Изобретение относится к получению гетероциклических соединений, в част7 ности к способу получения тиокетеновых производных пиперидина общей формулы
0 SR3
AxfS
N
I
R
i
где R - водород, С -С алкил,
С -С -алкенил, фенил или группа формулы B-Y, где Y пиридил, фенил или фенил, замещенный одной или двумя группами, выбранными из галогена, ,,-ал- кила, С,-С -алкокси, нит- ро, амино, бензилоксикар- бониламино; В килен{
КгиR,- С1 С4-алкил каждый, либо они вместе образуют группу формулы или -СН-СН-, R4 - водород, С,-С -алкил или
С -С -алкоксикарбонил; А - группа формулы -CHt- или
-СН(СООЯ5)-,где Ry- С,,-С4 -алкил,
обладающим гепатозащитными свойствами или их фармацевтически приемлемых кислотно-аддитивных солей,
Цель изобретения - создание на основе известных методов способа получения новых химических соединений, обладающих ценными фармакологическими свойствами.
Пример 1. 2,03 г 1-бензил- -2,4-диоксипиперидина и 760 мг сероуглерода растворяют в 20 мл диметил- сульфоксида и в раствор добавляют по каплям при комнатной температуре раствор 1,1 г гидрата окиси калия в 3 мл воды. Смесь перемешивают в течение 30 мин и затем в нее добавляют по каплям раствор 1,37 г 1,2 -дибромэтана в 2 мл диметилсуль- фоксида. Смесь перемешивают при комнатной температуре в течение 2 ч, а затем при 60°С в течение 1 ч. Реакционную смесь выливают в воду и подвергают экстракционной обработке смесью бензола с этилацетатом. Экстракт промывают водой, сушат и отгоняют растворитель. Остаток очищают в хроматографической колонке с сили- кагелем с использованием в качестве элюента смеси бензола с этилацетатом в соотношении 8:2 и перекристаллизо- вывают из этанола, в результате че
0
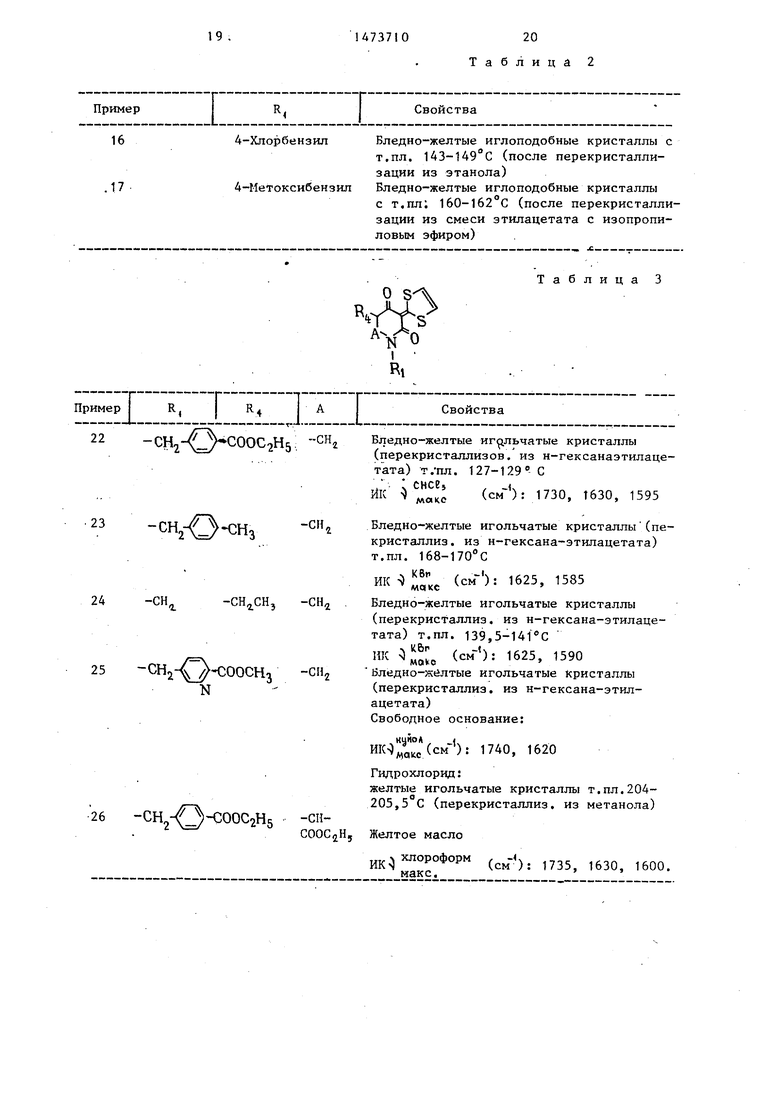
5
го в виде бледно-желтых иглоподобных кристаллов получают 1,08 г 1-бензил- -3-(1,3-дитиолан-2-илиден)-2,4-диок- сопиперидина с т.пл.148-150 С.
П р и м е р 2. Аналогично примеру 1 соответствующие исходные материалы, а именно 1-бензил-2,4-диоксопи- перидин, сероуглерод и метилйодид, подвергают обработке с получением соединения (I), в котором R% - бензил; RJ - СН3 ; R3 - СН3 ; R4-H; A-CH2. Получают бледно-желтый маслоподобный продукт,выход 59,3%; ИК-спектрограмма,
ХАОрОТврМ162() 1655 см
Приме р 3. 60%-ную дисперсию 432 мг гидрида натрия в масле суспендируют в 10 мл диметилсульфоксида и в эту суспензию по каплям добавляют раствор 1,0 г 1-бензил-2,4-диоксо- пиперидина и 0,33 мл сероуглерода в 20 мл диметилсульфоксида. Смесь перемешивают в течение 2 ч и затем добавляют в нее по каплям раствор 0,42 мл цис-1,2-дихлорэтилена в 5 мл диметилсульфоксида. Смесь перемешивают при комнатной температуре в течение 1,5 ч, после чего при 50° С в течение 1 ч. Реакционную смесь выливают в воду и подвергают экстракционной обработке бензолом. Экстракт промывают водой, сушат, а затем удаляют из него растворитель. Остаток перекристаллизовывают из изопропило- вого спирта, в результате чего в виде бледно-желтых иглоподобных кристаллов получают 486 мл 1-бензил-З- -(1-дитиол-2-илиден)-2,4-диоксопипе- ридина, с т.пл. 122-124°С,
Пример4. А. Смесь 15,1 г 2,4-диоксопиперидина, 30 мл метилор- тоформиата и 3,0 г п-толуолсульфокис- лоты в 300k мл метанола кипятят в течение 1 ч обратным холодильником.После выпаривания растворителя остаток растворяют в бензоле и кипятят с обратным холодильником в течение 5 ч. В раствор добавляют 9,0 г карбоната калия и смесь перемешивают в течение ночи при комнатной температуре, а затем фильтруют. Фильтрат перегоняют с целью удаления растворителя. Остаток перекристаллизовывают из смеси этилацетата с изопропиловым спиртом, в результате чего получают 12,3 г (вы- 5 ход 72,5%) 4-метокси-5,6-дигидро-2 (1Н)-пиридина в виде бесцветных призматических кристаллов с т.пл.115- 117°С.
