Изобретение относится к способу получения новых биологически активных соединений - производных пирими- до (5-4Ь)-(1,4)-оксазина или их ад-
дитивных солей с кислотами, которые могут найти применение в медицине.
Цель изобретения - получение новых соединений производных ряда пиримидо-
оксазина - малотоксичных и обладающих способностью повышать сократительную силу сердечной мышцы, сопровождающуюся незначительным повышени- ем частоты пульса.
Пример 1. 4-(4-Этоксикарбо- нил-1-пииеразинил)-2-метид-6,7-дигид- ро-81 -пиримидо(5 ,4-Ь)-( 1 ,4)-оксазин- 7-он.
Смесь, состоящую из 1 г 4-хлор-2- метил-6,7-дигидро-8Н-пиримидо(5,4-Ь)- (1,4)-оксазин-7-она, 1,58 г 1-этокси- карбонилниперазина и 10 мл н-бутило- вого спирта, в условиях, исключающих доступ влаги и двуокиси углерода, нагревают в течение 13 ч при температуре кипения реакционной смеси, а затем реакционную смесь упаривают. Остаток растирают с 20 мл воды, смесь выдер- живают в течение ночи, затем осадок отфильтровывают, промывают водой и диэтиловым эфиром, а затем сушат. В результате получают 1,4 г (87%) продукта, т.пл. 197-199°С.
II р и м е р 2 . 2 , 6-Диметил- 4- Ј4- (2-оксиэтил)-1-пиперазинио -6,7-ди- гидро-8Н-пиримидо(5,4-Ь)-(1,4)-окса- зин-7-он.
Смесь, состоящую из 0,86 г 4-хлор2,6-диметил-6,7 дигидро-8Н-пиримидо (5,4)-(1-4)оксазин-7-она, 1,04 г 1-оксиэтилпиперазина и 20 мл н-бутилового спирта, нагревают в течение 7 ч при температуре кипения реакционной смеси, после чего реакционную смесь упаривают. Остаток смешивают с 20 мл воды, после чего производят экстрагирование этиловым эфиром уксусной кислоты. Органическую фазу сушат и упаривают. После перекристаллизации остатка из этилового эфира уксусной кислоты получают 0,90 г продукта, т.пл. 160-161°С.
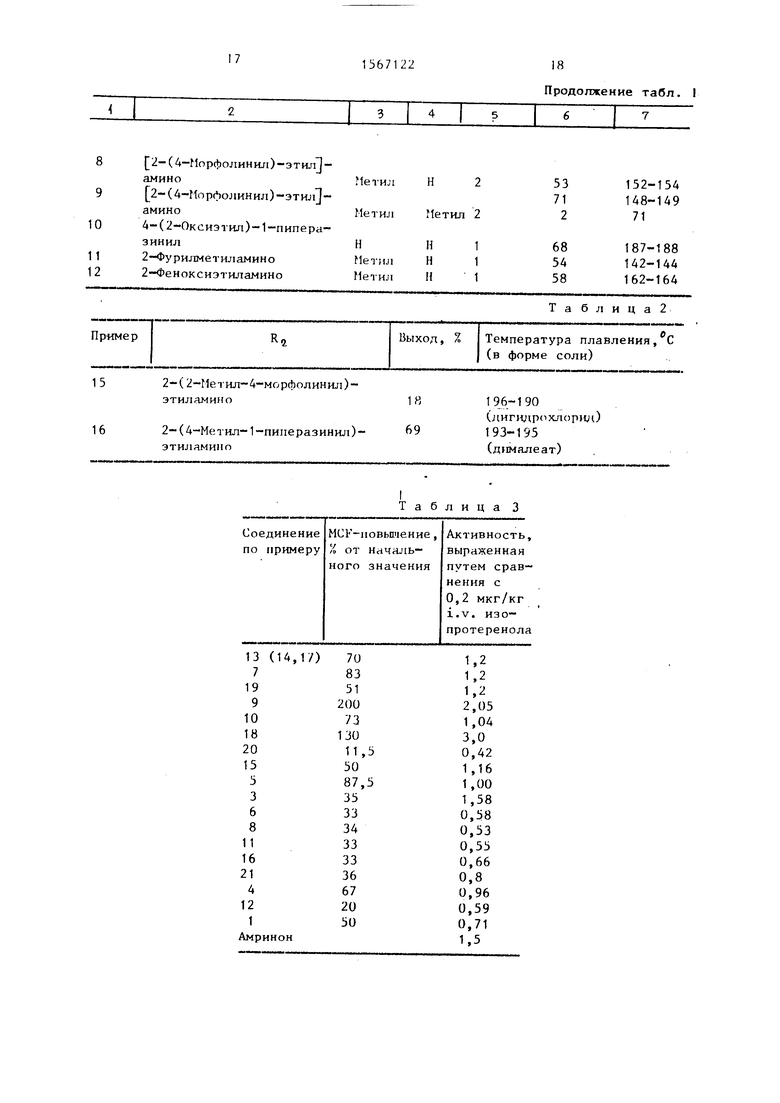
Соединения указанной формулы, в которой R, - метильный радикал, полу- чают аналогично примеру 1 или 2.
