Изобретение относится к химии олигомерных соединений, конкретно к совершенствованному способу получения низкомолекулярных ароматических олигоэфиров с двумя концевыми гидроксильными группами, которые являются необходимыми промежуточными продуктами для получения регулярно построенных полиэфиркарбонатов, полиэфиров: эфиров тере- или изофталевой кислоты.
Целью изобретения является интенсификация процесса и повышение степени конверсии бисфенола за счет изменения режимов проведения процесса.
При получении ароматических олигоэфиров с двумя концевыми гидроксильными группами конденсацией фенольного соединения и хлорангидрида ароматической декарбоновой кислоты в присутствии инертного органического растворителя предварительно проводят смещение фенольного соединения общей формулы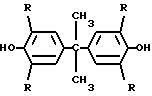 где R = H, Br, с водным раствором щелочи в молярном соотношении бисфенол: щелочь : вода 1: 1: 8,4-12,9. Смесь перемешивают и нагревают до 75-85оС до получения гомогенного раствора, потом раствор охлаждают до 20-30оС до образования порошкообразного продукта
где R = H, Br, с водным раствором щелочи в молярном соотношении бисфенол: щелочь : вода 1: 1: 8,4-12,9. Смесь перемешивают и нагревают до 75-85оС до получения гомогенного раствора, потом раствор охлаждают до 20-30оС до образования порошкообразного продукта 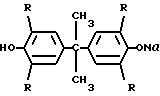 и подвергают конденсации в суспензии инертного органического растворителя с хлорангидридом тере- или изофталевой кислоты.
и подвергают конденсации в суспензии инертного органического растворителя с хлорангидридом тере- или изофталевой кислоты.
Это позволяет на 3,4-8,2% повысить конверсию фенольного соединения, ускорить проведение реакции в 2-15 раз и проводить процесс без сточных вод.
Порошкообразный продукт, образующийся при охлаждении гомогенного водно-щелочного раствора бисфенола, является гидратированной монозамещенной щелочной солью бисфенола. Это мелкокристаллический белый порошок, который может быть сразу использован для синтеза олигоэфиров либо после сушки в вакууме или токе инертного газа при температуре 25-30оС.
Идентификацию выделенного гидрата монозамещенной натриевой соли бисфенола А проводили следующим образом.
После приготовления соли и ее сушки в вакууме при 80оС методом потенциометрического титрования было определено соотношение Na - бисфенол, равное 1: 1. Затем соль промывают сухим ацетоном, сушат и снова титруют, соотношение Nа - бисфенол стало 1,06; 1. Бисфенол А хорошо растворяется а ацетоне, тогда как соли бисфенола в нем нерастворимы. Промывка сухим ацетоном позволяет отмыть соли бисфенола от свободного бисфенола. Таким образом, получаемое соединение содержит 88,6 мол. % (88,6 мол. % ) гидрата монозамещенной натриевой соли бисфенола А; 5,7 мол. % (4,3 мас. % ) бисфенола А и 5,7 мол. % (7,2 мас. % ) гидрата динатриевой соли бисфенола А.
Молекулярный вес соли равен 304,3. При нагревании соль начинает терять кристаллизационную воду, при 140оС темнеет и при 180-183оС разлагается.
Неожиданно было найдено, что, когда используется твердая монозамещенная соль бисфенола в отсутствие водной фазы, реакция между хлорангидридом и этой солью протекает с высокой скоростью (100% -ная конверсия хлорангидрида кислоты достигается за 30-60 мин) с образованием дигидроксиэфира низкой молекулярной массы с высоким выходом.
Этот результат невозможно было предсказать, основываясь на известных способах поликонденсации, поскольку процесс является межфазным с участием твердой фазы и общая продолжительность реакции определяется скоростью диффузии, которая в твердой фазе очень низка. Вследствие этих причин, например, поликонденсация в твердой фазе протекает на несколько порядков медленнее, чем в расплаве и растворе.
По предлагаемому способу гомогенный водно-щелочной расвор бисфенола получается при молярном сооношении бисфенол : щелочь : вода 1: 1: 8,4 - 12,9, при температуре 75-85оС. Столь узкие интервалы обусловлены тем, что при увеличении или уменьшении количества щелочи в соотношении невозможно получить монозамещенную соль бисфенола, при уменьшении количества воды гомогенный раствор при температуре 75-85оС не образуется, а при увеличении количества воды согласно диаграмме состояния бисфенол - щелочь - вода из раствора выделяется не монозамещенная соль, а смесь бисфенола, моно- и дизамещенной соли. Низкий температурный предел обусловлен температурой образования гомогенного раствора при данном соотношении реагентов, а верхний - тем, что дальнейшее повышение температуры экономически нецелесообразно.