5
0
5
0
514
Б.447 г гидрида натрия в форме 60%-ной дисперсии в масле суспендируют в 10 мл М,П-диметилформамида и добавляют в суспензию по каплям раствор 1,29 г 4-метокси-5,6-дигид- ро-2(1И)-пиридина в 30 мл N,N- -диметилформамида. Смесь перемешивают с охлаждением льдом в течение 30 мин, после чего в нее по каплям добавляют 0,97 мл йодистого этила. Эту смесь перемешивают с охлаждением льдом с течение 30 мин, а затем при комнатной температуре в течение еще 2,5 ч. Реакционную смесь выпивают в воду и подвергают экстракционной обработке хлороформом. Экстракт промывают насыщенным раствором хлористого натрия и сушат. После выпаривания растворителя остаток растворяют в 120 мл этанола и добавляют в раствор 60 мл 10%-ной соляной кислоты, а затем смеси дают постоять при 25°С в течение 5 ч. После отгонки под пониженным давлением растворителя оста- ток растворяют в хлороформе, промывают 5%-ным водным раствором бикарбоната натрия и водой с последующей сушкой. Растворитель отгоняют с получением 1,12 г (78%-ный выход) 1-этил- -2,4-диоксопиперидин а.
В. 1,06 г 1-этил-2,4-диоксопипери- дина обрабатывают по способу, описанному в примере 3, и получают в виде бледно-желтых иглоподобных кристаллов 1,10 г 1-этил-3-(1,3-дитиол-2- -илиден)-2,4-диоксопиперидина с т.пл. 114,5-117аС.
П р и м е р ы 5-12. А. Аналогично изложенному в примере 4 Б соответству- ющие исходные материалы обрабатывают с получением промежуточных соединений. Все полученные таким образом соединения представляют собой сырые продукты, которые, однако, могут быть использованы как таковые, т.е. без предварительной очистки, при осуществлении последующей стадии.
Б. Аналогично вышеизложенному в примере 3 каждое из соединений, полу- ченных по пункту 1, обрабатывают с получением соединений (1-j), как это показано в табл.1.
Пример 13. А. 121 г фенетил- амина растворяют в 500 мл метанола и в раствор по каплям добавляют при комнатной температуре 86 г метилакри- лата. Смесь перемешивают в течение 4ч, а затем перегоняют для удале
g 5 0 5 0
0 с
5
5
106
ния растворителя, получая 157,8 г (76%-ный выход) М-фенетил-бета-аланин- метилового эфира с т.кип. 124-127 С/ 1,2 мм рт.ст.
Б. 80,2 г N-фенетил-бета-аланин- метилового эфира растворяют в 150 мл метанола и в раствор по каплям с одновременным охлаждением льдом добавляют 32 мл дикетена. Далее смесь перемешивают при комнатной температуре в течение 1ч, после чего перегоняют для удаления растворителя. Остаток растворяют в 100 мл метанола и приготовленный раствор по каплям добавляют в раствор метоксида натрия, приготовленный с использованием 9,8 г металлического натрия и 150 мл метанола, после чего смесь перемешивают при комнатной температуре в течение 2 ч. После завершения реакции растворитель отгоняют и в остаток добавляют смесь воды со льдом. Смесь подвергают экстракционной обработке этилацетатом, водный слой подкисляют концентрированной соляной кислотой, а затем вновь подвергают экстракционной обработке этилацетатом. Этилаце- татный экстракт промывают водой, сушат, а затем перегоняют для удаления растворителя, в результате чего в виде бесцветного маслоподобного продукта получают 95,3 г (95%-ный выход) 1-фенетил-З-ацетил-2,4-диоксопипериди- на.
В. 5,0 г 1-фенетил-З-ацетил-2,4- -диоксопиперидина и 30 мл 10%-ной соляной кислоты добавляют в 50 мл этанола и смесь кипятят с обратным холодильником в течение 6ч. После отгонки растворителя остаток растворяют в хлороформе и раствор промывают насыщенным раствором хлористого натрия, сушат, а затем отгоняют для удаления растворителя, в результате чего в виде маслоподобного продукта
получают 3,62 г (выход 86,4%) 1-фене- тил-2,4-диоксопиперидина.
Г. 1- енетил-2,4-диоксопиперидин, полученный по пункту В, обрабатывают аналогично вышеизложенному в примере 3, в результате чего получают соединение (I-J), (где R,-фенетил), которое представляет собой бледно- желтые иглоподобные кристаллы с т, пл. 147-148°С (после перекристаллизации из изопропилового спирта); ИК-спектрограмма : 1535,
,jW Л -
1625 см
Пример 14. Способом, описанным в примере 3, обрабатывают 2,4-ди- оксопиперидин (200 мг). Получают в виде бледно-желтых иглоподобных кристаллов 284 мг (75%-ный выход) 3-(1,3- -дитиол-2-илиден)-2,4-диоксопипери- дина с-т.пл. 277-279аС.