Полученные соединения представлены
в табл. 1. it
Пример 13. Дигндрохлорид 2-ме-
тил-4- Ј2- (4-морфолинил) -этил -аминс- 6,7-дигидро-8Н-пиримидо(5,4-Ь)-(1, 4)- окспин- 7-она.
Способ осуществляют аналогично примеру 1, но применяют 1 г 4-хлор-2-ме- тил-b,7-дигидро-ОИ-пиримидо-(5,4-Ь)- (1 ,4)-оксазин-7-он, 1,01 г триэтил- амина и 0,65 г 2-(4-морфолинил)-этил- амина, причем смесь нагревают при
Q
,
Q
е
0
температуре кипения течение 40 ч,- В результате получают 0,46 г (25%) продукта, т.пл. 233-237°С (с разложением) .
П р и м е р 14. Дигидро 2-метил-4- 2 (4-морфолинил)- этил -амино- 6, 7- дигидро-8Н-пиримидо(5, 4-Ь)-(1,4)-ок- сазин-7-она.
Метод А. Смесь, состоящую из 50 г 2-метил-г 4-(4-толуол сульфо нил оке и) - 6,7-дигидро-8Н-пиримидо(5,4-Ь)-(1,4)- оксазин-7-она, 20,73 г безводного углекислого калия, 19,5 г 2-(4-морфо- линил)-этиламина и 1250 мл абсолютного этилового эфира уксусной кислоты, перемешивают в течение 72 ч при 20°С, затем отфильтровывают нерастворимую фракцию, после чего фильтрат экстрагируют 1 н. водным раствором соляной кислоты.
Значение рН водной фазы доводят до 5, после чего производят экстрагирование дихлорметаном. Значение рН водной фазы доводят затем до 10, после чего еще раз производят экстрагирование дихлорметаном. Полученную при последнем экстрагировании органическую фазу сушат и упаривают. Остаток, т.е. неочищенное основание, растворяют в ацетоне. Полученный раствор при охлаждении вводят во взаимодействие с раствором хлористого водорода в абсолютном этиловом спирте., Образовавшийся осадок отделяют фильтрованием и промывают ацетоном. В результате получают 27,31 г (50%) продукта, т.пл. 133-237 С (с разложением).
Метод В. Смесь, состоящую из 5 г 2-метил-4-(4-толуолсульфонилокси)- 6,7-дигидро-8Н-пиримидо(5,4-Ь)-(1,4)- оксазин-7-она, 2,99 г 2-(4-морфоли- нил)-этиламина, 9,11 г триэтиламина и 130 мл абсолютного этилового эфира уксусной кислоты, в течение 5 ч нагревают при температуре кипения, а затем фильтруют. Фильтрат упаривают, остаток смешивают с водой, после чего значение рН доводят до 2. Затем смесь экстрагируют этиловым эфиром уксусной кислоты, водную фазу отделяют и доводят ее значение рН до 5. Дальнейшую обработку проводят аналогично методу А. В результате получают 3,28 г (60%) продукта, т.пл. 233-237°С (с
разложением).
ii
Соль указанного основания с фума- ровой кислотой получ;шл по следующей методике.
D15671
К раствору 1 г неочищенного основания, полученного по методу А или 15, в 9 мл этилового спирта прибавляют при перемешивании и температуре 70°С нагретый до 70°С раствор 0,2 г фума- - ровой кислоты в 5 мл этилового спирта, затем смесь охлаждают и выдерживают в течение ночи при О-4°С. В результате почти с количественным выхо
дом получают фумарат, т.пл. 215- 216 С. Основание и фумаровая кислота содержатся в полученном фумарате в соотношении 2:2.
Вычислено, %: С 51,27; Н Ь,03; 19,93.
Ci0 0,o (м.масса 702,72).
Найдено, %: С 51,63; Н 5,96; 19,06.
Найденное содержание фумаровой кислоты 16,50% (вычислено 16,52%).
