ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к области фармацевтической химии, конкретно к ингибитору двойного целенаправленного действия на CDK6/DYRK2, способу его получения и его применению.
УРОВЕНЬ ТЕХНИКИ
Циклин-зависимая киназа 6 (CDK6) представляет собой серин/тирозинкиназу, которая регулирует переход клеточного цикла от G1 до S. В ранней фазе G1 клеточного цикла циклин D связывается с CDK6 и активирует его, а образовавшийся комплекс циклин D-CDK6 способствует фосфорилированию белка ретинобластомы (Rb). Фосфорилирование Rb приводит к высвобождению транскрипционного фактора E2F, который ускоряет прогрессирование клеточного цикла от G1 до S. Активация протоонкогена CDK6 приводит к ускоренному прогрессированию клеточного цикла от G1 до S, что приводит к ускоренному клеточному циклу и пролиферации клеток. Неконтролируемая пролиферация клеток является основным признаком рака. Следовательно, ингибирование CDK6 может замедлить процесс перехода клеточного цикла из фазы G1 в фазу S, вызывая антипролиферативный и противораковый эффекты. Однако представленные в настоящее время на рынке ингибиторы CDK6, палбоциклиб, рибоциклиб и абемациклиб, являются высокотоксичными и к ним уже выработана устойчивость.
Регулируемые фосфорилированием тирозина киназы с двойной специфичностью (DYRK) и CDK относятся к семейству CMGC и играют важную регулирующую роль в клеточном цикле и клеточной пролиферации. DYRK2 регулирует фосфорилирование Rpt3-T25, зависимого от клеточного цикла, и способствует деградации ингибиторов CDK, таких как p21 и p27, а также прогрессированию клеточного цикла от G1 до S. Ингибирование DYRK2 также может замедлить процесс перехода клеточного цикла из фазы G1 в фазу S, вызывая антипролиферативный и противораковый эффекты. До сих пор сообщалось только о нескольких ингибиторах DYRK2: первоначально было обнаружено, что акридиновое соединение, представляющее собой LDN192960, является ингибитором киназы Haspin с некоторыми терапевтическими эффектами при трижды негативном раке молочной железы и множественной миеломе. Также подтверждено, что другое лекарственное средство, куркумин, действует на DYRK2 и DYRK3, и может вызывать определенный эффект в отношении множественной миеломы при комбинировании с карфилзомибом. Однако противораковую активность и целенаправленную селективность существующих ингибиторов DYRK2 все еще необходимо оптимизировать, и особенно улучшению в дальнейшем подлежит свойство, касающееся возможности получения лекарственного средства.
Целенаправленные лекарственные средства демонстрируют высокую эффективность и надлежащую безопасность. Однако из-за сложности структуры и структурной целостности раковой опухоли, когда лекарственное средство, нацеленное на одну мишень, ингибирует один путь развития рака, связанные пути будут активироваться для компенсации ингибированных путей, что приведет к устойчивости к лекарственному средству.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Цель настоящего изобретения: с целью решения проблемы резистентности к лекарственным средствам, создаваемой существующими лекарственными средствами при лечении одной мишени, настоящее изобретение использует синергический эффект CDK6 и DYRK2, и предусматривает соединение или его фармацевтически приемлемую соль, которые могут быть одновременно нацелены на CDK6 и DYRK2, где соединение представляет собой ингибитор двойного целенаправленного действия на CDK6/DYRK2; и в тоже время подавляя DYRK2 и блокируя компенсаторный путь CDK6, улучшает противораковую активность соединения, снижает устойчивость к лекарственным средствам, легко вызываемую лекарственным средством, нацеленным на одну мишень CDK6. Настоящее изобретение также обеспечивает конкретный способ получения соединения и лекарственного препарата, предназначенных для предупреждения и/или лечения рака или связанных с опухолью заболеваний, в частности заболеваний, предусматривающих рак молочной железы, рак предстательной железы, рак легких, множественную миелому, лейкоз, рак желудка, рак яичников, рак толстой кишки, рак печени, рак поджелудочной железы, глиому человека и т. п., и ожидается, что настоящее изобретение будет использовано в качестве противоракового лекарственного препарата нового поколения.
Техническое решение: настоящее изобретение относится к соединению, представленному общей формулой (I), или его фармацевтически приемлемой соли,
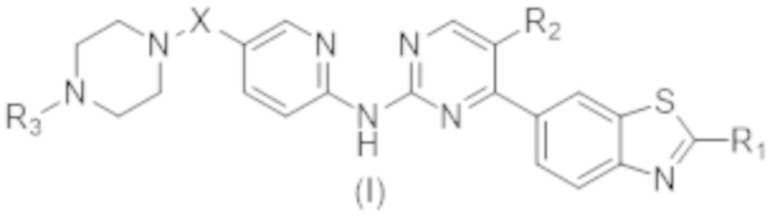 ,
,
где
X выбран из O, (CH2)n, C(O), NH или S(O)2, n равняется 0 или 1;
R1 выбран из водорода, дейтерия, галогена, гидроксигруппы, меркаптогруппы, цианогруппы, нитрогруппы, C1-C8алкильной группы, галоген-C1-C8алкильной группы, C1-C8алкоксигруппы, C3-C8циклоалкильной группы, C6-C10арильной группы, C3-C10гетероарильной группы, C4-C8гетероциклической группы, -C0-8-NR4R5;
R2 выбран из водорода, дейтерия, галогена, гидроксигруппы, меркаптогруппы, цианогруппы, нитрогруппы, C1-C8алкильной группы, C3-C8циклоалкильной группы;
R3 выбран из водорода, дейтерия, C1-C8алкильной группы, галоген-C1-C8алкильной группы, C1-C8алкоксигруппы, C3-C8циклоалкильной группы, -C0-8-S(O)2R6, -C0-8-C(O)OR7;
каждый из R4, R5 независимо выбран из водорода, дейтерия, C1-C8алкильной группы, галоген-C1-C8алкильной группы, C1-C8алкоксигруппы, C3-C8циклоалкильной группы;
каждый из R6, R7 независимо выбран из водорода, C1-C8алкильной группы, галоген-C1-C8алкильной группы, C3-C8циклоалкильной группы.
Предпочтительно
указанный X представляет собой C(O) или (CH2)n; n равняется 0 или 1;
указанный R1 представляет собой водород, C1-C8алкильную группу или -C0-8-NR4R5, где R4, R5 выбраны из водорода, C1-C8алкильной группы или C3-C8циклоалкильной группы;
R2 представляет собой водород или галоген;
R3 представляет собой водород, C1-C8алкильную группу, -C0-8-S(O)2R6 или -C0-8-C(O)OR7, где R6, R7 выбраны из C1-C8алкильной группы.
Предпочтительно
указанный X представляет собой C(O) или (CH2)n; n равняется 0 или 1;
указанный R1 представляет собой водород, C1-C3алкильную группу или -NR4R5, где R4, R5 выбраны из водорода, C1-C3алкильной группы, циклопентана или циклогексана;
R2 представляет собой водород или F;
R3 представляет собой водород, C1-C4алкильную группу, -S(O)2R6 или -C(O)OR7, где R6, R7 выбраны из C1-C4алкильной группы.
Предпочтительно
указанный X выбран из (CH2)n или C(O), n равняется 0 или 1;
указанный R1 выбран из водорода, метильной группы или -NR4R5, где R4, R5 выбраны из водорода, метильной группы, этильной группы или циклопентана;
указанный R2 представляет собой F;
указанный R3 выбран из водорода, этильной группы, изопропильной группы, -S(O)2R6 или -C(O)OR7, R6 представляет собой метильную группу, R7 выбран из трет-бутильной группы или этильной группы.
Предпочтительно
указанный X выбран из (CH2)n или C(O), n равняется 0 или 1;
указанный R1 выбран из водорода, метильной группы или -NR4R5, где R4, R5 выбраны из водорода, метильной группы, этильной группы или циклопентана;
указанный R2 представляет собой F;
указанный R3 выбран из водорода, этильной группы, изопропильной группы или -C(O)OR7, R7 выбран из трет-бутильной группы.
Предпочтительно
указанный X выбран из (CH2)n или C(O), n равняется 0 или 1;
указанный R1 выбран из водорода, метильной группы или -NR4R5, где R4 выбран из водорода, метильной группы, этильной группы, указанный R5 выбран из водорода, метильной группы, этильной группы или циклопентана;
указанный R2 представляет собой F;
указанный R3 выбран из водорода, этильной группы, изопропильной группы или -C(O)OR7, R7 выбран из трет-бутильной группы.
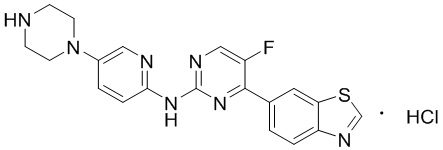
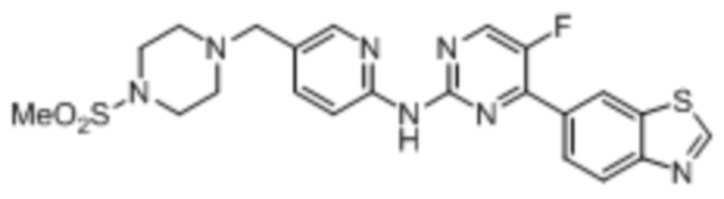
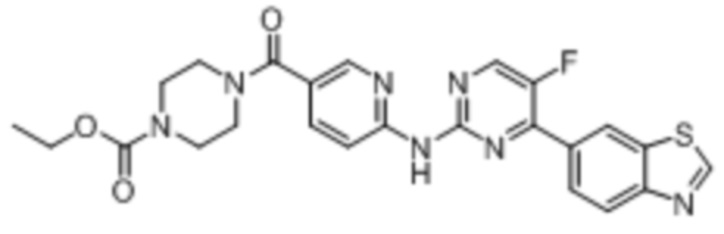
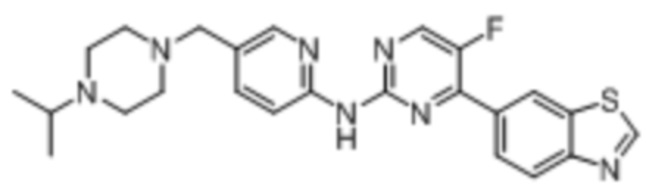
Предпочтительно соединение по настоящей заявке выбрано из I-1 – I-53:
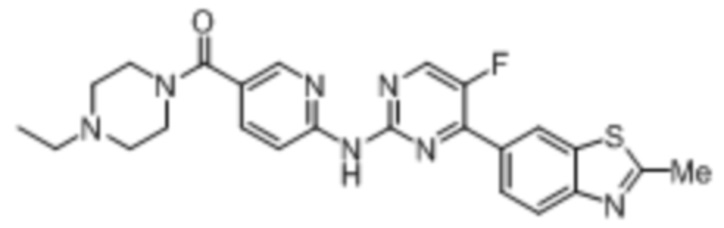
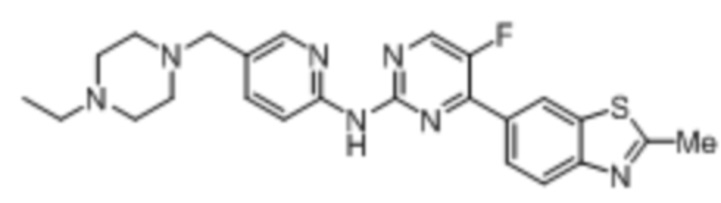
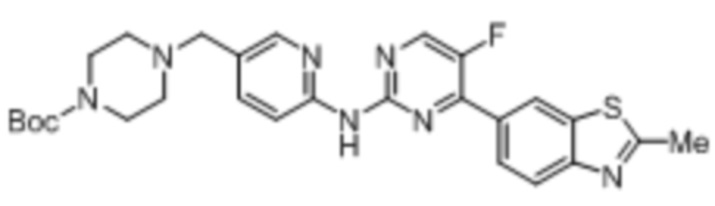

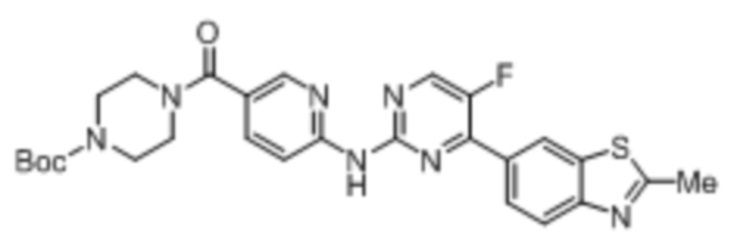
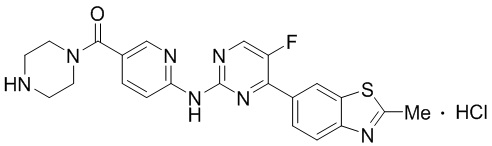
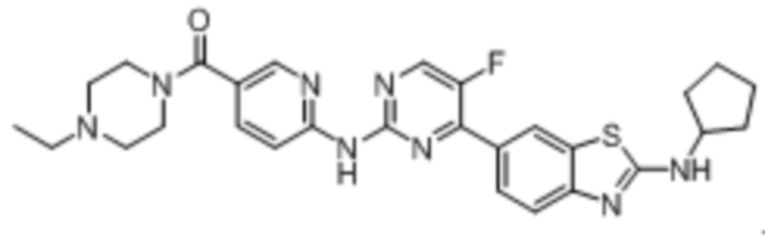
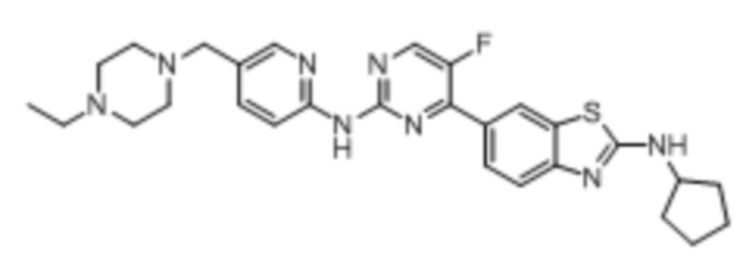
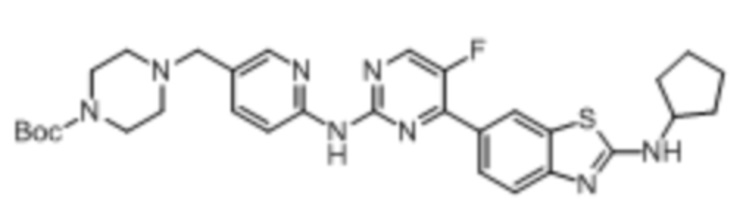
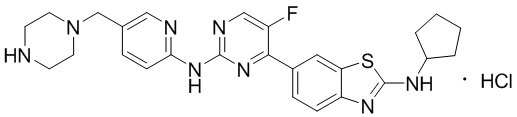

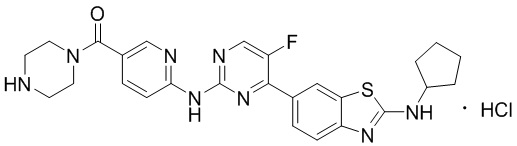

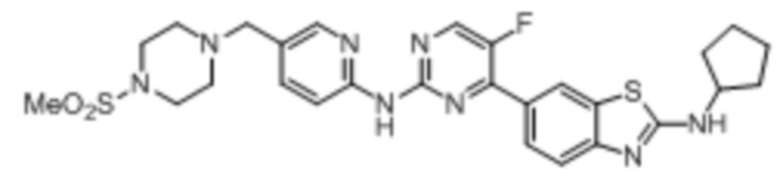
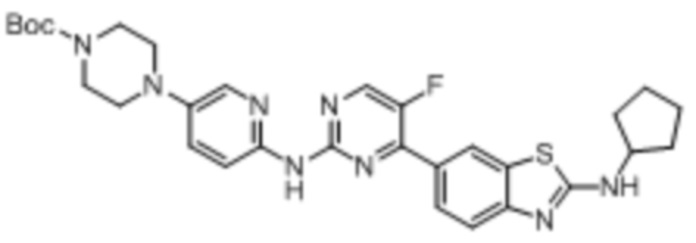
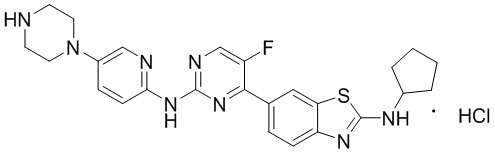
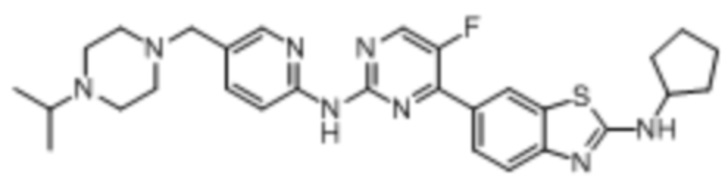
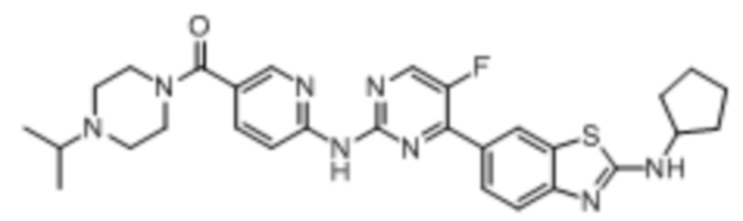
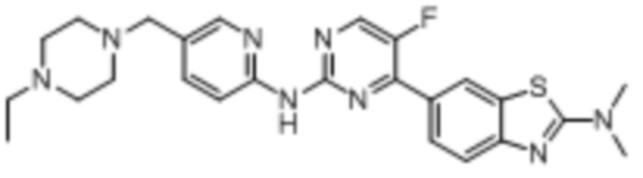
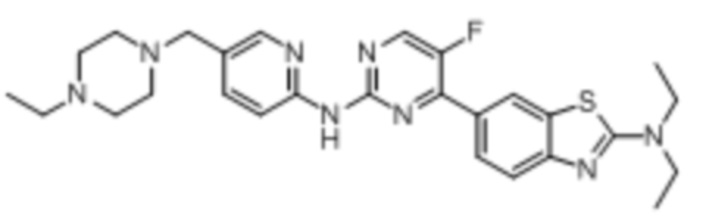
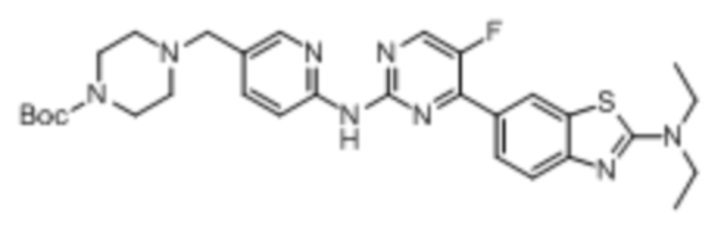
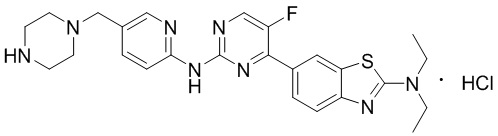
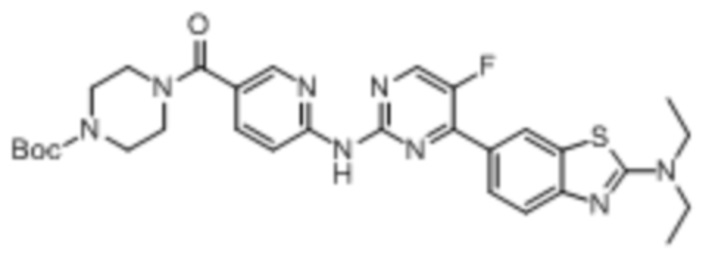
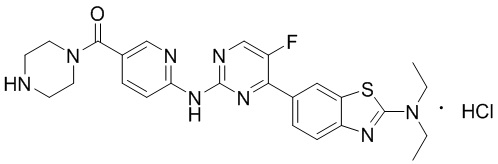
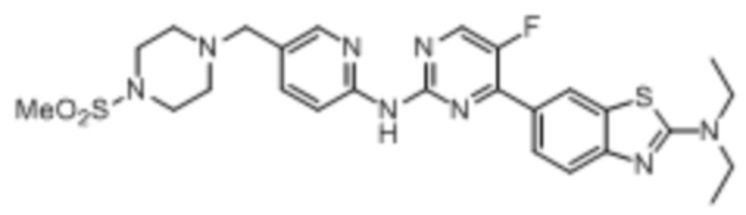
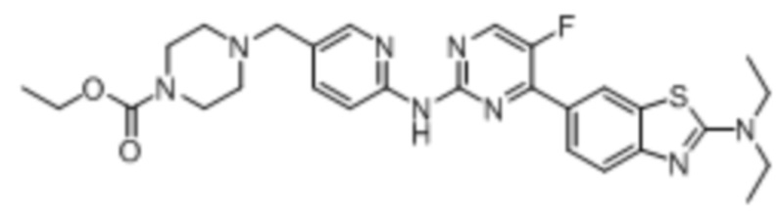
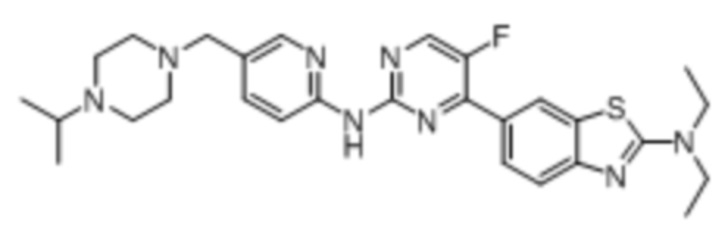
Предпочтительно
указанный X выбран из (CH2)n или C(O), n равняется 0 или 1;
указанный R1 выбран из водорода, метильной группы или -NR4R5, где каждый из R4 и R5 независимо выбран из водорода, метильной группы, этильной группы; или R4 представляет собой h, R5 представляет собой циклопентан;
указанный R2 представляет собой F;
указанный R3 выбран из водорода, этильной группы, изопропильной группы.
