теми же способами, что и стадии 1 и 2 получения в примере.
Стадия 3
В соединение, представленное химической формулой 1-4 (288,0 г, 1,0 экв.), полученное на стадии 2, добавляли 5760,0 мл ТГФ (количество (мл/г) в 20 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) и 1152,0 мл H2O (количество (мл/г) в 4 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) для получения раствора при комнатной температуре. Затем температуру внутри охлаждали до 0-5°С с помощью внешней бани с температурой 0-5°С (смесь льда и H2O) и затем добавляли NaHCO3 (186,0 г, 3,0 экв.) и растворяли. Раствор, в котором смешивали соединение, представленное химической формулой 1-5, (63,0 мл, 1,05 экв.) по каплям добавляли туда при температуре внутри 0-5°С. Смесь перемешивали при температуре 0-5°С в течение 0,5 ч для завершения реакции.
Стадия экстракции с этилацетатом
В реакционный сосуд, в котором были завершены реакции, добавляли 5760,0 мл этилацетата (количество (мл/г) в 20 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) и 2880,0 мл H2O (количество (мл/г) в 10 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) и затем проводили разделение слоев для выделения только органического слоя. Затем 1440,0 мл этилацетата (количество (мл/г) в 5 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) добавляли в водный слой, повторно экстрагировали и только органический слой собирали и концентрировали при пониженном давлении при температуре снаружи 40°С. Добавляли 5760,0 мл этилацетата (количество (мл/г) в 20 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) и 2880,0 мл H2O (количество (мл/г) в 10 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4), рН доводили до 7,0-7,5 1н раствором HCl и затем слои разделяли дважды. Выделенный органический слой собирали, сушили над Na2SO4 и затем концентрировали при пониженном давлении при температуре снаружи 40°С.
Стадия очистки фосфорной кислотой (Н3РО4)
Добавляли туда 5184,0 мл ТГФ (количество (мл/г) в 18 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) и проводили растворение. Затем по каплям добавляли туда раствор Н3РО4 (6,8 г, 0,095 экв.) в 288,0 мл ТГФ и затем перемешивали в течение 30 минут. Смесь отфильтровывали через Celite для удаления солей и затем раствор Н3РО4 (6,1 г, 0,085 экв.) в 288,0 мл ТГФ по каплям добавляли в фильтрат и перемешивали в течение 30 минут. Полученный остаток отфильтровывали через Celite для удаления солей и затем концентрировали при пониженном давлении при температуре снаружи 40°С.
Стадия кристаллизации с этилацетатом
Добавляли туда 5760,0 мл этилацетата (количество (мл/г) в 20 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) и затем, когда образовались кристаллы, их перемешивали при температуре 20-30°С в течение 2 ч и отфильтровывали. Фильтрат сушили при пониженном давлении при комнатной температуре в течение 12 ч с получением 195,5 г соединения, представленного химической формулой 1, (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинометил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она (выход: 60,0%).
1Н ЯМР (500 МГц, ДМСО): 10,57 (м, 1H), 6,91-6,90 (м, 1H), 6,80-6,85 (м, 0,5Н), 6,70-6,40 (м, 1,5Н), 6,10-5,96 (м, 3H), 5,65-5,63 (д, 0,5Н), 5,42-5,40 (д, 0,5Н), 4,42-4,40 (м, 0,5Н), 4,10-4,0 (м, 1H), 3,90-3,87 (м, 1,5Н), 3,56 (м, 4Н), 3,20 (с, 2Н), 3,14-3,10 (м, 1H), 2,68-2,63 (м, 0,5Н), 2,32 (м, 4Н), 2,19 (с, 3H), 1,90-2,0 (м, 1H), 1,80 (м, 1H), 1,50-1,40 (м, 2,5Н).
Сравнительный пример 5
Стадии 1 и 2
Соединение, представленное формулой 1-4, получали теми же способами, что и стадии 1 и 2 получения в примере.
Стадия 3
В соединение, представленное химической формулой 1-4 (15,0 г, 1,0 экв.), полученное на стадии 2, добавляли 270,0 мл ТГФ (количество (мл/г) в 18 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) и 60,0 мл H2O (количество (мл/г) в 4 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) для получения раствора при комнатной температуре. Затем температуру внутри снижали до 0-5°С с помощью внешней бани с температурой 0-5°С (смесь льда и Н2О) и затем добавляли К2СО3 (8,0 г, 1,5 экв.) и растворяли. Затем в ТГФ, охлажденный до 0-5°С, по каплям добавляли смешанный раствор N,N-диизопропилэтиламина (DIPEA) (334,4 мкл, 0,05 экв.) и соединения, представленного химической формулой 1-5, (3,3 мл, 1,05 экв.) при температуре внутри 0-5°С.Смесь перемешивали при температуре 0-5°С в течение 0,5 ч для завершения реакции.
