31505
в виде 1-изомера или смеси d- и1-изо меров, где Y и А имеют указанные значения;
L - йод или оксигруппа в виде
соли с п-толуолсульфокислотой подвергают взаимодействию с азидом щелочного металла при нагревании в среде инертного органического растворителя и целевой продукт, где В - азидогруппа, выделяют или, в случае необходимости, переводят в целевой ;: продукт, где В - аминогруппа, восстановлением и целевой продукт, где В - аминогруппа, в случае необходимости подвергают взаимодействию с кансульфонилхлоридом или с ангидридом, хлорангидридом или сложным эфиром карбоновой кислоты формулы
RjCOOH
где RJ имеет указанные для Rj значения, кроме аминометил или азидометил, и целевой продукт, где Rg - хлорме- тил, в случае необходимости подвергают взаимодействию с азидом щелочного металла при нагревании в среде инертного органического растворителя и це- левой продукт, где R - азидометил, в случае необходимости переводят в це
0
5
0
левой продукт, где R, - аминометил, восстановлением и, в случае необходимости, при наличии свободной аминогруппы целевой продукт вьщеляют в виде аддитивной соли с кислотой. Приоритет по признакам:
07.06.83при Y - водород; А находится в положении 4 бензольного кольца и означает С,-С,}.-алкилтиогруп- пу, С(-С4-алкилсульфинил или С, алкилсульфонил; В имеет указанные значения.
14.02.84при Y - водород; А находится в положении 3 бензольного кольца и означает С -С -ацил, С, алкил, метокси-, метилтио, трифтор- метил или нитрогруппу, или А находится в положении 4 бензольного кольца
и означает С -С -алкил, необязатель- но замещенный 1-3 атомами галогена, С -С -алкенил, циано-, С,-С -алкок- СИ-, группу формулы COR,, в которой водород, С,-С4.-алкил или цикло- гексил, или группу формулы
«к,
-С-СНз
где rt. 2 гидрокси- или метоксигруп- па, или Y и А всеете образуют 3,4- метилендиоксигруппу, В имеет указанные значения.





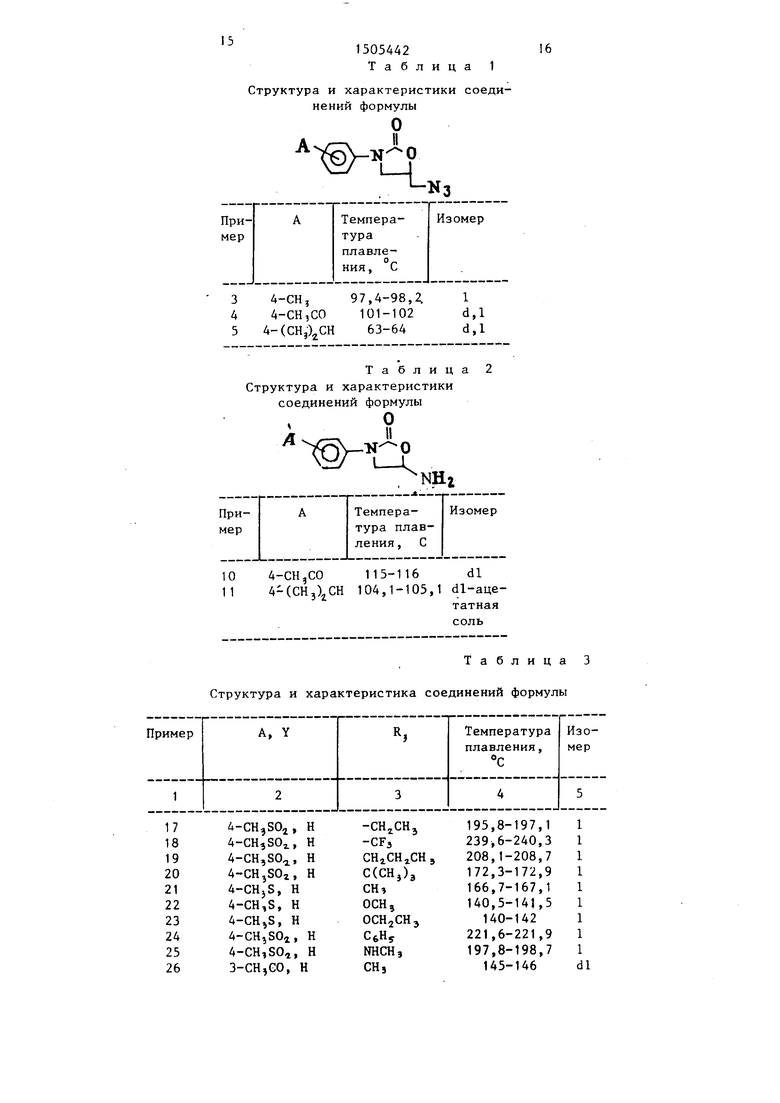





Способ получения производных оксазолидинона в виде 1-изомеров или смеси D- и 1-изомеров, или их аддитивных солей с кислотами.