Вышеуказанная фармацевтически приемлемая соль представляет собой соль присоединения кислоты соединения общей формулы (I), где солеобразующая кислота предусматривает неорганическую кислоту и органическую кислоту, причем указанная неорганическая кислота предусматривает хлористоводородную кислоту, серную кислоту, фосфорную кислоту и метансульфоновую кислоту, а указанная органическая кислота предусматривает уксусную кислоту, трихлоруксусную кислоту, пропионовую кислоту, масляную кислоту, малеиновую кислоту, п-толуолсульфоновую кислоту, яблочную кислоту, малоновую кислоту, коричную кислоту, лимонную кислоту, фумаровую кислоту, камфорную кислоту, диглюконовую кислоту, аспарагиновую кислоту и винную кислоту.
Предпочтительно фармацевтически приемлемая соль по настоящему изобретению представляет собой гидрохлорид.
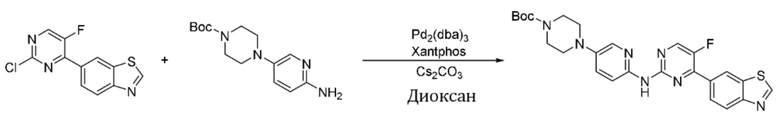
Настоящее изобретение относится к способу получения соединения общей формулы (I): получают соединение (I) из соединения (А) и соединения (В) посредством реакции сочетания под действием палладиевого катализатора:
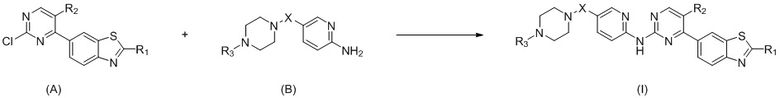
где
X выбран из O, (CH2)n, C(O), NH или S(O)2, n равняется 0 или 1;
R1 выбран из водорода, дейтерия, галогена, гидроксигруппы, меркаптогруппы, цианогруппы, нитрогруппы, C1-C8алкильной группы, галоген-C1-C8алкильной группы, C1-C8алкоксигруппы, C3-C8циклоалкильной группы, C6-C10арильной группы, C3-C10гетероарильной группы, C4-C8гетероциклической группы, -C0-8-NR4R5;
R2 выбран из водорода, дейтерия, галогена, гидроксигруппы, меркаптогруппы, цианогруппы, нитрогруппы, C1-C8алкильной группы, C3-C8циклоалкильной группы;
R3 выбран из водорода, дейтерия, C1-C8алкильной группы, галоген-C1-C8алкильной группы, C1-C8алкоксигруппы, C3-C8циклоалкильной группы, -C0-8-S(O)2R6, -C0-8-C(O)OR7;
каждый из R4, R5 независимо выбран из водорода, дейтерия, C1-C8алкильной группы, галоген-C1-C8алкильной группы, C1-C8алкоксигруппы, C3-C8циклоалкильной группы;
каждый из R6, R7 независимо выбран из водорода, C1-C8алкильной группы, галоген-C1-C8алкильной группы, C3-C8циклоалкильной группы.
Предпочтительно указанную реакцию проводят в защитной атмосфере аргона и температура реакции составляет 95–105oC, и предпочтительно температура реакции составляет 100oC.
Настоящее изобретение дополнительно раскрывает фармацевтическую композицию для ингибирования CDK6/DYRK2, содержащую вышеуказанное соединение общей формулы (I) или его фармацевтически приемлемую соль или его изомер, и фармацевтически приемлемый носитель.
Фармацевтически приемлемый носитель относится к вспомогательному веществу или разбавителю, не вызывающих значительного раздражения организма и не влияющих на биологическую активность и свойства вводимого соединения.
Настоящее изобретение относится к применению соединения или его фармацевтически приемлемой соли в изготовлении лекарственного препарата на основе ингибитора двойного целенаправленного действия на CDK6/DYRK2.
Лекарственный препарат на основе ингибитора двойного целенаправленного действия на CDK6/DYRK2 может использоваться для лечения рака или связанного с опухолью заболевания.
Настоящее изобретение относится к применению соединения или его фармацевтически приемлемой соли в изготовлении лекарственного препарата, предназначенного для предупреждения и/или лечения рака или связанного с опухолью заболевания. Рак или связанное с опухолью заболевание предусматривает рак молочной железы, рак предстательной железы, рак легких, множественную миелому, лейкоз, рак желудка, рак яичников, рак толстой кишки, рак печени, рак поджелудочной железы и глиому человека.
Настоящее изобретение относится к соединению общей формулы (I) или его фармацевтически приемлемой соли, обладающим ингибиторной активностью двойного целенаправленного действия на CDK6/DYRK2 и демонстрирующим терапевтическое действие на злокачественную пролиферацию клеток опухоли.
Термины в настоящем изобретении имеют следующие значения, если не указано иное.
Термин "алкильная группа" означает линейную или разветвленную насыщенную углеводородную группу, содержащую определенное количество атомов углерода.
Термин "C1-C8алкильная группа" относится к линейной или разветвленной насыщенной углеводородной группе, содержащей 1–8 атомов углерода. C1-C8алкильная группа включает без ограничения метильную группу, этильную группу, н-пропильную группу, изопропильную группу, н-бутильную группу, изобутильную группу, трет-бутильную группу, н-пентильную группу, изопентильную группу, неопентильную группу, н-гексильную группу, изогексильную группу, 2,2-диметилбутильную группу и 2,3-диметилбутильную группу и т. п. Термин "C1-C3алкильная группа" относится к линейной или разветвленной насыщенной углеводородной группе, содержащей 1–3 атома углерода.
Термин "алкоксигруппа" означает O-алкильную группу. Термин "C1-C8алкоксигруппа" относится к группе, содержащей O-C1-C8алкильную группу.
"C(O)" означает "-C (O)-", в частности, карбонильную группу. Термин "галоген" означает фтор, хлор, бром или йод. Предпочтительно это фтор, хлор, бром.
Термин "галоген-алкильная группа" означает алкильную группу, содержащую по меньшей мере один (включая один) галогеновый заместитель.
Термин "циклоалкильная группа" означает насыщенную моноциклическую или полициклическую кольцевую структуру, состоящую из атомов углерода.
Термин "C3-C8циклоалкильная группа" относится к насыщенной моноциклической или полициклической кольцевой структуре, содержащей 3–8 атомов. C3-C6циклоалкильная группа включает без ограничения циклопропильную группу, циклобутильную группу, циклопентильную группу, циклогексильную группу.
Термин "циклоалкенил" относится к моноциклическому или полициклическому алкильному заместителю, содержащему по меньшей мере одну циклическую углерод-углеродную двойную связь.
Термин "C3-C8циклоалкенил" относится к циклоалкенилу, содержащему 3–8 атомов углерода. C3-C8 циклоалкенил включает без ограничения циклопентенильную группу, циклобутенильную группу.
Термин "C2-C8алкенил" относится к линейной или разветвленной углеводородной группе, содержащей одну или несколько углерод-углеродных двойных связей и содержащей 2–8 атомов углерода.
Термин "C2-C8алкинил" относится к линейной или разветвленной углеводородной группе, содержащей одну или несколько углерод-углеродных тройных связей и содержащей 2–8 атомов углерода.
Термин "C6-C10арильная группа" означает моноциклическую или конденсированную полициклическую группу, состоящую из 6–10 атомов углерода, содержащую полностью сопряженную π-электронную систему. Как правило, она включает без ограничения фенильную группу, нафтильную группу.
Термин "гетероарильная группа" означает моноциклическую или конденсированную циклическую группу, содержащую один, два, три или четыре циклических гетероатома, выбраных из группы, состоящей из N, O или S, а остальные атомы в цикле представляют собой C, причем дополнительно содержащую полностью сопряженную π-электронную систему. Термин "C3-C10гетероарильная группа" относится к гетероарильной группе, содержащей 3–10 атомов углерода в своем кольце. C3-C10гетероарильная группа включает без ограничения пиррол, фуран, тиофен, имидазол, оксазол, тиазол, пиразол, пиримидин, пиридин.
Термин "гетероциклическая группа"представляет собой гетероциклоалкильную группу и означает моноциклическую или конденсированную циклическую группу, содержащую один или несколько гетероатомов N, O или S. Термин "C4-C8гетероциклическая группа" относится к гетероциклической группе, содержащей 4–8 атомов углерода в своем кольце. C4-C8гетероциклическая группа включает без ограничения пиперазинoгруппу, морфолинoгруппу, пиперидинoгруппу, пирролидинoгруппу и т. п.
Благоприятные эффекты: По сравнению с предшествующим уровнем техники настоящее изобретение имеет следующие существенные характеристики, а именно настоящее изобретение раскрывает новое соединение, представленное общей формулой (i), которое может одновременно ингибировать несколько путей развития рака, обладает хорошим лечебным эффектом, низкой токсичностью, хорошим метаболизмом лекарственного средства и характеризуется затруднением в отношении выработки резистентности к лекарственному средству, и может применяться для изготовления лекарственного препарата, предназначеного для лечения рака или связанных с опухолью заболеваний. Настоящее изобретение также раскрывает способ получения соединения общей формулы (I).
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На фигуре 1 показано изменение веса тела мыши в анализе острой токсичности по настоящему изобретению.
На фигуре 2 представлен график, демонстрирующий результаты окрашивания H&E в анализе острой токсичности по настоящему изобретению.
На фигуре 3 представлен график, касающийся результатов по объему опухоли рака предстательной железы согласно настоящему изобретению.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящая заявка описана подробно ниже со ссылкой на конкретные варианты осуществления.
I. Синтез промежуточных реагентов
Реагент (A) и реагент (B) могут быть заказаны напрямую или независимо разработаны, и расходы могут быть значительно снижены за счет независимой разработки. Независимо разработанные конкретные способы получения реагента (А) и реагента (В) следующие:
(1) Синтез 6-(2-хлор-5-фторпиримидин-4-ил)бензoтиазола (A-1)

Стадия 1. Синтез 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензoтиазола. 6-Бромбензoтиазол (0,43 г, 2,0 ммоль) растворяли в DMF (10 мл). Затем добавляли пинаколборат (0,53 г, 2,1 ммоль), Pd(dppf)Cl2 (22 мг, 0,06 ммоль), ацетат калия (0,59 г, 6,0 ммоль). Реакционную смесь продували аргоном три раза, нагревали до 80oC и оставляли на 24 ч. Смесь охлаждали, фильтровали и концентрировали, и очищали с помощью колоночной флэш-хроматографии на силикагеле с получением соединения, представляющего собой 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензoтиазол (0,47 г, выход 90%).
1H ЯМР (300 MГц, CDCl3) δ 9,07 (s, 1H), 8,46 (s, 1H), 8,14 (d, J = 8,2 Гц, 1H), 7,94 (dd, J = 8,2, 1,1 Гц, 1H), 1,38 (s, 12H).
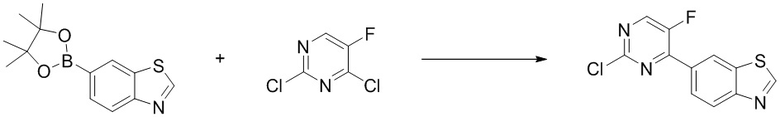
Стадия 2. Синтез 6-(2-хлор-5-фторпиримидин-4-ил)бензoтиазола (A-1). Отбирали навеску соединения, представляющего собой 2,4-дихлор-5-фторпиримидин (0,23 г, 1,4 ммоль), и добавляли в трехгорлую колбу объемом 250 мл. Затем добавляли Pd(PPh3)2Cl2(21 мг, 0,03 ммоль), карбонат натрия (0,27 г, 2,5 ммоль), диметоксиэтан (10 мл) и H2O (0,25 мл). Реакционную смесь продували аргоном три раза, нагревали до 80oC. Соединение, 6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензoтиазол (0,26 г, 1,0 ммоль), растворяли в диметоксиэтане (5 мл), добавляли по каплям в трехгорлую колбу и оставляли на 16 ч. Смесь охлаждали, фильтровали и концентрировали, и очищали с помощью колоночной флэш-хроматографии на силикагеле с получением соединения, представляющего собой 6-(2-хлор-5-фторпиримидин-4-ил)бензoтиазол (0,22 г, выход 82%). 1H ЯМР (300 MГц, CDCl3) δ 9,17 (s, 1H), 8,84 (d, J = 1,7 Гц, 1H), 8,58 (d, J = 3,1 Гц, 1H), 8,37 - 8,24 (m, 2H).
(2) Синтез 6-(2-хлор-5-фторпиримидин-4-ил)-2-метилбензoтиазола (A-2)
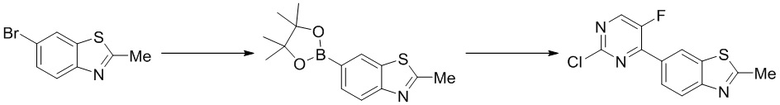
Ссылаясь на синтез соединения (A-1), выходы составляли 90% и 84% соответственно. 1H ЯМР (400 MГц, CDCl3) δ 8,70 (d, J = 1,9 Гц, 1H), 8,55 (d, J = 3,1 Гц, 1H), 8,28 - 8,25 (m, 1H), 8,07 (d, J = 8,6 Гц, 1H), 2,90 (s, 3H).
(3) Синтез 6-(2-хлор-5-фторпиримидин-4-ил)-N-циклопентилбензoтиазол-2-амина (A-3)

Стадия 1. Синтез 6-бром-N-циклопентилбензoтиазол-2-амина. 6-Бром-2-хлорбензoтиазол (0,50 г, 2,0 ммоль) растворяли в DMSO (10 мл) и добавляли циклопентиламин (0,19 г, 2,2 ммоль) и N-этилдиизопропиламин (0,39 г, 3,0 ммоль). Реакционную смесь продували аргоном три раза, нагревали до 80oC и оставляли на 12 ч. Смесь охлаждали, фильтровали и концентрировали, и очищали с помощью колоночной флэш-хроматографии на силикагеле с получением соединения, представляющего собой 6-бром-N-циклопентилбензoтиазол-2-амин (0,53 г, выход 90%). 1H ЯМР (400 MГц, CDCl3) δ 7,68 (d, J = 1,7 Гц, 1H), 7,38 - 7,33 (m, 2H), 6,28 (s, 1H), 3,98 - 3,93 (m, 1H), 2,14 - 2,04 (m, 2H), 1,72 - 1,54 (m, 6H).
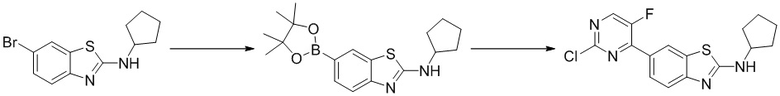
Стадия 2. Синтез 6-(2-хлор-5-фторпиримидин-4-ил)-N-циклопентилбензoтиазол-2-амина (A-3). Ссылаясь на синтез соединения (A-1), выходы составляли 88% и 83% соответственно. 1H ЯМР (400 MГц, CDCl3) δ 8,49 (d, J = 1,9 Гц, 1H), 8,46 (d, J = 3,5 Гц, 1H), 8,17 - 8,14 (m, 1H), 7,58 (d, J = 8,6 Гц, 1H), 5,88 (s, 1H), 4,13 - 4,07 (m, 1H), 2,19 - 2,11 (m, 2H), 1,77 - 1,61 (m, 6H).
(4) Синтез 6-(2-хлор-5-фторпиримидин-4-ил)-N,N-диметилбензoтиазол-2-амина (A-4)
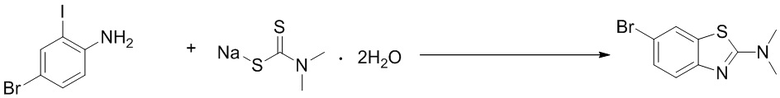
Стадия 1. Синтез 6-бром-N,N-диметилбензoтиазол-2-амина. Отбирали навеску 4-бром-2-йоданилина (0,60 г, 2,0 ммоль), дигидрата диметилдитиокарбамата натрия (0,72 г, 4,0 ммоль), ацетата меди (0,36 г, 2,0 ммоль) и карбоната калия (0,55 г, 4,0 ммоль) и растворяли в DMF (10 мл), нагревали до 120oC и оставляли на 6 ч. Смесь охлаждали, фильтровали и концентрировали, и очищали с помощью колоночной флэш-хроматографии на силикагеле с получением соединения, представлюящего собой 6-бром-N,N-диметилбензoтиазол-2-амин (0,44 г, выход 85%). 1H ЯМР (400 MГц, CDCl3) δ 7,69 (d, J = 1,9 Гц, 1H), 7,41 - 7,35 (m, 2H), 3,20 (s, 6H).
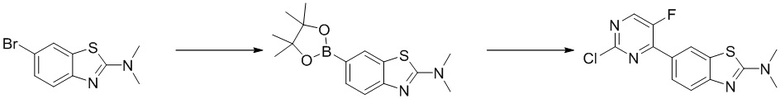
Стадия 2. Синтез 6-(2-хлор-5-фторпиримидин-4-ил)-N,N-диметилбензoтиазол-2-амина (A-4). Ссылаясь на синтез соединения (A-1), выходы составляли 88% и 80%. 1H ЯМР (400 MГц, CDCl3) δ 8,49 (d, J = 1,9 Гц, 1H), 8,45 (d, J = 3,6 Гц, 1H), 8,17 - 8,14 (m, 1H), 7,62 (d, J = 8,7 Гц, 1H), 3,27 (s, 6H).
(5) Синтез 6-(2-хлор-5-фторпиримидин-4-ил)-N,N-диэтилбензoтиазол-2-амина (A-5)
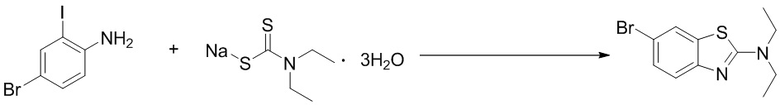
Стадия 1. Синтез 6-бром-N,N-диэтилбензoтиазол-2-амина. Отбирали навеску 4-бром-2-йоданилина (0,60 г, 2,0 ммоль), тригидрата диэтилдитиокарбамата натрия (0,90 г, 4,0 ммоль), ацетата меди (0,36 г, 2,0 ммоль) и карбоната калия (0,55 г, 4,0 ммоль) и растворяли в DMF (10 мл), нагревали до 120oC и оставляли на 6 ч. Смесь охлаждали, фильтровали и концентрировали, и очищали с помощью колоночной флэш-хроматографии на силикагеле с получением соединения, представлюящего собой 6-бром-N,N-диметилбензoтиазол-2-амин (0,46 г, выход 80%).
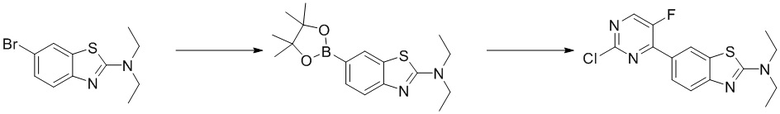
Стадия 2. Синтез 6-(2-хлор-5-фторпиримидин-4-ил)-N,N-диэтилбензoтиазол-2-амина (A-5). Ссылаясь на синтез соединения (A-1), выходы составляли 90% и 82%. 1H ЯМР (300 MГц, CDCl3) δ 8,44 (dd, J = 8,8, 2,7 Гц, 2H), 8,13 (d, J = 8,6 Гц, 1H), 7,58 (d, J = 8,7 Гц, 1H), 3,61 (q, J = 7,2 Гц, 4H), 1,32 (t, J = 7,2 Гц, 6H).
