Цриоритет по признакам: 02.02.78 лри г CHj- СО-ОСН.,, CH2-CO-CHJJC1, C%-CO-CHj.
04.07.78 npHZ- остальные зна-чения , перечисленные в формуле изобретения .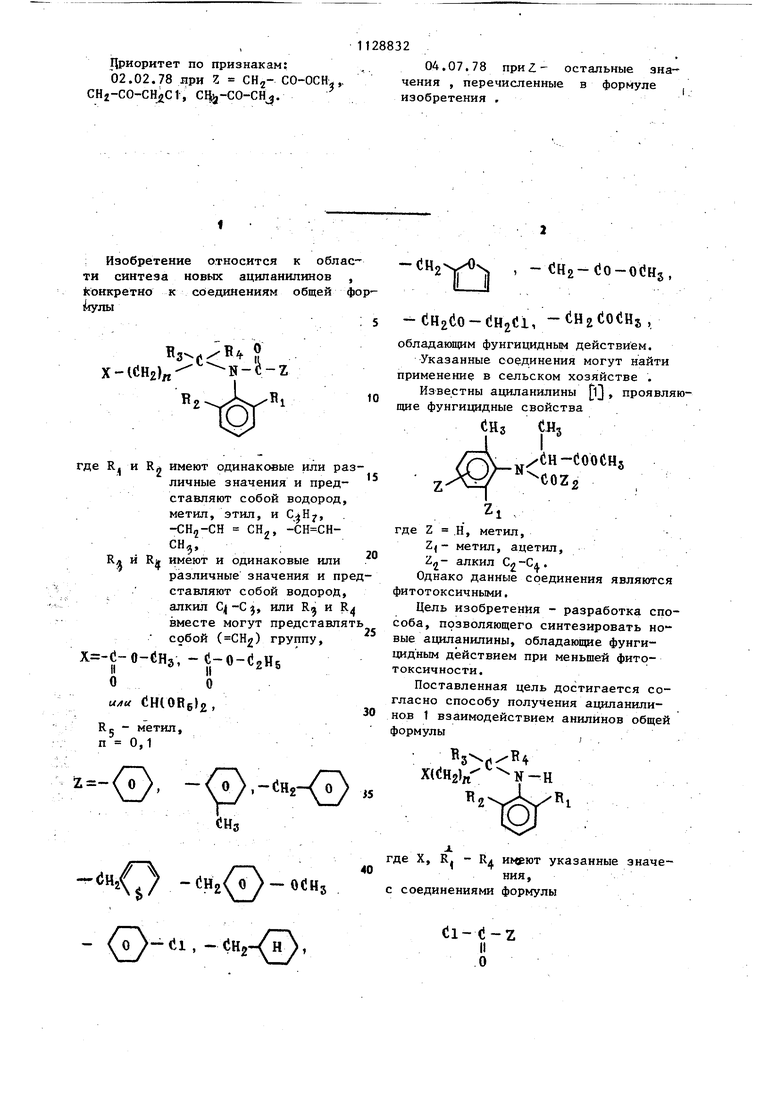










| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения диэфиров гидрохинона | 1981 |
|
SU1109048A3 |
| Способ получения триазолсодержа-щиХ эфиРОВ ТиОфОСфОРНыХ КиСлОТ | 1977 |
|
SU795485A3 |
| Катализатор для полимеризации @ -олефинов | 1976 |
|
SU1075949A3 |
| Способ получения эфиров 2,2-диметилциклопропанкарбоновых кислот | 1980 |
|
SU1053744A3 |
| Способ получения @ -арил- @ -аминокарбоксамидов или их солей с фармацевтически приемлемой кислотой или возможной стереохимической изомерной формы | 1984 |
|
SU1313344A3 |
| Способ получения производных алкилендиамина,их смесей,рацематов или солей | 1982 |
|
SU1246890A3 |
| Способ получения @ -арил- @ -ацил-3-аминооксазолидин-2-онов | 1981 |
|
SU1011049A3 |
| Способ получения производных 4-бензил-1(2Н)-фталазинона или их физиологически переносимых кислотно-аддитивных солей | 1986 |
|
SU1454251A3 |
| Способ получения производных гомосерина | 1980 |
|
SU1093242A3 |
| Способ получения бензимидазолкарбаматов и его вариант | 1979 |
|
SU1169532A3 |
СПОСОБ.ПОЛУЧЕНИЯ АЦИЛАНИЛИНОВ общей формулы 4cClvt Z х-1«нг)„ KI : Ы и R имеют одинаковые или различные значения и представляют собой водород, метил, этил и пропил, СН, - СН СН - СН R имеют одинаковые или различные значения и представляют собой Н, алкил C -С или R и R вместе могут представ-, лять собой (СН2) группу. X -C-OCH -C-O-Cj Hj или CHCOfej-tg ОО Re - метил, п 0,1 (-( .-ся,-, СНз 2- - Oti-Hj , i СО ЧНг-С-ойНз о -CHft-C-CHoiil, iH2-C- iH5 IIН оо .отличающийся тем, что аниг лины общей формулы -в В 3.(i ХССНг), N-H Чтде X, R, R, - имеют указанные значения, подвергают взаимодействию с соединением формулы Cl-t-Z где Z - имеет указанное значение, в среде инертного растворителя в присутствии основания.
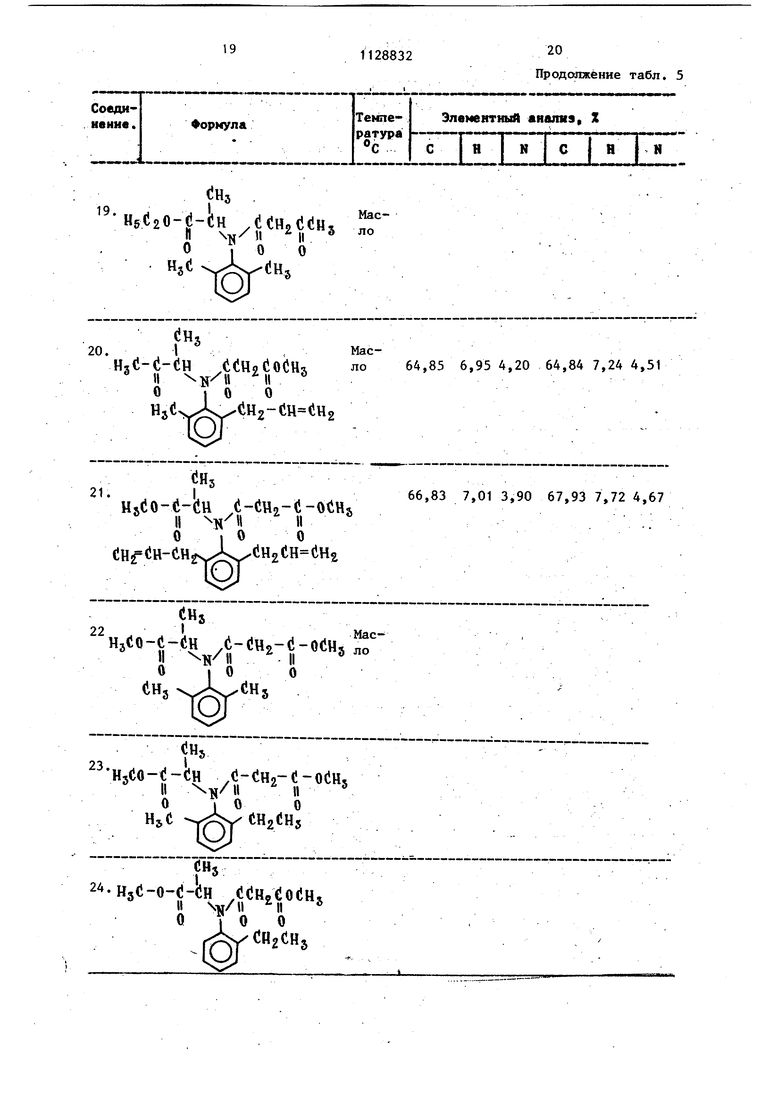
: Изобретение относится к обла ти синтеза новых аципаиилинов koHKpeTHO к соединениям общей Х-(СН2)л где R и RA имеют одинаковые или р личные значения и пред ставляют собой водород метил, этил, и СлНу, CHg, Кл и Rji имеют и одинаковые или различные значения и п ставляют собой водород алкил , или R и вместе могут представл собой (СН) группу. х -с-о- ;нз, -«l-o-UzHs или ЙШОЙб, RC - метил, п 0,1 , ,-CHj-0 ,( -СН20-ОСН5 -0 о - (ii, - Сн - (}н, (1Н2(10-(Н2С1, (iO(Hs, обладающим фунгицидным действием. Указанные соединения могут найти применение в сельском хозяйстве . Известны ациланилины ij , проявляющие фунгицидные свойства СИз СНз /(iH-COOCHj где Z .