2-Метил-4-(4-толуолсульфонилокси)- 6,7-дигидро-8Н-пиримидо-(5,4-Ь)-(1,4) оксазин-7-он, который является общим исходным веществом при осуществлении обоих способов А (и В), получают следующим образом.
Раствор 157,2 г 4-толуолсульфонил- хлорида в 450 мл ацетона в течение 3 ч прибавляют при комнатной темпе
ратуре и перемешивании к раствору 135,8 г 4-окси-2-метил-6,7-дигидро- 8Н-пиримидо(5,4-Ь)-(1,4)-оксазин-7- она в 1205 мл 1 н. водного раствора гидроокиси натрия, после чего смесь дополнительно перемешивают в течение 5 ч при комнатной температуре. Кристаллический продукт отфильтровывают, промывают водой, а затем растворяют в 1800 мл ацетона. Нерастворимую фракцию отделяют фильтрованием и промывают ацетоном. Ацетоновый раствор упаривают, осадок отфильтровывают, промывают небольшим количеством ацетона и сушат. В результате получают 123,2 г (49%) продукта, т.пл. 185- 186°С.
Соединения предлагаемой формулы, где R, - метильный радикал, а 1Ц, К атомы водорода, полученные по примеру 14, приведены в табл. 2.
Приме р 17. Дигидрохлорид 2- метил-4- 2-(4-морфолинил)-этил -ами- но-6, 7-дигидро-81 -пиримидо(5,4-Ь)- (1,4)-оксазин-7-она.
Смесь 0,34 г 4-метанеульфонилокси- 2-метил-6,7-дигидро-8Н-пиримидо - (5,4-Ь)-(1,4)-оксазин-7-она и 0,40 г 2-(4-морфолинил)-этиламина нагревают
0
5
0
5
0
5
0
5
0
5
22
в течение 10 мин при 75-80°0. После охлаждения смесь растворяют в 8 мл 1 н. раствора гидроокиси натрия, после чего производят полное экстрагирование дихлорметаном. Органический раствор встряхивают с водой, сушат, фильтруют и упаривают. В результате получают 0,21 г (52,5%) однородного неочищенного основания, которое превращают в дигидрохлорид способом, описанным в примере 14. Неочищенное основание может быть подвергнуто кристаллизации путем перемешивания с этиловым спиртом или ацетоном, т.пл. 143-145°С.
П-римененный в качестве исходного соединения 4-метансульфонил-2-метил- 6,7-дигидро-8Н-пиримидо(5,4-Ь)-(1,4)- оксазин-7-он получают следукл им образом.
2 мл 30%-ного раствора перекиси водорода прибавляют к смеси, состоящей из 0,83 г 2-метил-4-метилтио- 6,7-дигидро-8Н-пиримидо(5,4-Ь)-(1,4)- оксаэин-7-она и 8 мл уксусной кислоты, после чего реакционную смесь перемешивают в течение 3 ч при 60 С. Затем реакционный раствор упаривают досуха, после удаления следов уксусной кислоты кристаллический остаток растирают с 5 мл воды, после чего значение рН смеси доводят до 6,7. Образовавшийся осадок отделяют, фильтруют и сушат. В результате получают 0,36 г (38%) исходного соединения, т.пл. 211°С.
Пример 18. Дигидрат дигидрохло- рида 4- (4-бензил-2- морфолинил)- тшГ -амино-2-метил-6,7-дигидро-8Н- пиримидо(5,4-Ь)-(1,4)-океазин-7-она.
Метод А. Суспензию 16,1 г 2-метил- 4-(4-толуолсульфонилокси)-6,7-дигид- ро-8Н-пиримидо(5,4-Ь)-(1,4)-оксазин- 7-она (пример 23), 8,93 г 2-аминоме- тил-4-бензилморфолина и 6,6 г безводного углекислого калия в 400 мл абсолютного этилового эфира уксусной кислоты перемешивают в течение 80 ч при комнатной температуре. Затем нерастворимую фракцию отфильтровывают, раствор экстрагируют t н. раствором соляной кислоты. Значение рН водной фазы доводят до 7 посредством прибавления 1 н. раствора гидроокиси натрия, после чего производят экстрагирование диэтиловым эфиром. Эфирный раствор сушат и упаривают, остаток растворяют в абсолютном этиловом спир
те, а затем к приготовленному раст- гюру прибавляют по каплям рассчитанное количество раствора хлористого водорода в этиловом спирте. После охлаждения до 0-4°С образовывается кристаллический осадок, который отфильтровывают, промывают холодным этиловым спиртом и сушат. В результате получают 2,33 г (15%) продукта, т.пл. 196-199°С.