Изобретение относится к способу получения новых производных оксазоли- динона в виде 1-изомеров или смеси d- и и1-изомеров, или их аддитивных солей с кислотами, которые обладают антибактериальным действием и могут найти применение в медицине.
Цель изобретения - создание способа получения новых производных ок- сазолидинона, обладающих антибакте- риаль}1ым действием с увеличенной производительностью действия ин виво и ослабленными побочными эффектами.
Примеры, приведенные ниже, иллюст рируют изобретение.
В нижеследующих примерах значения А и В приведены для соединений формулы :
J.
40
5
-50
55
Пример 1. Получение (dl)-5- азидометил-3- 4-(метилсульфонил)фенил -2-оксазолидинона (I, В Н,).
Часть А.
Получение (dl)-5-йoдмeтил-3- 4- (метилсульфонил)фенил 2-оксазолицино- на.
Смесь 50 г (345 ммоль) (dl)-5- хлорметил-3- 4-(метилсульфонил)фенил 2-оксазолидинона и 100 г йодистого натрия в 300 мл 2-бутанола кипятят с обратным холодильником в течение ночи.. Затем ее охлаждают и выливают в 1 л смеси воды со льдом, сульфат натрия добавляют до полного исчезновения желтой окраски, смесь фильтруют и промывают водой с получением 61,7 г .1йодметильного соединения t т.пл. 175,5-177 С. Этот материал далее перекристаллизовывают из 370 мл ацетонитрила, в результате чего
получают 44,8 г продукта с т.пл. 177,5-179°С.
Часть В.
Смесь 7,6 г (20 ммоль) (dl)-5-fiOA метил-3- 4-(метилсульфонил)фенилТ-2- оксазолидинона и 4 г азида натрия в 150 мл сухого ДМАЦ выдерживают при 125°С в течение 3 ч. Затем ее выпивают в смесь воды со льдом.Про- дукт экстрагируют тремя порциями хлороформа,экстракты сушат над сульфатом натрия и концентрируют, получая полутвердую пасту, которую перемешивают с диэтиловым эфиром,фильт руют и сушат с получением 4,7 г материала. Последний перекристаллизовы- |Вают из 14 мл ацетонитрила, в результате чего получают 2,2 г азидометило вого соединения с т.пл. 152 , 3-153 , 5
Пример 2. Получение (1)-5- азидометил-3- 4- (метилсульфонил)- фенил -2-оксазолидинона (I; А 4 метил-SO ; В
N,).
Часть А.
Получение (1)-3-оксиметил-3- 4(метилсульфонил )-фeнил -2-oкcaзoлидин нa, 4-метилбензолсульфо 1ата (I; А 4-метил-50,, ; В OSO СеН -метип) .
Раствор 5,00 г (dl)-5-oкcимeтил- 3- L4- (метилсульфонил )фенил | -2-окса- золицинона в 30 мл сухогт пиридина перемешивают при и постепенно добавляют в него раствор 3,7 г п-то- луолсульфонилхлорида в 10 мл П1фиди- на. После добавления смесь перемеип - вают в тече}{ие еще 1 ч, а затем кристаллизуют до полутвердой массы, в которую добавляют несколько капель воды, в результате чего выделяется тепло. Смесь вьшивают в смесь воды со льдом, фильтруют и промывают водой. В результате получают 4,02 г продукта с т.пл. 187,1-188,6°С.
Часть В.
Смесь 3,5 г (1)-5-оксиметил-3- 4- (метилсульфоНИЛ)фенил -2-оксазолиди- нона, 4-метилбензолсульфоната, с 2 г азида натрия в 20 мл ДМФ нагребают до 90-100°С. Через 1 ч смесь охлаждают и разбавляют смесью воды со льдом, в результате чего продукт кристаллизуется. Затем его фильтруют и тш;ательнс промывают водой, бпагоца ря чему получают 1,25 г вещества с т.пл. 146,5-148,5°С. Продукт кристаллизуют из метанола с получением вещества с т.пл. 148,9-149,4 С.
Q 15 20
5
0
5
0
5
0
5
В соответствии с процедурами, которые описанг. в npHMtipax 1-2 получают азиды пpи fepoв 3-5, приведенные в табл. 1.
Пример 6. Получение (dl)-5- ам11нометил-3- 4-(метилсульфонш1)фе- нил -2-оксазолидиновой соли трифтор- уксус1гой кислоты (А 4-СН ) ; В - NHj.).
Раствор 1,1 г (dl)-3-азидометил-3- 4- (метилсульфонил)фенил 1 -2-окслзо- лидинона в 75 мл трифторуксусной кислоты и 0,5 г 10%-ного палладия на древесном угле встр)т 1вают в водороде при избыточном давлении приблизительно 50 (Ьунтов/кв . , 3,5 КГ/КБ,см в течение 1 ч. Смесь далее фильтруют и концентрируют, в результате чего получают 0,8 г продукта с т.пл. 158- 170°С (с разложен1 ем) .