(6) Синтез (6-аминопиридин-3-ил)(4-этилпиперазин-1-ил)кетона (B-1)
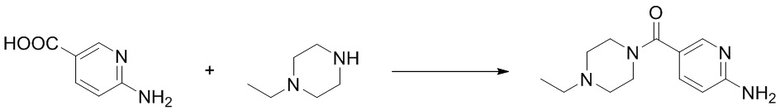
Отбирали навеску 6-аминоникотиновой кислоты (0,28 г, 2,0 ммоль), N,N'-карбонилдиимидазола (0,39 г, 2,4 ммоль), и растворяли в DMF(5 мл), и оставляли при 70oC на 10 мин. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. и добавляли N-этилпиперазин (0,46 г, 4,0 ммоль). Реакцию проводили в течение ночи при комнатной температуре, концентрировали и очищали с помощью колоночной флэш-хроматографии на силикагеле с получением соединения, представляющего собой (6-аминопиридин-3-ил)(4-этилпиперазин-1-ил)кетон (0,40 г, выход 85%). 1H ЯМР (300 MГц, CDCl3) δ 8,19 - 8,17 (m, 1H), 7,57 - 7,54 (m, 1H), 6,51 - 6,48 (m, 0,9 Гц, 1H), 4,79 (s, 2H), 3,73 - 3,60 (m, 4H), 2,49 - 2,42 (m, 6H), 1,13 - 1,08 (m, 3H).
(7) Синтез 5-((4-этилпиперазин-1-ил)метил)пиридин-2-амина (B-2)
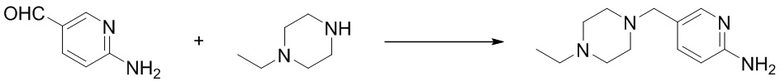
2-Амино-5-формилпиридин (0,32 г, 2,6 ммоль) и N-этилпиперазин (0,45 г, 3,9 ммоль) растворяли в 1,2-дихлорэтане (20 мл), перемешивали при комнатной температуре в течение 2 ч. Затем добавляли триацетилборгидрид натрия (0,87 г, 4,1 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 8 ч. Для гашения добавляли 1 М раствор NaOH (30 мл). Смесь экстрагировали с помощью DCM (20 мл × 3), высушивали безводным сульфатом натрия, концентрировали и затем подвергали колоночной хроматографии (DCM/MeOH = 10:1) с получением соединения, представляющего собой 5-((4-этилпиперазин-1-ил)метил)пиридин-2-амин (0,52 г, 91%). 1H ЯМР (300 MГц, CDCl3) δ 7,94 (d, J = 2,3 Гц, 1H), 7,40 (dd, J = 8,3, 2,4 Гц, 1H), 6,46 (d, J = 8,3 Гц, 1H), 4,57 (s, 2H), 3,36 (s, 2H), 2,47-2,37 (m, 10H), 1,07 (t, J = 7,2 Гц, 3H).
(8) Синтез трет-бутил-4-((6-аминопиридин-3-ил)метил)пиперазин-1-карбоксилата (B-3)

Ссылаясь на синтез соединения (B-2), выход составлял 89%. 1H ЯМР (300 MГц, CDCl3): δ 7,94 (d, J = 2,3 Гц, 1H), 7,40 (dd, J = 8,4, 2,3 Гц, 1H), 6,48 (d, J = 8,4 Гц, 1H), 4,54 (s, 2H), 3,40 (t, J = 5,1 Гц, 4H), 3,36 (s, 2H), 2,35 (t, J = 5,1 Гц, 4H), 1,45 (s, 9H).
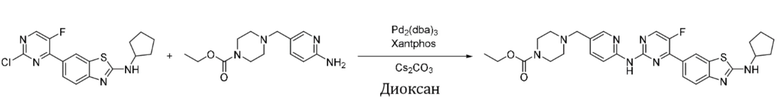
(9) Синтез трет-бутил-4-(6-аминоникотиноил)пиперазин-1-карбоксилата (B-4)
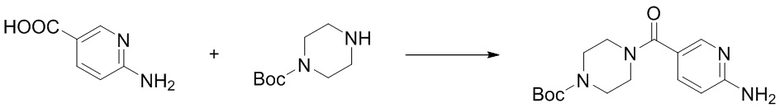
Ссылаясь на синтез соединения (B-1), выход составлял 87%. 1H ЯМР (300 MГц, CDCl3) δ 8,18 (d, J = 2,2 Гц, 1H), 7,56 (dd, J = 8,5, 2,2 Гц, 1H), 6,51 (d, J = 8,5 Гц, 1H), 4,76 (s, 2H), 3,65 - 3,56 (m, 4H), 3,48 - 3,42 (m, 4H), 1,48 (s, 9H).
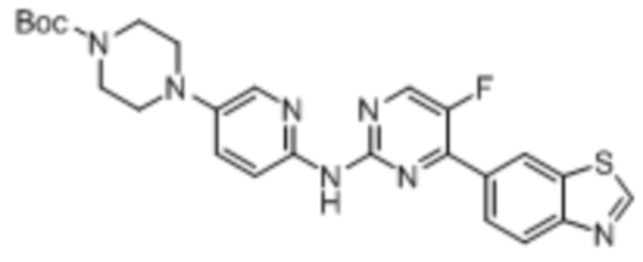
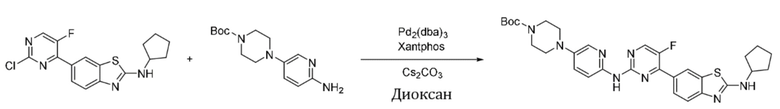
(10) Синтез трет-бутил-4-(6-аминопиридин-3-ил)пиперазин-1-карбоксилата (B-5)
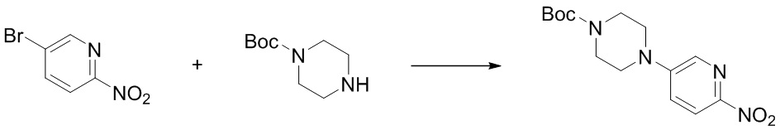
Стадия 1. Синтез трет-бутил-4-(6-нитропиридин-3-ил)пиперазин-1-карбоксилата. Отбирали навеску 5-бром-2-нитропиридина (0,41 г, 2,0 ммоль), трет-бутилпиперазин-1-карбоксилата (0,48 г, 2,6 ммоль) и триэтиламина (0,41 г, 4,0 ммоль) и растворяли в DMSO (5 мл), нагревали до 60oC и оставляли на 18 ч. Смесь охлаждали, фильтровали и концентрировали, и очищали с помощью колоночной флэш-хроматографии на силикагеле с получением соединения, представляющего собой трет-бутил-4-(6-нитропиридин-3-ил)пиперазин-1-карбоксилат (0,49 г, выход 80%). 1H ЯМР (400 MГц, CDCl3) δ 8,17 - 8,13 (m, 2H), 7,22 (dd, J = 9,2, 3,1 Гц, 1H), 3,66 - 3,64 (m, 4H), 3,49 - 3,46 (m, 4H), 1,49 (s, 9H).
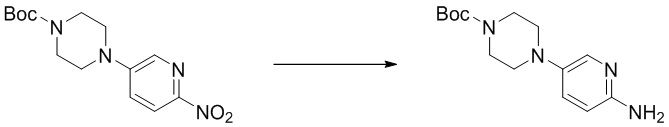
Стадия 2. Синтез трет-бутил-4-(6-аминопиридин-3-ил)пиперазин-1-карбоксилата. Отбирали навеску трет-бутил-4-(6-нитропиридин-3-ил)пиперазин-1-карбоксилата (0,31 г, 1,0 ммоль), порошка восстановленного железа (0,17 г, 3,0 ммоль) и хлорида аммония (0,49 г, 9,0 ммоль) и растворяли в 70% этаноле (10 мл), нагревали до 70oC и оставляли на 6 ч. Смесь охлаждали, фильтровали и концентрировали, и очищали с помощью колоночной флэш-хроматографии на силикагеле с получением соединения, представляющего собой трет-бутил-4-(6-аминопиридин-3-ил)пиперазин-1-карбоксилат (0,24 г, выход 85%). 1H ЯМР (300 MГц, CDCl3) δ 7,78 (d, J = 2,9 Гц, 1H), 7,17 (dd, J = 8,8, 2,9 Гц, 1H), 6,49 (d, J = 8,8 Гц, 1H), 4,19 (s, 2H), 3,59 - 3,55 (m, 4H), 2,98 - 2,94 (m, 4H), 1,48 (s, 9H).
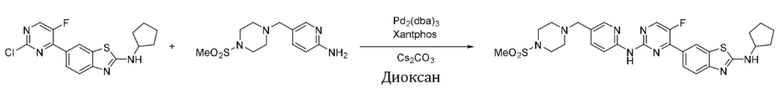
(11) Синтез 5-((4-(метансульфонил)пиперазин-1-ил)метил)пиридин-2-амина (B-6)
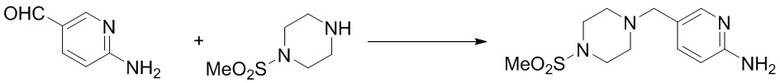
Ссылаясь на синтез соединения (B-2), выход составлял 90%. 1H ЯМР (400 MГц, CDCl3) δ 8,10 - 7,79 (m, 1H), 7,40 (d, J = 8,3 Гц, 1H), 6,57 (s, 1H), 4,55 (s, 2H), 3,41 (s, 2H), 3,22 (t, J = 4,9 Гц, 4H), 2,77 (s, 3H), 2,53 (t, J = 5,0 Гц, 4H).
(12) Синтез этил-4-((6-аминопиридин-3-ил)метил)пиперазин-1-карбоксилата (B-7)
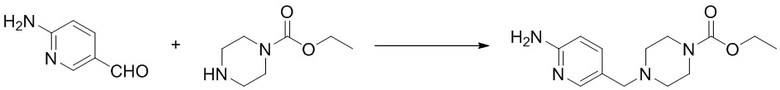
Ссылаясь на синтез соединения (B-2), выход составлял 86%. 1H ЯМР (400 MГц, CDCl3) δ 7,87 (d, J = 2,2 Гц, 1H), 7,43 (dd, J = 8,5, 2,3 Гц, 1H), 6,51 (d, J = 8,5 Гц, 1H), 5,71 (s, 2H), 4,12 (q, J = 7,1 Гц, 2H), 3,46 (t, J = 5,1 Гц, 4H), 3,36 (s, 2H), 2,37 (t, J = 5,1 Гц, 4H), 1,25 (t, J = 7,1 Гц, 3H).
(13) Синтез 5-((4-изопропилпиперазин-1-ил)метил)пиридин-2-амина (B-8)
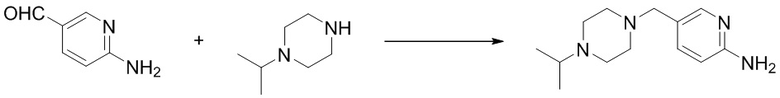
Ссылаясь на синтез соединения (B-2), выход составлял 81%. 1H ЯМР (400 MГц, CDCl3) δ 7,88 (d, J = 2,2 Гц, 1H), 7,43 (dd, J = 8,4, 2,2 Гц, 1H), 6,48 (d, J = 8,4 Гц, 1H), 4,95 (s, 2H), 3,39 (s, 2H), 2,88 - 2,80 (m, 1H), 2,72 - 2,51 (m, 8H), 1,10 (d, J = 6,6 Гц, 6H).
(14) Синтез (6-аминопиридин-3-ил)(4-изопропилпиперазин-1-ил)кетона (B-9)
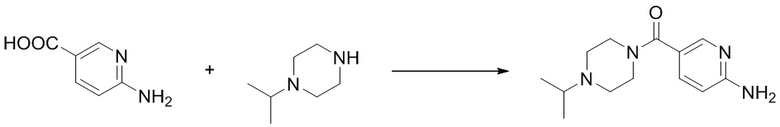
Ссылаясь на синтез соединения (B-1), выход составлял 85%. 1H ЯМР (300 MГц, CDCl3) δ 8,18 (s, 1H), 7,55 (d, J = 8,4 Гц, 1H), 6,49 (dd, J = 8,6, 2,1 Гц, 1H), 4,86 (s, 2H), 3,67 - 3,61 (m, 4H), 2,77 - 2,71 (m, 1H), 2,55 - 2,51 (m, 4H), 1,05 (d, J = 6,0 Гц, 6H).
II. Синтез соединения I-1 – I-53
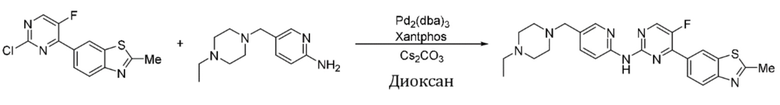
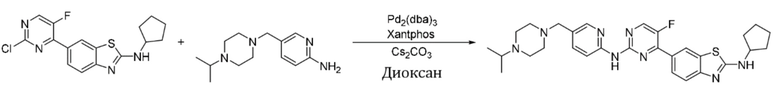
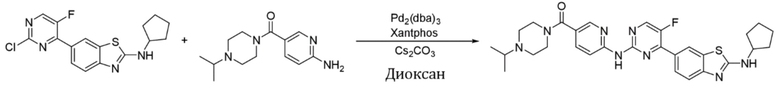
Пример 1.
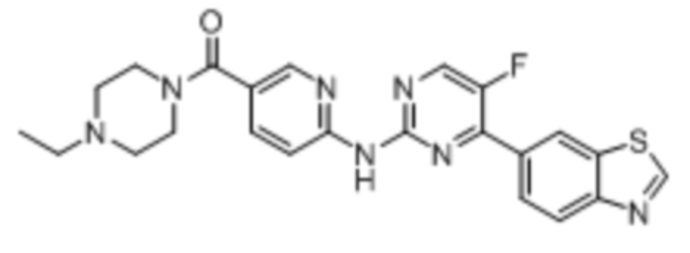
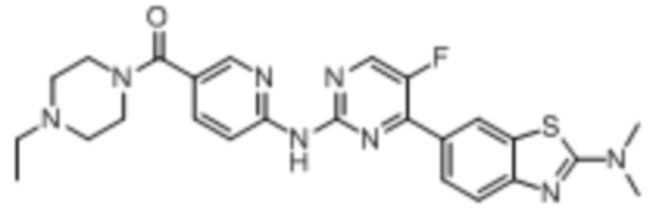
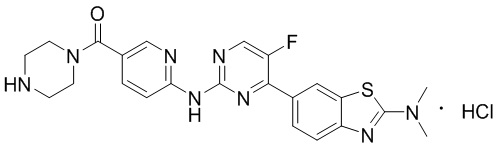
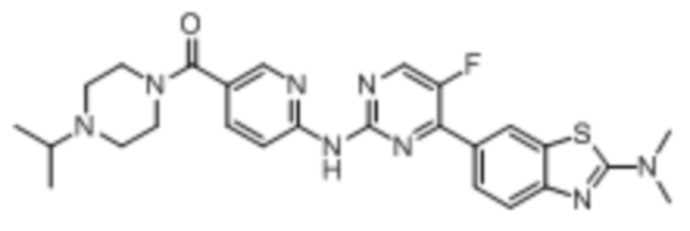
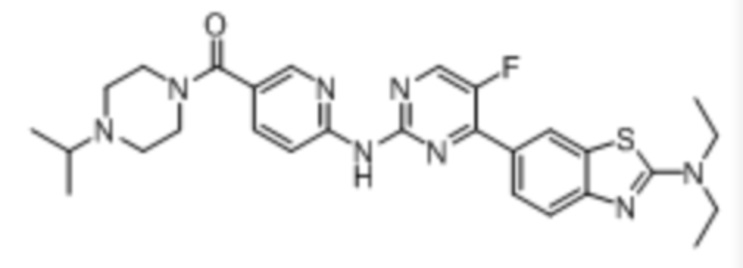
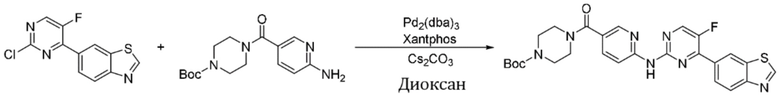
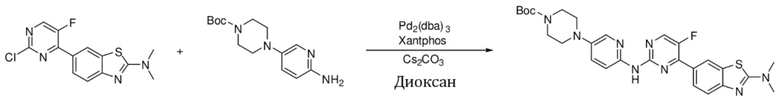
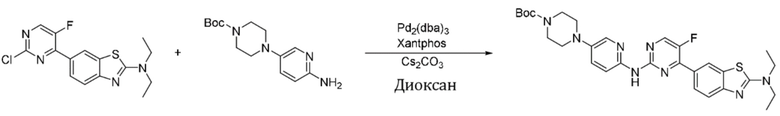
Синтез (6-((4-(бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)(4-этилпиперазин-1-ил)кетона (I-1).
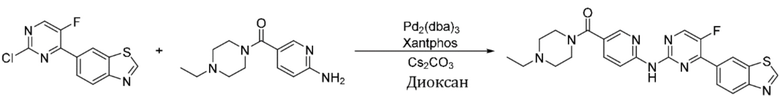
Соединение, 6-(2-хлор-5-фторпиримидин-4-ил)бензoтиазол (133 мг, 0,5 ммоль), и 6-аминопиридин-3-ил)(4-этилпиперазин-1-ил)кетон (141 мг, 0,6 ммоль) растворяли в диоксане (5 мл). Затем добавляли Pd2(dba)3 (23 мг, 0,025 ммоль), Xantphos (58 мг, 0,1 ммоль), карбонат цезия (326 мг, 1,0 ммоль). Реакционную смесь продували аргоном три раза, нагревали до 100oC и оставляли на 12 ч. Смесь охлаждали, фильтровали и концентрировали, и подвергали колоночной хроматографии (DCM~DCM/MeOH = 10:1) с получением соединения, представляющего собой (6-((4-(бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)(4-этилпиперазин-1-ил)кетон (88 мг, выход 38%). 1H ЯМР (300 MГц, CDCl3) δ 9,68 (s, 1H), 9,15 (s, 1H), 8,76 - 8,75 (m, 1H), 8,61 - 8,58 (m, 2H), 8,52 (dd, J = 8,7, 0,9 Гц, 1H), 8,33 - 8,26 (m, 2H), 7,86 (dd, J = 8,7, 2,3 Гц, 1H), 3,80 - 3,61 (m, 4H), 2,51 - 2,44 (m, 6H), 1,12 (t, J = 7,2 Гц, 3H).
Пример 2.
Синтез 4-(бензoтиазол-6-ил)-N-(5-((4-этилпиперазин-1-ил)метил)пиридин-2-ил)-5-фторпиримидин-2-амина (I-2).
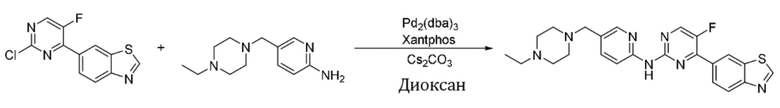
Ссылаясь на синтез соединения (I-1), выход составлял 43%. 1H ЯМР (300 MГц, CDCl3) δ 9,15 (s, 1H), 9,11 (s, 1H), 8,78 - 8,76 (m, 1H), 8,54 (d, J = 3,5 Гц, 1H), 8,41 (d, J = 8,6 Гц, 1H), 8,35 - 8,26 (m, 3H), 7,73 (dd, J = 8,6, 2,3 Гц, 1H), 3,52 (s, 2H), 2,58 - 2,47 (m, 10H), 1,14 (t, J = 7,2 Гц, 3H).
Пример 3.
