сн
Изобретение относится к способу получения новых феноксипроизводных, которые могут быть использованы для профилактики и лечения артериосклеро за, инфаркта мозга, сердечной недостаточности, грудной «абы и тромбоза.
Цель изобретения - получение новы феноксипроизводных, обладающих как л Ипйдопонижающей активностью,так и активностью в отношении блокирования гагрегирования тромбоцитов, что является не характерным для феноксипро- изводньк тиазольного ряда.
Получение исходных соединений.
Справочньй пример 1.
СНз
0(СН2)з- С-СООС2Н5 СНз
Смесь 30 г п-гидроксиацетофенона и 30 г карбоната калия перемешивают в 500 мл этанола в течение«30 мин при 60°С. После охлаждения до комнатной температурь. к реакционной смеси добавляют 52 г этилового эфира 5-бром 2,2-диметилпентановой кислоты. Результирующую смесь нагревают с обрат- ньм холодильником в течение ночи при перемешивании. Растворитель отгоняют . из реакционной смеси дистилляцией и к дстатку добавляют 500 мл хлороформа и 300 мл 5%-ного водного раствора кислого карбоната натрия. После перемешивания смеси хлороформовьй слой фракционируют и последовательно промыва- ют водой, а затем-насыщенным водным раствором хлористого Натрия с последующим осушением над безводным сульфа fOM натрия. Отгонка хлороформа дис- тилляцией дает остаточное маслянис- Тое вещество. Его подвергают хроматографии в колонке на силикагеле и элю- Ируют с использованием хлороформа в качестве элюента с получением 42 г этилового эфира п-ацетилфенокси-2,2-диметклпентановой кислоты в виде маслянистого вещества. Полученное та- КИМ путем маслянистое вещество раст- воряют в 1,6 л безводного метиленхло- рида и к раствору при охлаждении на льду каплями добавляют 24 г брома при перемешивании п реагировании в течение 3 ч. К реакционной смеси добавляют 1 л ледяной воды. После тщатель
с
0
S
0
О 5 0 5 Q
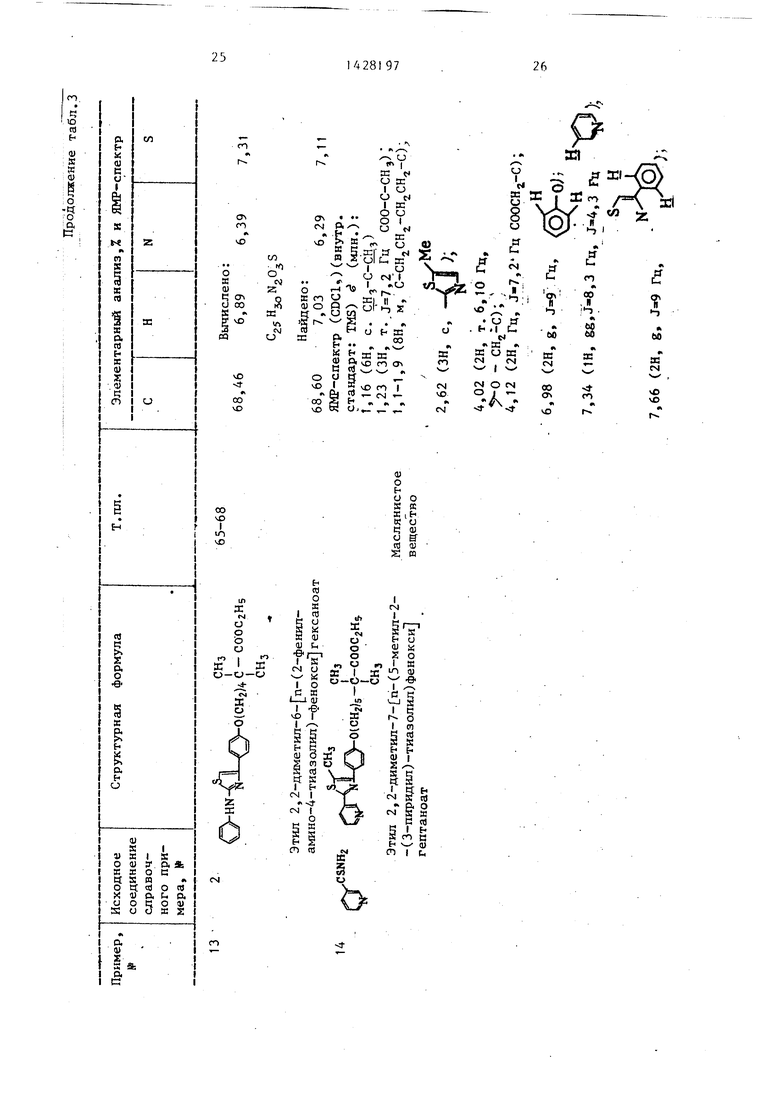
ного перемешивания органический слой последовательно промывают водой и пасьш;енным водным раствором хлористого натрия с последую1цим осушением над безводным сульфатом натрия. Отгонка метиленхлорида дистилляцией дает целевой продукт- этиловый эфир 5-(п-(бром- ацетил )фенокси)-2,2-диметилпентановой кислоты в виде маслянистого вещества. Справочный пример 2.