В качестве инертных органических растворителей могут быть использованы хлорированные углеводороды, такие, например, как хлорбензол, метиленхлорид и т. п. , а также алифатические или ароматические углеводороды, например гептан, толуол.
В качестве щелочи могут применяться гидроксиды натрия или калия.
Технологический процесс заключается в следующем.
Смешивают дифункциональное фенольное соединение, щелочь и воду в молярном соотношении 1: 1: 8,4-12,9. Через 5-10 мин образуется гомогенный водно-щелочной раствор бисфенола, который охлаждают при перемешивании до 20-30оС до образования порошкообразного продукта. Затем в полученную массу приливают инертный органический растворитель перемешивают до образования суспензии. Далее поднимают температуру системы до необходимой (30-70оС), в течение 3-10 с при перемешивании вводят раствор хлорангидрида кислоты и проводят реакцию конденсации в течение 30-60 мин.
Количество инертного органического растворителя в пределах 1,4-5,8 л на 1 моль исходного бисфенола.
Выделение олигомера производят известным методом, для чего реакционную массу слегка подкисляют (до рН 4-5), добавляя в нее при перемешивании раствор соляной, серной или фосфорной кислот, а затем добавляют высадитель (алифатический углеводород С5-С7).
В случае использования в качестве инертного органического растворителя гептана или толуола высаживание не требуется (в толуоле дигидроксиэфир также нерастворим).
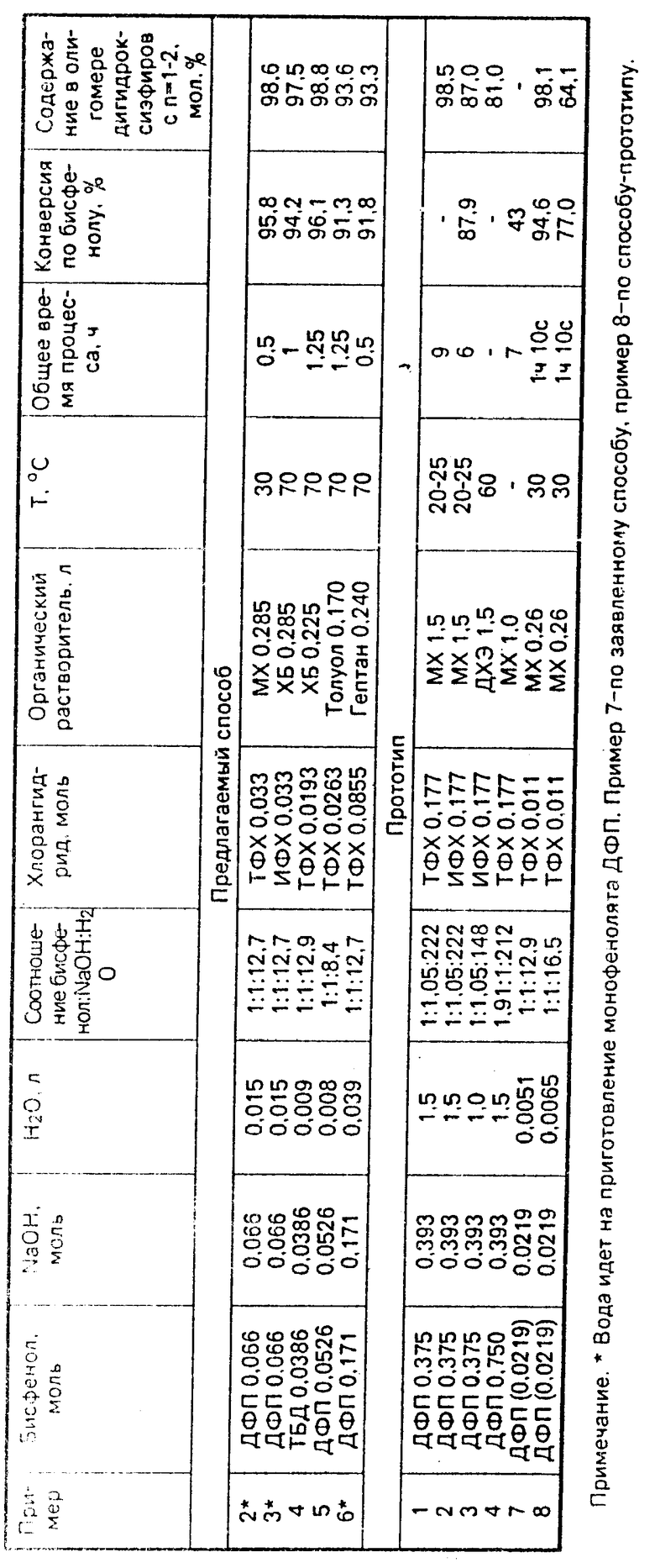
П р и м е р 1. К 100,0 г (0,438 моль) дифенилопропана (ДФП) при комнатной температуре добавляют 38,94 г 45% -ного водного раствора NaOH (0,438 моль NaOH) и 78,6 мл воды. Соотношение ДФП : NaOH : H2O 1: 1: 12,7. Смесь перемешивают и одновременно поднимают температуру до 75оС до получения гомогенного раствора. Затем при перемешивании охлаждают полученный раствор до 25оС. При этом образуется порошкообразный продукт, который сушат в вакууме до постоянной массы при 25-30оС. Выход готового продукта 130,6 г (98% от теор. ).