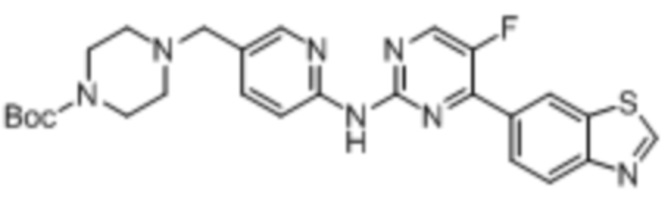
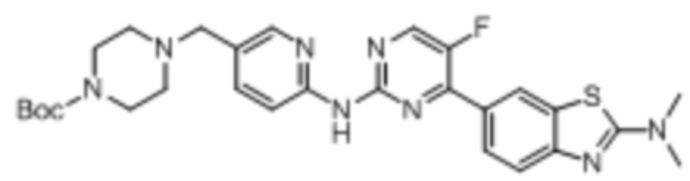
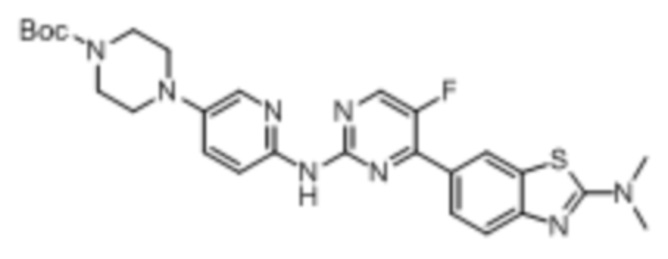
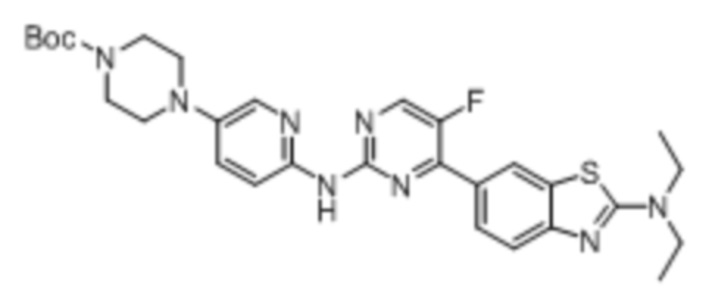
Синтез трет-бутил-4-((6-((4-(бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)метил)пиперазин-1-карбоксилата (I-3).
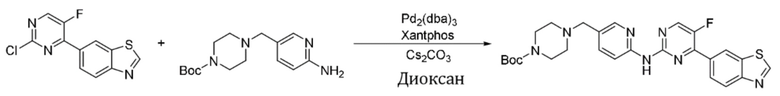
Ссылаясь на синтез соединения (I-1), выход составлял 52%. 1H ЯМР (400 MГц, CDCl3) δ 9,14 (s, 1H), 8,83 (s, 1H), 8,77 (d, J = 1,5 Гц, 1H), 8,52 (d, J = 3,5 Гц, 1H), 8,41 (d, J = 8,6 Гц, 1H), 8,34 - 8,27 (m, 3H), 7,75 - 7,73 (m, 1H), 3,50 (s, 2H), 3,45 - 3,43 (m, J = 5,2 Гц, 4H), 2,43 - 2,40 (m, 4H), 1,46 (s, 9H).
Пример 4.
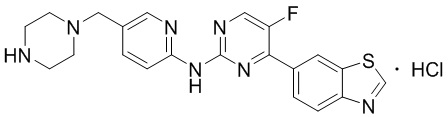
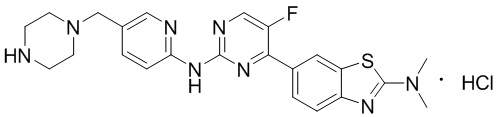
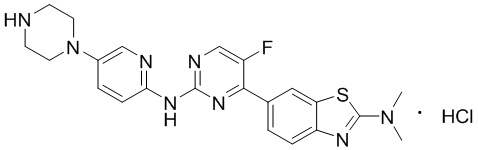
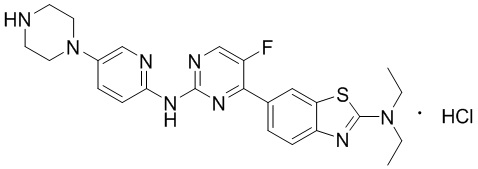
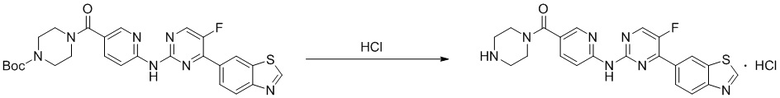
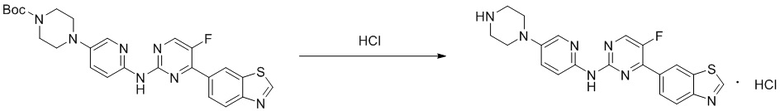
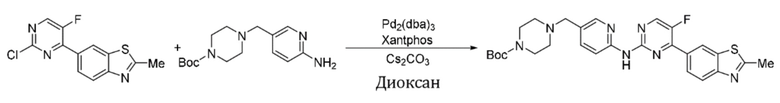
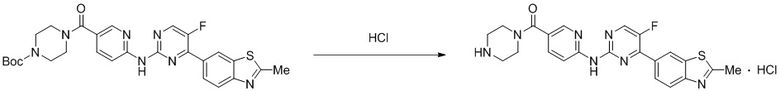
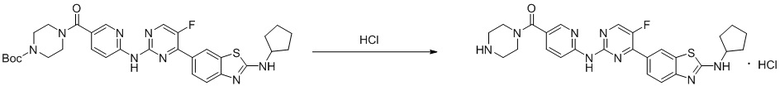
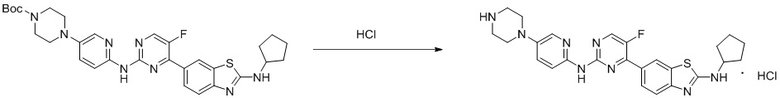
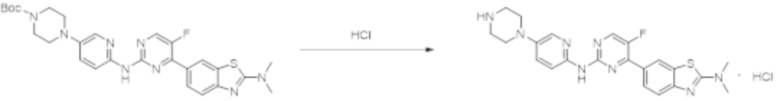
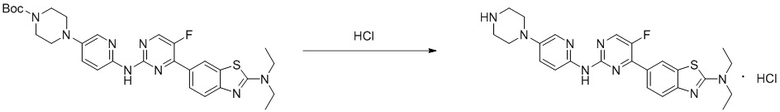
Синтез 4-(бензoтиазол-6-ил)-5-фтор-N-(5-(пиперазин-1-илметил)пиридин-2-ил)пиримидин-2-амина гидрохлорида (I-4).
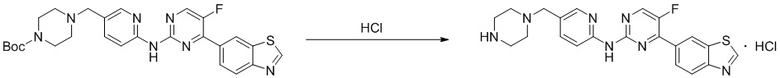
трет-Бутил-4-((6-((4-(бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)метил)пиперазин-1-карбоксилат растворяли в дихлорметане, и вводили газообразный HCl при 0oC, и оставляли на 2 ч. После завершения реакции смесь концентрировали с получением соединения, представляющего собой 4-(бензoтиазол-6-ил)-5-фтор-N-(5-(пиперазин-1-илметил)пиридин-2-ил)пиримидин-2-амин гидрохлорид, выход составлял 100%.
1H ЯМР (300 MГц, DMSO-d6) δ 12,17 (s, 1H), 10,08 (s, 1H), 9,62 (s, 1H), 8,95 (t, J = 2,7 Гц, 2H), 8,71 (s, 1H), 8,53 (d, J = 8,8 Гц, 1H), 8,30 (q, J = 8,7 Гц, 2H), 7,99 (d, J = 9,0 Гц, 1H), 4,57 (s, 2H), 3,54 - 3,42 (m, 10H).
Пример 5.
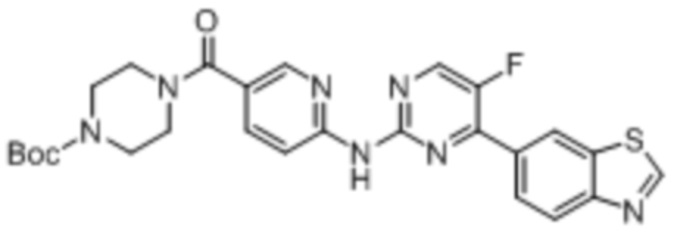
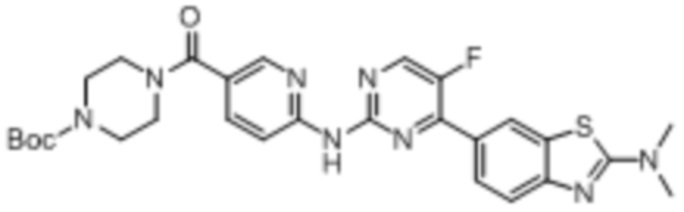
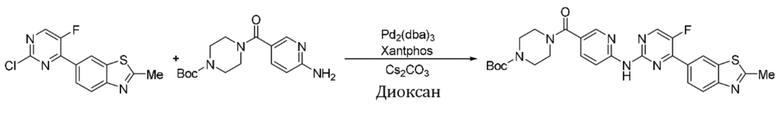
Синтез трет-бутил-4-(6-((4-(бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)никотиноил)пиперазин-1-карбоксилата (I-5).
Ссылаясь на синтез соединения (I-1), выход составлял 52%. 1H ЯМР (400 MГц, CDCl3) δ 9,16 (s, 1H), 8,78 - 8,78 (m, 1H), 8,52 - 8,49 (m, 2H), 8,44 (dd, J = 2,4, 0,8 Гц, 1H), 8,34 - 8,28 (m, 3H), 7,84 (dd, J = 8,7, 2,3 Гц, 1H), 3,70 - 3,44 (m, 8H), 1,48 (s, 9H).
Пример 6.
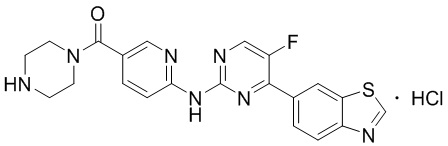
Синтез 6-((4-(бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)(пиперазин-1-ил)кетона гидрохлорида (I-6).
Способ получения 6-((4-(бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)(пиперазин-1-ил)кетона гидрохлорида ссылается на синтез соединения (I-4), и выход составлял 100%.
1H ЯМР (400 MГц, DMSO-d6) δ 11,25 (s, 1H), 9,60 - 9,58 (m, 3H), 8,93 (d, J = 1,6 Гц, 1H), 8,88 (d, J = 3,3 Гц, 1H), 8,51 (d, J = 2,2 Гц, 1H), 8,32 - 8,25 (m, 2H), 8,19 - 8,09 (m, 2H), 3,79 - 3,76 (m, 4H), 3,18 - 3,16 (m, 4H).
Пример 7.
Синтез трет-бутил-4-(6-((4-(бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)пиперазин-1-карбоксилата (I-7).
Ссылаясь на синтез соединения (I-1), выход составлял 47%. 1H ЯМР (400 MГц, CDCl3) δ 9,15 (s, 1H), 8,77 - 8,76 (m, 1H), 8,45 (d, J = 3,4 Гц, 1H), 8,33 - 8,26 (m, 3H), 8,04 - 8,02 (m, 2H), 7,38 (dd, J = 9,1, 3,0 Гц, 1H), 3,63 - 3,60 (m, 4H), 3,11 - 3,09 (m, 4H), 1,49 (s, 9H).
Пример 8.
Синтез 4-(бензoтиазол-6-ил)-5-фтор-N-(5-(пиперазин-1-ил)пиридин-2-ил)пиримидин-2-амина гидрохлорида (I-8).
Ссылаясь на синтез соединения (I-4), выход составлял 100%. 1H ЯМР (400 MГц, DMSO-d6) δ 11,43 (s, 1H), 9,60 (s, 1H), 9,33 (s, 2H), 8,91 (s, 1H), 8,87 (d, J = 3,3 Гц, 1H), 8,33 - 8,23 (m, 2H), 8,07 (d, J = 9,4 Гц, 1H), 8,01 (d, J = 2,9 Гц, 1H), 7,85 (d, J = 9,4 Гц, 1H), 3,44 (t, J = 5,1 Гц, 4H), 3,27 - 3,25 (m, 4H).
Пример 9.
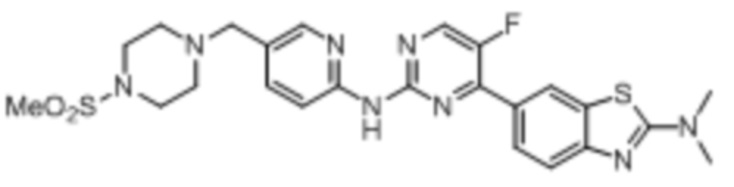
Синтез 4-(бензoтиазол-6-ил)-5-фтор-N-(5-((4-(метансульфонил)пиперазин-1-ил)метил)пиридин-2-ил)пиримидин-2-амина (I-9).
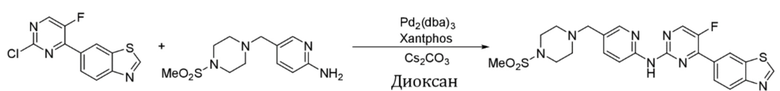
Ссылаясь на синтез соединения (I-1), выход составлял 45%. 1H ЯМР (400 MГц, CDCl3) δ 9,15 (s, 1H), 8,78 - 8,77 (m, 1H), 8,48 (d, J = 3,4 Гц, 1H), 8,40 (dd, J = 8,5, 0,8 Гц, 1H), 8,34 - 8,29 (m, 2H), 8,25 - 8,25 (m, 1H), 8,18 (s, 1H), 7,70 (dd, J = 8,6, 2,4 Гц, 1H), 3,53 (s, 2H), 3,25 (t, J = 4,9 Гц, 4H), 2,78 (s, 3H), 2,58 (t, J = 5,0 Гц, 4H).
Пример 10.
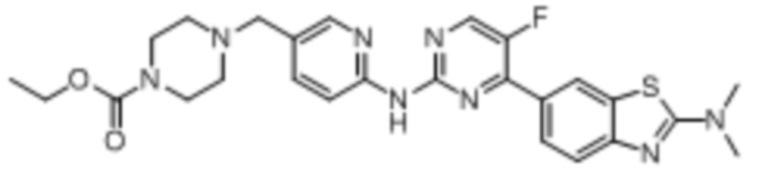
Синтез этил-4-((6-((4-(бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)метил)пиперазин-1-карбоксилата (I-10).
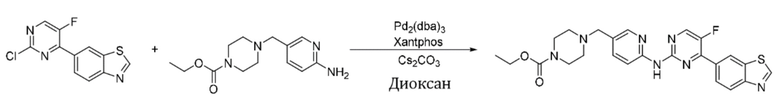
Ссылаясь на синтез соединения (I-1), выход составлял 55%. 1H ЯМР (400 MГц, CDCl3) δ 9,15 (s, 1H), 8,78 - 8,77 (m, 1H), 8,48 (d, J = 3,4 Гц, 1H), 8,39 (dd, J = 8,6, 0,8 Гц, 1H), 8,34 - 8,27 (m, 2H), 8,25 - 8,24 (m, 2H), 7,72 (dd, J = 8,6, 2,3 Гц, 1H), 4,13 (q, J = 7,1 Гц, 2H), 3,50 - 3,47 (m, 6H), 2,42 (t, J = 5,0 Гц, 4H), 1,26 (t, J = 7,1 Гц, 3H).
Пример 11.
Синтез 4-(бензoтиазол-6-ил)-5-фтор-N-(5-((4-изопропилпиперазин-1-ил)метил)пиридин-2-ил)пиримидин-2-амина (I-11).
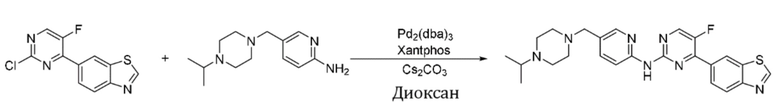
Ссылаясь на синтез соединения (I-1), выход составлял 48%. 1H ЯМР (400 MГц, CDCl3) δ 9,15 (s, 1H), 8,78 (d, J = 1,6 Гц, 1H), 8,48 (d, J = 3,5 Гц, 1H), 8,38 (dd, J = 8,5, 0,8 Гц, 1H), 8,35 - 8,32 (m, 1H), 8,30 - 8,27 (m, 1H), 8,25 - 8,23 (m, 2H), 7,73 (dd, J = 8,6, 2,3 Гц, 1H), 3,50 (s, 2H), 2,68 - 2,44 (m, 9H), 1,06 (d, J = 6,5 Гц, 6H).
Пример 12.
Синтез (4-этилпиперазин-1-ил)(6-((5-фтор-4-(2-метилбензoтиазол-6-ил)пиримидин-2-ил)амино)пиридин-3-ил)кетона (I-12).
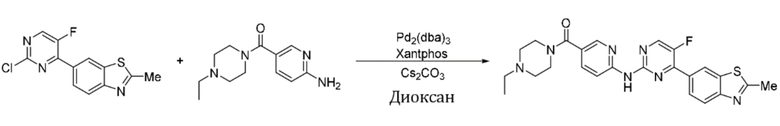
Ссылаясь на синтез соединения (I-1), выход составлял 40%. 1H ЯМР (400 MГц, CDCl3) δ 9,25 (s, 1H), 8,63 (d, J = 1,8 Гц, 1H), 8,56 - 8,54 (m, 2H), 8,51 (d, J = 8,7 Гц, 1H), 8,24 (dt, J = 8,7, 1,3 Гц, 1H), 8,09 (d, J = 8,6 Гц, 1H), 7,86 (dd, J = 8,7, 2,4 Гц, 1H), 3,78 - 3,64 (m, 4H), 2,90 (s, 3H), 2,51 - 2,46 (m, 6H), 1,13 (t, J = 7,1 Гц, 3H).
Пример 13.
Синтез N-(5-((4-этилпиперазин-1-ил)метил)пиридин-2-ил)-5-фтор-4-(2-метилбензoтиазол-6-ил)пиримидин-2-амина (I-13).
Ссылаясь на синтез соединения (I-1), выход составлял 52%. 1H ЯМР (300 MГц, CDCl3) δ 8,63 (d, J = 1,8 Гц, 1H), 8,49 - 8,47 (m, 2H), 8,38 (d, J = 8,6 Гц, 1H), 8,28 - 8,24 (m, 2H), 8,08 (d, J = 8,7 Гц, 1H), 7,72 (dd, J = 8,6, 2,3 Гц, 1H), 3,51 (s, 2H), 2,90 (s, 3H), 2,55 - 2,45 (m, 10H), 1,11 (t, J = 7,2 Гц, 3H).
Пример 14.
Синтез трет-бутил-4-((6-((5-фтор-4-(2-метилбензoтиазол-6-ил)пиримидин-2-ил)амино)пиридин-3-ил)метил)пиперазин-1-карбоксилата (I-14).
Ссылаясь на синтез соединения (I-1), выход составлял 55%. 1H ЯМР (300 MГц, CDCl3) δ 8,63 (s, 1H), 8,46 (d, J = 3,5 Гц, 1H), 8,39 (d, J = 8,6 Гц, 1H), 8,32 (s, 1H), 8,27 - 8,24 (m, 2H), 8,08 (d, J = 8,7 Гц, 1H), 7,72 (d, J = 8,4 Гц, 1H), 3,49 (s, 2H), 3,43 (t, J = 4,8 Гц, 4H), 2,91 (s, 3H), 2,42 - 2,39 (m, 4H), 1,46 (s, 9H).
Пример 15.
Синтез 5-фтор-4-(2-метилбензoтиазол-6-ил)-N-(5-(пиперазин-1-илметил)пиридин-2-ил)пиримидин-2-амина гидрохлорида (I-15).

Ссылаясь на синтез соединения (I-4), выход составлял 100%. 1H ЯМР (400 MГц, DMSO-d6) δ 11,67 (s, 1H), 9,90 (s, 2H), 8,89 (d, J = 3,3 Гц, 1H), 8,80 (d, J = 1,7 Гц, 1H), 8,64 (d, J = 2,2 Гц, 1H), 8,40 (dd, J = 9,0, 2,2 Гц, 1H), 8,21 (dt, J = 8,6, 1,3 Гц, 1H), 8,13 (d, J = 8,6 Гц, 1H), 8,04 (d, J = 8,9 Гц, 1H), 4,50 (s, 2H), 3,50 - 3,45 (m, 8H), 2,88 (s, 3H).
Пример 16.
Синтез трет-бутил-4-(6-((5-фтор-4-(2-метилбензoтиазол-6-ил)пиримидин-2-ил)амино)никотиноил)пиперазин-1-карбоксилата (I-16).
Ссылаясь на синтез соединения (I-1), выход составлял 40%. 1H ЯМР (400 MГц, CDCl3) δ 9,11 (s, 1H), 8,63 (s, 1H), 8,55 - 8,51 (m, 3H), 8,24 (d, J = 8,6 Гц, 1H), 8,09 (dd, J = 8,7, 3,3 Гц, 1H), 7,86 (dt, J = 8,9, 2,6 Гц, 1H), 3,67 - 3,46 (m, 8H), 2,91 (s, 3H), 1,48 (s, 9H).
Пример 17.
Синтез (6-((5-фтор-4-(2-метилбензoтиазол-6-ил)пиримидин-2-ил)амино)пиридин-3-ил)(пиперазин-1-ил)кетона гидрохлорида (I-17).