Пример 7. Получе1пте С1)-5- aNniHOMeT.4n-3- 4- (метилсульфонпл)фе- Hiij J-2-оксазолидинома (I; Л 4-ме- тил-SO ; В f. Hj),
Смесь 3,48 г (0,0117 моль) (1)-5- aзи;ш eтIгл-3- 4- (мети.чсу, 1ьфонил)фс- HKnj-2-i ксазо.чи,,и:юня, 11 t-vi 1,3-про- пан 1итиола и 15 мл триэти гл -шна в 30 мл метанола ni uepxaiBaiOT при 40- 5П С во время 15Ы,|;йлепич азота с ощутимой скоростью.После прекраисния вы- де:геилч азота раствор кон ;е: трирук Т под по)1иженньп-( давлением, остаток перемешивают с диэтилг-пым эфиром, ч вердьгй материал отфильтровывают и сушат, вследствие чег О получают 3,09 г вещества с т.пл. 137-142 С. Материал раствс-ряют приблизительно в 200 мл абсолютированного спирта при температуре деф1пегмации (некоторое количество коричневого твердого про- дук-Га остается нррагтроривш1гмся) и фильтруют. Продукт кристаллизуют с получением 2,46 г вещества с т.пл. 146,6-147,1°С.
Пример 8. Получение (1)-5- аминометил-3- 4- (метилсульфонил)фенил - 2-оксазолидинона (I; А 4-метиг1-50 ; В NHj) .
2,00 г (6,75 ммоль) (1)-5-азидо- метил-3- (метилсульфонил)фенил1 -2- оксазолидинона в 25 мл 1,2-диметокси- этана перемешивают в токе азота, добавив к ней 3,2 мл триметилфосфита в 5 мл 1,2-диметоксиэтана. Смесь теплеет и при этом наблюдается быстрое выделение азота. Затем смесь концентрируют с получением коричневой
смолы. Смолу перемешивают с водой, при этом твердый материал кристаллизуется. Его растворяют в воде, добавив разбавленной уксусной кислоты до величины рН, равной 4, фильтруют и воду подщелачивают добавлением концентрированного раствора гидрата окиси аммония. Получают 0,94 г продукта с т.пл. 129-132,8 С.
Пример 9. Получение (1)-5- аминометил-3- 4-(метилтио)фенил -2- оксазолидинона (I; А 4-метил-5; В амино).
Смесь 30,3 г (115 ммоль) (1)-5- азидометил-3- 4-(метилтио)фенил -2- оксазолидинона, 13,1 мл 1,3-пропан- дитиола и 18,2 мл триэтиламина в 150 мл метанола перемеишвают при
10
15 N
нием, перемешивают совместно с диэти ловым эфиром и фильтруют, получив 2,72 г материала с т.пл. 174,0 - 181,. Материал перемешивают с водой, подкисЛяют уксусной кислотой, фильтруют и промывают водой, получив 2,60 г материала с т.пл. 194,5 - 196,1°С. Его растворяют в кипящей смеси 70% этанола с водой, подкисляют уксусной кислотой, охлаждают и фильтруют, получив 1,65 г продукта с т.пл. 203,3-204,3°С.
Вычислено, %: С 40,95; Н 3,70; 7,35.
C,,H,C1.,,S
Найдено, %: С 40,82; Н 3,70; 7,10; 7,15. Пример 14. Получение (1)-N50°С в течение 8 ч. Затем ее концен- 20 (метилсульфонил)фенил -2-оксотрируют, остаток перемешивают с водным раствором лимонной кислоты, фильтруют и фильтрат подщелачивают добавлением концентрированного раствора гидрата окиси аммония. Жидкость фильтруют, получив 16,5 г продукта с т.пл. 160-162 С.
В соответствии с процедурами примеров 6-9 получают нижеследующие лг-и- амины примеров 10-11, приведеин -е в табл. 2.
30
оксазолидин-5-илметилj-ацетамида (I; А 4-метил-802;; -В NHCOCHj). 2,00 г (7,4 ммоль) (1)-5-аминоме- тил-3- 4-(метилсульфонил)фенил -2- 25 оксазолидинона в 10 мл пиридина охлаждают в смеси воды со льдом, доЪав- ляя 0,72 мл уксусного ангидрида. Смесь далее перемешивают в течение 10-20 мин, а затем разбавляют смесью воды со льдом. Твердый материал отфильтровывают и промывают водой, получают продукт с т.пл. 191,9 - 192,9°С. После перекристаллизации из ацетонитрила получают 1,01 г продукта с т.пл. 192,7-193,2 С.
П р и м е р 12. Получение (1)-N- (З- 4- (метилсульфонил)фенил 1 -2-оксо- оксазолидин-5-илметил -формамида (I; А 4-метил-30 ; В NHCHO).