Метод В. Суспензию, приготовленную из 3 г 2-метил-4-(4-толуолсульфо- нилокси)-6,/-дигидро-8Н-пиримидо- (5 ,4-Ь)-(1,4)-оксазин-7 она (пример 14), 1,85 г 2 аминометил-4-бен- зилморсЪолина, 5,43 г триэтиламина и 80 мл абсолютного этилового эфира уксусной кислоты, нагревают при перемешивании в течение 8 ч irpn температуре кипения смеси, а затем смесь упаривают. Остаток перемешивают с 60 мл воды, после чего значение рН смеси доводят до 1-2 посредством прибавления 1 н. раствора соляной кислоты. После экстрагирования этиловым эфиром уксусной кислоты значение рН водной фазы доводят до 7 посредством прибавления 1 н. раствора гидроокиси натрия, а затем проводят экстрагирование диэтиловым эфиром. Дальнейшую обработку проводят в соответствии с методом А, в результате чего получают 1,0 г (13%) продукта, т.пл. 195-198°С.
2 Аминометш1-4-бензилморфолин, являющийся обцим исходным веществом при осуществлении обоих методов А и В, получают, например, следующим образом.
a)4-Бензил-2-фталимидометилморфо лин.
iO,2 г Лталимида калия прибавляют к раствору Ь1,2 г 4-бензил-2-хлорме- тилморфолина в 150 мл абсолютного ди метилформамида, после чего смесь в течение 6 ч нагревают при перемешивании и температуре кипения. Образовавшийся после охлаждения кристаллический продукт отфильтровывают, промывают диметилЛормамидом, а затем водой и сушат. В результате получают 68,95 г (76%) продукта, т.пл. 130- 132°С.
b)Дигидрохлорид 4-бензил-2-амино мет илформолина.
Смесь 19,3 г продукта, полученног на стадии а, с 38 мл концентрированной соляной кислоты нагревают в тече
ние 14 ч при перемешивании и температуре кипения. Выделившуюся после охлаждения в осадок фталевую кислоту отфильтровывают, а водный раствор полностью освобождают от присутствия фталевой кислоты посредством экстрагирования этиловым эЛиром укгусной кислоты. Водную Лазу упаривают досуха, остаток растирают с абсолютным
5
0
5
5
5
0
0
5
этиловым спиртом, кристаллическое вещество отделяют Лильтрованием, промывают абсолютным этиловым спиртом и сушат. В результате получают 10,3 г (65%) продукта, т.пл. 244-246 С.
Свободное основание может быть выделено в свободном состоянии из ди- гидрохлорида. При этом соль растворяют в воде, раствор подцелачивают прибавлением раствора гидроокиси натрия, после чего основание экстрагируют хлороформом. После сушки органическую фазу упаривают и в виде остатка получают с количественным выходом неочищенное основание, которое в полученной форме может быть применено при осуществлении способов, указанных в примерах.
И р и м е р 19. 2-Метил-4- 3-(4- Q морфолинил)-1-пропил -амино-6,7-ди- гидро-8Н-пиримидо(,5,4-Ь)-(1 ,4)-окса зин-7-он.
Раствор, приготовленный из 0,7 мл триэтиламина и 1,44 г 3-(4-морфоли- нил)-1-пропиламина, в течение 8 ч прибавляют но каплям при перемешивании и температуре кипения к смеси, состоящей из 2 г 4-хлор-2-метил-6,7- дигидро-8Н-пиримидо(5,4-Ь)-(1,4)-ок- сазин-7-он; 1,4 мл триэтиламина и 5 мл абсолютного бензола. Затем реакционную смесь в течение 8 ч нагревают при перемешивании и температуре ее кипения. Непосредственно после этого реакционную смесь упаривают, остаток смешивают с 16 мл воды, твердое вещество отфильтровывают, промывают сначала водой, а затем диэтиловым эфиром. В результате послучают 1,1 г (36%) продукта, т.пл. 165-168°С.
Гидрохлорид получают посредством прибавления раствора хлористого водорода в этиловом спирте к раствору основания в этиловом эфире уксусной кислоты. Температура плавления гидро-
хлорида 250-251°С. i .i
Пример 20.. 4-(2-Оксиэтил)- амино-2,6,6-тримет ил-6,7-дигидро-8Н- пиримидо(5,4-Ь)-(1,4)-оксазин-7-рн.