Н, метил, Zj- метил, ацетил, Ъ„- алкил . Однако данные соединения являются фитотоксичными. Цель изобретения - разработка способа, позволяющего синтезировать но вые ациланилины, обладающие фунгидидным действием при меньшей фитотоксичности. Поставленная цель достигается согласно способу получения ациланилиНО.В 1 взаимодействием анилинов общей формулы XidHjIrt- N-H де X, R. - RX указанные значения, соединениями формулы di- (-Z II о где Z имеет указанное значение, .в среде инертного растворителя в присутствии основания. Способ основан на известной реакции взаимодействия азотсодержащих соединений с галоидангидридами кислот в среде инертного растворителя в присутствии соединений, связываници вьщеляющейся галоидводород 2j . Прим ер 1, Получение N(2,6-диметилфенил)-Н-ацетоацетил-о(, -амин метилпропионата (соединение 18, тйбл. 5). 7.06 г только что перегнанного ди кетена добавляют к 14,5 г метилового сложного эфира - Ы-(2,6-диметилфенил -2-аминопропионовой кислоты в 25 мл толуола. Реакционную смесь нагревают с обратным холодильником в течени 24 ч. После охлаждения и выпаривания растворителя остаток очищают хроматографически на колонке силикагеля, используя в качестве элюента хлороформ. Получают 20 г целевого продукта в виде масла, с выходом 98% по отношению, к теоретическому вькоду. Структура продукта подтверждена ЯМР-спектроскопией. Пример 2. Получение N-(2,6-диаллилфенил) -N-ацетоацетил-оС-аминометилпропионата (соединение 26, табл. 1), , 0,02 моль метилового сложного эфи ра N- С 2,6-диаллилфенил) -о6-аминопропи оновой кислоты растворяют в толуоле (10 мл) 0,025 моль только что перв гнанного дикетена добавляют к раство ру и всю смесь нагревают при темпера туре флегмы в течение 24 ч. После охлаждения и выпаривания растворителя остаток очищают хроматографией на колонке силикагеля, используя смесь гексана и этилацетата (4:1) в качест ве элюента. Получают 3 г целевого продукта в виде масла. Пример 3. Получение а-(2-ал лш1фенил)-Н-ацетоацетил-()6-аминометш1 пррпионата (соединение 30, табл. 5). О,02iмоль метилового сложного эфи ра