Раствор 1,00 г (3,70 ммоль) (1)-5- аминометил-3- 4-(метилсульфонил)фе- нил 1-2-оксазолидинона в 10 мл 2-про- панола, содержа1ций 2,5 мл этилформи- ата, выдерживают при температуре кипения с обратным холодильником в течение 24 ч. Далее смесь охлаждают и разбавляют диэтиловым эфиром с получением 0,96 г материала, который кристаллизуют из 9,5 мл ацетонитри- ла, в результате чего получают 0,65 г продукта с т.пл, 190-191,6 С.
Пример 13. Получение (1)- 2,2-дихло -К- (метилсульфонил)
оксазолидин-5-илметилj-ацетамида (I; А 4-метил-802;; -В NHCOCHj). 2,00 г (7,4 ммоль) (1)-5-аминоме- тил-3- 4-(метилсульфонил)фенил -2- 25 оксазолидинона в 10 мл пиридина охлаждают в смеси воды со льдом, доЪав- ляя 0,72 мл уксусного ангидрида. Смесь далее перемешивают в течение 10-20 мин, а затем разбавляют смесью воды со льдом. Твердый материал отфильтровывают и промывают водой, получают продукт с т.пл. 191,9 - 192,9°С. После перекристаллизации из ацетонитрила получают 1,01 г продукта с т.пл. 192,7-193,2 С.
Вычислено, %: С 49,99; Н 5,16; N 8,97.
Навдено, %: С 49,482; Н 5,17; N 8,93; 8,88.
Пример 15. Получение Cl)N- I 3-(4-метилсульфонил)фенил1 -2-оксо- оксазолидин-5-илметил7-метансульфон- амида (I; А 4-метил-30 ; В метил).
Раствор 1,00 г (3,70 ммоль) (L)-5- аминометил-3- 4-(метилсульфонил)фенил -2-оксазолидинона в 50 мл сухого пиридина перемешивают на ледяной ба35
40
фенил -2-оксооксазолидин-5-илметил - 50 постепенно добавляя в него 2,3мл
-ацетамида (I; А 4-метил-50,, ; В NHCOCnClj) .
Смесь 2,00 г (7,4 ммоль) (1)-5- аминометил-3- 4-(метилсульфонил)фе- нил1-2-оксазолидинона, 2 мл метилди- хлорацетата и 10 мл этанола кипятят с обратным холодильником в токе азота в течение 5 ч. Затем эту смесь концентрируют под пониже)шым давлеметансульфонилхлорида. После завершения операции добавления в этот раствор добавляют 3 капли воды и всю массу концентрируют. Остаток переме- сг шивают с Водой и добавляют в смесь несколько капель концентрированной соляной кислоты до придания всему раствору кислой реакции. Выпавший в осадок материал фильтруют, промывают
N
нием, перемешивают совместно с диэти- ловым эфиром и фильтруют, получив 2,72 г материала с т.пл. 174,0 - 181,. Материал перемешивают с водой, подкисЛяют уксусной кислотой, фильтруют и промывают водой, получив 2,60 г материала с т.пл. 194,5 - 196,1°С. Его растворяют в кипящей смеси 70% этанола с водой, подкисляют уксусной кислотой, охлаждают и фильтруют, получив 1,65 г продукта с т.пл. 203,3-204,3°С.
Вычислено, %: С 40,95; Н 3,70; 7,35.
C,,H,C1.,,S
Найдено, %: С 40,82; Н 3,70; 7,10; 7,15. Пример 14. Получение (1)(метилсульфонил)фенил -2-оксо0 (метилсульфонил)фенил -2-оксо0
оксазолидин-5-илметилj-ацетамида (I; А 4-метил-802;; -В NHCOCHj). 2,00 г (7,4 ммоль) (1)-5-аминоме- тил-3- 4-(метилсульфонил)фенил -2- 5 оксазолидинона в 10 мл пиридина охлаждают в смеси воды со льдом, доЪав- ляя 0,72 мл уксусного ангидрида. Смесь далее перемешивают в течение 10-20 мин, а затем разбавляют смесью воды со льдом. Твердый материал отфильтровывают и промывают водой, получают продукт с т.пл. 191,9 - 192,9°С. После перекристаллизации из ацетонитрила получают 1,01 г продукта с т.пл. 192,7-193,2 С.
Вычислено, %: С 49,99; Н 5,16; N 8,97.
Навдено, %: С 49,482; Н 5,17; N 8,93; 8,88.
Пример 15. Получение Cl)N- I 3-(4-метилсульфонил)фенил1 -2-оксо- оксазолидин-5-илметил7-метансульфон- амида (I; А 4-метил-30 ; В метил).
