Область применения способа
Изобретение относится к способам получения однофазного железоиттриевого граната Y3Fe5O12 с последующим применением его в устройствах с магнитным пузырьковым доменом для памяти интегральных схем и устройств с зарядовой связью, магнитных записывающих устройств, микроволновых устройств. Однофазность получаемого порошка определяет точность и воспроизводимость (независимо от времени синтеза) функциональных характеристик материала заявленного материала и определяет стабильность работы указанных устройств.
Уровень техники
Аналог 1. Известен способ получения ферритового материала при помощи измельчения исходной смеси компонентов в шаровых мельницах в течение 24 часов с последующим отжигом при 1200-1250 °С в течение 5 часов, затем смесь повторно измельчают в шаровых мельницах в течение 96 часов, после измельчения смесь высушивают сутки, затем вводят пластификатор, прессуют при 200 МПа и подвергают радиационно-термическому спеканию заготовок путем их нагрева до температуры спекания 1350-1450 °С облучением проникающим пучком быстрых электронов с выдержкой при температуре спекания в течение 30-90 минут под непрерывным электронным пучком. Итоговое суммарное время синтеза не менее 150 часов. Результатом синтеза является порошок железоиттриевого граната, итоговый продукт заявлен как однофазный. Недостатками данного способа синтеза является использование достаточно сложной техники для получения конечного продукта, длительный процесс получения и высокая температура синтеза [Способ получения поликристаллических ферритов-гранатов: пат. 2660493 RU, 03.11.2017].
Аналог 2. Известен способ получения наноразмерного порошка железоиттриевого граната, включающего приготовление водного раствора солей иттрия (III) и водного раствора солей железа (III). Сначала реагент-осадитель, в качестве которого используют сильноосновный гелевый анионит АВ-17-8 в гидроксидной форме, приводят в контакт с раствором солей иттрия (III) при комнатной температуре в течение 1 часа затем добавляют раствор солей железа (III). Из полученного раствора осаждают продукт-прекурсор, отделяют его от раствора, промывают водой, сушат и обжигают при температуре не менее 700 °C в течение 4 часов. Результатом синтеза является наноразмерный порошок железоиттриевого граната, итоговый продукт заявлен как однофазный, общее время синтеза не регламентировано. Недостатком данного метода является длительный процесс подготовки полупродукта к отжигу, включающего стадии осаждения, отделения раствора от осадка, промывание осадка и сушка осадка [Способ получения наноразмерного порошка железоиттриевого граната: пат. 2509625 RU, 28.12.2012].
Аналог 3. Известен способ получения наноструктурированных порошков ферритов со структурой перовскита или граната, включающий получение смеси соли азотной кислоты и по крайней мере одного оксидного соединения металла, ультразвуковую обработку, термообработку и фильтрацию. Получают смесь азотнокислого железа и по крайней мере одного оксида металла, выбранного из группы: марганец, висмут, литий, иттрий, или карбоната лития при мольном соотношении азотнокислое железо: оксиды металлов или карбонат лития равном (2-10):(1-5). Ультразвуковую обработку осуществляют с частотой 1,7 МГц при мощности излучения 20 Вт в течение 4 часов в потоке воздуха, который подают со скоростью 0,014-0,15 м/с. Термообработку осуществляют в три стадии: при 300-350 °С на первой стадии; при 700-900 °С на второй стадии и при 120-150 °С на третьей стадии. Фильтрацию осуществляют с использованием электрофильтра, на коронирующий электрод которого подают напряжение 6-9 кВ. Результатом синтеза является наноструктурированный порошок феррита, чистота итогового продукта не регламентирована. Недостатки данного метода состоят в использовании специальной, не серийной техники для получения конечного продукта [Способ получения наноструктурированных порошков ферритов и установка для его осуществления: пат. 2653824 RU, 30.03.2017].