Ы-(2-аллилфенил)- -аминопропионовой кислоты растворяют в 20 мп бензог ла. К раствору добавляют 0,5 моль пиридина и 0,25 моль только что пере. гнанного дикетена. Реакционную смесь нагревают под флегмой в течение 10 ч После охлаждения ее разбавляют бензолом, промывают раствором хлористого водорода (1%-ной концентрации) и водой. Органическую фазу отделяют, подкисляют Na2S04 и выпаривают растворитель. Остаток счищают хромато- графически на колонке силикагеля, используя в качестве элюента гексанэтилацетат (4:1). Получают 3 г целевого продукта (масло). Пример 4. По примерам 1,2 или 3 получают соединения 19, 27, 28, . 29, 31, 33 и 34 (табл, 5). П р и м ер 5. Получение N-(2-метил-6-аллилфенил)-Ы-(карбоксиметилацетил) -ц -аминометилпропионата (соединение 20, табл. 5). 5 г (0,021 моль) метилового сложного эфира N-(2-мeтил-6-aллил-фeнил)-oi-аминопропионовой кислоты растворяют в толуоле (120 мл). К раствору добавляют по каплям при перемешивании в течение 15 мин при комнатной температуре 3,5 г. (0,027 моль) однохлорзамещенного ме-., тилового сложного эфира малоновой кислоты (С1СО-СНл-СООСН). Реакционную смесь затем перемещивают при комнатной температуре в те сение I .ч, затем нагревают с обратным холодидьником в течение 5ч. После охлаждения раствор отфильтровывают и растворитель выпаривают. Маслянистый остаток очищают хроматогра ически на колонке силикагеля, используя в качестве элюента гексан-этилацетат (3:1). Получают 4,6 г целевого продукта в виде красного масла. Пример 6. Получение N-(2,6-диметилфенил)-N-(2,2-диметоксиэтшт)-кар.бометокриацетамида (соединение 32, табл. 5). К раствору N-(2,2-димeтoкcиэтшI)2,6-димeтилaнилинa (4,45 г, 0,02 моль), риэтиламина (2,76 мл, 0,02 моль) в иэтиловом эфире (25 мл) добавляют о каплям в течение 15 мин при переешивании при 0-5 С монохлорзамещеный метиловый сложный эфир малоновой ислоты (2,1 мл, 0,02 моль). Реакционную смесь затем перемешиают в течение 1 ч при и 10 мин ри комнатной температуре, затем ее тфильтровывают, дважды промывают 10 мл раствором хлористого водорода (5%), затем водой до нейтрального значения рН (3 х 10 мл). 