Стадия экстракции с этилацетатом
В реакционный сосуд, в котором были завершены реакции, добавляли 300,0 мл этилацетата при температуре 0-5°С (количество (мл/г) в 20 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) и 150,0 мл Н2О при температуре 0-5°С (количество (мл/г) в 10 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) и затем проводили разделение слоев для выделения только органического слоя. Затем 75,0 мл этилацетата (количество (мл/г) в 5 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) добавляли в водный слой, повторно экстрагировали и только органический слой собирали и концентрировали при пониженном давлении при температуре снаружи 40°С. Добавляли туда 300,0 мл этилацетата при температуре 0-5°С (количество (мл/г) в 20 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) и 150,0 мл H2O (количество (мл/г) в 10 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4), рН доводили до 7,0-7,5 1н раствором HCl и затем слои разделяли дважды. Выделенный органический слой собирали, сушили над Na2SO4 и затем концентрировали при пониженном давлении при температуре снаружи 35°С.
Стадия очистки фосфорной кислотой (Н3РО4)
270,0 мл ТГФ при температуре 0-5°С (количество (мл/г) в 18 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) добавляли туда и растворяли. Затем раствор Н3РО4 (0,36 г, 0,095 экв.), растворенный в 15,0 мл ТГФ, по каплям добавляли и перемешивали смесь в течение от 30 минут. Смесь отфильтровывали через Celite для удаления солей, а раствор Н3РО4 (0,32 г, 0,085 экв.), растворенный в 15,0 мл ТГФ, по каплям добавляли в фильтрат, и раствор перемешивали в течение 30 минут. Полученный остаток отфильтровывали через Celite для удаления солей и затем концентрировали при пониженном давлении при температуре снаружи 35°С.
Стадия кристаллизации с этилацетатом
Добавляли туда 225,0 мл этилацетата (количество (мл/г) в 15 раз (по объему) большее относительно массы соединения, представленного химической формулой 1-4) и затем, когда образовались кристаллы, их перемешивали при температуре 20-30°С в течение 2 ч и отфильтровывали. Фильтрат сушили при пониженном давлении при комнатной температуре в течение 12 ч с получением 10,2 г соединения, представленного химической формулой 1, (S)-1-(3-((6-((5-метилтиазол-2-ил)амино)-4-(морфолинометил)пиридин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-он (выход: 60,0%).
1Н ЯМР (500 МГц, ДМСО): 10,57 (м, 1H), 6,91-6,90 (м, 1H), 6,80-6,85 (м, 0,5Н), 6,70-6,40 (м, 1,5Н), 6,10-5,96 (м, 3H), 5,65-5,63 (д, 0,5Н), 5,42-5,40 (д, 0,5Н), 4,42-4,40 (м, 0,5Н), 4,10-4,0 (м, 1H), 3,90-3,87 (м, 1,5Н), 3,56 (м, 4Н), 3,20 (с, 2Н), 3,14-3,10 (м, 1H), 2,68-2,63 (м, 0,5Н), 2,32 (м, 4Н), 2,19 (с, 3H), 1,90-2,0 (м, 1H), 1,80 (м, 1H), 1,50-1,40 (м, 2,5Н).
Ингибиторная активность в отношении ВТК и ITK
Ингибиторную активность в отношении ВТК и ITK измеряли для соединения, представленного химической формулой 1, полученного в приведенных выше примерах, следующим образом.