Раствор 1,00 г (3,70 ммоль) (L)-5- аминометил-3- 4-(метилсульфонил)фенил -2-оксазолидинона в 50 мл сухого пиридина перемешивают на ледяной ба5
0
постепенно добавляя в него 2,3мл
метансульфонилхлорида. После завершения операции добавления в этот раствор добавляют 3 капли воды и всю массу концентрируют. Остаток переме- шивают с Водой и добавляют в смесь несколько капель концентрированной соляной кислоты до придания всему раствору кислой реакции. Выпавший в осадок материал фильтруют, промывают
водой и сушат, получают 0,77 г вещества с т.пл. 216,7-220,7 С. Его пере кристаллиэовывают из смеси ацетонит- рила с водой в соотноиении 4:1 и получают 0,51 г продукта с т.пл. 219,7-220,7°С.
Пример 16. Получение (1)-N- (метилсульфонил)фенил -2-оксо- оксазолидин-5-илметил1-карбаминовой кислоты, метилового эфира (I; А
i -мeтиsl-SO ; В
NHCOj-метил).
Смесь 5,41 г (0,02 моль) (1)-5- аминометил-3- 4-(метилсульфонил)феРаствор 5 г (16,1 ммопь) (dl)-2- хлор-Н-Гз- 4- (1 -метилэтил )фен11. 1 -2- гжсооксазолиднн-5- шметил i-пцетамад в 50 мл сухого диметилсульфоксида и 1,5 г азида натрия перемешивают, вы держивая его при 90°С в токе сухого азота в течение 5 ч. Далее смесь ко центрируют под пониженным давлением и остаток перемешивают с впдо11. Час тично кристаллизующийся тпсрдьи : мат
нил -2-оксазолидинона в 50 мл тетра- 5 риаЯ- отделяют и при стоянии он затг.идрофурана перемешивают на ледяной бане, добавляя в нее раствор 2 мл метилхлорформиата в 10 мл тетрагидро- фурана совместно с 2 и. раствором гидвердевает, в результате чего получа 5,8 г продукта. Последни перр.крис-- таллизовьшают из этилацетатл и полу чают 3,4 г продукта с т.пл, 122,425
рата окиси натрия для поддержания ве- 20 123,4 С (с разложением). Тонкослойна ч
личины рН в интервале 10-11. Затем
эту смесь перемешивают в течение
45 мин после того, как добавление
метилхлорформиата было закончено.
Органические растворители удаляют
под пониженным давлением,а остаток
разбавляют водой, доводя величину
рН до 7, фильтруют твердый материал
и промывают его водой, получают 6,5 г
продукта с т.пп. 210-211 С. Его пере- ЗО
кристаллизовывают из ацетонитрила
и получают 3,5 г продукта с т.пл.
214-215 с.
После дополнительной перекристаллизации температура плавления образца достигает 216 ,9-21 7 ,6 С.
Вычислено, %: С 47,55; Н 4,91; N 8,53.
35
хроматограмма на двуокиси кремния с использованием для элюирования сь:г:С1 хлороформа с метанолом в соотношении 9:1 показывает, что продукт представляет собой исходного соединения с целевым продуктом. Смесь используют на последующей стадии без предварительной ОЧИСТК1 .
Часть В.
Суспензию 3,4 г (dl) 2-азидо-|314- (1 -метилэтил )фенил1 -2-оксоок.сато1 - лидин-5-илмет1:Ш 1-ацетлмида в 50 мл
этанола, 5 мл воды и 5 мл уксусной кислоты, содержащую 0,5 г 10%-ного палладия на древесном угле, перемешивают, одновременно iiponycxaioT через раствор по подводящей трубке водород. Реакция продолжается в течение 3 ч, после чего раствор фильтруют и концентрируют. Оста т ок перемешивают с водой и пoдц eлa гивaют концентрированным раствором гидрата окиси аммония, в результате чего образуется смолоподобный материал. Его экстрагируют этилацетатоь:, сушат над сульфатом натрия и концентрируют раствор.Остаток перемешивают с диэти- ловым эфиром и фильтруют. Получают 1,4 г продукта с т.пл. 82-92°С. Последний перекристаллизовывают из 10 мл этилацетата, добавив в него несколько капель триэтитамина, и получают 0,84 г продукта с т. т. 105- 107 С.
C,,H,gObNjS
НаГщено, %: С 47,55; 47,46;Н4,88 4,81; N 8,73; 8,62.
ot -47,7±0,4° ( в ацетонит- риле).
Аналогично вышеизложенному путем проведения соответствующих ацилгапогенидов, изоцианатов, хлор- формиатного эфира или других сложных эфиров с аминами общей формулы
,
-f I-
L.:NHj
.55
получают соединения примеров 17-95,
приведенные в табл. 3. Пример97. Получение 1-2Пример 96. Получение (dl)-2-aзидo-N-| з- 4-(метилсульфонил)фенил - .
амино-N- З- 4-(1-метилэтил) -фенил-2-оксооксазолидин-5-илметил -ацет1 ,2-оксооксазолидин-5-илметил -ацет-амида (I; А 4-CHjSOi ; В NHCOCH N.).