5 Органическую фазу подкисляют безводным и растворитель выпаривают. Остаток (желтое масло) очищают хроматографически на колонке силикагеля, используя гексан-этилацетат (7:3) в качестве элюента. Получают 2,1 г целевого продукта (масло). Пример 7. По примерам 5 или 6 получают соединения 21-25. Пример 8. Получение N-(2,6-диметилфенил)-Н-( 1-карбометоксиэтил -фенилацетамида (соединение 4,табл.5 17 г (0,11 моль) фенилацетилхлорида добавляют по каплям в течение 30 мин и при комнатной температуре к раствору N-(1-кapбoмeтoкcиэтил)-2,6-димeтилaнилинa; (21,2 г при чистоте 95%, 0,1 моль) в толуоле (150мл и диметилфломамиде (.1 мл). Реакционную смесь перемешивают в течение 1 ч при комнатной температуре и 3 ч при температуре флегмы, затем ее охлаждают до комнатной температуре и промывают 5%-ным водным раствором NaHCO, и потом водой Органическую фазу отделяют и подкисляют безводным Na.SO. Растворитель выпаривают и полученный неочищенный продукт перекристаллизовывают из лигроина (75-120 С получив при этом 26 г целевого продукта (белое твердое вещество, т.пл. 78-80°С). 9. По примеру 8 nony Пример чают соединения 1,2,3,7-10, 12-17 и 34 (табл. 5), однако соединения 10, .,13 и 34 (масла при комнатной темпера туре) очищают хроматографией на колонке силикагеля (элюент гексан-этил ацетат (3:1) вместо перекристаллизации. Пример 10. Получение N-(2,2 .-димeтoкcиэтил)-N-(2,6-димeтИлфeнил) -бензамида (соединение 6, табл. 5). 2,81 г (0,02 моль) хлористого бен зоила добавляют по каплям в течение 20 мин и при к раствору N-(2,2 -диметоксиэтил)-2,6-диметиланилина (4,45 г, 0,02 моль) в этиловом эфире (20 мп), содержащем триэтиламин (2,76 МП, 0,02 моль). Реакционную смесь перемешивают при комнатной тем пературе в течение 15 мин. Полученну соль отфильтровывают и раствор промывают 8 мл водного раствора соляной кислоты при 5%, а затем водой до нейтрального значения рН. Органи32ческую фазу подкисляют безводным КалЗОл и выпаривают растворитель, получив таким образом 5,2 г белого твердого вещества, которое при кристаллизации Из петролейного простого эфира (25 мл) дает 4,5 г продукта (кислоты 91% по тонкослойной хроматографии) с выходом 65,5% (белое вещество, т,пл. . 58-59с). Пример 11. Действуя согласно, примеру 10 и исходя из N-(1-мeтил-2,2-димeтoкcиэтил)-2,6-димeтилaнилинa и из хлористого бензоила, получают в виде прозрачного масла N-(1-мeтил-2,2-димeтoкcиэтил) -N-( 2 , 6-диметилфенил) -бензамид (соединение 5, табл. 5). Пример 12. Получение Ы-(метилме.