Область техники, к которой относится изобретение
Настоящее изобретение относится к секвенированию следующего поколения и идентификации олигонуклеотидов в способах мультиплексирования.
Уровень техники
Индексные последовательности, также называемые штрихкодами, обычно используются как короткие последовательности нуклеотидов, которые добавляются к фрагментам в библиотеке таким образом, чтобы фрагменты из одного образца ассоциировались с уникальным непустым набором штрихкодов. Это позволяет смешивать и секвенировать многочисленные образцы вместе, снижая затраты на секвенирование и увеличивая производительность (параллельное секвенирование или мультиплексирование). Эта процедура представлена на фиг. 1. В левой части фиг. 1 показаны три образца (эллипсоиды), каждый из которых содержит набор фрагментов (фигурные линии). Во время мультиплексирования штрихкоды BC1, BC2 и BC3 добавляются к фрагментам в образце один, два и три, соответственно, и фрагменты штрихкода смешиваются вместе. Таким образом, на фиг. 1 непустой набор штрихкодов, ассоциированных с образцом, состоит из одного штрихкода. Это наиболее распространенная ситуация при мультиплексировании, в результате которой между штрихкодами и образцами возникает взаимно однозначное соотношение. После секвенирования мультиплексированной библиотеки исследуется последовательность штрихкода каждого фрагмента. Если последовательность соответствует нуклеотидной последовательности штрихкода BC1, BC2 или BC3, нештрихкодовая последовательность фрагмента относится к образцу один, два и три, соответственно. Этот процесс присвоения последовательностей фрагментов образцам в соответствии с их ассоциированной последовательностью штрихкода называется демультиплексированием.
Синтез штрихкода, подготовка библиотеки и секвенирование могут внести ошибки в последовательность штрихкода, и поэтому демультиплексирование может привести к неправильному присвоению фрагментов образцам. Во избежание загрязнения образцов последовательностями фрагментов из других образцов, штрихкоды обычно разрабатываются таким образом, чтобы свести к минимуму вероятность преобразования друг в друга. Этого можно добиться, максимизируя количество изменений, необходимых для преобразования одного штрихкода в другой, или, другими словами, максимизируя расстояние между штрихкодами. Так как достижимое расстояние между штрихкодами увеличивается с уменьшением количества образцов, набор штрихкодов для эксперимента должен быть оптимизирован в зависимости от количества образцов в эксперименте. Расстояние между штрихкодами можно дополнительно увеличить за счет увеличения длины штрихкода. Однако это происходит за счет уменьшения длины секвенируемого фрагмента, так как ограничено общее количество секвенированных нуклеотидов для штрихкода и фрагмента. Следовательно, длина штрихкода для эксперимента должна быть выбрана таким образом, чтобы требуемый уровень перекрестного загрязнения был достигнут без ненужной потери длины последовательности фрагментов.
Если расстояние между штрихкодами является достаточно большим, незначительные ошибки все же могут привести к присвоению, которое, вероятно, является правильным. Они называются штрихкодами с исправлением ошибок и обычно используют способ оценки расстояния, который очень напоминает количество изменений нуклеотидов, которые могут произойти в физическом образце (смотри, например, Buschmann et al. [1], Hawkins et al. [3], WO 2016/018960 А1). Другие подходы, которые решают другие проблемы, которые могут помешать правильному присвоению, такие как скачкообразный переход индекса, представляют собой использование двойных индексов (смотри MacConaill [5] и WO 2018/136248 A1).
В документе WO 2018/204423 A1 раскрыт цветовой баланс индексных последовательностей путем спаривания A и C с G и T (или U).
В документе WO 2011/100617 A1 раскрыты индексные последовательности, которые не имеют 4 или более смежных идентичных подблоков.
Сущность изобретения
Несмотря на различные попытки улучшить штрихкоды, остается потребность в создании олигонуклеотидов с улучшенной индексной последовательностью, которые имеют оптимальную различимость, позволяющую выполнять присвоение даже в случае ошибок. Эти штрихкоды должны максимизировать эту различимость для имеющегося образца, используемого практикующим специалистом, но при этом допускать компромисс с эффективностью, учитывая повышенные усилия и затраты на каждый нуклеотид, который необходимо секвенировать.
В настоящем изобретении предложен набор олигонуклеотидов, содержащих индексные последовательности, причем набор содержит множество поднаборов олигонуклеотидов с разными индексными последовательностями,
где индексные последовательности поднабора олигонуклеотидов отличаются друг от друга по меньшей мере ненулевым количеством изменений последовательностей;
и где набор содержит по меньшей мере 2 иерархических уровня поднаборов,
где индексные последовательности поднабора более высокого уровня являются элементами поднабора более низкого уровня, и где индексные последовательности поднабора более низкого уровня отличаются друг от друга меньшим минимальным количеством изменений последовательностей по сравнению с индексными последовательностями поднабора более высокого уровня; и где олигонуклеотиды присваиваются одному или нескольким поднаборам.
Настоящее изобретение дополнительно предоставляет способ выработки набора олигонуклеотидов, содержащего множество поднаборов олигонуклеотидов с поднабором индексных последовательностей, содержащий этапы выработки первого поднабора олигонуклеотидов с индексными последовательностями с первым секвенциальным расстоянием друг от друга в пределах первого поднабора, где секвенциальное расстояние представляет собой количественную величину изменений последовательностей, которая преобразует одну последовательность в другую, или монотонно убывающую функцию вероятности изменений последовательностей, которая преобразует одну последовательность в другую, вырабатывая второй поднабор путем включения первого поднабора и добавления дополнительных олигонуклеотидов с индексными последовательностями со вторым секвенциальным расстоянием друг от друга в пределах второго поднабора, где второе секвенциальное расстояние является меньшим секвенциальным расстоянием, чем первое секвенциальное расстояние.
Настоящее изобретение дополнительно предоставляет способ присвоения секвенирующих прочтений образцу олигонуклеотидов, содержащий этапы
а) получение образцов олигонуклеотидов из множества образцов,
b) выбор поднабора олигонуклеотидных индексных последовательностей из набора согласно изобретению, где поднабор выбирается вместо другого поднабора на основе большего секвенциального расстояния между индексными последовательностями друг от друга в пределах выбранного поднабора; где секвенциальное расстояние представляет собой количественную величину изменений последовательностей, которая преобразует одну последовательность в другую, или монотонно убывающую функцию вероятности изменений последовательностей, которая преобразует одну последовательность в другую, и где выбранный поднабор имеет по меньшей мере такое же количество различных индексных последовательностей, как и количество образцов на этапе а), с) добавление индексных последовательностей из упомянутого поднабора в каждый олигонуклеотид образца, где индексные последовательности указывают на образец, d) определение последовательности образцовых олигонуклеотидов или фрагментов образцовых олигонуклеотидов и определение индексной последовательности, e) присвоение последовательности с полученным прочтением образцу на основе определенной индексной последовательности или на основе индексной последовательности, которая имеет наименьшее секвенциальное расстояние до определенной индексной последовательности, где, если две или более индексных последовательностей имеют одинаковое наименьшее расстояние, то упомянутое полученное прочтение отбрасывается; где при необходимости секвенциальное расстояние не превышает заданного значения согласно критерию.
Последующее подробное описание и предпочтительные варианты осуществления применимы ко всем аспектам изобретения и могут комбинироваться друг с другом без ограничений, за исключением случаев, когда это явно указано. Например, набор согласно изобретению может быть получен способом выработки; набор может быть подходящим для способа присвоения секвенирующих прочтений. Предпочтительные варианты осуществления и аспекты определены в формуле изобретения.
Краткое описание чертежей
Фиг. 1 - мультиплексирование, секвенирование и демультиплексирование. Фрагменты (фигурные линии) в трех образцах (эллипсоиды) отмечены индексными последовательностями BC1, BC2 и BC3.
Фиг. 2 - вложенные наборы штрихкодов. Меньшие наборы индексных последовательностей (поднаборы более высокого уровня) содержатся в более крупных наборах индексных последовательностей (поднаборах более низкого уровня). Увеличение размера набора штрихкодов уменьшает расстояние между штрихкодами.
Фиг. 3 - вложенные последовательности штрихкодов. Расширение индексных последовательностей увеличивает расстояние между штрихкодами и сохраняет вложенную структуру наборов индексных последовательностей.
Фиг. 4 - схема алгоритма динамического программирования для вычисления расстояний Левенштейна.
Фиг. 5 - схема вычисления обратной вероятности.
Фиг. 6 - распределение B1, B2, B3, B4 на 8x12-луночном планшете.
Фиг. 7 - схема последовательности считывания и индексирования для двойной индексации (i7/i5).
Фиг. 8 - позиционное распределение нуклеотидов для B1C |B1| = 4.
Фиг. 9 - позиционное распределение нуклеотидов для B2C |B2| = 8.
Фиг. 10 - позиционное распределение нуклеотидов для B3C |B3| = 16.
Фиг. 11 - позиционное распределение нуклеотидов для B4C |B4| = 24.
Фиг. 12 - матрица подсчета для эксперимента с двойным индексом, в ходе которого измеряется перекрестное загрязнение, зависящее от поставщика синтеза.
Подробное описание изобретения
Используемый в данном документе термин «штрихкод» относится к «индексной последовательности», которая представляет собой последовательность нуклеотидов, способную и используемую для идентификации последовательностей (обычно в олигонуклеотидах или их секвенирующих прочтениях), которые помечены этими индексными последовательностями. В наборах и поднаборах согласно изобретению эти индексные последовательности включены в олигонуклеотиды, и, таким образом, олигонуклеотиды имеют нуклеотидную последовательность упомянутой индексной последовательности. Олигонуклеотиды могут содержать дополнительные нуклеотиды или не содержать их. Обычно олигонуклеотиды используются для мечения других нуклеиновых кислот образца путем присоединения, и, таким образом, полученный олигонуклеотид имеет больше нуклеиновых кислот. Кроме того, можно также метить другие фрагменты, такие как белки, такие как антитела или ферменты, или гранулы или частицы, такие как наночастицы, или клетки или химические соединения, такие как лекарства, путем присоединения к ним.
Настоящее изобретение предоставляет набор олигонуклеотидов, содержащих индексные последовательности, причем набор содержит множество поднаборов олигонуклеотидов с разными индексными последовательностями. Индексные последовательности поднабора олигонуклеотидов отличаются друг от друга по меньшей мере ненулевым количеством изменений последовательностей. Такие изменения последовательности можно оценивать секвенциальным расстоянием, как будет более подробно описано ниже. Используя терминологию секвенциального расстояния, можно также утверждать, что секвенциальное расстояние индексных последовательностей не равно нулю. Это расстояние может быть равным 1 или более в расстояниях, которые указаны как целые числа или ненулевая дробь или функция дроби (например, вероятности изменения последовательности). Набор содержит по меньшей мере 2 (то есть 2 или более) иерархических уровня поднаборов, где индексные последовательности поднабора более высокого уровня являются элементами поднабора более низкого уровня. Это означает, что набор содержит первый поднабор и по меньшей мере один дополнительный (второй или более) поднабор, который содержит элементы первого поднабора. Первый поднабор считается поднабором более высокого уровня, и когда термин «первый» представляет собой первый из всех поднаборов, даже самый высокий поднабор. Это означает, что поднаборы более низкого уровня содержат больше элементов (индексов последовательностей), чем поднаборы более высокого уровня. При включении большего количества элементов расстояние между всеми этими элементами (минимальное расстояние или наименьшее расстояние) уменьшается, если длина индексной последовательности остается неизменной. Соответственно, в наборе согласно изобретению индексные последовательности поднабора более низкого уровня отличаются друг от друга меньшим минимальным количеством изменений последовательностей по сравнению с индексными последовательностями поднабора более высокого уровня.
Термин «минимальное количество изменений последовательностей» относится к наименьшему количеству изменений последовательностей, которое присутствует для всех возможных изменений последовательностей между любыми двумя элементами поднабора.
Олигонуклеотиды набора присваиваются одному или нескольким поднаборам. Это означает, что пользователь знает, к какому поднабору принадлежит каждая индексная последовательность (или олигонуклеотид). Такое присвоение можно осуществить физически, например, путем помещения олигонуклеотидов в контейнеры, которые помечены или упорядочены в соответствии с присвоением поднабора.
Структура поднабора согласно изобретению также называется «вложенными наборами», так как один поднабор вложен в другой поднабор (или является его элементом). Например, индексные последовательности первого поднабора могут содержаться в упомянутом первом поднаборе, а также во втором поднаборе, к которому также принадлежат дополнительные индексные последовательности, не найденные в первом поднаборе.
Эта вложенная иерархия поднаборов позволяет предоставлять несколько поднаборов индексных последовательностей, которые имеют разные размеры. Под «размером поднабора» понимается количество различных индексных последовательностей в упомянутом поднаборе. Эти поднаборы различных размеров позволяют избежать множественных наборов физических штрихкодов для различных их применений в зависимости от потребности в разных размерах. Практикующий специалист, который использует вложенные наборы согласно изобретению, может выбирать из ряда поднаборов, чтобы соответствовать требованиям практикующего специалиста к размеру, например количеству образцов, которые должны быть индивидуально помечены индексными последовательностями. Выбирая поднабор более высокого уровня - насколько это возможно в зависимости от требований к размеру из-за количества образцов - практикующий специалист может оптимизировать расстояние между индексными последовательностями и, таким образом, повысить качество присвоения помеченных объектов, таких как прочтения или фрагменты последовательности, образцу.
Качество присвоения по существу означает конфиденциальность присвоения и возможность присвоить определенную индексную последовательность образцу, даже если эта определенная последовательность не идентична индексным последовательностям упомянутого образца, например, путем присвоения упомянутой расходящейся определенной последовательности образцу, если он имеет наименьшее расстояние до правильной индексной последовательности этого образца (исправление ошибок) по сравнению с другими индексными последовательностями других образцов. Этот тип исправления ошибок известен в технике - смотри ссылку [1]. Термин «индексная последовательность этого образца» означает безошибочную индексную последовательность, которая была присвоена образцу практикующим специалистом, например, путем связывания олигонуклеотидов с индексной последовательностью с образцовыми нуклеиновыми кислотами. Таким образом, качество присвоения является свойством для оценки неправильного присвоения и перекрестного загрязнения.
Еще одним аспектом, влияющим на качество присвоения, помимо предоставления размеров поднаборов для соответствия потребностям образца, является длина индексной последовательности. Для некоторых простых образцов или надежных измерительных установок требуется лишь небольшое расстояние между индексными последовательностями, в то время как для других более сложных образцов или подверженных ошибкам измерительных установок требуются большие расстояния. Так как определение каждого нуклеотида индексной последовательности увеличивает стоимость (особенно в способах крупномасштабного мультиплексирования), поэтому желательно измерять только такое количество нуклеотидов индексной последовательности, которое необходимо или приемлемо для данного применения. Чтобы также удовлетворить потребность в гибком выборе длин индексных последовательностей, в предпочтительных вариантах осуществления изобретения предусмотрены индексные последовательности, которые также полезны тогда, когда для присвоения используется только часть индексной последовательности или усеченная индексная последовательность. Для решения этой задачи эти усеченные индексные последовательности корректируются в пределах поднабора, чтобы поддерживать надежное расстояние.
Усеченные индексные последовательности представляют собой части индексных последовательностей, которые подходят для поддержания желаемого расстояния друг от друга усеченными индексными последовательностями одного и того же поднабора. Это свойство, присущее части более крупной последовательности, также называется «вложенной последовательностью», относящейся к последовательности внутри последовательности. Это не следует путать с упомянутыми выше вложенными поднаборами, которые относятся к поднаборам внутри других поднаборов.
Свойства усеченной, вложенной индексной последовательности позволяют использовать всю индексную последовательность в экспериментах, которые могут быть удовлетворены более короткими индексными последовательностями, а также использовать в экспериментах, которым требуются более длинные индексные последовательности. Таким образом, практикующему специалисту нужен только один такой универсальный набор. На практике для эксперимента пользователь обычно выбирает штрихкоды из наименьшего из вложенных наборов, превышающих количество образцов, и секвенирует столько нуклеотидов штрихкодов (индексных последовательностей), сколько необходимо для достижения требуемого (низкого) уровня перекрестного загрязнения. Вложенные наборы штрихкодов получают увеличение расстояния между штрихкодами для меньших наборов и для более длинных последовательностей. Это гарантирует то, что пользователь всегда выберет оптимальную конфигурацию среди всех возможных комбинаций вложенных наборов и последовательностей.
Соответственно, в вариантах изобретения каждая индексная последовательность поднабора содержит усеченную индексную последовательность, и усеченные индексные последовательности по меньшей мере одного поднабора отличаются по меньшей мере ненулевым количеством изменений друг от друга усеченной индексной последовательности (расстоянием между штрихкодами) в упомянутом поднаборе.
Предпочтительно, чтобы минимальное количество изменений последовательностей между усеченными индексными последовательностями поднабора было больше, чем минимальное количество изменений последовательностей индексных последовательностей в поднаборе за вычетом разности между индексными последовательностями и усеченными индексными последовательностями, или в другой более общей формулировке предпочтительно, чтобы секвенциальное расстояние (которое поясняется в данном документе) между усеченными индексными последовательностями поднабора было больше, чем секвенциальное расстояние индексных последовательностей в поднаборе, за вычетом разности между длиной индексных последовательностей и усеченными индексными последовательностями. Эта формулировка по существу означает, что нуклеотиды, которые не учитываются в индексной последовательности для получения усеченной индексной последовательности (выраженной в виде разности длин), не должны быть сильными определителями секвенциального расстояния, и это означает, что оставшиеся нуклеотиды в усеченных индексных последовательностях имеют сильное влияние на секвенциальное расстояние. Обычно такая структура в пределах (вложенной) индексной последовательности устанавливается заранее и сообщается практикующему специалисту, чтобы практикующий специалист знал, какие нуклеотиды должны быть определены в качестве усеченной индексной последовательности. Предпочтительно, чтобы усеченная индексная последовательность состояла из непрерывных нуклеотидов индексной последовательности. Особенно предпочтительно, чтобы укороченная индексная последовательность содержала конец 3' или 5' индексной последовательности.
Что касается вложенных поднаборов, концепция усеченных индексных последовательностей может применяться несколько раз, предоставляя несколько последовательностей вложенных индексов. Это означает, что можно получить более одного уровня усечения. В случае нескольких этапов усечения каждая усеченная индексная последовательность имеет определенное расстояние друг от друга с другой усеченной индексной последовательностью того же самого уровня в пределах поднабора. Может быть 1, 2, 3, 4, 5 или более уровней усеченных индексных последовательностей, из которых 2 являются предпочтительными, так как они могут быть хорошо приспособлены к обычным длинам индексных последовательностей.
Конечно, вложенные последовательности могут быть объединены со структурой вложенного набора. Структура уровней для поднаборов остается прежней. Таким образом, усеченные индексные последовательности поднабора более высокого уровня являются элементами усеченных индексных последовательностей поднабора более низкого уровня. Из-за различий в размерах поднаборов усеченные индексные последовательности поднабора более низкого уровня могут отличаться меньшим минимальным количеством изменений последовательностей друг от друга, чем усеченные индексные последовательности поднабора более высокого уровня.
Существуют различные способы определения секвенциального расстояния, как описано в ссылках, упомянутых выше в разделе «Уровень техники». Можно использовать любой из этих способов. В частности, согласно изобретению изменения последовательности предпочтительно выбирают из нуклеотидных замен, делеций и вставок. Минимальное количество изменений последовательностей соответствует минимальному количеству этих изменений последовательностей, необходимых для замены любой индексные последовательности на другую индексную последовательность. Может существовать множество путей для замены одной последовательности на другую, тогда как «расстояние» относится к кратчайшим путям, то есть с наименьшими (минимальными) изменениями. Это может быть один путь или несколько путей, если несколько путей имеют одинаковое минимальное расстояние. Дополнительный вариант расстояния, который можно использовать согласно изобретению для количественной оценки величины изменений последовательностей, которая преобразует одну последовательность в другую, представляет собой сумму отдельных расстояний отдельных путей изменений, которые преобразуют одну последовательность в другую. Такую сумму можно использовать для всех путей данного изменения. Пути должны быть прямыми путями от одной последовательности к другой без обходных путей, таких как изменения, которые нейтрализуют друг друга.
Секвенциальные расстояния, описанные в данной области техники (смотри раздел «Уровень техники»), представляют собой, например, расстояние Хэмминга, расстояние Левенштейна или расстояние Левенштейна между последовательностями (Sequence-Levenshtein distance). Эти расстояния можно использовать согласно изобретению для количественной оценки расстояния или определения величины (или количества) изменений последовательностей, которая преобразует одну последовательность в другую. Расстояние Хэмминга - это по существу подсчет замен. Расстояние Левенштейна рассчитывается с использованием вставок, делеций (вместе «инделов») и замен. Предпочтительно использовать расстояние Левенштейна между последовательностями (ссылка [1]). Расстояние Левенштейна между последовательностями - это вариант расстояния Левенштейна, который также учитывает вставки и замены, но сохраняет длину индекса всякий раз, когда происходит вставка или делеция. Это означает, что вставка и делеция будут считаться не более чем одним изменением. Делеция может и не привести к изменению в том случае, если удален последний нуклеотид в последовательности, и следующий нуклеотид вне рамки, который теперь перемещается в рамку, идентичен удаленному нуклеотиду. Аналогичным образом, вставка идентичного нуклеотида в последний нуклеотид в последовательности может не проявляться как изменение и не приводить к появлению расстояния. В отличие от этого, расстояние Левенштейна рассматривает делецию в контексте олигонуклеотидной последовательности, где за индексной последовательностью следуют другие нуклеотиды (такие как адаптер или произведенное прочтение) как два изменения: первое - удаление удаленного нуклеотида, и второе - смещение следующего нуклеотида в рамку индекса последовательности, так как сравнивается вся длина индекса последовательности, и это смещение считается еще одним различием между сравниваемыми последовательностями (смотри [3], фиг. 1 для различий между расстоянием Хэмминга, расстоянием Левенштейна и расстоянием Левенштейна между последовательностями). Другими терминами для расстояния Левенштейна между последовательностями являются расстояние FREE Левенштейна, модифицированное расстояние Левенштейна или расстояние Левенштейна с фиксированной рамкой (расстояние Левенштейна ff). Например, ссылаясь на пример в дополнении к ссылке [3], в котором расстояние Левенштейна между последовательностями называется «FREE-дивергенция», последовательности TAGA и ACGC имеют расстояние, равное 3, в соответствии со следующими изменениями:
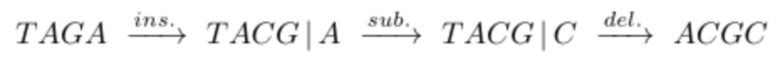
где «ins.» - вставка, «sub.» - замена и «del.» -делеция (каждое из которых также упоминается как «редакции» или «изменения»); вертикальные черты («|») показывают конец рамки штрихкода, хотя этап усечения не произойдет до тех пор, пока не будут внесены все фактические изменения. Эти смещения по длине рамки приводят к нарушению неравенства треугольника. Способы определения расстояния, которые не учитывают эти смещения за пределы и внутрь рамки индексной последовательности (или усеченной индексной последовательности), могут привести к определению расстояния, которое не отражает фактические изменения при преобразовании одной последовательности в другую. В этом примере расстояние между TAGA и TACG будет равно 1 (вставка C); как и расстояние между TACG и ACGC (делеция T со смещением 3'C в рамку). Однако расстояние между TAGA и ACGC не равно 1+1=2, а равно 3, как показано выше (нарушение неравенства треугольника). Здесь вне рамки происходит подстановка, которая может учитываться в одних способах определения расстояния, но не учитываться в других. Хотя работают оба типа измерений расстояния, так как они дают сравнимое указание расстояния между последовательностями, некоторые оценки расстояния, используемые согласно изобретению, используют изменения последовательности вне рамки индексной последовательности (или усеченной индексной последовательности), которые смещаются в рамку индексной последовательности (или усеченной индексной последовательности) для более близкого сходства с естественными процессами преобразования одной последовательности в другую (по разным причинам, таким как вставки, делеции и замены во время выполнения способов секвенирования). Это было бы дополнительным этапом для вышеупомянутого расстояния Хэмминга, расстояния Левенштейна и расстояния Левенштейна между последовательностями. С другой стороны, расстояние Левенштейна между последовательностями (фиксированная рамка) имеет процедурные преимущества и является предпочтительным способом. Затем на этапах исправления ошибок обычно рассматривается возможное нарушение неравенства треугольника (что означает, что сумма частичных расстояний не обязательно равна полному расстоянию). Другой последовательностью вне рамки индексной последовательности, которую можно считать аналогичной нуклеотидам со смещением рамки самой индексной последовательности, являются нуклеотиды или последовательности, следующие за индексной последовательностью. Они могут быть известны, например, в случае последовательности адаптера, которая следует за индексной последовательностью.
В общем, во всех вариантах осуществления изобретения изменения последовательности могут быть количественно определены как секвенциальное расстояние, которое представляет собой количество замен нуклеотидов или вероятность изменений. Каждое возможное изменение можно рассчитать либо как целое число, либо как его вероятность. Такая вероятность может зависеть от платформы, или может использоваться заданная вероятность, например, исходя из средних значений. Например, вероятность может быть выведена из частот естественных мутаций, которые, например, происходят в секвенаторе. Например, вероятности замен, вставок и делеций могут составлять 0,002, 0,00002 и 0,0005, соответственно, в этом порядке.
В предпочтительных вариантах осуществления изобретения вероятность изменений равна максимальной вероятности или сумме вероятностей. В некоторых случаях несколько серий изменений (называемых также «путями») могут привести к преобразованию одной (индексной) последовательности в другую. В таком случае путь с наибольшей (максимальной) вероятностью может предоставить подходящую оценку в качестве секвенциального расстояния. В качестве альтернативы, можно сложить вероятности нескольких путей, чтобы получить сумму вероятностей, которая также является подходящей оценкой для использования в качестве секвенциального расстояния. Предпочтительно использовать сумму вероятностей изменений нуклеотидов, которые преобразуют одну последовательность в другую.
Следует отметить, что взаимность между сравнением вероятностей и целочисленным количеством изменений последовательностей является обратной, тогда как большое количество изменений последовательностей соответствует большому расстоянию; в этом случае низкая вероятность коррелирует с большим расстоянием (а высокая вероятность коррелирует с маленьким расстоянием). Соответственно, ссылаясь на взаимосвязь уровней, как упомянуто выше, индексные последовательности поднабора более низкого уровня отличаются более высокой вероятностью изменения последовательностей друг от друга, чем индексные последовательности поднабора более высокого уровня. Кроме того, усеченные индексные последовательности поднабора более низкого уровня могут также отличаться более высокой вероятностью изменения последовательности, чем усеченные индексные последовательности поднабора более высокого уровня.
Конечно, для сохранения одного и того же направления соотношения (выше-выше; ниже-ниже) можно использовать функцию вероятности, которая меняет порядок или направленность вероятности. Такие функции являются монотонно убывающими функциями вероятности. Конечно, это просто еще одно представление вероятности, и соотношения основных вероятностей (или средних или сумм) остаются прежними. Тем не менее, в предпочтительных вариантах осуществления вероятность изменений количественно определяется с помощью монотонно убывающей функции вероятности. Такой функцией является, например, отрицательный логарифм или отрицательная вероятность (меняющая свой знак, порядок или направление), например, в 1-P (где P - вероятность, включающая в себя среднее или максимальное значение, как указано выше). Предпочтительно вероятность оценивается как такая монотонно убывающая функция от максимальной вероятности или суммы вероятностей, предпочтительно суммы вероятностей, изменений нуклеотидов, которые преобразуют одну последовательность в другую. Такие изменения нуклеотидов могут представлять собой серию изменений, если для преобразования одной последовательности в другую требуется более одного изменения.
В таком случае изменения соотношений уровней с индексными последовательностями поднабора более низкого уровня отличаются более низкой монотонно убывающей функцией вероятности изменений последовательностей друг от друга, чем индексные последовательности поднабора более высокого уровня. Кроме того, усеченные индексные последовательности поднабора более низкого уровня могут также отличаться более низкой монотонно убывающей функцией вероятности изменения последовательности, чем усеченные индексные последовательности поднабора более высокого уровня.
Набор согласно изобретению (и то, как он выбирается в способе согласно изобретению) предпочтительно определяется соотношением расстояний между индексными последовательностями поднабора, где расстояние Левенштейна между последовательностями, то есть расстояние между индексными последовательностями поднабора более высокого уровня, больше не менее чем на 1, предпочтительно на 2, 3, 4, 5, 6, 7 или более, чем расстояние Левенштейна между последовательностями, то есть расстояние между индексными последовательностями поднабора более низкого уровня.
При использовании других расстояний можно также указать, что расстояние Левенштейна между индексными последовательностями поднабора более высокого уровня больше не менее чем на 1, предпочтительно на 2, 3, 4, 5, 6, 7 или более, чем расстояние Левенштейна между индексными последовательностями поднабора более низкого уровня; или расстояние Хэмминга между индексными последовательностями поднабора более высокого уровня больше не менее чем на 1, предпочтительно на 2, 3, 4, 5, 6, 7 или более, чем расстояние Хэмминга между индексными последовательностями поднабора более низкого уровня.
При использовании суммы вероятностей или максимальной вероятности (со значениями в диапазоне от 0 до 1) предпочтительно, чтобы сумма вероятностей или максимальная вероятность преобразования одного индекса последовательности в другой в поднаборе более низкого уровня была больше не менее чем на 0,00001, предпочтительно не менее 0,0001, или не менее 0,001, или более, чем вероятность между индексными последовательностями поднабора более высокого уровня. Эта разность суммы вероятностей или максимальной вероятности между уровнями может зависеть от используемой платформы и может составлять от 0,00001 до 0,9. Если логарифм по основанию «е» (натуральный логарифм) используется для того, чтобы -log(P) использовался для определения разности расстояний между уровнями, то значение предпочтительно находится в диапазоне от 0,1 до 10.
Для абсолютных расстояний в пределах уровня предпочтительно, чтобы расстояние Левенштейна между последовательностями между индексными последовательностями поднабора самого высокого уровня составляло не менее 4, например 4, 5, 6, 7, 8 или более. Затем следующий более низкий уровень будет в том случае, когда разность между уровнями, равная 1, будет иметь расстояние Левенштейна между последовательностями между индексными последовательностями не менее 3 и так далее для следующих уровней. То же самое относится и к другим целочисленным расстояниям (Левенштейна, Хэмминга). Предпочтительно, чтобы поднабор самого низкого уровня в наборе имел расстояние Левенштейна между последовательностями, Левенштейна или Хэмминга между его индексными последовательностями не менее 1, предпочтительно 2 или 3.
Так как более длинные индексные последовательности допускают большие расстояния, предпочтительно обеспечивать минимальную длину. Конечно, более короткие индексные последовательности также имеют преимущество, то есть более низкие затраты, как упоминалось выше. Таким образом, выбирается компромисс. Предпочтительно индексные последовательности имеют длину не менее 4, например 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16 нуклеотидов в смежной последовательности. Особенно предпочтительной является длина нуклеотидов, равная не менее 6. Поднабор самого высокого уровня также является самым маленьким (наименьшее количество элементов). Каждый последующий поднабор более низкого уровня имеет больше элементов, но обычно меньшие расстояния. В предпочтительных вариантах осуществления поднабор самого высокого уровня содержит не менее 2, например 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 или более различных индексных последовательностей. Предпочтительно он содержит не менее 4-х различных индексных последовательностей.
Важно, чтобы структура поднабора была видна практикующему специалисту, то есть, чтобы индексные последовательности относились к поднабору, которому они принадлежат. Например, олигонуклеотиды (с индексной последовательностью) могут быть отнесены к поднабору путем помещения в контейнер, который помечен идентификатором поднабора. Идентификатор может быть размещен на контейнере или на носителе информации, например, в руководстве, в электронном или физическом виде. Контейнер может представлять собой лунку в луночном планшете.
В дополнительных предпочтительных вариантах осуществления, последовательности индексных последовательностей могут быть оптимизированы, например, для лучшей стабильности или возможности секвенирования. Распространенными концепциями являются оптимизация содержания GC и/или избежание нуклеотидных повторов. Особенно предпочтительной является балансировка распределения всех нуклеотидов генетического кода по различным индексным последовательностям в пределах поднабора. Нуклеотидами генетического кода являются A, T или U, G, C. Обычно используется один из T и U, причем преимущественно используется T, поэтому «T или U» также записывается как «T (U)». Таким образом, в индексной последовательности обычно находятся 4 различных типа нуклеотидов. T находится в DNA, U в RNA. Олигонуклеотиды могут представлять собой, например, ДНК или РНК и/или содержать модифицированные нуклеотиды, такие как LNA.
Предпочтительно индексные последовательности имеют содержание G/C от 20% до 80%, или от 30% до 70% или даже от 40% до 60%.
Предпочтительно индексные последовательности не содержат повторов одного и того же нуклеотида длиной не менее 3, то есть не содержат гомополимерных троек.
Предпочтительно избегать последовательности GGC в некоторых установках, особенно для секвенирования на основе Illumina, так как это мотив ошибки на основе Illumina (ссылка [3]).
