рига щелочного металла на первой стадии процесса используют нитрит натрия.
Д. Спр.соб по пп. 1 и 2, от л ич ающи и ся тем, что в качестве минеральной кислоты на первой стадий процесса используют соляную кислоту,
4.Способ поп,1, отл,и чающий с я тем, что последнюю стадию процесса осуществляют в среде инертного органического растворителя, когда используют соединение (1), где У - водород, R им. указанные значения.
5.Способ ПОП.1, отличающ и и с я тем, что используют соединение формулы {I), где Y - окси или галоид, R имеет указанные значения.
6.Способ по пп. 1 и 5, о т л ичающийся тем, что в качестве конденсирующего агента используют дициклогексилкарбодиимид.
7.Crtoco6 по п.1, отличающий с я тем, что в качестве акцептора кислоты используют третичное органическое основание или неорганическое основание.
8.Способ по пп. 1и 7, отличающийся тем, что в качестве акцептора кислоты используют окись магния.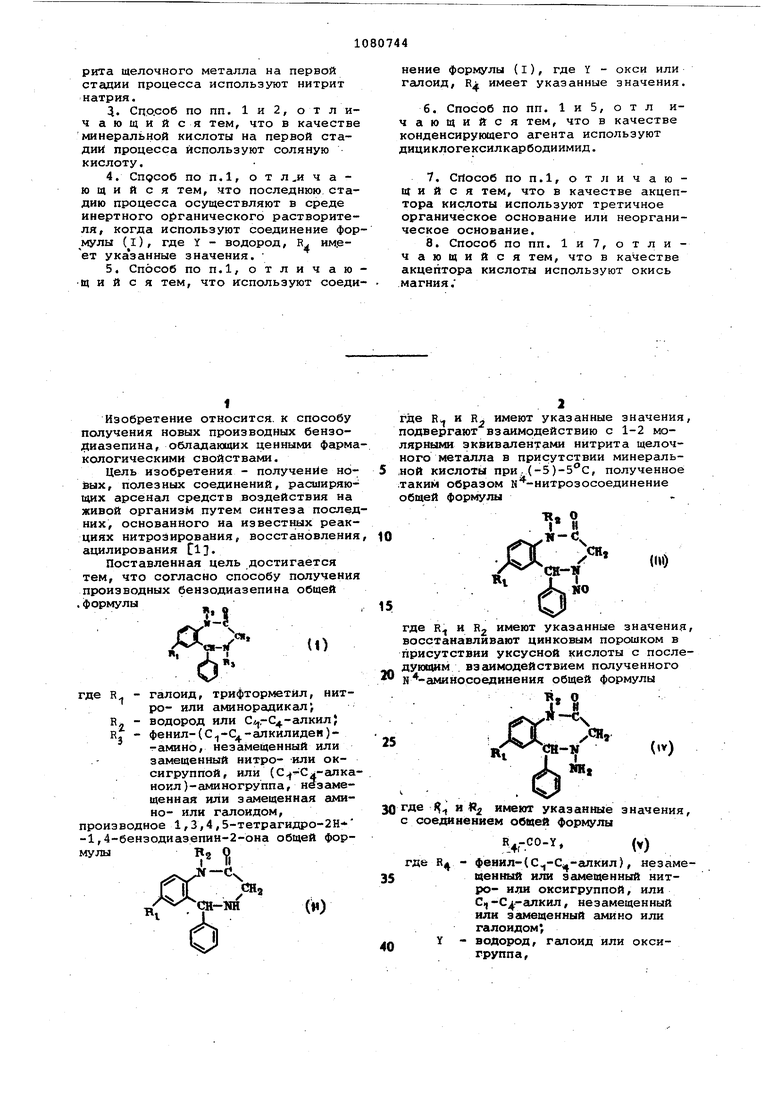





1. СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ БЕНЗОДИАЗЕПИНА общей форму-
Изобретение относится, к способу получения новых производных бензо иазепина, обладающих ценными фармакологическими свойствами.