Аналог 4. Известен способ приготовления иттрий-замещенного граната. Растворы нитрата или сульфата железа (III), нитрата иттрия медленно смешиваются с осадителем (в течение не менее 1 часа). В качестве осадителя может выступать аммиак, гидроксид тетраметиламмония. Полученный осадок после дозревания (не менее 2 часов) отделяют от маточного раствора и многократно промывают. После этого осадок сушат (время и температура сушки не регламентированы), прокаливают при температуре 600-1300 °С не менее 4 часов. Недостатком данного способа является несоответствие исходных реактивов (солей металлов) к требованиям, предъявляемым для весовых форм (точное соответствие состава весовой формы ее химической формуле и достаточная химическая устойчивость во времени), т.к. указанные нитраты или сульфаты металлов являются кристаллогидратами, вследствие чего могут возникать отклонения от расчетного соотношения металлов, что может привести к получению неоднофазного продукта, проявляющего в результате нерегулярные функциональные характеристики (магнитные, диэлектрические, электродинамические) [Method of manufacturing shaped body made of ferrite crystals of garnet polycrystal structure: пат. 2015606 CA, 27.04.1990].
Аналог 5 (прототип). Известен способ получения железоиттриевого граната с помощью метода, обозначенного как solution combustion synthesis (метод сжигания раствора). Растворы исходных веществ получали из гексагидрата нитрата иттрия Y(NO3)3*6H2O, 9-водного нитрата железа Fe(NO3)3*9H2O в качестве прекурсоров и аминоуксусную кислоту C2H5NO2 в качестве органического восстановителя. Глицин брали в количестве, двукратно превышающем стехиометрическое для целенаправленного получения гелеобразного полупродукта. Растворы прекурсоров нитратов металлов смешивали, при нагреве вносили в реакционную ёмкость глицин. В результате реакции получали гелеобразный полупродукт, при дальнейшем нагреве происходило возгорание смеси с образованием сухого неорганического полупродукта, обогащённого органическими остатками. Полученный порошок переносился в тигель и подвергался высокотемпературному отжигу при 800, 900, 1000, 1100 и 1200 °С в течение 4 часов при каждой температуре (не менее 20 часов общего времени отжига). После 1200 °С получали конечный продукт - железоиттриевый гранат, аттестованный методом рентгенофазового анализа, однако приведённая авторами дифрактограмма содержит малоинтенсивные рефлексы, не относящиеся к фазе железоиттриевого граната, что может свидетельствовать о недостаточной чистоте продукта [Size-Dependent Structural, Magnetic and Magnetothermal Properties of Y3Fe5O12 Fine Particles Obtained by SCS // Nanomaterials, 2022, 12, 2733].
Техническая задача заключается в создании способа получения однофазного железоиттриевого граната, обладающего регулярными воспроизводимыми функциональными характеристиками, с целью расширения средств, лишенного недостатков аналогов, а именно с использованием только доступного, типового лабораторного оборудования, посуды и реактивов, без использования дополнительных установок (например, источника быстрых электронов или специальных установок на базе ультразвуковых диспергаторов) или дорогостоящего оборудования (например, шаровых или планетарных лабораторных мельниц), с относительно быстрым получением полупродукта (не более 3 часов) и конечного материала со структурой граната (суммарное время синтеза не более 35 часов).
Технический результат поставленной задачи достигается в предлагаемом способе получения однофазного железоиттриевого граната, заключающемся в глицин-нитратном синтезе граната и двухстадийной термической обработке.
Частицы порошка граната приготавливали с использованием прекурсора из оксида иттрия, азотнокислого железа (III), n-водного. Содержание основного вещества по металлическим примесям составило не менее 99,9% для обоих реактивов. Фактическое содержание воды (n) в кристаллогидрате азотнокислого железа (III) Fe(NO3)3×nH2O, предварительно определяли гравиметрически, путем термического разложения известного количества указанной соли до оксида Fe2O3 (отжиг при 800 °C в течение 5 часов в воздушной атмосфере). Число n рассчитывали по формуле (1):

где M - молярные массы соответствующих соединений,
 - масса вещества до термического разложения,
- масса вещества до термического разложения,
 - масса вещества после термического разложения. Анализ проводили не менее чем в пяти параллелях, полученное значение n усредняли и учитывали в расчете с точностью до тысячных долей после запятой.