Пример 15. 60%-ную дисперсию 226 мг гидрида натрия в масле суспендируют в 10 мл М,М-диметилформамида и в эту суспензию по каплям добавляют раствор 1,0 г 3-(1,З-дитиол-2-или- ден)-2,4-диоксопиперидина,(который готовят по примеру 14) в 100 мл М,К-диметилформамида при одновременном охлаждении до 0-5 С. Смесь далее перемешивают в течение 10 мин при той же температуре, после чего в нее по каплям добавляют 861 мг йодистого метила и конечную смесь вновь перемешивают при 0-2°С в течение 1,5 ч. После отгонки растворителя под пониженным давлением остаток очищают хроматографической обработкой в колонке с силикагелем с использованием в качестве элюента смеси н-гек- сана с этилацетатом в соотношении 1:1 и перекристаллизовывают с получением в виде бледно-желтых иглоподобных кристаллов 634 мг (выход,59,5%) 3-(1,З-дитиол-2-илиден)-2,4-диоксопиперидина с т.пл. 148,5-153 С.
Пример ы1би 17. Проводят реакцию 3-(1 , 3- ДИтиол-2-илиден)- -2,4-диоксопиперидина, полученного согласно изложенному в примере 14, с соответствующим исходным материалом аналогично изложенному в примере 15, в результате чего получают соединения (1-j), как это показано в табл.2.
П р и м е р 18. 60%-ную дисперсию 47 мг гидрида натрия в масле суспендируют в 2 мл N,N-диметилформами- да и в эту суспензию по каплям добавляют раствор 200 мг 3-(1,3-дитиол -2-илиден)-2,4-диоксопиперидина в 16 мл М,Я-диметилформамида. Эту смес перемешивают в течение 10 мин, после чего в нее добавляют 0,12 мл бен- зилбромида, а затем смесь подвергают дальнейшему перемешиванию в течение 1 ч. После завершения реакции реакционную смесь перегоняют для удалени растворителя под пониженным давлени- ем, а остаток растворяют в хлороформе Хлороформовый раствор промывают насыщенным водным раствором хлористо
5
0
5
5
го натрия, сушат и затем отгоняют растворитель. Остаток перекристалли- зовывагот из изопропилового спирта с получением в виде бледно-желтых иглоподобных кристаллов 176 мг (62%-ный выход) 1-бензил-3-(1,З-дитиол-2-или- ден)-2,4-диоксопиперидина с т.пл.122- 124°С.
Прим ер 19. А. 10,81 г3-пири- дилметиламина растворяют в 50 мл метанола и в раствор по каплям добавляют 8, 60 г метилакрилата при охлаждении льдом. Эту смесь перемешивают при комнатной температуре в течение 18 ч, перегоняют для удаления растворителя, а затем остаток перегоняют под пониженным давлением с получением 16,02 г (выход 82,5%) метил-3-(3-пиридилметиламино)-про- пионата с т.кип. 11 9-120°С/0,4 мм рт.ст.
Б. Смесь 28,72 г м,етил-3-(3-пири- дилметиламино)-пропионата с 22 мл триэтиламина и 240 мл хлористого метилена охлаждают до 5-10 С и добавляют в нее по каплям раствор 23,38 г этоксикарбонилацетилхлорида в 60 мл хлористого метилена, перемешивают при комнатной температуре в течение 1 ч,затем фильтруют для удаления осадка (триэтиламингидрохлорид). Фильтрат промывают водой, сушат и затем перегоняют для удаления растворителя . Остаток очищают в хроматографической колонке с силикагелем с использованием в качестве растворителя этилацетата, в результате чего в виде маслоподобного продукта получают 35,22 г (выход 77,3%) этил- N- -(3-пиридилметил)-N-(метоксикарбонил- этил) -карбамошт -ацетата .
В. 0,77 г натрия растворяют в 30 мл метанола и в этот раствор добавляют по каплям при 3-8 С раствор 10,0чг этил- М-( 3-пиридилметил)- -Н-(метоксикарбонилэтил)-карбамоилЗ -ацетата в 170 мл бензола. Смесь перемешивают при той же температуре в течение 2 ч. Далее смесь перегоняют для выделения растворителя и остаток растворяют в воде. Водный раствор подвергают экстракционной обработке бензолом и водный слой нейтрализуют 10%-ной соляной кислотой, после чего вновь подвергают экстракционной обработке хлороформом. Экстракт сушат и перегоняют для удаления растворителя, в результате чего в виде маслоподобного продукта получают
0
5
0
0
8,50 г (количественный выход) метил- -1-(3-пиридилметил)-2,4-диоксопипери- дин-3-карбоксилата.
Г. Смесь 8,50 г метил-1-(3-пиридил метил)-2,4-диоксопиперидин-З-карбо- ксилата, 100 мл ацетонитрила и 1,4мл воды кипятят с обратным холодильником в течение 1 ч. Эту смесь перегоняют для удаления растворителя, в ре- зультате чего получают 6,66 г (количественный выход) кристаллического 1-(3-пиридилметил)-2,4-диоксопипери- дина с т.пл. 55-60°С.
Д. 2,01 г 1-(3-пиридилметил)-2,4- -диоксопиперидина обрабатывают аналогично вышеизложенному в примере- 1В, в результате чего в виде желтых пластинчатых кристаллов получают 1,50 г (выход 50,1%) 1-(3-пиридилметил)-3- -(1,3-дитиол-2-илиден)-2,4-диоксопи- перидина, т.пл. 131,5-133°С.
Соединение, полученное выше, обрабатывают соляной кислотой, простым эфиром и получают соответствующий гидрохлорид в виде бледно-желтых игольчатых кристаллов, т.пл. 243,5 С (разлож., перекристаллизованный из метанола).
П р и м е р 20 А. 93 г анилина растворяют в 500 мл этанола и в этот раствор по каплям с охлаждением льдом добавляют 100 г этилацетата. Смесь перемешивают при 50 С в течение 3 ч, а затем кипятят с обратным холодишь- ником в течение 8 ч. Далее смесь перегоняют для удаления из нее растворителя. Остаток перегоняют под пониженным давлением, в результате чего получают 26,6 г N-фенил-бета-аланин- этилового эфира с т.кип. 119-120 С/ 2 мм рт.ст.
жидкость -, ПК-спектрограмма (-5 „„„ , см J:
МЭ.КС
1725.