Цель изобретения - получение новых, полезных соединений, расширяющих арсенал средств воздействия на живой организм путем синтеза последних, основанного иа известных реакциях нитрозирования, восстановления ацилирования fl.
Поставленная цель достигается тем, что согласно способу получения производных бензодиазепина общей .формулы ж о
Л
U)
6
- галоид, трифторметйл, нитR,
ро- или аминорадикал, R - водород или Сл,,-С4-алкил; R. - фенил-СС -С -алкилиден)тамино, незамещенный или замещенный нитро- или оксигруппой, или (С Сл-алканоил )-аминогруппа , не замещенная или замещенная амино- или галоидом,
зводное 1,3,4,5-тетрагидро-2Н -бензодиазепин-2-она общей форВ О I (1
-е
сн
3
(И)
CH-NH
где R и R имеют указанные значения, подвергают взаимодействию с 1-2 молярными эквивалентами нитрита щелочного мет аипл а в присутствии минеральной кислоты при, (-5), полученное таким образом к -нитрозосоединение общей формулы
((И)
где R и R2 имеют указанные значения, восстанавливают цинковым порошком в присутствии уксусной кислоты с последующим взаимодействием полученного Ы -аминосоединения общей формулы
-К,
(,у)
0 где f и « имеют указанные значения, с соединением общей формулы
.о.у. (V)
где R - фенил-((,-апкил), незамещенный или замещенный нитро- или оксигруппой, или С,|-С -алкил, незамещенный или замещенный амино или галоидом,
Y - водород, галоид или оксигруппа. в случае необходимости в присутствии конденсирующего агента или акцептора кислоты. . Все соединения, полученные предлагаемым способом, являются новыми Прямое нитрозирование соединений, Которые отвечают общей формуле (II), проводят обычно с использо ванием нитрита натрия в присутствии минеральной кислоты. В качестве кислоты предпочтитель но использование соляной кислоты. Согласно способу соединения общей формулы (II) суспендируют в концентрированной соляной кислоте, по каплям добавляют в суспензию вод ный раствор нитрита натрия при температуре приблизительно 0°С и после завершения реакции смесь подщелачивают. Таким образом получают соотве ствующие соединения общей формулы (III), где значения каждого из символов R и R идентичны изложенным. Реакционную смесь можно подщелачивать любым сильным основанием, в частности водным раствором гидрата окиси щелочного металла, напри мер гидрата окиси калия, однако для этого предпочтительно использовать концентрированный раствор; амми ка. Нейтрализацию смесь предпочтительно проводить при интенсивном охлаждении.. . Реакцию восстановления осуществляют следующим образом. N -нитрозотетрагидро-1,4-бензодиазёпиновое производное общей формулы (III) суспендируют в уксусной кислоте, а затем в перемешиваемую суспензию небольшими порциями добавляют порошкообразньлй цинк. В ходе проведения этой операции смесь охлаждают водой. После завершения реакции смесь подщелачивают с вьщелением полученного продукта общей (IV). в соответствии с предпочтительным вариантом осуществления последней стадии данного способа Н -аминотетрагидро-1,4-бензодиазепиновое производное общей формулы (IV) раст воряют в инертном органическом раст ворителе, в частности в бензоле, и в раствор добавляют альдегид общей формулы (v), где значения символа Rj( идентичны определенным выше, после чего смесь перемеигаваюг при комнатной температуре в течение ночи. . . - ; ; -, . . Если соединение общей форк1ул1Ы (V где символом Y обозначен гидрбксил используют в качестве исходного вёщества, .