- масса вещества после термического разложения. Анализ проводили не менее чем в пяти параллелях, полученное значение n усредняли и учитывали в расчете с точностью до тысячных долей после запятой.
Взятие навесок стехиометрических количеств исходных веществ осуществляли на аналитических весах, затем оксид иттрия и азотнокислое железо n-водное растворяли в стехиометрическом количестве азотной кислоты концентрацией 3-3,5М и в воде соответственно. Далее растворы помещали в один сосуд и перемешивали в течение 30 минут. Затем реакционную смесь переносили в широкий термостойкий (фарфоровый или кварцевый) реакционный сосуд и добавляли аминоуксусную кислоту (глицин) с целью получения нитрат-органического прекурсора.
Аминоуксусную кислоту брали в стехиометрическом количестве из расчета на полное восстановление нитрат-ионов в реакционной смеси до газообразного азота, согласно уравнению реакции (2), а именно брали 2,87 г аминоуксусной кислоты на 1 г оксида иттрия.

Затем полученный нитрат-органический прекурсор упаривали при интенсивном кипении (не более двух часов), вплоть до начала самопроизвольного пиролиза реакционной смеси. В результате пиролиза образовался ультрадисперсный порошок, являющийся полупродуктом. Данный полупродукт помещали в тигель для дальнейшей двухстадийной термической обработки: на воздухе при 750 °C (отжиг) в течение 8 часов с последующим перемешиванием в ступке, а затем на воздухе при 1200 °C (спекание) в течение 20 часов. Согласно результатам рентгенофазового анализа и сканирующей электронной микроскопии, полученный продукт представляет собой однофазный железоиттриевый гранат, размер частиц около 2 мкм, форма частиц округлая, стержнеобразная. Скорость нагрева образцов при термической обработке составляла 8 °С/мин.
Эффект (свойство), которое проявляется при осуществлении способа
Методы пиролиза нитрат-органических прекурсоров основаны на термообработке смесей необходимых исходных компонентов и органического восстановителя, в качестве которого в глицин-нитратном методе используют аминоуксусную кислоту C2H5NO2. Аминоуксусная кислота выступает в роли бидентантного лиганда, способного образовывать устойчивые комплексы, что повышает растворимость нитратов и предотвращает выпадение осадков при испарении воды. С другой стороны, аминоуксусная кислота в процессе пиролиза формирует наивысшую температуру пламени (до 1500 °С), что способствует снижению конечной температуры синтеза. Большое количество газов, выделяющихся во время реакции пиролиза, приводит к диспергированию твердого продукта, и, соответственно, к равномерному распределению иттрия и железа по всему объему и увеличению реакционной площади частиц полупродукта. Поэтому получение порошкообразного материала после стадии пиролиза позволяет сократить количество стадий термической обработки, измельчения и перемешивания, понизить температуру конечного отжига, и использовать только доступное, типовое лабораторное оборудование, посуду и реактивы без использования дополнительных установок (источник быстрых электронов или специальных установок на базе ультразвуковых диспергаторов, шаровых или планетарных лабораторных мельниц), и относительно быстро получить полупродукт (менее чем за 3 часа) и конечный о материал железоиттриевого граната (суммарное время синтеза не более 35 часов).