Осуществляют способ аналогично примеру 2, в результате чего получают продукт с выходом 83%, причем температура плавления продукта 168-170°С. Гидрохлорид имеет т.пл. 190-191°С.
П р и м е р 21. 4-Гидразино-2-ме 1ил-b,7-дигидро-8Н-пиримидо(Ь,4-Ь)- (1 ,4)-оксазин-7-он.
Раствор 12,8 мл 98%-ного гидразин- гидрата в 0 мл н-бутилового спирта в течение 30 мин прибавляют но каплям при перемешивании и температуре 117°С к раствору 20 г 4-хлор-2-метил-6,7-ди гидро-8Н-пиримидо(Ь,4-Ь)-(1,4)-окса- зин-7-она в 320 мл н-бутилового спирта. Затем реакционную смесь перемешивают в течение 4Ь мин при указанной температуре и непосредственно после этого охлаждают до комнатной темпера- туры. Осадок отфильтровывают и промывают водой. В результате получают 11,66 г (56,3%) продукта, т.пл. 262- 264°С.
Пример 22. Получение таблеток
Состав (в расчете на 100 таблеток), г: Лумарат основания по примеру 2 10; лактоза 18Ъ; микрокристаллическая целлюлоза 23; тальк Ь; кукурузный крахмал 73; стеарат магния 2. Всего 300 г.
I Компоненты смешивают друг с другом смесь гомогенизируют, после чего из смеси отпрессовывают таблетки, которые содержат по 10 мг биологически активного вещества.
Приме р 23. Получение раствора для инъекций.
Состав (в расчете на 2 л раствора) фумарат основания по примеру 2 2 г, хлористый натрии 20 г, вода для инъекционных целей до 2000 мл.
Из указанных компонентов приготавливают раствор, который помещают в ампулы и стерилизуют. Одна ампула со- держит 2 мл раствора.
Проведены биологические испытания соединений, полученных описываемым способом.
Повышающий сократительную силу сердца эффект (Myocardial Contractile Force, далее сокращено MCF) определяли в проведенных на живых организмах опытах по следующим методикам.
А) Испытание на подвергнутых нар- козу кошках.
Животных наркотизировали внутривенным (i.v.) введением 30 мг/кг натриевой соли пентобарбитала, после чего при искусственном дыхании раскрывали их грудные клетки. После раскрытия перикарда прикрепляли strain gauge к эпикардиальной поверхности левого желудочка сердца. Кровяное давление измеряли посредством того, что катетер, соединенный с передатчиком давления и электроманометром, вводили в бедренную артерию. Биологически активное вещество вводили либо внутривенно через введенную в бедренную вену канюлю, либо интрадуоденаль- но (i.d,) через i.d. канюлю. В начале -опыта вводили 0,2 мкг/кг изопроте- ренола (IS) в качестве внутреннего стандарта для того, чтобы проконтролировать миокардиальную реактивность а именно наибольший peak) MCF-от- вет мог измеряться при всех положительных инотропных средствах в соответствии с личностью животного. Так изопротеренол применяли не в качестве подлинного вещества для сравнения
MCF-ответ выражали в процентном изменении начального значения: действие Ь мг/кг исследуемого вещества, введенного внутривенно, сравнивали с действием 0,2 мкг/кг изопротерено- ла, введенного внутривенно, и выражали отношением.
Полученные результаты представлены в табл. 3.
i
Но сравнению с изопротеренолом соответствующие изобретению соединения проявляют аналогичное или более сильное действие, проявляющееся в повышении сократительной силы сердца. В качестве более сильного/3-ядрено- цептор-антагониста изопротеренол вызывает измеримое MCF-повышение уже при вводимой внутривенно дозе 0,2 мкг/кг, но этот эффект краткосрочен, полностью обратим и не оказывает влияния на параметры кровообращения. При действии соединений, указанных в табл. 3, частота пульса повышается незначительно или совериен- но не повышается.
Действие некоторых соединений также исследовали после интрадеуденально- го введения. Соединение 18 обладает сильным положительным инотропным действием в дозе 20 мг/кг или даже после 5 мг/кг. Вещество 13 проявляет сильное и сохраняющееся продолжительное время действие ухе при введенной внутривенно дозе 1 мг/кг.
B)Испытание на подвергнутых наркозу собаках.