токсикарбонилметилен)-2,6-ди- . метиланилинаi К раствору 2,6 диметиланилина (37,2 МП, О,3 моль) в бензоле (200 мя) добавляют 0,5 г ZnCl2 при комнатной температуре, а затем 33,2 мл , (0,33 моль) метилового эфира пировиноградной кислоты. Реакционную смесь нагревают под флегмой в течение 7 ч при одновременной азеотропной отгонке воды, образующейся во время реакции, затем растворитель выпаривают, получив 65 г вещества в виде масла, которое перегоняют, собирают фракции,, кипящие при 87-8.8 С под давлением 0,07 мм рт.ст. Таким образом, получают 42,5 г продукта, имеющего чистоту 92% по тонкослойной хроматографии (выход |бЗ,5%). Пример 13. Получение N-(2,6-димeтилфeнил)-N-(1-карбометоксивинил) фенилацетамида (соединение Т1, табл. 5). 4,35 МП фенилацетил хлорида (0,033 моль) добавляют по каплям и при комнатной температуре к раствору 6,7 г (0,03 моль) Н-(метилметоксикарбонилметилен)-2,6-диметиланилина (полученного так, как описано в примере 12, частотой 92%) в толуоле (100 мп). ; 71 Реакционную смесь нагревают под флегмой и вьщерживают в потоке азота в течение 3 ч, досле чего выпаривают растворитель, получив 10,8 г свет ло-желтого масла, которое затвердевает при трении. Полученный неочищенный продукт перекристаллизовывают из петролейного эфира, получив 2 г продукта (более твердое вещество, чистое согласно тонкослойной хроматографии), выход составляет 21% , В табл. 1 приведена целебная активность, при дозе 0,01% против Р. viticola на винограде, выраженная в процентах уменьшения заражения В табл. 2 приведена активность при дозе 0,01% против Р. tabacina Н.N2,6-диметилфенил-Ы-( 1 карбометоксиэтил)-2- фуроиламид. (2,6-диметилфенил)-N-(1-карбометоксиэтил)-метоксиацетамид. 32,8 на растениях табака , выраженная в процентах. уменьшения заражения . В табл. 3 приведена профилактическая активность при дозе 0,01% против Р. viticola, на винограде, выраженная в процентах ингибирования заражения. В табл. 4 приведен индекс фитотоксичности при дозе 0,3% на винограде, вьфаженный шкалой величин от О (отсутствие фитотоксичности, здоровое растение) до tOO (полная фитотоксичАость, растение полностью повреждено или уничтожено).
Т
Активность,%
Соединение
90 100 100 100 ТОО 80 IQO . 100 100 100 100
too
100
100
100
100
100
100
100
100
100
90
100
Таблица 2
Активность, %
Соедииение
100
18 22 23 25 26
100
100
100
100
100 27 31 32 33 35
100
100
100
100
uralaxyl
100
100
Таблица 3
100 100 100 100
, 90 . 100 100 100 100 100
100
100
100
95
100
100
100
100
jj
Zineb - цинк-этилен-бис-дитиокарбамат.