Особенно предпочтительно, чтобы индексные последовательности поднабора имели сбалансированное распределение нуклеотидов, где количество совместно используемых нуклеотидов в одной и той же позиции в индексных последовательностях между различными индексными последовательностями не превышает 0,5-кратного количества индексных последовательностей в упомянутом поднаборе. В этом критерии используется сумма (количество совместно используемых нуклеотидов на позицию), и она сравнивается, соответственно, ее с кратным (например, 0,5) числом индексных последовательностей в поднаборе (размером поднабора). Количество совместно используемых нуклеотидов в одной и той же позиции означает, что для каждой позиции, например, нуклеотида (nt) 1, nt 2, nt 3 и т.д., тип нуклеотида (A, T(U), G или C) подсчитывается по всем индексным последовательностям. Таким образом, когда рассматривается больше индексных последовательностей, число увеличивается. Следовательно, значение критерия (0,5 или ниже, например, от 0 до 0,5) также умножается на количество рассматриваемых индексных последовательностей. Это эквивалентно использованию средних значений, соответствующих частотам, которые сравниваются со значением 0,5 в качестве предпочтительной максимальной частоты. Это означает, что количество совместно используемых нуклеотидов в одной и той же позиции затем делится на количество рассматриваемых индексных последовательностей. Это среднее значение также называется частотой нуклеотидов (в расчете на одну позицию). Примеры таких частот для каждого нуклеотида показаны на фиг. 8-11. Идеально сбалансированные нуклеотиды означают, что каждый нуклеотид, выбранный из A, T(U), G, C, распределен равномерно, то есть частота составляет одну четверть или 0,25 для всех позиций. Однако такая оптимальная балансировка не всегда возможна, так как должен также выполняться критерий для секвенциального расстояния. Следовательно, необходимы отклонения купола от идеальной балансировки. Это значение может быть высоким для поднаборов с маленькими размерами, так как отклонение одной индексной последовательности от среднего значения может означать большее отклонение от 0,25 (например, на фиг. 8 показано распределение в поднаборе из 4 индексных последовательностей). Для больших поднаборов, как правило, можно приблизиться к желаемому значению 0,25. В предпочтительных вариантах осуществления это значение критерия или частота составляет 0,4 или менее, например, в диапазоне от 0,1 до 0,4, особенно предпочтительно для поднабора с размером 8 или более.
В качестве дополнения или альтернативы предпочтительно, когда при не менее 50% позиций индексных последовательностей частота нуклеотидов для всех индексных последовательностей поднабора для каждого типа нуклеотидов составляет 0,5 или менее, например, от 0 до 0,5, предпочтительно составляет 0,4 или менее, например от 0,1 до 0,4.
Конкретные предпочтительные варианты осуществления наборов согласно изобретению содержат индексные последовательности (или олигонуклеотиды, содержащие эти индексные последовательности), выбранные из любой из SEQ ID NO: 1-784, предпочтительно из SEQ ID NO: 1-208. В наборе содержатcя предпочтительно не менее 10, предпочтительно не менее 15, не менее 20, не менее 30, не менее 40, не менее 50, не менее 60, не менее 70, не менее 80 из SEQ ID NO: от 1 до 784, предпочтительно из SEQ ID NO: от 1 до 208.
Настоящее изобретение дополнительно предоставляет способ выработки набора олигонуклеотидов согласно изобретению, содержащего множество поднаборов олигонуклеотидов с поднабором индексных последовательностей. Все, что раскрыто для набора, также применимо к способу, например, получен набор с этими параметрами или параметры используются и выбираются в способе, таком как раскрытые способы определения секвенциального расстояния.
Способ содержит этапы выработки поднабора олигонуклеотидов первого или более высокого уровня с индексными последовательностями с секвенциальным расстоянием первого или более высокого уровня применительно друг к другу в пределах поднабора первого или более высокого уровня, где секвенциальное расстояние представляет собой количественную величину изменений последовательностей, которая преобразует одну последовательность в другую, или монотонно убывающая функция вероятности изменений последовательностей, которая преобразует одну последовательность в другую, как указано выше, вырабатывая поднабор второго или более низкого уровня путем включения поднабора первого или более высокого уровня и добавления дополнительных олигонуклеотидов с индексными последовательностями с секвенциальным расстоянием другу в пределах поднабора второго или более низкого уровня, где секвенциальное расстояние второго или более низкого уровня меньше, чем секвенциальное расстояние первого или более высокого уровня.
Термины «поднабор более высокого уровня» и «первый поднабор» могут использоваться как синонимы и относиться к относительной взаимосвязи между поднаборами. Преимущество использования числовых значений состоит в том, что они также относятся к уровням, которые находятся ниже второго поднабора, например к третьему поднабору, который содержит индексы последовательности второго поднабора (и, следовательно, также первого) и дополнительные индексы последовательности. Следовательно, требования к их секвенциальному расстоянию, вероятно, будут ниже, чем для второго уровня. Эта установка соответствует набору, содержащему по меньшей мере 3 иерархических уровня поднаборов, что является предпочтительным вариантом осуществления для всех аспектов изобретения. 3 иерархических уровня по терминологии «более высоки-более низкий» означают, что существует первая взаимосвязь между более высоким (1-м) и более низким (2-м) уровнями, как уже отмечалось, и затем еще одна вторая взаимосвязь, где этот более низкий уровень (2-й уровень) становится более высоким уровнем для следующего более низкого уровня (3-го уровня).
Набор согласно изобретению может иметь 2, 3, 4, 5, 6, 7, 8 или более иерархических уровней, то есть первый, второй, третий, четвертый, пятый, шестой, седьмой, восьмой или дополнительные уровни, где каждый поднабор уровней в этом порядке содержит индексные последовательности уровня поднабора до и после индексных последовательностей, как уже отмечалось для первого и второго уровня (или более высокого и более низкого уровня), соответственно.
В предпочтительных вариантах осуществления способ содержит выработку поднабора более низкого уровня путем включения поднабора более высокого уровня и добавления дополнительных олигонуклеотидов с индексными последовательностями с меньшим секвенциальным расстоянием друг от друга, чем для поднабора более высокого уровня в поднаборе более низкого уровня. Аналогичным образом, способ может содержать выработку третьего поднабора путем включения второго поднабора и добавления дополнительных олигонуклеотидов с индексными последовательностями с третьим секвенциальным расстоянием в пределах третьего поднабора, где третье секвенциальное расстояние является меньшим секвенциальным расстоянием, чем второе секвенциальное расстояние. По мере необходимости этот способ может применяться к любому другому иерархическому уровню поднаборов.
Этап выработки первого, второго (или следующего) поднабора индексных последовательностей может содержать для одного, или нескольких или каждого поднабора этап выбора индексных последовательностей из пула различных индексных последовательностей-кандидатов. Согласно данному варианту осуществления пул индексных последовательностей вырабатывается в качестве кандидатов для включения в поднаборы. Эти кандидаты обычно имеют желаемую длину индексных последовательностей, но не имеют выбранных секвенциальных расстояний в пуле кандидатов. Упомянутый пул содержит несколько индексных последовательностей-кандидатов в количестве, достаточном для заполнения поднабора. Обычно число по меньшей мере в два раза превышающее размер поднабора, предоставляется в качестве пула, чтобы гарантировать то, что доступно достаточное количество вариантов индексных последовательностей для обеспечения необходимых секвенциальных расстояний и при необходимости других критериев, описанных в данном документе, для поднабора. Предпочтительно пул индексных последовательностей имеет по меньшей мере в 2 раза, более предпочтительно в 3, 4, 5, 6, 7, 8, 9, 10 или более раз больше элементов, чем поднабор. Индексные последовательности пула могут быть случайными или могут удовлетворять некоторым другим критериям, таким как выбранное содержание GC, например, отсутствие тройных гомополимеров. Затем кандидаты добавляются в поднабор во время его построения, где соблюдаются критерии для секвенциальных расстояний (и другие критерии, такие как балансировка, если это необходимо). Если критерии не выполняются, то из пула выбираются другие индексы-кандидаты последовательности. Если этого недостаточно, могут быть выработаны и, соответственно, использованы новые индексы-кандидаты последовательности и/или новые пулы.
Предпочтительно выработка первого и/или второго поднабора (или дополнительных аналогичных поднаборов) содержит выбор индексных последовательностей, которые содержат усеченные индексные последовательности, и усеченные индексные последовательности по меньшей мере одного поднабора отличаются по меньшей мере ненулевым количеством изменений последовательностей от каждой другой усеченной индексной последовательности в упомянутом поднаборе. Ненулевое количество изменений последовательностей может представлять собой секвенциальное расстояние, равное 1, 2, 3, 4, 5, 6, 7, 8 или более, особенно предпочтительно расстояние Хэмминга, Левенштейна или Левенштейна между последовательностями, или вероятность изменений, как это указано выше. Предпочтительно усеченные индексные последовательности по меньшей мере одного поднабора отличаются по меньшей мере на количество изменений последовательностей, превышающее 1, от каждой другой усеченной индексной последовательности в упомянутом поднаборе. В отношении усеченных поднаборов применяется то же самое, как описано выше. Они позволяют использовать в способе присвоения секвенирующих прочтений только частичную последовательность (соответствующую усеченной последовательности), в то время как заданное требование к секвенциальному расстоянию между всеми усеченными последовательностями поднабора все еще выполняется, как описано выше.
В дополнительных предпочтительных вариантах осуществления корректируемые последовательности вырабатываются для индексной последовательности поднабора, где упомянутые корректируемые последовательности имеют секвенциальное расстояние, которое составляет менее половины секвенциального расстояния между индексными последовательностями упомянутого поднабора, и где корректируемые последовательности различных индексных последовательностей в упомянутом поднаборе не перекрываются. Такие корректируемые последовательности в индексных последовательностях также присутствуют в наборе согласно изобретению. Корректируемые последовательности - это последовательности, которые могут ассоциироваться только с одной индексной последовательностью. Это делает последовательность «корректируемой». Таким образом, корректируемая последовательность является представлением ошибочно определенной последовательности, которая имеет одну или несколько ошибок секвенирования, но когда эта последовательность является корректируемой, ее по-прежнему можно присвоить («декодировать») одной индексной последовательности. В способе выработки индексных последовательностей для поднабора в данном документе учитывается то, что вокруг каждой индексной последовательности существует множество корректируемых последовательностей, которые по-прежнему приводят к одному присвоению при использовании набора согласно изобретению. Это множество также называется «сферой декодирования», используя аналогию объема последовательностей с заданным расстоянием до индексной последовательности в центре сферы. Для того, чтобы выполнить присвоение одной (и только одной) индексной последовательности, расстояние должно быть меньше половины секвенциального расстояния между индексными последовательностями упомянутого поднабора. Это не всегда будет так, учитывая возможность нарушения упомянутого выше неравенства треугольника. Соответственно, поднабор может учитывать эту возможность отдельно для критерия расстояния между индексными последовательностями и максимизировать количество корректируемых последовательностей или уменьшать количество последовательностей, которые могут быть присвоены более чем одной, например, двум или более индексным последовательностям с равным (не корректируемым) расстоянием. Это также называется оптимизацией сферы декодирования, что означает уменьшение или минимизацию перекрытия двух или более таких сфер. Это можно сделать, выбрав различные индексные последовательности для данного поднабора.
В предпочтительном варианте выработка поднабора содержит выбор индексных последовательностей путем добавления индексной последовательности-кандидата и оценки секвенциального расстояния индексного расстояния-кандидата до всех других ранее существовавших индексных последовательностей в поднаборе. Индексная последовательность-кандидат добавляется к индексным последовательностям поднабора, если он удовлетворяет заданному требованию к секвенциальному расстоянию, такому как любое свойство секвенциального расстояния, как обсуждалось выше. Индексная последовательность-кандидат может быть или не может быть из вышеупомянутого пула. В общем, в данном варианте осуществления утверждается, что индексные последовательности-кандидаты добавляются поэтапно во время построения поднабора, где индексные последовательности добавляются одна за другой. Индексная последовательность-кандидат сравнивается с другими ранее существовавшими индексными последовательностями в поднаборе, если они существуют (очевидно, это не делается для первой индексной последовательности, добавленной в поднабор). Когда сравнение приводит к выполнению требования к расстоянию и, возможно, других требований, в поднабор добавляется кандидат индексные последовательности. Этот процесс может быть выполнен для других поднаборов или даже для поднаборов-кандидатов. Поднабор-кандидат рассматривается как поднабор, но может быть не включен в набор, если также вырабатываются другие поднаборы-кандидаты одного и того же размера. Затем обычно поднабор-кандидат добавляется в поднабор, если он лучше другого поднабора-кандидата. Улучшением может быть любой критерий, упомянутый выше, например улучшенная балансировка.
Такое требование к балансировке, которое предпочтительно выполняется, является любым из упомянутых выше, где индексная последовательность-кандидат предпочтительно содержит не менее 50% своих позиций типа нуклеотидов генетического кода с наименьшей частотой в соответствующей позиции в ранее существовавших индексных последовательностях поднабора. Этот критерий предпочтительно применяется по меньшей мере к 25% индексных последовательностей-кандидатов, которые добавляются в поднабор последними. Как упоминалось выше, оценка частоты не имеет смысла при рассмотрении только одной индексной последовательности и имеет небольшое значение для небольших поднаборов в процессе выработки, к которым добавляются дополнительные индексные последовательности или кандидаты. Балансировка лучше всего достигается тогда, когда поднабор имеет почти желаемый размер, например, когда он составляет 75% или более от своего размера, то есть на этом этапе оцениваются оставшиеся 25%. Особенно предпочтительно, когда по этому критерию оценивается последняя индексная последовательность, добавленная в поднабор.
В предпочтительных вариантах осуществления индексная последовательность-кандидат выбирается из пула индексных последовательностей-кандидатов, где элементы пула индексных последовательностей-кандидатов выполняют заданное требование к секвенциальному расстоянию для каждого другого элемента пула. Кроме того, индексная последовательность-кандидат пула добавляется к индексным последовательностям поднабора тогда, когда сумма расстояний частоты каждого типа нуклеотидов генетического кода до 0,25 в каждой позиции является наименьшей для индексной последовательности-кандидата по сравнению с другими индексными последовательностями-кандидатами в пуле. Расстояние частоты каждого типа нуклеотида генетического кода до 0,25 в каждой позиции может быть измерено как сумма абсолютных значений разности, или, что предпочтительнее, квадрата или возведенной в степень разности между частотой каждого нуклеотида и 0,25 в каждой позиции, или как мера вероятностного расстояния между частотой каждого нуклеотида и 0,25 в каждой позиции, где возможными мерами вероятностного расстояния будет дивергенция Кульбака-Лейблера или Дженсена-Шеннона. Это абсолютное значение разностей является еще одним предпочтительным вариантом балансировки, как обсуждалось выше. Частота 0,25 была бы оптимальной балансировкой (при выполнении для каждой позиции), но она редко достигается. Чем ближе частоты нуклеотидов индекса последовательности к 0,25, тем лучше сбалансирован поднабор.
Еще один предпочтительный критерий балансировки, используемый в способе (и обнаруженный в наборе), состоит в том, что по меньшей мере в 50% позиций индексных последовательностей частота нуклеотидов для всех индексных последовательностей поднабора для каждого типа нуклеотидов составляет 0,5 или менее. Предпочтительные варианты вариантов балансировки описаны выше.
В предпочтительных вариантах осуществления способ выработки набора согласно изобретению содержит выработку множества поднаборов-кандидатов, каждый из которых имеет заданное количество элементов (индексные последовательности). Эти конкурирующие поднаборы-кандидаты с одинаковым размером сравниваются друг с другом, и один из них выбирается для включения в набор, называемый поднабором. Способ предпочтительно содержит выбор поднабора-кандидата в качестве поднабора для набора, когда упомянутый поднабор-кандидат имеет наименьшее среднее значение по всем индексным последовательностям для соответствующего поднабора-кандидата суммы абсолютных значений разностей частот каждого типа нуклеотидов генетического кода в каждой позиции до 0,25. Так, для каждого поднабора-кандидата средние абсолютные значения разностей частот каждого типа нуклеотидов генетического кода до 0,25 для каждой позиции суммируются для всех его индексных последовательностей. Поднабор-кандидат, который имеет более низкое значение (то есть меньшее различие означает лучшую сбалансированность - смотри выше), выбирается для включения в поднабор. Предпочтительно, чтобы поднабор-кандидат выбирался с наименьшим значением. Если учитывать и другие критерии, балансировка может оказаться самой низкой или даже наихудшей. Предпочтительно один выбранный поднабор-кандидат находится среди лучшей половины (в соответствии с меньшим значением в этой формулировке) рассматриваемых поднаборов-кандидатов. Выбор может применяться для полных поднаборов-кандидатов, но он также может быть очевиден во время построения, например, когда индексы-кандидаты последовательности добавляются последовательно, как упомянуто выше, когда во время упомянутого построения становится очевидным, что данный поднабор-кандидат не приведет к хорошему значению. Такие поднаборы-кандидаты с худшими характеристиками могут быть исключены из дальнейшего рассмотрения.
В качестве альтернативы или в комбинации, способ может содержать выработку множества поднаборов-кандидатов, каждый из которых имеет заданное количество элементов (индексные последовательности), и выбор поднабора-кандидата в качестве поднабора для набора, где упомянутый поднабор-кандидат выбирается путем исключения других поднаборов-кандидатов,
где поднабор-кандидат исключается тогда, когда в способе, который содержит добавление кандидатов-индексных последовательностей из пула индексных последовательностей-кандидатов к поднабору-кандидату и при необходимости дополнительное добавление сравнительных индексных последовательностей, кандидат-поднабор имеет более высокое среднее значение во всех своих суммах индексных последовательностей абсолютных значений разностей частот каждого типа нуклеотидов генетического кода в каждой позиции до 0,25 по сравнению с другим поднабором или поднабором-кандидатом.
Такой выбранный поднабор затем добавляется к набору. Фраза «поднабор-кандидат имеет более высокую среднюю во всех своих суммах индексных последовательностей абсолютных значений разностей частот каждого типа нуклеотидов генетического кода в каждой позиции до 0,25 по сравнению с другим поднабором или поднабором-кандидатом» пояснена выше. Сравнение со сравнительными индексными последовательностями означает, что, когда поднабор-кандидат вырабатывается путем последовательного добавления индексов последовательностей и индексов-кандидатов последовательностей, то этот поднабор или поднаборы-кандидаты приведут к полному поднабору желаемого размера только тогда, когда последний индекс последовательности или индекс-кандидат последовательности будет добавлен для рассмотрения. Для лучшей оценки промежуточных добавленных индексов-кандидатов последовательности могут быть добавлены дополнительные сравнительные индексные последовательности для заполнения поднабора или поднабора-кандидата до его желаемого размера. Критерии, особенно критерии балансировки, затем рассчитываются для индекса-кандидата последовательности применительно к каждому другому индексу последовательности и сравнительному индексу последовательности. Таким образом, эти сравнительные индексные последовательности позволяют смоделировать полный поднабор для поднабора-кандидата без использования в поднаборе или поднаборе-кандидате. Конечно, они могут быть добавлены к нему, если они выбраны в качестве индекса-кандидата последовательности на следующем этапе. Способ может содержать удаление поднаборов-кандидатов из дальнейшего рассмотрения на каждом этапе последовательного построения поднабора-кандидата, если критерий балансировки хуже, чем у других поднаборов-кандидатов или ранее существовавших поднаборов. Предпочтительно по меньшей мере один поднабор-кандидат исключается на каждом этапе добавления одного индекса последовательности к поднабору-кандидату.
Настоящее изобретение дополнительно предоставляет способ использования набора согласно изобретению для мечения фрагментов, таких как олигонуклеотид, белок, частица, такая как наночастица, химических соединений, особенно низкомолекулярных соединений размером 5 кДа или меньше и т.д. Настоящее изобретение предоставляет способ идентификации меченых фрагментов путем определения последовательности индексной последовательности, которая была присоединена к ним, и сопоставления определенной последовательности с известной индексной последовательностью набора. В частности, настоящее изобретение предоставляет способ присвоения секвенирующих прочтений (то есть определенных последовательностей) образцу олигонуклеотидов, содержащий этапы:
а) получения образцовых олигонуклеотидов из множества образцов,
b) выбора поднабора олигонуклеотидных индексных последовательностей из набора согласно изобретению, где поднабор выбирается вместо другого поднабора на основе большего секвенциального расстояния между индексными последовательностями друг от друга в пределах выбранного поднабора; где секвенциальное расстояние представляет собой количественную величину изменений последовательностей, которая преобразует одну последовательность в другую, или монотонно убывающую функцию вероятности изменений последовательностей, которая преобразует одну последовательность в другую, и где выбранный поднабор имеет по меньшей мере такое же количество различных индексных последовательностей, как и количество образцов на этапе а),
c) добавления индексных последовательностей из упомянутого поднабора к каждому его образцовому олигонуклеотиду (который может быть фрагментом или продуктом фрагментации), где индексные последовательности указывают образец,
d) определения последовательности образцовых олигонуклеотидов или фрагментов образцовых олигонуклеотидов и определение индексной последовательности,
e) присвоения последовательности с полученным прочтением образцу на основе определенной индексной последовательности или на основе индексной последовательности, которая имеет наименьшее секвенциальное расстояние в определенной индексной последовательности, где, если две или более индексных последовательностей имеют одинаковое наименьшее расстояние, то упомянутое полученное прочтение отбрасывается; где при необходимости секвенциальное расстояние не превышает заданного значения согласно критерию.
Способ направлен на сохранение в образце ассоциации олигонуклеотидов, последовательность которых определена. Таким образом, индексные последовательности являются метками, идентифицирующими выборочную ассоциацию. Это позволяет одновременно определять последовательности многих олигонуклеотидов из нескольких образцов параллельно (мультиплекс), так как ассоциация образца поддерживается информацией метки (определяющей индексную последовательность). Разумеется, способ применим к любым меченым фрагментам, и не только к олигонуклеотидам. Ассоциация прочтений олигонуклеотидов является наиболее распространенной для поднаборов согласно изобретению.
Нет необходимости использовать весь набор, но можно использовать только один из его поднаборов до тех пор, пока поднабор имеет необходимое количество индексных последовательностей (размер). Конечно, можно использовать весь набор, который по существу представляет собой поднабор самого низкого уровня с наибольшим размером, доступным в наборе. На этапе а) определяется количество образцов, которые должны быть помечены по-разному. Ссылка на образцы означает, конечно, образцы, которые должны быть выделены в способе. На этапе b) выбирается поднабор из набора, который может вмещать в себя это количество образцов, то есть размер поднабора равен по меньшей мере количеству образцов. Для наилучшего использования предложенной в изобретении структуры поднабора и для оптимизации секвенциального расстояния между индексными последовательностями поднабора поднабор выбирается вместо другого поднабора на основе большего секвенциального расстояния между индексными последовательностями друг от друга в пределах выбранного поднабора. Секвенциальное расстояние для наборов определено и описано выше. Этот этап означает, что, если возможно, то есть если позволяет размер поднабора, поднабор с большими секвенциальными расстояниями между его элементами выбирается вместо другого поднабора с меньшим секвенциальным расстоянием между его элементами. В предпочтительных вариантах осуществления этап b) содержит выбор олигонуклеотидов с индексными последовательностями из набора согласно изобретению, где выбирается поднабор олигонуклеотидов с наибольшим секвенциальным расстоянием индексных последовательностей в поднаборе. То есть выбирается наилучший поднабор с наибольшим расстоянием, если позволяет размер поднабора. Выбранный поднабор должен иметь по меньшей мере такое же количество различных индексных последовательностей, как и количество образцов на этапе а), которые необходимо идентифицировать или различить. Другой поднабор может использоваться в других экспериментах или оставаться избыточным.
Этап с) содержит добавление индексных последовательностей из упомянутого поднабора к каждому его образцовому олигонуклеотиду. «Добавление» означает присоединение, которое присоединяет индексные последовательности (в виде олигонуклеотидов) к образцовым олигонуклеотидам или фрагментам, поэтому это присоединение поддерживается для присвоения данных секвенирования. Обычно используется ковалентное присоединение. В случае олигонуклеотидов это может содержать лигирование. Образцовый олигонуклеотид может быть фрагментом или продуктом фрагментации более крупного полинуклеотида. Возможен любой способ подготовки образца. Ради простоты изобретение относится только к продукту приготовления, который будет идентифицирован, например, на этапе секвенирования. Этот этап секвенирования может быть этапом мультиплексирования, как указано выше, когда многие олигонуклеотиды из разных образцов объединяются вместе, и, следовательно, на этом этапе необходимо мечение. Любое приготовление образцовых фрагментов на этапах, когда образцы все еще хранятся отдельно, не требует мечения, специфичного для образца. Например, дополнительная фрагментация без меток (индексных последовательностей) может выполняться отдельно для каждого образца.
Этап d) содержит определение последовательности образцовых олигонуклеотидов или фрагментов образцовых олигонуклеотидов и определение индексной последовательности. Эти последовательности (индексная последовательность и последовательность образцового олигонуклеотида) обычно определяются вместе, так как обычно они находятся после этапа с) на одной и той же объединенной молекуле олигонуклеотида. Определенная последовательность, которая соответствует «последовательности образцового олигонуклеотида», также упоминается как «прочтение» или «секвенирующее прочтение». Помимо ошибок секвенирования или повреждений нуклеотидов во время приготовления, эта определенная последовательность должна соответствовать последовательности образцового олигонуклеотида из образца на этапе а).
Этап e) содержит присвоение полученной/определенной последовательности прочтения образцу на основе определенной индексной последовательности или на основе индексной последовательности, которая имеет наименьшее секвенциальное расстояние в определенной индексной последовательности, где, если две или более индексных последовательностей имеют одинаковое наименьшее расстояние, после чего упомянутое полученное прочтение отбрасывается. Определенная индексная последовательность может идеально безошибочно соответствовать индексной последовательности известного поднабора. Преимущество набора согласно изобретению состоит в том, что даже в случае разностей с ошибками, такими как ошибки секвенирования или повреждения во время приготовления, определенная индексная последовательность, полученная на этапе d), может быть присвоена известной индексной последовательности поднабора и, следовательно, образцу, который помечает ее с использованием «исправления ошибок», как описано выше. То есть из-за больших секвенциальных расстояний между индексными последовательностями поднабора во время сингамии и большой сферы декодирования, многочисленные различные определенные последовательности могут быть присвоены индексной последовательности, несмотря на различия (то есть по существу также расстояниям до индексной последовательности). Это присвоение обычно выбирает ближайшую индексную последовательность, то есть ту, которая находится на наименьшем расстоянии от определенной индексной последовательности. Если более чем одна индексная последовательность показывает ближайшее расстояние, то есть однозначное присвоение невозможно, то прочтение может оказаться непригодным для использования и может быть отброшено. Предпочтительно это присвоение отличающейся определенной последовательности имеет значение отсечки, означающее, что секвенциальное расстояние не превышает заданного значения согласно критерию. Если расстояние превышает такую отсечку, то прочтение также может быть отброшено. Такой отсечкой может быть расстояние 3, 4, 5, 6 или 7 согласно любому способу измерения расстояния, как раскрыто выше, таким как расстояние Хэмминга, расстояние Левенштейна или расстояние Левенштейна между последовательностями.
Предпочтительно секвенируемые олигонуклеотиды содержат по меньшей мере индексную последовательность, последовательность образцового олигонуклеотида и при необходимости дополнительно последовательность адаптера и при необходимости универсальный идентификатор. Адаптер может представлять собой последовательность, которая используется для гибридизации праймеров с олигонуклеотидом. Обычно эта последовательность является одной и той же для всех олигонуклеотидов. Универсальный идентификатор может идентифицировать эксперимент по секвенированию или запуск мультиплексирования и может быть специфичным для него, но по-прежнему может быть универсальным для всех олигонуклеотидов, секвенируемых вместе. Предпочтительно олигонуклеотид содержит по меньшей мере две индексные последовательности. Данный вариант осуществления также называется двойной индексацией (когда используются две индексные последовательности) или множественной индексацией. Двойное или множественное индексирование позволяет дополнительно идентифицировать или исправлять ошибки, в частности, оно позволяет идентифицировать ошибки из-за скачка индекса (который также называется «скачком штрихкода»), то есть когда одна индексная последовательность присоединяется к олигонуклеотиду неправильного образца, который она не должна метить (смотри ссылку [5] в перечне ссылок). Когда используются две или более индексных последовательностей, они обычно выбираются из разных групп индексных последовательностей, таких как наборы или поднаборы. Обычно эти группы обозначаются как «i7» и «i5» или «левый штрихкод» и «правый штрихкод». Например, согласно изобретению индексные последовательности «i7» могут быть выбраны из SEQ ID NO: 1-104 и SEQ ID NO: 209-496, и индексные последовательности «i5» могут быть выбраны из SEQ ID NO: 105-208 и SEQ ID NO: 497-784 или наоборот.
В дополнительных предпочтительных вариантах осуществления определение последовательности нуклеотидов индексной последовательности содержит определение последовательности всей индексной последовательности или ее части, где предпочтительно определяется частичная индексная последовательность в случае, если секвенциальное расстояние от частичной индексной последовательности до других частичных индексных последовательностей в одном и том же поднаборе больше, чем ненулевое значение согласно критерию. Части индексных последовательностей могут находиться на достаточном расстоянии друг от друга, чтобы можно было выполнить присвоение - конечно, определенной последовательности без ошибок, но в некоторых случаях также и последовательности с исправлением ошибок, как упомянуто выше. Частичная индексная последовательность предпочтительно представляет собой последовательность смежных нуклеотидов индексной последовательности. Она может быть на 1, 2, 3, 4, 5, 6 или более нуклеотидов короче индексной последовательности. Предпочтительно она по-прежнему имеет длину не менее 4, 5, 6, 7, 8, 9, 10 или более нуклеотидов. Присвоение с частичными последовательностями работает так же, как и для полных индексных последовательностей, в том смысле, что частичная последовательность сравнивается с соответствующей частью индексной последовательности. В предпочтительных вариантах осуществления индексные последовательности имеют усеченные последовательности, которые имеют концептуальное секвенциальное расстояние, как описано выше. Как было отмечено, усеченные индексные последовательности индексных последовательностей также имеют отрегулированное секвенциальное расстояние, которое поддерживается для всех усеченных индексных последовательностей поднабора, и это означает то, что во время использования набора можно определить или учесть только ту частичную последовательность, которая соответствует усеченной индексной последовательности. Соответственно, в особенно предпочтительных вариантах осуществления частичная индексная последовательность имеет свойства секвенциального расстояния усеченной индексной последовательности, как описано выше.
Настоящее изобретение также выигрывает от использования компьютеров. Любой способ может быть выполнен на компьютере, особенно при проектировании индексных последовательностей, усеченных индексных последовательностей, поднаборов и набора и дальнейшем его использовании, например, при присвоение определенных последовательностей индексным последовательностям и поднаборам, как это описано в данном документе. Таким образом, любой способ изобретения может быть реализован на компьютере. Настоящее изобретение также предоставляет компьютерный программный продукт, содержащий инструкции, которые, при выполнении программы компьютером, предписывают компьютеру выполнять любой способ изобретения или его этапы, в частности те, которые указаны в этом абзаце. Настоящее изобретение также предоставляет машиночитаемый носитель информации, содержащий эти инструкции.
В последующем описании изобретения используется подробная практическая терминология. Конечно, это описание и его части могут быть объединены с любым из общих элементов, описанных выше.
1. Вложенные наборы штрихкодов
Вложенный набор B штрихкодов содержит S ≥ 1 вложенных поднаборов B1⊂B2⊂…⊂BS, так что расстояние между штрихкодами внутри Bi увеличивается для меньших наборов штрихкодов. Если расстояние между штрихкодами b,b' задано в виде d(b,b') и di = d(Bi) = minb,b'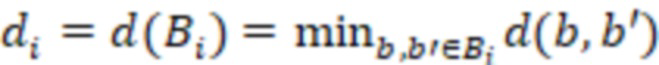 Bid(b,b'), то d1 > d2 > …> dS. Общая схема того, как может быть выработан такой вложенный набор штрихкодов, представлена ниже. Допустим, что
Bid(b,b'), то d1 > d2 > …> dS. Общая схема того, как может быть выработан такой вложенный набор штрихкодов, представлена ниже. Допустим, что  является длиной штрихкодов и выбрана такая последовательность расстояний, что d1 > d2 > …> dS. Начнем с выработки набора штрихкодов B1 с минимальным расстоянием между штрихкодами, равным d1. В некоторых случаях это может быть достигнуто с использованием лексикографического поиска [2]. Если d1 выбрано слишком большим, может оказаться невозможным найти непустое, B1 состоящее из штрихкодов длиной n. Затем выбирается другое d1. Однако в дальнейшем предполагается, что последовательность d1, …, dS была помечена таким образом, что d1 является первым расстоянием, для которого можно найти непустое B1. Так как, d1 > d2 набор B1 штрихкодов может использоваться в качестве начального набора при поиске B2, который, опять же, может использоваться в качестве начального набора при поиске для B3 и т.д. Этот процесс показан на фиг. 2. В данном документе B1 состоит из 4 штрихкодов с меткой 1 с минимальным расстоянием между штрихкодами d1. B2 получается путем использования B1 в качестве начального набора и добавления 4 штрихкодов с меткой 2. Для B2 имеем d1 > d2. Наконец, B3 получается путем использования B2 в качестве начального набора и добавления 16 штрихкодов с меткой 3. Это дает d1 > d2 > d3 и B1 ⊂ B2 ⊂ B3. Точный способ получения Bi+1 из Bi зависит от меры расстояния штрихкода и желаемых свойств наборов штрихкодов Bi. Чтобы гарантировать определенные уровни перекрестного загрязнения, может также потребоваться проверка других предпочтительных свойств Bi в дополнение к d(Bi) = di. Подробности относительно этого будут обсуждены в разделе 4.1.