Б. 24,45 г М-фенил-бета-аланинэти- лового эфира растворяют в 60 мл бензола, после чего в раствор добавляют по каплям при 70°С 12 мл дикетена. Смесь кипятят с обратным холодильником в течение ночи. Далее смесь перегоняют для удаления растворителя, в результате чего получают 37,23 г сырого N-фенил-М-метоксикарбонил- метилкарбонил-бета-аланинэтилового эфира. Указанный сырой продукт растворяют в 70 мл метанола и смесь добавляют по каплям в раствор метоксигJQ
15 20
25
30 -,с 0
5
0
да натрия (приготовленный с использованием 3,90 г натрия) в 70 мл метанола при 50 С. Смесь перемешивают при 50 С в течение 2 ч и перегоняют для удаления растворителя. Остаток растворяют в воде и водный раствор подвергают экстракционной обработке этилацетатом. Водный слой нейтрализуют 10%-ной соляной кислотой и подвергают экстракционной обработке этилацетатом. Экстракт промывают насыщенным водным раствором хлористого натрия, сушат, а затем перегоняют для удаления растворителя. Остаток очищают в хроматографической колонке с силикагелем с использованием в качестве растворителя смеси этилацетата с н-гексаном в соотношении 1:2, в результате чего в виде маслоподоб- ного продукта получают 23,87 г (выход 81,6%) 1-фенил-3-ацетил-2,4-ди- оксопиперидина.
В. 2,0 г 1-фенил-3-ацетил-2,4- -диоксопипериднна растворяют в 60 мл этанола и в этот раствор добавляют 30 мл 10%-ной соляной кислоты. Смесь кипятят с обратным холодильником в течение 7 ч. Далее эту смесь перегоняют для удаления растворителя, а полученный остаток растворяют в этил- ацетате. Раствор промывают насыщенным водным раствором хлористого натрия, сушат, а затем перегоняют для удаления растворителя, получая 1,12 г сырого 1-фенил-2,4-диоксопиперидина. 1,12 г указанного сырого соединения обрабатывают по способу, описанному в примере 3, и после чего перекрис- таллизовывают из смеси этилацетата с н-гексаном, получая в результате в виде желтых иглоподобных кристал- - лов 354 мг 1-фенил-3(1,3-дитиол-2- -илиден)-2,4-диоксопиперидина с т.пл. 150,5-152°С.
Пример21 А. Смесь 1,51 г п-нитробензальдегида, 1,40 г гидрохлорида бета-аланинметилового эфира, 1,6 мл триэтиламина и 20 мл метанола перемешивают при комнатной температуре в течение 25 мин. Эту смесь охлаждают льдом и добавляют в нее 807 мг боргидрида натрия. Далее смесь перемешивают при той же температуре в течение 10 мин. Смесь подкисляют 10%-ной соляной кислотой и подвергают экстракционной обработке этилацетатом. Водный слой нейтрализуют 10%-ной гидроокисью натрия
и подвергают экстракционной обработке этилацетатом. Экстракт промывают насыщенным раствором хлористого натрия, сушат, а затем перегоняют для удаления растворителя. Остаток растворяют в диэтиловом эфире и добавляют в этот раствор соляной кислоты в диэтиловом эфире. Осадок собирают, перекристаллизовывают из метанола с получением в виде бледно-желтых пластинчатых кристаллов 1,95 г (выход 71,1%) N-(п-нитробензил)-бета-ала- нинметилового эфира, гидрохлорида с т.пл, 219,5-220°С (с разложением).
Б. 32,66 г N-(п-нитробензил)-бе- та-аланинметилового эфира, гидрохлорида и 38 мл триэтиламина растворяют в 200 мл хлористого метилена и в этот раствор в условиях охлаждения льдом по каплям добавляют раствор 20,0 г этоксикарбонилхлорида в 100мл хлористого этилена. Смесь перемешивают при той же температуре в течение 1 ч, после чего эту смесь фильтруют для удаления осадка (триэтиламино- гидрохлорида). Фильтрат промывают 2%-ной соляной кислотой, 2%-ным раствором гидроокиси натрия и водой, сушат, а затем перегоняют для удаления растворителя, в результате чего в виде желтого маслоподо.бного продукта получают 40,4 г (выход 96,4%) этил- -Гн-(п-нитробензил)-М- етоксикарбо- нилэтил)-карбамоил -ацетата.
.„,, жидкость -(ч
ИК-спектрограмма (s ,см )
макс.
1735, 1650.
В. 690 мг натрия растворяют в 25 мл этанола, после чего в раствор по каплям добавляют раствор 10,0 г этил-LN -(п-нитробензил)-N-(метокси- карбонилэтил)-карбамошт -ацетата в 160 мл бензола при комнатной темпера туре. Смесь перемешивают при той же температуре в течение 1,5 ч. Эту смесь выливают в воду. Водный слой отделяют, нейтрализуют 10%-ной соляной кислотой и подвергают экстракционной обработке этилацетатом.Экст- ракт промывают насыщенным раствором хлористого натрия, сушат, а затем перегоняют для удаления растворителя в результате чего в виде желтого маслоподобного продукта получают 8,03 г (выход 88,4%) этил-1-(п-нитробензил) -2,4-диоксопиперидин-З-кар- боксилата.
, л жидкость -1.
ИК-спектрограмма ts , см )
макс.
1720, 1645.
Этил-1-(п-нитробензил)-2,4-диоксо пиперидин-3-карбоксилата обрабатывают аналогично вышеизложенному в примере 19 Г, в результате чего в виде сырого продукта получают 608 мг (количественный выход) 1-(п-нитробен- зил)-2,4-диоксопиперидина.
Mace-спектрограмма 243 (M 562 мг полученного таким образом продукта обрабатывают аналогично вышеизложенному в примере 3, в результате чего в виде желтых иглоподобных кристаллов получают 446 мг (выход 56,9%) 1-(п-нитробензил)-3-(1,3-ди- тиол-2-илиден)-2,4-диоксопиперидина с т.пл. 192-193°С (после перекристаллизации из этилацетата).
вазелин овое
масло
ИК спектрограмма (
макс
см 1): 1620, 1580.