для повьаиения скорости протекания или полного завершения реак ции в реакционную смесь можно добавить обычный конденсирующий агент, в частности дициклогексилкарбодиимид. Исходные вещества предпочтительно использовать приблизительно в эквимолярных количествах. Конденсационный агент предпочтительно добавлять в 5-15%-ном. избытке. Если в качестве .ацилирующего: агента исйользуют соё;Ьгинение общей формулы- (V), где символом У обозначен атом галогена, для повышения скорости протекания или завершения реакции в смесь можно добавлять связывающий кислоту агент, например окись щелочноземельного элемента, в частности окись магния. Однако если индивидуальные компоненты оказываются, достаточно реакционными, описанная реакция протекает с приемлемой скоростью даже без использования конденсирующего или связывающего кислоту агента. Любое из соединений, которое отвечает общей формуле (I), где значения каждого из символов R и R идентичны определенным, а символом R обозначен водородный атом, можно алкилировать, если это желательно, с получением соответствующих гшкильных производных. В ходе проведения этой реакции можно использовать обычные алкилнрующие агенты, в частности алкилгалогеннды, предпочтительно алкилиодиды, или дналкилсульфаты. Процесс можно проводить, в частности , вначале путем конверсии соедиг нения общей формулы (I) в его производное щелочного металла, а затем путем реакции полученного таким обраг зом производного щелочного металла с соответствующим алкилирующим агентом. Произвсъаное щелочного металла можно получить, например, реакцией соответствующего соединения общей формулы fl), где R - водородный атом, а значения каждого из символов R, и Н,. идентичны определенным вьхае, со щелочным металлом, гидридом щелочного металла или .гмидом щелочного металла, в частности с натрием или С соединением натрия, при температуре от О до в среде инертного растворителя, в частности диоксана дИметилформёЦхшда, , бензола или толуола . Реакционные смеси можно подверг гать обработке способами, известными в данной области технологии. Выбор конкретного спо.соба пррведения процесса зависит art природы исходного вещества, конечного продукта и растворителя. Когда продукт выделяют из реакционной смеси, ее .просто фильтруют, причем в этой случае продукт остается в растворе, после чего продукт можно высадить соотвеТствукш|им растворителем или .же выпарить раствор после удаления твердых побочных продуктов. В .ходе проведения процесса обработки реакционной смеси конечный продукт обычно получают в кристалли ческой форме. Однако если получают маслянистое вещество, его можно подвергать кристаллизации обычно без каких- либо затруднений с использованием обычных .растворителей, например алифатических или цикли- . ческих простых эфиров, в частности диэтилового э.фира, диоксана, тетрагидрофурана и т.д. При необходимости соединения общей формулы (I), где значения символов R, R и Rj идентичны определенным, можно подвергать дополнител ной очистительной обработке, в част ности перекристаллизацией. 