является длиной штрихкодов и выбрана такая последовательность расстояний, что d1 > d2 > …> dS. Начнем с выработки набора штрихкодов B1 с минимальным расстоянием между штрихкодами, равным d1. В некоторых случаях это может быть достигнуто с использованием лексикографического поиска [2]. Если d1 выбрано слишком большим, может оказаться невозможным найти непустое, B1 состоящее из штрихкодов длиной n. Затем выбирается другое d1. Однако в дальнейшем предполагается, что последовательность d1, …, dS была помечена таким образом, что d1 является первым расстоянием, для которого можно найти непустое B1. Так как, d1 > d2 набор B1 штрихкодов может использоваться в качестве начального набора при поиске B2, который, опять же, может использоваться в качестве начального набора при поиске для B3 и т.д. Этот процесс показан на фиг. 2. В данном документе B1 состоит из 4 штрихкодов с меткой 1 с минимальным расстоянием между штрихкодами d1. B2 получается путем использования B1 в качестве начального набора и добавления 4 штрихкодов с меткой 2. Для B2 имеем d1 > d2. Наконец, B3 получается путем использования B2 в качестве начального набора и добавления 16 штрихкодов с меткой 3. Это дает d1 > d2 > d3 и B1 ⊂ B2 ⊂ B3. Точный способ получения Bi+1 из Bi зависит от меры расстояния штрихкода и желаемых свойств наборов штрихкодов Bi. Чтобы гарантировать определенные уровни перекрестного загрязнения, может также потребоваться проверка других предпочтительных свойств Bi в дополнение к d(Bi) = di. Подробности относительно этого будут обсуждены в разделе 4.1.
2. Вложенные последовательности штрихкодов
Выбор поднабора соответствующего размера делает вложенный набор штрихкодов адаптируемым к количеству образцов в эксперименте. Чтобы сделать набор штрихкодов адаптируемым к требуемому уровню перекрестного загрязнения, мы, кроме того, разработали наши наборы штрихкодов таким образом, чтобы последовательность штрихкода могла быть расширена до определенной длины с гарантированным увеличением минимального расстояния между штрихкодами. Затем нерасширенная последовательность соответствует усеченной или частичной индексной последовательности, а расширенная последовательность -индексной последовательности. В дополнение к этому, расширенные наборы штрихкодов сохраняют вложенную структуру, где поднаборы в расширенном наборе штрихкодов состоят из расширенных штрихкодов в поднаборах исходного набора штрихкодов. Что касается вложенных поднаборов, то процесс расширения штрихкода можно применять несколько раз для получения вложенных последовательностей штрихкода. Это означает, что может существовать более одного уровня усечения. В случае нескольких этапов усечения, каждая усеченная индексная последовательность имеет определенное расстояние для каждой другой усеченной индексной последовательностью одного и того же уровня в пределах поднабора. Общая структура вложенного набора штрихкодов с вложенными последовательностями штрихкодов показана на фиг. 3. Исходный набор штрихкодов в левой части графика, обозначенный B и с минимальным расстоянием d(B) между штрихкодами, имеет структуру, аналогичную набору B3, показанному на фиг. 2. На фиг. 3 показано, что расширение последовательностей B с помощью последовательности нуклеотидов на концах штрихкода, обозначенных стрелкой, помеченной как «EXT» для расширения, сохраняет штрихкодов 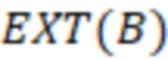 вложенную структуру поднабора в новом наборе при минимальном расстоянии между штрихкодами
вложенную структуру поднабора в новом наборе при минимальном расстоянии между штрихкодами 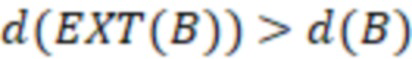 . В общем, имеем
. В общем, имеем 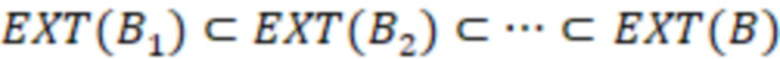 с
с 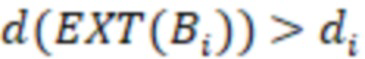 . Шаблон для получения вложенных наборов штрихкодов с вложенными последовательностями штрихкодов приведен ниже. Во-первых, мы выбираем количество поднаборов
. Шаблон для получения вложенных наборов штрихкодов с вложенными последовательностями штрихкодов приведен ниже. Во-первых, мы выбираем количество поднаборов  и количество подпоследовательностей
и количество подпоследовательностей  . Последнее равно количеству расширений плюс один. Затем мы определяем длины
. Последнее равно количеству расширений плюс один. Затем мы определяем длины 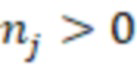 штрихкодов для
штрихкодов для 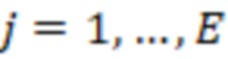 . Так как последующие расширения увеличивают длину штрихкода, нам требуется
. Так как последующие расширения увеличивают длину штрихкода, нам требуется 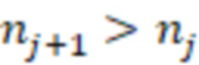 . Далее мы определяем расстояния
. Далее мы определяем расстояния 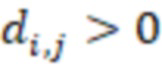 между штрихкодами для поднаборов
между штрихкодами для поднаборов 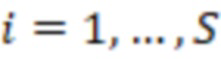 и подпоследовательностей . Нам требуется
и подпоследовательностей . Нам требуется 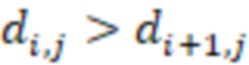 , так как увеличение размера набора штрихкодов уменьшает расстояние между штрихкодами, и нам требуется
, так как увеличение размера набора штрихкодов уменьшает расстояние между штрихкодами, и нам требуется 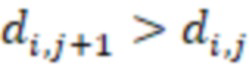 , так как расширение последовательностей штрихкодов увеличивает расстояние между штрихкодами. Мы ищем набор штрихкодов
, так как расширение последовательностей штрихкодов увеличивает расстояние между штрихкодами. Мы ищем набор штрихкодов 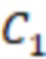 длиной
длиной 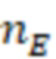 , которая удовлетворяет равенству
, которая удовлетворяет равенству 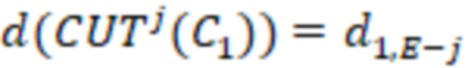 для
для  . Здесь функция
. Здесь функция 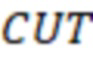 является противоположной функции
является противоположной функции  , то есть
, то есть 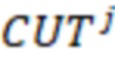 удаляет последние
удаляет последние 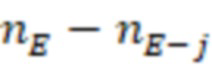 нуклеотиды из последовательностей длиной с
нуклеотиды из последовательностей длиной с 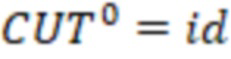 , где
, где 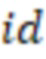 - оператор тождественности. Затем мы ищем набор штрихкодов
- оператор тождественности. Затем мы ищем набор штрихкодов 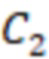 длиной с
длиной с 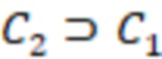 и
и 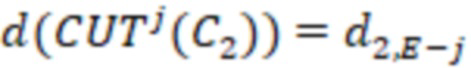 для
для  . Продолжаем таким образом, пока не найдем все
. Продолжаем таким образом, пока не найдем все 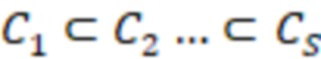 . Этот поиск похож на тот, который обсуждался в разделе 1, за исключением того, что мы ищем последовательности штрихкодов длиной , а не , которые должны удовлетворять равенству
. Этот поиск похож на тот, который обсуждался в разделе 1, за исключением того, что мы ищем последовательности штрихкодов длиной , а не , которые должны удовлетворять равенству 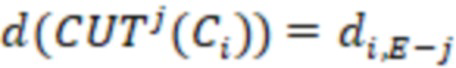 не только для
не только для 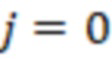 , но и для всех . Из
, но и для всех . Из 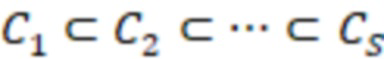 можно вывести
можно вывести 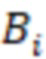 и
и 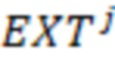 , полагая
, полагая 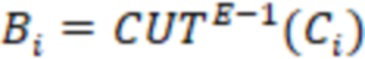 и
и 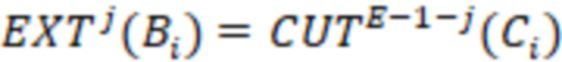 для . В дальнейшем мы будем обозначать вложенный набор штрихкодов с вложенными последовательностями как
для . В дальнейшем мы будем обозначать вложенный набор штрихкодов с вложенными последовательностями как 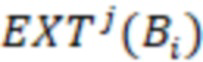 , где и .
, где и .
3. Меры секвенциального расстояния
Расстояние 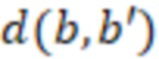 между штрихкодами должно отражать частоту, с которой штрихкод
между штрихкодами должно отражать частоту, с которой штрихкод 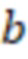 меняется на штрихкод
меняется на штрихкод 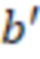 . Так как это связано со подобием последовательностей и , секвенциальное расстояние часто выбирается равным минимальному количеству операций, преобразующих последовательность в последовательность . Операции, рассматриваемые на таком секвенциальном расстоянии, зависят от типов ошибок, ожидаемых при обработке штрихкода. Если ожидаются только замены, то является расстоянием Хэмминга, смотри раздел 3.1. Если дополнительно учитываются вставки и удаления, то является расстоянием Левенштейна или связанным с ним расстоянием, смотри разделы 3.2 и 3.3. Так как совпадения между последовательностями также могут быть подсчитаны с учетом секвенциального расстояния, мы в дальнейшем будем включать совпадения в качестве операций, когда будем ссылаться на типы ошибок или классы ошибок.
. Так как это связано со подобием последовательностей и , секвенциальное расстояние часто выбирается равным минимальному количеству операций, преобразующих последовательность в последовательность . Операции, рассматриваемые на таком секвенциальном расстоянии, зависят от типов ошибок, ожидаемых при обработке штрихкода. Если ожидаются только замены, то является расстоянием Хэмминга, смотри раздел 3.1. Если дополнительно учитываются вставки и удаления, то является расстоянием Левенштейна или связанным с ним расстоянием, смотри разделы 3.2 и 3.3. Так как совпадения между последовательностями также могут быть подсчитаны с учетом секвенциального расстояния, мы в дальнейшем будем включать совпадения в качестве операций, когда будем ссылаться на типы ошибок или классы ошибок.
В разделе 3.4 - вероятность 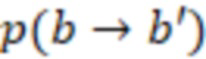 того, что преобразуется в . В отличие от секвенциального расстояния, не является минимальным количеством операций преобразования в . Скорее, это сумма вероятностей всех преобразований, которые заменяют на . В качестве альтернативы можно также установить
того, что преобразуется в . В отличие от секвенциального расстояния, не является минимальным количеством операций преобразования в . Скорее, это сумма вероятностей всех преобразований, которые заменяют на . В качестве альтернативы можно также установить 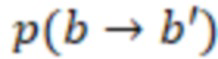 на среднее или максимальное значение вероятности всех преобразований, заменяющих на . Преимущество использования в качестве расстояния между штрихкодами состоит в том, что высокая/малая вероятность соответствует высокой/малой частоте преобразования. Это не всегда имеет место в случае секвенциального расстояния, так как штрихкоды с большим расстоянием могут заменяться друг на друга чаще, чем штрихкоды с малым расстоянием, например, если типы ошибок имеют разные вероятности.
на среднее или максимальное значение вероятности всех преобразований, заменяющих на . Преимущество использования в качестве расстояния между штрихкодами состоит в том, что высокая/малая вероятность соответствует высокой/малой частоте преобразования. Это не всегда имеет место в случае секвенциального расстояния, так как штрихкоды с большим расстоянием могут заменяться друг на друга чаще, чем штрихкоды с малым расстоянием, например, если типы ошибок имеют разные вероятности.
3.1. Расстояние Хэмминга
Расстояние Хэмминга между двумя последовательностями равно количеству замен, которые преобразуют в . Это идентично количеству позиций, в которых различаются последовательности и . Штрихкоды 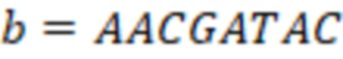 и
и 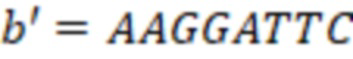 , например, различаются в позициях 3 и 7, и поэтому их расстояние Хэмминга равно 2. Расстояние Хэмминга является в правильном математическом смысле расстоянием, то есть оно обладает симметрией
, например, различаются в позициях 3 и 7, и поэтому их расстояние Хэмминга равно 2. Расстояние Хэмминга является в правильном математическом смысле расстоянием, то есть оно обладает симметрией 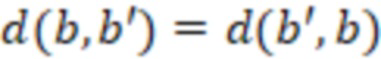 , подчиняется неравенству треугольника
, подчиняется неравенству треугольника 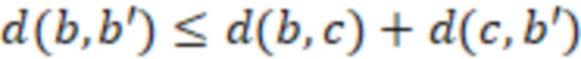 , и равенство
, и равенство 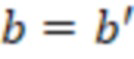 эквивалентно равенству
эквивалентно равенству 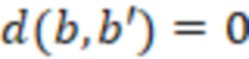 .
.
3.2. Расстояние Левенштейна
Расстояние Левенштейна между и равно минимальному количеству замен, вставок и делеций, необходимых для преобразования в . Это количество можно рассчитать с помощью алгоритма динамического программирования, как показано на фиг. 4. Здесь  и
и 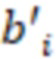 обозначают i-й нуклеотид последовательности и , обозначающие строки и столбцы матрицы
обозначают i-й нуклеотид последовательности и , обозначающие строки и столбцы матрицы 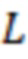 . Имеется дополнительная строка и столбец, помеченные
. Имеется дополнительная строка и столбец, помеченные  перед строкой
перед строкой 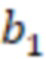 и столбцом
и столбцом 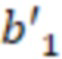 . Далее мы будем индексировать строки и столбцы как с помощью,
. Далее мы будем индексировать строки и столбцы как с помощью,  , так и с помощью
, так и с помощью  . Следовательно, мы имеем
. Следовательно, мы имеем 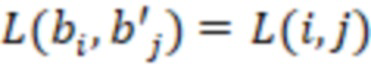 . Изначально содержит одно значение
. Изначально содержит одно значение  . График в середине фиг. 4 показывает, что
. График в середине фиг. 4 показывает, что 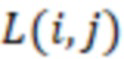 получено из
получено из 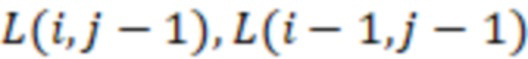 и
и 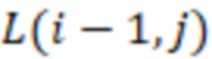 . Переходы в из
. Переходы в из 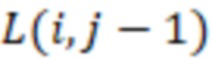 и соответствуют вставке и делеции, соответственно. Переход из
и соответствуют вставке и делеции, соответственно. Переход из 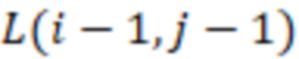 в соответствует совпадению, если
в соответствует совпадению, если 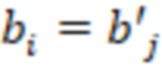 , и в противном случае замене. Матричный элемент можно рассчитать следующим образом
, и в противном случае замене. Матричный элемент можно рассчитать следующим образом
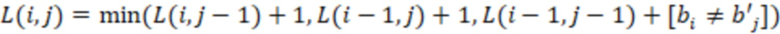 (1)
(1)
где  - скобка Айверсона, равная
- скобка Айверсона, равная  , если утверждение внутри истинно, и на противном случае. Аргументы
, если утверждение внутри истинно, и на противном случае. Аргументы 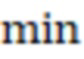 в (1) с несуществующими элементами в , то есть для
в (1) с несуществующими элементами в , то есть для 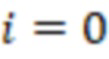 и , удаляются из уравнения. Алгоритм динамического программирования, представленный в уравнении (1), выполняется построчно с начала и до конца. Это делается для всех строк в последовательности, начиная со строки . Для строки и столбца это означает, что
и , удаляются из уравнения. Алгоритм динамического программирования, представленный в уравнении (1), выполняется построчно с начала и до конца. Это делается для всех строк в последовательности, начиная со строки . Для строки и столбца это означает, что 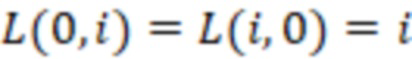 . После завершения выполнения алгоритма расстояние Левенштейна содержится в
. После завершения выполнения алгоритма расстояние Левенштейна содержится в 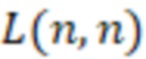 . Уравнение (1) показывает, что штрафы за вставки, делеции и замены равны . Эти штрафы могут быть изменены, если определенные типы ошибок являются более дорогостоящими или частыми, чем другие. Если вставки и делеции имеют одинаковый вес, расстояние Левенштейна является симметричным, в противном случае оно является несимметричным. Остальные свойства расстояния в математическом смысле всегда выполняются.
. Уравнение (1) показывает, что штрафы за вставки, делеции и замены равны . Эти штрафы могут быть изменены, если определенные типы ошибок являются более дорогостоящими или частыми, чем другие. Если вставки и делеции имеют одинаковый вес, расстояние Левенштейна является симметричным, в противном случае оно является несимметричным. Остальные свойства расстояния в математическом смысле всегда выполняются.
3.3. Расстояние Левенштейна с фиксированной рамкой
Обычное расстояние Левенштейна не является идеальным для измерения расстояния между штрихкодами, так как рамка последовательности для штрихкодов имеет постоянную ширину. Это означает, что если ожидается штрихкод длиной n секвенсор всегда будет считывать n нуклеотидов. В результате, если штрихкод имеет вставку, последний нуклеотид штрихкода смещается за пределы рамки последовательности штрихкода и, следовательно, не записывается. Если отсутствующий последний нуклеотид штрихкода считается ошибкой делеции, как в случае с расстоянием Левенштейна, то каждая вставка, которая не компенсируется делецией, будет считаться как 2 ошибки. Аналогично, делеция, не смещенная вставкой, будет считаться как 2 ошибки, так как нуклеотид, который входит в рамку в конце, будет интерпретирован как вставка. Это искусственное увеличение расстояния между штрихкодами может привести к неправильному выводу о том, что два штрихкода не похожи друг на друга и, следовательно, маловероятно, что они превратятся друг в друга, хотя на самом деле они похожи и вероятность скачкообразного изменения штрихкода является высокой. Таким образом, более подходящим расстоянием ошибки является вариант расстояния Левенштейна, в котором учитывается, что размер рамки последовательности штрихкода является фиксированным. Это расстояние, которое по-разному называют FREE-дивергенцией [3], расстоянием Левенштейна между последовательностями [1] или просто модифицированным расстоянием Левенштейна [4, 6], можно получить, присвоив вес 0 вставкам и делециям в последней строке и столбце матрицы , показанной на фиг. 4. Следовательно, вставки, входящие в рамку секвенирования после окончания штрихкода, не считаются ошибками, как и делеции, происходящие из-за того, что был достигнут конец рамки секвенирования. Это расстояние Левенштейна с фиксированной рамкой (ff-Levenshtein) не является правильной метрикой, так как оно не удовлетворяет неравенству треугольника. Это означает, что если два штрихкода имеют расстояние 3, может существовать другой штрихкод с расстоянием 1 от них обоих [3]. В этом случае расстояние между штрихкодами, равное 3, не гарантирует, что набор штрихкодов может исправить одну ошибку.
3.4. Вероятностное расстояние перехода последовательности
Подобие последовательностей, измеряемое по минимальному количеству операций, необходимых для преобразования последовательностей друг в друга, не всегда прямо коррелирует с частотой, с которой последовательности заменяются друг на друга. Последовательности с большим расстоянием Левенштейна, например, могут чаще преобразовываться друг в друга, чем последовательности с малым расстоянием Левенштейна, если операции, влияющие на преобразование в первом случае, происходят чаще, чем операции во втором случае. Поэтому вместо того, чтобы выработать наборы штрихкодов на основе минимального количества операций, целесообразно оптимизировать штрихкоды в отношении частоты или вероятности выполнения этих операций. Этот подход будет реализован в этом разделе путем исследования расстояний между штрихкодами на основе вероятностей переходов последовательностей (STP).
Далее мы будем использовать сокращения 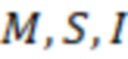 и
и 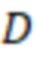 для совпадений, замен, вставок и делеций, соответственно. Учитывая распределение
для совпадений, замен, вставок и делеций, соответственно. Учитывая распределение 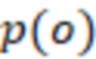 с вероятностей при
с вероятностей при 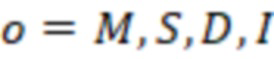 , вероятность того, что последовательность изменится на последовательность , может быть рассчитана путем модификации алгоритма, показанного на фиг. 4. В этом случае инициируется при
, вероятность того, что последовательность изменится на последовательность , может быть рассчитана путем модификации алгоритма, показанного на фиг. 4. В этом случае инициируется при 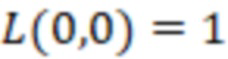 , и получается следующим образом:
, и получается следующим образом:
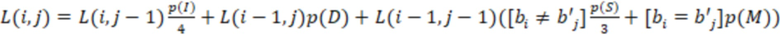 (2)
(2)
Как и прежде, члены в правой части (2) с неопределенным значением в , то есть для и , игнорируются. После завершения алгоритма имеем 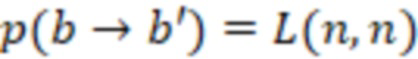 . В дальнейшем
. В дальнейшем  будет обозначать одно из:
будет обозначать одно из: 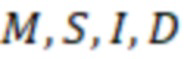 и ассоциированный с ним переход между элементами . Таким образом,
и ассоциированный с ним переход между элементами . Таким образом, 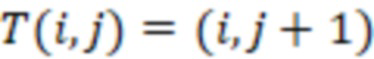 и
и 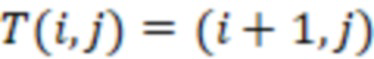 для
для 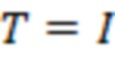 и
и 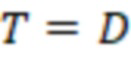 , тогда как
, тогда как 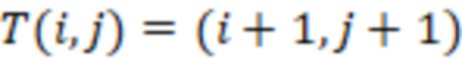 для
для 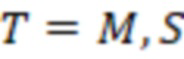 .
. 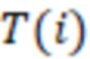 и
и 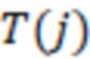 будет обозначать первый и второй компоненты
будет обозначать первый и второй компоненты 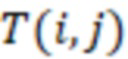 . В (2)
. В (2) 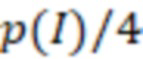 - вероятность вставки
- вероятность вставки 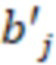 после , и
после , и 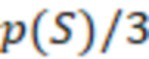 - вероятность замены на . Таким образом, из уравнения (2) получаем
- вероятность замены на . Таким образом, из уравнения (2) получаем
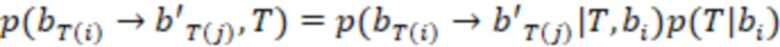 (3)
(3)
где 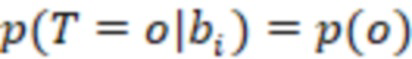 не зависит от , и
не зависит от , и 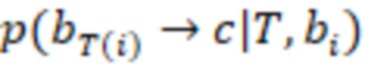 является однородным по всем возможным
является однородным по всем возможным  заданным нуклеотидам и . Например, для все 4 нуклеотида могут быть вставлены после и, таким образом,
заданным нуклеотидам и . Например, для все 4 нуклеотида могут быть вставлены после и, таким образом, 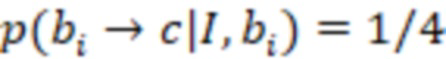 . Для
. Для 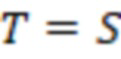 , с другой стороны,
, с другой стороны, 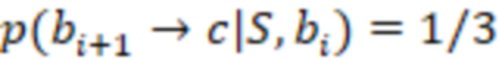 для
для 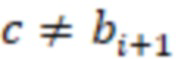 и
и 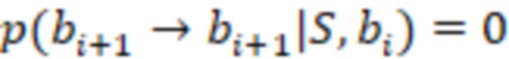 . Если (3) не выполняется или множители в правой части (3) неравномерны, то
. Если (3) не выполняется или множители в правой части (3) неравномерны, то 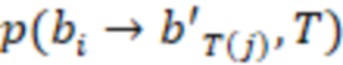 необходимо заменить на более подходящее распределение вероятностей. Аналогично расстоянию Левенштейна с фиксированной рамкой, можно избежать наказания за вставки и делеции вне рамки последовательности, установив
необходимо заменить на более подходящее распределение вероятностей. Аналогично расстоянию Левенштейна с фиксированной рамкой, можно избежать наказания за вставки и делеции вне рамки последовательности, установив 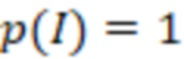 и
и 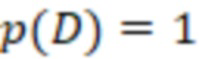 в последней строке и столбце . Если за штрихкодом всегда следует определенная последовательность
в последней строке и столбце . Если за штрихкодом всегда следует определенная последовательность  , например, последовательность адаптера, то можно добавить в , чтобы получить комбинированную последовательность
, например, последовательность адаптера, то можно добавить в , чтобы получить комбинированную последовательность 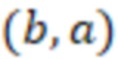 , для которой может быть рассчитана вероятность
, для которой может быть рассчитана вероятность 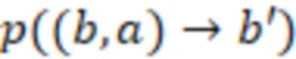 перехода, как и раньше. Однако необходимо отметить, что матрица для вычисления не является квадратной, и что конечным результатом является элемент в последней строке и столбце , то есть
перехода, как и раньше. Однако необходимо отметить, что матрица для вычисления не является квадратной, и что конечным результатом является элемент в последней строке и столбце , то есть 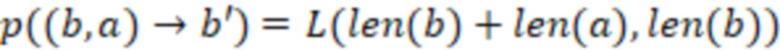 . Используя STP , мы определяем расстояние между и , которое должно удовлетворять равенству
. Используя STP , мы определяем расстояние между и , которое должно удовлетворять равенству 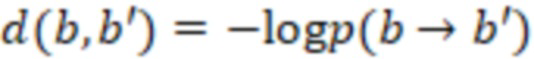 . Это гарантирует, что увеличение расстояния всегда соответствует уменьшению вероятности, которая преобразуется в . По сравнению с секвенциальным расстоянием значения для являются не целыми числами, и действительными числами, большими или равными нулю. Расстояние является симметричным, если
. Это гарантирует, что увеличение расстояния всегда соответствует уменьшению вероятности, которая преобразуется в . По сравнению с секвенциальным расстоянием значения для являются не целыми числами, и действительными числами, большими или равными нулю. Расстояние является симметричным, если 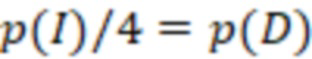 . Эквивалентность
. Эквивалентность 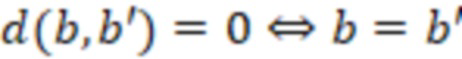 обычно не будет истинной, так как
обычно не будет истинной, так как 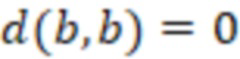 только в случае, если
только в случае, если 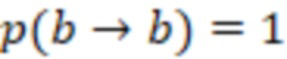 , что требует того, чтобы
, что требует того, чтобы 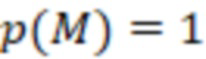 . В дополнение к этому, неравенство треугольника является неверно, так как
. В дополнение к этому, неравенство треугольника является неверно, так как 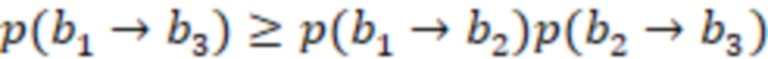 не выполняется в общем случае.
не выполняется в общем случае.
Оценка вероятностей классов ошибок
В этом разделе описывается оценка вероятностей 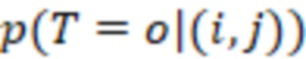 класса ошибок для записей
класса ошибок для записей  в матрице с учетом набора последовательностей
в матрице с учетом набора последовательностей 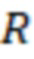 , полученных путем секвенирования штрихкодов в наборе
, полученных путем секвенирования штрихкодов в наборе 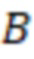 штрихкодов. Для этой цели показана вероятность выравниваний с . Выравнивание с представляет собой путь через матрицу, начинающийся в
штрихкодов. Для этой цели показана вероятность выравниваний с . Выравнивание с представляет собой путь через матрицу, начинающийся в 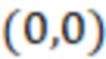 и заканчивающийся в
и заканчивающийся в 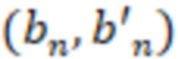 , так что за элементом
, так что за элементом 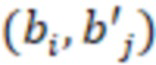 по пути следует
по пути следует 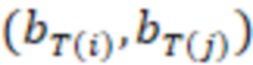 , где
, где 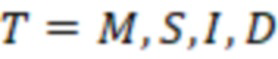 . В дальнейшем
. В дальнейшем 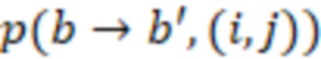 и
и 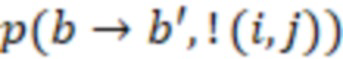 будут обозначать вероятность того, что преобразуется в с выравниванием, содержащим или не содержащим , соответственно. Далее мы будем обозначать
будут обозначать вероятность того, что преобразуется в с выравниванием, содержащим или не содержащим , соответственно. Далее мы будем обозначать 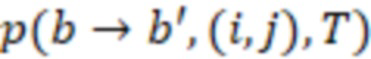 вероятность того, что преобразуется в с выравниванием, содержащим с последующей операцией . Эти вероятности будут рассчитаны с использованием следующей факторизации.
вероятность того, что преобразуется в с выравниванием, содержащим с последующей операцией . Эти вероятности будут рассчитаны с использованием следующей факторизации.
 (4)
(4)
где 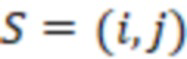 означает, что выравнивание
означает, что выравнивание 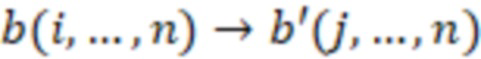 начинается с . Для расчета
начинается с . Для расчета 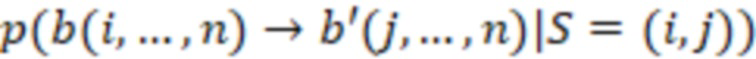 обратимся к алгоритму расчета . Для этого матрица
обратимся к алгоритму расчета . Для этого матрица 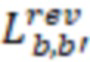 размером
размером 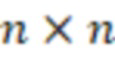 инициируется
инициируется 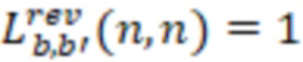 , и
, и 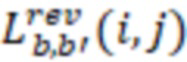 рассчитывается следующим образом:
рассчитывается следующим образом:
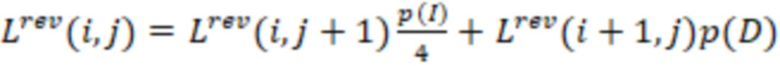 (5)
(5)
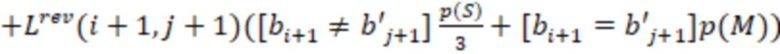
Этот алгоритм действует построчно справа налево и от последней к первой строке. Эта процедура показана на фиг. 5. После завершения алгоритма имеем
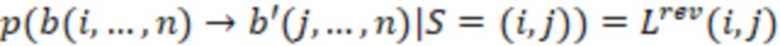 (6)
(6)
Так как все пути в начинаются при , из этого следует, что 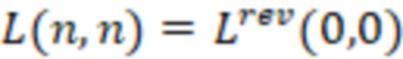 . Из (4) следует, что
. Из (4) следует, что
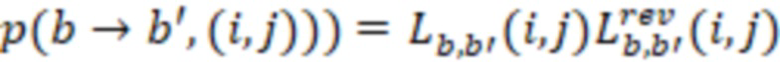 (7)
(7)
и, кроме того,
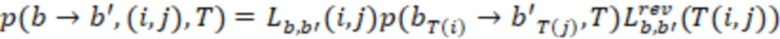 (8)
(8)
Мы используем (7) и (8) для оценки вероятности вслед за . Для этой цели предположим, что - это набор последовательностей, полученных путем упорядочивания штрихкодов в наборе штрихкодов. Мы начинаем с начальной оценки 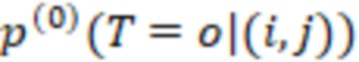 и вычисляем итеративно
и вычисляем итеративно
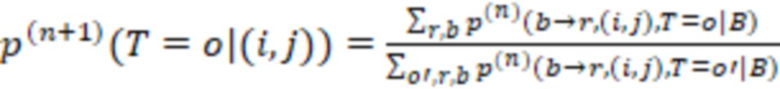 (9)
(9)
Мы обнаружили, что эта процедура сходится к правильному решению, если не слишком далеко от решения. Уравнение (9) вычисляет вероятность 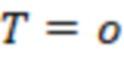 для каждой комбинации . Для расчета вероятности после
для каждой комбинации . Для расчета вероятности после 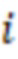 -ой позиции используем следующую итерационную схему.
-ой позиции используем следующую итерационную схему.
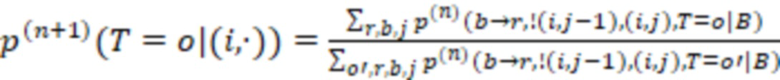 (10)
(10)
Как и в уравнении (9), мы обнаружили, что (10) сходится к правильному решению, если 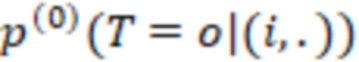 не слишком далеко от него. Наконец, для расчета общей вероятности наблюдения при выравнивании с мы использовали следующую итеративную процедуру.
не слишком далеко от него. Наконец, для расчета общей вероятности наблюдения при выравнивании с мы использовали следующую итеративную процедуру.
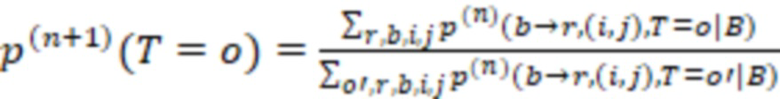 (11)
(11)
Как и в (9) и (10), мы также обнаружили, что (11) сходится к правильному решению. Так как (10) и (11) накапливают данные для нескольких комбинаций , этим процедурам для сходимости требуется меньше данных, чем в (9). Процедура (11) нужна меньше всего, так как она накапливает данные для всех комбинаций .