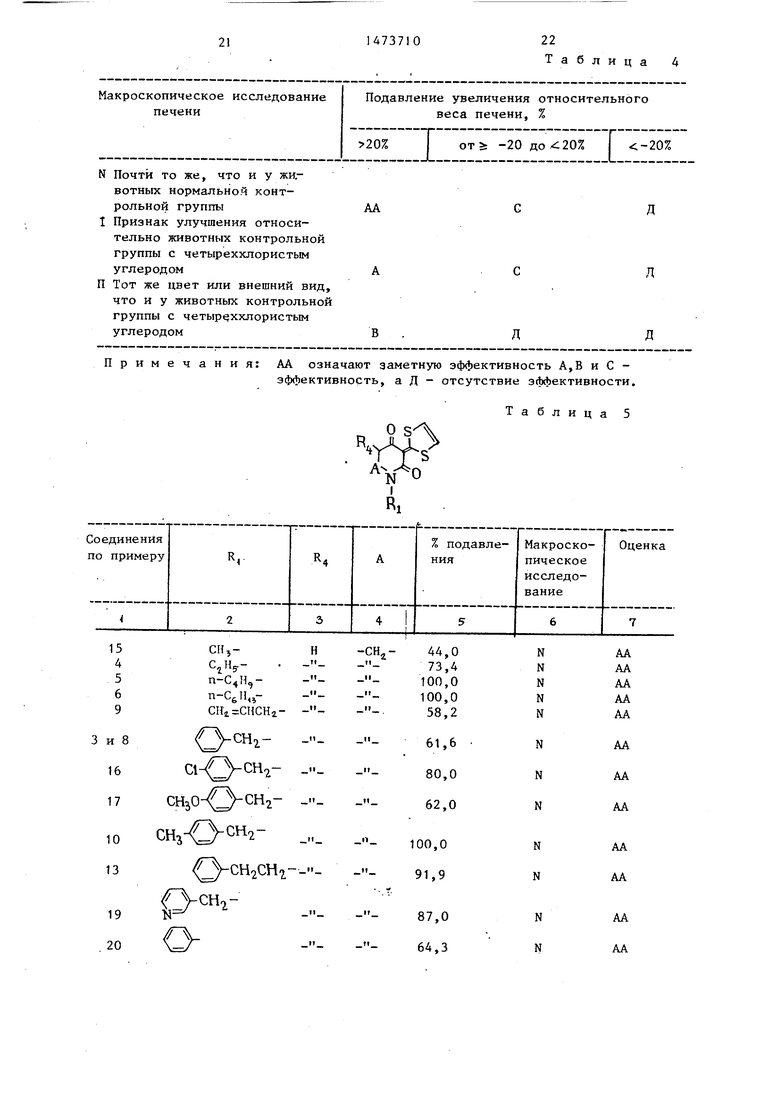
Примеры 22-26. Из соответствующих исходных соединений способом, описанным в примере 21, получают соединения, свойства которых приведены в табл.3.
Пример 27. 100 мг (п-нитробензил) -3-(1,З-дитиол-2-илиден)-2,4- -диоксопиперидина растворяют в 20 мл этанола и в этот раствор добавляют 100 мл 10%-ного палладия на угле. Смесь подвергают каталитическому восстановлению под атмосферным давлением. Далее эту смесь фильтруют для удаления катализатора. Фильтрат перегоняют для удаления растворителя, в результате чего в виде кристаллов получают 92 мг (количественный выход) 1-(п-аминобензил)-3-(1,3-ди- тиол-2-илиден)-2,4-лиоксопиперидина с т.пл. 156,5-158,5°С (после перекристаллизации из этанола).
Гидрохлорид; бледно-желтые игло- подобные кристаллы, т.пл. 212°С (с разложением, после перекристаллизации из смеси метанола с диэтило- вым эфиром).
П р и м е р 28 А. 1,01 г этил- м- -(п-нитробензил)-N-(метоксикарбонил- этил)-карбамогаГ -ацетата,полученного по примеру 21 Б, 10 мл этанола и 150 мг 10%-ного палладия на угле обрабатывают аналогично изложенному в примере 27, в ре/ 5
10
зультате чего в виде бледно-желтого маслоподобного продукта получают 810 мг (выход 87,5%) этил к-(п-ами- нобензил)-1 -метоксикарбонилэтил)- -карбамоил ацетата.
Б. Раствор 539 мг бензилоксикар- бонилхлорида в 1,5 мл бензола по каплям добавляют в смесь 810 мг этил- -Г;;т-(п-амннобензил)-И-(метоксикарбо- нилэтил)-карбамоил -ацетата с 595 мг карбоната калия, 10 мл бензола и 10 мл воды при 8 С с одновременным перемешиванием. Далее смесь перемешивают при той же температуре в тече- 15 ние 30 мин, затем ее подвергают экстракционной обработке этилацетатом и экстракт промывают водой, сушат, а затем перегоняют для удаления растворителя. Остаток очищают в хроматогра- 20 фической колонке с силикагелем с использованием в качестве элюента этил- ацетата с бензолом в соотношении 3:7, в результате чего в виде бесцветМасс-спектрограмма т/еЗ:352.
660 мг полученного таким образо продукта обрабатывают аналогично и ложенному в примере 3, в результат чего в виде бледно-желтых пластинч тых кристаллов получают 518 мг (вы 62,0%) 1-Јл-(N-бензилоксикарбонил- амино)-бензил -3-(1,З-дитиол-2-или ден)-2,4-диоксопиперидина с т,пл.1 176,5°С (после перекристаллизации из этилацетата).
П р и м е р 29. 100 мг (N -бензилкарбамоиламино)-бензил 1 -3- -(1,3-дитиол-2-илиден)-2,4-диоксоп перидина растворяют в 2 мл уксусно кислоты и в этот раствор добавляют 0,5 мл 25%-ного раствора бромистог водорода в уксусной кислоте. Смесь перемешивают при комнатной темпер туре в течение 40 мин, а затем при 40-50 С в течение 1 ч. После охлаж дения смеси в нее добавляют диэтил вый эфир. Выпавшие в осадок крист
ного маслоподобного продукта получа- 25 лы собирают фильтрованием и пере30
ют 1,08 г (выход 94,2%)этил-JN n- -(N-бензоилоксикарбониламино)-бензил - -Н-(метоксикарбонилэтил)-карбамоил1 - -ацетата.