СНз
OlCH2)г C-COOC2H5 СНз
I .
Этиловый эфир 6-бром-2,2-диметил- гексановой кислоты используют в качестве исходного материала вместо этилового эфира 5-бром-2,2-диметилпентановой кислоты справочного примера 1 . Исходный материал вводят в реакцию и обрабатывают по методике справочного примера 1 с получением целевого продукта - этилового эфира 6- п-бромацетилфенокси -2,2-ди- метилгексановой кислоты в виде маслянистого вещества.
Справочный пример 3. .
ОСНз
BPCH2C-Q-0(CH2)5-C-COOC2H5
СНз
Этиловьй,эфир 7-бром-2,2-диметил- гептановой кислоты: используют в Качестве исходного материала вместо этилового эфира 5-бром-2,2-диметилпентановой кислоты справочного примера 1. Исходный материал вводят в реакцию и обрабатывают по методике справочного примера 1 с получением целевого продукта - этилового эфира 7- п-бромацетилфенокси -2,2-ди- метилгептановой кислоты в виде маслянистого вещества.
Справочный пример 4.
СНз01СН2Ь-С-СОрС2Нд CHj
-ЗО 35 40 45 JQ
55
в качестве исходного материала вместо п-гидроксиацетофенона справочного примера 1 используют п-гид ксипропиофенон. Исходный материал
1,8 Г этилового эфира (2- -бромацетил)фенокси -2,2-диметилпентановой кислоты 1,8 г, полученного по методике справочного примера 1 и 0,7 г З-пиридилтиоамида перемешивают в 20 мл метанола при комнатной температуре в течение 5 ч. Растворитель отгоняют из реакционной смеси дистилляцией и к остатку добавляют 30 мл хлороформа и 30 мл 5%-ного водного раствора кислого карбоната натрия. После перемешивания смеси хлороформо- вый слой фракционируют и последовательно промывают водой и насыщенным водным раствором хлорис гого натрия с последуницим осушением над безводным сульфатом натрия.Хлороформ отгоняют с получением маслянистого остатка.
Маслянистый остаток подвергают хроматографии в колонке на силикаге- ле и целевой продукт элюируют, используя хлороформ,метанол (10:1 по объему) в качестве элюента. Растворитель удаляют из элюата дистилляцией при пониженном давлении с получением целевого продукта - этилового эфира 2,,90 (2Н, g, ,3 Гц
35
); 8,20 (1Н, 8, J-1,8 Гц
40Н
45
р и м е р 2.
SO
СНз
0-{СН2)5-С-СООС2Н5 СНз
gg Этиловый эфир (бромацетил)фе- нокси -2,2-диметилгептановой кислоты, полученный по методике справочного примера 3, использ уют в качестве ис- хрдного материала вместо этилового
514
эфира S-fn-(2-бромацетил)фенокси - -2,2-диметилпентановой кислоты согласно примеру 1. Исходный материал вводят в реакцию и обрабатьгеают по методике примера 1. Результирунлций остаток перекристаллизовывают из пет- ролейного эфира с получением целевого продукта - этилового эфира 2,2 диме- -тил-7- п-2-(3-пиридил)-4-тиазолил - фенокси гептановой кислоты в виде белых кристаллов.
Т.пл. 41-42 С.
Вычислено, %: С 68,46; Н 6,89; N 6,39; S 7,31.
C gHjoNiO S
Найдено, %: С 68,60; Н 6,87; .N 6,36-, S 7,П.
П р и м е р 3.
СНз 0(СН2)з-С-СООС2Н5
сн.
После перемешивания 3,6 г этилового эфира 5-Гп(бромацетш1)фенокси - -2,2-диметилцентановой кислоты, полученной в справочном примере 1, и 1 г 4-имИдазолилтиоамида в 40 мл метанола при комнатной температуре в течение 15 мин смесь перемешивают при нагревании с обратным холодильником при 70 С в течение 2 ч. Затем раст- зоритель удаляют из реакционной смеси дистилляцией и к остатку добавляют 200 мл хлороформа и 100 мл 5%-ного водного раствора кислого карбоната натрия. После перемешивания смеси xno роформовый слой фракционируют и nor следовательно промывают водой, а затем насыщенным водным раствором хлористого натрия с последующим осзтпени- ем над безводным сульфатом натрия. Хлороформ удаляют дистилляцией с получением маслянистого вещества. Его подвергают хроматографии в колонке на силикагеле. Целевой продукт элюи- руют с использованием хлороформа - метанола (20:1 по объему) в качестве элюента. Растворитель удаляют из элюата дистилляцией при пониженном давлении. Остаток перекристаллизовывают из этилацетата с получением целевого продукта - этилового эфира (4-имидазолил)-4-тиазолил фенокси J-2 ,2-диметилпентановой кислоты в виде белых кристаллов.