Животных наркотизировали внутривенным введением 35 мг/кг натриевой соли пентобарбитала. После раскрытия грудной клетки и перикарда препарировали нижнюю восходящую часть Arteria coronaria circumflexa и снабжали электромагнитным кровоточным зондом для того, чтобы измерять в коронарной артерии ток крови. На снабженную этим сосудом поверхность, помещали strain gauge для того, чтобы измерять повышающий сократительную силу сердца эффект. Для измерения общего артериального давления крови применяли введенную в бедренную артерию канюлю, а для внутривенного введения биологически активного вещества (исследуемого вещества) использовали введенную в бедренную вену канюлю. dp/dt-значение рассчитывали из давления, которое измеряли с помощью введенного в левый желудочек сердца передатчика давления типа Millar. Все параметры регистрировали на приборе Beckraann R12 Dynograph (с 8 каналами) .
C)Испытание на бодрствующих кошках.
В случае этих животных применяли метод Раблоцки и Иадера или некоторую модификацию этого метода. В аорту и легочную артерию вводили на постоянное время катетеры для измерения давления крови. В соответствии с модификацией катетер также вводили и в правый желудочек сердца для того, чтобы определить dp/dtMal(C-значение.
Положительное инотропное и расширяющее коронарные сосуды действие соответствующих изобретению соединений показано на полученных при проведении испытаний соединений 13, 18 резуль- татах, причем испытания осуществляли по описанным методикам.
Испытание соединения 13.
Положительное инотропное действие этого вещества в случае подвергнутых наркозу собак с раскрытой грудной клеткой наблюдалось уяе при дозах 0,025-1,6 мг/кг. Сила и продолжительность повышения сократительной силы сердца зависят от дозы. Положительное инотропное и расширяющее коронарные сосуды действие при введенных внутривенно дозах 0,2, 0,4 и 0,8 мг/кг этого соединения сравнимо с действием
0
5
5
0
0
5
0
0
5
амрйнона (5-амино-З,4 -биииридин-6- 1Н-он) в дозах 0,5, 1,0 и 2,0 мг/кг.
В этом интервале доз соединение 13 вызывает зависимое от дозы повышение сократительной силы сердца. Действие введенного внутривенно амрйнона в дозе 2 мг/кг не было более сильным, чем действие соединения 13 в доте 0,4 мг/кг. Очевидно, что на положи- тельное инотроиное действие обоих соединений оказывает благоприятное влияние расширяющий коронарные сосуды эффект. Однако повышающее сократительную силу сердца действие не
было более продолжительным, чем действие на коронарные сосуды.
Благоприятное действие соединения 13 на сердечную мышцу также было обна ружено после интрадуоденального введения. После дозы 1 мг/кг сократительная сила сердца увеличивалась на 35-40% без изменения кровотока в коронарных сосудах. Новышаюций сократительную силу сердца эффект амрйнона был по меньшей мере в пять раз слабее,.чем действие вецества 13.
На подвергнутых наркозу собаках с раскрытой грудной клеткой после острой ишемии значительно уменьшается положительное инотропное действие ам- ринона, но действие соединения 13 остается статистически неизменным.
Отрицательное инотропное действие Вазопрессина в дозе 0,2 Ш7кг значительно уменьшается благодаря применению соединения 13 в дозе 0,8 мг/кг, введенной внутривенно. Сосудосуживающее действие этих соединений является прямо противоположным. Таким образом, соединение 13 может предотвращать развитие вызванной химическим путем ишемии.
Если в бодрствующих кошек с постоянным катетером вводят орально соединение 13 в дозах 0,5, 2,0, 4,0 и 8,0 мг/кг, то общее артериальное давление крови и частота пульса практически не изменяются, но сократительная сила сердца после любой дозы значительно повышается. Чем больше доза, тем более продолжительным является промежуток времени, при котором наблюдается положительное инотропное действие. Максимальное (peak) увеличение dp/dt-значения 60%.
Положительное инотропное действие соединения 13 остается неизменным и при предварительном введении й-адре
ноцептор-антагонистов , гистамин И-2у антагонистов или резерпина.
Испытания соединения 18.
Это соединение вводили в подвергнутых наркозу собак с раскрытой грудной клеткой внутривенно в дозе 1 мг/кг или интрадуоденально в дозе 5 мг/кг.
После внутривенного введения дозы значение, характеризующее сократи- тельную силу сердца, повышается на 40%, циркуляция крови в коронарных сосудах увеличивается на 20%, частот пульса увеличивается в очень незначительной степени (на 10%), общее артериальное систолическое давление крови уменьшается на 10%, а диастоли ческое давпение крови падает на 20%. Период уменьшения повышаюцего сократительную силу сердца эффекта в два раза оказался равным 10 мин. Почти неизменный ответ коронарной артерии длится в течение 20 минут.