П р и м е р 2. К 200 мл метиленхлорида (МХ) при комнатной температуре добавляют 20,1 г (0,066 моль) тригидрата монозамещенной натриевой соли ДФП, полученной по примеру 1, смесь перемешивают и одновременно поднимают температуру до 30оС. В полученную суспензию в течение 10 с вводят раствор 6,70 г (0,033 моль) терефталилхлорида (ТФХ) в 85,0 мл МХ. Реакцию проводят при перемешивании в течение 30 мин при 30оС.
Результаты приведены в таблице.
П р и м е р 3. Аналогично примеру 2 за исключением того, что вместо ТФХ используется изофталилхлорид (ИФХ), вместо МХ - хлорбензол (ХБ), температура 70оС, время реакции 1 ч.
Результаты приведены в таблице.
П р и м е р 4. К 21,0 г (0,0386 моль) тетрабромдифенилолпропана (ТБД) при комнатной температуре добавляют 3,43 г 45% -ного водного раствора NaOH (0,0386 моль) и 7,1 мл Н2О. Соотношение ТБД : NaOH : H2O 1: 1: 12,9. При перемешивании поднимают температуру до 85оС до получения гомогенного раствора. Затем полученный раствор охлаждают при перемешивании до 20оС до образования порошкообразного продукта, к которому приливают 18,0 мл хлорбензола и при перемешивании поднимают температуру до 70оС. В реакционную массу в течение 3 с вводят раствор 3,95 г (0,0193 моль) ТФХ в 45,0 мл ХБ. Соотношение ТБД : ТФХ 2: 1. Реакцию проводят при перемешивании в течение 1 ч при 70оС.
П р и м е р 5. К 12,0 г (0,0526 моль) ДФП при комнатной температуре добавляют 4,68 г 45% -ного водного раствора NaOH (0,0526 моль NaOH) и 5,4 мол Н2О. Соотношение ДФП : NaOH : H2O 1: 1: 8,4. При перемешивании поднимают температуру до 80оС до получения гомогенного раствора. На эту операцию затрачивается 7-10 мин. После этого раствор охлаждают при перемешивании в течение 2-5 мин до 25-30оС до образования порошкообразного продукта, к которому добавляют 100,0 мл толуола, перемешивают и в течение 10 с вводят в суспензию раствор 5,37 г (0,0263 моль) ТФХ в 7,0 мл толуола. Соотношение ДПФ : ТФХ 2: 1. Реакцию проводят в течение 1 ч при 70оС.
П р и м е р 6. К 52,08 г (0,171 моль) тригидрата монозамещенной натриевой соли ДФП, полученной по примеру 1, при комнатной температуре добавляют 200,0 мл гептана, перемешивают и одновременно поднимают температуру до 70оС. В полученную суспензию в течение 5 с вводят раствор 17,36 г (0,0855 моль) ТФХ в 40,0 мл гептана. Реакцию проводят при перемешивании в течение 30 мин при 70оС.
