Изобретение относится к биотехнологии получения ферментных препаратов из микробиологического сырья и может быть использовано в ферментной промышленности, а получаемый комплекс - для количественного определения уровня тиаминдифосфата (ТДФ) в
крови или другой биологической жидкости с целью диагностики ранней 1 Bi-недостаточности при обследовании 1 различных слоев населения в стационарных условиях клиники или в обычных условиях, а также в животноводстве при выращивании молодняка на животноводческих комплексах и в зверо- хозяйствах.
Определение общего ТДФ в биологических объектах осуществляют ферментативном методом, который основан на способности дифосфорного эфира как кофермента рекомбинировать в апо пируватдекарбоксилазой. Образующаяся при этом активная холоформа белка окисляет пируват до уксусного альдегида, который в сопряженной системе с алкогольдегидрогеназой и восстановленным никотинамидадениндинуклеоти- дом (НАДН) превращается в этиловый спирт. О скорости реакции судят по снижению поглощения НАДН при 340 нм. Количественное содержание ТДФ, пропорциональное акт внести образовавшейся холопируватдекарбоксилазы и убыли восстановленного НАД, находят по калибровочному --рафику с известными концентрациями кофермента. Основ - ными компонентами системы, обеспечивающими рекомбинацию тиаминдифосфата и последующее окисление пирувата, а затем и НАДН, служат алкогольдегид- рогеназа и апопируватдекарбоксилаза.
Цель изобретения - расширение технологических возможностей способа, ускорение процесса и удешевление целевого продукта.
Способ осуществляется следующим образом.
Берут свежую пастообразную массу пивных дрожжей Saccharomyces carls- bergensis 7-9 регенераций и готовят суспензию для экстрагирования белкового препарата. Приготовленную суспезию обрабатывают ультразвуком с частотой 20-22 кГц в течение 18-25 мин и проводят двухэтапное фракционирование ацетоном и сульфатом аммония. На пзрвом этапе ацетонового фракционирования на каждые 100 мл дрожжевог экстракта добавляют 55 мл ацетона, а на втором - 10 мл ацетона. Затем выполняют адсорбционную хроматографию на гидроксиапатите элюцией комплекса 0,12 М фосфатным буфером до соотношения пируватдекарбоксилазной и алкогольдегидрогеназной активносте в полученном ферментном комплексе 1:(0,15-0,5), причем пируватдекарбок силазная активность должна составлят не менее 14 2д„акТо/мг белка.
После заключительной очистки на гидроксиапатите белкового препарата получают апопируватдекарбоксилазу в
составе комплекса с последующей его стабилизацией, так как ферментативный метод определения ТДФ в биологи- ческих объектах с использованием
ферментного комплекса основан на способности дифосфорного эфира как ко фермента рекомбинировать с апопиру- BL декарбоксилазой.
Наилучший эффект от использования . способа наблюдается при проведении ацетонЬвого фракционирования на первом этапе добавлением 55 мл ацетона на каждые 100 мл дрожжевого экстракта
и заключительной очистки целевого продукта элюцией комплекса 0,12 М фосфатным буфером до соотношения пируватдекарбоксилазной и алкогольдегидрогеназной активностей в ферментном
0 комплексе 1:(0,15-0,5), при пируватдекарбоксилазной активности не менее 14,0 ед.акт./мг белка.
Увеличение количест ва вносимого ацетона свыше 55 мл на каждые 100 мл
5 дрожжевого экстракта на первом этапе ацетонового фракционирования ведет к удалению из ферментного комплекса одного из двух компонентов - алкоголь- дегидрогеназы. При повышении первоначального объема ацетона от 58 до 60 мл, добавляемого на каждые 100 мл дрожжевого экстракта, ферментный комплекс содержит остаточные концентрации (следы) алкогольдегидрогеназы, а увеличении количества ацетона свыше 60 мл - алкогольдегидрогеназа в комплексе не обнаруживается. Уменьшение объема ацетона ниже 55 мл, добавляемого на каждые дрожжевого экстракта, резко снизит содержание пируватдекарбоксилазы в ферментном комплексе. При добавлении 52-53 мл ацетона на каждые 100 мл дрожжевого экстракта ферментный комплекс содержит следы пируватдекарбоксилазы, при внесении 54 мл ацетона - активность пируватдекарбоксилазы снижена.