0
амида (I; А -(СН-) СЕ; B NHCOCK - амин).
Часть А.
Раствор 5 г (16,1 ммопь) (dl)-2- хлор-Н-Гз- 4- (1 -метилэтил )фен11. 1 -2- гжсооксазолиднн-5- шметил i-пцетамада в 50 мл сухого диметилсульфоксида и 1,5 г азида натрия перемешивают, выдерживая его при 90°С в токе сухого азота в течение 5 ч. Далее смесь концентрируют под пониженным давлением и остаток перемешивают с впдо11. Частично кристаллизующийся тпсрдьи : матс5 риаЯ- отделяют и при стоянии он затвердевает, в результате чего получают 5,8 г продукта. Последни перр.крис-- таллизовьшают из этилацетатл и получают 3,4 г продукта с т.пл, 122,45
О
5
0
5
0
хроматограмма на двуокиси кремния с использованием для элюирования сь:г:С1 хлороформа с метанолом в соотношении 9:1 показывает, что продукт представляет собой исходного соединения с целевым продуктом. Смесь используют на последующей стадии без предварительной ОЧИСТК1 .
Часть В.
Суспензию 3,4 г (dl) 2-азидо-|314- (1 -метилэтил )фенил1 -2-оксоок.сато1 - лидин-5-илмет1:Ш 1-ацетлмида в 50 мл
этанола, 5 мл воды и 5 мл уксусной кислоты, содержащую 0,5 г 10%-ного палладия на древесном угле, перемешивают, одновременно iiponycxaioT через раствор по подводящей трубке водород. Реакция продолжается в течение 3 ч, после чего раствор фильтруют и концентрируют. Оста т ок перемешивают с водой и пoдц eлa гивaют концентрированным раствором гидрата окиси аммония, в результате чего образуется смолоподобный материал. Его экстрагируют этилацетатоь:, сушат над сульфатом натрия и концентрируют раствор.Остаток перемешивают с диэти- ловым эфиром и фильтруют. Получают 1,4 г продукта с т.пл. 82-92°С. Последний перекристаллизовывают из 10 мл этилацетата, добавив в него несколько капель триэтитамина, и получают 0,84 г продукта с т. т. 105- 107 С.
.55
Используя 1-2-хлор-Н- 3- 4-(метил с,ульфонил)фенил -2-оксооксазолидин- 5-илметил1-ацетамид, аналогично примеру 96 часть А, получают предлагаемое соединение с т.пл. 188,8-189,8 С
Пример 98. N- -(4-ацетил- фенил)-2-оксооксазолидин-5-илметил - ацетаминоксим (1; А ,
В NHCOCH).
3,16 г (4-ацетилфенил)-2-ок- сооксазолидин-5-илметил -ацетамида растворяют в смеси 20 мл пиридина с 20 мл этанола, добавляют в раствор 5 г гидроксиламингидрохлорида.Смесь выдерживают при температуре кипения с обратным холодильником в течение 2 ч. После ее самопроизвольного охлаждения до комнатной температуры растворители вьтаривают и остаток растирают в разбавленном водном растворе соляной кислоты. Твердый продук отделяют и промьюают водой. Перекристаллизацией из водного этанола получают 1,6 г чистого (Д-ацетилфе- нил)-2-оксооксазолидин-5-илметил - ацетамидоксима с т.пл. 213-215°С.
Пример 99. (4-ацетилфе нил)-2-оксооксазолидин-5-илметил - ацетамидоксим, метиловьш эфир (I; А CHjC; В NHCOCH,).
МОСНг Используя метоксиламингидрохлорид
вместо гидроксиламингидрохлорида и работая аналогично примеру 98, получают 1,8 г метилового эфира N-L3-(4- ацетилфенил)-2-оксооксазолидин-5-ил- метил -ацетамидоксима с т.пл. 208- 211°С.
Дозировочные формы.
Антибактериальные предлагаемые соединения формулы I можно вводить в организм с помощью любых средств, которые обеспечивают введение актив- нодействующего вещества в контакт с местом действия этого вещества в организм млекопитающего. Их можно вводить в сочетании с фармацевтическими средствами либо в виде индивидуальных терапевтических агентов, либо в сочетанки с терапевтическими агентами. Их можно вводить индивидуально, однако обычно вводят в организм в сочетании с фармацевтическим носителем, выбираемым с учетом выбранного пути введе- ния в организм и сложившейся фармацевтической практики.
Вводимые в организм дозировки изменяются в зависимости от таких фак
0
5
5
п с
торов, как фармакодинамические характеристики конкретно используемого «агента, метод и путь его введения в организм; возраст, состояние здоровья и вес пациента; природа и степень проявления симптомов заболевания, вид параллельного лечения, частота лечения и целевого эффекта. Обычно ежедневная дозировка активнодействуняце- го вещества составляет приблизительно 5-20 мг/кг живого веса. Обычно, когда применяют более сильнодействующее предлагаемое соединение, желае- мый эффект достигается при введении 5-15, предпочтительно 5-7,5 мг/кг живого веса в день, причем эти дозы делят на дробные дозировки для ежедневного приема от 2.до 4 раз или же употребляют в такой форме, которая обеспечивает постоянное выделение активнодействующего вещества. Такие лекарства можно также вводить в организм парентерально.