Новизна предлагаемого способа
В сравнении с указанным прототипом [Kiseleva T. et al. Size-Dependent Structural, Magnetic and Magnetothermal Properties of Y3Fe5O12 Fine Particles Obtained by SCS // Nanomaterials, 2022, 12, 2733], авторы настоящей заявки использовали в качестве исходного реагента оксид иттрия вместо нитрата иттрия, аминоуксусная кислота взята в количестве, строго отвечающем окислительно-восстановительной реакции (2), а также проводилась предварительная аттестация состава кристаллогидратных форм солей. В прототипе количество аминоуксусной кислоты было намеренно кратно (вдвое) увеличено относительно расчетных значений для образования гелеобразного полупродукта; это одновременно приводило к снижению температуры пиролиза, наблюдалась частичная карбонизация полупродукта и появление примесных фаз. В результате для удаления примесей в прототипе использовали пять последовательных отжигов с промежуточными перетираниями образца общей продолжительностью не менее 20 часов. Напротив, в предполагаемом способе можно получить более чистый продукт со сравнительно меньшими трудовыми и временными затратами. Первая стадия термической обработки - отжиг при 750 °С необходим в предлагаемом способе для полного удаления остатков органических веществ и формирования промежуточных фаз Y3Fe5O12, YFeO3 и Fe2O3. Последующая стадия термической обработки - спекание при 1200 °С приводит к образованию однофазного продукта Y3Fe5O12. Основной особенностью предлагаемого метода является использование оптимального количества аминоуксусной кислоты и оптимальных режимов температурной обработки, за счет чего получают диспергированный твердый продукт, в котором элементы железо и иттрий равномерно распределены по всему объему, а природа восстановителя и размер зерен полупродукта, который обеспечивает большую реакционную площадь частиц, приводят к минимизации количества стадий термической обработки - его снижение до двух. Кроме того, предполагаемый метод позволяет уменьшить стоимость синтеза за счет использования оксида иттрия вместо более дорогостоящего нитрата иттрия, использовать только типовое, доступное лабораторное оборудование, посуду и реактивы, а также уменьшить общие трудозатраты химика-синтетика. Предлагаемый метод позволяет получить однофазный железоиттриевый гранат, проявляющий регулярные, воспроизводимые физико-химические и функциональные характеристики.
Общие технические характеристики способа
Осуществление заявляемого способа получения однофазного граната Y3Fe5O12 при помощи глицин-нитратного пиролиза не превышает 35 часов: 1 час занимает получение ультрадисперсного порошка (полупродукта), 8 часов занимает первая стадия термической обработки (отжиг) при 750 °С, вторая стадия (спекание) при температуре 1200 °С длится 20 часов, включая процессы нагрева и охлаждения, после чего, по данным рентгенофазового анализа, формируется однофазный железоиттриевый гранат Y3Fe5O12. Расход сырья осуществлялся в соответствии с необходимым молярным соотношением реактивов оксида иттрия Y2O3 и азотнокислого железа n-водного (III) Fe(NO3)3×nH2O. По данным рентгеновской дифракции выход продукта составляет 100%.
Общая характеристика продукции, полученной заявляемым способом
Образцы, изготовленные по заявляемому способу приобретают кристаллическую структуру граната Y3Fe5O12. Примесных фаз не обнаружено.
Примеры осуществления способа
Осуществления способа синтеза железоиттриевого граната рассмотрены на нескольких примерах, основанных на изменении условий различных этапов синтеза (таблица 1).
Пример 1
Синтез проводили по способу, описанному в заявке. После отжига при 750 °C в течение 8 часов с последующим перетиранием в ступке и спеканием при 1200 °C в течение 20 часов образовался однофазный железоиттриевый гранат Y3Fe5O12; размер частиц около 2 мкм; форма частиц округлая, стержнеобразная; частицы образуют агломераты (фиг. 1 и 2).