4. Выработка набора штрихкодов
4.1. Сведение к минимуму перекрестного загрязнения
Перекрестное загрязнение происходит тогда, когда последовательность 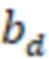 , считанная для штрихкода , совпадает с последовательностью другого штрихкода , или когда считывается последовательность, не являющаяся штрихкодом, которая исправлена на неправильный штрихкод. Первый тип перекрестного загрязнения, также называемый скачком штрихкода, особенно проблематичен, так как его невозможно обнаружить. Штрихкод, который преобразуется в другой штрихкод, кажется пользователю неотличимым от штрихкода, в который он преобразовался. Скачкообразное изменение штрихкода можно уменьшить путем поиска штрихкодов, находящихся на большом расстоянии друг от друга. В случае секвенциального расстояния большое расстояние гарантирует, что штрихкоды непохожи и требуют большого количества ошибок для преобразования друг в друга. В случае расстояния STP большое расстояние между штрихкодами напрямую связано с низкой вероятностью скачкообразного изменения штрихкода. Как правило, перед выработкой набора штрихкодов указывается минимальное расстояние между штрихкодами (MIB). Затем выработка начинается с начального набора штрихкодов , который может состоять из одного случайного штрихкода или заданного набора штрихкодов, смотри раздел 4.2. Исходный набор штрихкодов расширяется за счет добавления штрихкода при
, считанная для штрихкода , совпадает с последовательностью другого штрихкода , или когда считывается последовательность, не являющаяся штрихкодом, которая исправлена на неправильный штрихкод. Первый тип перекрестного загрязнения, также называемый скачком штрихкода, особенно проблематичен, так как его невозможно обнаружить. Штрихкод, который преобразуется в другой штрихкод, кажется пользователю неотличимым от штрихкода, в который он преобразовался. Скачкообразное изменение штрихкода можно уменьшить путем поиска штрихкодов, находящихся на большом расстоянии друг от друга. В случае секвенциального расстояния большое расстояние гарантирует, что штрихкоды непохожи и требуют большого количества ошибок для преобразования друг в друга. В случае расстояния STP большое расстояние между штрихкодами напрямую связано с низкой вероятностью скачкообразного изменения штрихкода. Как правило, перед выработкой набора штрихкодов указывается минимальное расстояние между штрихкодами (MIB). Затем выработка начинается с начального набора штрихкодов , который может состоять из одного случайного штрихкода или заданного набора штрихкодов, смотри раздел 4.2. Исходный набор штрихкодов расширяется за счет добавления штрихкода при 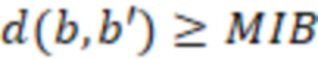 и
и 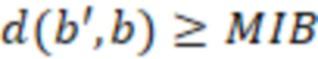 для всех
для всех 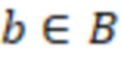 . Эти два неравенства будем называть условием MIB. Если расстояние является симметричным, необходимо проверить только одно из неравенств в условии MIB. Чтобы найти следующий элемент для добавления к , последовательно проверяются элементы в
. Эти два неравенства будем называть условием MIB. Если расстояние является симметричным, необходимо проверить только одно из неравенств в условии MIB. Чтобы найти следующий элемент для добавления к , последовательно проверяются элементы в 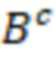 - дополнение к . В данном документе дополнением называется множество всех последовательностей одинаковой длины, элементы которых не содержатся в . Порядок, в котором проверяются элементы в , может быть случайным или следовать особому порядку , такому как лексикографический порядок [2], или быть комбинацией случайного и упорядоченного. Если все в рассмотрены, последовательность, в которой обрабатываются, не имеет значения. Если удовлетворяет условию MIB, он добавляется в набор потенциальных штрихкодов
- дополнение к . В данном документе дополнением называется множество всех последовательностей одинаковой длины, элементы которых не содержатся в . Порядок, в котором проверяются элементы в , может быть случайным или следовать особому порядку , такому как лексикографический порядок [2], или быть комбинацией случайного и упорядоченного. Если все в рассмотрены, последовательность, в которой обрабатываются, не имеет значения. Если удовлетворяет условию MIB, он добавляется в набор потенциальных штрихкодов 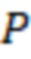 . Как только удовлетворяет требуемым свойствам, например, то, что он не пустой или что его нельзя увеличить в размерах, то
. Как только удовлетворяет требуемым свойствам, например, то, что он не пустой или что его нельзя увеличить в размерах, то 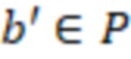 выбирается с помощью другой, возможно, случайной, процедуры и добавляется в . На этом этапе могут уже быть выполнены все требования к , предъявляемые к набору штрихкодов, например достаточный размер, в случае которого поиск будет завершен. Этот поиск штрихкода можно кратко изложить следующим образом.
выбирается с помощью другой, возможно, случайной, процедуры и добавляется в . На этом этапе могут уже быть выполнены все требования к , предъявляемые к набору штрихкодов, например достаточный размер, в случае которого поиск будет завершен. Этот поиск штрихкода можно кратко изложить следующим образом.
Алгоритм 1 поиска
1. Указать длину штрихкода , и 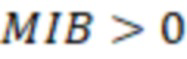 .
.
2. Инициировать набор штрихкодов со случайным штрихкодом или с заданным набором штрихкодов.
3. Инициировать , набор возможных штрихкодов, при 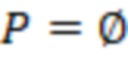 .
.
4. Исследовать последовательности 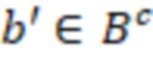 , где - дополнение в множестве последовательностей длиной , то есть набор всех последовательностей длиной , не содержащихся в . Если и для всех , добавить в набор потенциальных штрихкодов . Повторять этот этап до тех пор, пока не будет удовлетворять требуемым свойствам или не будут проверены все элементы .
, где - дополнение в множестве последовательностей длиной , то есть набор всех последовательностей длиной , не содержащихся в . Если и для всех , добавить в набор потенциальных штрихкодов . Повторять этот этап до тех пор, пока не будет удовлетворять требуемым свойствам или не будут проверены все элементы .
5. Если не удовлетворяет требуемым свойствам, завершить процедуру.
6. Выбрать и добавить в . Если удовлетворяет требуемым свойствам, завершить процедуру, в противном случае перейти к этапу 3.
Если мы ищем вложенные наборы штрихкодов с вложенными последовательностями, вышеупомянутая процедура должна быть изменена. В данном документе мы будем использовать обозначения из раздела 2, то есть - количество поднаборов, и - количество подпоследовательностей. В предыдущем алгоритме длина штрихкода заменяется на длины  штрихкодов, и минимальное расстояние между штрихкодами (MIB) заменяется на расстояния
штрихкодов, и минимальное расстояние между штрихкодами (MIB) заменяется на расстояния 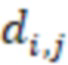 между штрихкодами, где , и . Кроме того, условие MIB должно выполняться для всех
между штрихкодами, где , и . Кроме того, условие MIB должно выполняться для всех 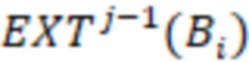 и . Как отмечалось в разделе 2, поиск вложенных наборов штрихкодов осуществляется путем поиска наборов
и . Как отмечалось в разделе 2, поиск вложенных наборов штрихкодов осуществляется путем поиска наборов 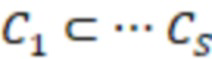 штрихкодов длиной . В частности, последовательность
штрихкодов длиной . В частности, последовательность 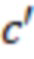 считается потенциальным штрихкодом-кандидатом для
считается потенциальным штрихкодом-кандидатом для 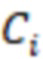 только в том случае, если
только в том случае, если 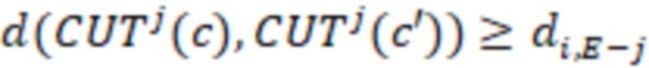 и
и 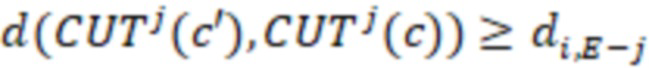 для всех
для всех 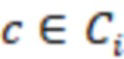 и . Модифицированная версия описанного выше поиска штрихкодов для вложенных наборов штрихкодов с вложенными последовательностями теперь выглядит следующим образом.
и . Модифицированная версия описанного выше поиска штрихкодов для вложенных наборов штрихкодов с вложенными последовательностями теперь выглядит следующим образом.
Алгоритм 2 поиска
1. Указать количество поднаборов 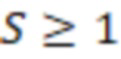 , количество подпоследовательностей и
, количество подпоследовательностей и 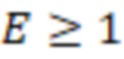 длиной штрихкодов и для и при , и расстояния
длиной штрихкодов и для и при , и расстояния 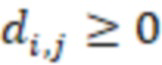 между штрихкодами при и .
между штрихкодами при и .
2. Установить 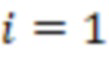 и инициировать набор штрихкодов
и инициировать набор штрихкодов 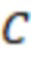 со случайным штрихкодом длиной или с заданным набором штрихкодов длиной .
со случайным штрихкодом длиной или с заданным набором штрихкодов длиной .
3. Инициировать , набор возможных штрихкодов, при .
4. Исследовать последовательности 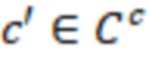 , где
, где 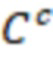 - дополнение в множестве последовательностей длиной . Если и для и все
- дополнение в множестве последовательностей длиной . Если и для и все 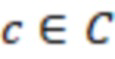 , добавить в набор потенциальных штрихкодов . Повторять этот этап до тех пор, пока не будет выполнены требуемые условия или не будут проверены все элементы .
, добавить в набор потенциальных штрихкодов . Повторять этот этап до тех пор, пока не будет выполнены требуемые условия или не будут проверены все элементы .
5. Если не удовлетворяет требуемым условиям, перейти к этапу 7.
6. Выбрать 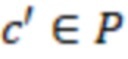 и добавить в . Если удовлетворяет требованиям для набора , перейти к этапу 7. В противном случае перейти к этапу 3.
и добавить в . Если удовлетворяет требованиям для набора , перейти к этапу 7. В противном случае перейти к этапу 3.
7. Присвоить 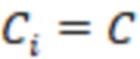 и: если
и: если 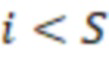 , установить
, установить 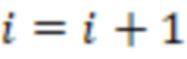 и перейти к этапу 3, если
и перейти к этапу 3, если 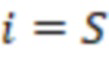 , установить и для , затем завершить процедуру.
, установить и для , затем завершить процедуру.
Следует отметить, что наборы штрихкодов, произведенные алгоритмом 2 поиска, содержат, для 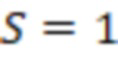 и
и 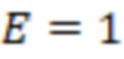 , наборы штрихкодов, произведенные алгоритмом 1 поиска. Для и
, наборы штрихкодов, произведенные алгоритмом 1 поиска. Для и 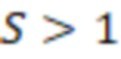 алгоритм 2 поиска производит вложенные наборы штрихкодов без вложенных последовательностей, и для алгоритм 2 поиска
алгоритм 2 поиска производит вложенные наборы штрихкодов без вложенных последовательностей, и для алгоритм 2 поиска 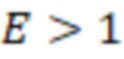 производит вложенные последовательности без вложенных поднаборов. Следовательно, алгоритм 2 поиска можно использовать для выборочной выработки наборов штрихкодов с несколькими вложенными поднаборами и/или последовательностями.
производит вложенные последовательности без вложенных поднаборов. Следовательно, алгоритм 2 поиска можно использовать для выборочной выработки наборов штрихкодов с несколькими вложенными поднаборами и/или последовательностями.
Далее мы будем ссылаться на набор вложенных штрихкодов с поднаборами, подпоследовательностями с длинами , где является расстоянием типа DTYPE, как DTYPE-S(). Следовательно, если был разработан для расстояния ff-Levenshtein при 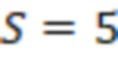 и
и 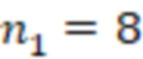 ,
, 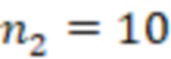 и
и 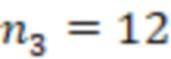 , вложенный набор штрихкодов будет упоминаться как ff-Levenshtein-5(8,10,12). Если , мы будем использовать DNAME(
, вложенный набор штрихкодов будет упоминаться как ff-Levenshtein-5(8,10,12). Если , мы будем использовать DNAME(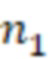 ), а не DNAME-1(), чтобы обратиться к набору штрихкодов с длиной . Следовательно, Hamming(6) относится к набору штрихкодов длиной 6, который был разработан для расстояния Хэмминга.
), а не DNAME-1(), чтобы обратиться к набору штрихкодов с длиной . Следовательно, Hamming(6) относится к набору штрихкодов длиной 6, который был разработан для расстояния Хэмминга.
Второй тип перекрестного загрязнения, упомянутый в начале этого раздела, является результатом ложного исправления ошибок. Здесь считанная последовательность (определенная последовательность) для штрихкода неверна и не совпадает с другим штрихкодом. Ошибка возникает тогда, когда нештрихкодовая последовательность назначается неправильному штрихкоду с помощью процедуры исправления ошибок. Набор последовательностей, скорректированных в штрихкоде b, является сферой декодирования штрихкода. Следовательно, чтобы гарантировать надлежащее исправление по меньшей мере минимального количества ошибок (MEC), необходимо проверить, что последовательности, выработанные с ошибками вплоть до MEC из штрихкода b, лежат в сфере b декодирования, и что сферы декодирования для разных штрихкодов не перекрываются. Нештрихкодовые последовательности c обычно исправляются на штрихкод с минимальным расстоянием. Если расстояние d подчиняется неравенству треугольника и 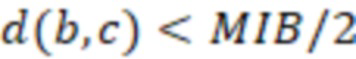 , подразумевается, что
, подразумевается, что 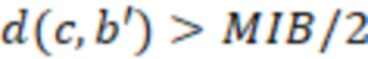 . Следовательно, если d является симметричным, и c был выработан из b с не более чем
. Следовательно, если d является симметричным, и c был выработан из b с не более чем 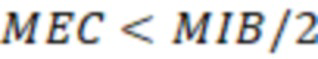 ошибками, то c лежит только в сфере b декодирования. Если расстояние d не симметрично, как в случае расстояния Левенштейна с неравными весами вставок и делеций, то необходимо дополнительно проверить то, выполняется ли неравенство
ошибками, то c лежит только в сфере b декодирования. Если расстояние d не симметрично, как в случае расстояния Левенштейна с неравными весами вставок и делеций, то необходимо дополнительно проверить то, выполняется ли неравенство 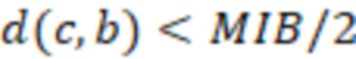 . Это подразумевает то, что
. Это подразумевает то, что 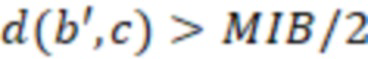 , и, таким образом, лежит только в сфере декодирования. Для расстояния, которое не подчиняется неравенству треугольника, этого обычно недостаточно, чтобы гарантировать, что было получено из с не более чем ошибками. Например, для расстояния Левенштейна с фиксированной рамкой можно найти последовательности
, и, таким образом, лежит только в сфере декодирования. Для расстояния, которое не подчиняется неравенству треугольника, этого обычно недостаточно, чтобы гарантировать, что было получено из с не более чем ошибками. Например, для расстояния Левенштейна с фиксированной рамкой можно найти последовательности 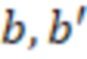 и , такие что
и , такие что 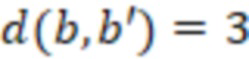 , но
, но 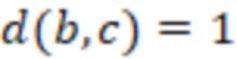 и
и 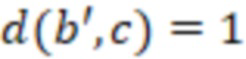 , смотри [3]. Таким образом, в этом случае при
, смотри [3]. Таким образом, в этом случае при 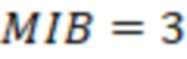 ,
, 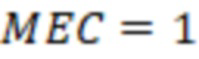 и сферы декодирования перекрываются. Следовательно, если неравенство треугольника не выполняется, обычно необходимо непосредственно проверять, что сферы декодирования не перекрываются. В дальнейшем мы всегда будем предполагать, что нештрихкодовая последовательность исправляется на с наименьшим расстоянием. Далее мы будем обозначать
и сферы декодирования перекрываются. Следовательно, если неравенство треугольника не выполняется, обычно необходимо непосредственно проверять, что сферы декодирования не перекрываются. В дальнейшем мы всегда будем предполагать, что нештрихкодовая последовательность исправляется на с наименьшим расстоянием. Далее мы будем обозначать 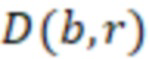 сферой с радиусом
сферой с радиусом  вокруг , которая является набором
вокруг , которая является набором 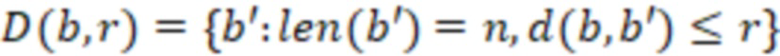 , и писать
, и писать 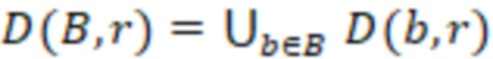 для сферы с радиусом вокруг набора штрихкодов. Поиск наборов штрихкодов в [3] осуществляется путем поиска с и , таких что
для сферы с радиусом вокруг набора штрихкодов. Поиск наборов штрихкодов в [3] осуществляется путем поиска с и , таких что 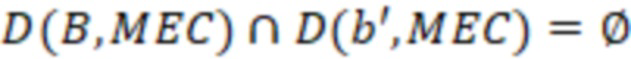 . Эту процедуру можно кратко изложить следующим образом.
. Эту процедуру можно кратко изложить следующим образом.
Алгоритм 3 поиска
1. Указать длину штрихкода 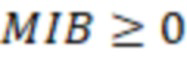 и
и 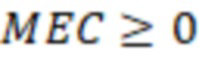 при
при 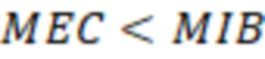 .
.
2. Инициировать набор штрихкодов со случайным штрихкодом или с заданным набором штрихкодов. Если сфера 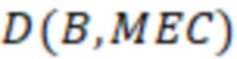 декодирования неизвестна, вычислить сферу .
декодирования неизвестна, вычислить сферу .
3. Инициировать , набор возможных штрихкодов, при .
4. Исследовать последовательности , где -дополнение в множестве последовательностей длиной . Если и для всех и , добавить в набор потенциальных штрихкодов . Повторять этот этап до тех пор, пока не будет удовлетворять требуемым свойствам или не будут проверены все элементы .
5. Если не удовлетворяет требуемым свойствам, завершить процедуру.
6. Выбрать и добавить в . Если удовлетворяет требуемым свойствам, завершить процедуру, в противном случае перейти к этапу 3.
Этот поиск штрихкода в вычислительном отношении более затратен, чем алгоритм 1 поиска. Это связано с тем, что вычисление 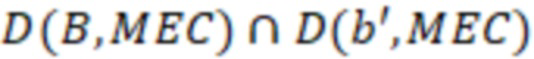 требует вычисления расстояния для всех последовательностей в . Для вложенных наборов штрихкодов с вложенными последовательностями приведенный выше алгоритм необходимо адаптировать следующим образом. Во-первых, необходимо определить расстояния
требует вычисления расстояния для всех последовательностей в . Для вложенных наборов штрихкодов с вложенными последовательностями приведенный выше алгоритм необходимо адаптировать следующим образом. Во-первых, необходимо определить расстояния 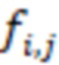 таким образом, чтобы
таким образом, чтобы 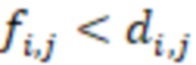 для и . Для этих расстояний мы требуем, чтобы исправление ошибок отображало последовательность длиной
для и . Для этих расстояний мы требуем, чтобы исправление ошибок отображало последовательность длиной 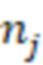 в
в 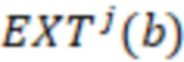 , если
, если 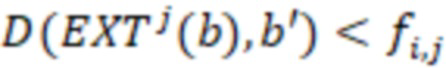 и
и 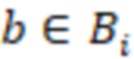 . В более общем случае мы требуем, чтобы
. В более общем случае мы требуем, чтобы 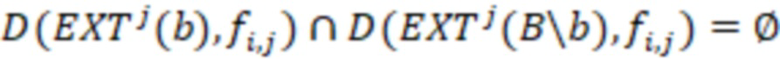 . Полный алгоритм приведен ниже.
. Полный алгоритм приведен ниже.
Алгоритм 4 поиска
1. Указать количество поднаборов , количество подпоследовательностей и с длинами штрихкодов для и при , и расстояния между штрихкодами при и . Кроме того, указать расстояния 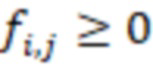 исправления ошибок при .
исправления ошибок при .
2. Установить и инициировать набор штрихкодов со случайным штрихкодом длиной или с заданным набором штрихкодов длиной . Вычислить сферы 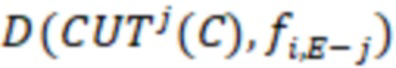 декодирования для .
декодирования для .
3. Инициировать , набор возможных штрихкодов, при .
4. Исследовать последовательности , где -дополнение в множестве последовательностей длиной . Если и для и все , и если, кроме того, 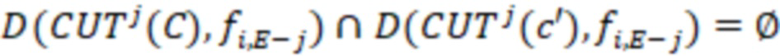 для , добавить в набор потенциальных штрихкодов . Повторять этот этап до тех пор, пока не будет удовлетворять требуемым условия или не будут проверены все элементы .
для , добавить в набор потенциальных штрихкодов . Повторять этот этап до тех пор, пока не будет удовлетворять требуемым условия или не будут проверены все элементы .
5. Если не удовлетворяет требуемым условиям, перейти к этапу 7.
6. Выбрать и добавить в . Если удовлетворяет требованиям для набора , перейти к этапу 7. В противном случае перейти к этапу 3.
7. Присвоить и: если , установить и перейти к этапу 3, если , установить 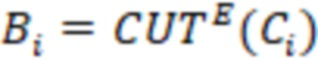 и
и 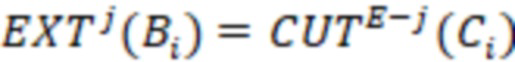 для , затем завершить процедуру.
для , затем завершить процедуру.
Аналогично алгоритму 2 поиска алгоритм поиска 4 можно также использовать для выборочной выработки наборов штрихкодов с несколькими вложенными поднаборами и/или последовательностями путем соответствующего выбора параметров и .
Вложенные наборы штрихкодов при , и 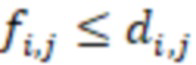 могут быть выработаны без точного определения и на этапе 1 в алгоритмах 2 и 4 поиска. Соответствующий алгоритм поиска приведен ниже, где мы будем использовать обозначения
могут быть выработаны без точного определения и на этапе 1 в алгоритмах 2 и 4 поиска. Соответствующий алгоритм поиска приведен ниже, где мы будем использовать обозначения 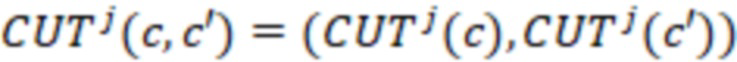 и
и 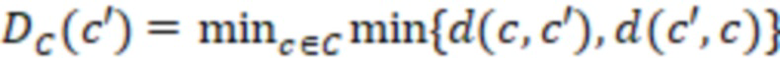 . Последнее можно интерпретировать как расстояние от до .
. Последнее можно интерпретировать как расстояние от до .
Алгоритм 5 поиска
1. Указать количество поднаборов 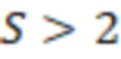 , количество расширений
, количество расширений 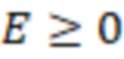 и для и
и для и  длины штрихкодов при .
длины штрихкодов при .
2. Установить и инициировать набор штрихкодов со случайным штрихкодом длиной 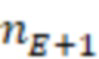 . Инициировать
. Инициировать 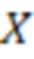 , набор штрихкодов, которые необходимо исключить из поиска, при
, набор штрихкодов, которые необходимо исключить из поиска, при 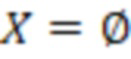 .
.
3. Присвоить 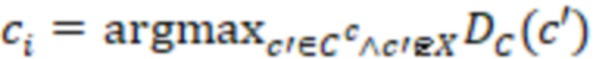 .
.
4. Если , перейти к этапу 6.
5. Установить 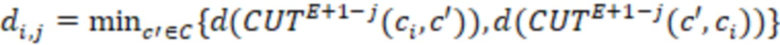 . Если
. Если 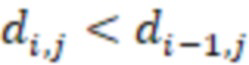 и
и 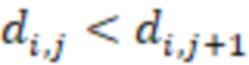 для , перейти к этапу 6, в противном случае установить
для , перейти к этапу 6, в противном случае установить 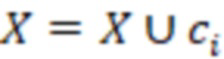 и перейти к этапу 3, если только
и перейти к этапу 3, если только 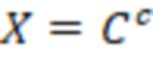 .
.
6. Установить 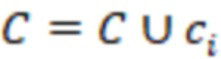 , ,
, , 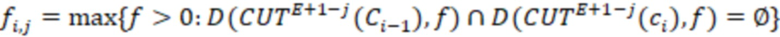 и . Если , установить , и перейти к этапу 3, в противном случае завершить процедуру.
и . Если , установить , и перейти к этапу 3, в противном случае завершить процедуру.
Описанная выше процедура вырабатывает последовательность вложенных наборов штрихкодов при 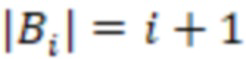 . Максимум на этапе 3 вышеприведенного алгоритма может быть не уникальным. В этом случае
. Максимум на этапе 3 вышеприведенного алгоритма может быть не уникальным. В этом случае 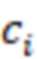 необходимо выбрать из множества всех с максимальным расстоянием от . Это происходит, в частности, когда расстояние
необходимо выбрать из множества всех с максимальным расстоянием от . Это происходит, в частности, когда расстояние 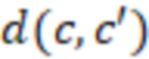 , такое как секвенциальное расстояние, принимает конечное число значений. Если значения являются непрерывными, как для расстояния STP, максимум на этапе 3 и, таким образом, , будут в общем уникальны.
, такое как секвенциальное расстояние, принимает конечное число значений. Если значения являются непрерывными, как для расстояния STP, максимум на этапе 3 и, таким образом, , будут в общем уникальны.
4.2. Предотвращение нежелательных штрихкодовых последовательностей
Существуют различные ситуации, в которых может потребоваться удалить последовательности из набора возможных штрихкодов. Это, например, относится к случаю, когда последовательность имеет низкую эффективность амплификации. Штрихкодирование образца с такой последовательностью может привести к тому, что образец получит значительно меньшую долю полосы, чем другие мультиплексные образцы. Во избежание такой последовательности, поиск штрихкода в разделе 4.1 должен быть немного изменен. Вместо того, чтобы проверять все последовательности , или в или , соответственно, следует проверять только или , а не те последовательности, которые должны быть исключены. Другая ситуация, вызывающая проблемы при демультиплексировании, возникает тогда, когда секвенирование вырабатывает неправильные, но частые последовательности, не ассоциированные ни с одним штрихкодом. Такие последовательности могут, например, появляться тогда, когда индекс не связан должным образом с фрагментом. В этом случае некоторые секвенсоры часто вырабатывать последовательность, почти полностью состоящую из G. Такие искусственные последовательности, не ассоциированные со штрихкодом, будут иметь негативное влияние на исправление ошибок, если они будут присвоены ближайшему штрихкоду. Во избежание этой проблемы, такие искусственные последовательности не должны содержаться в сфере декодирования любого штрихкода. Если искусственные последовательности сами по себе могут иметь варианты, то сфера вокруг искусственных последовательностей, содержащих эти варианты, не должна пересекаться со сферой декодирования любого штрихкода. Эту проблему можно решить, добавив искусственные последовательности (последовательности сравнительных индексов) к начальному набору штрихкодов, из которых поиск в разделе 4 вырабатывает полные наборы штрихкодов. Это гарантирует, что сферы декодирования результирующих штрихкодов не перекрываются сферами декодирования искусственных последовательностей. После завершения поиска штрихкода искусственные последовательности удаляются из окончательного набора штрихкодов. В качестве искусственных последовательностей перед началом поиска штрихкода можно добавлять последовательности, полностью состоящие из A, C, G или T, к набору 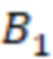 штрихкодов.
штрихкодов.
4.3. Позиционное балансирование нуклеотидов
Набор штрихкодов, используемый для мультиплексирования образцов при прогоне RNA-Seq, должен иметь сбалансированное распределение нуклеотидов в каждой позиции штрихкода. Неравномерное распределение может привести к низким показателям качества или низкой скорости полосовой фильтрации. Для достижения сбалансированного распределения нуклеотидов для  образцов при прогоне RNA-Seq с вложенным набором
образцов при прогоне RNA-Seq с вложенным набором 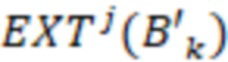 штрихкодов необходимо выбрать соответствующий набор штрихкодов при
штрихкодов необходимо выбрать соответствующий набор штрихкодов при 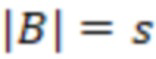 из
из 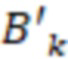 , где
, где 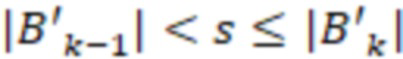 . Для получения такого набора можно использовать следующий способ выбора. Этот способ позволяет оценить все возможные поднаборы,
. Для получения такого набора можно использовать следующий способ выбора. Этот способ позволяет оценить все возможные поднаборы, 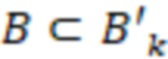 удовлетворяющие с помощью поиска A
удовлетворяющие с помощью поиска A . Для часто используемых номеров
. Для часто используемых номеров 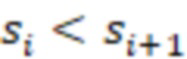 образцов, мультиплексированных при прогоне RNA-Seq, мы использовали этот способ выбора для получения вложенного набора штрихкодов при
образцов, мультиплексированных при прогоне RNA-Seq, мы использовали этот способ выбора для получения вложенного набора штрихкодов при 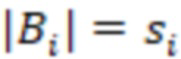 . Начнем с выбора из , где
. Начнем с выбора из , где 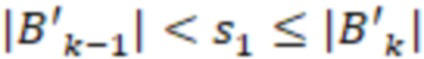 . Далее выбираем
. Далее выбираем 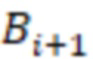 из , где таким образом, чтобы
из , где таким образом, чтобы 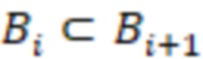 . Так как многочисленные могут удовлетворять выражению
. Так как многочисленные могут удовлетворять выражению 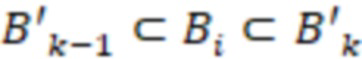 , возможно, что
, возможно, что 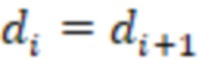 для некоторых . Таким образом, такой вложенный набор штрихкодов представляет собой небольшую вариацию концепции, рассмотренной до сих пор.
для некоторых . Таким образом, такой вложенный набор штрихкодов представляет собой небольшую вариацию концепции, рассмотренной до сих пор.
Распределение нуклеотидов набора штрихкодов в позиции  задано в виде
задано в виде 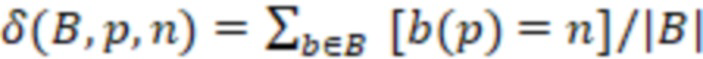 , где
, где 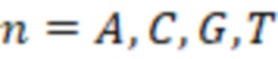 . Если
. Если  не используются, то это распределение будет записано просто как
не используются, то это распределение будет записано просто как 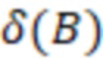 . Мы измеряем расстояние
. Мы измеряем расстояние 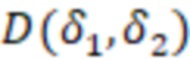 между двумя позиционными распределениями
между двумя позиционными распределениями 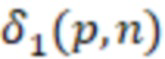 и
и 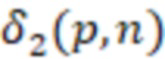 нуклеотидов следующим образом:
нуклеотидов следующим образом: 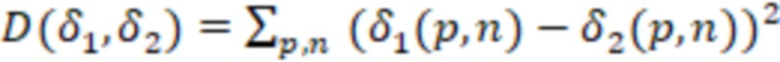 и обозначаем
и обозначаем 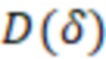 расстоянием
расстоянием 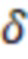 до равномерного позиционного распределения нуклеотидов (UPND), то есть
до равномерного позиционного распределения нуклеотидов (UPND), то есть 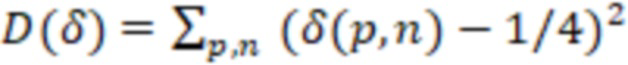 . Чтобы найти набор штрихкодов размером в другом наборе
. Чтобы найти набор штрихкодов размером в другом наборе 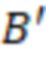 штрихкодов, мы используем следующий поиск A. Предположим, что поднабор
штрихкодов, мы используем следующий поиск A. Предположим, что поднабор 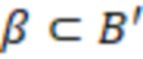 при
при  уже выбран. Мы хотим найти нижнюю границу для
уже выбран. Мы хотим найти нижнюю границу для 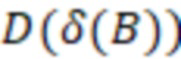 , учитывая, что
, учитывая, что 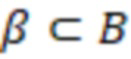 . Штрихкодовая последовательность , дающая наименьшее значение
. Штрихкодовая последовательность , дающая наименьшее значение 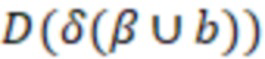 , содержит в каждой позиции нуклеотид с наименьшей частотой в
, содержит в каждой позиции нуклеотид с наименьшей частотой в 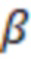 , то есть
, то есть 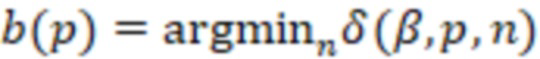 . Если не является единственно возможным, то он выбирается случайным образом из всех , минимизирующих
. Если не является единственно возможным, то он выбирается случайным образом из всех , минимизирующих 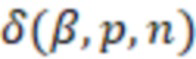 . Нуклеотидная последовательность не обязательно содержится в . Следовательно, добавление одного штрихкода из в позволяет потенциально получить расстояние от UPND, которое больше, чем . Если установить
. Нуклеотидная последовательность не обязательно содержится в . Следовательно, добавление одного штрихкода из в позволяет потенциально получить расстояние от UPND, которое больше, чем . Если установить 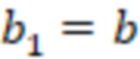 ,
, 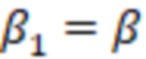 и
и 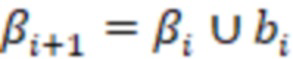 , то повторное применение этой конструкции дает последовательности
, то повторное применение этой конструкции дает последовательности 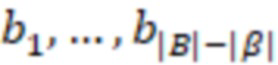 штрихкодовых последовательностей. Мы используем
штрихкодовых последовательностей. Мы используем  в качестве нижней границы для расстояния от UPND для поднабора
в качестве нижней границы для расстояния от UPND для поднабора 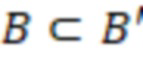 при . В нашем поиске A мы используем первый подход для глубины. Чтобы найти размер с минимальным расстоянием
при . В нашем поиске A мы используем первый подход для глубины. Чтобы найти размер с минимальным расстоянием 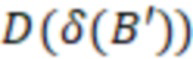 , мы последовательно вырабатываем все поднаборы штрихкодов размером . Поднаборы размера сами по себе вырабатываются путем добавления одного штрихкода за другим. Когда новый штрихкод добавляется к поднабору , мы вычисляем нижнюю границу выше для , заданного таким образом, чтобы
, мы последовательно вырабатываем все поднаборы штрихкодов размером . Поднаборы размера сами по себе вырабатываются путем добавления одного штрихкода за другим. Когда новый штрихкод добавляется к поднабору , мы вычисляем нижнюю границу выше для , заданного таким образом, чтобы 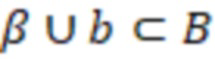 . Если эта оценка лежит выше или равна расстоянию для набора размером , для которого расстояние до UPND уже рассчитано, то
. Если эта оценка лежит выше или равна расстоянию для набора размером , для которого расстояние до UPND уже рассчитано, то 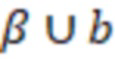 и все поднаборы, содержащие , удаляются из поиска. Это значительно сокращает количество наборов штрихкодов, которые необходимо проверить, и позволяет во многих случаях найти набор штрихкодов при и минимальное значение .