После интрадуоденальной дозы зна- чение, характеризующее сократительную силу сердца, увеличивается приблизительно на 30% и остается неизменным в течение 30 мин.
Биохимические фармакологические испытания.
Приводимые испытания проводили для того, чтобы прояснить действие соединения 13 на различные ферменты и, прежде всего, на связанные с мембраной ферменты клеток сердечной мышцы.
Действие соединения 13 на ферменты Na-K-ATP-азу и NADH-оксидазу.
Сарколеммный препарат получали из клеток сердечной мышцы по методу Д.М.Берса.
NADH-дихлорфенол-редуктазу определяли с помощью анализирующего ферменты прибора (Centrif i-СНШт).
Активность Na-K-ATP-азы измеряли в 50 мМ растворе трис-HCl при рН 7,5 Уабаин, который применяли в качестве вещества для сравнения, сильно инги- бировал Na-K-ATP-азу и лишь незначительно уменьшая активность NADH-индо фенол-редуктазы. На активность этого фермента соединение 13 не влияло даже при концентрации 10 М.
Действие соединения 13 на фермент Са-АТР-азу, регулирующую выведение Са -ионов.
Активность связанной с мембраной Са-АТР-азы клеток сердечной мышцы определяли по методу Me Namara. В хо-
Q
де in vitro опытов активность фермента под действием соединения 13 концентрации 10 М изменялась от контрольного значения ,83 мМ до Кд 1,8 мМ. Это означало низкий эффект.
При осуществлении другого опыта соединение 13 в течение 5 сут вводили i.p. при суточной дозе 2 мг/кг. По отобранным из обработанных животных ex vivo пробам находили, что ак- тивность Са-АТР-азы в результате указанной обработки уменьшается на 30% (скорректировано на 1 г сердечной ткани).
Действие соединения 13 на прием
15
Са-ионов в Sarcoplasma-Reticulun
0
5
Q
0
5
0
5
0
в присутствии АТР.
Применяли метод Харигайа. Прием Са2 -ионов в Sarcoplasna-Reticulum уменьшался на 25% в присутствии соединения 13 в концентрации 5х1СГЭМ.
Это соответствовало такому действию, которое вызывает кофеин концентрации .
Действие соединения 13 на содержание циклического аденозинмонофосфата .(аАМР) в клетках сердечной мышцы.
Соединение 13 вводили i.p. в крыс в дозе 5 мг/кг. Через 0,2,5,15 и 30 мин после этой обработки животных , умерщвляли. Их сердце гомогенизировали с трихлоруксусной кислотой, после чего с помощью специфического связующего белка определяли радиоактивным методом содержание циклического адено- зинмонофосфата. Соединение 13 не оказывало существенного влияния на содержание циклического аденозинмонофосфата в сердце.
Действие соединения 13 на активность фосфодиэстеразы (фДЭ) сердечной мышцы.
Выделение ФДЭ( -фермента и определение активности проводили по методу Шарма и Ванга. Соединение 13 концентрации 100 мкМ не оказывало влияния на активность ФДЭ-ферментов.
На основании результатов, получен - ных в описанных опытах, можно сделать вывод, что соединение 13 не оказывает характерного и типичного для сердечных гликозидов действия на ферменты (на ферменты Na-K-ATP-азу или NADH- оксидазу) и не влияет на образ действия вегетативных рецептор-агонистов (т.е. на функции фермента аденилат циклаза).
1515
В отличие от некоторых ксантиновых производных или от новых положительных инотропных соединений типа бипи-. ридина (например, амринона) соедине ние 13 не оказывает влияния на функции ФДЭ-иэоферментов.
Острую токсичность соединения 13 изучали на мышах и крысах.
Значения представлены в табл. 4.
Из табл. 4 видно, что испытанное соединение обладает низкой токсичностью.
Проведенные испытания показали, что производные пиримидооксазина, полученные в условиях описываемого способа, обладают другим спектром биологического действия, чем соединения этого ряда, они малотоксичны и повы- шают работу сердечной мышцы более чем на 30%.