Результаты приведены в таблице.
П р и м е р 7. Аналогично примеру 5 за исключением того, что загрузка ДФП 5,0 г (0,0219 моль); 45% -ного раствора NaOH 1,947 г (0,0219 моль); воды 4,0 мл. Соотношение ДПФ : NaOH : H2O 1: 1: 12,9. В качестве растворителя используют метиленхлорид в количестве 260,0 мл, половина которого идет на приготовление суспензии монозамещенной соли ДФП, а половина - на приготовление раствора ТФХ. Загрузка ТФХ 2,238 г (0,011 моль). Реакцию проводят в течение 45 мин при 30оС.
Результаты приведены в таблице.
П р и м е р 8 (по описанию прототипа). Водно-щелочной раствор готовят, смешивая 1,947 г (0,0219 моль) 45% -ного раствора NaOH и 4,4 мл воды. Добавляют к нему 5,0 г (0,0219 моль) ДФП и смесь перемешивают. Соотношение ДФП : NaOH : H2O 1: 1: 16,5. К этой водной фазе добавляют 130,0 мл метиленхлорида и опять перемешивают. К полученной дисперсии за 10 с добавляют раствор 2,238 г (0,011 моль) ТФХ в 30,0 мл метиленхлорида. После этого смесь перемешивают в течение 1 ч. Соотношение ДФП : ТФХ 2: 1. Температура реакции 30оС. Результаты приведены в таблице.
Для сравнения эффективности предложенного способа синтеза олигоэфиров со способом синтеза, описанным в известном способе (прототип) были проведены опыты в максимально сопоставимых условиях.
В качестве растворителя использовался метиленхлорид, температура 30оС. Время ввода раствора хлорангидрида терефталевой кислоты в систему NaOH - ДФП - Н2О 10 с, т. е. минимальное по прототипу. Соотношение NaOH : ДПФ : Н2О взято с максимальным количеством воды по заявляемому способу 1: 1: 12,9 и с минимальным по прототипу 1: 1: 16,5. Соотношение ДФП : ХА в обоих опытах равно 2: 1. Общее время процесса одинаково 1 ч 10 с. Процесс по прототипу состоит из времени ввода раствора хлорангидрида 10 с и времени конденсации 1 ч; по заявляемому способу приготовление монозамещенной соли ДФП 15 мин, ввод раствора хлорангидрида 10 с и время конденсации 45 мин.
Получены следующие результаты:
Прототип Предла-
гаемый
способ
Конверсия исходного ДФП, - 77,0 94,6
Содержание
дигидроксиэфиров с
n = 1-2 в олигомере (селективность), мол. % 64,1 98,1
Содержание дигидро-
оксиэфиров с n = 1-2 в
продуктах реакции (выход), мас. % 28,7 89,4
Приведенные данные свидетельствуют о том, что предварительная обработка исходного бисфенола щелочью в соотношении бисфенол : щелочь : вода 1: 1: 8,4 - 12,9 с образованием монозамещенной соли бисфенола с последующей ее суспензионной конденсацией с хлорангидридом кислоты существенно повышает конверсию мономера и выход целевого продукта. Кроме того, заявляемый способ позволяет снизить расход воды по крайней мере на 22% по сравнению с прототипом. Если проводить сравнение с примерами, представленным в прототипе, эта цифра возрастает до 2000% . Использовани сухой монозамещенной соли бисфенола, как описано в примерах 2, 3, 6, вообще исключает наличе воды в синтезе, а также необходимость разделения эмульсии высаливанием или центрифугированием.
Из сопоставления результатов, представленных в таблице, следует, что основные цели представленного изобретения достигнуты.
Интенсификация процесса достигается за счет сокращения времени синтеза олигоэфиров. Так минимальное время процесса у заявителе составляет 0,5 ч, максимальное 1,25 ч, тогда как из примеров, представленных в прототипе, минимальное время процесса, необходимое для достижения конверсии по бисфенолу порядка 90% составляет 6 ч, максимальное 10 ч. Сокращение времени, необходимого для проведения синтеза, согласно описанию, представленному в прототипе, приводит к значительному понижению конверсии по бисфенолу и выхода целевого продукта (пример 8). Процесс в этом случае не представляет практического интереса. Таким образом, время процесса в предлагаемом способе сокращается в 5-12 раз.