Увеличение мо рности фосфатного буфера свыше 0,12 М ведет к снижению активности ферментного комтекса за счет элюции примесных белков, а сни- жение молярности этого же буфера ниже 0,12 М способствует разделению алкогольдегидрогеназы и пируватдекарбоксилазы (алкогольдегидрогеназа ос5 тается на колонке с адсорбентом). Для количественного определения тиаминдифосфата в биологических объектах ферментативным методом необходимо,,
0
5
0
5
0
чтобы соотношение пируватдекарбокси- лазной и алкогольдегидрогеназной активностей в ферментном комплексе составляло 1:(0,15-0,5) при пируват- декарбоксилазной активности не менее 14,0 ед.акт./мг белка.
Превышение указанных верхних пределов не приводит к существенному повышению чувствительности ферментативного способа определения ТДФ с использованием ферментного комплекса в биологических объектах по сравнени
с применяемым соотношением пируват- декарбоксилазной и алкогольдегидрогеназной активностей, а снижение указанных пределов способствует резкому ослаблению чувствительности. Кроме того, уменьшение пируватдекарбоксила ной активности ниже 14,0 ед.акт./мг белка также ведет к значительному ослаблению чувствительности ферментативного способа определения ТДФ с использованием данного комплекса в биологических объектах.
Изобретение иллюстрируется примерами, i
Пример
1. Подготовка исходного материала, выделение и очистка комплекса пируватдекарбоксилазы и ал когольдегидрогеназы. Берут 5 кг свежих пивных дрожжей Saccharomyces carlsbergensis 7-9 регенерации. Свеж дрожжевые клетки многократно промывают холодной водой и центрифугируют 15 минут при 5000 g. Полученную пастобразную массу используют для приготовления суспензии и экстрагирования белкового препарата. Для этого 400 г дрожжевых клеток смешивают с 1750 мл водного раствора, содержащего 67 мл глицерина, 0,668 г ЭДТА, 13,2 г и 2,8 г (. Приготовленную суспензию интенсивно перемешивают в течение 10 мин. Смесь охлаждают до -2°С и обрабатывают ультразвуком при частоте 22 кГц в течение 20 мин, добиваясь полного разрушения клеток, способствующего максимальному выходу конечного продукта. Полученный дрожжевой экстракт центрифугируют в течение 30 мин при 4000 g, а затем 1 ч при 105000 g. Осевшие после центрифугирования субклеточные фракции удаляют, а на каждые 100 мл надосадоч- ной жидкости при непрерывном перемешивании приливают по 55 мл ацетона, охлажденного до -20°С, не допуская нагревания раствора выше 0°С. После
0
5
15-минутного выдерживания при этой температуре суспензию центрифугируют в течение 5 мин при 4000 g и осадок отбрасывают. На каждые 100 мл надо- садочной жидкости повторно приливают по 10 мл ацетона, снижая температуру раствора до -2°С. Через 15 мин экстракт центрифугируют, а конечный осаQ док быстро суспендируют в 400 мл 0,02 М трис-HCl буфера, рН 7,3, с 1 М ЭДТА и 5 мМ цистеин-гидрохлоридом. После 30-минутного перемешивания при О С однородный белковый раст-
5 вор подвергают фракционированию сульфатом аммония, добавляя 108 г сухого измельченного (К1Ц)г.504. Через 1 ч смесь центрифугируют, осадок отбрасывают, а на каждые 100 мл надосздоч0 ной жидкости повторно вносят по 7 г сульфата аммония в течение 20 мин при перемешивании. Высоленный белок собирают центрифугированием.