Ингибиторную активность в отношении ВТК оценивали с помощью набора ферментативной системы ADP-Glo™+ВТК киназа (от Promega Corporation). В белом 96-луночном планшете 10 мкл фермента БТК, приготовленного так, чтобы получить конечную концентрацию 10 нг/мл, смешивали с 5 мкл соединений, имеющих конечную концентрацию 1 мкМ в случае оценки соединений с одной концентрацией, и концентрацию 1000, 200, 40, 8, 1,6 и 0,32 нМ в случае оценки IC50, и затем проводили реакцию при комнатной температуре в течение 15 мин. В планшет, на котором реакции были завершены, добавляли 5 мкл субстрата и 5 мкл АТФ, приготовленных так, чтобы получить конечную концентрацию 10 мкМ, и оставляли реагировать при температуре 30°С в течение 1 ч. Все лунки планшета, на которых реакции были завершены, обрабатывали 25 мкл реагента ADP-Glo™ и оставляли реагировать при температуре 30°С в течение 40 мин. После этого все лунки обрабатывали 50 мкл буфера для обнаружения киназ, после чего оставляли реагировать в течение 30 минут в условиях защиты от света. Для планшета, на котором были завершены все реакции, измеряли люминесценцию и проводили математическую обработку результатов. Оценку проводили на двух повторах, а отрицательный контроль и положительный контроль обсчитывали в зависимости от того, добавлялся ли фермент или нет без обработки соединения. IC50 рассчитывали на основе вычисленных значений.
Кроме того, ингибиторную активность в отношении ITK оценивали при использовании набора ферментативной системы ADP-Glo™+ITK киназа (от Promega Corporation). В белом 96-луночном планшете 10 мкл фермента ITK, приготовленного так, чтобы получить конечную концентрацию 4 нг/мл, смешивали с 5 мкл соединений, имеющих конечную концентрацию 1 мкМ в случае оценки соединений с одной концентрацией, и концентрацию 1000, 200, 40, 8, 1,6 и 0,32 нМ в случае оценки IC50, и затем проводили реакцию при комнатной температуре в течение 15 мин. В планшет, на котором все реакции были завершены, добавляли 5 мкл субстрата и 5 мкл АТФ, приготовленных так, чтобы получить конечную концентрацию 25 мкМ, и оставляли реагировать при температуре 30°С в течение 1 ч. Все лунки планшета, на которых реакции были завершены, обрабатывали 25 мкл реагента ADP-Glo™ и оставляли реагировать при температуре 30°С в течение 40 мин. После этого все лунки обрабатывали 50 мкл буфера для обнаружения киназ, после чего оставляли реагировать при температуре 30°С в течение от 30 минут в условиях защиты от света. Для планшета, на котором были завершены все реакции, измеряли люминесценцию и проводили математическую обработку результатов. Оценку проводили на двух повторах, а отрицательный контроль и положительный контроль обсчитывали в зависимости от того, добавлялся ли фермент или нет без обработки соединения. IC50 рассчитывали на основе вычисленных значений.
В результате вычислений ингибиторная активность в отношении ВТК (ВТК IC50) соединения, представленного химической формулой 1, полученного в примере, составила от 0,4 нМ до 1,4 нМ, а ингибиторная активность в отношении ITK (ITK IC50) составила от 1,0 нМ до 1,7 нМ. Из этого можно заключить, что соединение, представленное химической формулой 1, проявляет превосходный ингибирующий эффект двойной активности в отношении ВТК и ITK.
Сравнение примеров и сравнительных примеров 2-5
Чтобы подтвердить наличие примесей, образующихся совместно в процессе получения соединения, представленного химической формулой 1, которое является конечным соединением, полученным в способах получения в примере по изобретению и сравнительных примерах 2-5, примеси выделяли с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ) в соответствии с относительным временем удерживания (RRT) в условиях, показанных в таблице 1 ниже, а результаты показаны в таблице 2 ниже вместе с выходом на стадии 3.