N
Т.пл. 122-1234. Вычислено, %: С 63,14; Н 6,31; 10,52; S 8,02. Cj,,0,S
Найдено, %: С 63,21; Н 6,25; N 10,46; S 8,14. П р и м е р 4.
СНз
0(СН2)5-С- ООС2Н5
СНз
Этиловый эфир (бромацетил)- феноксиЗ-2,2-диметилгептановой кислоты, полученный по методике справочного примера 3, используют в качестве исходного соединения вместо этилового эфира (бромацетил)феноксиЗ- -2,2-диметилпентановой кислоты согласно примеру 3. Исходное соединение вводят в реакцию и обрабатывают по методике примера 3 (за исключением того, что перекристаллизацию проводят из этилацетата/гексана) с получением целевого продукта - этилового эфира 5- п-Г2-(4-имидазолйл)-4-тиазолил фе- нокси J-2,2-диметилгептановой кислоты в виде белых кристаллов.
Т.пл. 114-115°С.
Вычислено, %: С 64,61; Н 6,84; N 9,83; S 7,50.
С„Н„Ыз
OgS,
Найдено, %:С 64,69; Н 7,02; N 9,75; S 7,40. Пример 5.
СНз
0(СН2)з С-СООС2Н5 СНз
Тиоацетамид используют в качестве исходного соединения вместо 4-имида- золилтиоамида в примере 3. Исходное соединение вводят в реакцию и обрабатывают по методике примера 3. Результирующее маслянистое вещество подвергают хроматографии в колонке на силикагеле. Целевой продукт элюируют с использованием хлороформа в качестве элюента и растворитель удаляют из элюата дистилляцией при пониженном давлении с получением целевого продукта - этилового эфира (2-метил-4-тиа-, золил)фенокси}-2,2-диметилпентановой кислоты в виде маслянистого вещества.
,3 Гц
П p и м e p 6.
Примеры 7-9.
HzN
снз .
0(CH2)m-C-COOC2Hs CHj







| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения производных имидазола | 1985 |
|
SU1530093A3 |
| Способ получения феноксисоединений или их солей | 1985 |
|
SU1380609A3 |
| Способ получения феноксиалкиловых эфиров или их солей | 1985 |
|
SU1422998A3 |
| Способ получения производных 1,4-дигидропиридина | 1985 |
|
SU1342413A3 |
| Способ получения гетероциклических соединений | 1986 |
|
SU1438610A3 |
| Способ получения гетероциклических соединений или их фармацевтически приемлемых солей щелочного металла | 1986 |
|
SU1454249A3 |
| Способ получения гетероциклических соединений | 1985 |
|
SU1470186A3 |
| Способ получения производных катехина | 1984 |
|
SU1424729A3 |
| Способ получения гетероциклических соединений | 1986 |
|
SU1491337A3 |
| Способ получения гетероциклических соединений | 1986 |
|
SU1452481A3 |