и все поднаборы, содержащие , удаляются из поиска. Это значительно сокращает количество наборов штрихкодов, которые необходимо проверить, и позволяет во многих случаях найти набор штрихкодов при и минимальное значение .
5. Размещение штрихкодов на луночных планшетах.
Как упоминалось во введении, наборы 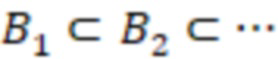 штрихкодов используются для экспериментов с различным количеством образцов. Если пользователь имеет не более
штрихкодов используются для экспериментов с различным количеством образцов. Если пользователь имеет не более 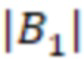 образцов, он будет использовать штрихкоды из набора . Если имеется более чем и не более
образцов, он будет использовать штрихкоды из набора . Если имеется более чем и не более 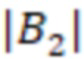 образцов, будут использоваться штрихкоды из набора
образцов, будут использоваться штрихкоды из набора 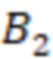 . Минимальное количество штрихкодов, которое может, по меньшей мере теоретически, иметь UPND, равно 4. Следовательно, должно содержать не менее 4 штрихкодов. Так как количество образцов в эксперименте часто кратно 8, разумно требовать, чтобы
. Минимальное количество штрихкодов, которое может, по меньшей мере теоретически, иметь UPND, равно 4. Следовательно, должно содержать не менее 4 штрихкодов. Так как количество образцов в эксперименте часто кратно 8, разумно требовать, чтобы 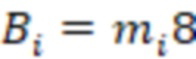 для
для 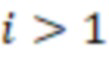 , где
, где 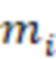 - целое положительное число. Чтобы сделать пипетирование таких наборов штрихкодов более удобным или легко автоматизируемым, целесообразно размещать их на луночных планшетах таким образом, чтобы штрихкоды были сгруппированы вместе. Возможное размещение на луночном планшете размером 8x12, где наборы штрихкодов размером 4, 8, 16, 24, 96 сгруппированы в столбцы, показано на фиг. 6. Здесь лунки A1-D1 содержат штрихкоды в , лунки A1-H1 содержат штрихкоды в , столбцы 1 и 2 содержат штрихкоды в
- целое положительное число. Чтобы сделать пипетирование таких наборов штрихкодов более удобным или легко автоматизируемым, целесообразно размещать их на луночных планшетах таким образом, чтобы штрихкоды были сгруппированы вместе. Возможное размещение на луночном планшете размером 8x12, где наборы штрихкодов размером 4, 8, 16, 24, 96 сгруппированы в столбцы, показано на фиг. 6. Здесь лунки A1-D1 содержат штрихкоды в , лунки A1-H1 содержат штрихкоды в , столбцы 1 и 2 содержат штрихкоды в 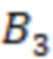 , и столбцы 1, 2 и 3 содержат штрихкоды в
, и столбцы 1, 2 и 3 содержат штрихкоды в 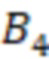 . Полный набор штрихкодов во всех лунках составляет
. Полный набор штрихкодов во всех лунках составляет 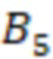 . Если наборы штрихкодов могут быть расширены до
. Если наборы штрихкодов могут быть расширены до 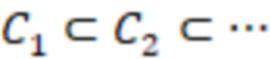 , то штрихкоды длиной
, то штрихкоды длиной 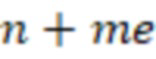 в содержатся в лунках луночного планшета и сгруппированы таким образом, как это описано для .
в содержатся в лунках луночного планшета и сгруппированы таким образом, как это описано для .
6. Уменьшение и количественная оценка перекрестного загрязнения с помощью двойных индексов.
Штрихкоды могут использоваться как одиночные или двойные индексы на секвенаторе Illumina [5]. При одноиндексном прогоне RNA-Seq штрихкод 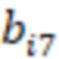 получается из набора
получается из набора 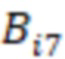 штрихкодов (например, выбранный из SEQ ID NO: 1-104 и SEQ ID NO: 209-496), «индекс i7», перед адаптером P7. При двухиндексном прогоне второй штрихкод
штрихкодов (например, выбранный из SEQ ID NO: 1-104 и SEQ ID NO: 209-496), «индекс i7», перед адаптером P7. При двухиндексном прогоне второй штрихкод 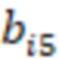 получается из другого набора
получается из другого набора 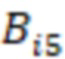 штрихкодов (например, выбранного из SEQ ID NO: 105-208 и SEQ ID NO: 497-784), «индекс i5», перед адаптером P5. Эта настройка показана на фиг. 7. Здесь штрихкодовые последовательности - и наборы штрихкодов могут быть идентичными. На фиг. 7 показано, что и секвенируются в одном и том же направлении, как прочтение 1 и прочтение 2, производя последовательности
штрихкодов (например, выбранного из SEQ ID NO: 105-208 и SEQ ID NO: 497-784), «индекс i5», перед адаптером P5. Эта настройка показана на фиг. 7. Здесь штрихкодовые последовательности - и наборы штрихкодов могут быть идентичными. На фиг. 7 показано, что и секвенируются в одном и том же направлении, как прочтение 1 и прочтение 2, производя последовательности 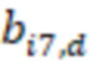 и
и 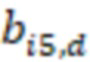 , где потенциально
, где потенциально 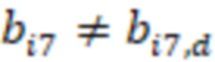 и
и 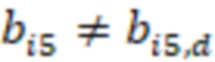 . Доля
. Доля 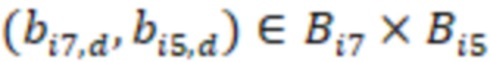 будет называться чистотой прогона RNA-Seq. Для мультиплексирования образцов при двухиндексном прогоне RNA-Seq необходимо выбрать поднабор кортежей
будет называться чистотой прогона RNA-Seq. Для мультиплексирования образцов при двухиндексном прогоне RNA-Seq необходимо выбрать поднабор кортежей 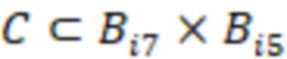 штрихкодов того же размера, что и количество образцов. Затем каждый образец при прогоне RNA-Seq помечается уникальной комбинацией
штрихкодов того же размера, что и количество образцов. Затем каждый образец при прогоне RNA-Seq помечается уникальной комбинацией 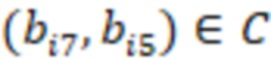 штрихкодов. Чтобы свести к минимуму перекрестное загрязнение, следует выбирать таким образом, чтобы и
штрихкодов. Чтобы свести к минимуму перекрестное загрязнение, следует выбирать таким образом, чтобы и 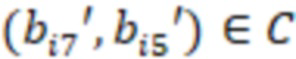 , при этом подразумевается, что
, при этом подразумевается, что 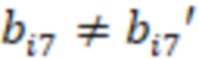 и
и 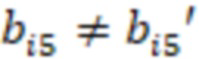 . Такой набор будет называться уникальным компонентным двухиндексным набором штрихкодов (UCDI). UCDI гарантирует, что скачкообразный переход штрихкода либо в , либо в приведет к выработке кортежа штрихкодов, не содержащегося в . Таким образом, для UCDI можно обнаружить скачкообразный переход одного штрихкода. Это является преимуществом по сравнению с одноиндексными прогонами RNA-Seq, в которых скачкообразные переходы штрихкода не обнаруживаются. Однако UCDI не может скорректировать скачкообразный переход одного штрихкода, так как для
. Такой набор будет называться уникальным компонентным двухиндексным набором штрихкодов (UCDI). UCDI гарантирует, что скачкообразный переход штрихкода либо в , либо в приведет к выработке кортежа штрихкодов, не содержащегося в . Таким образом, для UCDI можно обнаружить скачкообразный переход одного штрихкода. Это является преимуществом по сравнению с одноиндексными прогонами RNA-Seq, в которых скачкообразные переходы штрихкода не обнаруживаются. Однако UCDI не может скорректировать скачкообразный переход одного штрихкода, так как для 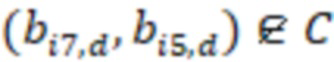 и он имеет место, и в общем случае невозможно утверждать то, выполняются ли условия
и он имеет место, и в общем случае невозможно утверждать то, выполняются ли условия 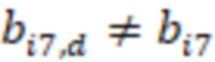 или
или 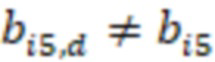 . Как следствие, фрагменты с ассоциированными и должны быть удалены из дальнейшего последующего анализа. Кортеж получается в любом из трех взаимоисключающих случаев,
. Как следствие, фрагменты с ассоциированными и должны быть удалены из дальнейшего последующего анализа. Кортеж получается в любом из трех взаимоисключающих случаев,
1. 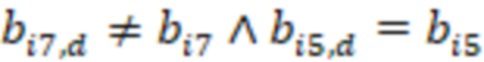
2. 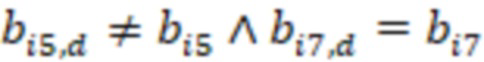
3. 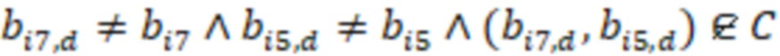
Следовательно, вероятность наблюдения представляет собой сумму вероятностей описанных выше случаев. В достаточно чистом прогоне RNA-Seq одновременный скачкообразный переход двух штрихкодов крайне маловероятен, и поэтому можно пренебречь вероятностью того, что 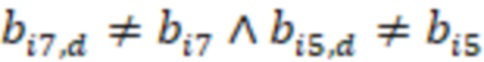 . Аналогичным образом, подразумевает, что
. Аналогичным образом, подразумевает, что 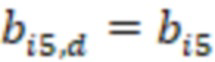 , и подразумевает, что
, и подразумевает, что 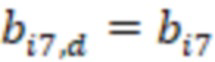 . Следовательно, для достаточно чистого прогона RNA-Seq вероятность того, что , приблизительно определяется следующей суммой:
. Следовательно, для достаточно чистого прогона RNA-Seq вероятность того, что , приблизительно определяется следующей суммой:
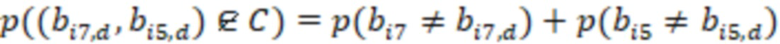 (12)
(12)
Следовательно, верхний порог для вероятности скачкообразного перехода штрихкода в и определяется выражением
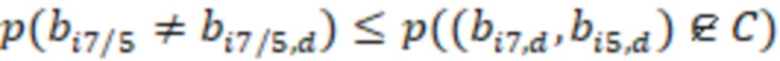 (13)
(13)
Здесь 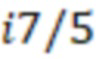 означает, что
означает, что 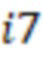 и
и 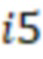 могут использоваться взаимозаменяемо. Верхняя граница в (13) является точной, только в том случае, если одно из
могут использоваться взаимозаменяемо. Верхняя граница в (13) является точной, только в том случае, если одно из 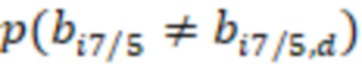 равно нулю. Это означает, что за все ошибки отвечает только один набор
равно нулю. Это означает, что за все ошибки отвечает только один набор 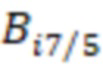 штрихкодов. Однако на практике более вероятно то, что оба набора штрихкодов вносят одинаковый вклад в наблюдаемые ошибки, и поэтому вероятности в левой части (13) будут ближе к половине правой части (13). Для уравнение (13) принимает вид:
штрихкодов. Однако на практике более вероятно то, что оба набора штрихкодов вносят одинаковый вклад в наблюдаемые ошибки, и поэтому вероятности в левой части (13) будут ближе к половине правой части (13). Для уравнение (13) принимает вид:
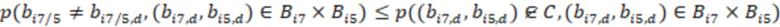 (14)
(14)
Левая часть (14) представляет собой вероятность того, что скачкообразный переход штрихкода происходит в или , в то время как правая часть (14) может быть оценена путем демультиплексирования прогона RNA-Seq по отношению ко всем комбинациям штрихкодов в  и вычисления соотношения в кортежах при . Следовательно, двухиндексный прогон RNA-Seq можно использовать для получения верхней границы вероятности скачкообразного перехода штрихкода в эксперименте с одним индексом. Вероятность наблюдения скачкообразного перехода штрихкода в двухиндексном прогоне RNA-Seq с использованием штрихкодов из является произведением вероятностей , удовлетворяющих уравнению (12). Это произведение становится максимальным в том случае, если
и вычисления соотношения в кортежах при . Следовательно, двухиндексный прогон RNA-Seq можно использовать для получения верхней границы вероятности скачкообразного перехода штрихкода в эксперименте с одним индексом. Вероятность наблюдения скачкообразного перехода штрихкода в двухиндексном прогоне RNA-Seq с использованием штрихкодов из является произведением вероятностей , удовлетворяющих уравнению (12). Это произведение становится максимальным в том случае, если 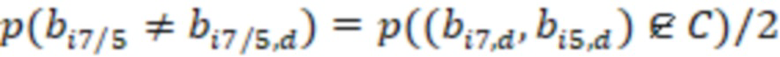 . Следовательно, вероятность наблюдения скачкообразного перехода штрихкода при двухиндексном прогоне RNA-Seq ограничивается выражением:
. Следовательно, вероятность наблюдения скачкообразного перехода штрихкода при двухиндексном прогоне RNA-Seq ограничивается выражением:
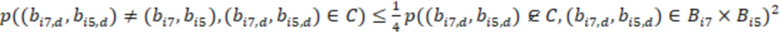 (15)
(15)
Если за демультиплексированием (то есть присвоением секвенирующих прочтений образцам или индексным последовательностям) следует исправление ошибок, вероятность одновременного скачкообразного перехода штрихкода и увеличивается. Таким образом, уравнение (12), где 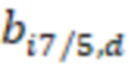 обозначают последовательности, полученные исправлением ошибок, будет неточным. Однако, так как верхняя граница для скачкообразного перехода штрихкода с одним индексом является довольно консервативной, мы используем (14) в этом случае в качестве верхней границы. Таким образом, прогон RNA-Seq с UCDI можно использовать для:
обозначают последовательности, полученные исправлением ошибок, будет неточным. Однако, так как верхняя граница для скачкообразного перехода штрихкода с одним индексом является довольно консервативной, мы используем (14) в этом случае в качестве верхней границы. Таким образом, прогон RNA-Seq с UCDI можно использовать для:
1. Обнаружения, но не исправления скачкообразного перехода штрихкода одного штрихкода после секвенирования или исправления ошибок.
2. Получения верхней границы для вероятности скачкообразного перехода штрихкода с одним индексом (14) и с двойным индексом (15) после секвенирования или исправления ошибок. Правая часть неравенства (14) получена путем демультиплексирования по отношению ко всем кортежам штрихкодов в и вычисления соотношения в наборе всех , где представляют собой индексы i7/5 после упорядочивания или исправления ошибок.
Предпочтительно настоящее изобретение определяется следующими пронумерованными вариантами осуществления, которые, конечно, могут быть дополнительно объединены с любым аспектом или вариантом осуществления или вариантами, описанными в данном документе:
1. Набор олигонуклеотидов, содержащий индексные последовательности, и где набор содержит множество поднаборов олигонуклеотидов с разными индексными последовательностями, где индексные последовательности поднабора олигонуклеотидов отличаются друг от друга по меньшей мере ненулевым количеством изменений последовательностей; и где набор содержит не менее 2-х иерархических уровней поднаборов, где индексные последовательности поднабора более высокого уровня являются элементами поднабора более низкого уровня, и где индексные последовательности поднабора более низкого уровня отличаются друг от друга меньшим минимальным количеством изменений последовательностей, чем индексные последовательности поднабора более высокого уровня; и где олигонуклеотиды присваиваются одному или нескольким поднаборам.
2. Набор по п. 1, в котором каждая индексная последовательность поднабора содержит усеченную индексную последовательность, и усеченные индексные последовательности по меньшей мере одного поднабора отличаются по меньшей мере ненулевым количеством изменений последовательностей от каждой другой усеченной индексной последовательности в упомянутом поднаборе; предпочтительно, когда минимальное количество изменений последовательностей между усеченными индексными последовательностями поднабора больше, чем минимальное количество изменений последовательностей индексных последовательностей в поднаборе за вычетом разности между длиной индексных последовательностей и усеченных индексных последовательностей.
3. Набор по п. 2, в котором усеченные индексные последовательности поднабора более высокого уровня являются элементами усеченных индексных последовательностей поднабора более низкого уровня, и где усеченные индексные последовательности поднабора более низкого уровня отличаются друг от друга меньшим минимальным количеством последовательности по сравнению с усеченными индексными последовательностями поднабора более высокого уровня.
4. Набор по любому из пп. 1-3, где изменения последовательностей выбираются из нуклеотидных замен, делеций и вставок, и где минимальное количество изменений последовательностей соответствует минимальному количеству, необходимому для замены любой индексной последовательности на другую индексную последовательность.
5. Набор по любому из пп. 1-4, в котором изменения последовательности количественно оцениваются как секвенциальное расстояние, которое представляет собой количество замен нуклеотидов или вероятность изменений.
6. Набор по п. 5, в котором величина секвенциального расстояния представляет собой расстояние Хэмминга, расстояние Левенштейна или расстояние Левенштейна между последовательностями, предпочтительно расстояние Левенштейна между последовательностями.
7. Набор по п. 5, в котором вероятность изменений представляет собой максимальную вероятность или сумму вероятностей, предпочтительно сумму вероятностей нуклеотидных изменений, которые преобразуют одну последовательность в другую.
8. Набор по п. 5 или 7, где индексные последовательности поднабора более низкого уровня отличаются друг от друга более высокой вероятностью изменения последовательностей по сравнению с индексными последовательностями поднабора более высокого уровня, и предпочтительно, где в зависимости от варианта осуществления 2 усеченные индексные последовательности поднабора более низкого уровня отличаются более высокой вероятностью изменения последовательности, чем усеченные индексные последовательности поднабора более высокого уровня.
9. Набор по п. 5, где вероятность изменений количественно определяется монотонно убывающей функцией вероятности, предпочтительно отрицательным логарифмом или отрицательной вероятностью, где вероятность предпочтительно оценивается по максимальной вероятности или сумме вероятностей, предпочтительно по сумме вероятностей, изменений нуклеотидов, которые преобразуют одну последовательность в другую.
10. Набор по п. 6, в котором расстояние Левенштейна между последовательностями между индексными последовательностями поднабора более высокого уровня больше не менее чем на 1, предпочтительно на 2, чем расстояние Левенштейна между последовательностями между индексными последовательностями поднабора более низкого уровня.
11. Набор по п. 6 или 10, в котором расстояние Левенштейна между последовательностями между индексными последовательностями поднабора самого высокого уровня равно не менее 4.
12. Набор по любому из пп. 1-11, в котором индексные последовательности имеют длину не менее 4, предпочтительно не менее 6, нуклеотидов, и/или поднабор самого высокого уровня содержит не менее 2, предпочтительно не менее 4, различных индексных последовательностей.
13. Набор по любому из пп. 1-12, в котором олигонуклеотиды присваиваются поднабору путем помещения их в контейнер, который помечается идентификатором поднабора; предпочтительно, когда контейнер представляет собой лунку в луночном планшете.
14. Набор по любому из пп. 1-13, в котором индексные последовательности имеют содержание G/C от 30% до 70%; и/или где индексные последовательности не содержат повторов одного и того же нуклеотида длиной не менее 3; и/или где индексные последовательности поднабора имеют сбалансированное распределение нуклеотидов, где количество совместно используемых нуклеотидов в одной и той же позиции в индексных последовательностях между различными индексными последовательностями не более чем в 0,5 раза превышает количество индексных последовательностей в упомянутом поднаборе, или где в не менее 50% позиций индексных последовательностей, частота для всех индексных последовательностей поднабора для каждого типа нуклеотидов составляет 0,5 или менее.
15. Способ выработки набора олигонуклеотидов, содержащего множество поднаборов олигонуклеотидов с поднабором индексных последовательностей, содержащий этапы:
выработку первого поднабора олигонуклеотидов с индексными последовательностями с первым секвенциальным расстоянием друг от друга в пределах первого поднабора, где секвенциальное расстояние представляет собой количественное количество изменений последовательностей, которые преобразуют одну последовательность в другую, или монотонно убывающую функцию вероятности изменений последовательности, которая преобразует одну последовательность в другую,
выработку второго поднабора путем включения первого поднабора и добавления дополнительных олигонуклеотидов с индексными последовательностями со вторым секвенциальным расстоянием друг от друга во втором поднаборе, где второе секвенциальное расстояние является меньшим секвенциальным расстоянием, чем первое секвенциальное расстояние.
16. Способ по п. 15, в котором этап формирования первого и второго поднабора индексных последовательностей содержит для каждого поднабора выбор набора индексных последовательностей из пула различных индексных последовательностей.
17. Способ по п. 15 или 16, в котором выработка первого и/или второго поднабора содержит выбор индексных последовательностей, которые содержат усеченные индексные последовательности, и усеченные индексные последовательности по меньшей мере одного поднабора отличаются по меньшей мере ненулевым количеством изменений последовательностей, предпочтительно отличаются по меньшей мере количеством изменений последовательностей, превышающим 1, от каждой другой последовательности с усеченным индексом в упомянутом поднаборе.
18. Способ по любому из пп. 15-17, в котором корректируемые последовательности вырабатываются для индексной последовательности поднабора, где упомянутые корректируемые последовательности имеют секвенциальное расстояние, которое составляет менее половины секвенциального расстояния между индексными последовательностями упомянутого поднабора, и где корректируемые последовательности различных индексных последовательностей в упомянутом поднаборе не перекрываются.
19. Способ по любому из пп. 15-18, содержащий выработку поднабора более низкого уровня путем включения поднабора более высокого уровня и добавления дополнительных олигонуклеотидов с индексными последовательностями с меньшим секвенциальным расстоянием, чем для поднабора более высокого уровня, друг к другу в пределах поднабора более низкого уровня.
20. Способ по любому из пп. 15-19, содержащий выработку третьего поднабора путем включения второго поднабора и добавления дополнительных олигонуклеотидов с индексными последовательностями с третьим секвенциальным расстоянием друг от друга в пределах третьего поднабора, где третье секвенциальное расстояние является меньшим секвенциальным расстоянием, чем второе секвенциальное расстояние.
21. Способ по любому из пп. 15-20, в котором выработка поднабора содержит выбор индексных последовательностей путем добавления индексной последовательности-кандидата и оценку секвенциального расстояния индексного расстояния-кандидата во всех других ранее существовавших индексных последовательностях в поднаборе; и добавление индексной последовательности- кандидата к индексным последовательностям поднабора, если она удовлетворяет заданному требованию к секвенциальному расстоянию.
22. Способ по п. 21, в котором индексная последовательность-кандидат содержит не менее 50% своих позиций типа нуклеотидов генетического кода с наименьшей частотой в соответствующей позиции в ранее существовавших индексных последовательностях поднабора.
23. Способ по п. 21 или 22, в котором индексная последовательность-кандидат выбирается из пула индексных последовательностей-кандидатов, где элементы пула индексных последовательностей-кандидатов выполняют заданное требование к секвенциальному расстоянию для каждого другого элемента пула, и где индексная последовательность-кандидат пула добавляется к индексным последовательностям поднабора тогда, когда сумма абсолютных значений разностей частот каждого нуклеотидного типа генетического кода в каждой позиции до 0,25 является наименьшей для индексной последовательности-кандидата по сравнению с другими кандидатами индексной последовательности пула.
24. Способ по п. 23, в котором критерии, изложенные в п. 22, применяются по меньшей мере к 25% индексных последовательностей-кандидатов, которые добавляются в поднабор последними.
25. Способ по п. 23 или 24, в котором пул индексных последовательностей содержит по меньшей мере в 2 раза больше элементов, чем поднабор.
26. Способ по любому из пп. 21-25, в котором по меньшей мере в 50% позиций индексных последовательностей частота для всех индексных последовательностей поднабора для каждого типа нуклеотидов составляет 0,5 или менее.
27. Способ по любому из пп. 21-26, содержащий выработку множества поднаборов-кандидатов, каждый из которых имеет заданное количество элементов, как в способах по любому из пп. 21-26, и выбор поднабора-кандидата в качестве поднабора, когда упомянутый поднабор-кандидат имеет наименьшее среднее значение по всем индексным последовательностям для соответствующего поднабора-кандидата суммы абсолютных значений разностей частот каждого нуклеотидного типа генетического кода в каждой позиции до 0,25.
28. Способ по любому из пп. 21-27, содержащий выработку множества поднаборов-кандидатов, каждый из которых имеет заданное количество элементов, как в способах по любому из пп. 21-27, и выбор поднабора-кандидата в качестве поднабора, где упомянутый поднабор-кандидат выбирается путем исключения других поднаборов-кандидатов,
где поднабор-кандидат исключается тогда, когда в способе, который содержит добавление кандидатов-индексных последовательностей из пула индексных последовательностей-кандидатов к поднабору-кандидату и при необходимости дополнительное добавление сравнительных индексных последовательностей, кандидат-поднабор имеет более высокое среднее значение во всех своих суммах индексных последовательностей абсолютных значений разностей частот каждого нуклеотидного типа генетического кода в каждой позиции до 0,25 по сравнению с другим поднабором или поднабором-кандидатом
29. Способ присвоения секвенирующих прочтений образцу олигонуклеотидов, содержащий этапы
а) получение образцов олигонуклеотидов из множества образцов,
b) выбор поднабора последовательностей олигонуклеотидных индексов из набора по любому из пп. 1-14 или из набора, который может быть получен или получен способом по любому из пп. 15-28, где поднабор выбирается из другого поднабора на основе большего секвенциального расстояния между индексными последовательностями друг от друга в пределах выбранного поднабора; где секвенциальное расстояние представляет собой количественную величину изменений последовательностей, которая преобразует одну последовательность в другую, или монотонно убывающую функцию вероятности изменений последовательностей, которая преобразует одну последовательность в другую, и где выбранный поднабор имеет по меньшей мере такое же количество различных индексных последовательностей, как и количество образцов на этапе а),
c) добавление индексных последовательностей из упомянутого поднабора в каждый олигонуклеотид образца, где индексные последовательности указывают образец,
d) определение последовательности образцовых олигонуклеотидов или фрагментов образцовых олигонуклеотидов и определение индексной последовательности,
e) присвоение последовательности с полученным прочтением образцу на основе определенной индексной последовательности или на основе индексной последовательности, которая имеет наименьшее секвенциальное расстояние до определенной индексной последовательности, где, если две или более индексных последовательностей имеют одинаковое наименьшее расстояние, то упомянутое полученное прочтение отбрасывается; где при необходимости секвенциальное расстояние не превышает заданного значения согласно критерию.
30. Способ по п. 29, в котором этап b) содержит выбор олигонуклеотидов с индексными последовательностями из набора по любому из пп. 1-14 или набора, который может быть получен или получен с помощью способа по любому из пп. 15-28, где выбирается поднабор олигонуклеотидов с наибольшим секвенциальным расстоянием между индексными последовательностями в поднаборе, который имеет по меньшей мере такое же количество различных индексных последовательностей, как и количество образцов на этапе а).
31. Способ по п. 29 или 30, в котором определение последовательности нуклеотидов индексной последовательности содержит определение последовательности всей индексной последовательности или ее части, где предпочтительно определяется частичная индексная последовательность в случае, если секвенциальное расстояние от одной частичной индексной последовательности до другой частичной индексной последовательности в одном том же поднаборе больше, чем ненулевое значение согласно критерию.
32. Способ по п. 31, в котором частичная индексная последовательность имеет свойства секвенциального расстояния усеченной индексной последовательности согласно варианту осуществления 2 или 3.
Настоящее изобретение далее определяется следующими примерами, не ограничиваясь этими вариантами осуществления изобретения.
Примеры
Пример 1. Выработка сбалансированных по нуклеотидам наборов штрихкодов ff-Levenshtein-7(8,10,12)
Для получения наборов штрихкодов в этом разделе мы использовали алгоритм 4 поиска, описанный выше в разделе 4.1, с фиксированным расстоянием Левенштейна в качестве секвенциального расстояния. На этапе 1 алгоритма мы устанавливаем , 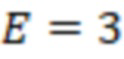 и , и , в результате чего получается набор штрихкодов ff-Levenshtein-5(8,10,12). Далее, мы выбрали для расстояний между штрихкодами
и , и , в результате чего получается набор штрихкодов ff-Levenshtein-5(8,10,12). Далее, мы выбрали для расстояний между штрихкодами 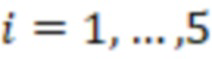 и
и 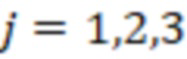 при
при 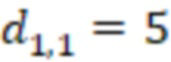 ,
, 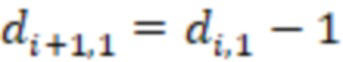 и
и 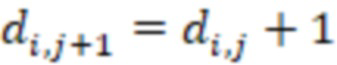 и расстояния исправления ошибок
и расстояния исправления ошибок 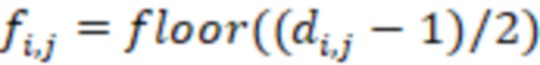 , где
, где  возвращает наибольшее целое число, меньшее
возвращает наибольшее целое число, меньшее 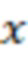 . Эти расстояния между штрихкодами и исправления ошибок можно найти в таблице 1. Набор потенциальных штрихкодов в алгоритме 4 поиска требовался для выполнения
. Эти расстояния между штрихкодами и исправления ошибок можно найти в таблице 1. Набор потенциальных штрихкодов в алгоритме 4 поиска требовался для выполнения 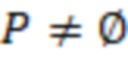 . Следовательно, первая последовательность, удовлетворяющая требованиям расстояния на этапе 4, была добавлена к набору штрихкодов на этапе 6. Так как расстояние Левенштейна с фиксированной рамкой и позиционное распределение нуклеотидов не зависят от алфавита последовательности, мы первоначально создали наборы штрихкодов из алфавита 0, 1, 2, 3. Во избежание последовательностей штрихкодов, состоящих полностью из одного нуклеотида, а также подобных последовательностей, мы инициировали наш набор штрихкодов при
. Следовательно, первая последовательность, удовлетворяющая требованиям расстояния на этапе 4, была добавлена к набору штрихкодов на этапе 6. Так как расстояние Левенштейна с фиксированной рамкой и позиционное распределение нуклеотидов не зависят от алфавита последовательности, мы первоначально создали наборы штрихкодов из алфавита 0, 1, 2, 3. Во избежание последовательностей штрихкодов, состоящих полностью из одного нуклеотида, а также подобных последовательностей, мы инициировали наш набор штрихкодов при 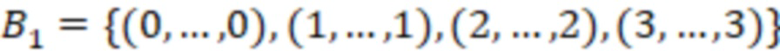 . После завершения поиска эти последовательности были удалены из набора штрихкодов. Это привело к вложенным наборам штрихкодов, из которых мы выбрали вложенные наборы штрихкодов со сбалансированным распределением нуклеотидов и поднаборами размеров 4, 8, 16, 24, 96, 768, 9216, таким образом, получив набор штрихкодов ff-Levenshtein-7(8,10,12). Выбор производился по алгоритму, описанному в разделе 4.3. Порядок, в котором элементы дополнения
. После завершения поиска эти последовательности были удалены из набора штрихкодов. Это привело к вложенным наборам штрихкодов, из которых мы выбрали вложенные наборы штрихкодов со сбалансированным распределением нуклеотидов и поднаборами размеров 4, 8, 16, 24, 96, 768, 9216, таким образом, получив набор штрихкодов ff-Levenshtein-7(8,10,12). Выбор производился по алгоритму, описанному в разделе 4.3. Порядок, в котором элементы дополнения 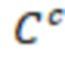 обрабатывались на этапе 4 алгоритма 4 поиска, выбирался случайным образом. Таким образом, повторения алгоритма давали разные результаты. Таким образом, мы выработали 245 наборов штрихкодов, из которых был выбран окончательный набор. Среди при
обрабатывались на этапе 4 алгоритма 4 поиска, выбирался случайным образом. Таким образом, повторения алгоритма давали разные результаты. Таким образом, мы выработали 245 наборов штрихкодов, из которых был выбран окончательный набор. Среди при 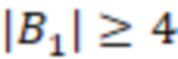 мы выбрали вложенный набор штрихкодов, чьи распределения
мы выбрали вложенный набор штрихкодов, чьи распределения 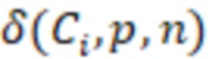 нуклеотидов имели минимальное расстояние
нуклеотидов имели минимальное расстояние  от равномерного позиционного распределения нуклеотидов. Как и прежде, мы установили
от равномерного позиционного распределения нуклеотидов. Как и прежде, мы установили 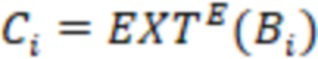 . На последнем этапе мы рассмотрели пары различных отображений из алфавита
. На последнем этапе мы рассмотрели пары различных отображений из алфавита 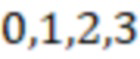 в
в 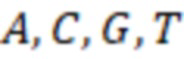 , получив пары наборов штрихкодов, и рассчитали температуру плавления всех гомо- и димеров. В итоге мы выбрали пару наборов штрихкодов с наименьшими температурами плавления и выбрали один из наборов штрихкодов как «индексы i7», и другой как «индексы i5». Обозначения i5 и i7 относятся к двойным индексам, добавленным к разным участкам олигонуклеотида, который необходимо пометить (смотри фиг. 7 и ссылку [5]). Количество элементов и расстояние между штрихкодами вложенных поднаборов в конечном наборе ff-Levenshtein-7(8,10,12) приведены в таблице 1 для вложенных последовательностей длиной 8, 10 и 12 нуклеотидов. Длины 8 и 10 вложены в большую(ие) последовательность(и).