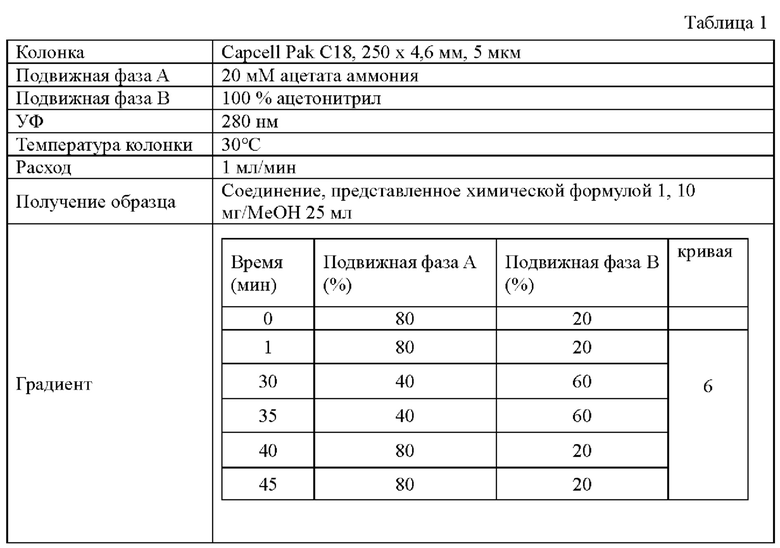
1) Примесь, имеющая RRT менее 1,0 (RRT<1,0)
2) Примесь А, имеющая RRT 1,17 (ImpA): соединение, в котором связаны соединение, представленное химической формулой 1, и соединение, представленное химической формулой 1-4
3) Примесь В, имеющая RRT 1,37 (ImpB): димер соединения, представленного химической формулой 1
4) Примесь С (ImpC) с RRT 1,18: соединение, в котором НСl присоединилась по двойной связи соединения, представленного химической формулой 1, и соединение, в котором HCl присоединилась по двойной связи акриламида
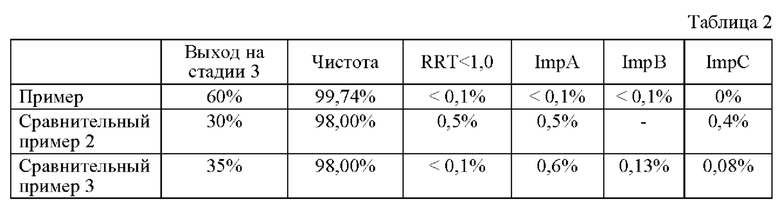

Как показано в таблице 2 выше, было подтверждено, что, когда соединение получают способом из примера по изобретению, можно получить с высоким выходом соединение высокой чистоты, в котором снижено количество всех видов примесей, по сравнению со способом из сравнительного примера 2, в котором соединение выделяют и очищают путем первичной и вторичной кристаллизации, дополнительно к способу из сравнительного примера 1, описанного выше.
Кроме того, было подтверждено, что, когда соединение получают способом из примера по изобретению, могут быть получены высококачественные соединения с пониженным содержанием примесей по сравнению со способами из сравнительных примеров 3-5, в которых соединение, представленное химической формулой 1, получают путем замены некоторых процессов на стадии 3 в способе из примера по изобретению.
В частности, может быть подтверждено, что соединение, полученное согласно способу из примера по изобретению, в котором (1) N,N-диизопропилэтиламина (DIPEA) используют в реакции на стадии 3, (2) температуру реакции на стадии 3 доводят до от -8 до -3°С, (3) конечное соединение выделяют путем кристаллизации с этилацетатом (ЭА), имеет следующие особенности:
по сравнению с соединением, полученным согласно способу из сравнительного примера 3, в котором (1) N,N-диизопропилэтиламин (DIPEA) не используют в реакции на стадии 3, (2) температуру реакции на стадии 3 доводят до от 0 до 5°С, и (3) конечное соединение выделяют путем кристаллизации с этилацетатом (ЭА) и диметоксиэтаном (DME), содержание примеси A (ImpA), примеси В (ImpB) и примеси С (ImpC) низкое;
по сравнению с соединением, полученным согласно способу из сравнительного примера 4, в котором (1) N,N-диизопропилэтиламин (DIPEA) не используют в реакции на стадии 3, (2) температуру реакции на стадии 3 доводят до от 0 до 5°С, и (3) конечное соединение выделяют путем кристаллизации с этилацетатом (ЭА), содержание примеси В (ImpB) и примеси С (ImpC) низкое; и
по сравнению с соединением, полученным согласно способу из сравнительного примера 5, в котором (1) N,N-диизопропилэтиламин (DIPEA) используют в реакции на стадии 3, но (2) температуру реакции на стадии 3 доводят до от 0 до 5°С, и (3) конечное соединение выделяют путем кристаллизации с этилацетатом (ЭА), содержание примеси В (ImpB) низкое.
Сравнение примера, сравнительного примера 1 и сравнительного примера 2 Выходы на каждой стадии способов получения из примера по изобретению, сравнительного примера 1 и сравнительного примера 2 показаны в таблице 3 ниже.