Пример 2
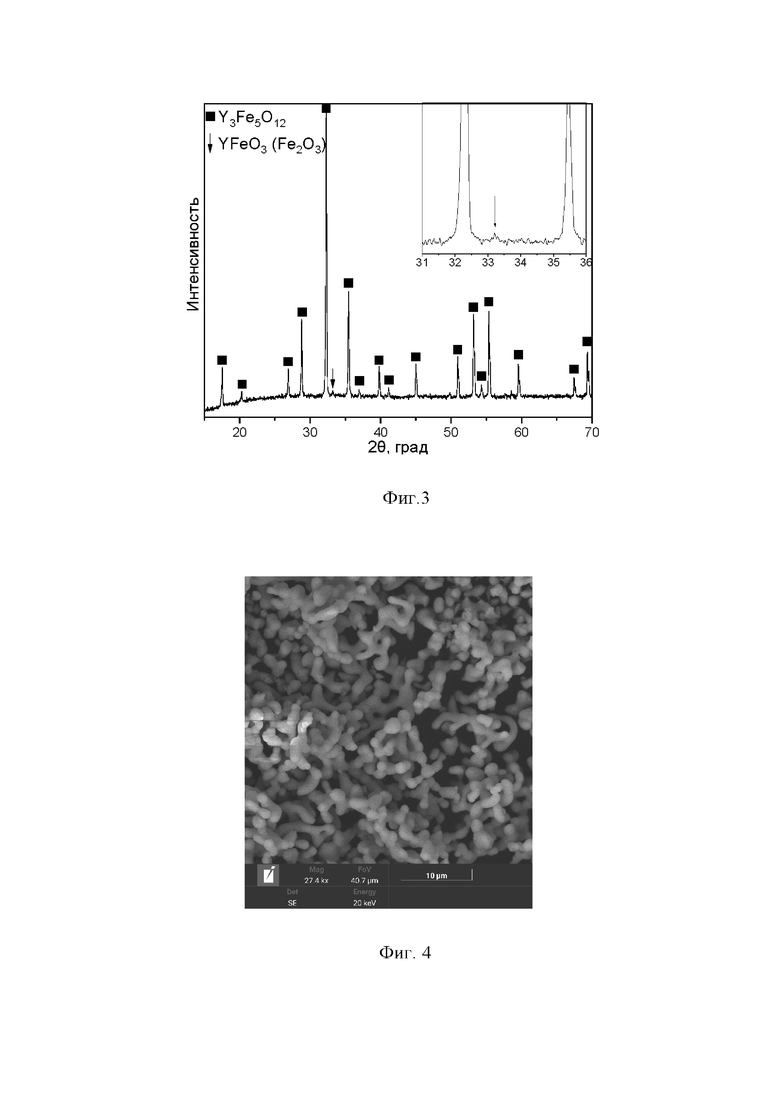
Синтез проводили по способу, описанному в заявке, однако вместо глицина в качестве органического восстановителя использовалась лимонная кислота. После получения полупродукта и проведения двухстадийной термической обработки был получен железоиттриевый гранат с наличием примесной фазы ортоферрита иттрия YFeO3 (возможно оксида железа Fe2O3). Выход железоиттриевого граната составил 98,5-99 %; размер частиц около 1,5-2 мкм; форма частиц округлая, стержнеобразная; частицы формируют агломераты (фигуры 3 и 4).
Пример 3
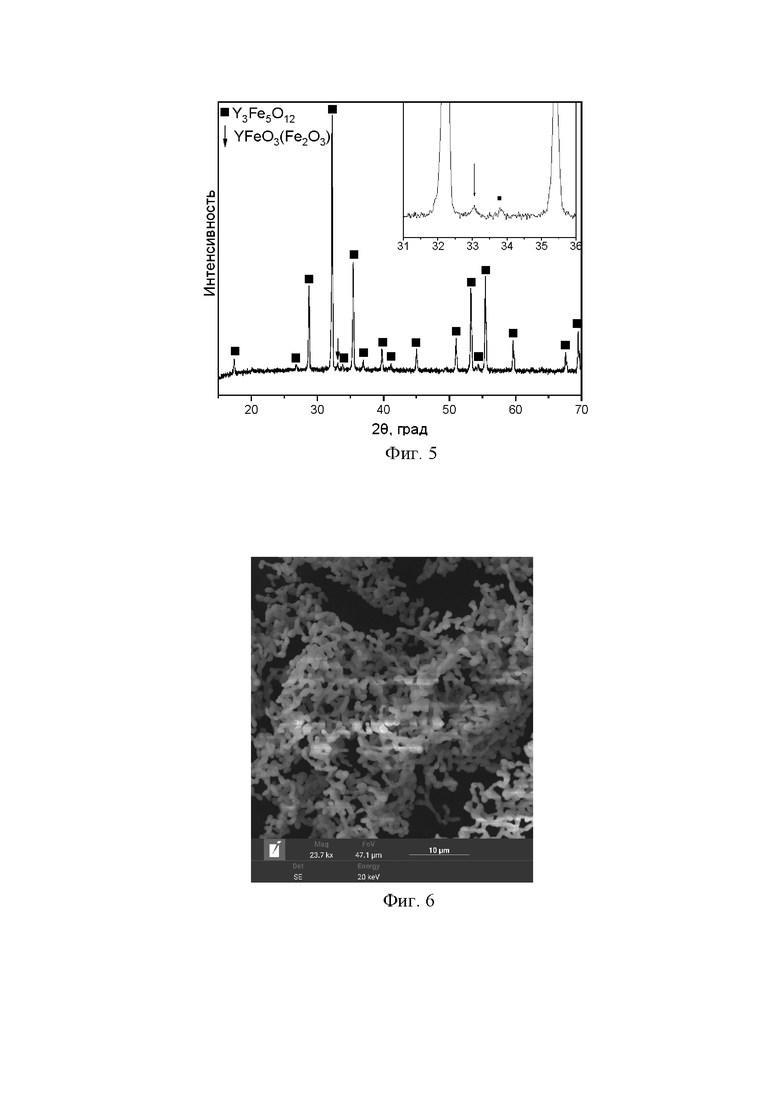
Синтез проводили по способу, описанному в заявке, однако была изменена температура спекания: она была снижена до 1100 °C, при этом продолжительность спекания составляла 20 часов. В результате в образце присутствовала примесная фаза ортоферрита иттрия YFeO3 (возможно оксида железа Fe2O3). Выход железоиттриевого граната составил 98-98,5 %; размер частиц около 1-1.4 мкм; форма частиц округлая, стержнеобразная; частицы образуют агломераты (фигуры 5 и 6).
Пример 4
Синтез проводили по способу, описанному в заявке, однако продолжительность спекания была понижена до 12 ч, его температура была 1200 °C. В результате в образце присутствовала примесная фаза ортоферрита иттрия YFeO3 (возможно оксида железа Fe2O3). Выход железоиттриевого граната составил 98 %; размер частиц около 1-1.8 мкм; форма частиц округлая, стержнеобразная; частицы образуют агломераты (фигуры 7 и 8).