Заключительную очистку осуществ- 5 ляют методом адсорбционной хроматографии на гидроксиапатите. Экспериментально подобрано значение рН и ионной силы уравновешивающего буфера, при котором ферментный комплекс не связывается адсорбентом и обнаруживается в первых же фракциях элюирую- щего раствора, тогда как сопутствующие примеси все еще эффективно удерживаются на носителе. С этой целью белок (3 г) сульфо-аммонийной пасты диализуют в течение 4 ч против 20-кратного объема 0,12 М натрий-фосфатного буфера, рН 6,8, с 2 мМ Mg2, 1 мМ ЭДТА, 0,5 мМ ТДФ и 5 мМ цистеин
гидрохлоридом, а выпавший осадок удаляют центрифугированием. Надосадоч- ную жидкость разбавляют исходным 0,12 М натрий-фосфатным буфером до концентрации белка в растворе 10 мг/мл, смешивают с 3-кратным объемом уплотненного осадка гидроксиапатита, уравновешенного этим же буфером, и после 10 мин экспозиции при 4°С центрифугируют 10 мин при 12000g. Надосадоч- ная жидкость содержит смесь алкоголь- дегидрогеназы и пирувзтдекарбоксила- - зы. Для полноты экстракции адсорбент повторно промывают, белок надосадоч- ной жидкости объединяют, осаждают (NH4)SO (полное высаливание) и собирают центрифугированием. На данном этапе очистки (табл. 1) удельная активность пируватдекарбоксилазы возрастает в 2,9 раза (с 4,8 ед. акт./мг
71
,0 ед.акт./мг), а алкогольдегид рогеназы - в 1,75 раза (с 1 ,2 ед.акт./ до 2, ед.акт./мг) и является приемлемой для ферментативного определе- ния содержания ТДФ. Соотношение пиру ватдекарбоксилазной (14 ед.акт./мг белка) и алкогольдегчдрогеназной (2,1 ед о акт ,,/мг белка) активностей в ферментном комплексе составляет 1:0,15 (табл. 1).
Процесс выделения занимает 12 ч.
Пример 2. Получение апопиру ватдекарбоксилазы в составе комплекса и его стабилизация.
Получение апопируватдекарбоксила- зы основано на способности холоферме ча легко диссоциировать в щелочной среде с освобожде -ем ТДФ и ионов
л ji
Mg . Для удаления кофермента осажденный после фракционирования на гидроксияпатите ферментный комплекс (680 мг) растворяют в 5 мл 0,01 М трис-HCl буфера, рН 950, с 1 мМ ЭДТА и 5 мМ цистеин-гидрохлоридом, выдер- живают для полноты диссоциации при данном рН в течение 15 мин и наносят на колонку (5,4X70 см) с декстрановым гелем ЭД-1,0 (фракция до 50 мкМ), уравновешенную исходным буфером. По
мере выхода белка раствор собирают в объеме 30 мл. ТДФ элюируется позже начиная с 40 мл, В таком состоянии без кофермента апофермент чувствителен к наличию с льфгидрильных реагентов, ионов металла, воздействию рН, температуры и ионной силы буферного раствора, поэтому скорость тока жидкости через колонку должна быть достаточно высокой (36-48 мп/ч), а элюируемый белок после выхода немедленно стабилизирован (способность к рекомбинации апопируватдекарбокси- лазы дрожжей сохраняется в 0,01 М трис-HCl буфере, рН 9,0 в течение ч), Все операции по выделению апофермента и последующей защите от инактивации проводят при .
Стабилизацию комплекса апопируват декарбоксилазы и алкогольдегидрогена зы осуществляют глицерином или сульфатом аммония в присутствии ионов Mg2-4. В первом случае значение рН элюируемого с колонки раствора апофермента снижают 0,05 М натрий-фосфатным буфером до 6,8, а затем сюда же вносят глицерин и 0,5 М MgSO$ до конечной концентрации компонентов 30 и 0,02 М соответственно. Во вто
0
5
0
5
483
(
0
5
0
8
ром - к белковому элюату при непрерывном перемешивании приливают равный объем 5,4 М сульфата аммония в 0,1 М натрий-фосфатном буфере, ,8, с 0,04 М MgS04, после чего белок лиофилизируют на установке для сублимационной сушки при температуре
и давлении б.. В 30%-ном
г.