ормула изобретения
Способ получения производных пири- до (574Ь)-(1,4)-оксазина общей форлы
2
е
R - RtжУ
BrSr
1Н
С,-С4-алкил;
5 Ь
группа общей формулы , где Ну - водород, С,-С4- алкил; Kg - аминогруппа, незамещенный или замещенный ал- кильный (С,-С4) радикал, у которого замест ит елями могут быть гидроксил, меркаптогруп16
па, аминокарбонил, фурил, фе- нокси- или бонзильная, или
морфолинильная группы, незамещенные или С -алкилом;
замененные С(
бензил;
гидроксиалкил ();
V
R4NK-Ug - пиперазинил, замещенный алк (Cj-С4)оксикарбонилом или оксиалкилом (.); одинаковые или различные и независимо друг от друга водород или С -С4 алкил;
или их аддитивных солей с кислотами,
отличающийся тем, что
соединение общей формулы
П|0
Sfc
Н
5
где К,,к, и К4. имеют указанные значения;
хлор, метансульфонилокси- или 4-толуол-сульфонилокси- группы,
вводят во взаимодействие с амином,
общей формулы
HMjRj,
где RSH R6 имеют указанные значения, и в случае необходимости полученные основания переводят в аддитивную соль с кислотой и/или в случае необходимости из соли выделяют свободное основание.
Т а б л и ц а 1







| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения производных пиримидо[5,4- @ ] [1,4]оксазина или их кислотно-аддитивных солей | 1988 |
|
SU1567123A3 |
| 6,7-Диэтокси-1-(4-этокси-3-оксибензил)-3,4-дигидроизохинолин,обладающий действием на сердечно-сосудистую систему | 1982 |
|
SU1097621A1 |
| Способ получения 3-замещенных тетрагидропирроло/1,2- @ / пиримидинов,их кислотно-аддитивных или четвертичных солей | 1980 |
|
SU1048986A3 |
| Способ получения производных @ -(3,3-дифенилпропил)-пропилендиамина или их солей | 1973 |
|
SU1014468A3 |
| Способ получения геминальных дигалоидных производных конденсированных пиримидин-4-онов,рацематов или оптически активных антиподов | 1980 |
|
SU1151210A3 |
| Способ получения замещенных ксилита или гексита | 1980 |
|
SU1075975A3 |
| Способ получения производных арилоксиаминобутанола или их солей | 1975 |
|
SU1025327A3 |
| Способ получения производных пиридо (1,2- @ ) пиримидина или их кислотно-аддитивных солей | 1978 |
|
SU1022659A3 |
| Способ получения конденсированных производных пиримидина или их солей | 1980 |
|
SU1082324A3 |
| Способ получения производных пиридо/1,2-а/пиримидинов или их солей,или их оптически активных изомеров | 1978 |
|
SU906379A3 |
Изобретение относится к гетероциклическим соединениям, в частности к получению производных пиримидо /5-4B/-/1,4/-оксазина ф-лы 1 N=C(R1)-N=C(R2)-C=C-NH-C(O)-C(R3)(R4)-O, где R1 - C1-C4-алкил
R2 - группа ф-лы NR5R6, где R5 = H, C1-C4-алкил, R6-аминогруппа, незамещенный или замещенный алкильный /C1-C4/ радикал, у которого заместителями могут быть гидроксил, меркаптогруппа, аминокарбонил, фурил, фенокси или бензильная, или морофолинильная группы, незамещенные или замещенные алкилом /C1-C4/, или R5-бензил, R6-гидроксиалкил /C1-C4/, или NR5R6 может представлять собой пиперазинил, замещенный алк(C2-C4)оксикарбонилом или оксиалкилом (C1-C4)
R3 и R4 - одинаковые или различные и независимо друг от друга обозначают H или алкил C1-C4-алкил, или их аддитивных солей с кислотами, которые обладают способностью повышать сократительную силу сердечной мышцы. Цель изобретения - разработка способа получения соединений, обладающих указанным действием. Получение ведут реакцией соединения ф-лы 1, где R1, R3 и R4 - указано выше, а R2 - хлор, метансульфонил окси или 4-толуолсульфонилокси группы с амином HNR5R6, где R5 и R6 указано выше. В случае необходимости из соли выделяют свободное основание. Новые соединения повышают работу сердечной мышцы более чем на 30%. 4 табл.
52-Меркаптоэтиламино
6Н-Метнл-М-(аминокарбо- нилметил)-амино
7N-Бензил-N-(2-оксиэтнл)- амино
2-( 2-Метил-4-морфолинил)- этиламино18
2-(4-Метил-1-пииеразинил)-69
этиламино
Продолжение табл. 1
Таблица2
196-190
(дигидрохлорид) 193-195 (дималеат)
I
Таблица 3
| Вейганд-Хильгетаг | |||
| Современные методы эксперимента в органической химии | |||
| - М.: Химия, 1968, с | |||
| Электромагнитный счетчик электрических замыканий | 1921 |
|
SU372A1 |