, получив пары наборов штрихкодов, и рассчитали температуру плавления всех гомо- и димеров. В итоге мы выбрали пару наборов штрихкодов с наименьшими температурами плавления и выбрали один из наборов штрихкодов как «индексы i7», и другой как «индексы i5». Обозначения i5 и i7 относятся к двойным индексам, добавленным к разным участкам олигонуклеотида, который необходимо пометить (смотри фиг. 7 и ссылку [5]). Количество элементов и расстояние между штрихкодами вложенных поднаборов в конечном наборе ff-Levenshtein-7(8,10,12) приведены в таблице 1 для вложенных последовательностей длиной 8, 10 и 12 нуклеотидов. Длины 8 и 10 вложены в большую(ие) последовательность(и).
Таблица 1. Размер и расстояние между штрихкодами поднаборов в ff-Leveshtein-7(8,10,12).
104 индексные последовательности i7 и i5 в 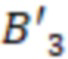 длиной 12 содержатся в списке последовательностей. SEQ ID NO: от 1 до 104 представляют собой индексные последовательности i7, и SEQ ID NO: от 105 до 208 представляют собой индексные последовательности i5. Ассоциация между порядковыми номерами и поднаборами ,
длиной 12 содержатся в списке последовательностей. SEQ ID NO: от 1 до 104 представляют собой индексные последовательности i7, и SEQ ID NO: от 105 до 208 представляют собой индексные последовательности i5. Ассоциация между порядковыми номерами и поднаборами , 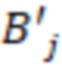 штрихкодов представлена в таблице 2. Это ясно показывает вложенную структуру поднаборов. Подпоследовательностями в этих индексах являются первые 8 и 10 нуклеотидов, что демонстрирует вложенную структуру подпоследовательностей в ff-Levenshtein-7(8,10,12).
штрихкодов представлена в таблице 2. Это ясно показывает вложенную структуру поднаборов. Подпоследовательностями в этих индексах являются первые 8 и 10 нуклеотидов, что демонстрирует вложенную структуру подпоследовательностей в ff-Levenshtein-7(8,10,12).
Таблица 2. Порядковые номера штрихкодов в поднаборах.
Для набора i7:
Для набора i5:
На фиг. 7, 8, 9 и 10 показано позиционное распределение нуклеотидов для , , и . Здесь ось x представляет позицию штрихкода, , и ось y представляет , долю нуклеотида в позиции . Каждая из 4-х линий на этих фигурах представляет долю одного из 4 нуклеотидов . Для , который содержит 4 штрихкода, является однородным, если в каждой позиции каждый нуклеотид встречается ровно в одном штрихкоде. Если 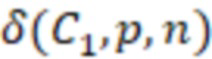 распределен неравномерно по позиции , то по меньшей мере один из нуклеотидов не содержится ни в одном из 4-х штрихкодов в позиции и, следовательно, в
распределен неравномерно по позиции , то по меньшей мере один из нуклеотидов не содержится ни в одном из 4-х штрихкодов в позиции и, следовательно, в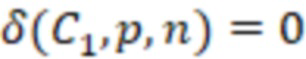 . Следовательно, на фиг. 8 показано, что распределен неравномерно в 8 позициях. Таким образом, в этих позициях присутствуют только один нуклеотид и остальные нуклеотиды. Этого достаточно для получения хороших показателей качества на двухцветных секвенаторах Illumina, которые требуют наличия A или C и дополнительно G или T [5] в каждой позиции. На фиг. 8, 9 и 10 показано, что для , и все нуклеотиды присутствуют во всех позициях, так как линии на этих графиках никогда не равны нулю. Для , который содержит 8 штрихкодов, на фиг. 9 показано, что в 8 позициях один нуклеотид встречается только один раз. Для , при
. Следовательно, на фиг. 8 показано, что распределен неравномерно в 8 позициях. Таким образом, в этих позициях присутствуют только один нуклеотид и остальные нуклеотиды. Этого достаточно для получения хороших показателей качества на двухцветных секвенаторах Illumina, которые требуют наличия A или C и дополнительно G или T [5] в каждой позиции. На фиг. 8, 9 и 10 показано, что для , и все нуклеотиды присутствуют во всех позициях, так как линии на этих графиках никогда не равны нулю. Для , который содержит 8 штрихкодов, на фиг. 9 показано, что в 8 позициях один нуклеотид встречается только один раз. Для , при 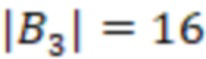 , на фиг. 10 показано одна позиция, в котором один нуклеотид встречается только дважды. На фиг. 11 показаны для при
, на фиг. 10 показано одна позиция, в котором один нуклеотид встречается только дважды. На фиг. 11 показаны для при 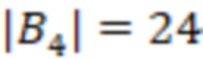 две позиции, в которых один нуклеотид встречается четыре раза, и другая позиция, в которой два нуклеотида встречаются четыре раза. Эти позиции являются позициями , где наиболее сильно отклоняется от равномерного распределения. В целом это показывает, что распределение приближается к равномерному распределению с увеличением .
две позиции, в которых один нуклеотид встречается четыре раза, и другая позиция, в которой два нуклеотида встречаются четыре раза. Эти позиции являются позициями , где наиболее сильно отклоняется от равномерного распределения. В целом это показывает, что распределение приближается к равномерному распределению с увеличением .
Пример 2/ Перекрестное загрязнение в прогонах RNA-Seq с наборами штрихкодов Hamming(6) и ff-Levenshtein-7(8,10,12)
Для экспериментов в этом разделе мы синтезировали 96 штрихкодов длиной 12 в из сбалансированного по нуклеотидам набора штрихкодов ff-Levenshtein-7(8,10.12) из примера 1. Далее мы синтезировали 96 штрихкодов длиной 6, которые имели минимальное расстояние Хэмминга, равное 3. Следовательно, этот набор может исправить одну замену. Мы синтезировали оба набора штрихкодов как индексы i5 и i7 и использовали их как уникальные двойные индексы (UDI) для мечения 96 образцов коммерчески доступной универсальной эталонной RNA человека (UHRR) в двух прогонах RNA-Seq с двойным индексом. UDI - это UCDI с дополнительным требованием относительно того, чтобы файлы 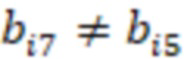 . Впоследствии мы демультиплексировали каждый прогон по отношению ко всем 96 кортежам штрихкодов i5/i7 соответствующих UDI и оценили частоту скачкообразных переходов штрихкодов, а также скорости перекрестного загрязнения после исправления ошибок. Далее мы рассчитали чистоту, то есть долю . В случае набора ff-Levenshtein-7(8,10,12) мы провели этот анализ для всех длин штрихкода 8, 10 и 12. Результаты можно найти в таблице 3. Это показывает, что без исправления ошибок частота появления ошибок для ff-Levenshtein-7(8,10,12) является почти одинаковой для длины 8, 10 и 12 и составляет 0,01%, в то время как чистота является самой высокой для длины 8 при 93,028% и самой низкой для длины 12 при 90,913%. Снижение чистоты с увеличением длины штрихкода ассоциировано с тем, что более длинные последовательности имеют больше шансов содержать ошибку. По сравнению с ff-Levenshtein-7(8,10,12), для Hamming(6) частота появления ошибок значительно выше и составляет 0,244%. Это увеличение по сравнению с ff-Levenshtein-7(8,10,12) в 24 раза является не только результатом более короткой длины штрихкода, но и из-за другого расстояния, используемого для дизайна штрихкода. По сравнению с расстоянием ff-Levenshtein наборы штрихкодов, разработанные с учетом расстояния Хэмминга, не гарантируют разумного расстояния между штрихкодами после вставок и делеций. Это также можно увидеть при исправлении одиночной ошибки для Хэмминга (6), что увеличивает частоту появления ошибок примерно от 7 до 1,5%. Для сравнения, при исправлении одной ошибки для ff-Levenshtein-7(8,10,12) частота появления ошибок на длине 12 остается неизменной на уровне 0,01%, и чистота увеличивается до 97,013%. Для ff-Levenshtein-7(8,10,12) и длины 10 частота появления ошибок немного увеличивается, и для ff-Levenshtein-7(8,10,12) и длины 8 частота появления ошибок увеличивается в 10 раз до 0,1%. Это показывает, что если необходимо исправить одну ошибку, рекомендуется использовать штрихкод длиной не менее 10 для ff-Levenshtein-7(8,10,12). С другой стороны, если необходимо выбрать длину штрихкода 12, рекомендуется выполнить исправление ошибок, так как это повысит чистоту до того же уровня, что и для длины штрихкода 8 и 10. Исправление 2 ошибок, что возможно только для ff- Левенштейна (12), приводит лишь к небольшому повышению чистоты за счет более чем двукратного увеличения частоты появления ошибок до 0,024%. Следовательно, исправление двух ошибок с ff-Levenshtein-7(8,10,12) и длиной 12 является нецелесообразным. В целом результаты экспериментов в таблице 3 показывают, что перекрестное загрязнение для ff-Levenshtein-7(8,10,12) значительно ниже, чем для Hamming(6), в то время как чистота увеличивается. Кроме того, результаты показывают, что все длины штрихкода ff-Levenshtein-7(8,10,12) могут использоваться для мультиплексирования образцов при прогоне RNA-Seq.
. Впоследствии мы демультиплексировали каждый прогон по отношению ко всем 96 кортежам штрихкодов i5/i7 соответствующих UDI и оценили частоту скачкообразных переходов штрихкодов, а также скорости перекрестного загрязнения после исправления ошибок. Далее мы рассчитали чистоту, то есть долю . В случае набора ff-Levenshtein-7(8,10,12) мы провели этот анализ для всех длин штрихкода 8, 10 и 12. Результаты можно найти в таблице 3. Это показывает, что без исправления ошибок частота появления ошибок для ff-Levenshtein-7(8,10,12) является почти одинаковой для длины 8, 10 и 12 и составляет 0,01%, в то время как чистота является самой высокой для длины 8 при 93,028% и самой низкой для длины 12 при 90,913%. Снижение чистоты с увеличением длины штрихкода ассоциировано с тем, что более длинные последовательности имеют больше шансов содержать ошибку. По сравнению с ff-Levenshtein-7(8,10,12), для Hamming(6) частота появления ошибок значительно выше и составляет 0,244%. Это увеличение по сравнению с ff-Levenshtein-7(8,10,12) в 24 раза является не только результатом более короткой длины штрихкода, но и из-за другого расстояния, используемого для дизайна штрихкода. По сравнению с расстоянием ff-Levenshtein наборы штрихкодов, разработанные с учетом расстояния Хэмминга, не гарантируют разумного расстояния между штрихкодами после вставок и делеций. Это также можно увидеть при исправлении одиночной ошибки для Хэмминга (6), что увеличивает частоту появления ошибок примерно от 7 до 1,5%. Для сравнения, при исправлении одной ошибки для ff-Levenshtein-7(8,10,12) частота появления ошибок на длине 12 остается неизменной на уровне 0,01%, и чистота увеличивается до 97,013%. Для ff-Levenshtein-7(8,10,12) и длины 10 частота появления ошибок немного увеличивается, и для ff-Levenshtein-7(8,10,12) и длины 8 частота появления ошибок увеличивается в 10 раз до 0,1%. Это показывает, что если необходимо исправить одну ошибку, рекомендуется использовать штрихкод длиной не менее 10 для ff-Levenshtein-7(8,10,12). С другой стороны, если необходимо выбрать длину штрихкода 12, рекомендуется выполнить исправление ошибок, так как это повысит чистоту до того же уровня, что и для длины штрихкода 8 и 10. Исправление 2 ошибок, что возможно только для ff- Левенштейна (12), приводит лишь к небольшому повышению чистоты за счет более чем двукратного увеличения частоты появления ошибок до 0,024%. Следовательно, исправление двух ошибок с ff-Levenshtein-7(8,10,12) и длиной 12 является нецелесообразным. В целом результаты экспериментов в таблице 3 показывают, что перекрестное загрязнение для ff-Levenshtein-7(8,10,12) значительно ниже, чем для Hamming(6), в то время как чистота увеличивается. Кроме того, результаты показывают, что все длины штрихкода ff-Levenshtein-7(8,10,12) могут использоваться для мультиплексирования образцов при прогоне RNA-Seq.
Таблица 3. Ошибка и чистота (%) для Hamming(6) (H(6)) и ff-Levenshtein-7(8,10,12) с длиной 8 (ff-L(8)), 10 (ff-L(10)) и 12 (ff-L(12)) без исправления ошибок и с исправлением 1 ошибки (1c) и 2 ошибок (2c).
Пример 3. Количественная оценка различных типов перекрестного загрязнения с низким уровнем ff-Levenshtein-5(8,10,12)
В этом разделе представлены результаты экспериментов по оценке перекрестного загрязнения на разных этапах синтеза индекса, подготовки библиотеки и секвенирования. Чтобы измерить ожидаемые низкие уровни перекрестного загрязнения, мы выбрали 12 штрихкодов из 25 штрихкодов в 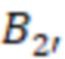 ff-Levenshtein-5(8,10,12) из примера 1. Выбранный набор штрихкодов содержал все штрихкоды в
ff-Levenshtein-5(8,10,12) из примера 1. Выбранный набор штрихкодов содержал все штрихкоды в 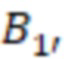 , и в результате 6 штрихкодов имели расстояние ff-Levenshtein, равное 7, для длины штрихкода 12, в то время как все 12 штрихкодов имели расстояние ff-Levenshtein, равное 6. Мы разделили этот набор штрихкодов на 3 набора из 4 штрихкодов, которые были синтезированы 3 поставщиками синтеза олигонуклеотидов как штрихкоды i5 и i7. Мы использовали штрихкоды как уникальные двойные индексы при
, и в результате 6 штрихкодов имели расстояние ff-Levenshtein, равное 7, для длины штрихкода 12, в то время как все 12 штрихкодов имели расстояние ff-Levenshtein, равное 6. Мы разделили этот набор штрихкодов на 3 набора из 4 штрихкодов, которые были синтезированы 3 поставщиками синтеза олигонуклеотидов как штрихкоды i5 и i7. Мы использовали штрихкоды как уникальные двойные индексы при 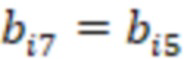 . Такие наборы уникальных двойных индексов также называются уникальными двойными индексами (UDMI) [5]. В нашем эксперименте мы пометили 9 образцов UHRR в разделе 3.2 с 3 UDMI каждого поставщика синтеза. Остальные 3 UDMI, по одному для каждого поставщика, никогда не трогались. Этот экспериментальный дизайн позволяет оценить перекрестное загрязнение на сайте поставщика синтеза, так как обнаружение пропущенного штрихкода после демультиплексирования показывает, что этот штрихкод оказался в неправильной пробирке перед доставкой. Как и в примере 2, мы демультиплексировали по отношению ко всем кортежам из 96 штрихкодов в ff-Levenshtein-7(8,10,12). Это дало нам счетную матрицу со строками и столбцами, помеченными и . Различные типы перекрестного загрязнения соответствуют разным областям в этой матрице, которые показаны на фиг. 12. Подсчеты в области C соответствуют кортежам
. Такие наборы уникальных двойных индексов также называются уникальными двойными индексами (UDMI) [5]. В нашем эксперименте мы пометили 9 образцов UHRR в разделе 3.2 с 3 UDMI каждого поставщика синтеза. Остальные 3 UDMI, по одному для каждого поставщика, никогда не трогались. Этот экспериментальный дизайн позволяет оценить перекрестное загрязнение на сайте поставщика синтеза, так как обнаружение пропущенного штрихкода после демультиплексирования показывает, что этот штрихкод оказался в неправильной пробирке перед доставкой. Как и в примере 2, мы демультиплексировали по отношению ко всем кортежам из 96 штрихкодов в ff-Levenshtein-7(8,10,12). Это дало нам счетную матрицу со строками и столбцами, помеченными и . Различные типы перекрестного загрязнения соответствуют разным областям в этой матрице, которые показаны на фиг. 12. Подсчеты в области C соответствуют кортежам 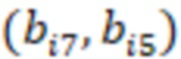 штрихкодов, где по меньшей мере один из , не появился ни на одном этапе эксперимента. Таким образом, подсчеты в области C количественно определяют частоту, с которой прогон RNA-Seq случайным образом вырабатывает индекс i5 или i7 в наборе штрихкодов. Область B содержит кортежи штрихкодов, где оба и были синтезированы, но по меньшей мере один из них никогда не использовался в эксперименте. Таким образом, подсчеты в области B количественно определяют кумулятивное случайное перекрестное загрязнение и перекрестное загрязнение на сайте поставщика синтеза. Область A содержит кортежи , где как , так и были синтезированы и использовались в эксперименте. Таким образом, недиагональные элементы в области А количественно определяют кумулятивное случайное, зависящее от места, и экспериментальное перекрестное загрязнение. Экспериментальное перекрестное загрязнение содержит, среди прочего, перекрестное загрязнение из-за ошибок при обращении, лабораторных условий и экспериментальных ошибок, зависящих от поставщика. Последнее может быть, например, результатом нестабильности синтезированных последовательностей во время секвенирования. Различия в экспериментальной ошибке, зависящей от поставщика синтеза, отражаются различиями в недиагональных подсчетах в областях P1, P2 и P3, которые содержат кортежи штрихкодов, выработанные поставщиком 1, 2 и 3. Из областей в матрице подсчета на фиг. 12 мы получили количественные значения перекрестного загрязнения в таблице 4. Из-за небольших уровней перекрестного загрязнения значения в таблице 4 даны в частях на миллион. Строки в таблице 4 помечены областью, в которой было измерено перекрестное загрязнение. Метка строки «Cdiag» означает количество перекрестного загрязнения по диагонали области «C». Метка «A-nonP» означает независимое от поставщика экспериментальное перекрестное загрязнение. Мы оцениваем последнее путем вычитания поставщика, зависящего от общего экспериментального перекрестного загрязнения. Во избежание недооценки независимого от поставщика экспериментального перекрестного загрязнения, мы предполагаем, что наименьшее перекрестное загрязнение, измеренное для любого поставщика, является полностью результатом независимых от поставщика факторов. С другой стороны, мы предполагаем, что различия между экспериментальным перекрестным загрязнением, зависящим от поставщика, полностью являются результатом факторов, зависящих от поставщика. Таким образом, в таблице 4 мы вычисляем независимое от поставщика экспериментальное перекрестное загрязнение как
штрихкодов, где по меньшей мере один из , не появился ни на одном этапе эксперимента. Таким образом, подсчеты в области C количественно определяют частоту, с которой прогон RNA-Seq случайным образом вырабатывает индекс i5 или i7 в наборе штрихкодов. Область B содержит кортежи штрихкодов, где оба и были синтезированы, но по меньшей мере один из них никогда не использовался в эксперименте. Таким образом, подсчеты в области B количественно определяют кумулятивное случайное перекрестное загрязнение и перекрестное загрязнение на сайте поставщика синтеза. Область A содержит кортежи , где как , так и были синтезированы и использовались в эксперименте. Таким образом, недиагональные элементы в области А количественно определяют кумулятивное случайное, зависящее от места, и экспериментальное перекрестное загрязнение. Экспериментальное перекрестное загрязнение содержит, среди прочего, перекрестное загрязнение из-за ошибок при обращении, лабораторных условий и экспериментальных ошибок, зависящих от поставщика. Последнее может быть, например, результатом нестабильности синтезированных последовательностей во время секвенирования. Различия в экспериментальной ошибке, зависящей от поставщика синтеза, отражаются различиями в недиагональных подсчетах в областях P1, P2 и P3, которые содержат кортежи штрихкодов, выработанные поставщиком 1, 2 и 3. Из областей в матрице подсчета на фиг. 12 мы получили количественные значения перекрестного загрязнения в таблице 4. Из-за небольших уровней перекрестного загрязнения значения в таблице 4 даны в частях на миллион. Строки в таблице 4 помечены областью, в которой было измерено перекрестное загрязнение. Метка строки «Cdiag» означает количество перекрестного загрязнения по диагонали области «C». Метка «A-nonP» означает независимое от поставщика экспериментальное перекрестное загрязнение. Мы оцениваем последнее путем вычитания поставщика, зависящего от общего экспериментального перекрестного загрязнения. Во избежание недооценки независимого от поставщика экспериментального перекрестного загрязнения, мы предполагаем, что наименьшее перекрестное загрязнение, измеренное для любого поставщика, является полностью результатом независимых от поставщика факторов. С другой стороны, мы предполагаем, что различия между экспериментальным перекрестным загрязнением, зависящим от поставщика, полностью являются результатом факторов, зависящих от поставщика. Таким образом, в таблице 4 мы вычисляем независимое от поставщика экспериментальное перекрестное загрязнение как 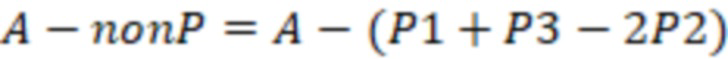 .
.
Таблица 4. Количество (частей на миллион) различных типов перекрестного загрязнения, зависящего от поставщика синтеза
В таблице 4 показано, что общее перекрестное загрязнение значительно увеличивается при уменьшении длины штрихкода до 8. Для длины 10 и 12 штрихкодов общее перекрестное загрязнение меньше приблизительно в 5 и 6 раз, соответственно. Основной вклад в это увеличение вносит большая случайная ошибка (C) для длины 8 штрихкода. В таблице 4 также показано, что общее перекрестное загрязнение на сайте поставщика (B) является незначительным. Однако экспериментальное перекрестное загрязнение заметно различается между поставщиками. По сравнению с поставщиком 2 (P2) экспериментальное перекрестное загрязнение для поставщиков 1 (P1) и 3 (P3) выше примерно в 4 и 11 раз, соответственно. Независимое от поставщика экспериментальное перекрестное загрязнение (A-nonP) для всех длин штрихкодов близко к 69% от общей суммы за вычетом случайного перекрестного загрязнения, что указывает на то, что 69% неслучайного перекрестного загрязнения в этом эксперименте вносится источниками, независящими от поставщика.
В целом, результаты, приведенные в этом примере, показывают, что набор штрихкодов ff-Levenshtein-5(8,10,12) можно использовать с длинами 8, 10 и 12 штрихкодов для количественной оценки низких уровней перекрестного загрязнения. Это является необходимой предпосылкой для выявления и уменьшения различных источников перекрестного загрязнения.
Список литературы
[1] Buschmann and Bystrykh. BMC Bioinformatics, 14:272, 2013
[2] Conway and Sloane. IEEE Trans. Inf. Theor., 32(3):337-348, 1986
[3] Hawkins et al. PNAS, 115(27): E6217-E6226, 2018
[4] WO 2018/204423 A1
[5] MacConaill et al. BMC Genomics, 19(1):30-30, 2018
[6] WO 2018/136248 A1
[7] WO 2018/204423 A1
[8] WO 2011/100617 A1
Все ссылки включены в настоящее описание путем ссылки.