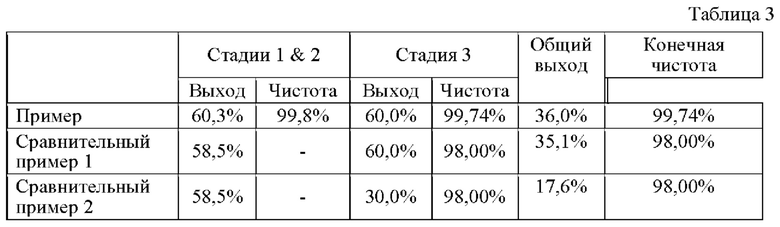
Как показано в таблице 3 выше, было подтверждено, что, когда соединение, представленное химической формулой 1, получают способом из примера по изобретению, стадию 1 и стадию 2 проводят in-situ, а выделение и очистку соединения получают путем кристаллизации, что является преимуществом для промышленного производства, в отличие от способа сравнительного примера 1, в котором требуется микроволновое устройство, а выделение и очистку полученного соединения осуществляют с помощью колонки, и, кроме того, нет снижения выхода по сравнению со способом из сравнительного примера 1.
Кроме того, может быть подтверждено, что способ из примера по изобретению может давать конечное соединение с очень высоким выходом по сравнению со способом сравнительного примера 2, в котором выделение и очистку соединения дополнительно проводят путем первичной и вторичной кристаллизации после способа сравнительного примера 1 для повышения конечной чистоты соединения.
Таким образом, было подтверждено, что соединение, представленное химической формулой 1, имеющее высокое качество, можно производить массово в промышленном масштабе с помощью способа из примера по настоящему изобретению.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ ПРОИЗВОДНОЕ ГЕТЕРОЦИКЛИЧЕСКОГО АМИНА И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2019 |
|
RU2760184C1 |
| НОВОЕ ПРОИЗВОДНОЕ ФЕНИЛПИРИДИНА И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2018 |
|
RU2748945C1 |
| КРИСТАЛЛ ПИРРОЛОПИРИМИДИНА ДЛЯ ПОЛУЧЕНИЯ JAK-ИНГИБИТОРА | 2017 |
|
RU2746045C2 |
| ПРОИЗВОДНЫЕ (АЗЕТИДИН-1-ИЛАЛКИЛ)ЛАКТАМОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ ЧЕЛОВЕКА, ПРИ КОТОРОМ ЗАБОЛЕВАНИЕ ЛЕЧАТ ПОСРЕДСТВОМ ПРОДУЦИРОВАНИЯ АНТАГОНИСТИЧЕСКОГО ДЕЙСТВИЯ НА ТАХИКИНИН, ДЕЙСТВУЮЩИЙ В ЧЕЛОВЕЧЕСКОМ NК-, NК- И NК-РЕЦЕПТОРЕ ИЛИ В ИХ СОЧЕТАНИИ | 1995 |
|
RU2150468C1 |
| АЗОТСОДЕРЖАЩЕЕ СПИРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2010 |
|
RU2630694C2 |
| ПРИМЕНЕНИЕ МОНОЦИКЛИЧЕСКОГО β-ЛАКТАМНОГО СОЕДИНЕНИЯ В ФАРМАЦИИ | 2019 |
|
RU2785715C1 |
| НОВОЕ КОНДЕНСИРОВАННОЕ ПИРИМИДИНОВОЕ СОЕДИНЕНИЕ ИЛИ ЕГО СОЛЬ | 2014 |
|
RU2666349C2 |
| НОВЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРА ГИСТОНДЕАЦЕТИЛАЗЫ 6 И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2021 |
|
RU2814288C1 |
| ИНГИБИТОРЫ КАСПАЗ | 1999 |
|
RU2274642C2 |
| ИНГИБИТОР FGFR И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2018 |
|
RU2771311C2 |
Настоящее изобретение относится к способу получения соединения, представленного химической формулой 1, которое проявляет ингибирующий эффект в отношении ITK (интерлейкин-2 тирозинкиназа) и BTK (тирозинкиназа Брутона). Предлагаемый способ включает стадии 1)-3). На стадии 1) получают соединение, представленное химической формулой 1-3, путем взаимодействия соединения, представленного химической формулой 1-1, с соединением, представленным химической формулой 1-2, в присутствии палладиевого катализатора и основания. На стадии 2) получают соединение, представленное химической формулой 1-4, путем взаимодействия соединения, представленного химической формулой 1-3, в присутствии кислоты. На стадии 3) получают соединение, представленное химической формулой 1, путем взаимодействия соединения, представленного химической формулой 1-4, с соединением, представленным химической формулой 1-5, в присутствии основания. Предлагаемый способ позволяет получать соединение, представленное химической формулой 1, с высоким выходом и пониженным содержанием примесей. 16 з.п. ф-лы, 3 табл., 7 пр.