25
О
5
10
40
36
30
8
18
10
29
6
100
95
100
100
100
30
Таблица 4
2
25
38
29
;
38
30
25
15
laxyl
100
myl
100
c-deHs
, nI 0
2-((1НзНн-0-С-(
ОI о
Ю)
97-100 74,31 7,42 4,13 74,45 7,77 4,25
Ъа
3,
CH,o-j-da /(
О
114-115 Н5 Н,й-0-е- 1Г е-ЙН,, «: : / о H,((H3
1Нз
f -M
о
Нз6-о/ VH dHjv I iH
73,37 7,70 4,18 75,4 8,4 4,3
58-59 72,81 7,40 4,47 73,14 7,66 4,69 ;« 7,12 4.30 ,75,34 7,47 4,67 л
15
СоедиФормуланение .
Hjii-o-t-CH ,(,Н5
W II О I о
(1Н2(1Н(1Н2
(iHj
-о
«н,(,-«-Сн Д-«н,8.ОI О
СН. . , - Cfl / ОI
/-V
,0 (tH3)i-(( Ht-{e} СНз
;н,
ft
H3e-o-|-c t-CH,-0
16
1128832
Продолжение табл. 5
74,28 6,54 4,33 73,6 6,6 4,6
63-64 65,23 6,39 4,23 66,53 6,74 4,54
Мас74,76 7,70 3,96 73,84 7,91 3,09 ло
56-57
07 - .; - ,..:;,:. : -/Л)(;Н ° 70,36 6,79 4,10 70,5 6,9 4,0 О 74,31 7,424,13 74,037,524,
СоедиФормуланение.
dHs
3-H5(i,0-j4il -(lH2-{o)
О СН5
(iH.
йНг
14
5 iN II W
Элементный анализ, Z
т 1г ----j
СНN I СИ I N
Масло74,31 7,424,13 72,967,164,34
69-70 73,82 .7,12 4,30 73,41 7,28 4,31
н,Со-е(1н S «
0 ,
Hgri
97-10065,99 5,83 4,06 67,0 5,9 3,7.
f HS
II 1J/1I
0,0
(JHj
Hxd
60-64 72,47 8,82 4,22 71,75 9,12 3,81 Hid-o-C-dH (J- lH2-/oVo H,, II u/«Xl/ 0 70 90-93 70,96 7,09 3,94 70,59 7,28 3,70
dHs
Hjd-o-d-KlH /d-dHgII -N/ll 00
ХЧ. ,
о
Масло
19
fHs . н.йгО-й-Сн /fdHjdciHs
И 1/ II и
о 1 о о - .,,Л.(н,
(н
II ll/U 11
о I о о
н jd ,,Л,(1Н2 - сн(1н 2
dHs I
ii-du /-Cu2-C-o(iH5
II Н
о I о о
о о йн
HS
-li-4,
о I о о HsC ,
CH-t
I.
НзС-о-{-(н /ОНгйойн,
и 5i.. 5
II
Q О О
СН2СН,
20
1128832 Продолжение табл. 5
64,85 6,95 4,20 64,84 7,24 4,51
66,83 7,01 3,90 67,93 7,72 4,67
Масло
21
1Н5 25I
НтйО-С-йн .(i((li
II Ж и и
о 1 0 о
HsCo-C-dH /-dHo-diiHs II -H/tt II о 1 о о (.хСН2СН СН2
22
1128832 Продолжение, табл. .5
105-110 ло 69,95 7,34 4,08 68,60 7,30 4,32
(iHs 27.I.H5((iH .(iiiHodtHa II N/11 и
Я 1 J о М --,(iH(iHCH5 , 28.I Мас CHjIaCH-O-d-XlH i-iiHgtiOls ло H/-JJcH/ HjC-J-CH C-dHg-jjdUg ло68,12 ОI Оо (lH2
- --- J-liH , II /ii
о о
О
(1Иг-(
68,12 7,30 4,41 65,85 7,49 4,35
4,62 66,2 6,90 4,90 67,69 7,89 4,38 67,718,324,40 7,30 4,41 67,707,334,36
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| КЕРАМИЧЕСКАЯ МАССА ДЛЯ ПРОИЗВОДСТВА КИРПИЧА | 2013 |
|
RU2513789C1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| К | |||
| Бншер, Д | |||
| Пирсон | |||
| Органические синтезы | |||
| Ч | |||
| П, М., Мир, 1973, с | |||
| Уровень с пузырьком | 1922 |
|
SU388A1 |