Дозировочные формы, приемлемые для введения внутрь, содержат приблизительно 1,0-500 мг активнодействующего вещества на единицу (дозировку). В этих фармацевтических композициях активнодействукяций компонент обычно присутствует в количестве приблизительно 0,5-95 мас.% в пересчете на общее весовое количество композиции.
Полезные фармацевтические дозировочные формы, предназначенные для введения предлагаемых соединений в организм, могут быть проиллюстрированы следующими примерами.
Капсулы.
Целый ряд единичных капсул готовят наполнением стандартных двухком- понентных твердых желатиновых капсул, в каждую из которых вводят по 75 мл порошкообразного активнодействующего вещества, 150 мг лактозы, 24 мг талька и 6 мг стеарата магния.
Мягкие желатиновые капсулы.
Готовят смесь активнодействующего вещества с соевым маслом, после чего эту смесь с помощью нагнетательного поршневого насоса вводят в желатиновый контейнер с получением мягких желатиновых капсул, содержащих по 75 мг активнодействующего вещества. Далее такие капсулы промывают и сушат.
0
5
0
Таблетки.
Целый ряд таблеток готовят таким образом, что дозировочная единица
включает по го вещества,
1315
75 мг активиодействующе- 0,2 мг коллоидной двуокиси кремния, 5 мг стеарата магния 250 мг микрокристаллической целлюлозы, 11 мг кукурузного крахмала и 98,8 мг лактозы. Для повышения аппетитности или отсрочки момента усвоения можно наносить соответствующие покрытия.
Препараты для инъекций.
Парентеральную композицию, приемлемую для введения в организм путем инъекции, готовят смешением 1,5мас. активнодействующего компонента с 10 об.7, пропиленгликоля и водой. Добавлением хлористого натрия раство делают изотоническим и стерилизуют.
Суспензия.
Боднуто суспензию готовят для введения в организм через рот таким образом, что каждые ее 5 мл содержат по 75 мг тонкодиспергированного ак- |тивнодействующего вещества, 200 мг |карбоксиметилцеллюлозы натрия, 5 мг бензоата натрия, 1,0 г раствора аор- бита и 0,025 мл ванилина.
Испытания.
Результаты испытаний показывают, что предлагаемые соединения формулы I являются биологически активными против грамположительных и грамотри- цательных бактерий, включая культуры Staphylococcus aureus, продуцирующие й-лактамазу. Эти агенты потенциально полезны при лечении заразных заболеваний как у человека, так и у животных, включая заболевания дыхательных путей, желудочно-кишечные заболевания, мочеполовые заболевания и болезни центральной нервной систе
мы, крови,интерстициальных жидкостей, мягких тканей и костей.
Соединения формулы I проявляют антибактериальное действие ин витро. Для определения 24-часовьгх минимальных ингибирующих концентраций (МИК)
14
0
5
0
в отношении подопытных штаммов микроорганизмов Staphylococcus epidermidis и Escherichia coli используют жидкую среду Мюллера-Хйнтона в соответствии со стандартной методикой микроразбавления.
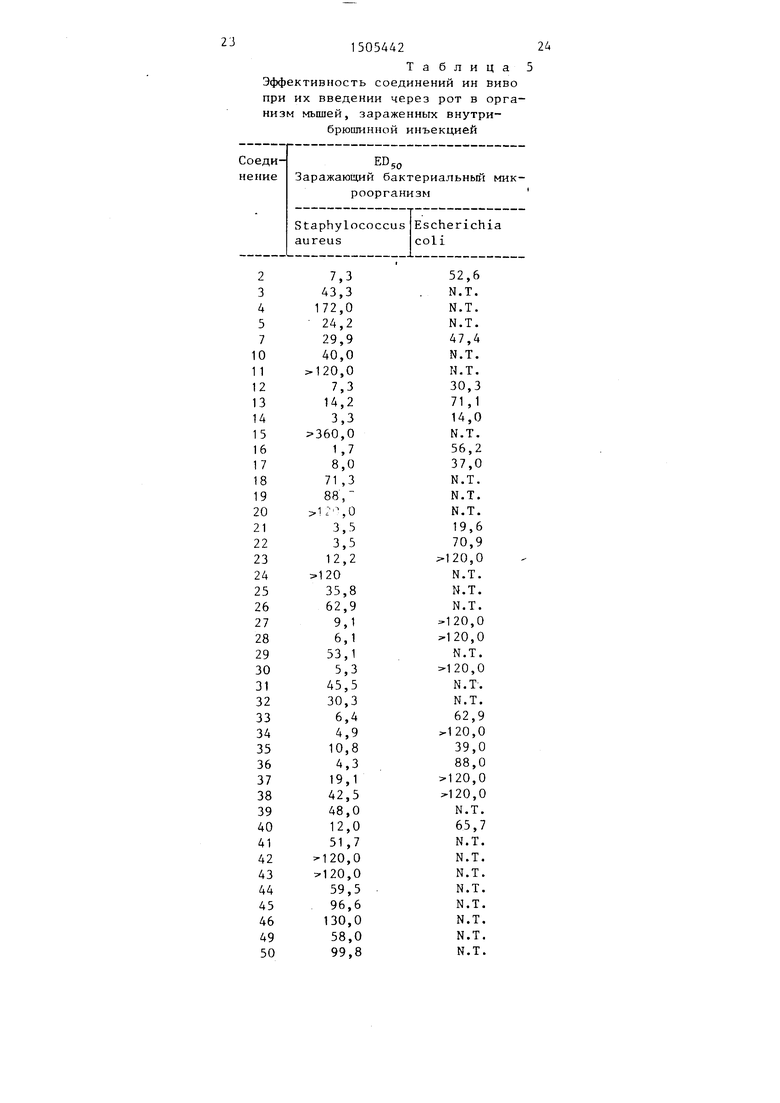
Результаты приведены в табл. 4.