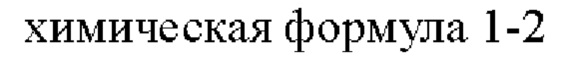
1. Способ получения соединения, представленного химической формулой 1, включающий следующие стадии:
1) получение соединения, представленного химической формулой 1-3, путем взаимодействия соединения, представленного химической формулой 1-1, с соединением, представленным химической формулой 1-2, в присутствии палладиевого катализатора и основания;
2) получение соединения, представленного химической формулой 1-4, путем взаимодействия соединения, представленного химической формулой 1-3, в присутствии кислоты; и
3) получение соединения, представленного химической формулой 1, путем взаимодействия соединения, представленного химической формулой 1-4, с соединением, представленным химической формулой 1-5, в присутствии основания:
2. Способ по п. 1, в котором на стадии (1) соединение, представленное химической формулой 1-1, и соединение, представленное химической формулой 1-2, используют в молярном отношении от 1:0,1 до 1:2.
3. Способ по п. 1, в котором на стадии (1) палладиевый катализатор представляет собой по меньшей мере один, выбранный из группы, состоящей из трис(дибензилиденацетон)дипалладия (0), тетракис(трифенилфосфин)палладия (0), бис[трис(2-метилфенил)фосфин]палладия и ацетата палладия (II).
4. Способ по п. 1, в котором на стадии (1) основание представляет собой по меньшей мере одно, выбранное из группы, состоящей из карбоната цезия, карбоната калия, карбоната натрия, трет-бутоксида натрия и трет-бутоксида калия.
5. Способ по п. 1, в котором реакцию на стадии (1) проводят при температуре от 80 до 150°С в течение от 3 до 15 ч.
6. Способ по п. 1, в котором на стадии (2) кислота представляет собой соляную кислоту.
7. Способ по п. 1, в котором стадия (2) также включает стадию кристаллизации продукта реакции.
8. Способ по п. 7, в котором стадию кристаллизации проводят путем первичной кристаллизации с водой и вторичной кристаллизации с метанолом.
9. Способ по п. 1, в котором на стадии (3) соединение, представленное химической формулой 1-4, и соединение, представленное химической формулой 1-5, используют в молярном отношении от 1:0,5 до 1:2,0.
10. Способ по п. 1, в котором на стадии (3) основание представляет собой по меньшей мере одно, выбранное из группы, состоящей из карбоната калия, гидроксида натрия, гидроксида лития, гидроксида калия, триэтиламина, диизопропиламина, диизопропилэтиламина, гидрокарбоната натрия, гидрокарбоната калия, карбоната цезия, карбоната натрия, метилата натрия и бутирата калия.
11. Способ по п. 1, в котором на стадии (3) соединение, представленное химической формулой 1-5, добавляют в виде смеси с N,N-диизопропилэтиламином или триэтиламином.
12. Способ по п. 1, в котором реакцию на стадии (3) проводят при температуре от -10 до 10°С.
13. Способ по п. 1, в котором стадия (3) включает также стадию экстракции продукта реакции с использованием этилацетата.
14. Способ по п. 13, в котором стадию экстракции проводят при рН от 6,5 до 7,5.
15. Способ по п. 1, в котором стадия (3) также включает стадию растворения продукта реакции в тетрагидрофуране, смешивания раствора, содержащего фосфорную кислоту, с полученным раствором и фильтрации полученной смеси с последующим концентрированием фильтрата при пониженном давлении.
16. Способ по п. 15, в котором раствор, содержащий фосфорную кислоту, представляет собой раствор, в котором в тетрагидрофуране растворено от 0,05 до 0,5 моль фосфорной кислоты на 1 моль соединения, представленного химической формулой 1-4.
17. Способ по п. 1, в котором стадия (3) также включает стадию кристаллизации продукта реакции с этилацетатом.
| J | |||
| D | |||
| HARLING et al., Discovery of Novel Irreversible Inhibitors of Interleukin (IL)-2-inducible Tyrosine Kinase (Itk) by Targeting Cysteine 442 in the ATP Pocket, J | |||
| BIOL | |||
| CHEM., 2013, vol | |||
| ДВОЙНОЙ ГАЕЧНЫЙ КЛЮЧ | 1920 |
|
SU288A1 |
| Машина для изготовления проволочных гвоздей | 1922 |
|
SU39A1 |
| ШЛИХТОВАЛЬНАЯ МАШИНА | 1931 |
|
SU28195A1 |
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| CN 104628657 A, 20.05.2015 | |||
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| RU | |||