В. Раствор 967 мг этил-JN -Јn-(N- -бензилоксикарбониламино)-бензил - М-(метоксикарбонилэтил)-карбамогоп - -ацетата в 15 мл бензола при комнатной температуре добавляют по каплям в раствор этоксида натрия, приготовленный с использованием 60 мг натрия и 2 мл этанола. Приготовленную смесь перемешивают при той же температуре в течение 30 мин. Далее ее выливают в воду. Водный слой нейтрализуют 10%-ной соляной кислотой и подвергают экстракционной обработке этилацетатом. Экстракт промывают насыщенным раствором хлористого натрия, сушат, а затем перегоняют для удале-45 ния растворителя, в результате чего в виде бесцветного маслоподобного продукта получают 841 мг (выход 93,5%) этил-1- п-(М-бензилоксикарбонилами- но)-бензил -2,4-диоксопиперидин-З- -карбоксилата.
35
40
50
кристаллизовывают из смеси метанол с диэтиловым эфиром, в результате чего в виде бледно-желтых иглоподо ных кристаллов получают 70 мг (вых 79,3%) 1-(п-аминобензил)-3-(1,3- -дитиол-2-илиден)-2,4-диоксопипери дин гидробромида с т.пл. 210-214°С (с разложением).
Фармакологические испытания. Эксперимент 1. Защита от остро гепатитного повреждения, вызываемо го действием четыреххлористого угл рода.
Методика. Испытываемые соединен суспендируют в 0,5%-ном растворе ка оксиметилцеллюлозы и суспензию (с с держанием испытываемого соединения 100 мг/10 мл/кг) через рот вводят самцам мышей разновидности dd у (возраст 5-6 недель, масса 25-30 г состав каждой группы: 3 особи), по ле чего животным не дают корма. По истечении 3 ч животным через рот вводят раствор четыреххлористого у лерода в оливковом масле в дозировке по 50 мкл/5 мл оливкового масла По истечении 3 ч через рот животным вводят испытываемое соединение в той же дозировке, что указана выше Массу животных измеряют по истечени 24 ч после введения четыреххлористо го углерода, после чего животных убивают. Сразу же после этого у них удаляют печень, взвешивают ее и под
Г. 823 мг этил-1- л-(М-бензилокси- карбониламино)-бензил -2,4-диоксопи- перидин-3-карбоксилата обрабатывают „ аналогично примеру 19 Г, в результате чего в виде сырого продукта получают 693 мг (М-бензилоксикарбонил- амино)-бензил -2,4-диоксопиперидина.
0
5 0
Масс-спектрограмма т/еЗ:352.
660 мг полученного таким образом продукта обрабатывают аналогично изложенному в примере 3, в результате чего в виде бледно-желтых пластинчатых кристаллов получают 518 мг (выход 62,0%) 1-Јл-(N-бензилоксикарбонил- амино)-бензил -3-(1,З-дитиол-2-или- ден)-2,4-диоксопиперидина с т,пл.175- 176,5°С (после перекристаллизации из этилацетата).
П р и м е р 29. 100 мг (N- -бензилкарбамоиламино)-бензил 1 -3- -(1,3-дитиол-2-илиден)-2,4-диоксопи- перидина растворяют в 2 мл уксусной кислоты и в этот раствор добавляют 0,5 мл 25%-ного раствора бромистого водорода в уксусной кислоте. Смесь перемешивают при комнатной температуре в течение 40 мин, а затем при 40-50 С в течение 1 ч. После охлаждения смеси в нее добавляют диэтило- вый эфир. Выпавшие в осадок кристал0
5
5
0
0
кристаллизовывают из смеси метанола с диэтиловым эфиром, в результате чего в виде бледно-желтых иглоподоб- ных кристаллов получают 70 мг (выход 79,3%) 1-(п-аминобензил)-3-(1,3- -дитиол-2-илиден)-2,4-диоксопипери- дин гидробромида с т.пл. 210-214°С (с разложением).
Фармакологические испытания. Эксперимент 1. Защита от острого гепатитного повреждения, вызываемого действием четыреххлористого углерода.
Методика. Испытываемые соединения суспендируют в 0,5%-ном растворе карб- оксиметилцеллюлозы и суспензию (с содержанием испытываемого соединения 100 мг/10 мл/кг) через рот вводят самцам мышей разновидности dd у (возраст 5-6 недель, масса 25-30 г, состав каждой группы: 3 особи), пос- ле чего животным не дают корма. По истечении 3 ч животным через рот вводят раствор четыреххлористого углерода в оливковом масле в дозировке по 50 мкл/5 мл оливкового масла. По истечении 3 ч через рот животным вводят испытываемое соединение в той же дозировке, что указана выше. Массу животных измеряют по истечении 24 ч после введения четыреххлористого углерода, после чего животных убивают. Сразу же после этого у них удаляют печень, взвешивают ее и подвергают макроскопическому исследованию. Что касается животных контрольной группы, то вместо суспензии испытываемого соединения и раствора четыреххлористого углерода им вводят 0,5%-ный раствор карбоксиметил- целлюлозы и оливковое масло (перо- рально). Кроме того, используют контрольную группу животных, которым вво- дят раствор четыреххлористого углерода и 0,5%-ный раствор карбоксиме- тилцеллюлозы.