3 качест ве растворителя для перекристаллиза ции можно использовать, например, алифатический спирт, в частности метанол или этанол, ароматический углеводород, в частности бензол, кетон, в частности ацетон, сложныйалифатический эфир, в частности алканкарбоксилат, в особенности этилацетат, алифатический углеводород, в особенности насыщенный алифа тический углеводород с 5-10 углерод ными атомами, в частности н-гексан, простой эфир, в особенности дигшкиловый эфир, в частности диэтиловый эфир, насыщенный циклический простой эфир, в частности тетрагидрофуран, а также ацетонитрил и их смеси (например, смесь тетparидрофурана с гексаном или смесь этилацетата с простым эфиром). Осуществление предлагаемогоспотсоба позволяет получать соедщнения общей формулы (I) с достижением высоког.о выхода конечного продукта и в легко идентифицированном состоянии. Данные элементарного анализа полученных веществ хорошо согласуются с расчетными величинами. Новые тетрагидро-1,4-бензодиаэепиновые производные общей формулы (1) показывают энзиминдицирующие эффекты и оказывают лишь умеренное воздействие на центральную нервную. систему.; Энзиминдуцирующее действие данных новых соединений проверяли в условиях ин виво путем определения активности гексабарбиталоксидазы. Активнодействующие веществ общей формулы (I) вводили в организм больных крыс Wistar, вес которых находился в интервале от 50 до 60 г, через рот в дозировке 40 мг/кг живого веса. Эффективность проявлялась по истечении 24 ч после введения энзиминдицирующйх веществ. В это время определили степень инактивации гексабарбитала путем внутреннего введения в дозировке 40 мг/кг живого веса гексабарбитала натрия и определения времени, прошедшего с момента введени и до вторичного появления рефлекса выпрямления. Продолжительност сна, которую наблюдали у животных; использованных в опытах по введению в их организм новых соединений, сопоставили с той, которую наблюдали у контрольной группы животных (в организм животных этой контрольной группы ввели только вещество наполнителя или носителя) . По истечении 24 ч после введения в организм энзиминдицирукяцих веществ ииактивация ин виво гексабарбитала ускоряется, что подтверждается сокращением продолжительности сна. Результаты были выргикены в процентной разнице по сравнению с контрольным опытом. На основании предварительных экспериментов разницу, которая превьаиала 25%, рассмат{жвали как биологически заметную. . Результаты испытаний сведены та табл. i. Таблица 1 Биологический период полуинактивации гексабарбитала определили путем УФ-спектрофотометрии . Результаты испытания, которое провели с использованием соединения, полученного в соответствии с пряма ром 5 (самый активный представител новых соединений в соответствии с изобретением) сведены в табл. 2. Из оценки данных табл. 2 совершенн очевидно, что соединение, полученное в соответствии с примером 5, эффективно сокращает период полуинактивации гексабарбитала в орга низме крыс. Т а блиц а йериод полуинактив Вещество ции , мин Контрольный опыт По примеру 5 Степень чистоты полученных веществ определяли путем тонкослойного хроматографического ангшиза. Величины R определилис примене нием силикагелевой пластины типа , Шталь С (фирма Мерк), а в качестве растворителя для элюирования использовали, одну из следующих систем: смесь и-гексана с уксусной кис лотой и хлороформом в соотношении 1:1:8 (1), смесь бензола с метанолом в соотношении 95:5 (2), смесь хлороформа с метанолом и уксусной кислотой в соотношении 75:20:5 (3) смесь н-гексана с этилацетатом и хлороформом в соотношении 1:4:8 (4 смесь н-бутанола с уксусной кислотой и водой в соотношении 4:1:1 (5 Пятна проявляли путем осуществления хлортолидиновой технологии. Темпера гуру плавления определяли в приборе типа д-ра Тотолли (температура плав ления приведена в примерах в некорректированных величинах).