Таблица 1. Описание особенностей синтеза граната по примерам
спекания, ч
Чертежи, поясняющие материалы и их описание.
Фигура 1. Дифрактограмма продукта, полученного в рамках примера 1. Фиксируется только фаза железоиттриевого граната Y3Fe5O12. Здесь и на фигурах 3, 5, 7 черные квадраты - рефлексы Y3Fe5O12; на врезке - увеличенный в 3 раза по оси интенсивности фрагмент дифрактограммы в диапазоне 2θ=31-36о; порошковый дифрактометр XRD-7000, излучение Cu Ka.
Фигура 2. SE-изображения продукта, полученного в рамках примера 1. Размер частиц около 2 мкм, форма частиц округлая, стержнеобразная. Масштаб 5 мкм, увеличение *3000, ускоряющее напряжение U=10 кВ. Здесь и на фигурах 4, 6, 8 сканирующий электронный микроскоп JSM 6390LV.
Фигура 3. Дифрактограмма продуктов, полученных в рамках примера 2. Наряду с фазой граната Y3Fe5O12 фиксируется и посторонняя фаза. Здесь и на фигурах 5, 7 черная стрелка - рефлекс примесной фазы ортоферрита иттрия YFeO3 (возможно оксида железа Fe2O3).
Фигура 4. SE-изображения продукта, полученного в рамках примера 2. Масштаб 10 мкм; увеличение *27000; U=20 кВ.
Фигура 5. Дифрактограмма продуктов, полученных в рамках примера 3. Наряду с фазой граната Y3Fe5O12 фиксируется и посторонняя фаза.