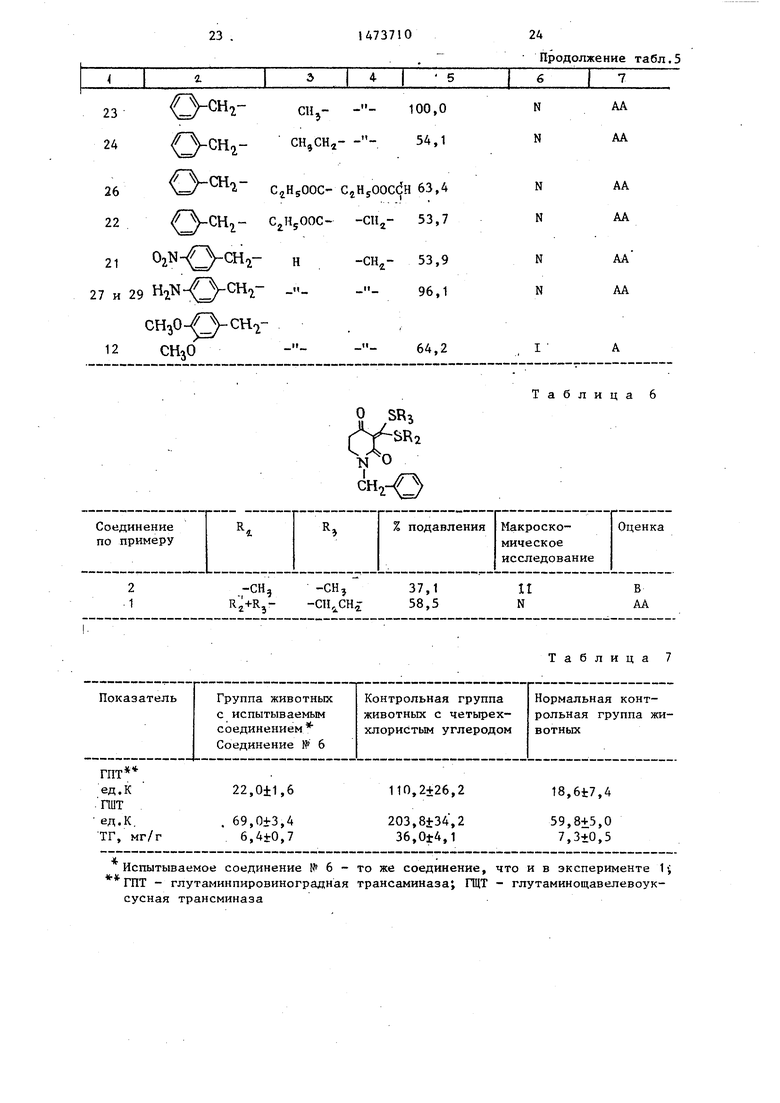
Терапевтическое действие испытываемых соединений на эффект повреж- дения печени оценивают как степень подавления в процентах относительного увеличения веса печени, рассчитанного с помощью нижеследующего уравнения и основанного на макроскопи- ческом исследовании печени (термин относительный вес печени обозначает вес (в граммах) печени/100 г живого веса животного).
Подавпение в процентах увеличения относительного веса печени:
Средний относитель-Средний отноный вес печени всительный вес
группе животных,печени у живот-
получивших испыты-ных нормальной
ваемое соединениеконтрольной
группы j о
Средний относитель- Средний относи- ный вес печени у тельный вес пе- животных контроль- чеки у животных ной группы, полу- нормальной конт чивших четыреххло- рольной группы ристый углерод
Критерии оценки действия соединений приведены в табл.4.
Результаты эксперимента приведены в табл.5 и 6.
Эксперимент 2. Действие против острого гепатитного повреждения, вызванного четыреххлористым углеродом.
Методика. Раствор четыреххлористого углерода в оливковом масле подкожно вводят самцам крыс разновидности SD/(возраст 6 недель, масса 170- 210 г, состав группы: 5 особей) в дозировке 1 мл/кг (четыреххлористый углерод: 5 мг/кг) по одному разу в течение 4 дней подряд. В то же время через рот животным вводят суспензию испытываемого соединения в 0,5%-ном растворе карбоксиметилцеллюлозы по одному разу в течение четырех дней
(испытываемое соединение: 100 мг/ 10 мг/кг). Спустя 24 ч после последнего введения в организм суспензии испытываемого соединения из нижней полой вены животных собирают кровь. Непосредственно после этого удаляют печень. Из крови выделяют плазму и определяют активность ГПТ и ГЩТ в плазме. Определяют также содержани триглицерида (ТГ) в печени. Животным нормальной контрольной группы вместо раствора четыреххлористого углерода и суспензии испытываемого соединения дают оливковое масло, 0,5%-ный раствор карбоксиметилцеллю- лозы. Кроме того, животным контрольной группы с четыреххлористым углеродом дают раствор четыреххлористого углерода и 0,5%-ный раствор карб- oKCHMefилцеллюлозы.
Результаты этих испытаний приведены в табл.7.
Свойства приведены в табл.8. Формула изобретения
Способ получения тиокетеновых производных пиперидина общей формулы I
О SR3
yV-sfy
AN 0 I
R
где Rj - водород, С4-С,0-алкил,
С -Сц-алкенил, фенил или группа общей формулы -B-Y, где Y - пиридил, фенил или фенил, имеющий один или два заместителя, выбранных из галогена, Cf-С -алкил, С С -алкокси, нитро, амино и бензилоксикарбониламино, В - С,| -С -алкилен; R4 и R} - С4-Сф-алкил каждый либо
вместе образуют группу формулы или , Кц. - водород, С,-С4-алкил или
С -С -алкоксикарбонил, А - группа формулы -СНа- или
-CH(COORS)-, где R,-- С,-С4- -алкил,
или их кислотно-аддитивных солей, отличающийся тем, что,соединение общей формулы II
Ч1
RI
171
где R,,R4 и А имеют указанные значения,
подвергают взаимодействию с сероуглеродом и соединением, выбранным из С -С тал оидалкил а, 1,2-дигалоид- этана и 1,2-дигалоидэтилена, и целевой продукт выделяют или когда R - водород, соединения общей формулы I подвергают взаимодействию с соедине- нием общей формулы R6-X, где X - галоген; Rg-C -Сед-алкил, С4-С4-алкэ- нил, фенил или группа общей формулы - B-Y, где В и Y имеют указанные зна- че ния, или восстанавливают соединение формулы I, 1-де R, - группа формулы В - Y, где Y - нитрофенил, а В имеет указанные значения, или удаляют бензилоксикарбонильную группу в соединении общей формулы I, когда
О
х
N °
10
18
R - группа формулы В -Y, где Y - бензилоксикарбониламинофенил, В имеет указанное значение, и выделяют целевой продукт или соединение формулы I,- где R - группа; B-Y, где Y - пиридил или аминофенил, обрабатывают кислотой и выделяют целевой продукт в виде кислотно-аддитивной соли. Приоритет по признакам:
10
14.01.84 при R f-Н, С4-С10-алкил, С2-С4-ал- кенил;В -С,-С4 алкилен, Y -фенил, содержащий один или два
15заместителя,выбранные из галогена, С4-С -алкила, -алкокси; R и Rj-CH CH-, или каждый из них. С -Сф-алкил; R4- H;A--CH -.