Ссылаясь на синтез соединения (I-4), выход составлял 100%. 1H ЯМР (300 MГц, DMSO-d6) δ 12,04 (s, 1H), 9,85 (s, 2H), 8,92 (d, J = 2,9 Гц, 1H), 8,81 (s, 1H), 8,60 (s, 1H), 8,28 (d, J = 9,1 Гц, 1H), 8,21 (d, J = 8,7 Гц, 1H), 8,13 (d, J = 8,6 Гц, 1H), 8,06 (d, J = 9,0 Гц, 1H), 3,83 - 3,77 (m, 4H), 3,20 - 3,14 (m, 4H), 2,88 (s, 3H).
Пример 18.
Синтез (6-((4-(2-(циклопентиламино)бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)(4-этилпиперазин-1-ил)кетона (I-18).
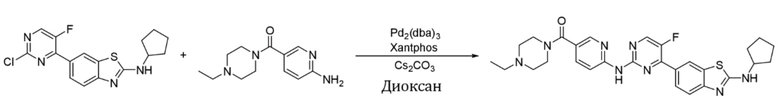
Ссылаясь на синтез соединения (I-1), выход составлял 45%. 1H ЯМР (400 MГц, CDCl3) δ 8,90 (s, 1H), 8,50 (d, J = 8,7 Гц, 1H), 8,46 (d, J = 2,2 Гц, 1H), 8,43 (d, J = 3,6 Гц, 1H), 8,40 (d, J = 1,8 Гц, 1H), 8,07 (dd, J = 8,7, 1,8 Гц, 1H), 7,84 (dd, J = 8,7, 2,3 Гц, 1H), 7,64 (d, J = 8,5 Гц, 1H), 6,08 (s, 1H), 3,80 - 3,65 (m, 4H), 2,52 - 2,47 (m, 6H), 2,18 - 2,12 (m, 2H), 1,81 - 1,64 (m, 6H), 1,13 (t, J = 7,2 Гц, 3H).
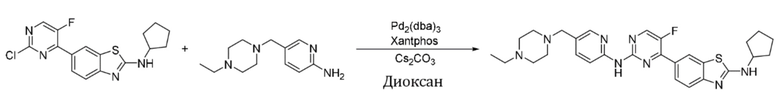
Пример 19.
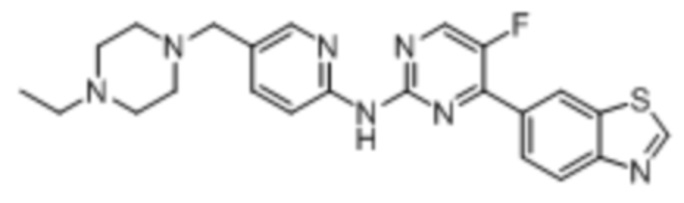
Синтез N-циклопентил-6-(2-((5-((4-этилпиперазин-1-ил)метил)пиридин-2-ил)амино)-5-фторпиримидин-4-ил)бензoтиазол-2-амина (I-19).
Ссылаясь на синтез соединения (I-1), выход составлял 51%. 1H ЯМР (400 MГц, CDCl3) δ 8,59 (s, 1H), 8,43 - 8,35 (m, 3H), 8,23 (d, J = 2,2 Гц, 1H), 8,08 - 8,05 (m, 1H), 7,71 (dd, J = 8,6, 2,3 Гц, 1H), 7,64 (d, J = 8,6 Гц, 1H), 6,12 (d, J = 6,7 Гц, 1H), 4,08 - 4,04 (m, 1H), 3,50 (s, 2H), 2,54 - 2,42 (m, 10H), 2,19 - 2,11 (m, 2H), 1,79 - 1,64 (m, 6H), 1,10 (t, J = 7,2 Гц, 3H).
Пример 20.
Синтез трет-бутил-4-((6-((4-(2-(циклопентиламино)бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)метил)пиперазин-1-карбоксилата (I-20).
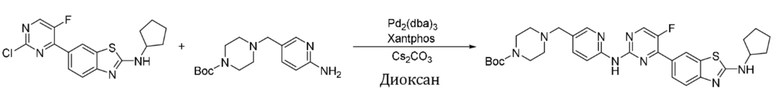
Ссылаясь на синтез соединения (I-1), выход составлял 57%. 1H ЯМР (400 MГц, CDCl3) δ 8,43 - 8,40 (m, 2H), 8,38 (d, J = 3,7 Гц, 1H), 8,22 (s, 2H), 8,11 (d, J = 7,8 Гц, 1H), 7,79 (s, 1H), 7,63 (d, J = 8,4 Гц, 1H), 5,71 (s, 1H), 4,13 - 4,05 (m, 1H), 3,60 - 3,45 (m, 6H), 2,57 - 2,43 (m, 4H), 2,20 - 2,12 (m, 2H), 1,80 - 1,61 (m, 6H), 1,46 (s, 9H).
Пример 21.
Синтез N-циклопентил-6-(5-фтор-2-((5-(пиперазин-1-илметил)пиридин-2-ил)амино)пиримидин-4-ил)бензoтиазол-2-амина гидрохлорида (I-21).

Ссылаясь на синтез соединения (I-4), выход составлял 100%. 1H ЯМР (400 MГц, DMSO-d6) δ 11,31 (s, 1H), 9,80 (s, 2H), 9,40 (s, 1H), 8,79 (d, J = 3,6 Гц, 1H), 8,60 (d, J = 2,2 Гц, 1H), 8,53 (d, J = 1,8 Гц, 1H), 8,32 (dd, J = 9,1, 2,2 Гц, 1H), 8,10 - 8,06 (m, 2H), 7,65 (d, J = 8,6 Гц, 1H), 4,44 (s, 2H), 4,29 - 4,24 (m, 1H), 3,46 - 3,38 (m, 8H), 3,17 (s, 1H), 2,06 - 1,99 (m, 2H), 1,73 - 1,59 (m, 6H).
Пример 22.
Синтез трет-бутил-4-(6-((4-(2-(циклопентиламино)бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)никотиноил)пиперазин-1-карбоксилата (I-22).
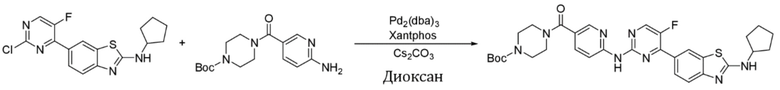
Ссылаясь на синтез соединения (I-1), выход составлял 53%. 1H ЯМР (400 MГц, CDCl3) δ 8,82 (s, 1H), 8,51 (d, J = 8,4 Гц, 1H), 8,46 - 8,41 (m, 3H), 8,09 (d, J = 8,3 Гц, 1H), 7,85 - 7,82 (m, 1H), 7,63 (d, J = 8,3 Гц, 1H), 6,20 (s, 1H), 4,09 - 4,03 (m, 1H), 3,68 - 3,45 (m, 8H), 2,18 - 2,12 (m, 2H), 1,80 - 1,62 (m, 6H), 1,48 (s, 9H).
Пример 23.
Синтез (6-((4-(2-(циклопентиламино)бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)(пиперазин-1-ил)кетона гидрохлорида (I-23).
Ссылаясь на синтез соединения (I-4), выход составлял 100%. 1H ЯМР (400 MГц, DMSO-d6) δ 11,15 (s, 1H), 9,70 (s, 1H), 9,52 (s, 2H), 8,80 (s, 1H), 8,56 - 8,50 (m, 2H), 8,15 - 8,09 (m, 3H), 7,69 (d, J = 8,6 Гц, 1H), 4,32 - 4,25 (m, 1H), 3,80 - 3,74 (m, 4H), 3,33 (s, 1H), 3,20 - 3,14 (m, 4H), 2,05 - 1,99 (m, 2H), 1,76 - 1,59 (m, 6H).
Пример 24.
Синтез этил-4-((6-((4-(2-(циклопентиламино)бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)метил)пиперазин-1-карбоксилата (I-24).
Ссылаясь на синтез соединения (I-1), выход составлял 56%. 1H ЯМР (400 MГц, CDCl3) δ 8,82 (s, 1H), 8,42 - 8,38 (m,3H), 8,25 - 8,24 (m, 1H), 8,05 (d, J = 8,3 Гц, 1H), 7,73 (d, J = 8,4 Гц, 1H), 7,64 (d, J = 8,3 Гц, 1H), 6,25 (s, 1H), 4,14 (q, J = 7,0 Гц, 2H), 4,08 - 4,02 (m,1H), 3,54 - 3,47 (m, 6H), 2,46 - 2,42 (m, 4H), 2,19 - 2,11 (m, 2H), 1,79 - 1,61 (m, 6H), 1,26 (t, J = 7,0 Гц, 3H).
Пример 25.
Синтез N-циклопентил-6-(5-фтор-2-((5-((4-(метансульфонил)пиперазин-1-ил)метил)пиридин-2-ил)амино)пиримидин-4-ил)бензoтиазол-2-амина (I-25).
Ссылаясь на синтез соединения (I-1), выход составлял 53%. 1H ЯМР (400 MГц, CDCl3) δ 8,54 (s, 1H), 8,42 - 8,39 (m, 3H), 8,24 (d, J = 2,3 Гц, 1H), 8,10 - 8,07 (m, 1H), 7,69 (dd, J = 8,6, 2,3 Гц, 1H), 7,64 (d, J = 8,6 Гц, 1H), 5,96 (d, J = 6,8 Гц, 1H), 4,09 - 4,04 (m, 1H), 3,52 (s, 2H), 3,25 (t, J = 4,9 Гц, 4H), 2,79 (s, 3H), 2,58 (t, J = 4,9 Гц, 4H), 2,19 - 2,11 (m, 2H), 1,80 - 1,70 (m, 6H).
Пример 26.
Синтез трет-бутил-4-(6-((4-(2-(циклопентиламино)бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)пиперазин-1-карбоксилата (I-26).
Ссылаясь на синтез соединения (I-1), выход составлял 44%. 1H ЯМР (400 MГц, CDCl3) δ8,48 (s, 1H), 8,39 (d, J = 1,8 Гц, 1H), 8,36 (d, J = 3,7 Гц, 1H), 8,31 (d, J = 9,1 Гц, 1H), 8,08 - 8,05 (m, 1H), 8,03 (d, J = 2,9 Гц, 1H), 7,63 (d, J = 8,5 Гц, 1H), 7,37 (dd, J = 9,1, 3,0 Гц, 1H), 6,10 (d, J = 6,6 Гц, 1H), 4,08 - 4,03 (m, 1H), 3,61 (t, J = 5,1 Гц, 4H), 3,09 (t, J = 5,0 Гц, 4H), 2,17 - 2,11 (m, 2H), 1,77 - 1,64 (m, 6H), 1,49 (s, 9H).
Пример 27.
Синтез N-циклопентил-6-(5-фтор-2-((5-(пиперазин-1-ил)пиридин-2-ил)амино)пиримидин-4-ил)бензoтиазол-2-амина гидрохлорида (I-27).
Ссылаясь на синтез соединения (I-4), выход составлял 100%. 1H ЯМР (400 MГц, DMSO-d6) δ 11,97 (s, 1H), 10,53 (s, 1H), 9,86 (s, 2H), 8,83 (d, J = 3,3 Гц, 1H), 8,57 (d, J = 1,7 Гц, 1H), 8,27 (dd, J = 9,6, 2,6 Гц, 1H), 8,11 - 8,09 (m, 1H), 8,05 (d, J = 2,6 Гц, 1H), 7,86 (d, J = 9,5 Гц, 1H), 7,77 (d, J = 8,6 Гц, 1H), 4,41 - 4,35 (m, 1H), 3,52 - 3,50 (m, 4H), 3,26 - 3,23 (m, 4H), 2,08 - 2,02 (m, 2H), 1,76 - 1,60 (m, 6H).
Пример 28.
Синтез N-циклопентил-6-(5-фтор-2-((5-((4-изопропилпиперазин-1-ил)метил)пиридин-2-ил)амино)пиримидин-4-ил)бензoтиазол-2-амина (I-28).
Ссылаясь на синтез соединения (I-1), выход составлял 50%. 1H ЯМР (400 MГц, CDCl3) δ8,78 (s, 1H), 8,40 - 8,38 (m, 3H), 8,23 (d, J = 2,2 Гц, 1H), 8,05 (dd, J = 8,5, 1,8 Гц, 1H), 7,71 (dd, J = 8,6, 2,3 Гц, 1H), 7,65 (d, J = 8,6 Гц, 1H), 6,32 (d, J = 6,2 Гц, 1H), 4,07 - 4,02 (m, 1H), 3,49 (s, 2H), 2,70 - 2,55 (m, 9H), 2,18 - 2,11 (m, 2H), 1,80 - 1,64 (m, 6H), 1,06 (d, J = 6,5 Гц, 6H).
Пример 29.
Синтез (6-((4-(2-(циклопентиламино)бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)(4-изопропилпиперазин-1-ил)кетона (I-29).
Ссылаясь на синтез соединения (I-1), выход составлял 48%. 1H ЯМР (400 MГц, CDCl3) δ 9,04 (s, 1H), 8,52 - 8,39 (m, 4H), 8,05 (dd, J = 8,4, 1,9 Гц, 1H), 7,85 (dt, J = 8,6, 2,2 Гц, 1H), 7,64 (dd, J = 8,6, 1,8 Гц, 1H), 6,19 (s, 1H), 4,09 - 4,04 (m, 1H), 3,73 - 3,62 (m, 4H), 2,79 - 2,75 (m, 1H), 2,60 - 2,55 (m, 4H), 2,18 - 2,13 (m, 2H), 1,78 - 1,64 (m, 6H), 1,08 (d, J = 6,6 Гц, 6H).
Пример 30.
Синтез 6-(2-((5-((4-этилпиперазин-1-ил)метил)пиридин-2-ил)амино)-5-фторпиримидин-4-ил)-N,N-диметилбензoтиазол-2-амина (I-30).
Ссылаясь на синтез соединения (I-1), выход составлял 42%. 1H ЯМР (400 MГц, CDCl3) δ 8,45 (d, J = 1,8 Гц, 1H), 8,38 - 8,36 (m, 2H), 8,21 (d, J = 2,3 Гц, 1H), 8,15 (dt, J = 8,7, 1,2 Гц, 1H), 7,94 (s, 1H), 7,71 (dd, J = 8,6, 2,3 Гц, 1H), 7,65 (d, J = 8,6 Гц, 1H), 3,49 (s, 2H), 3,27 (s, 6H), 2,60 - 2,36 (m, 10H), 1,09 (t, J = 7,2 Гц, 3H).
Пример 31.
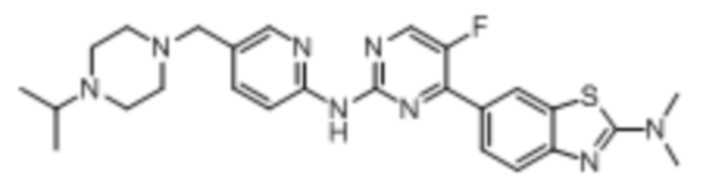
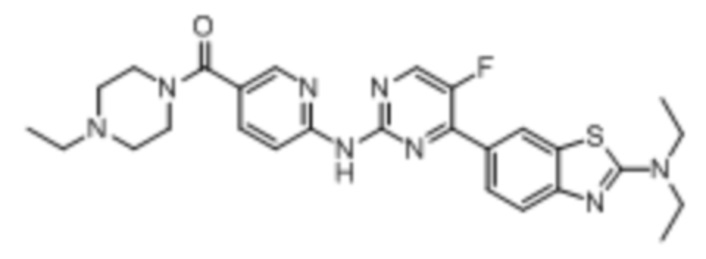
Синтез (6-((4-(2-(диметиламино)бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)(4-этилпиперазин-1-ил)кетона (I-31).
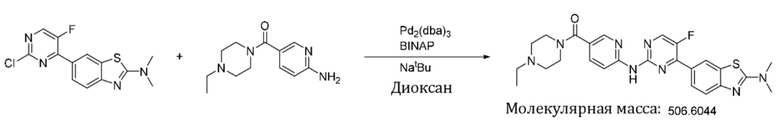
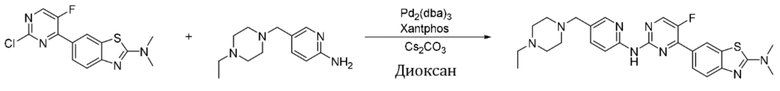
6-(2-Хлор-5-фторпиримидин-4-ил)-N,N-диметилбензoтиазол-2-амин (154 мг, 0,5 ммоль) и 6-аминопиридин-3-ил)(4-этилпиперазин-1-ил)кетон (141 мг, 0,6 ммоль) растворяли в диоксане (5 мл). Затем добавляли Pd2(dba)3 (23 мг, 0,025 ммоль), BINAP (31 мг, 0,05 ммоль), трет-бутоксид натрия (96 мг, 1,0 ммоль). Реакционную смесь продували аргоном три раза, нагревали до 100oC и оставляли на 12 ч. Смесь охлаждали, фильтровали и концентрировали, и подвергали колоночной хроматографии (DCM~DCM/MeOH = 10:1) с получением соединения, представляющего собой (6-((4-(2-(диметиламино)бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)(4-этилпиперазин-1-ил)кетон (139 мг, выход 55%). 1H ЯМР (400 MГц, DMSO-d6) δ 10,27 (s, 1H), 8,69 (d, J = 3,8 Гц, 1H), 8,52 (d, J = 2,0 Гц, 1H), 8,35 (d, J = 2,4 Гц, 1H), 8,31 (d, J = 8,7 Гц, 1H), 8,06 (d, J = 8,6 Гц, 1H), 7,86 (dd, J = 8,6, 2,4 Гц, 1H), 7,59 (d, J = 8,6 Гц, 1H), 3,59 - 3,47 (m, 4H), 3,21 (s, 6H), 2,44 - 2,34 (m, 6H), 1,01 (t, J = 7,1 Гц, 3H).
Пример 32.
Синтез трет-бутил-4-((6-((4-(2-(диметиламино)бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)метил)пиперазин-1-карбоксилата (I-32).
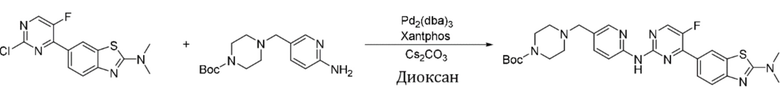
Ссылаясь на синтез соединения (I-1), выход составлял 48%. 1H ЯМР (400 MГц, CDCl3) δ 8,45 (d, J = 1,8 Гц, 1H), 8,40 - 8,36 (m, 2H), 8,20 (d, J = 2,2 Гц, 1H), 8,15 (dt, J = 8,8, 1,2 Гц, 1H), 7,94 (s, 1H), 7,71 (dd, J = 8,6, 2,3 Гц, 1H), 7,65 (d, J = 8,6 Гц, 1H), 3,48 (s, 2H), 3,45 - 3,42 (m, 4H), 3,27 (s, 6H), 2,41 - 2,39 (m, 4H), 1,46 (s, 9H).
Пример 33.
Синтез 6-(5-фтор-2-((5-(пиперазин-1-илметил)пиридин-2-ил)амино)пиримидин-4-ил)-N,N-диметилбензoтиазол-2-амина гидрохлорида (I-33).

Ссылаясь на синтез соединения (I-4), выход составлял 100%. 1H ЯМР (300 MГц, DMSO-d6) δ 11,44 (s, 1H), 9,83 (s, 2H), 8,79 (d, J = 2,9 Гц, 1H), 8,59 (d, J = 11,9 Гц, 2H), 8,35 (d, J = 8,9 Гц, 1H), 8,07 (dd, J = 14,1, 8,8 Гц, 2H), 7,65 (d, J = 8,6 Гц, 1H), 4,46 (s, 2H), 3,49 - 3,41 (m, 8H), 3,24 (s, 6H).
Пример 34.
Синтез трет-бутил-4-(6-((4-(2-(диметиламино)бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)никотиноил)пиперазин-1-карбоксилата (I-34).
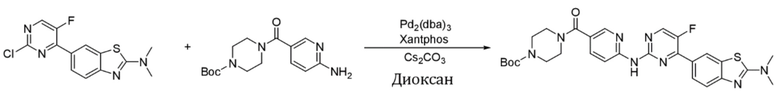
Ссылаясь на синтез соединения (I-1), выход составлял 50%. 1H ЯМР (400 MГц, CDCl3) δ 8,91 (s, 1H), 8,53 (dd, J = 8,6, 0,8 Гц, 1H), 8,50 (dd, J = 2,4, 0,8 Гц, 1H), 8,45 (d, J = 3,8 Гц, 1H), 8,44 (d, J = 1,8 Гц, 1H), 8,16 - 8,13 (m, 1H), 7,84 (dd, J = 8,7, 2,4 Гц, 1H), 7,66 (d, J = 8,6 Гц, 1H), 3,70 - 3,59 (m, 4H), 3,51 - 3,46 (m, 4H), 3,27 (s, 6H), 1,48 (s, 9H).