Фигура 6. SE-изображения продукта, полученного в рамках примера 3. Масштаб 10 мкм; увеличение *24000; U=20 кВ.
Фигура 7. Дифрактограмма продуктов, полученных в рамках примера 4. Наряду с фазой граната Y3Fe5O12 фиксируется и посторонняя фаза.
Фигура 8. SE-изображения продукта, полученного в рамках примера 4. Масштаб 10 мкм; увеличение *29000; U=20 кВ.
Результаты получены в ЦКП «Геоаналитик» ИГГ УрО РАН в рамках госзадания № 123011800012-9, дооснащение ЦКП поддержано Минобрнауки РФ, соглашение № 075-15-2021-680
Литература
1. Способ получения поликристаллических ферритов-гранатов: пат. 2660493 Рос. Федерация МПК B22F 3/16, H01F 1/10, C04B 35/40 / Костишин В.Г., Щербаков С.В., Налогин А.Г., Мезенцева М.П., Михайленко М.А., Коробейников М.В., Брязгин А.А.; заявитель и патентообладатель Федеральное государственное автономное образовательное учреждение высшего образования "Национальный исследовательский технологический университет "МИСиС". - №2017138381; заявл. 03.11.2017; опубл. 06.07.2018, Бюл. № 19
2. Способ получения наноразмерного порошка железоиттриевого граната: пат. 2509625 Рос. Федерация МПК B22F 9/16, B82Y 30/00 / Пашков Г.Л., Сайкова С.В., Пантелеева М.В., Линок Е.В.; заявители и патентообладатели Федеральное государственное автономное образовательное учреждение высшего профессионального образования «Сибирский федеральный университет», Федеральное государственное бюджетное учреждение науки Институт химии и химической технологии Сибирского отделения РАН. - №2012158138/02; заявл. 28.12.2012; опубл. 20.03.2014, Бюл. № 8.
3. Способ получения наноструктурированных порошков ферритов и установка для его осуществления: пат. 2653824 Рос. Федерация МПК C01G 49/00, B82B 3/00, B82Y 30/00, C01G 29/00, C01G 45/00, C01F 17/00 C01D 15/00 C01B 12/18 B22F 9/14 B01J 19/10 / Дмитриев А.В., Владимирова Е.В., Кандауров М.В., Подгорбунских Д.Е.; заявитель и патентообладатель Федеральное государственное бюджетное учреждение науки Институт химии твердого тела Уральского отделения Российской академии наук. - №2017110533; заявл. 30.03.2017; опубл. 14.05.2018, Бюл. №14.
4. Method of manufacturing shaped body made of ferrite crystals of garnet polycrystal structure: patent №2015606 CA IPC C01G49/00, C04B35/26, C04B35/645, H01F1/34 / Imaeda M., Asai E., Okamoto K.; applicant NGK INSULATORS, LTD. - № CA2015606C; application 27.04.1990; publication 04.08.1998.