Изобретение касается производных гетероциклических соединений, в частности способа получений фенокси- производных ФП общей формулы I B-C-N-CA-CR -S, где В -. имидазолнп, пиридил, низший алкил, NHj, Гона может быть замещена фенилом, низшим алки- лом, карбамоил он может быть замещен имидазолилалкилом или циклоал- киламиногруппой , A -n-CjH4-0- LCHj - -CRjUj-Cfni-R,, при , или низший алкил, RjH R - низший алкил, R, - низший алкоксил, ОН или NH, или их солей, которые могут быть ис- пользованы в медицине для лечения и профилактики артериосклероза, инфаркта мозга, сердечной недостаточности, стенокардии и тромбоза. Цель изобретения - создание веществ с активностью, не характерной для данного класса. Синтез ведут из тиоамида и У ало- генида формул (II) и (III): B-cfs - -NH (II) и X-CHR -cfol-A (III) , где А, В без карбомоила} R указаны с последующим выделением целевого продукта. Для получения соединения I с В - соответствующий карбамоил синтез ведут превращением группы обработкой либо соответствуилцим изоцианатом в среде органического растворителя при кипячении, либо ими- дазолилкарбоновой кислотой в среде N,N-димeтилфopмaмидa в присутстЕ11Ш конденсирующего агента. Целевой продукт выделяют в виде феноксипроизводного, который либо гидролИзуют с получением кислоты или ее соли, либо обрабатывают NHj с получением в целевом соединении группы . Новые ФП снижают общее количество холестегг рина на 36-69% и уменьшают агрегацию тромбоцитов ЮО мкм на 16-100% при токсичности г/кг. 3 табл. V О) с
СНз
0(CH2)5- f-COOC2H5 CHj
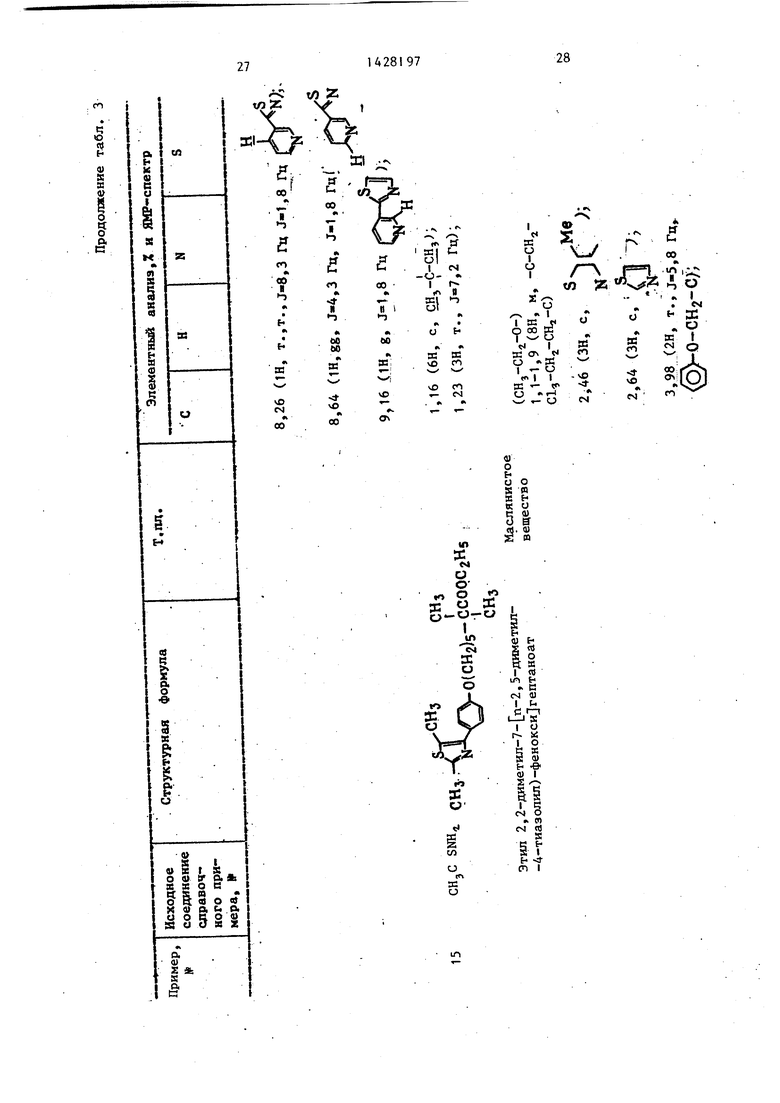
. Тиоацетамид используют в качестве исходного соединения вместо 3-пиридил ацетамида в примере 2. Исходное соединение вводят в реакцию и обрабатывают по методике примера 1. Результи- рующее маслянистое вещество подвергают хроматографии в колонке на сили- кагеле. Целевой продукт элюируют с использованием смеси н-гексан/этил- ацетат (8:1 по объему) в качестве элюента, и растворитель удаляют из элюата дистилляцией при пониженном давлении с получением целевого продукта - этилового эфира (2-метил-4-тиазолил)фенокси -2,2-диметш1геп- тановой кислоты в виде маслянистого вещества.
51МР-спектр (CDC1,) (внутренний стандарт: ТМС) 5 ): 1,16 (6Н, с. CHj-C-CH,) 1,23 (ЗН, T.,,2rq/
Me
сн,); 1,1-1,9 (н, м - j:-CH4. ме
0
5
5
.
(),
Этиповые эфиры бромацетилфенокси- карбоновых кислот, полученные в справочных примерах 1-3, используют в качестве исходных соединений в сочетании с тиомочевиной. Исходные соединения вводят в реакцию и обрабатывают по методике примера 1 с получением соединений примеров 7-9.
Примеры 10 и 11.
СНз 0(СН2)т-С-СООС2Н5
СНз
)Этиловые эфиры бромацетилфенокси- карбоновых кислот, полученные в справочных примерах 1 и 2,. используют в качестве исходных соединений в сочетании с метилтиомочевиной. Исходные соединения вводят в реакцию и обрабатывают по методике примера 1 с получением соединений примеров 10 и 11.