Пример 35.
Синтез (6-((4-(2-(диметиламино)бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)(пиперазин-1-ил)кетона гидрохлорида (I-35).
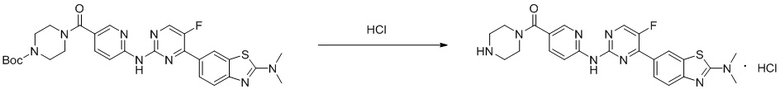
Ссылаясь на синтез соединения (I-1), выход составлял 100%. 1H ЯМР (300 MГц, CDCl3) δ 11,70 (s, 1H), 9,84 (s, 1H), 9,71 (s, 1H), 8,83 (s, 1H), 8,58 (d, J = 17,9 Гц, 2H), 8,21 (d, J = 8,9 Гц, 1H), 8,14 - 8,05 (m, 2H), 7,70 (d, J = 8,6 Гц, 1H), 3,35 - 3,32 (m, 4H), 3,28 (s, 6H), 3,20 - 3,15 (m, 4H).
Пример 36.
Синтез 6-(5-фтор-2-((5-((4-изопропилпиперазин-1-ил)метил)пиридин-2-ил)амино)пиримидин-4-ил)-N,N-диметилбензoтиазол-2-амина (I-36).
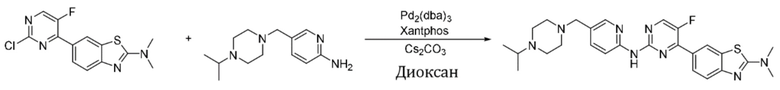
Ссылаясь на синтез соединения (I-1), выход составлял 52%. 1H ЯМР (400 MГц, CDCl3) δ 8,45 (d, J = 1,8 Гц, 1H), 8,38 (dd, J = 6,3, 2,4 Гц, 2H), 8,23 (d, J = 2,3 Гц, 1H), 8,16 - 8,14 (m, 2H), 7,71 (dd, J = 8,6, 2,3 Гц, 1H), 7,65 (d, J = 8,6 Гц, 1H), 3,49 (s, 2H), 3,27 (s, 6H), 2,68 - 2,54 (m, 9H), 1,05 (d, J = 6,5 Гц, 6H).
Пример 37.
Синтез 6-(5-фтор-2-((5-((4-(метансульфонил)пиперазин-1-ил)метил)пиридин-2-ил)амино)пиримидин-4-ил)-N,N-диметилбензoтиазол-2-амина (I-37).
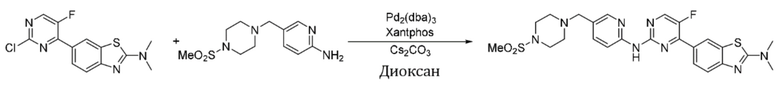
Ссылаясь на синтез соединения (I-1), выход составлял 43%. 1H ЯМР (400 MГц, CDCl3) δ 8,44 - 8,35 (m, 3H), 8,21 (d, J = 7,7 Гц, 1H), 8,16 - 8,07 (m, 2H), 7,71 (s, 1H), 7,66 - 7,61 (m, 1H), 3,55 (s, 2H), 3,29 - 3,25 (m, 10H), 2,79 (s, 3H), 2,63 - 2,57 (m, 4H).
Пример 38.
Синтез этил-4-((6-((4-(2-(диметиламино)бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)метил)пиперазин-1-карбоксилата (I-38).
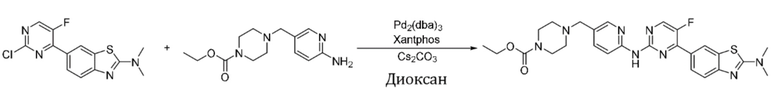
Ссылаясь на синтез соединения (I-1), выход составлял 53%. 1H ЯМР (400 MГц, CDCl3) δ 8,44 - 8,36 (m, 3H), 8,31 - 8,23 (m, 2H), 8,15 (d, J = 8,3 Гц, 1H), 7,73 (s, 1H), 7,65 (d, J = 8,3 Гц, 1H), 4,13 (q, J = 7,1 Гц, 2H), 3,52 - 3,49 (m, 6H), 3,27 (s, 6H), 2,46 - 2,41 (m, 4H), 1,26 (t, J = 7,3 Гц, 3H).
Пример 39.
Синтез (6-((4-(2-(диметиламино)бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)(4-изопропилпиперазин-1-ил)кетона (I-39).
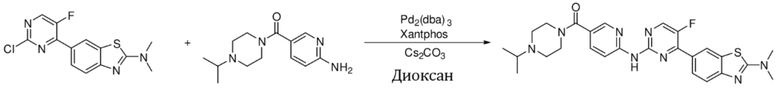
Ссылаясь на синтез соединения (I-1), выход составлял 44%. 1H ЯМР (400 MГц, CDCl3) δ 8,73 (s, 1H), 8,52 - 8,48 (m, 2H), 8,45 - 8,44 (m, 2H), 8,16 - 8,13 (m, 1H), 7,85 (dd, J = 8,7, 2,4 Гц, 1H), 7,65 (d, J = 8,6 Гц, 1H), 3,75 - 3,63 (m, 4H), 3,27 (s, 6H), 2,79 - 2,75 (m, 1H), 2,62 - 2,55 (m, 4H), 1,08 (d, J = 6,5 Гц, 6H).
Пример 40.
Синтез трет-бутил-4-(6-((4-(2-(диметиламино)бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)пиперазин-1-карбоксилата (I-40).
Ссылаясь на синтез соединения (I-1), выход составлял 40%. 1H ЯМР (400 MГц, CDCl3) δ 8,44 (s, 1H), 8,36 - 8,33 (m, 2H), 8,25 (s, 1H), 8,14 (dd, J = 8,7, 1,6 Гц, 1H), 8,00 (d, J = 2,8 Гц, 1H), 7,64 (dd, J = 8,6, 1,2 Гц, 1H), 7,40 (dd, J = 9,3, 2,9 Гц, 1H), 3,61 (t, J = 5,0 Гц, 4H), 3,27 (s, 3H), 3,09 (t, J = 5,0 Гц, 4H).
Пример 41.
Синтез 6-(5-фтор-2-((5-(пиперазин-1-ил)пиридин-2-ил)амино)пиримидин-4-ил)-N,N-диметилбензoтиазол-2-амина гидрохлорида (I-41).
Ссылаясь на синтез соединения (I-4), выход составлял 100%. 1H ЯМР (400 MГц, DMSO-d6) δ 11,99 (s, 1H), 9,77 (s, 2H), 8,81 (d, J = 3,5 Гц, 1H), 8,57 (d, J = 1,8 Гц, 1H), 8,28 (dd, J = 9,7, 2,8 Гц, 1H), 8,12 - 8,05 (m, 1H), 8,02 (d, J = 2,9 Гц, 1H), 7,82 (d, J = 9,6 Гц, 1H), 7,69 (d, J = 8,6 Гц, 1H), 3,49 (t, J = 5,1 Гц, 4H), 3,27 (s, 6H), 3,25 - 3,23 (m, 4H).
Пример 42.
Синтез (6-((4-(2-(диэтиламино)бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)(4-этилпиперазин-1-ил)кетона (I-42).
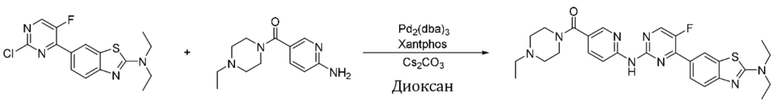
Ссылаясь на синтез соединения (I-1), выход составлял 44%. 1H ЯМР (300 MГц, CDCl3) δ 8,56 (s, 1H), 8,50 (d, J = 8,7 Гц, 1H), 8,47 (d, J = 2,3 Гц, 1H), 8,43 - 8,41 (m, 2H), 8,13 (dd, J = 8,7, 1,7 Гц, 1H), 7,84 (dd, J = 8,7, 2,4 Гц, 1H), 7,62 (d, J = 8,6 Гц, 1H), 3,80 - 3,60 (m, 8H), 2,54 - 2,47 (m, 6H), 1,33 (t, J = 7,1 Гц, 6H), 1,13 (t, J = 7,1 Гц, 3H).
Пример 43.
Синтез N,N-диэтил-6-(2-((5-((4-этилпиперазин-1-ил)метил)пиридин-2-ил)амино)-5-фторпиримидин-4-илбензoтиазол-2-амина (I-43).
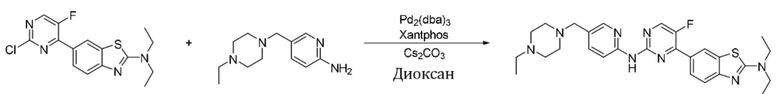
Ссылаясь на синтез соединения (I-1), выход составлял 38%. 1H ЯМР (400 MГц, CDCl3) δ 8,69 (s, 1H), 8,43 - 8,39 (m, 3H), 8,29 (d, J = 2,3 Гц, 1H), 8,15 - 8,12 (m, 1H), 7,71 (dd, J = 8,6, 2,3 Гц, 1H), 7,62 (d, J = 8,6 Гц, 1H), 3,63 (q, J = 7,2 Гц, 4H), 3,50 (s, 2H), 2,53 - 2,42 (m, 10H), 1,33 (t, J = 7,1 Гц, 6H), 1,10 (t, J = 7,2 Гц, 3H).
Пример 44.
Синтез трет-бутил-4-((6-((4-(2-(диэтиламино)бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)метил)пиперазин-1-карбоксилата (I-44).
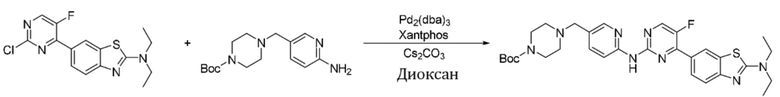
Ссылаясь на синтез соединения (I-1), выход составлял 48%. 1H ЯМР (400 MГц, CDCl3) δ 8,45 (s, 1H), 8,43 - 8,39 (m, 3H), 8,26 (d, J = 2,2 Гц, 1H), 8,15 - 8,12 (m, 1H), 7,73 (d, J = 8,2 Гц, 1H), 7,62 (d, J = 8,6 Гц, 1H), 3,63 (q, J = 7,1 Гц, 4H), 3,51 (s, 2H), 3,46 - 3,44 (m, 4H), 2,44 - 2,41 (m, 4H), 1,46 (s, 9H), 1,33 (t, J = 7,1 Гц, 6H).
Пример 45.
Синтез N,N-диэтил-6-(5-фтор-2-((5-(пиперазин-1-илметил)пиридин-2-ил)амино)пиримидин-4-ил)бензoтиазол-2-амина гидрохлорида (I-45).
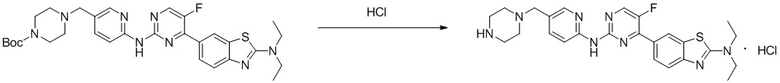
Ссылаясь на синтез соединения (I-4), выход составлял 100%. 1H ЯМР (400 MГц, DMSO-d6) δ 11,71 (s, 1H), 9,94 (s, 2H), 8,80 (d, J = 3,6 Гц, 1H), 8,65 (d, J = 2,2 Гц, 1H), 8,56 (d, J = 1,8 Гц, 1H), 8,42 (dd, J = 8,9, 2,2 Гц, 1H), 8,11 - 8,08 (m, 1H), 8,01 (d, J = 8,9 Гц, 1H), 7,65 (d, J = 8,6 Гц, 1H), 4,51 (s, 2H), 3,63 (q, J = 7,1 Гц, 4H), 3,50 - 3,44 (m, 8H), 1,26 (t, J = 7,1 Гц, 6H).
Пример 46.
Синтез трет-бутил-4-(6-((4-(2-(диэтиламино)бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)никотиноил)пиперазин-1-карбоксилата (I-46).
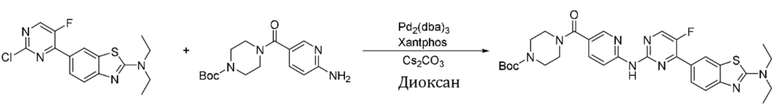
Ссылаясь на синтез соединения (I-1), выход составлял 40%. 1H ЯМР (400 MГц, CDCl3) δ 8,99 (s, 1H), 8,53 (dd, J = 8,7, 0,8 Гц, 1H), 8,51 (dd, J = 2,4, 0,9 Гц, 1H), 8,45 (d, J = 3,8 Гц, 1H), 8,42 (d, J = 1,9 Гц, 1H), 8,15 - 8,12 (m, 1H), 7,84 (dd, J = 8,8, 2,4 Гц, 1H), 7,64 (d, J = 8,6 Гц, 1H), 3,67 - 3,61 (m, 8H), 3,50 - 3,47 (m, 4H), 1,48 (s, 9H), 1,34 (t, J = 7,1 Гц, 6H).
Пример 47.
Синтез (6-((4-(2-(диэтиламино)бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)(пиперазин-1-ил)кетона (I-47).
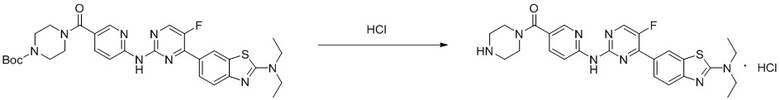
Ссылаясь на синтез соединения (I-4), выход составлял 100%. 1H ЯМР (400 MГц, DMSO-d6) δ 11,78 (s, 1H), 10,03 (s, 1H), 9,88 (s, 2H), 8,83 (d, J = 3,6 Гц, 1H), 8,60 (d, J = 1,8 Гц, 1H), 8,58 (d, J = 2,1 Гц, 1H), 8,23 (dd, J = 8,9, 2,2 Гц, 1H), 8,13 - 8,07 (m, 2H), 7,71 (d, J = 8,6 Гц, 1H), 3,86 - 3,78 (m, 3H), 3,67 (q, J = 7,1 Гц, 4H), 3,19 - 3,15 (m, 3H), 1,27 (t, J = 7,1 Гц, 6H).
Пример 48.
Синтез N,N-диэтил-6-(5-фтор-2-((5-((4-(метансульфонил)пиперазин-1-ил)метил)пиридин-2-ил)амино)пиримидин-4-ил)бензoтиазол-2-амина (I-48).
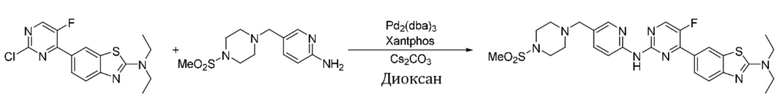
Ссылаясь на синтез соединения (I-1), выход составлял 44%. 1H ЯМР (300 MГц, CDCl3) δ 8,92 (s, 1H), 8,44 - 8,42 (m, 3H), 8,32 - 8,21 (m, 1H), 8,13 (d, J = 8,6 Гц, 1H), 7,70 (dd, J = 8,6, 2,3 Гц, 1H), 7,62 (d, J = 8,7 Гц, 1H), 3,63 (q, J = 7,2 Гц, 4H), 3,52 (s, 2H), 3,25 (t, J = 4,8 Гц, 3H), 2,78 (s, 3H), 2,58 (t, J = 4,7 Гц, 4H), 1,33 (t, J = 7,1 Гц, 6H).
Пример 49.
Синтез этил-4-((6-((4-(2-(диэтиламино)бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)метил)пиперазин-1-карбоксилата (I-49).
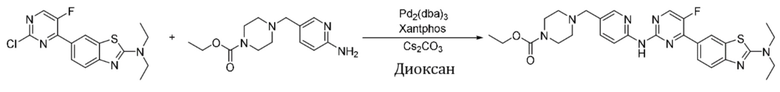
Ссылаясь на синтез соединения (I-1), выход составлял 50%. 1H ЯМР (300 MГц, CDCl3) δ 8,82 (s, 1H), 8,45 - 8,42 (m, 3H), 8,30 (d, J = 2,2 Гц, 1H), 8,13 (d, J = 8,6 Гц, 1H), 7,74 (d, J = 8,6 Гц, 1H), 7,62 (d, J = 8,6 Гц, 1H), 4,13 (q, J = 7,2 Гц, 2H), 3,63 (q, J = 7,2 Гц, 4H), 3,53 - 3,49 (m, 6H), 2,46 - 2,42 (m, 4H), 1,33 (t, J = 7,1 Гц, 6H), 1,26 (t, J = 7,1 Гц, 3H).
Пример 50.
Синтез N,N-диэтил-6-5-фтор-2-((5-((4-изопропилпиперазин-1-ил)метил)пиридин-2-ил)амино)пиримидин-4-ил)бензoтиазол-2-амина (I-50).
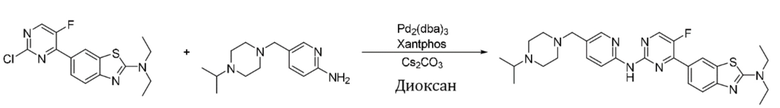
Ссылаясь на синтез соединения (I-1), выход составлял 48%. 1H ЯМР (300 MГц, CDCl3) δ 9,05 (s, 1H), 8,45 - 8,40 (m, 3H), 8,32 (d, J = 2,3 Гц, 1H), 8,14 (d, J = 8,6 Гц, 1H), 7,71 (dd, J = 8,7, 2,3 Гц, 1H), 7,61 (d, J = 8,7 Гц, 1H), 3,62 (q, J = 7,2 Гц, 4H), 3,49 (s, 2H), 2,68 - 2,55 (m, 9H), 1,32 (t, J = 7,1 Гц, 6H), 1,05 (d, J = 6,4 Гц, 6H).
Пример 51.
Синтез (6-((4-(2-(диэтиламино)бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)(4-изопропилпиперазин-1-ил)кетона (I-51).
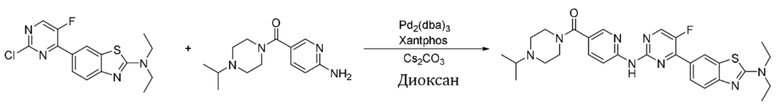
Ссылаясь на синтез соединения (I-1), выход составлял 47%. 1H ЯМР (300 MГц, CDCl3) δ 9,69 (s, 1H), 8,60 (d, J = 2,3 Гц, 1H), 8,54 (d, J = 8,7 Гц, 1H), 8,50 (d, J = 3,7 Гц, 1H), 8,41 (s, 1H), 8,12 (d, J = 8,6 Гц, 1H), 7,86 (dd, J = 8,7, 2,3 Гц, 1H), 7,62 (d, J = 8,6 Гц, 1H), 3,77 - 3,59 (m, 8H), 2,78 - 2,72 (m, 1H), 2,59 - 2,54 (m, 4H), 1,32 (t, J = 7,1 Гц, 6H), 1,07 (d, J = 6,4 Гц, 6H).
Пример 52.
Синтез трет-бутил-4-(6-((4-(2-(диэтиламино)бензoтиазол-6-ил)-5-фторпиримидин-2-ил)амино)пиридин-3-ил)пиперазин-1-карбоксилата (I-52).
Ссылаясь на синтез соединения (I-1), выход составлял 49%. 1H ЯМР (300 MГц, CDCl3) δ 8,58 (s, 1H), 8,41 (d, J = 1,8 Гц, 1H), 8,37 (d, J = 4,0 Гц, 1H), 8,34 (d, J = 9,1 Гц, 1H), 8,14 - 8,10 (m, 1H), 8,06 (d, J = 2,9 Гц, 1H), 7,61 (d, J = 8,6 Гц, 1H), 7,38 (dd, J = 9,1, 3,0 Гц, 1H), 3,66 - 3,58 (m, 8H), 3,09 (t, J = 5,1 Гц, 4H), 1,49 (s, 9H), 1,32 (t, J = 7,1 Гц, 6H).
Пример 53.
Синтез N,N-диэтил-6-(5-фтор-2-((5-(пиперазин-1-ил)пиридин-2-ил)амино)пиримидин-4-ил)бензoтиазол-2-амина гидрохлорида (I-53).
Ссылаясь на синтез соединения (I-4), выход составлял 100%. 1H ЯМР (300 MГц, DMSO-d6) δ 12,15 (s, 1H), 10,04 (s, 2H), 8,79 (s, 1H), 8,57 (s, 1H), 8,34 (d, J = 9,3 Гц, 1H), 8,08 - 8,05 (m, 2H), 7,87 (d, J = 8,9 Гц, 1H), 7,71 (d, J = 8,4 Гц, 1H), 3,69 - 3,67 (m, 4H), 3,56 - 3,53 (m, 4H), 3,27 - 3,23 (m, 4H), 1,30 - 1,25 (m, 6H).