В некоторых примерах строение молекул конеч ных продуктов идентифицировали с помощью спектрометрии УФ, ЯМР или , масс-спектрометрии. Пример 1. 1-Метил-4-нитрозо-5-фенил-7-хлор-1,3,5-тетрагидро-2Н-1,4-§ензодиазепин-2-он. 16,0 г (55,6 моль) 1-метил-5-феНШ1-7-ХЛОР-1,3,4,5-тетрагидро-2Н-1,4-бензодиазепин-2-она суспендируют в 56 мл концентрированной соля ной кислоты. Эту суспензию охлаждают до температуры и по каплям добавляют в нее раствор 3,9 г (55,6 ммоль) нитрита натрия в 20 мл воды. Смесь перемешивают при ОС в течение 2 ч, после чего по аналогии с вышеизложенным в нее добавляют дополнительно 3,9 г (55,6 ммоль) нитрита натрия. После завершения реакции суспензию нейтрализуют концентрированным водньгм раствором аммиака. При интенсивном охлаждении осторожно добавляют щелочной раствор. Выделенный сырой продукт отфильтровывают, промывают водой и перекристаллизовывают из этанола. Получают 15,6 г (88,6%) 1-метил-4-нитрозо-5-фенил-7-ХЛОР-1,3,4,5-тетрагидро-2Н-1,4-бензодйазепин-2-она. Т.пл. 180-182 с, R/,8. Вычислено,%: С 60,8, Н 4,5, N 13,3.. С К ( MB 315,76) Найдено,: С 60,8, Н 4,7, N 13,3. Аналогичным образом из соответствующих исходных продуктов получают соединение по примеру 2. Пример2. 4-Нитрозо-5-фенил-1,3,4,5-тетрагидро-2Н-1,4-бензодиазепин-2-он. Выход 80,5%. Т.пл. 211-212 с (после-151ерекристаллизации из эта.нола), ,75. Вычислено,%: С57,7, Н3,9, N 17,95 С.5 1 2N404 (MB 312,29) Найдено,%: С 57,6, Н 4,4, N 17,95 Пример 3. 1-Метил-4-амин-З-фенил-7-хлор 1,3,4,5-тетрагидро-2Н -1,4-бензодиазепин-2-он, 30 г цинковой пыли добавляют в суспензию 9,0 г (28,4 ммоль) 1-метил-4-нитрозо-5-фенил-7-хлор-1,3,4,5-тетрагидро-2Н-1,4-бензодиазепин-2-она в 60 мл уксусной кислоты при охлаждении водой и перемешивании. В процессе этого добавления твердый материал растворяют постепенно. По истечении 15 мин перемешивания цинковую пыль отфильтровывают и .фильтрат нейтрализуют водным насыщенным раствором бикарбоната натрия. Выделенное вещество, фильтрование которого сопряжено со значительными затруднениями, экстрагируют несколькими порциями хлороформа. Хлороформный раствор сушат и выпаривают до конечного объема приблизительно 16 мл. В концентрат добавляют 30 мл 2 н. раствбра соляной кислоты и смес оставляют на неЬколько минут. Отделившиеся примеси отфТсльтровывают, а полученный двухфазный фильтрат нейтрализуют насыщенным водным paci BopoM бикарбоната -натрия. Хлороформную фазу отделяют, а водную фазу подвергают экстрагированию двумя порциями по 100 мл хлороформа. Органические фазы объединяют, сушат и выпаривают до сухого состояния при пониженном давлении. Маслянистый остаток кристаллизуют из изопропанола с получением 5,1 г (59,3%) 1-метил-4-амин-5-фенил-7-хлор-1,3,4,5-тетрагидро-2Н-1,4-бензодиаэепин-2-она. Т,пл. 147-148Ь, С П и , 1Э , . Вычислено,: С 63,6, Н 5,3, N 13,9. С-,ьН :, ONj С1 (MB 301,79) Найдено,: С 63,7, Н 5,6, N 14, Аналогичным образом из соответс вунадих исходных веществ получают следующее соединение. Пример 4. 