--->
СПИСОК ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> LEXOGEN GMBH
<120> Index sequences for multiplex parallel sequencing
<130> R 77289
<150> EP19214355.0
<151> 2019-12-09
<160> 784
<210> 1
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 1
aaacgttcat cc 12
<210> 2
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 2
ttgtccgata tg 12
<210> 3
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 3
cgggaacccg ca 12
<210> 4
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 4
gtttaaaggc ag 12
<210> 5
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 5
tcctctcttc ta 12
<210> 6
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 6
ccaaagaggg at 12
<210> 7
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 7
gaagggtaaa gc 12
<210> 8
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 8
agtctcagca aa 12
<210> 9
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 9
gcactgacgc ta 12
<210> 10
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 10
cccaattttg cc 12
<210> 11
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 11
cagataatac gt 12
<210> 12
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 12
aggtggttct ac 12
<210> 13
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 13
agaggccgaa ca 12
<210> 14
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 14
cttaccgggt ac 12
<210> 15
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 15
tgctaaatta gt 12
<210> 16
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 16
tacgcccacg tg 12
<210> 17
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 17
atcgacttgt gt 12
<210> 18
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 18
ctatgcaagc tg 12
<210> 19
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 19
aaccctggga ag 12
<210> 20
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 20
tattggcggc ct 12
<210> 21
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 21
ccgggcgtca tg 12
<210> 22
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 22
gatttccccc ga 12
<210> 23
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 23
attatatctg aa 12
<210> 24
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 24
tcaacaaccg gt 12
<210> 25
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 25
tggagactgg gc 12
<210> 26
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 26
ctgtagtcgc ca 12
<210> 27
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 27
acaggactct gg 12
<210> 28
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 28
atttttaggg cc 12
<210> 29
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 29
ttatcactcc tt 12
<210> 30
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 30
cactagtttc gt 12
<210> 31
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 31
gcctaataca ac 12
<210> 32
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 32
acgatacgcc aa 12
<210> 33
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 33
ccgacggacc at 12
<210> 34
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 34
gaattcgtat ac 12
<210> 35
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 35
gacccgtctt ga 12
<210> 36
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 36
caccagagat at 12
<210> 37
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 37
aaaatcccag tt 12
<210> 38
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 38
tacggtatag aa 12
<210> 39
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 39
gcatccatgc at 12
<210> 40
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 40
gagtcggtgg ca 12
<210> 41
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 41
caccttcggt tg 12
<210> 42
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 42
ctctttaaac aa 12
<210> 43
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 43
gcgtgttaac gc 12
<210> 44
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 44
gtgagtagta gt 12
<210> 45
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 45
tgccatgttc gg 12
<210> 46
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 46
aaatgtagtg ag 12
<210> 47
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 47
tgtggggtga tt 12
<210> 48
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 48
gagcacgcga gc 12
<210> 49
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 49
taattacaaa ga 12
<210> 50
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 50
aagttgcggg ta 12
<210> 51
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 51
ccgttgaagg gg 12
<210> 52
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 52
aaactaactg tc 12
<210> 53
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 53
gctagctcag at 12
<210> 54
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 54
cgagtttatc ag 12
<210> 55
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 55
agcaaaggat gt 12
<210> 56
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 56
tcgagtcccg ga 12
<210> 57
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 57
ttccaaaaaa tg 12
<210> 58
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 58
tcctagcgat tt 12
<210> 59
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 59
taaccagcac tt 12
<210> 60
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 60
tttgtggaca cg 12
<210> 61
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 61
ttgcgttctc aa 12
<210> 62
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 62
atcggaaaat tc 12
<210> 63
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 63
actaagcgcg tg 12
<210> 64
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 64
ccgccctatt tc 12
<210> 65
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 65
cttaatgata tc 12
<210> 66
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 66
tgttttgcta ac 12
<210> 67
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 67
gaaaatttac gc 12
<210> 68
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 68
ttgacagcgt cg 12
<210> 69
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 69
cctggtactt tc 12
<210> 70
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 70
gtcaggctgc gt 12
<210> 71
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 71
ctctccatcg aa 12
<210> 72
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 72
gcgccgggtc cc 12
<210> 73
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 73
tataagggaa tg 12
<210> 74
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 74
attcctgagt ta 12
<210> 75
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 75
cccaccgtaa gc 12
<210> 76
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 76
aatagctttt tc 12
<210> 77
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 77
gtaccgaacc cg 12
<210> 78
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 78
ttccccgttt ag 12
<210> 79
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 79
acccgaacga gc 12
<210> 80
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 80
aagccacccc cg 12
<210> 81
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 81
atgcattgcc ct 12
<210> 82
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 82
aggcttaatc gg 12
<210> 83
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 83
ttaggacgca aa 12
<210> 84
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 84
cgaccactac cg 12
<210> 85
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 85
cgggtagggc gt 12
<210> 86
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 86
aatgaccgta gg 12
<210> 87
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 87
attcaacctc ta 12
<210> 88
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 88
caaggtcccc tt 12
<210> 89
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 89
gatagaaaca cc 12
<210> 90
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 90
gtggcaccac tt 12
<210> 91
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 91
agcttctttt cc 12
<210> 92
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 92
tcgtctggcc gt 12
<210> 93
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 93
atccgccagg at 12
<210> 94
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 94
tgaggcattt gg 12
<210> 95
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 95
gacattattc tt 12
<210> 96
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 96
taagatcgat ta 12
<210> 97
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 97
gtttgacttt at 12
<210> 98
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 98
aaaacatgcg tt 12
<210> 99
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 99
caaattggaa cg 12
<210> 100
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 100
atgggctaga ca 12
<210> 101
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 101
gcgcgaagtt ga 12
<210> 102
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 102
ccattgtcta aa 12
<210> 103
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 103
tcccggctaa aa 12
<210> 104
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 104
gtcaaatgtc ct 12
<210> 105
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 105
cccagttact aa 12
<210> 106
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 106
ttgtaagctc tg 12
<210> 107
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 107
agggccaaag ac 12
<210> 108
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 108
gtttcccgga cg 12
<210> 109
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 109
taatatatta tc 12
<210> 110
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 110
aacccgcggg ct 12
<210> 111
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 111
gccgggtccc ga 12
<210> 112
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 112
cgtatacgac cc 12
<210> 113
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 113
gacatgcaga tc 12
<210> 114
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 114
aaaccttttg aa 12
<210> 115
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 115
acgctcctca gt 12
<210> 116
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 116
cggtggttat ca 12
<210> 117
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 117
cgcggaagcc ac 12
<210> 118
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 118
attcaagggt ca 12
<210> 119
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 119
tgatcccttc gt 12
<210> 120
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 120
tcagaaacag tg 12
<210> 121
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 121
ctagcattgt gt 12
<210> 122
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 122
atctgaccga tg 12
<210> 123
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 123
ccaaatgggc cg 12
<210> 124
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 124
tcttggagga at 12
<210> 125
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 125
aagggagtac tg 12
<210> 126
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 126
gctttaaaaa gc 12
<210> 127
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 127
cttctctatg cc 12
<210> 128
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 128
taccaccaag gt 12
<210> 129
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 129
tggcgcatgg ga 12
<210> 130
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 130
atgtcgtaga ac 12
<210> 131
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 131
cacggcatat gg 12
<210> 132
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 132
ctttttcggg aa 12
<210> 133
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 133
ttctacataa tt 12
<210> 134
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 134
acatcgttta gt 12
<210> 135
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 135
gaatcctcac ca 12
<210> 136
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 136
cagctcagaa cc 12
<210> 137
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 137
aagcaggcaa ct 12
<210> 138
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 138
gccttagtct ca 12
<210> 139
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 139
gcaaagtatt gc 12
<210> 140
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 140
acaacgcgct ct 12
<210> 141
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 141
cccctaaacg tt 12
<210> 142
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 142
tcaggtctcg cc 12
<210> 143
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 143
gactaactga ct 12
<210> 144
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 144
gcgtaggtgg ac 12
<210> 145
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 145
acaattaggt tg 12
<210> 146
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 146
atatttccca cc 12
<210> 147
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 147
gagtgttcca ga 12
<210> 148
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 148
gtgcgtcgtc gt 12
<210> 149
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 149
tgaactgtta gg 12
<210> 150
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 150
ccctgtcgtg cg 12
<210> 151
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 151
tgtggggtgc tt 12
<210> 152
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 152
gcgacagagc ga 12
<210> 153
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 153
tccttcaccc gc 12
<210> 154
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 154
ccgttgaggg tc 12
<210> 155
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 155
aagttgccgg gg 12
<210> 156
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 156
cccatccatg ta 12
<210> 157
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 157
gatcgatacg ct 12
<210> 158
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 158
agcgtttcta cg 12
<210> 159
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 159
cgacccggct gt 12
<210> 160
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 160
tagcgtaaag gc 12
<210> 161
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 161
ttaacccccc tg 12
<210> 162
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 162
taatcgagct tt 12
<210> 163
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 163
tccaacgaca tt 12
<210> 164
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 164
tttgtggcac ag 12
<210> 165
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 165
ttgagttata cc 12
<210> 166
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 166
ctaggcccct ta 12
<210> 167
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 167
catccgagag tg 12
<210> 168
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 168
aagaaatctt ta 12
<210> 169
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 169
attcctgctc ta 12
<210> 170
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 170
tgttttgatc ca 12
<210> 171
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 171
gcccctttca ga 12
<210> 172
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 172
ttgcacgagt ag 12
<210> 173
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 173
aatggtcatt ta 12
<210> 174
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 174
gtacggatga gt 12
<210> 175
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 175
atataactag cc 12
<210> 176
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 176
gagaagggta aa 12
<210> 177
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 177
tctccgggcc tg 12
<210> 178
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 178
cttaatgcgt tc 12
<210> 179
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 179
aaacaagtcc ga 12
<210> 180
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 180
cctcgatttt ta 12
<210> 181
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 181
gtcaagccaa ag 12
<210> 182
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 182
ttaaaagttt cg 12
<210> 183
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 183
caaagccagc ga 12
<210> 184
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 184
ccgaacaaaa ag 12
<210> 185
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 185
ctgacttgaa at 12
<210> 186
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 186
cggattccta gg 12
<210> 187
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 187
ttcggcagac cc 12
<210> 188
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 188
agcaacatca ag 12
<210> 189
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 189
agggtcggga gt 12
<210> 190
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 190
cctgcaagtc gg 12
<210> 191
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 191
cttaccaata tc 12
<210> 192
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 192
accggtaaaa tt 12
<210> 193
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 193
gctcgcccac aa 12
<210> 194
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 194
gtggacaaca tt 12
<210> 195
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 195
cgattatttt aa 12
<210> 196
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 196
tagtatggaa gt 12
<210> 197
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 197
ctaagaacgg ct 12
<210> 198
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 198
tgcggacttt gg 12
<210> 199
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 199
gcacttctta tt 12
<210> 200
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 200
tccgctagct tc 12
<210> 201
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 201
gtttgcattt ct 12
<210> 202
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 202
ccccactgag tt 12
<210> 203
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 203
acccttggcc ag 12
<210> 204
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 204
ctgggatcgc ac 12
<210> 205
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 205
gagagccgtt gc 12
<210> 206
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 206
aacttgtatc cc 12
<210> 207
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 207
taaaggatcc cc 12
<210> 208
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 208
gtaccctgta at 12
<210> 209
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 209
agtattatgc cc 12
<210> 210
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 210
agcacactta ca 12
<210> 211
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 211
agaatgtcga ca 12
<210> 212
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 212
aagtaacagc tt 12
<210> 213
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 213
tagactctga ct 12
<210> 214
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 214
atagacgatc cc 12
<210> 215
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 215
tcatgccgaa ct 12
<210> 216
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 216
cgtcgaatat gg 12
<210> 217
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 217
atgacccaga tg 12
<210> 218
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 218
atttggccga tt 12
<210> 219
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 219
agtggcgcaa gc 12
<210> 220
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 220
cgccttttag gg 12
<210> 221
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 221
ctcggtgatc gt 12
<210> 222
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 222
ccgtgtgctg aa 12
<210> 223
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 223
atcctgtgcc ta 12
<210> 224
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 224
tctaatctga cg 12
<210> 225
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 225
agtggataag tt 12
<210> 226
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 226
tgccctcaaa cc 12
<210> 227
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 227
ctcccctgtg ac 12
<210> 228
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 228
gtatgcggta gc 12
<210> 229
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 229
ccactacatc tt 12
<210> 230
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 230
ctaaacttct tg 12
<210> 231
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 231
gcagatatgg ta 12
<210> 232
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 232
tctgcttgag gt 12
<210> 233
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 233
gaaatgtgaa gg 12
<210> 234
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 234
caaacgctga gg 12
<210> 235
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 235
gctactttgg gg 12
<210> 236
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 236
agaaattggc at 12
<210> 237
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 237
ctgccaacac ga 12
<210> 238
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 238
ccaaatcctt cg 12
<210> 239
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 239
cgcccaaata tc 12
<210> 240
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 240
tttatcgtta at 12
<210> 241
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 241
acgtcaacgt cc 12
<210> 242
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 242
tcaggtaaac tt 12
<210> 243
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 243
agagtattag ag 12
<210> 244
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 244
tacatggcca ct 12
<210> 245
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 245
gcaggttctc gt 12
<210> 246
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 246
cgatgataac gg 12
<210> 247
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 247
agttctacgg ac 12
<210> 248
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 248
tctcagattc at 12
<210> 249
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 249
actgtcccgc ta 12
<210> 250
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 250
atgacggtga gc 12
<210> 251
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 251
gaccgtgcgc aa 12
<210> 252
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 252
tactcgtgct gt 12
<210> 253
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 253
gttcataatc ac 12
<210> 254
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 254
agagaagcgt ta 12
<210> 255
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 255
aggacatcgg ac 12
<210> 256
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 256
tgcgttaact ct 12
<210> 257
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 257
aacttgtaaa ta 12
<210> 258
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 258
tatcctactc at 12
<210> 259
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 259
ccacgagcac tg 12
<210> 260
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 260
gcgttcgatg aa 12
<210> 261
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 261
ttagctatct tg 12
<210> 262
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 262
aggtcctggg ga 12
<210> 263
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 263
acttcgtcca gt 12
<210> 264
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 264
tagtgcttct aa 12
<210> 265
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 265
gccggaggtc tg 12
<210> 266
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 266
gttcgatcag ta 12
<210> 267
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 267
ccagaagtta tt 12
<210> 268
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 268
catagggagg gg 12
<210> 269
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 269
caggcgcgaa ga 12
<210> 270
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 270
cccgatagta ca 12
<210> 271
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 271
agcccgcgtc gt 12
<210> 272
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 272
tggccatacg ta 12
<210> 273
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 273
aagcggcaga gg 12
<210> 274
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 274
tacgcagtac aa 12
<210> 275
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 275
tctgtaattg ca 12
<210> 276
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 276
taccattcgc ag 12
<210> 277
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 277
cgcaagccct cg 12
<210> 278
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 278
cacgcgatgg gc 12
<210> 279
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 279
cacacaaggg ag 12
<210> 280
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 280
ttagtgttaa aa 12
<210> 281
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 281
taggggacat ca 12
<210> 282
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 282
gaaagatcgc cg 12
<210> 283
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 283
gaacgagaat gt 12
<210> 284
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 284
gcctcctccg tg 12
<210> 285
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 285
ctgatgtgag ag 12
<210> 286
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 286
attgatcaag ct 12
<210> 287
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 287
agacgggcac aa 12
<210> 288
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 288
ccctgcttac ga 12
<210> 289
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 289
ttaatgcgtg at 12
<210> 290
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 290
gtgctaatgg ga 12
<210> 291
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 291
cttaacacat aa 12
<210> 292
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 292
aaaaacggca gg 12
<210> 293
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 293
tgtttgagtc gg 12
<210> 294
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 294
tgcccgacag gt 12
<210> 295
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 295
aacgaatctg ta 12
<210> 296
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 296
gtcatccgtt tt 12
<210> 297
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 297
ataactctcc tc 12
<210> 298
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 298
tagaatacgg tt 12
<210> 299
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 299
acgttttgat tc 12
<210> 300
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 300
cgacctgaac gg 12
<210> 301
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 301
agtgtgaccg gt 12
<210> 302
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 302
ttataagccg tc 12
<210> 303
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 303
tattgaactc ca 12
<210> 304
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 304
cagttcctaa cc 12
<210> 305
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 305
acattcaagt gc 12
<210> 306
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 306
gcgacgctcg ta 12
<210> 307
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 307
cgtgatttaa ag 12
<210> 308
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 308
acctacagac ct 12
<210> 309
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 309
gtttattcag tg 12
<210> 310
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 310
gctcaccctc tt 12
<210> 311
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 311
acctgatgta tt 12
<210> 312
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 312
agttatgtgc ag 12
<210> 313
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 313
tcggctcaca ag 12
<210> 314
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 314
gctgcataca ag 12
<210> 315
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 315
gccccacatg at 12
<210> 316
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 316
aaagcctatc ac 12
<210> 317
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 317
ctatgttttt gc 12
<210> 318
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 318
cacgggtcag aa 12
<210> 319
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 319
gcgtaggcaa ga 12
<210> 320
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 320
tgtcgcaacg gc 12
<210> 321
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 321
atctcattat gg 12
<210> 322
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 322
gagatcagtc ag 12
<210> 323
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 323
ttcgaaccga ga 12
<210> 324
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 324
ctcactccgc cc 12
<210> 325
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 325
tttgcaaagc cg 12
<210> 326
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 326
cttgtaacga gt 12
<210> 327
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 327
gagtgtctga gg 12
<210> 328
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 328
acggcgccag gg 12
<210> 329
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 329
gtaagcacgt at 12
<210> 330
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 330
aagtctttgt cg 12
<210> 331
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 331
gacttcaatc ga 12
<210> 332
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 332
tctagacaaa gt 12
<210> 333
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 333
gatgcgctcc tt 12
<210> 334
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 334
gtactcccaa tg 12
<210> 335
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 335
tagcgaatat ct 12
<210> 336
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 336
gcattgcgaa tt 12
<210> 337
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 337
atggctggct ca 12
<210> 338
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 338
tatcaatacc cg 12
<210> 339
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 339
tcataaaggc ca 12
<210> 340
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 340
tgtgccctat aa 12
<210> 341
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 341
aggcaagaac gt 12
<210> 342
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 342
gtgcggccga gt 12
<210> 343
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 343
gcggagcacc ca 12
<210> 344
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 344
cgctcggtgg ga 12
<210> 345
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 345
accctagatc gc 12
<210> 346
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 346
cagaacgaat cc 12
<210> 347
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 347
ccgcaatgaa ct 12
<210> 348
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 348
tgagaggggt tc 12
<210> 349
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 349
catacttatt tg 12
<210> 350
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 350
catcgacgta cg 12
<210> 351
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 351
gccatctata ga 12
<210> 352
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 352
gcctctgaac ta 12
<210> 353
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 353
cccaggcagc tt 12
<210> 354
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 354
tagcccgggg ca 12
<210> 355
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 355
cacgtaccag tc 12
<210> 356
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 356
aatccggcgc ac 12
<210> 357
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 357
cctctcgaca tt 12
<210> 358
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 358
actgcgatcg tc 12
<210> 359
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 359
gtccaactca aa 12
<210> 360
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 360
tttcttcctc gc 12
<210> 361
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 361
tagccgttgt ac 12
<210> 362
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 362
gacgatcctc ac 12
<210> 363
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 363
gtgcttgcaa ac 12
<210> 364
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 364
cagtgaggta ct 12
<210> 365
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 365
tagtttaggg tg 12
<210> 366
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 366
ttttcgggat gg 12
<210> 367
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 367
gattacatga ga 12
<210> 368
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 368
ttcaacggta ag 12
<210> 369
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 369
aaggtagatg cg 12
<210> 370
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 370
ttcacgaatg ct 12
<210> 371
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 371
attcccttgg ta 12
<210> 372
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 372
agctccgatg cc 12
<210> 373
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 373
cgcaactaga ag 12
<210> 374
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 374
tatggttacc ag 12
<210> 375
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 375
gctacgagag ag 12
<210> 376
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 376
tggcggcggc ga 12
<210> 377
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 377
agatggcaac cc 12
<210> 378
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 378
agtaaccaca gc 12
<210> 379
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 379
ccttttgtac cc 12
<210> 380
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 380
ataagaagtg cc 12
<210> 381
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 381
aatcggagta ga 12
<210> 382
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 382
gcagtaagct gt 12
<210> 383
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 383
cgcggcacga ta 12
<210> 384
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 384
ctgcagctac tt 12
<210> 385
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 385
acagggggtg tc 12
<210> 386
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 386
gatccaagag gg 12
<210> 387
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 387
gacagtcgag ag 12
<210> 388
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 388
gttggagaaa ta 12
<210> 389
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 389
tcactctagg at 12
<210> 390
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 390
ttaataaacg ta 12
<210> 391
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 391
ttgatattcg ca 12
<210> 392
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 392
cgttggctcg cg 12
<210> 393
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 393
cgtctgtctt ca 12
<210> 394
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 394
aacagtgtca cc 12
<210> 395
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 395
gcgattaaat ca 12
<210> 396
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 396
cggagccgac cg 12
<210> 397
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 397
gtatagttgc ga 12
<210> 398
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 398
cgagcatcgg gt 12
<210> 399
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 399
tgactaccgt aa 12
<210> 400
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 400
ctttcatgtc tc 12
<210> 401
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 401
gcacctagcg gc 12
<210> 402
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 402
ctccgtctag ag 12
<210> 403
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 403
gtcgctagcc ag 12
<210> 404
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 404
ttcggttgta ta 12
<210> 405
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 405
gcggtatcat ca 12
<210> 406
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 406
taaagcgtac gc 12
<210> 407
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 407
ttcttacgca ag 12
<210> 408
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 408
acgcgcggac ta 12
<210> 409
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 409
agctataaga tc 12
<210> 410
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 410
gtaatggcaa ca 12
<210> 411
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 411
tccatttgat gc 12
<210> 412
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 412
cagggatttc ca 12
<210> 413
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 413
caatcaatgg ac 12
<210> 414
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 414
cgattctgat tg 12
<210> 415
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 415
tgggtcttcg cc 12
<210> 416
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 416
gtcgtcgaac gc 12
<210> 417
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 417
gaagtgcccc ca 12
<210> 418
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 418
gccgggaata ag 12
<210> 419
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 419
actttggaat ag 12
<210> 420
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 420
ttctggtccc ag 12
<210> 421
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 421
aacttaattt ct 12
<210> 422
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 422
tcgaagtgct gt 12
<210> 423
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 423
tcgcaacacg ct 12
<210> 424
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 424
gtatcttgtc ac 12
<210> 425
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 425
catgaagagg cg 12
<210> 426
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 426
tacgagctgg tt 12
<210> 427
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 427
cccttactgt ga 12
<210> 428
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 428
tccggtgcat tt 12
<210> 429
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 429
gccaaggtgc ta 12
<210> 430
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 430
cttccgcaaa ct 12
<210> 431
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 431
cggaagggaa cc 12
<210> 432
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 432
cgccgcgtta ag 12
<210> 433
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 433
ccttctagtt at 12
<210> 434
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 434
ttgtgaaaca tt 12
<210> 435
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 435
ccagtcctga ct 12
<210> 436
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 436
tccttaaccc gt 12
<210> 437
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 437
tgaaacgcgc aa 12
<210> 438
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 438
cgtccttgat cc 12
<210> 439
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 439
actggcagcg gg 12
<210> 440
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 440
cgttcacagc cg 12
<210> 441
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 441
tgaatcctcg ag 12
<210> 442
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 442
tgcgtcgcct ta 12
<210> 443
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 443
tctgaccaga aa 12
<210> 444
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 444
tattccgtcc aa 12
<210> 445
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 445
agatttcgct ac 12
<210> 446
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 446
caatgctcaa tt 12
<210> 447
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 447
tctttgcccc at 12
<210> 448
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 448
ctatatacgc gg 12
<210> 449
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 449
ccttagcaaa tc 12
<210> 450
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 450
tcgtactaat cg 12
<210> 451
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 451
gagcccctgc tc 12
<210> 452
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 452
gttgctgtcc ac 12
<210> 453
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 453
cgtgggaatg tc 12
<210> 454
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 454
atcatggata gg 12
<210> 455
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 455
gacttagccc ct 12
<210> 456
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 456
tcgtgtatga ct 12
<210> 457
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 457
aatgtcatgc at 12
<210> 458
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 458
ccccgttctt at 12
<210> 459
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 459
tgggatattt ac 12
<210> 460
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 460
tccctgtcat cg 12
<210> 461
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 461
ttaaagtaga gt 12
<210> 462
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 462
gcacttgtat cc 12
<210> 463
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 463
ctagttgcgc at 12
<210> 464
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 464
gttctttgct ga 12
<210> 465
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 465
tatatttctt at 12
<210> 466
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 466
ctgaaacagg gg 12
<210> 467
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 467
gagggttgct aa 12
<210> 468
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 468
ctagggtgcc ag 12
<210> 469
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 469