5
0
5
глицерине в присутствии Mg реактивация апопируватдекарбоксилазы с низкими концентрациями ТДФ и алкоголь- дегидрогеназная активность остаются высокими в пределах 1,0-1,5 недель, позволяя проводить серийные исследования уровня ТДФ в биологических объектах без ежедневной практики подбора необходимой концентрации белка- апофермента, как это наблюдается в iлучае работы с сульфо-аммонийными препаратами.
Лиофилизированный из 2,7 М ( 0,02 М MgSOq. комплекс стабилен в течение года (табл. 2).
Пример 3. Модельная система количественного определения ТДФ в биологических объектах: в животных тканях, крови.
С целью диагностики В -недостаточ- ности с использованием ферментного комплекса животных за 12 ч до забоя лишают пищи. После декапитации животного исследуемые ткани быстро извлекают, очищают от оболочек: продавливают через пресс с диаметром пор 1 мм и гомогенизируют в гомогенизаторе с тефлоновым пестиком (7-9 движений) с 5 объемами охлажденной до 0 - 2°С 17%-ной трихлоруксуеной кислоты. Гомо- генат центрифугируют 10 мин при
3000 об/мин, а осадок повторно перемешивают с 5 объемами 7%-ной трихлоруксуеной кислоты при 0 - 2 С с по- - следующим центрифугированием. Полученную от двух центрифугирований на- досадочную жидкость дважды обрабатывают 3 объемами насыщенного водного раствора эфира для удаления трихлоруксуеной кислоты. Конечное разведение для тканей печени, почек, сердца, равное 1:20, а мозга и мышц 1:10 достигается внесением к безбелковому экстракту 0,05 М натрий-фосфатного буфера рН 6,8, после чего отбирают аликвоты по 0,1 мл для рекомбинации содержащегося в тканях ТДФ с апрпи- руватдекарбоксилазой фурментного комплекса. Рекомбинацию проводят при комнатной температуре в течение 30 мин.
Для этого к анализируемым аликвотам добавляют по 0,1 мл 0,02 М MgSO$ и 0,1 мл раствора очищенного ферментного комплекса апопируватцекарбокси- лазы с алкогольдегидрогеназой (200- 600 мкг белка), разбавленного до необходимой концентрации 0,05 М натрий- фосфатным буфером, рН 6,8. Общий
оси абсцисс используемые концентрации ТДФ, а по оси ординат соответствующие им значения экстннкции за вычетом контроля, фиксирующего степень самопроизвольного окисления. НАД- Н в отсутствие (см. фиг. 1-3) . Ь ри ферментативной детекции общего ТДФ в анализируемых пробах крови или тка




| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения пируватдекарбоксилазы из пивных дрожжей | 1988 |
|
SU1541255A1 |
| СПОСОБ ПРОИЗВОДСТВА МУЛЬТИФЕРМЕНТНОГО ПРЕПАРАТА | 2009 |
|
RU2437935C2 |
| Способ получения @ - @ -галактозидазы | 1982 |
|
SU1082812A1 |
| Субстанция протеолитического фермента на основе Протосубтилина ГЗХ, иммобилизованного на хитозане, и композиция для лечения гнойно-некротических ран | 2016 |
|
RU2630668C1 |
| Способ получения комплексного ферментного препарата ацетокиназы и аденилаткиназы из биомассы Е.coLI | 1979 |
|
SU837066A1 |
| СПОСОБ ПОЛУЧЕНИЯ ДРОЖЖЕВОЙ АЛКОГОЛЬОКСИДАЗЫ | 1990 |
|
RU2032743C1 |
| Способ получения сорбента | 1981 |
|
SU979354A1 |
| Способ получения рестриктазы, способной узнавать и расщеплять последовательность нуклеотидов GTCGAC | 1989 |
|
SU1752769A1 |
| Способ получения протеолитического препарата для медицинского применения | 2015 |
|
RU2610669C1 |