Примеры 12и.13.
« «Ч оHNCHj
OiCH2)m-C-COOC2He СНз
(,4)
Этиловые эфиры бромацетилфенокси- карбоновых кислот, полученные в справочных примерах 1 и 2, используют в качестве исходных соединений в сочетании с 1-фенил-2-тиомоче1виной. Исходите соединения вводят в реакцию и обрабатывают По методике примера 1 с получением соединений примеров 12 и 13.
П р и м е р ы 14-16.
сн, ,,
0(СН2)5-С-СООС2Н5 СНз
(-gr
- СНз, -NHz)
Этиловый эфир (-бромпропио- нил)фенокси -2,2-диметш1гептановой кислоты, полученный в справочном примере 5, используют в сочетании с 3-пиридилтиоамидом, тиоацетамидом и тиомочевиной в качестве исходных соединений. Исходные соединения вводят в реакцию и обрабатывают по методике примера 3 с получением соединений примеров 14-16. Пример17..
н,с
CHj
0(СН2)з-С СООС2 СНз
Этиловый эфир 2,2-диметил-5- п-(2- -бромпропионил)фенокси -пентановой кислоты, полученньй в справочном примере 4, используют в качестве исходного соединения вместо этилового эфира (-бромпропионил)фенокси -2,2- -д 1метилгептановой кислоты в приме- ре 15. Исходное соединение вводят в реакцию и обрабатьшают по методике примера 15. Результирующее маслянистое вещество подвергают хроматографии в колонке на силикагеле. Растворитель удаляют из элюата дистилляцией при пониженном давлении с получением целевого продукта - этилового эфира (2,5-диметил-4-тиазолил)- -фенокси -2,2-диметйлпентановой кислоты в виде маслянистого вещества .
ЯМР-йпектр (CDCl,) (внутренний стандарт: ТМС): 1,20 (6Н, с, -СН,); 1.22 (ЗН, т, J.7,2 Гц, -СООС- -СН,); 1,-5-1,8 (4Н, м, C-CH CHi-f-);
/J-СНз 2,48 (ЗН, с, ij;; 2,65 (ЗН,с,
сн.
-{Р-:
3,98 (2Н, КБ, - С);
4,12 (2Н, KB, ,2 Гц, СООСН,С), 6,91 (2Н, g, ,3 Гц
-0-j; 7,52 (2Н, g, J-8,3
Гц
o-i;;
Пример 18.
-NHCONH
loIfH, o(cHj)4- -eoociH8
CHj
в .15 мл тетрагидрофурана, 1,13 г этиловогр эфира (2-амино-4-тиа- золил)фенокси -2,2-диметилгептановой кислоты, полученного в примере 9,вводят в реакцию с 0,38 г циклогексилизоцианата на протяжении ночи при нагревании с обратным холодильником.
В реакционную смесь выливают ледяную воду, после чего проводят экстрагирование метиленхлоридом. После осушения метипенхлоридного слоя над безводньм сульфатом магния растворитель удаляют отгонкой при пониженном давлении. Результирующий остаток подвергают хроматографии в колонке на силикагеле. Продукт элюируют с использованием смеси н-гексан/этипацетат (4:1 по объему) в качестве элюента. После отгонки растворителя в вакууме
результирующие кристаллы перекристал лизовывают из н-гексан/эфирной смеси растворителей с получением этилового эфира (3-циклогексилуреи- до)-4-тиазолил/фенокси1-2,2-диметил- гептановой кислоты в виде белых кристаллов.
Т.пл. 106-108 С.
Вычислено, %: С 64,64, Н 7,84; N 8,38; S 6,39.
Cj7H,,N,04S
Найдено, %: С 64,43} Н 8,04; N 8,24; S 6,40.
Пример 19.
:н ннсомн
а .,сн5
iidСНз-0(CH2l9- f-COOCiHs
CHj
Этиловый зфир (2-амино-5-ме- тил-4-тиазолип)фенокси |-2,2-диметил- гептановой кислоты, полученный в примере 16, используют в качестве не- ходкого соединения вместо этилового эфира (2-амино-4-тиазолил)фенок си1-2,2-диметилгептановой кислоты согласно примеру 18. Исходное соединение вводят в реакцию и обрабаты- вают по методике примера 18 с получением целевого продукта - этилового эфира 7- п-/2-(3-циклогексш1уреидо)- -5-метил-4-тиазолил/фенокси -2,2- -диметилгептановой кислоты в виде белых кристаллов.
Т.пл. 79-8Гс.
Вычислено, %: С 65,21; Н 8,01; N 8,15; S 6,22.