20 11.04.84 при R4- алкоксикарбонил. Q $sbТаблица
-й
N О









Изобретение относится к гетероциклическим соединениям , в частности, к способу получения тиокетеновых производных пиперидина формулы 1 CHR4-C(O)-C(M)-C(O)-NR1-A при M-ΣR3, где R - водород, C1-C10-алкил, C2-C4 - алкенил, фенил или группа формулы В-У, где У-пиридил, фенил или фенил, имеющий один или 2 заместителя, выбранных из галогена, C1-C4 - алкил, C1-C4 - алкокси, нитро, амино и бензилоксикарбониламино
B-C1-C4 -алкилен
R2 и R3-C1-C4 - алкил, каждый либо они вместе образуют группу формулы CH2CH2- или -CH=CH-
R4 - водород, C1-C4- алкил или C1-C4- алкокси карбонил
A-группа формулы -CH2- или -CH(COOR5)-, где R5-C1-C4- алкил, или их кислотно-аддитивных солей, которые обладают гепатозащитными свойствами. Получение целевых соединений ведут введением группы M в соответствующие производные пиперидина при взаимодествии их с сероуглеродом и соединением, выбранным из C1-C4-галоидалкила, 1, 2-дигалоидэтана и 1, 2-дигалоидэтилена с выделением целевого продукта, или когда R1-H, соединение 1 подвергают взаимодействию с соединением R6-X, где X-галоген
R6-C1-C10- алкил, C1-C4- алкенил, фенил или группа формулы В-У, где В и У имеют указанные значения или восстанавливают соединение 1, где R1- группа формулы В-У и У - нитрофенил, а В- указано выше, или удаляют бензилоксикарбонильную группу в соединении 1, когда R1- группа формулы В-У, где У- бензилоксикарбониламинофенил, а В указано выше, и выделяют целевой продукт, или соединение 1, где R1 - группа В-У, где У- пиридил или аминофенил, обрабатывают кислотой и выделяют целевой продукт в виде кислотноаддитивной соли. 8 табл.
н-Гексил
н-Октил
н-Децил
Алл ил
10
4-Метил бензил
11
3,4-Ди- хлорбензил
12
3,4-Димет- оксибенэил
с т.пл. 102-104 С (после перекристаллизации из смеси этилацетата с н-гек- саном)
Бледно-желтые иглоподобные кристаллы с т.пл. 88-89,5°С (после перекристаллизации из смеси этилацетата с н-гексаном) Бледно-желтые иглоподобные кристаллы с т.пл. 86-87,5°С (после перекристаллизации из смеси этилацетата с н-гексаном) Бледно-желтые иглоподобные кристаллы с т.пл. 93,5-94,5°С (после перекристаллизации из смеси этилацетата с н-гексаном)
Бледно-желтые иглоподобные кристаллы с т.пл. 133-134,5°С (после перекристаллизации из смеси этилацетата с н-гексаном)
Бледно-желтые иглоподобные кристаллы с т.пл. 137-138°€ (после перекристаллизации из изопропилового спирта)
Бледно-желтые иглоподобные кристаллы с т.пл.175-176,5°С (после перекристаллизации из этилацетата)
ледно-желтые иглоподобные кристаллы с т.пл. 195-196,5 С (после перекристализации из смеси.этилацетата с н-гекса- ом) .
ПримерR4 I Свойства
164-Хлорбензил Бледно-желтые иглоподобные кристаллы с
т.пл. 143-149°С (после перекристаллизации из этанола) .174-Метоксибензил Бледно-желтые иглоподобные кристаллы
с т.пл; 160-162°С (после перекристаллизации из смеси этилацетата с изопропи- ловым эфиром)
„....в,я,.и.......«..«.... .
Таблица 3 О S
VSV
I
Rt .
Пример R4 R4 Свойства
22 - CH / COOC-)Ht; CH2 Бледно-желтые игольчатые кристаллы
(перекристаллизов. из н-гексанаэтилацетата) т/пл. 127-129° С
йк (см(): 1730, 1630, 1595
23
-сн2-О сн3
-сн,
24
-СН,
-снгсн,
-CIL
25 -СН2 СООСН3 СИ2
N
26
//Л
°н7- )-соос2н5 -сисоосан.
Таблица 2
Бледно-желтые игольчатые кристаллы (пе- кристаллиз. из н-гексана-этилацетата) т.пл. 168-170°С
ИК Пкс (): 1625, 1585
Бледно-желтые игольчатые кристаллы (перекристаллиэ. из н-гексана-этилацетата) т.пл. 139,5-141°С
нк So 1625 159° Бледно-желтые игольчатые кристаллы
(перекристаллиз. из н-гексана-этилацетата) Свободное основание:
ИК-)
купол wane
-1
():
Гидрохлорид:
желтые игольчатые кристаллы т.пл.204205,5 С (перекристаллиз. из метанола)
-4
(см): 1735, 1630, 1600.
Почти то же, что и у животных нормальной контрольной группыАА
Признак улучшения относительно животных контрольной группы с четыреххлористым углеродомА
Тот же цвет или внешний вид, что и у животных контрольной группы с четыре.ххлористым углеродомВ
Примечания:
АА означают заметную эффективность А,В и С - эффективность, а Д - отсутствие эффективности.
Д
Д
Д
Д
лЛ
Таблица 5
24
26 22
21
, ,1
Nsr/ CH i сгн5оос- сгн5оосс н 63,4
C2HSOOC- -CH2- 53,7
02N Q-CH2- н-CH,- 53,9
..
96,1
7 и 29 H - Q-CH-T ....
CH30
12
n
64,2
iy
fV-bRa
Vo Я-О
П-.-.у- г-J
ГПТ - глутаминпировиноградная трансаминаза ПЦТ - глутаминощавелевоук- сусная трансминаза
Продолжение табл.5
AA
AA AA
AA AA
n
64,2
Таблица 6
Таблица 7
SCH3
22,
SCH3
23 LSCH3
VS
24
U4g
N S-J
25
26
27
Таблица -8
-7,5
И
-28,3
II
D
11,0
II
7,1
II
D
-36,4
II
-55,1
II
D
| Мищенко Г.Л., Вацуро К.В | |||
| Синтетические методы органической химии | |||
| М.: Химия, 1982, с | |||
| Способ приготовления искусственной массы из продуктов конденсации фенолов с альдегидами | 1920 |
|
SU360A1 |
| Bull Boc | |||
| Chim | |||
| Fr, 1968, 4477. | |||