Определение эффективности ин вино проводя 1 интраперитонеальным введением в организм мьш1ей культур заражающих микроорганизмов, разбавленных с учетом достижения 90-100%-ной смертности у контрольных животных в течение Се MJi дней. В качестве разбавителей используют триптиказную соевую жидкую среду для микроорганизмов Е coli и 5%-ный водный раствор муцина желудка свиньи для микроорганизмов Staphylococcus aureus. Испытываемые соединения растворяют или суспендируют в 0,25%-ном водном растворе продукта Метосел R (Метосел Кгокси- пропилметилцеллюлоза Е15 , 5 фирма Дау кемикал компани) для введения через рот или в стерильной дистиллированной воде, содержащей 5% , диметилсульфоксида (фирма Фишер сайертифик компани, Фейрлоун, штат Нью-Джерси) для подкожного введения. Эти соединения в определенных дозировках вводят мьшгам во время их заражения и вновь по истечении 4 ч заражения. Смертность фиксировали ежедневно до завершения испытаний, а 50%-ную эффективную дозировку, ED рассчитывают по методике Рида-Мьюнка,
Предлагаемые терапевтические уровни для человека должны быть достигнуты при введении через рот в количестве 5-20 мг/кг живого веса в виде дробных дозировок, принимаемых ежедневно от 2 до 4 раз. При серьезных или угрожающих жизни заражениях указанные дозировки могут быть увеличены.
Результаты представлены в табл.5 и 6.
0
5
0
5
15
Структура и характеристики соединений формулы
Таблица
Структура и характеристики соединений формулы
Таблица 3 Структура и характеристика соединений формулы
1505442 Таблица
16
1
L
О
L-k
NHi
татная соль
Продолжение табл. 3
19
Таблица 4
Минимальная ингибируклцая концентрация, разбавление жидкой среды при испытаниях ин витро
6,3
200,0
200,0
100,0
50,0
100,0
200,0
200,0
6,3
2,4
3,2
100,0
6,3
6,3
12,5
12,5
200,0
150544220
Продолжение табл.3
100,0 200,0 200,0 .200,0 100,0 100,0 200,0
,0
100,0
9,4
25,0
100,0
100,0
50,0
5070
100,0
200,0
150544224
Таблица 5
Эффективность соединений ин виво при их введении через рот в организм мышей, зараженных внутри- брюшинной инъекцией
25
Е0„
Заражакяций .бактериальный микроорганизм
Staphylococcus aureus
64,0
47,9
2,7
9,6
11,0
11,3
50,7
31,0
120,0
30,2
10,9
7,9
0,70
3,0
4,4
N.T.
58-, 9
11,4
6,5
5,1
Примечание. NT - не тестировано.
Таблицаб
Эффективность соединений ин виво при их подкожном введении в орга- кизм мьшей, зараженных внутрибрю- шинной инъекцией
1505442 Продолжение табл.
26
Eachjerichia coli
N.T. 120,0
65,9
120,0
138,0
98,4
N.T.
N.T.
N.T.
76,8 120,0
83,2
13,6
17,9
46,9 100,0
N.T.
56,5
71,5 105,3
27
Г имечание. NT-не тестировано.
Составитель 3. Латыпова Редактор Н. Киштулинец Техред М.Моргентал Корректор О.Кравцова
Заказ 5272/58
Тираж 352
ВНИИПИ Государственного комитета по изобретениям и открытиям при ГКНТ СССР 113035, Москва, Ж-35, Раушская наб., д. Д/5
Производственно-издательский комбинат Патент, г.Ужгород, ул. Г лгарина,101
I505AA228
Продолжение табл. 6
Подписное
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