Соответствующие фармацевтически приемлемые соли в приведенных выше примерах получали растворением основного продукта в дихлорметане и введением газообразного HCl при 0oC в течение 2 ч. После завершения реакции гидрохлоридную соль получали путем концентрирования.
III. Биологический анализ
(1) Анализ активности и способ выявления киназы CDK6
В данном эксперименте для выявления применяли способ Lance Ultra от PerkinElmer Co., Ltd. На тестовом планшете смешивали следующие соединения: протеинкиназу, Ulight-меченный полипептидный субстрат, ATP и инкубировали реакционную смесь. Затем для остановки реакции добавляли EDTA, а для выявления добавляли меченное хелатом европия (Eu) антитело. Анализ в данном эксперименте проводили с применением прибора Envision от PerkinElmer Co., Ltd. в режиме TR-FRET. После возбуждения при длине волны, составляющей 320/340 нм, можно было обеспечивать излучение сигнала флуоресценции при длинах волн, составляющих 665 нм и 615 нм. Eu мог быть перенесен на смежный рецептор флуоресцентного вещества ULight путем переноса энергии, а затем выявляли испускаемый свет.
Измеренные значения IC50 показаны в таблице 1 ниже. Из результатов эксперимента можно увидеть, что соединения из примеров по настоящему изобретению обладают сильной ингибирующей активностью в отношении активности киназы CDK6.
Таблица 1. Измеренные значения IC50 в отношении активности киназы CDK6 с помощью соединений по настоящему изобретению
(2) Анализ активности и способ выявления киназы DYRK2
Измеряли ингибирующую активность соединений по настоящему изобретению в отношении киназы DYRK2. Способ был вкратце описан следующим образом (для конкретных способов, см.: Banerjee S, Wei T, Wang J, et al. Inhibition of dual-specificity tyrosine phosphorylation-regulated kinase 2 perturbs 26S proteasome-addicted neoplastic progression[J]. Proceedings of the National Academy of Sciences, 2019, 116(49): 24881-24891):
1) Соединения с различными концентрациями добавляли в 384-луночный планшет и повторно растворяли с последующим добавлением белка DYRK2, субстрата Woodtide (KKISGRLSPIMTEQ) и 33P-γATP и смесь равномерно перемешивали.
2) Смесь инкубировали в течение 30 минут при комнатной температуре;
3) Для прекращения реакции добавляли 0,5 М (3%) раствор ортофосфорной кислоты, и затем смесь переносили на планшет Р81, и промывали с помощью 50 мМ раствора ортофосфорной кислоты.
4) Результаты IC50 рассчитывали с использованием программного обеспечения GraphPad Prism.
Измеренные значения IC50 показаны в таблице 2 ниже. Из результатов эксперимента можно увидеть, что соединения из примеров по настоящему изобретению обладают сильной ингибирующей активностью в отношении активности киназы DYRK2.
Таблица 2. Измеренные значения IC50 в отношении активности киназы DYRK2 с помощью соединений по настоящему изобретению
(3) Определение ингибирования в отношении пролиферации различных раковых клеток.
Ингибирующую активность соединений в отношении пролиферации 14 клеток, включая рак молочной железы человека (MCF-7), клеточную линию трижды негативного рака молочной железы (MDA-MB-231), клеточную линию множественной миеломы (RPMI8226), клеточную линию лейкоза (K562), клеточную линию рака желудка (MGC-803), клеточную линию рака яичников (SK-OV-3), клеточную линию рака толстой кишки (HT-29), клеточную линию рака печени (HepG2), клеточную линию рака поджелудочной железы (Panc-1), клеточную линию глиомы человека (U251), рак легкого (A-549), клеточную линию немелкоклеточного рака легкого (NCI-H1299) и клеточную линию рака предстательной железы (PC-3, Du-145), определяли следующим способом.
Протокол эксперимента:
Ингибирование соединением пролиферации различных раковых клеток определяли в соответствии со способом МТТ и получали IC50, концентрацию полумаксильного ингибирования, соединения в отношении пролиферативной активности клеток.
1) Клетки в логарифмической фазе инокулировали в 96-луночных планшетах в количестве 1×105 клеток/лунка и культивировали при 37oC и 5% CO2 до слияния 90% клеток. После этого клетки инкубировали в течение 2 ч. в бессывороточной среде DMEM, среде RPMI-1640, среде L-15, среде F12K, среде MEM или среде F-12 или среде IMDM для синхронизации клеток (соответствующую среду использовали для каждой клетки).
2) 100 мкл градиентно разбавленного раствора тестируемого соединения с различными концентрациями добавляли в культуральный планшет и культуральный планшет инкубировали в течение 72 часов при 37oC в инкубаторе с 5% CO2.
3) за 4 ч. до окончания инкубации в каждую лунку добавляли 20 мкл раствора МТТ (5 мг/мл). После инкубации супернатант из каждой лунки удаляли и в каждую лунку добавляли 150 мкл DMSO. Раствор осциллировали на клеточном осцилляторе в течение 10 мин. После полного растворения кристаллов измеряли OD570 с помощью считывающего устройства для микропланшетов. Скорость ингибирования = (значение OD контрольной группы - значение OD экспериментальной группы)/значение OD контрольной группы × 100%.
4) После получения данных их аппроксимировали с помощью GraphPad Prism 6 для получения IC50.
Соединение из примера 31 (I-31) и представленное на рынке лекарственное средство, ингибитор CDK4/6, представляющий собой палбоциклиб, тестировали на различные виды пролиферативной активности раковых клеток и измеренные значения IC50 показаны в таблице 3. Можно увидеть, что соединение I-31 проявляет ингибирующую активность в отношении пролиферации 14 клеток, включая рак молочной железы человека (MCF-7), клеточную линию трижды негативного рака молочной железы (MDA-MB-231), клеточную линию множественной миеломы (RPMI8226), клеточную линию лейкоза (K562), клеточную линию рака желудка (MGC-803), клеточную линию рака яичников (SK-OV-3), клеточную линию рака толстой кишки (HT-29), клеточную линию рака печени (HepG2), клеточную линию рака поджелудочной железы (Panc-1), клеточную линию глиомы человека (U251), рак легкого (A-549), клеточную линию немелкоклеточного рака легкого (NCI-H1299) и клеточную линию рака предстательной железы (PC-3, Du-145), а ингибирующая активность в отношении пролиферации 14 клеток была значительно сильнее, чем у представленного на рынке лекарственного средства, ингибитора CDK4/6, представляющего собой палбоциклиб.
Таблица 3. Ингибирующая активность IC50 соединения (I) в отношении пролиферации различных раковых клеток
(4) Определение острой токсичности соединений.
Подопытные животные: Мыши аутбредной популяции ICR; 18-22 г; половина группы самцы и половина группы самки; всего 40 особей.
Схемы дозировок для групп: (1) Контрольная группа: Крысам вводили через желудочный зонд одинаковое количество физиологического раствора однократно для 10 мышей, каждая группа была разделена поровну на самцов и самок; (2) группа 2500 мг/кг: Препарат вводили через желудочный зонд 10 мышам, половине самцов и половине самок, однократно. (3) группа 5000 мг/кг: Препарат вводили через желудочный зонд 10 мышам, половине самцов и половине самок, однократно. (4) 10000 мг/кг группа: Препарат вводили через желудочный зонд 10 мышам, половине самцов и половине самок, однократно.
Таблица 4. Схема дозировок для групп
[Концентрация состава (мг/мл)]
62,5 мг/мл
125 мг/мл
250 мг/мл
Лабораторные условия: комнатная температура 24±2oC, относительная влажность 60~70%. Объекты наблюдения: Тестируемое лекарственное средство (соединение, полученное в примере 31) вводили однократно в соответствии с дозой, указанной в таблице 4, и регистрировали симптомы токсичности и смерть мышей. Мертвых животных подвергали вскрытию. Период наблюдения составлял 14 дней. Результаты показали, что никаких отклонений от нормы не было обнаружено в течение 12 ч. после введения во всех группах. Ни одно животное не погибло в течение 24 ч. после введения дозы и ни одно животное не погибло спустя 14 дней после введения дозы. Других явных отклонений не наблюдалось.
Изменения веса тела показаны на фигуре 1. При внутрижелудочном введении дозировок, составляющих 2500 мг/кг, 5000 мг/кг или 10000 мг/кг, значительных токсических эффектов не наблюдалось по сравнению с контрольной группой.
Как показано на фиг. 2 по результатам окрашивания H&E соединение (I-31), полученное в примере 31, не показало значительной токсичности для сердца, печени, селезенки, легких, почек и других основных органов.
(5) Определение фармакокинетики соединения
Отбирали навеску тестируемого соединения, и помещали в стерильный флакон, и добавляли 250 мкл DMSO, а затем 10 мкл метансульфоновой кислоты. После растворения добавляли 4,78 мл 5% раствора глюкозы для инъекций и равномерно перемешивали с помощью ультразвука и встряхивания для приготовления раствора тестируемого соединения с концентрацией, составляющей 2 мг/мл, который применяли в качестве лекарственного средства для введения через желудочный зонд. Кроме того, 0,5 мл тестируемого раствора с концентрацией, составляющей 2 мг/мл, добавляли к 4,5 мл 5% раствора глюкозы для инъекций и смешивали с помощью встряхивания для приготовления тестируемого раствора с концентрацией, составляющей 0,2 мг/мл, который использовали в качестве препарата для внутривенного введения.
Шесть крыс линии SD были разделены на две группы. Одной группе соединение из примера 31 вводили через хвостовую вену (1 мг/кг), а другой вводили через желудочный зонд (10 мг/кг). Образцы крови объемом приблизительно 0,25 мл отбирали из заднего глазничного венозного сплетения, в группе внутривенных инъекций отбирали через 2 мин., 5 мин., 15 мин., 30 мин., 1 ч., 2 ч., 4 ч., 6 ч., 8 ч. и 12 ч. после введения, а в группе введения через желудочный зонд отбирали через 5 мин., 15 мин., 30 мин., 1 ч., 2 ч., 4 ч., 6 ч., 8 ч., 12 ч. и 24 ч. после введения. Концентрации соединения из примера 31 в образцах плазмы крыс линии SD определяли с помощью LC-MS/MS, а фармакокинетические параметры рассчитывали с применением программного обеспечения WinNolin, и результаты представлены в таблице 5.
Результаты демонстрируют, что соединение (I-31) из примера 31 по настоящему изобретению обладает хорошим метаболизмом у крыс, хорошим всасыванием и степенью воздействия и высокой биологической доступностью.
Таблица 5. Данные фармакокинетических параметров
(ч.*нг/мл)
(6) Определение активности соединения в отношении рака легких.
Лекарственные средства представляли собой соединение (I-31), полученное из примера 31, и представленное на рынке лекарственное средство, ингибитор CDK4/6, представлюящий собой палбоциклиб. Клеточную линию, представляющую собой клеточную линию немелкоклеточного рака легкого человека А-549, культивировали в среде RPMI-1640, содержащей 10% фетальной бычьей сыворотки. Подопытными животными были бестимусные мыши линии BALB/c класса SPF; самцы; по пять в каждой группе. Схемы дозирования лекарственного средства показаны в таблице 6.
Таблица 6. Схемы дозирования лекарственного средства
(мг/кг)
(мл/20 г вес тела)
дочное введение
дочное введение
льного контроля, получаю-
щая лекарственное средство
дочное введение
Способ получения лекарственного средства:
Пример 31 (150 мг/кг): Отбирали навеску порошка тестируемого соединения в количестве 30 мг, растворяли в 2 мл физиологического раствора, готовили в виде лекарственного средства с концентрацией, составляющей 15 мг/мл, и вводили перорально через желудочный зонд в объеме 0,2 мл/20 г.
Пример 31 (300 мг/кг): Отбирали навеску порошка тестируемого соединения в количестве 60 мг, растворяли в 2 мл физиологического раствора, готовили в виде лекарственного средства с концентрацией, составляющей 30 мг/мл, и вводили перорально через желудочный зонд в объеме 0,2 мл/20 г.
Палбоциклиб (150 мг/кг): Отбирали навеску порошка тестируемого соединения в количестве 30 мг, растворяли в 2 мл физиологического раствора и готовили в виде лекарственного средства с концентрацией, составляющей 15 мг/мл, для перорального введения через желудочный зонд в объеме 0,2 мл/20 г.
Экспериментальный способ: Модель ксенотрансплантатов рака легкого человека у бестимусных мышей была создана путем инокуляции клеточной линии рака легких человека A549 в подмышечной области бестимусных мышей. Клетки A549 в логарифмической фазе инокулировали подкожно в правой подмышечной области 30 бестимусных мышей в стерильных условиях и количество клеток для инокуляции составляло 5×106 клеток/мышь. Диаметр ксенотрансплантатов измеряли штангенциркулем. Когда опухоль выросла приблизительно до объема, составляющего 80 мм3, отобрали 20 бестимусных мышей с опухолью в хороших условиях для роста и с одинаковым размером опухоли и рандомно разделили на четыре группы, по пять в каждой группе, т.е. модельная группа, группа с низкой дозой соединения из примера 31 (150 мг/кг), группа с высокой дозой соединения из примера 31 (300 мг/кг) и группа положительного контроля, получающая лекарственное средство, представляющее собой палбоциклиб (150 мг/кг). Тестируемое лекарственное средство из примера 31 и палбоциклиб вводили внутрижелудочно группам с низкой и высокой дозой и группе положительного контроля, получающей лекарственное средство, однократно каждые 2 дня. Модельной группе внутрижелудочно вводили равный объем контрольной среды-носителя. Противоопухолевый эффект тестируемого вещества динамически наблюдали путем измерения диаметра опухоли. Диаметры опухолей измеряли через день и бестимусных мышей взвешивали при измерении диаметров опухолей. Мышей умерщвляли в день 22, а кусочки опухоли, удаленные хирургическим путем, фиксировали 10% формальдегидом и хранили в жидком азоте для дальнейшего использования.
Результаты эксперимента показали, что по сравнению с модельной группой относительные скорости пролиферации опухоли T/C(%) в группе с низкой дозой соединения из примера 31 (150 мг/кг) и в группе с высокой дозой соединения из примера 31 (300 мг/кг) составили 44,8% и 35,9% соответственно, а показатели ингибирования роста опухоли составили 55,2% и 64,1% соответственно. Когда группе положительного контроля, получающей лекарственное средство, представляющее собой палбоциклиб, вводили через желудочный зонд в дозе, составляющей 150 мг/кг, относительная скорость пролиферации опухоли T/C(%) и скорость ингибирования опухоли составляли 39,6% и 60,4% соответственно.
Следовательно, тестируемое лекарственное средство, полученное из примера 31, обладало значительным ингибирующим эффектом на рост ксенотрансплантатов рака легкого человека А549 у бестимусных мышей, и этот эффект был лучше, чем у группы положительного контроля, получающей лекарственное средство, представляющее собой палбоциклиб.
(7) Определение активности соединения в отношении рака предстательной железы (PC3).
Лекарственные средства представляли собой соединение (I-31), полученное из примера 31, и представленное на рынке лекарственное средство, ингибитор CDK4/6, представлюящий собой палбоциклиб. Штамм клеток представлял собой клетку рака РС-3 предстательной железы человека. Подопытными животными были бестимусные мыши линии BALB/c класса SPF; самцы; по восемь в каждой группе. Схемы дозирования лекарственного средства показаны в таблице 7.
Таблица 7. Схемы дозирования лекарственного средства
(мг/кг)
(мл/20 г вес тела)
Способ получения лекарственного средства:
Пример 31 (100 мг/кг): Отбирали навеску порошка тестируемого соединения в количестве 20 мг, растворяли в 2 мл физиологического раствора, готовили в виде лекарственного средства с концентрацией, составляющей 10 мг/мл, и вводили перорально через желудочный зонд в объеме 0,2 мл/20 г.
Пример 31 (200 мг/кг): Отбирали навеску порошка тестируемого соединения в количестве 40 мг, растворяли в 2 мл физиологического раствора, готовили в виде лекарственного средства с концентрацией, составляющей 20 мг/мл, и вводили перорально через желудочный зонд в объеме 0,2 мл/20 г.
Палбоциклиб (100 мг/кг): Отбирали навеску порошка тестируемого соединения в количестве 20 мг, растворяли в 2 мл физиологического раствора и готовили в виде лекарственного средства с концентрацией, составляющей 10 мг/мл, для перорального введения через желудочный зонд в объеме 0,2 мл/20 г.
Экспериментальный способ: Модель ксенотрансплантатов рака предстательной железы человека у бестимусных мышей была создана путем инокуляции рака предстательной железы человека PC-3 в подмышечной области бестимусных мышей. Клетки PC-3 в логарифмической фазе инокулировали подкожно в правой подмышечной области 40 бестимусных мышей в стерильных условиях и количество клеток для инокуляции составляло 5×106 клеток/мышь. Диаметр ксенотрансплантатов измеряли штангенциркулем. Когда опухоль выросла приблизительно до объема, составляющего 90 мм3, отобрали 32 бестимусных мыши с опухолью в хороших условиях для роста и с одинаковым размером опухоли и рандомно разделили на четыре группы, по восемь в каждой группе, т.е. модельная группа, группа с низкой дозой соединения из примера 31 (100 мг/кг), группа с высокой дозой соединения из примера 31 (200 мг/кг) и группа положительного контроля, получающая лекарственное средство, представляющее собой палбоциклиб (100 мг/кг). Тестируемое лекарственное средство из примера 31 и палбоциклиб вводили внутрижелудочно группам с низкой и высокой дозой и группе положительного контроля, получающей лекарственное средство, однократно каждый день. Модельной группе внутрижелудочно вводили равный объем контрольной среды-носителя. Противоопухолевый эффект тестируемого вещества динамически наблюдали путем измерения диаметра опухоли. Диаметры опухолей измеряли через день и бестимусных мышей взвешивали при измерении диаметров опухолей. Мышей умерщвляли в день 29, а кусочки опухоли, удаленные хирургическим путем, фиксировали 10% формальдегидом и хранили в жидком азоте для дальнейшего использования.
Результаты эксперимента показали, что по сравнению с модельной группой относительные скорости пролиферации опухоли T/C(%) в группе с низкой дозой соединения из примера 31 (100 мг/кг) и в группе с высокой дозой соединения из примера 31 (200 мг/кг) составили 35,7% и 23,4% соответственно, а показатели ингибирования роста опухоли составили 64,3% и 76,6% соответственно. Когда группе положительного контроля, получающей лекарственное средство, представляющее собой палбоциклиб, вводили через желудочный зонд в дозе, составляющей 100 мг/кг, относительная скорость пролиферации опухоли T/C(%) и скорость ингибирования опухоли составляли 35,5% и 64,5% соответственно.
Следовательно, тестируемое лекарственное средство, полученное из примера 31, обладало значительным ингибирующим эффектом на рост ксенотрансплантатов рака предстательной железы человека PC3 у бестимусных мышей, и этот эффект был лучше, чем у группы положительного контроля, получающей лекарственное средство, представляющее собой палбоциклиб.
(8) Определение активности соединения в отношении рака предстательной железы (Du-145)
Лекарственные средства представляли собой соединение (I-31), полученное из примера 31, и представленное на рынке лекарственное средство, ингибитор CDK4/6, представлюящий собой палбоциклиб, и лекарственное средство первой линии для лечения рака предстательной железы, представляющее собой энзалутамид. Штамм клеток представляет собой клетки рака предстательной железы человека Du-145. Подопытными животными были бестимусные мыши линии BALB/c класса SPF; самцы; по десять в каждой группе. Схемы дозирования лекарственного средства показаны в таблице 8.
Таблица 8. Схемы дозирования лекарственного средства
(мг/кг)
(мл/20 г вес тела)
ский раствор
дочное введение
дочное введение
дочное введение
дочное введение
дочное введение
Способ получения лекарственного средства:
Пример 31 (100 мг/кг): Отбирали навеску порошка тестируемого соединения в количестве 20 мг, растворяли в 2 мл физиологического раствора, готовили в виде лекарственного средства с концентрацией, составляющей 10 мг/мл, и вводили перорально через желудочный зонд в объеме 0,2 мл/20 г.
Пример 31 (200 мг/кг): Отбирали навеску порошка тестируемого соединения в количестве 40 мг, растворяли в 2 мл физиологического раствора, готовили в виде лекарственного средства с концентрацией, составляющей 20 мг/мл, и вводили перорально через желудочный зонд в объеме 0,2 мл/20 г.
Энзалутамид (100 мг/кг): Отбирали навеску порошка тестируемого соединения в количестве 20 мг, растворяли в 2 мл физиологического раствора и готовили в виде лекарственного средства с концентрацией, составляющей 10 мг/мл, для перорального введения через желудочный зонд в объеме 0,2 мл/20 г.