cggtcattcg cg 12
<210> 470
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 470
gatgtattgg tt 12
<210> 471
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 471
cctagagtag gt 12
<210> 472
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 472
caagaaacca cg 12
<210> 473
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 473
tgtggaagga at 12
<210> 474
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 474
tgcgtatggt tg 12
<210> 475
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 475
acgggtgatc ac 12
<210> 476
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 476
accgttggtg ac 12
<210> 477
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 477
atcagctgat aa 12
<210> 478
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 478
gctatcggct gg 12
<210> 479
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 479
agacagctaa ag 12
<210> 480
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 480
gaggtccaca ta 12
<210> 481
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 481
gtcctgcact gg 12
<210> 482
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 482
cttgagcctt aa 12
<210> 483
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 483
acacacacta gc 12
<210> 484
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 484
agggactccc tt 12
<210> 485
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 485
aaggacaata tt 12
<210> 486
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 486
atacatatat at 12
<210> 487
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 487
cctatgcttc ct 12
<210> 488
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 488
cggctggact gc 12
<210> 489
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 489
accctcataa gg 12
<210> 490
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 490
tagtagccgc ac 12
<210> 491
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 491
gatcatctgg aa 12
<210> 492
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 492
gagttataaa tt 12
<210> 493
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 493
gtgtccttac at 12
<210> 494
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 494
cgaatgcaga aa 12
<210> 495
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 495
cgagagattg at 12
<210> 496
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 496
accggctcga cc 12
<210> 497
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 497
cgtcttctga aa 12
<210> 498
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 498
cgacacattc ac 12
<210> 499
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 499
cgcctgtagc ac 12
<210> 500
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 500
ccgtccacga tt 12
<210> 501
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 501
tcgcatatgc at 12
<210> 502
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 502
ctcgcagcta aa 12
<210> 503
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 503
tactgaagcc at 12
<210> 504
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 504
agtagcctct gg 12
<210> 505
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 505
ctgcaaacgc tg 12
<210> 506
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 506
ctttggaagc tt 12
<210> 507
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 507
cgtggagacc ga 12
<210> 508
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 508
agaattttcg gg 12
<210> 509
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 509
ataggtgcta gt 12
<210> 510
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 510
aagtgtgatg cc 12
<210> 511
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 511
ctaatgtgaa tc 12
<210> 512
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 512
tatcctatgc ag 12
<210> 513
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 513
cgtggctccg tt 12
<210> 514
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 514
tgaaataccc aa 12
<210> 515
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 515
ataaaatgtg ca 12
<210> 516
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 516
gtctgaggtc ga 12
<210> 517
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 517
aacatcacta tt 12
<210> 518
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 518
atcccattat tg 12
<210> 519
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 519
gacgctctgg tc 12
<210> 520
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 520
tatgattgcg gt 12
<210> 521
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 521
gccctgtgcc gg 12
<210> 522
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 522
acccagatgc gg 12
<210> 523
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 523
gatcatttgg gg 12
<210> 524
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 524
cgcccttgga ct 12
<210> 525
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 525
atgaaccaca gc 12
<210> 526
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 526
aaccctaatt ag 12
<210> 527
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 527
agaaaccctc ta 12
<210> 528
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 528
tttctagttc ct 12
<210> 529
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 529
cagtaccagt aa 12
<210> 530
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 530
tacggtccca tt 12
<210> 531
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 531
cgcgtcttcg cg 12
<210> 532
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 532
tcactggaac at 12
<210> 533
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 533
gacggttata gt 12
<210> 534
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 534
agctgctcca gg 12
<210> 535
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 535
cgttatcagg ca 12
<210> 536
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 536
tatacgctta ct 12
<210> 537
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 537
catgtaaaga tc 12
<210> 538
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 538
ctgcaggtgc ga 12
<210> 539
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 539
gcaagtgaga cc 12
<210> 540
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 540
tcatagtgat gt 12
<210> 541
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 541
gttactccta ca 12
<210> 542
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 542
cgcgccgagt tc 12
<210> 543
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 543
cggcactagg ca 12
<210> 544
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 544
tgagttccat at 12
<210> 545
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 545
ccattgtccc tc 12
<210> 546
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 546
tctaatcata ct 12
<210> 547
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 547
aacagcgaca tg 12
<210> 548
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 548
gagttagctg cc 12
<210> 549
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 549
ttcgatctat tg 12
<210> 550
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 550
cggtaatggg gc 12
<210> 551
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 551
cattagtaac gt 12
<210> 552
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 552
tcgtgattat cc 12
<210> 553
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 553
gaaggcggta tg 12
<210> 554
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 554
gttagctacg tc 12
<210> 555
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 555
aacgccgttc tt 12
<210> 556
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 556
actcgggcgg gg 12
<210> 557
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 557
acggagagcc gc 12
<210> 558
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 558
aaagctcgtc ac 12
<210> 559
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 559
cgaaagagta gt 12
<210> 560
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 560
tggaactcag tc 12
<210> 561
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 561
ccgaggacgc gg 12
<210> 562
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 562
tcagacgtca cc 12
<210> 563
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 563
tatgtccttg ac 12
<210> 564
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 564
tcaacttaga cg 12
<210> 565
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 565
agaccgaaat ag 12
<210> 566
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 566
acagagctgg ga 12
<210> 567
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 567
acacaccggg cg 12
<210> 568
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 568
ttcgtgttcc cc 12
<210> 569
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 569
tcggggcact ac 12
<210> 570
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 570
gcccgctaga ag 12
<210> 571
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 571
gccagcgcct gt 12
<210> 572
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 572
gaataataag tg 12
<210> 573
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 573
atgctgtgcg cg 12
<210> 574
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 574
cttgctaccg at 12
<210> 575
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 575
cgcagggaca cc 12
<210> 576
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 576
aaatgattca gc 12
<210> 577
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 577
ttcctgagtg ct 12
<210> 578
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 578
gtgatcctgg gc 12
<210> 579
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 579
attccacact cc 12
<210> 580
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 580
cccccaggac gg 12
<210> 581
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 581
tgtttgcgta gg 12
<210> 582
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 582
tgaaagcacg gt 12
<210> 583
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 583
ccagcctatg tc 12
<210> 584
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 584
gtactaagtt tt 12
<210> 585
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 585
ctccatataa ta 12
<210> 586
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 586
tcgcctcagg tt 12
<210> 587
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 587
cagttttgct ta 12
<210> 588
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 588
agcaatgcca gg 12
<210> 589
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 589
cgtgtgcaag gt 12
<210> 590
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 590
ttctccgaag ta 12
<210> 591
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 591
tcttgccata ac 12
<210> 592
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 592
acgttaatcc aa 12
<210> 593
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 593
cacttaccgt ga 12
<210> 594
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 594
gagcagatag tc 12
<210> 595
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 595
agtgctttcc cg 12
<210> 596
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 596
caatcacgca at 12
<210> 597
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 597
gtttcttacg tg 12
<210> 598
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 598
gatacaaata tt 12
<210> 599
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 599
caatgctgtc tt 12
<210> 600
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 600
cgttctgtga cg 12
<210> 601
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 601
taggatacac cg 12
<210> 602
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 602
gatgactcac cg 12
<210> 603
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 603
gaaaacactg ct 12
<210> 604
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 604
cccgaatcta ca 12
<210> 605
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 605
atctgttttt ga 12
<210> 606
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 606
acagggtacg cc 12
<210> 607
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 607
gagtcggacc gc 12
<210> 608
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 608
tgtagaccag ga 12
<210> 609
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 609
ctatacttct gg 12
<210> 610
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 610
gcgctacgta cg 12
<210> 611
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 611
ttagccaagc gc 12
<210> 612
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 612
atacataaga aa 12
<210> 613
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 613
tttgacccga ag 12
<210> 614
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 614
attgtccagc gt 12
<210> 615
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 615
gcgtgtatgc gg 12
<210> 616
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 616
caggagaacg gg 12
<210> 617
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 617
gtccgacagt ct 12
<210> 618
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 618
ccgtatttgt ag 12
<210> 619
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 619
gcattaccta gc 12
<210> 620
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 620
tatcgcaccc gt 12
<210> 621
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 621
gctgagataa tt 12
<210> 622
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 622
gtcataaacc tg 12
<210> 623
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 623
tcgagcctct at 12
<210> 624
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 624
gacttgagcc tt 12
<210> 625
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 625
ctggatggat ac 12
<210> 626
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 626
tctacctcaa ag 12
<210> 627
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 627
tactcccgga ac 12
<210> 628
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 628
tgtgaaatct cc 12
<210> 629
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 629
cggaccgcca gt 12
<210> 630
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 630
gtgaggaagc gt 12
<210> 631
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 631
gaggcgacaa ac 12
<210> 632
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 632
agataggtgg gc 12
<210> 633
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 633
caaatcgcta ga 12
<210> 634
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 634
acgccagcct aa 12
<210> 635
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 635
aagacctgcc at 12
<210> 636
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 636
tgcgcggggt ta 12
<210> 637
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 637
actcattctt tg 12
<210> 638
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 638
actagcagtc ag 12
<210> 639
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 639
gaactatctc gc 12
<210> 640
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 640
gaatatgcca tc 12
<210> 641
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 641
aaacggacga tt 12
<210> 642
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 642
tcgaaagggg ac 12
<210> 643
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 643
acagtcaacg ta 12
<210> 644
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 644
cctaaggaga ca 12
<210> 645
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 645
aatatagcac tt 12
<210> 646
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 646
catgagctag ta 12
<210> 647
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 647
gtaaccatac cc 12
<210> 648
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 648
tttattaata ga 12
<210> 649
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 649
tcgaagttgt ca 12
<210> 650
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 650
gcagctaata ca 12
<210> 651
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 651
gtgattgacc ca 12
<210> 652
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 652
acgtgcggtc at 12
<210> 653
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 653
tcgtttcggg tg 12
<210> 654
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 654
ttttagggct gg 12
<210> 655
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 655
gcttcactgc gc 12
<210> 656
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 656
ttaccaggtc cg 12
<210> 657
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 657
ccggtcgctg ag 12
<210> 658
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 658
ttacagcctg at 12
<210> 659
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 659
cttaaattgg tc 12
<210> 660
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 660
cgataagctg aa 12
<210> 661
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 661
agaccatcgc cg 12
<210> 662
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 662
tctggttcaa cg 12
<210> 663
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 663
gatcagcgcg cg 12
<210> 664
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 664
tggaggagga gc 12
<210> 665
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 665
cgctggacca aa 12
<210> 666
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 666
cgtccaacac ga 12
<210> 667
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 667
aattttgtca aa 12
<210> 668
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 668
ctccgccgtg aa 12
<210> 669
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 669
cctaggcgtc gc 12
<210> 670
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 670
gacgtccgat gt 12
<210> 671
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 671
agaggacagc tc 12
<210> 672
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 672
atgacgatca tt 12
<210> 673
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 673
cacgggggtg ta 12
<210> 674
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 674
gctaaccgcg gg 12
<210> 675
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 675
gcacgtagcg cg 12
<210> 676
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 676
gttggcgccc tc 12
<210> 677
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 677
tacatatcgg ct 12
<210> 678
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 678
ttcctcccag tc 12
<210> 679
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 679
ttgctcttag ac 12
<210> 680
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 680
agttggatag ag 12
<210> 681
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 681
agtatgtatt ac 12
<210> 682
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 682
ccacgtgtac aa 12
<210> 683
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 683
gagcttccct ac 12
<210> 684
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 684
aggcgaagca ag 12
<210> 685
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 685
gtctcgttga gc 12
<210> 686
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 686
agcgactagg gt 12
<210> 687
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 687
tgcatcaagt cc 12
<210> 688
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 688
atttactgta ta 12
<210> 689
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 689
gacaatcgag ga 12
<210> 690
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 690
ataagtatcg cg 12
<210> 691
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 691
gtagatcgaa cg 12
<210> 692
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 692
ttaggttgtc tc 12
<210> 693
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 693
gaggtctact ac 12
<210> 694
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 694
tcccgagtca ga 12
<210> 695
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 695
ttattcagac cg 12
<210> 696
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 696
cagagaggca tc 12
<210> 697
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 697
cgatctccgc ta 12
<210> 698
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 698
gtcctggacc ac 12
<210> 699
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 699
taactttgct ga 12
<210> 700
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 700
acgggcttta ac 12
<210> 701
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 701
acctacctgg ca 12
<210> 702
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 702
agcttatgct tg 12
<210> 703
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 703
tgggtattag aa 12
<210> 704
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 704
gtagtagcca ga 12
<210> 705
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 705
gccgtgaaaa ac 12
<210> 706
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 706
gaagggcctc cg 12
<210> 707
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 707
catttggcct cg 12
<210> 708
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 708
ttatggtaaa cg 12
<210> 709
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 709
ccattccttt at 12
<210> 710
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 710
tagccgtgat gt 12
<210> 711
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 711
tagaccacag at 12
<210> 712
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 712
gtctattgta ca 12
<210> 713
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 713
actgccgcgg ag 12
<210> 714
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 714
tcagcgatgg tt 12
<210> 715
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 715
aaattcatgt gc 12
<210> 716
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 716
taaggtgact tt 12
<210> 717
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 717
gaaccggtga tc 12
<210> 718
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 718
attaagaccc at 12
<210> 719
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 719
aggccgggcc aa 12
<210> 720
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 720
agaagagttc cg 12
<210> 721
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 721
aattatcgtt ct 12
<210> 722
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 722
ttgtgcccac tt 12
<210> 723
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 723
aacgtaatgc at 12
<210> 724
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 724
taattccaaa gt 12
<210> 725
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 725
tgcccagaga cc 12
<210> 726
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 726
agtaattgct aa 12
<210> 727
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 727
catggacgag gg 12
<210> 728
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 728
agttacacga ag 12
<210> 729
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 729
tgcctaatag cg 12
<210> 730
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 730
tgagtagaat tc 12
<210> 731
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 731
tatgcaacgc cc 12
<210> 732
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 732
tcttaagtaa cc 12
<210> 733
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 733
cgctttagat ca 12
<210> 734
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 734
acctgatacc tt 12
<210> 735
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 735
tatttgaaaa ct 12
<210> 736
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 736
atctctcaga gg 12
<210> 737
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 737
aattcgaccc ta 12
<210> 738
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 738
tagtcatcct ag 12
<210> 739
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 739
gcgaaaatga ta 12
<210> 740
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 740
gttgatgtaa ca 12
<210> 741
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 741
agtgggcctg ta 12
<210> 742
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 742
ctactggctc gg 12
<210> 743
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 743
gcattcgaaa at 12
<210> 744
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 744
tagtgtctgc at 12
<210> 745
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 745
cctgtactga ct 12
<210> 746
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 746
aaaagttatt ct 12
<210> 747
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 747
tgggctcttt ca 12
<210> 748
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 748
taaatgtact ag 12
<210> 749
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 749
ttcccgtcgc gt 12
<210> 750
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 750
gacattgtct aa 12
<210> 751
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 751
atcgttgaga ct 12
<210> 752
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 752
gttatttgat gc 12
<210> 753
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 753
tctctttatt ct 12
<210> 754
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 754
atgcccacgg gg 12
<210> 755
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 755
gcgggttgat cc 12
<210> 756
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 756
atcgggtgaa cg 12
<210> 757
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 757
aggtacttag ag 12
<210> 758
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 758
gctgtcttgg tt 12
<210> 759
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 759
aatcgcgtcg gt 12
<210> 760
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 760
accgcccaac ag 12
<210> 761
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 761
tgtggccggc ct 12
<210> 762
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 762
tgagtctggt tg 12
<210> 763
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 763
cagggtgcta ca 12
<210> 764
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 764
caagttggtg ca 12
<210> 765
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 765
ctacgatgct cc 12
<210> 766
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 766
gatctaggat gg 12
<210> 767
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 767
cgcacgatcc cg 12
<210> 768
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 768
gcggtaacac tc 12
<210> 769
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 769
gtaatgacat gg 12
<210> 770
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 770
attgcgaatt cc 12
<210> 771
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 771
cacacacatc ga 12
<210> 772
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 772
cgggcataaa tt 12
<210> 773
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 773
ccggcacctc tt 12
<210> 774
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 774
ctcactctct ct 12
<210> 775
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 775
aatctgatta at 12
<210> 776
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 776
aggatggcat ga 12
<210> 777
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 777
caaatactcc gg 12
<210> 778
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 778
tcgtcgaaga ca 12
<210> 779
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 779
gctactatgg cc 12
<210> 780
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 780
gcgttctccc tt 12
<210> 781
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 781
gtgtaattca ct 12
<210> 782
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 782
agcctgacgc cc 12
<210> 783
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 783
agcgcgcttg ct 12
<210> 784
<211> 12
<212> DNA
<213> Artificial Sequence
<220>
<223> index sequence
<400> 784
caaggatagc aa 12
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| АНТИСМЫСЛОВЫЕ ОЛИГОНУКЛЕОТИДЫ К АЛЬФА-СИНУКЛЕИНУ И ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2773197C2 |
| Набор синтетических олигонуклеотидов для одновременного генотипирования 63 ДНК-маркеров, ассоциированных с группой крови АВ0, основными гаплогруппами Y-хромосомы, цветом радужной оболочки глаза, волос, кожи и половой принадлежностью, методом ПЦР с последующей гибридизацией | 2020 |
|
RU2740575C1 |
| КОМПОНЕНТЫ СИСТЕМЫ CRISPR-CAS, СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ МАНИПУЛЯЦИИ С ПОСЛЕДОВАТЕЛЬНОСТЯМИ | 2013 |
|
RU2796549C2 |
| СПОСОБ АМПЛИФИКАЦИИ И ИДЕНТИФИКАЦИИ НУКЛЕИНОВЫХ КИСЛОТ | 2019 |
|
RU2811465C2 |
| Способ идентификации личности и установления родства с помощью InDel полиморфизмов и набор синтетических олигонуклеотидов для их генотипирования | 2020 |
|
RU2738752C1 |
| ВЫСОКОПРОИЗВОДИТЕЛЬНОЕ СЕКВЕНИРОВАНИЕ ОДИНОЧНОЙ КЛЕТКИ СО СНИЖЕННОЙ ОШИБКОЙ АМПЛИФИКАЦИИ | 2019 |
|
RU2744175C1 |
| НАПРАВЛЯЕМАЯ РНК РЕГУЛЯЦИЯ ТРАНСКРИПЦИИ | 2014 |
|
RU2756865C2 |
| ОПРЕДЕЛЕНИЕ НУКЛЕИНОВЫХ КИСЛОТ ПУТЕМ АМПЛИФИКАЦИИ, ОСНОВАННОЙ НА ВСТРАИВАНИИ В ЦЕПЬ | 2014 |
|
RU2694976C1 |
| КОМПОЗИЦИИ ОЛИГОНУКЛЕОТИДОВ И СПОСОБЫ С НИМИ | 2016 |
|
RU2830607C2 |
| ГЕНОМНАЯ ИНЖЕНЕРИЯ | 2021 |
|
RU2812848C2 |
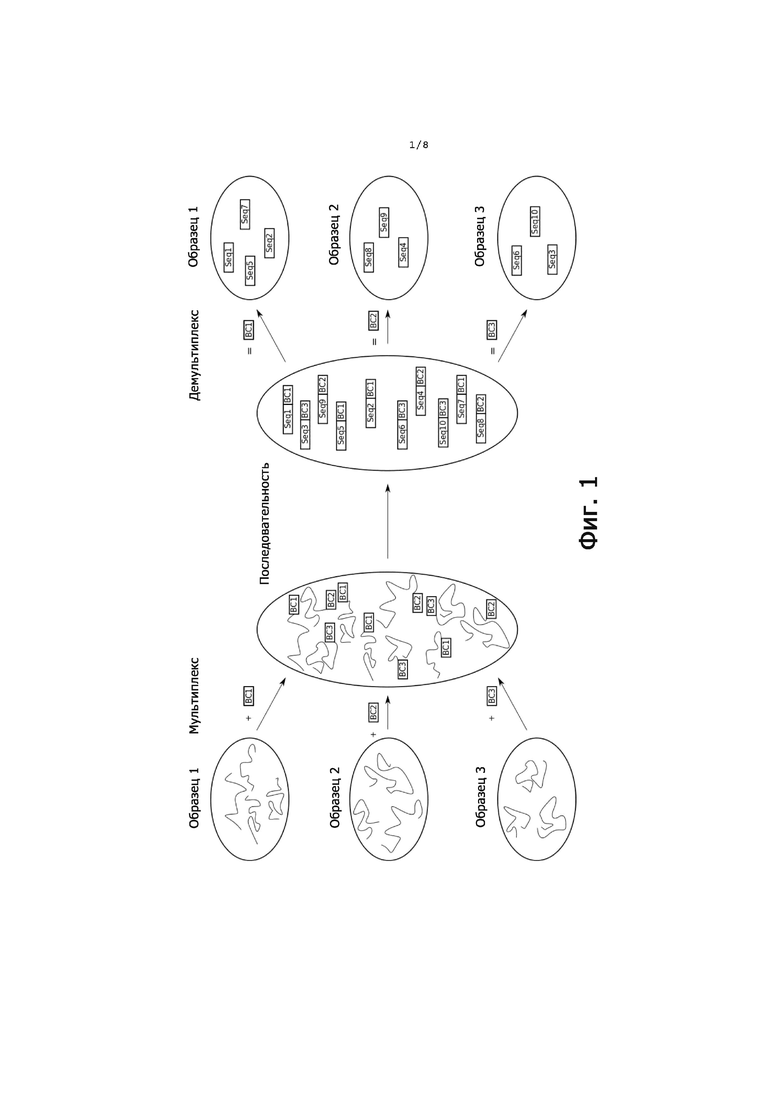
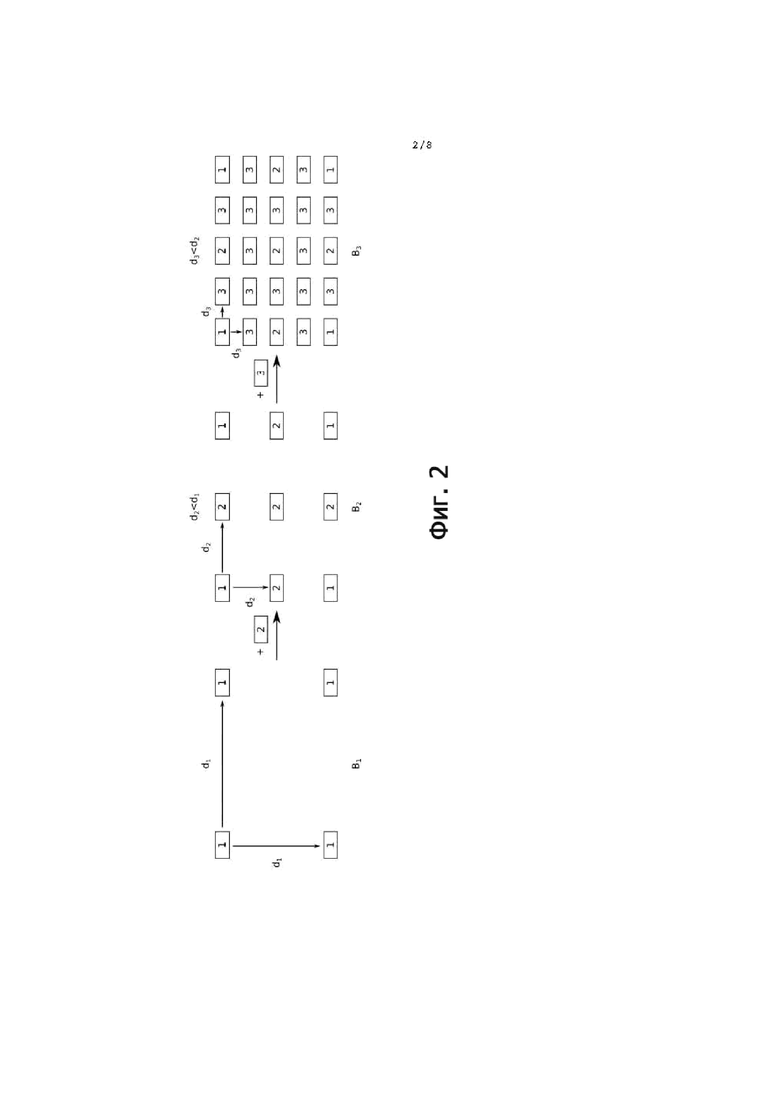
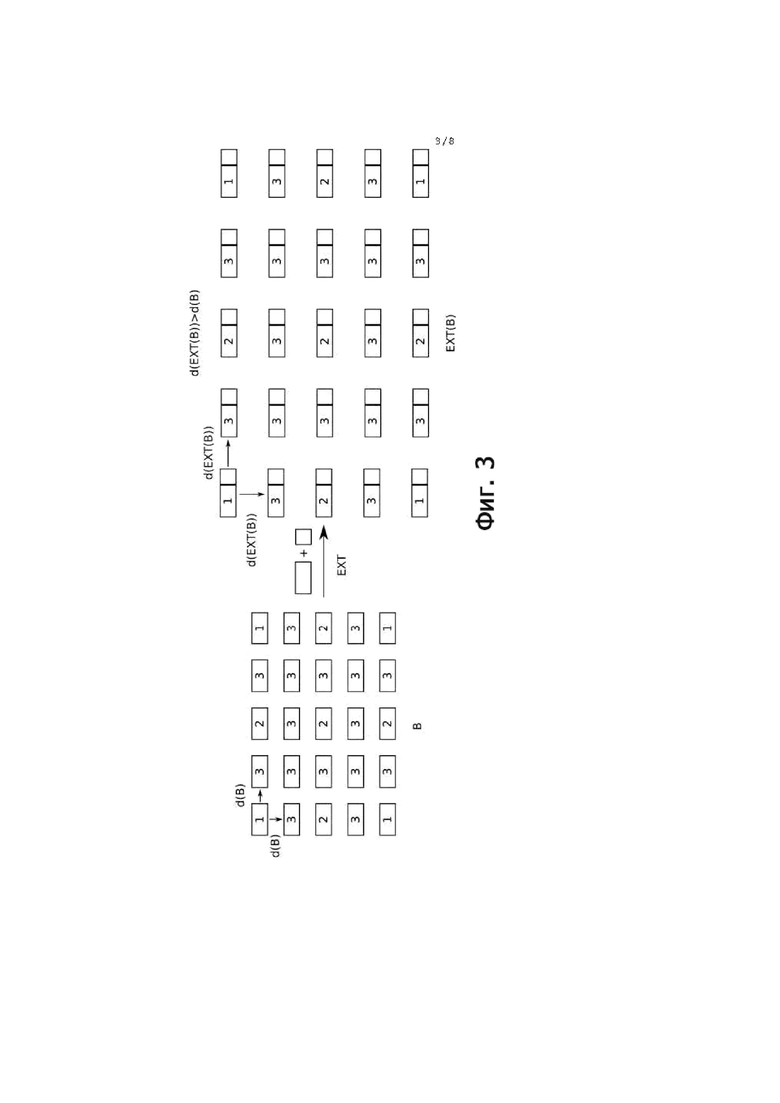
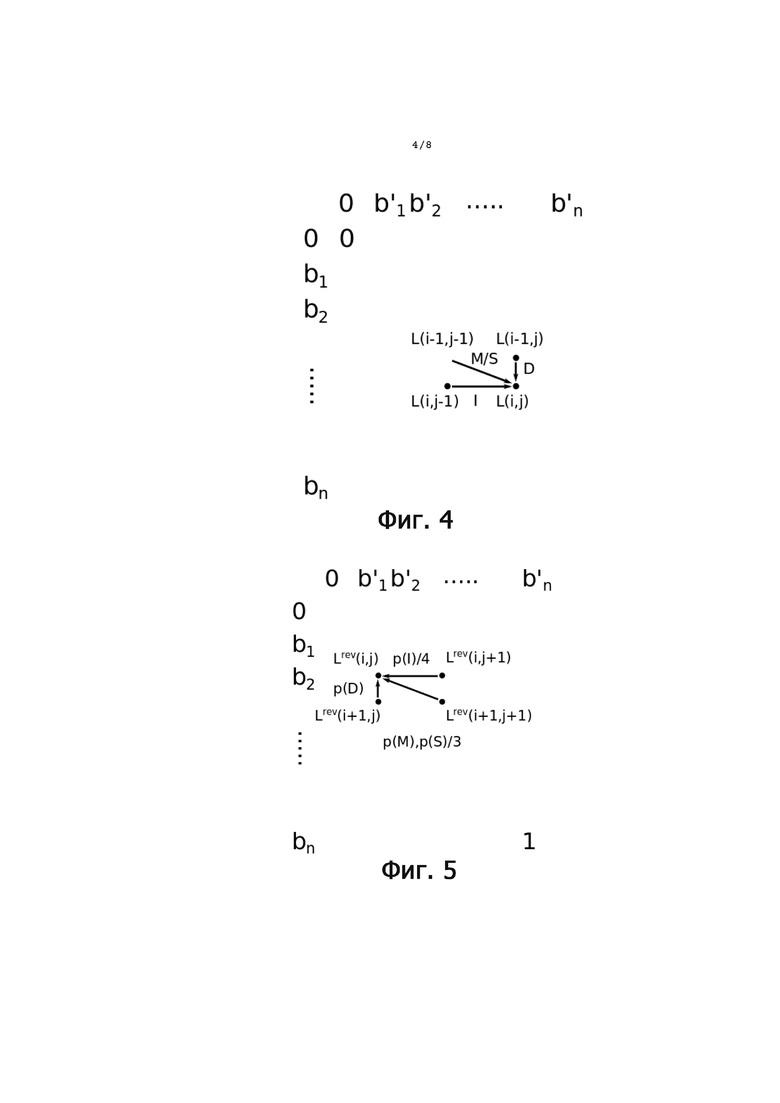
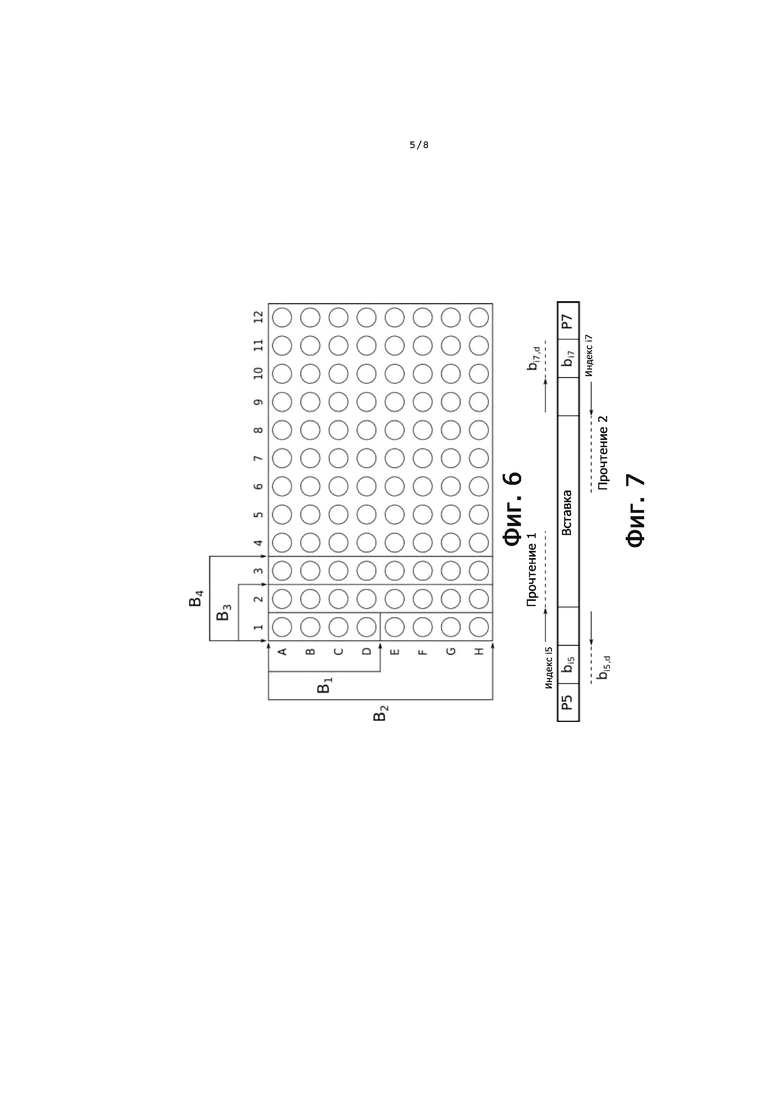
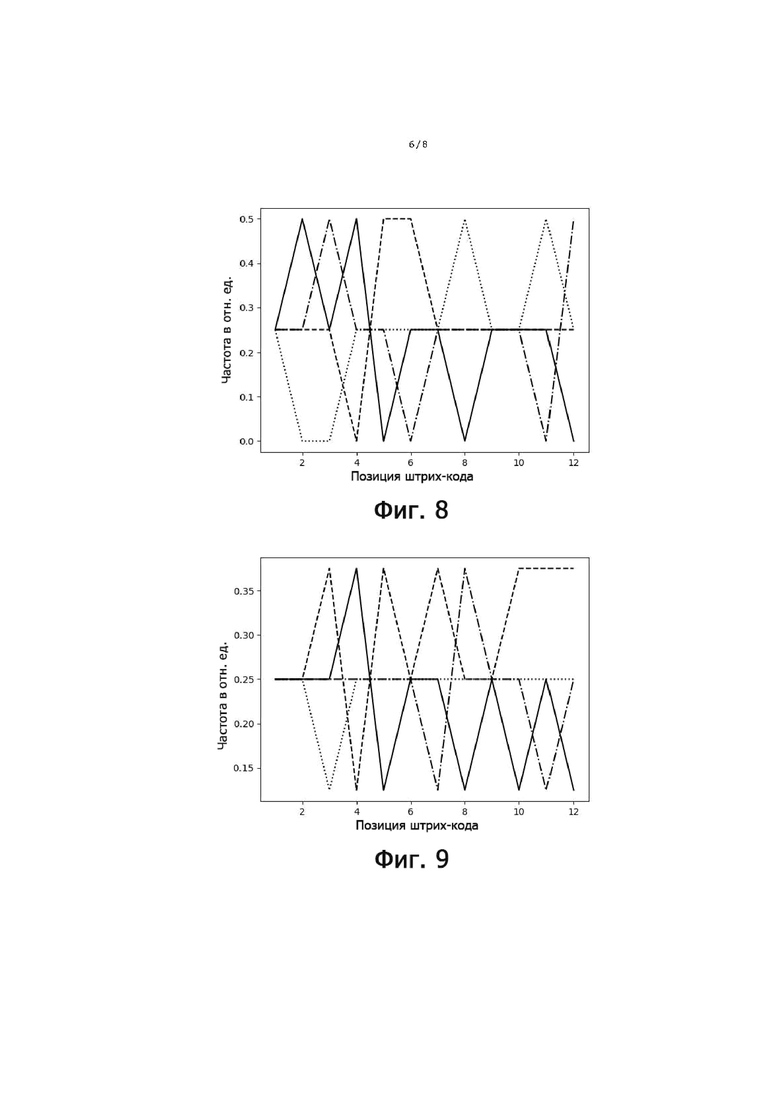
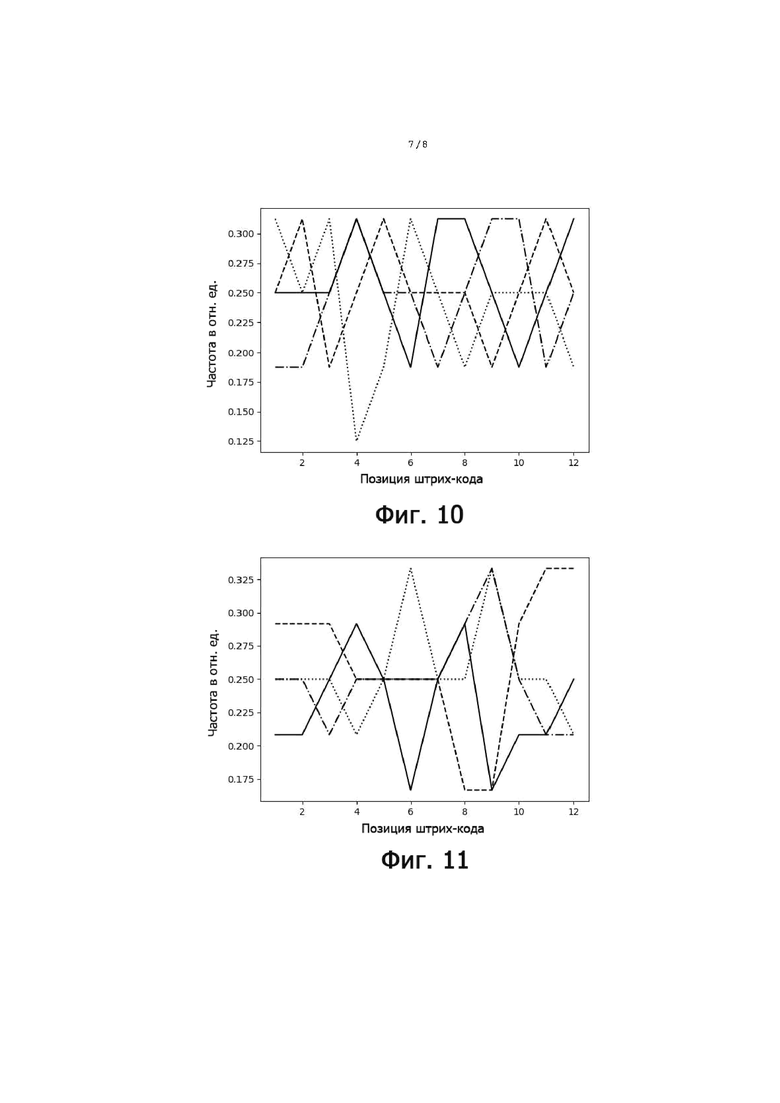
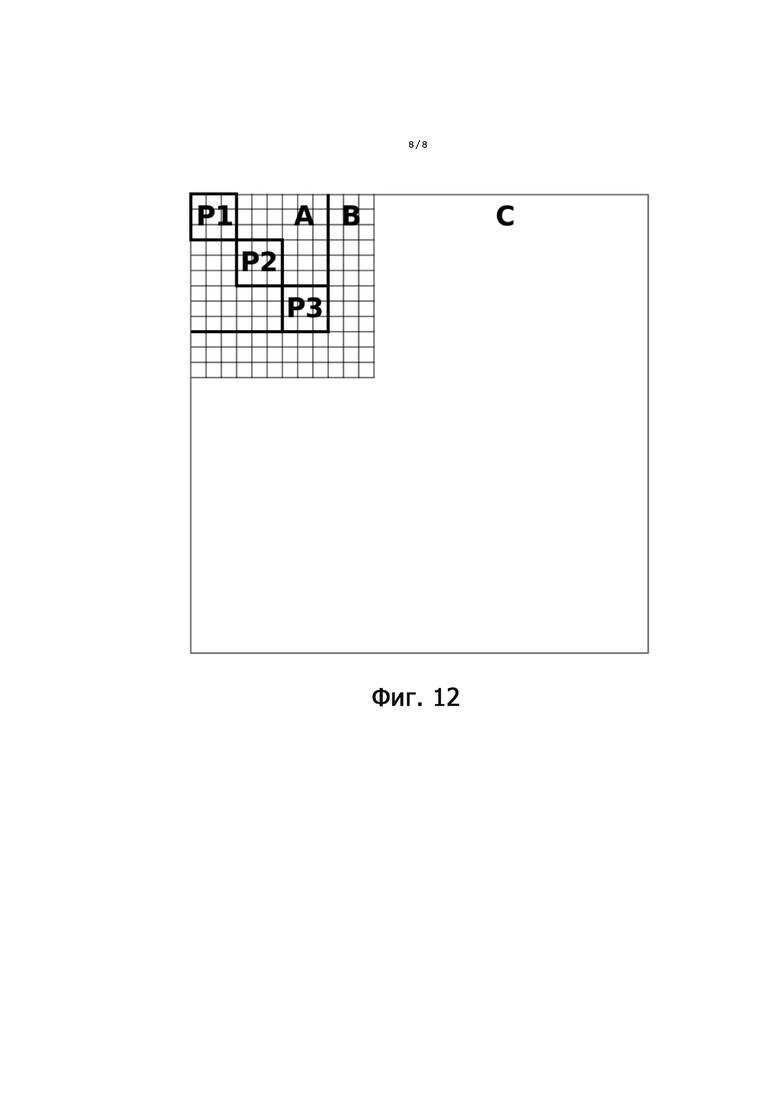
Изобретение относится к области молекулярной биологии. Описан набор олигонуклеотидов, содержащий индексные последовательности для мультиплексного параллельного секвенирования. Также описаны способ получения указанного набора олигонуклеотидов и способ присвоения секвенирующих прочтений образцу олигонуклеотидов. Технический результат заключается в максимальной различимости индексных последовательностей при компромиссе с эффективностью с учётом усилий и затрат для каждого секвенируемого нуклеотида. 3 н. и 12 з.п. ф-лы, 12 ил., 4 табл., 3 пр.
1. Набор олигонуклеотидов для идентификации олигонуклеотидов, содержащий индексные последовательности, причем набор содержит множество поднаборов олигонуклеотидов с разными индексными последовательностями,
где индексные последовательности поднабора олигонуклеотидов отличаются друг от друга по меньшей мере ненулевым количеством изменений последовательностей;
и где набор содержит не менее 2-х иерархических уровней поднаборов,
где индексные последовательности поднабора более высокого уровня являются элементами поднабора более низкого уровня, и где поднабор более низкого уровня содержит больше индексных последовательностей, чем поднабор более высокого уровня,
и где индексные последовательности поднабора более низкого уровня отличаются друг от друга меньшим минимальным количеством изменений последовательностей по сравнению с индексными последовательностями поднабора более высокого уровня;
и где олигонуклеотиды присваиваются одному или нескольким поднаборам.
2. Набор по п. 1, в котором каждая из индексных последовательностей поднабора содержит усеченную индексную последовательность, и усеченные индексные последовательности по меньшей мере одного поднабора отличаются по меньшей мере ненулевым количеством изменений последовательностей от каждой другой усеченной индексной последовательности в пределах упомянутого поднабора; предпочтительно, когда минимальное количество изменений последовательностей между усеченными индексными последовательностями поднабора больше, чем минимальное количество изменений последовательностей индексных последовательностей в поднаборе за вычетом разности между длиной индексных последовательностей и усеченных индексных последовательностей.
3. Набор по п. 2, в котором усеченные индексные последовательности поднабора более высокого уровня являются элементами усеченных индексных последовательностей поднабора более низкого уровня, и где усеченные индексные последовательности поднабора более низкого уровня отличаются друг от друга меньшим минимальным количеством изменений последовательностей по сравнению с усеченными индексными последовательностями поднабора более высокого уровня.
4. Набор по любому из пп. 1-3, в котором изменения последовательности выбираются из нуклеотидных замен, делеций и вставок, и где минимальное количество изменений последовательностей соответствует минимальному количеству, необходимому для замены любой индексной последовательности на другую индексную последовательность.
5. Набор по любому из пп. 1-4, в котором изменения последовательности количественно оцениваются как секвенциальное расстояние, которое представляет собой количество изменений нуклеотидов или вероятность изменений; предпочтительно, где величина секвенциального расстояния представляет собой расстояние Хэмминга, расстояние Левенштейна или расстояние Левенштейна между последовательностями, предпочтительно расстояние Левенштейна между последовательностями; или предпочтительно, где вероятность изменений является максимальной вероятностью или суммой вероятностей, такой как сумма вероятностей нуклеотидных изменений, которые преобразуют одну последовательность в другую.
6. Набор по п. 5, в котором изменения последовательности количественно определены как расстояние Левенштейна между последовательностями, и расстояние Левенштейна между последовательностями между индексными последовательностями поднабора самого высокого уровня равно не менее 4.
7. Набор по любому из пп. 1-6, в котором индексные последовательности имеют длину не менее 4 нуклеотида, и/или поднабор самого высокого уровня содержит не менее 2 различных индексных последовательностей.
8. Набор по любому из пп. 1-7, в котором олигонуклеотиды присваиваются к поднабору путем помещения в контейнер, который помечается идентификатором поднабора; предпочтительно, когда контейнер представляет собой лунку в луночном планшете.
9. Набор по любому из пп. 1-8, в котором индексные последовательности имеют содержание G/C от 30% до 70%; и/или где индексные последовательности не содержат повторов одного и того же нуклеотида длиной не менее 3; и/или где индексные последовательности поднабора имеют сбалансированное распределение нуклеотидов, где количество совместно используемых нуклеотидов в одной и той же позиции в индексных последовательностях между различными индексными последовательностями не превышает 0,5 кратного количества количество индексных последовательностей в упомянутом поднаборе, или где в не менее 50% позиций индексных последовательностей частота для всех индексных последовательностей поднабора для каждого типа нуклеотидов составляет 0,5 или менее.
10. Способ получения набора олигонуклеотидов, содержащего множество поднаборов олигонуклеотидов с поднабором индексных последовательностей, содержащий этапы
создание первого поднабора олигонуклеотидов с индексными последовательностями с первым секвенциальным расстоянием друг от друга в пределах первого поднабора, где секвенциальное расстояние представляет собой количественную величину изменений последовательностей, которая преобразует одну последовательность в другую, или монотонно убывающую функцию вероятности изменений последовательностей, которая преобразует одну последовательность в другую,
создание второго поднабора путем включения первого поднабора и добавления дополнительных олигонуклеотидов с индексными последовательностями со вторым секвенциальным расстоянием друг к другу во втором поднаборе, причем второе секвенциальное расстояние является меньшим секвенциальным расстоянием, чем первое секвенциальное расстояние, в результате чего вторая подгруппа содержит олигонуклеотиды с индексными последовательностями, которые не являются частью первого поднабора.
11. Способ по п. 10, в котором создание первого и/или второго поднабора содержит выбор индексных последовательностей, которые содержат усеченные индексные последовательности, и усеченные индексные последовательности по меньшей мере одного поднабора отличаются по меньшей мере ненулевым количеством изменений последовательностей от каждой другой усеченной индексной последовательности в упомянутом поднаборе.
12. Способ по п. 10 или 11, в котором создание поднабора содержит выбор индексных последовательностей путем добавления индексной последовательности-кандидата и оценки секвенциального расстояния кандидата расстояния индекса до всех других ранее существовавших индексных последовательностей в поднаборе; и добавление индексной последовательности-кандидата к индексным последовательностям поднабора, если выполнено заданное требование к секвенциальному расстоянию.
13. Способ по пп. 10, 11 или 12, в котором индексная последовательность-кандидат выбирается из пула индексных последовательностей-кандидатов, где элементы пула индексных последовательностей-кандидатов выполняют заданное требование к секвенциальному расстоянию для каждого другого элемента пула, и где индексная последовательность-кандидат пула добавляется к индексным последовательностям поднабора тогда, когда сумма абсолютных значений разностей частот каждого нуклеотидного типа генетического кода в каждой позиции до 0,25 является наименьшей для индексной последовательности-кандидата по сравнению с другими кандидатами индексной последовательности пула.
14. Способ по любому из пп. 10-13, содержащий создание множества поднаборов-кандидатов, каждый из которых имеет заданное количество элементов, как в способах по пп. 10-13, и выбор поднабора-кандидата в качестве поднабора тогда, когда упомянутый поднабор-кандидат имеет наименьшее среднее значение по всем индексным последовательностям для соответствующего поднабора кандидата суммы абсолютных значений разностей частот каждого нуклеотидного типа генетического кода в каждой позиции до 0,25; или
содержащий создание множества поднаборов-кандидатов, каждый из которых имеет заданное количество элементов, как в способах по пп. 10-13, и выбор поднабора-кандидата в качестве поднабора, где упомянутый поднабор-кандидат выбирается путем исключения других поднаборов-кандидатов,
где поднабор-кандидат исключается тогда, когда в способе, который содержит добавление индексных последовательностей-кандидатов из пула индексных последовательностей-кандидатов к поднабору-кандидату и при необходимости дополнительное добавление сравнительных индексных последовательностей, кандидат-поднабор имеет более высокое среднее значение во всех своих суммах индексных последовательностей абсолютных значений разностей частот каждого нуклеотидного типа генетического кода в каждой позиции до 0,25 по сравнению с другим поднабором или поднабором-кандидатом.
15. Способ присвоения секвенирующих прочтений образцу олигонуклеотидов, содержащий этапы
а) получение из множества образцов олигонуклеотидов образцов,
b) выбор поднабора олигонуклеотидных индексных последовательностей из набора по любому из пп. 1-9 или набора, который получен способом по любому из пп. 10-14, где поднабор выбирается вместо другого поднабора на основе большего секвенциального расстояния между индексными последовательностями друг от друга в выбранном поднаборе; где секвенциальное расстояние представляет собой количественную величину изменений последовательностей, которая преобразует одну последовательность в другую, или монотонно убывающую функцию вероятности изменений последовательностей, которая преобразует одну последовательность в другую, и где выбранный поднабор имеет по меньшей мере такое же количество различных индексных последовательностей, как и количество образцов на этапе а),
c) добавление индексных последовательностей из упомянутого поднабора в каждый олигонуклеотид образца, где индексные последовательности указывают образец,
d) определение последовательности олигонуклеотидов образцов или фрагментов олигонуклеотидов образцов и определение индексной последовательности,
e) присвоение последовательности с полученным прочтением образцу на основе определенной индексной последовательности или на основе индексной последовательности, которая имеет наименьшее секвенциальное расстояние до определенной индексной последовательности, где, если две или более индексных последовательностей имеют одинаковое наименьшее расстояние, то упомянутое полученное прочтение отбрасывается; где при необходимости секвенциальное расстояние не превышает заданного значения согласно критерию.
| WO 2018204423 A1, 08.11.2018 | |||
| US 2019085384 A1, 21.03.2019 | |||
| WO 2018226293 A1, 13.12.2018 | |||
| СПОСОБ ВЫЯВЛЕНИЯ МУТАЦИЙ В СЛОЖНЫХ СМЕСЯХ ДНК | 2014 |
|
RU2613489C2 |
| TILO BUSCHMANN et al., "Levenshtein error-correcting barcodes for multiplexed DNA sequencing", BMC BIOINFORMATICS, v | |||
| Паровоз для отопления неспекающейся каменноугольной мелочью | 1916 |
|
SU14A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Паровоз с приспособлением для автоматического регулирования подвода и распределения топлива в его топке | 1919 |
|
SU272A1 |