4,7-Диамин-5-фе нил-1,3,4,5-тетрагидро-2Н-1,4-бензоазепин-2-он. Выход 56,6%.-Т.пл. 218-221 С (после перекристаллизации из ацето нитрила), , 4. Вычислено,%: С 67,1, Н 6,0, N 20,9 (MB 268,33) Найдено,%: С 66,8, Н 6,3, N 20, П р и м е р 5. 1-Метил-4-бенза амин-5-фенил-7-хлор-1,3,4,5-тетрагидро-2Н-1,4-бензодиазепин-2-он. 1 МП (9,1 ммоль) бензальдегида добавляют в раствор 2,0 г (6,6 ммо 1-метил-4-с1МИн-5-фенил-7-хлор-1 3,4,5-тетрагидро-2Н-1,4-бензоди азепин-2-она в 20 мл бензола и сме перемешивают при комнатной темпера туре в течение ночи. В конце реакции смесь выпаривают до сухого состояния при пониженном давлении, ост.аток растирают в порошок с испо зованием 20 мл серного эфира/ а твердое вещество отфильтровывают. Получают 2,1 г (80,7%) 1-метил-4-бензальамино-5-фенил-7-хлор-1,3,4 -тетрагидро-2Н-1,4 бензодиазепин-2-она. Т.пл. 157-158 С (после перекристаллизации из этанола), Вычислено,%: С 7078, Н 5,2, И 10,8. CgjH gONjCl (MB 389,89) Найдено,: С 70,8, Н 5,35, N10 Аналогичным образом из соответствующих исходных веществ получают следующие соединения. П р и м е р 6. 1-Метил-4-(о-хл бензиламино)-5-фенил-7-хлор-1 ,.3,4, -тетрагидро-2Н-1,4-бензодиазепин-2-он.. Выход 91,2%, Т.пл. 185-187 С (после перекристаллизации из этано ла), R,7. Вычислено,%: С 65,1, Н 4,5, N . ,Cl2(MB 424,34) Найдено,: С 65,0, Н 4,9, N 9,8 Пример 7. 1-Метил-4-(н-ни робеН3альамин)-5-фенил-7-хлор-1,3,4,5-тетрагидро-2Н-1,4-бензоди азепин-2-он. . Выход 87,9%, Т.пл. 202-204 С (после перекристаллизации из этг1но ла), К|0,65. Вычислено,%: С 63,6, Н 4,4, N 12,75 (MB 434,89) Найдено,%: С 63,3, Н 4,4, Н 12,3. П р и м е р 8. 1-Метил-4-(м-нитробензальамино)-5-Фенил-7-хлор-1,3,4,5-тётрагидро-2Н-1,4-бензодиазепин-2-он. Выход 93,5%, Т..ПЛ. 224-226 С (после перекристаллизации из этанола), ,65. Вычислено,%: С 63,6, Н 4,4, N 12,75 (MB 434,89) Найдено,%: С 63,4, Н 4,4, N 12,6 - V ,., П р-и м е р 9. 1-Метил-1-(о-нитробензальамини)5-фенил-7-хлор-1,3,4,5-тетрагидро-2Н-1,4-бензодиазепин-2-он. выход 90,2%, Т.пл. 187-188 С (после перекристаллизации из этанола), Rfp-0,7. Вычислено,%: С 63,6, Н 4,4, Н 12,75 C2,H.O,N4.C1 (MB 434,89) Найдено;%: С 63,6, Н 4,7, N 12,2 Пример 10. 1-Метил-4-(п-оксибензальамино)-5-фенил-7-хлор-1/3,4,5-тетрагидрЬ-2Н-1,4-бензодиазепин-2-он. Выход 89,5%, Т.пл. 135-137 0 (после перекристёшлизации из бензола), iff 0,5. Вычислено,%1 С 68,06, Н 4,95, N 10,3 , CjjHjoOiNjCl (MB 405,89) Найдено,%: С 68,3, Н 5,0, N 10,2 Пример 11. 1-Метил-4-глициламино-5-фенил-7-хлор-1,3,4,5-тетрагидро-2Н-1,4-бензодиазепин-2-он. 4,2 г (0,02 моль) бензилоксикарбоннлглицина и 4,0 г (0,02 моль) дициклогексилкарбодиимида добавляют в раствор 6,0 г (0,02 моль) 1-метил-4-амин-5-фенил-7-хлор-1,3,4,5-тетраг1адро-2Н-1,4-бензодиазепин-2-она в 200 мл этилацетата. Реакционную смесь перемешивают при комнатной xeikBiepaxj pe в течение ночи, после чего вьэделившуюся дициклогексилмоче 9ииу отфильтровывают и фильтрат выпаривают при пониженном давлении. Остаток смешивают с ацетонитрилом, растдор кипятят, выделившуюся дйцикяогексил14очевину отфильтровывают из горячего раствора и фильтрату дают остыть. Вьщелишаийся СЕфой продукт отфил&тровывают и кристаллизуют ИЗ этанола. Получают 7,1 г (72%) 1-мвтил-4(н-бензилоксикарбонилглициламин)-5-феиил-7-хлор-1,3,4,5-твтрагидро-2Н-1,4 бензодиазепин-2-она. Т.пл. 184-190 с, .З.