Экспериментальный способ: Модель ксенотрансплантатов рака предстательной железы человека у бестимусных мышей была создана путем инокуляции рака предстательной железы человека Du-145 в подмышечной области бестимусных мышей. Клетки Du-145 в логарифмической фазе инокулировали подкожно в правой подмышечной области 60 бестимусных мышей в стерильных условиях и количество клеток для инокуляции составляло 5×106 клеток/мышь. Диаметр ксенотрансплантатов измеряли штангенциркулем. Когда опухоль выросла приблизительно до объема, составляющего 90 мм3, отобрали 50 бестимусных мышей с опухолью в хороших условиях для роста и с одинаковым размером опухоли и рандомно разделили на пять групп, по десять в каждой группе, т.е. модельная группа, группа с низкой дозой соединения из примера 31 (100 мг/кг), группа с высокой дозой соединения из примера 31 (200 мг/кг), группа положительного контроля, получающая лекарственное средство, представляющее собой палбоциклиб (100 мг/кг), и группа положительного контроля, получающая лекарственное средство, представляющее собой энзалутамид (100 мг/кг). Тестируемое лекарственное средство из примера 31, палбоциклиб и энзалутамид вводили внутрижелудочно группам с низкой и высокой дозой и группе положительного контроля, получающей лекарственное средство, однократно каждый день. Модельной группе внутрижелудочно вводили равный объем контрольной среды-носителя. Противоопухолевый эффект тестируемого вещества динамически наблюдали путем измерения диаметра опухоли. Диаметры опухолей измеряли через день и бестимусных мышей взвешивали при измерении диаметров опухолей. Мышей умерщвляли в день 35, а кусочки опухоли, удаленные хирургическим путем, фиксировали 10% формальдегидом и хранили в жидком азоте для дальнейшего использования. Оставшихся мышей умерщвляли в день 49, а кусочки опухоли, удаленные хирургическим путем, фиксировали 10% формальдегидом и хранили в жидком азоте для дальнейшего использования.
Результаты эксперимента показаны на фиг. 3: группа с низкой дозой тестируемого лекарственного средства из примера 31 (100 мг/кг) демонстрировала лучшее ингибирование роста опухоли, чем группа положительного контроля, получающая лекарственное средство, представляющее собой палбоциклиб (100 мг/кг); группа с низкой дозой тестируемого лекарственного средства из примера 31 (100 мг/кг) и группа положительного контроля, получающая лекарственное средство, представляющее собой энзалутамид (100 мг/кг) оказывали сходное ингибирующее действие на рост опухоли. Пример 31 (200 мг/кг): группа с высокой дозой ингибировала рост опухоли значительно лучше, чем группа положительного контроля, получающая лекарственное средство, представляющее собой палбоциклиб (100 мг/кг), и группа положительного контроля, получающая лекарственное средство, представляющее собой энзалутамид (100 мг/кг), и начинала уменьшать объем опухоли в день 31.
Следовательно, тестируемое лекарственное средство, полученное из примера 31 обладало значительным ингибирующим эффектом на рост ксенотрансплантатов рака предстательной железы человека Du-145 у бестимусных мышей, и этот эффект был лучше, чем у группы положительного контроля, получающей ингибитор CDK4/6, представляющий собой палбоциклиб, и лекарственное средство первой линии для лечения рака предстательной железы, представляющее собой энзалутамид.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТА АДЕНОЗИНОВОГО РЕЦЕПТОРА A2a И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2022 |
|
RU2840068C2 |
| ПИПЕРИДИН- И ПИПЕРАЗИНЗАМЕЩЕННЫЕ N-ГИДРОКСИФОРМАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ МЕТАЛЛОПРОТЕИНАЗ | 2001 |
|
RU2283306C2 |
| ИНГИБИТОРЫ МУТАЦИИ HER2 | 2022 |
|
RU2834124C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗО[4,5-С]ПИРИДИНА В КАЧЕСТВЕ АГОНИСТОВ TOLL-ПОДОБНОГО РЕЦЕПТОРА | 2020 |
|
RU2785124C1 |
| ТРИФТОРМЕТИЛЗАМЕЩЕННЫЙ СУЛЬФОНАМИД В КАЧЕСТВЕ СЕЛЕКТИВНОГО ИНГИБИТОРА BCL-2 | 2019 |
|
RU2815486C2 |
| КЛАСС БИФУНКЦИОНАЛЬНЫХ ХИМЕРНЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ДЛЯ НАПРАВЛЕННОГО РАЗРУШЕНИЯ АНДРОГЕННЫХ РЕЦЕПТОРОВ И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2825000C2 |
| ФЕНИЛ/ПИРИДИЛ-N-ФЕНИЛ/ПИРИДИЛЬНЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ РНК-ВИРУСНОЙ ИНФЕКЦИИ | 2019 |
|
RU2803216C2 |
| КОНДЕНСИРОВАННЫЕ ГЕТЕРОАРИЛЫ И ИХ ПРИМЕНЕНИЕ | 2011 |
|
RU2552114C2 |
| ПИРРОЛО[2,1-f][1,2,4]ТРИАЗИНОВОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2013 |
|
RU2589053C1 |
| СОЕДИНЕНИЕ ДЛЯ РАЗРУШЕНИЯ АНДРОГЕНОВОГО РЕЦЕПТОРА И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2021 |
|
RU2817356C1 |
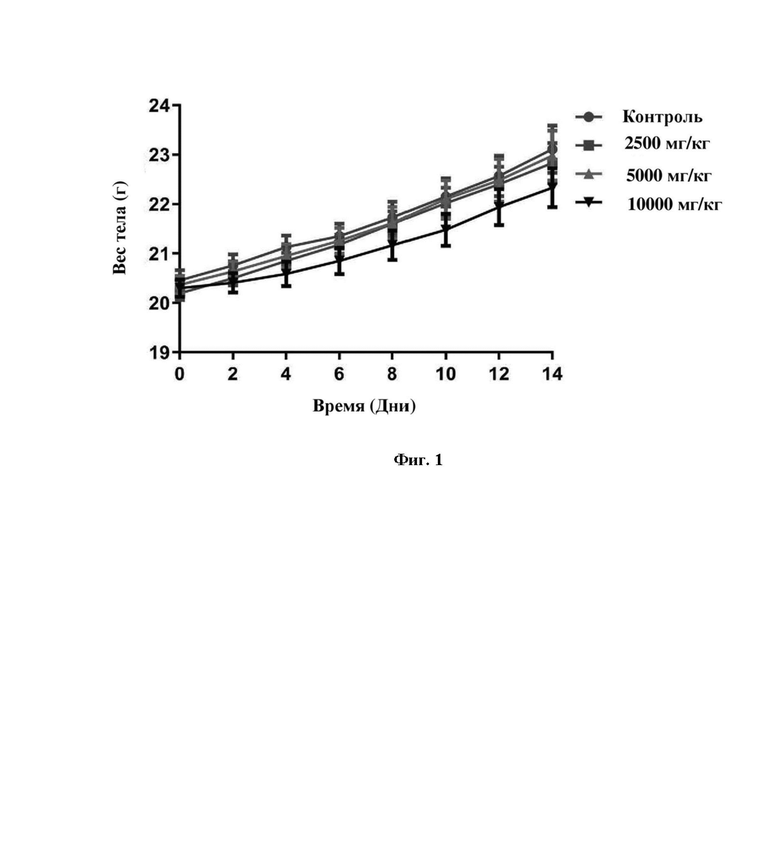

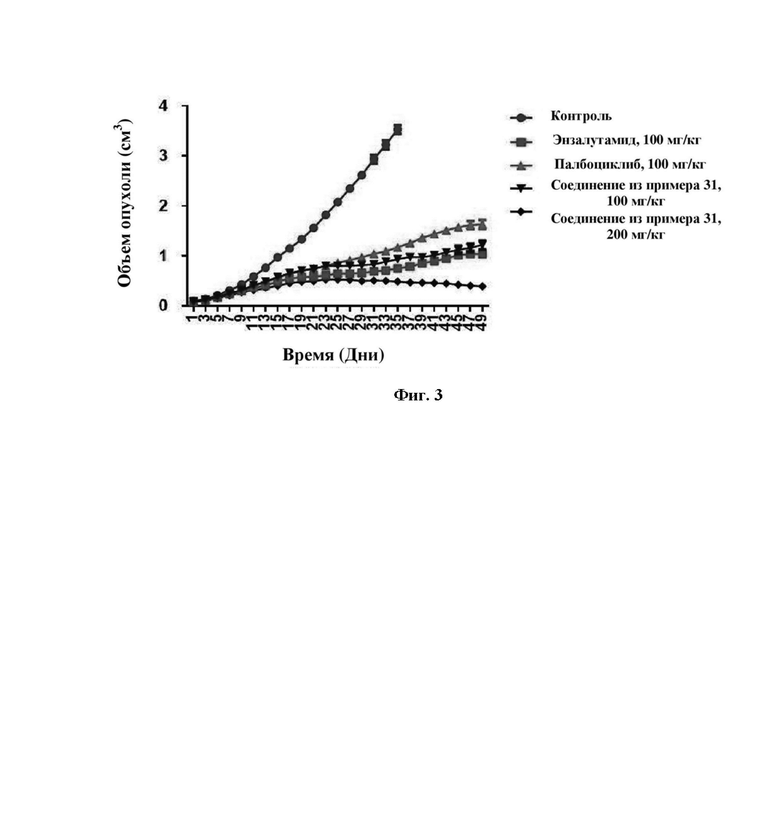
Настоящее изобретение относится к области фармацевтической химии, а именно к соединению, представленному общей формулой (I), или его фармацевтически приемлемой соли, где X представляет собой C(O) или (CH2)n; n равняется 0 или 1; R1 представляет собой водород, C1-C8алкильную группу или -NR4R5, где R4, R5 выбраны из водорода, C1-C8алкильной группы или C3-C8циклоалкильной группы; R2 представляет собой F; R3 представляет собой водород или C1-C8алкильную группу. Изобретение также относится к способу получения указанного соединения, а также фармацевтической композиции и применению на его основе. Технический результат заключается в получении альтернативных соединений, которые являются ингибиторами двойного целенаправленного действия на CDK6/DYRK2, которые могут найти применение в медицине для лечения рака, в частности рака молочной железы, рака предстательной железы, рака легких, множественной миеломы, лейкоза, рака желудка, рака яичников, рака толстой кишки, рака печени, рака поджелудочной железы и глиомы человека. 4 н. и 6 з.п. ф-лы, 3 ил., 8 табл., 31 пр.

1. Соединение, представленное общей формулой (I), или его фармацевтически приемлемая соль,
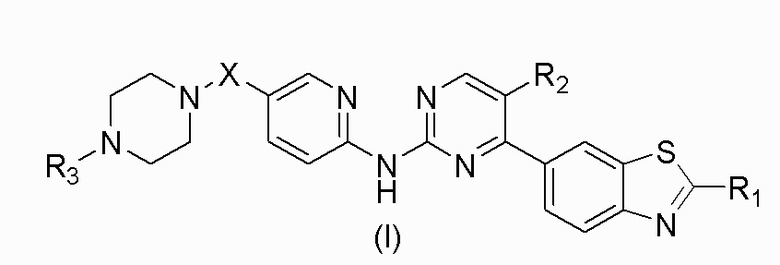 ,
,
где X представляет собой C(O) или (CH2)n; n равняется 0 или 1;
R1 представляет собой водород, C1-C8алкильную группу или -NR4R5, где R4, R5 выбраны из водорода, C1-C8алкильной группы или C3-C8циклоалкильной группы;
R2 представляет собой F;
R3 представляет собой водород или C1-C8алкильную группу.
2. Соединение или его фармацевтически приемлемая соль по п. 1, где
указанный X представляет собой C(O) или (CH2)n; n равняется 0 или 1;
указанный R1 выбран из водорода, C1-C3алкильной группы или -NR4R5, где R4, R5 выбраны из водорода, C1-C3алкильной группы, циклопентана или циклогексана;
R2 представляет собой F;
R3 представляет собой водород или C1-C4алкильную группу.
3. Соединение или его фармацевтически приемлемая соль по п. 1, где
указанный X представляет собой C(O) или (CH2)n; n равняется 0 или 1;
указанный R1 выбран из водорода, C1-C3алкильной группы или -NR4R5, где R4, R5 выбраны из водорода, метильной группы, этильной группы, циклопентана или циклогексана;
R2 представляет собой F;
R3 представляет собой водород или C1-C4алкильную группу.
4. Соединение или его фармацевтически приемлемая соль по п. 1, где
указанный X выбран из (CH2)n или C(O), n равняется 0 или 1;
указанный R1 выбран из водорода, метильной группы или -NR4R5, где R4 выбран из группы, состоящей из водорода, метильной группы, этильной группы, указанный R5 выбран из водорода, метильной группы, этильной группы или циклопентана;
указанный R2 представляет собой F;
указанный R3 выбран из водорода, этильной группы, изопропильной группы.
5. Соединение или его фармацевтически приемлемая соль по п. 1, где указанное соединение представляет собой любое соединение, выбранное из следующих соединений:
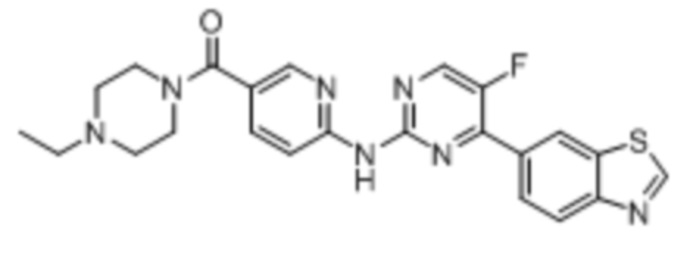
I-1,
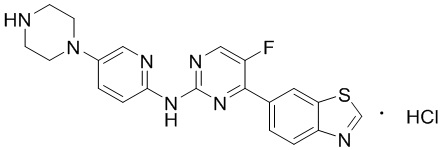
I-8,
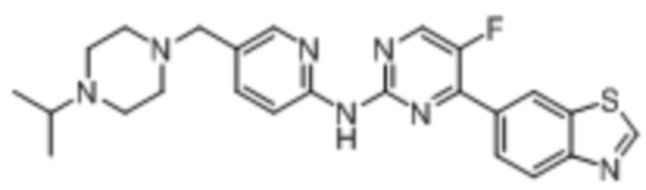
I-11,
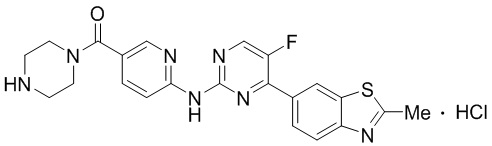 I-17,
I-17,
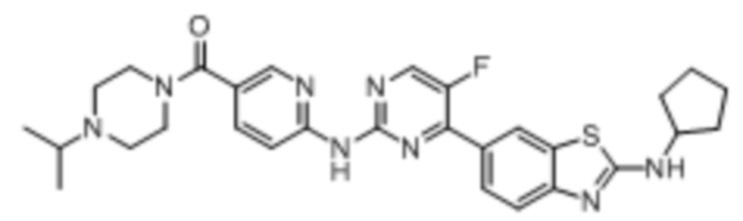
I-29,
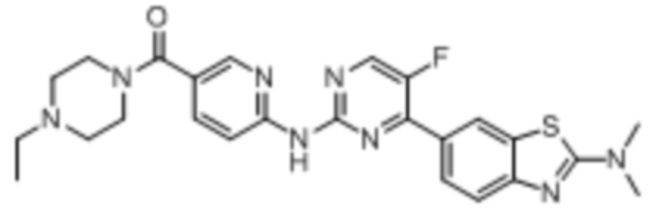
I-31,
I-39,
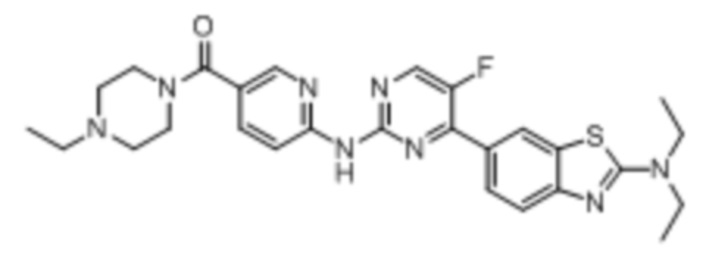
I-42,
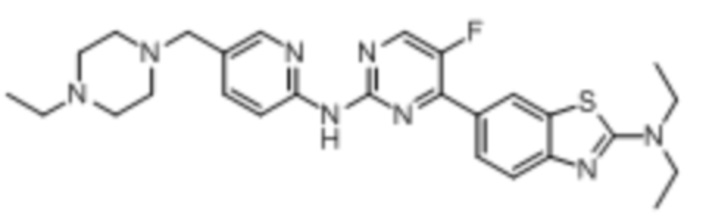
I-43,
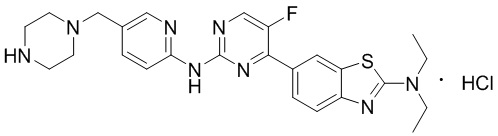
I-45,
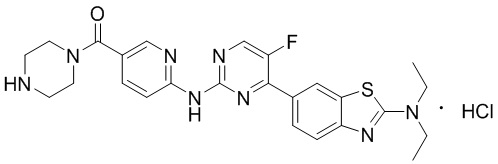
I-47,
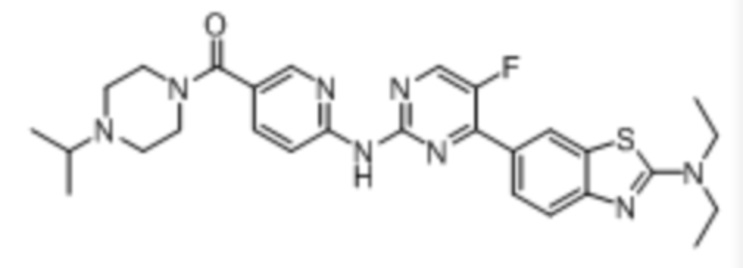
I-51.
6. Соединение или его фармацевтически приемлемая соль по п. 1, где указанное соединение представляет собой любое соединение, выбранное из следующих соединений:
I-31,
I-42,
I-51.
7. Соединение или его фармацевтически приемлемая соль по п. 1, где указанная фармацевтически приемлемая соль представляет собой соль присоединения кислоты соединения общей формулы (I), причем солеобразующая кислота предусматривает неорганическую кислоту и органическую кислоту, при этом указанная неорганическая кислота предусматривает хлористоводородную кислоту, серную кислоту, фосфорную кислоту и метансульфоновую кислоту, а органическая кислота предусматривает уксусную кислоту, трихлоруксусную кислоту, пропионовую кислоту, масляную кислоту, малеиновую кислоту, п-толуолсульфоновую кислоту, яблочную кислоту, малоновую кислоту, коричную кислоту, лимонную кислоту, фумаровую кислоту, камфорную кислоту, диглюконовую кислоту, аспарагиновую кислоту и винную кислоту.
8. Способ получения соединения общей формулы (I) по п. 1, где соединение (I) получают из соединения (А) и соединения (В) посредством реакции сочетания под действием палладиевого катализатора:
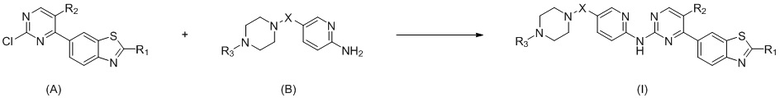
где X представляет собой C(O) или (CH2)n; n равняется 0 или 1;
R1 представляет собой водород, C1-C8алкильную группу, -NR4R5, где R4, R5 выбраны из водорода, C1-C8алкильной группы или C3-C8циклоалкильной группы;
R2 представляет собой F;
R3 представляет собой водород, C1-C8алкильную группу.
9. Фармацевтическая композиция для ингибирования CDK6/DYRK2, где композиция содержит соединение общей формулы (I) или его фармацевтически приемлемую соль по п. 1 и фармацевтически приемлемый носитель.
10. Применение соединения или его фармацевтически приемлемой соли по п. 1 в изготовлении лекарственного препарата, предназначенного для лечения и/или предупреждения рака или связанного с опухолью заболевания, где указанный рак или связанное с опухолью заболевание предусматривает рак молочной железы, рак предстательной железы, рак легких, множественную миелому, лейкоз, рак желудка, рак яичников, рак толстой кишки, рак печени, рак поджелудочной железы и глиому человека.
| CN 102264725 A, 30.11.2011 | |||
| CN 107266421 A, 20.10.2017 | |||
| CN 105294683 B, 23.01.2018 | |||
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИНДОЛ-5-ОЛА И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2013 |
|
RU2674249C2 |
| WO 2017062688 A1, 13.04.2017. | |||