CjgH.
Найдено, %: С 65,33; Н 8,22; N 8,03; S 6,20.
П р и м е р 20.
- «i lOoicH.Vs-pcooc.Hs
CHj
в 15 мл сухого К,Н-диметилформами- да (ДМФА) раств оряют 0,39 г 4-(1- -имида.золил)бутировой кислоты .и 0,37 г 1-гидроксибензотриазола. В ходе перемешивания к раствору при 40- 45 С добавляют 0,57 г дициклогекснл- карбодиимида. После осаждения из реQ
5
0
0 5
акционно1 г смеси дициклогексилмочеви- ны к смеси при той же температуре каплями добавляют раствор 0,94 г этилового эфира (2-амино-4-тиазо- лил)фенокси -2,2-диметилгептановой кислоты в 5 мл ДМФА смесь перемешивают при этой температуре в течение ночи. После удаления дициклогексил- мочевины Фильтрованием фильтрат кон- денсируют при пониженном давлении. К остатку добавляют ледяную воду с последующим экстрагированием хлороформом. После последовательной промыяки хлороформового слоя насьщенным водным бикарбонатом натрия и водой-хлоро- формовый слой сушат над безводным сульфатом магния. Хлороформ удаляют дистилляцией при пониженном давлении и результирующий остаток подвергают хроматографии в колонке на силикаге- ле. Продукт элюируют с использованием смеси хлороформ/метанол (40:1 по объему) в качестве элюента. После удаления растворителя дистилляцией результирующие кристаллы перекристал:Й13о- вьшают из растворительной смеси эфир/ бензол с получением этилового эфира 7-fn- 2-/4-(1-имидазолил)бутиламидо/- -4-тиазолил феноксиЯ 2,2-диметилгептановой кислоты в виде белых кристаллов. Т.пл. 148-150°С.
Вычислено, %: С 63,26, Н 7,08, .N 10,93; S 6,25.
Найдено, %: С 63,Of, Н 7,08; К 10,85; S 6,13.
П р и м е р 21.
ONH
/Н, СН, |-ПХ) С-ОМОН, .HJ . CHj
5
0
Этиловый эфир (2-амино-5-ме- тшт-4-тиазолил)фенокси -2,2-диметилгептановой кислоты, полученный в примере 16, используют в качестве исходного соединения вместо этилового эфира (2-амино-4-тиазолил)фенокси - -2,2-диметилгептановой кислоты согласно примеру 20. Исходный материал вводят в реакцию и обрабатывают по методике примера 20 с Получением целевого продукта - этилового эфира 7-Гп- 2-/4-(1-имидазолил)-бутирамидо/- -5-метил-4-тиазолш1 феноксиЯ-2,2-диметилгептановой кислоты.в виде белых кристаллов.
Х.пл. 85-87 С.
Вычислено, %: С 63,85; Н 7,27; N 10,64; S 6,09.
Найдено, %: С 63,87; Н 7,47;
N 10,62; S 6,25. Ц р и м е р 22.
СНз
0(СН2)з-С- COON СНз
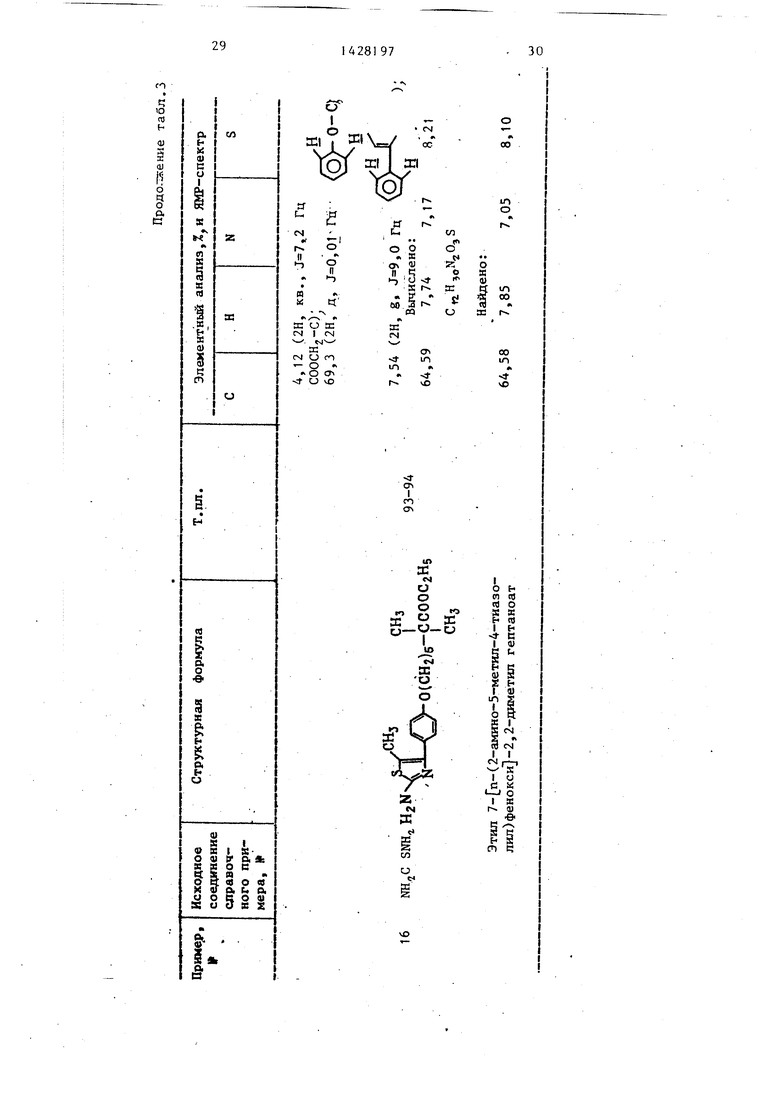
в 5 мл этанола растворяют 1,40 г этилового эфира 2,2-диметил-5- п-(2- -метил-4-тиазолил)феноксиЗпентановой кислоты, полученного в примере 5. Раствор добавляют к водному раствору гидроокиси натрия (0,4 г гидроокиси натрия в 5 мл воды), смесь кипятят с обратным холодильником 10 ч. После охлащ;ения выпавшие кристаллы промывают водой с получением целевого продукта - 2,2-диметил-5- п-(2-метил -4-тиазолил)фенокси пентаноата натри в виде белых кристаллов.