5. Kiseleva T., Abbas R., Martinson K., Komlev A., Lazareva E., Tyapkin P., Solodov E., Rusakov V., Pyatakov A., Tishin A., et al. Size-Dependent Structural, Magnetic and Magnetothermal Properties of Y3Fe5O12 Fine Particles Obtained by SCS // Nanomaterials, 2022, 12, 2733.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения высокоэнтропийного железоредкоземельного граната состава (Ln1Ln2Ln3Ln4Ln5)FeO с эквимолярным соотношением редкоземельных компонентов | 2023 |
|
RU2822522C1 |
| Способ получения однофазного циркона | 2023 |
|
RU2819814C1 |
| СПОСОБ ПОЛУЧЕНИЯ НАНОРАЗМЕРНОГО ПОРОШКА ЖЕЛЕЗОИТТРИЕВОГО ГРАНАТА | 2012 |
|
RU2509625C1 |
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНООКСИДНЫХ МАТЕРИАЛОВ | 2019 |
|
RU2733966C1 |
| Однофазный поликристаллический иттрий-алюминиевый гранат, активированный эрбием, иттербием, и способ его получения | 2018 |
|
RU2705848C1 |
| Электродный материал для электрохимических устройств | 2020 |
|
RU2749746C1 |
| Способ получения маггемита | 2020 |
|
RU2732298C1 |
| Комплексный способ получения малоагломерированных высокостехиометричных наноразмерных порошков прекурсора на основе иттрий-алюминиевого граната с оксидами редкоземельных элементов | 2019 |
|
RU2721548C1 |
| СПОСОБ ПОЛУЧЕНИЯ НАНОДИСПЕРСНОГО ПОРОШКА ОКСИДА ЦИРКОНИЯ, СТАБИЛИЗИРОВАННОГО ОКСИДОМ ИТТРИЯ И/ИЛИ СКАНДИЯ | 2011 |
|
RU2492157C2 |
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНЫХ ОКСИДОВ МЕТАЛЛОВ | 2023 |
|
RU2833013C1 |


Изобретение относится к химической промышленности и может быть использовано при изготовлении интегральных схем, устройств с зарядной связью, а также магнитных записывающих и микроволновых устройств. Однофазный железоиттриевый гранат Y3Fe5O12 получают с использованием технологии глицин-нитратного пиролиза. В качестве исходных реагентов используют оксид иттрия Y2O3, растворённый в азотной кислоте, и азотнокислое железо (III) n-водное Fe(NO3)3×nH2O, где число n определяют гравиметрическим анализом, в качестве органического восстановителя, необходимого для пиролиза – аминоуксусную кислоту C2H5NO2. Нитрат-органический прекурсор, полученный смешиванием указанных реагентов, упаривают до начала самопроизвольного пиролиза. Полупродукт, полученный в результате пиролиза, подвергают двухстадийной термической обработке: сначала отжигают при 750 °С в течение 8 ч, затем спекают при 1200 °С в течение 20 ч. Изобретение обеспечивает небольшую длительность синтеза, а однофазность получаемого граната определяет точность и воспроизводимость его функциональных характеристик. 8 ил., 1 табл., 4 пр.
Способ получения однофазного железоиттриевого граната, включающий получение сначала прекурсора, его пиролиз с образованием продукта сжигания, содержащего иттрий и железо, и последующую термическую обработку, отличающийся тем, что используют нитрат-органический прекурсор, при этом в качестве иттрийсодержащего соединения в нем используют оксид иттрия, растворённый в азотной кислоте, в качестве железосодержащего соединения – азотнокислое железо (III) n-водное, где число n определяют гравиметрическим анализом, в качестве органического восстановителя, необходимого для пиролиза, – аминоуксусную кислоту, термическую обработку проводят в две стадии: отжиг при температуре 750 °С в течение 8 часов и спекание при температуре 1200 °С в течение 20 часов.
| TATIANA KISELEVA et al | |||
| Size-Dependent Structural, Magnetic and Magnetothermal Properties of Y3Fe5O12 Fine Particles Obtained by SCS, Nanomaterials, 2022, 12, 2773 | |||
| ЛИДИН Р.А | |||
| Реакции неорганических веществ: справочник, Москва, "Дрофа", 2007, с | |||
| НОЖЕВАЯ ПОЧВООБРАБАТЫВАЮЩАЯ ФРЕЗА С ПРИСОБЛЕНИЕМ ДЛЯ ИЗВЛЕЧЕНИЯ СОРНЫХ ТРАВ И КАМНЕЙ | 1922 |
|
SU611A1 |
| СПОСОБ ПОЛУЧЕНИЯ НАНОРАЗМЕРНОГО ПОРОШКА ЖЕЛЕЗОИТТРИЕВОГО ГРАНАТА | 2012 |
|
RU2509625C1 |
| US 5256242 A, 26.10.1993 | |||
| ГЕРАСИМОВА Н.С., ЛОГИНОВА А.Ю | |||