| Способ получения 7-( - -аминофенилацетамидо)-дезацетоксицефалоспорановой кислоты (цефалексина) | 1972 |
|
SU579901A3 |

Изобретение относится к способам получения ферментных препаратов из микробиологического сырья и может быть использовано в ферментной промышленности, а получаемый комплекс - для количественного определения уровня тиаминдифосфата (ТДФ). Цель изобретения - расширение технологических возможностей способа, ускорение процесса и удешевление целевого продукта, Способ предусматривает использование свежей пастообразной массы пивных дрожжей 7-9 регенераций. Готовят суспензию для экстрагирования белкового препарата, которую обрабатывают ультразвуком с частотой 20-22 кГц в течение 18-25 мин с последующим двух- этапным фракционированием ацетоном и сульфатом аммония. На первом этапе ацетонового фракционирования на каждые 1UU мл дрожжевого экстракта добавляют 55 мл ацетона, а на втором - 10 мл. Заключительную очистку выполняют адсорбционной хроматографией на аксиапатите элюцией комплекса 0,12 М фосфатным буфером до соотношения пи- руватдекарбоксилазной и алкогольде- гидрогеназной активностей в ферментном комплексе 1 : (0,15-0,5) при пи- руватдекарбоксилазной активности не менее 14 ед. акт./мг белка. После заключительной очистки белкового препарата получают апопируватдекарбокси- лазу в составе комплекса с последующей его стабилизацией. 3 ил. 3 табл. а SS О5 ГС О Ј 00 со
15
20
25
30
объем доводят исходным 0,05 М натрий- JQ ней регистрируемые в эксперименте фосфатным буфером до 0,8 мл. По истечении времени инкубации в каждую пробу вносят по 2,0 мл 0,15 М натрий- цитратного буфера, рН 5,9 и 0,1 мл 0,2 М пировинограднокислого натрия. Реакцию запускают добавлением 0,1 мл 0,5 мМ НАД Н, измеряя изменение оптической плотности раствора первые 3 мин приД 340 нм. По суммарной величине спектрофотометрически регистрируемой оптической плотности (OD) находят соответствующее ей значение точки на калибровочной кривой. Ее параметры, умноженные -на исходное разведение ткани, соответствуют искомому значению концентрации общего ТДФ.
Кровь отбирают из безымянного пальца обследуемых пациентов, в течение 8-10 ч до анализа не принимавших пищу. Для определения уровня ТДФ 0,2 мл капиллярной крови смешивают с 0,4 мл охлажденного до 4°С 0,05 М натрий-фосфатного буфера, рН 6,8, содержащего 2%-ный гепарин. После 15-минутной экспозиции при 4°С смесь кипятят 2 мин, центрифугируют 10 мин при 3000 об/мин и отсюда отбирают аликвоты по 0,2 мл для рекомбинации ТДФ исследуемых проб с апопируватдекарбоксилазой. Условия проведения рекомбинации идентичны таковым для животных тканей.
Искомые значения точек для калибровочного графика находят в аналогичных для рекомбинации исследуемых образцов крови или тканей условиях, используя вместо 0,1 мл тканевого
экстракта или 0,2 мл крови известные концентрации (0,005-0,2 мкг) хрома- тографически чистого препарата ТДФ. По получаемым при этом разностям экстишсции восстановленного и окисленного НАД, регистрируемым первые 3 мин протекания сопряженной ферментативной реакции реактивированной холопи- руватдекарбоксилазы и алкогольдегид- рогеназы дрожжей при 340 нм, строят калибровочную кривую, откладывая по
35
40
45
величины экстраполируют на калибро вочную кривую, а затем на ось абсц с известными концентрациями ТДФ.