Вычислено, %: С 59,81; Н 5,90; N 4,10; S 9,39.
С„
Найдено, %: С 58,72; Н 5,73; N 4,12, S 9,34.
Пример 23.
СНз .-0-1СН2)з-С-СОО
СНз
в 50 мл воды растворяют 1,0 г 2,2-диметш1-5- п-(2-метил-4-тиазо- .лил)фенокси пентаноата натрия, полученного в примере 22. Раствор нейтрализуют добавлением 10%-ной хлористоводородной кислоты. Осажденные кристаллы отбирают фильтрованием и промывают водой с получением целевого продукта- 2,2-димeтил-5- n-(2-мe- тил-4-тиaзoлшl) фенокси пентановой кислоты в виде белых кристаллов.
Т.пл. 150-153°С.
Вычислено, %: С 63,92; Н 6,63; N 4,39; S 10,04..
С„Н,,Н,05
Найдено, %:С 63,67; Н 6,65; N 4,13; S 9,64.
Пример 24.
снсн.
0{CH2)3-C-CONH2 СН
0
5
0
5
5
в 10 мл дихлорметана растворяют 0,7 г 2,2-диметил-5- п-(,2-метил-4- -тиазолип)фенокси1пентановой кислоты, -подученной в примере 23, и 0,22 г триэтилвмина. Раствор охлаждают до при перемешивании и добавляют каплями 0,3 г изобутилхлорформи- ата. После перемешивания в течение 30 мин при той же температуре к смеси добавляют 0,6 г концентрированного аммиака в воде (29% аммиака).После окончания капельного.добавления температуру повьппают до комнатной, перемешивание продолжают еще 13 мин.
В реакционный раствор выливают ледяную воду и органический слой фрак- ционируют. Органический слой последо- вательно промывают насыщенным водным раствором бикарбоната натрия и водой, а затем осушают над безводным сульфатом магния. После удаления растворителя отгонкой при пониженном давлении результирующий остаток подвергают хроматографии в колонке на сили- кагеле. Продукт элюируют с использованием хлороформа/метанола (50:1 по объему) в качестве злюента. После отгонки растворителя в вакууме образованные кристаллы перекристаллизовыва- ют из растворительной смеси эфир/бензол с получением целевого продукта- 2,2-диметил-5- п-(2-мвтил-4-тиазо- лил)фенокси пентамида в виде белых кристаллов.
Т.пл. 148-150 с.
Вычислено,: С 64,12; Н 6,96; N 8,80; S 10,07.
C1,,H,N.,.
Найдено, %:С 64,20; Н 7,04; N 8,78; S 9,95.
I :
Фармакологическое действие соединений согласно изобретению подтверждается следующими данными.
Лишадопонижающая активность. . Рацион питания, содержащий 1,5% холестерина и 0,5% желчной кислоты, давали крысам-самцам породы Sprague Dawley четыре,хнедепьного возраста в течение 7 дней. Суспензию соединения согласно изобретению в 0,5%-ном водном растворе метилцеллюлозы вводили ежедневно орально через катетер в течение 4 дней. После лишения возможности двигаться в течение ночи под эфирной анестезией собирали кровь для определения количества общего ЛВП и холестерина в сыворотке. Измерение холестерина и ЛВП осуществляли по описанной методике. Липидопонижающая активность характерньгх соединений согласно изобретению приведена в табл.1 и 2 .