Вычислено,%: С 63,3, Н 5,1, N 11., 4
26 25 4Ч (MB 492,95) Найдено,%: С 63,6, Н 4,8, N 11,4 Смесь 4,6 г (9,35 ммоль) упомянутого продукта с 25 мл. 3 н.бромистоводородной кислоты в ледяной уксусной кислоте перемешивают в течение 0,5 ч без доступа воздуха, после чего в этот раствор добавляют 100 МП сухого эфира. Зьщелившееся вещество отфильтровывают и промывают сухим серным эфиром. 4,78 г полученного таким образом 1 метил-4-глициламино-5-фенил-7-хлор-1,3,4,5-тетрагидро-2Н-1,4-бензодиазепина-2-он гидробромида суспендируют в 20 мл воды, после чего величину рН этой смеси доводят до 9-10 добавлением концентрированного водного раствора аммиака. Эту водную суспензию насыщают хлористым натрием и подвергают экстрагированию 3 -порциями по 100 мл хлороформа. Хлороформный раствор сушат, выпаривают до сухого состояния при пониженном давлении и остаток перекристаллизовывают из этилацетата. Получают 2,6 г (76,4%) 1-метил-4-глициламин-5-ф,енил-7-хлор-1,3,4 ,5-тетрагидро-2Н-1,4-бензодиазепин-2-она. Т.пл. .173-175«С, ,35.
Вычислено,%: С 60,2, Н 5,3, N 15,6
C.AH.gO-N.Cl (МБ 358,82) Найдено,%: С 60,1, Н 5,6, N 15,6 Пример 12. 1-Метил-4-хлорацетамини-5-фенил-7-хлор-1,3,4,5-тетрагидро-2Н-1,4-бензодиазепин-2-он.
3 г окиси магния добавляют в раствор 3,0 г (0,01 моль) 1-метил-4-амино-5-фенил-7-хлор-1,3,4,5-тетрагид1 о-1,4-бензодиазепин-2-она в 30 МП хлороформа и в перемещиваемую смесь по каплям добавляют раствор 0,82 мл хлорацетилхлорида в 4 мл хлороформа. По истечении 2 ч перемешивания добавляют раствор 0,3--мл хлорацетилхлорида в 2 мл хлороформа, после чего смесь перемешивают при комнатной температуре в
0 течение ночи. Соль магния отфильтровывают и промывают хлороформом. Фильтрат и промывную жидкость ос ъединяют, промывают 20 мл воды, сушат и выпаривают растворитель. Получают 3,5 г (92,5%) белого кристалличес5кого остатка. Т.пл. 226-228 С. Этот сырой продукт перекристаллизовываиот из ацетонитрила с получением очищенного 1-метил-4-хлорацетиламино0 5-фенил-7-хлор-1,3,4,5-тетрагидро-2Н-1,4-бензрдиазепин-2-она. Т.пл. 227-229 с., RfPo,3.
Зычислено,%: С 57,2, Н 4,5, N 11,1
5 С.дН.-02НзС1 (MB 378,25)
Найдено,%: С 57,1, Н 4,4, N 11,2
Аналогичным образом из соответствующих исходных продуктов получают д следующее соединение.
Пример 13. 1-Метил-4-ацетиламино-5-фенил-7-хлор-1,3,4,5-тетрагидро-2Н-1,4-бензодиазепин-2-он.
Выход 74,5%, Т.пл. 198-20бс (после перекристаллизации из ацетонитрила), Кфо,25.
Вычислена,%; С 62,9, Н 5,3, N 12,2
C gH -O-N-Cl (MB. 343,81) 0 .Найдено;%: С 62,5, Н 4,6, N 11,65.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Каррер П | |||
| Курс.органической хиьши | |||
| Л., Госхимиэдат, 1960, с | |||
| Вага для выталкивания костылей из шпал | 1920 |
|
SU161A1 |