Одной из важнейших характеристик процесса является степень превращения исходных мономеров. Как видно из таблицы, конверсия по бисфенолу в предлагаемом способе колеблется в пределах 91,3-96,1% , тогда как у прототипа этот показатель составляет всего 43-87,9% . (56) Патент США N 4388455, кл. C 08 G 63/64, 1983.
Заявка ФРГ N 3026937, кл. C 07 C 69/773, 1981.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИКАРБОНАТА | 1991 |
|
RU2010810C1 |
| Способ получения поликарбоната | 1991 |
|
SU1837061A1 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИКАРБОНАТОВ | 1977 |
|
SU672872A1 |
| СПОСОБ ПЕРЕРАБОТКИ ОТРАБОТАННОГО ВАНАДИЕВОГО КАТАЛИЗАТОРА СЕРНОКИСЛОТНОГО ПРОИЗВОДСТВА | 1986 |
|
SU1385353A1 |
| Способ получения полиамидоэфиров | 1975 |
|
SU533142A1 |
| ГАЛОГЕНСОДЕРЖАЩИЕ ПРОСТЫЕ АРОМАТИЧЕСКИЕ ОЛИГОЭФИРЫ | 2010 |
|
RU2445304C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1-(3- АЛКОКСИ-2- КАРБАМОИЛОКСИПРОПИЛ) -(1H, 3H, 5H) -ПИРИМИДИН- 2,4,6-ТРИОНА И ПРОИЗВОДНЫЕ (1H, 3H, 5H)- ПИРИМИДИН -2,4,6-ТРИОНА | 1991 |
|
RU2050353C1 |
| Способ получения привитых и блок-сополимеров | 1960 |
|
SU138381A1 |
| Способ получения полиарилатов | 1977 |
|
SU729208A1 |
| Способ получения эпоксидиановых смол | 1977 |
|
SU732288A1 |
Изобретение касается эфиров ароматических кислот, в частности получения эфиров тере- или изофталевой кислоты (ЭТФК и ЭИФК), используемых в производстве полиэфиров. Для интенсификации процесса и повышения степени конверсии бисфенола используют другие условия и режимы. Синтез ведут из бисфенола (БФ) формулы C(CH3)2(R)2, где 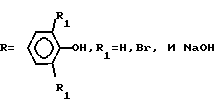 в среде воды с последующей конденсацией с хлорангидридом тере- или изофталевой кислоты в среде органического растворителя. Молярное соотношение БФ : NaOH : вода 1 : 1 : 8,4 - 12,9, процесс ведут при 75 - 85С до получения гомогенного раствора. После охлаждения последнего до порошкообразного состояния проводят конденсацию в суспензии хлорангидридов кислот. Время процесса сокращается в 2 - 15 раз без образования сточных вод. Конверсия БФ увеличивается на 3,4 - 8,2% . 1 табл.
в среде воды с последующей конденсацией с хлорангидридом тере- или изофталевой кислоты в среде органического растворителя. Молярное соотношение БФ : NaOH : вода 1 : 1 : 8,4 - 12,9, процесс ведут при 75 - 85С до получения гомогенного раствора. После охлаждения последнего до порошкообразного состояния проводят конденсацию в суспензии хлорангидридов кислот. Время процесса сокращается в 2 - 15 раз без образования сточных вод. Конверсия БФ увеличивается на 3,4 - 8,2% . 1 табл.
СПОСОБ ПОЛУЧЕНИЯ ЭФИРОВ ТЕРЕ- ИЛИ ИЗОФТАЛЕВОЙ КИСЛОТЫ путем взаимодействия бисфенола общей формулы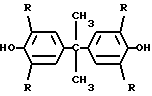
где R = H, Br,
с гидроксидом натрия в среде воды с последующим взаимодействием полученного монобисфенолята натрия с хлорангидридом тере- или изофталевой кислоты в среде инертного органического растворителя, отличающийся тем, что, с целью интенсификации процесса и повышения степени конверсии бисфенола, процесс взаимодействия бисфенола с гидроксидом натрия ведут при молярном соотношении бисфенол : щелочь : вода, равном 1 : 1 : 8,4 - 12,9, при температуре 75 - 85oС до получения гомогенного раствора, который затем охлаждают до образования порошкообразного продукта с последующей конденсацией в суспензии с хлорангидридом тере- или изофталевой кислоты.