Активность в отношении блокирования агрегирования тромбоцитов.
Обогащенную и обедненную тромбоцитами плазмы получали из венозной крови белых японских кроликов. Тромбоци- то-агрегирующую активность измеряли по известному методу, а активность в отношении блокирования агглютинации тромбоцитов для соединения в случае агрегирования тромбоцитов, вызываемого арахидоновой кислотой (конечная , концентрация 0,2 моль), определяли с помощью прибора Аггригометр.
Ингибирующая агрегирование тромбоцитов активность характерных соединений согласно изобретению приведена в табл„1 и 2.
Лекарственные препараты, содержащие соединения, представленные общей формулой 1, или их соли в качестве активных ингредиентов, могут быть приготовлены с использованием фар- мадевтических носит елей и разбавителей, традиционно используемых в рассматриваемой области техники обычным образом.
Способом введения может быть ораль ньй прием в виде порошков, гранул, капсул или парентеральный, например инъекция и,т.п., но предпочтителен оральньй прием.
Дозировка может быть определена подходящим образом в зависимости от условий, возраста и пола, от цели введения препарата, однако в случае орального приема дозировка может составлять от 1 до 100 мг/кг в сутки для взрослых, предпочтительно 5-25 мг/кг, что может быть принято за один раз или поделено на .2-4 приема.
Данные по полученным соединениям сведены в табл.3.
Формула изобретения
Способ получения феноксипроизвод- ных общей формулы (I)
В
S-i 4 2
- L1 0tCH2)m- -CO-1 b
5
Q
j
0 5
0
5
где В
- имидазолилгруппа, пиридил- группа, низшая алкильная группа, аминогруппа, которая может быть замещена фе- нильной группой или низшей алкильной группой, или кар- бамоильная группа, замещенная циклоалкиламиногруппой или имидазолилалкильной группой; га - 3-5; .
RJ- водород или низшая алкильная группа-,
низшая алкильная группа, RJ- низшая алкоксигруппа, оксигруппа или аминогруппа, или их солей,
отличающийся тем, что тио- амид общей формулы (II)
Х
D-c(
NH2,
где D - имидазолилгруппа, пиридилгруппа, низшая алкильная группа или аминогруппа, которая может быть замещена феНильной группой или низшей алкильной группой,
подвергают взаимодействию с галогенидом общей формулы (III)
о
с
0
5
X-CH-R
oi
В0(СН2)тп-С-СО-Ки R3
где X - галоген;
R,- m имеют указанные значения с выделением целевого продукта или для получения соединений общей формулы (I ), где В означает карбамо- ильную группу, замещенную циклоалкиламиногруппой или имидазолилалкильной группой, соединение общей формулы (1);, где В - свободная аминогруппа, подвергают взаимодействию с циклоалкилизо- цианатом при кипячении с обратным холодильником реакционной массы в органическом растворителе или.с имида- золилалкилкарбоновой кислотой в N,N-димeтшlфopмaмидe в присутствии конденсирующего агента с выделением целевого продукта в.виде феноксипроизводного, или с последующим гидроли- соли, или с последующим взаимодейст- зом и вьщелением целевого продукта в вием с аммиаком и вьщелением целевого виде кислоты или ее кислой аддитивной продукта, где R - аминогруппа.
)s-i -COOC2H9 ,
ХЛ11 - яипопротеин - высокой плотности;
ХДЛ-С- липолротеин - холистарнн высокой плотности; ЛДП-С- липопротеин - хопейтерин низкой плотности; (олестйрин,
ЛЛ( г/кг в рот крыс.
Таблиц) I
Оч
tn
«ч
00
о ю
00
го Г-
ш ш
го tN
to
|Л
о
«4
г ч р
а; ffи
гО
о
0)
о
S
ю
о
1Л
Л
ю
о
N
IK « м
а: о е«
и
t о
г
О
а:
ч}- 1Л
со
N
чО
о
00
fO vD
1
r
n
o
«Ti
I
r
ГО
Сч1
о. ш
es
со
глЗК
V) Z
Д
я
еч
1Л
.
о
О
i-От
in
ж и
4) О
И о
S W
к U
5 I S
rt
X о
ел
О (П
о
| Эльдерфилд Р | |||
| Гетероциклические .соединения | |||
| Т | |||
| V | |||
| Устройство для автоматического пуска в ход регистрирующих механизмов в самопишущих приборах | 1925 |
|
SU1954A1 |