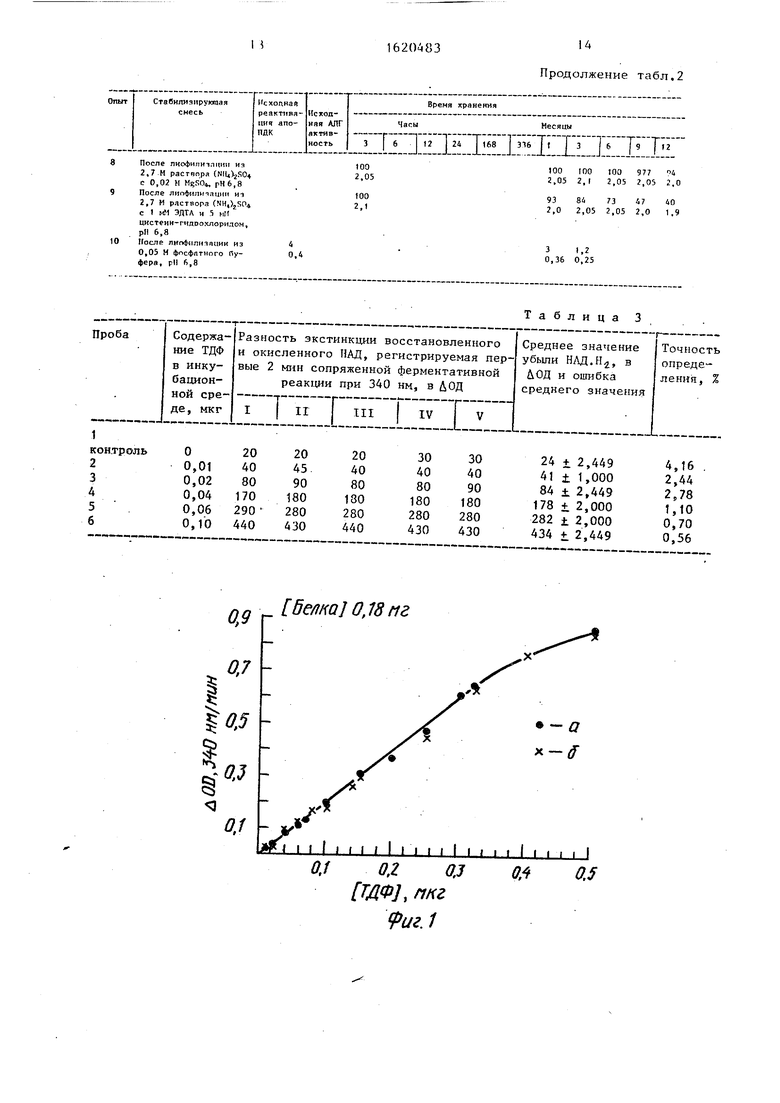
Пример 4. Интервал концен раций ТДФ, в котором сохраняется л нейность сопряженного процесса с и пользованием комплекса.
Н№ фиг. 1-3 представлены зависи мости суммарной скорости сопряженн ферментативного процесса от концен рации ТДФ при различных полярных к центрациях предлагаемого комплекса в отсутствие (фиг. 1-2 а) и при до бавочном внесении (фиг. 1 и 2 б) к мерческой алкогольдегидрогеназы. К видно из графиков, в исследуемом и тервале концентраций кофермента, п содержании белка 0,18-0,60 мг, удо летворительная линейность суммарно скорости процесса сохраняется до 0,3 мкг ТДФ. При более низком чем 0,18 мг содержании комплекса в сил недостаточной разности экстинкции м ду контрольной и опытной пробами в случае малых концентраций ТДФ снижа ется точность ферментативного определения (см. таблицу 3). Концентрация комплекса 0,6 мг практически об печивает белковое насыщение и ее ув личение способствует непроизводител ным затратам комплекса. Дополнитель ное внесение коммерческой алкогольдегидрогеназы (50 мкг на пробу) к комплексу не ведет к увеличению ско рости (см. фиг. 1-2) сопряженного п цесса, свидетельствуя тем самым о достаточности ферментативной активности алкогольдегидрогеназы дрожжей в составе комплекса.
50
и
55
Пример 5. Чувствительность точность определения.
Согласно данным табл. 3 ферментативный метод количественного опре деления общего ТДФ с использованием комплекса позволяет фиксировать кон центрации кофермента в дозе 0,005 мк а точность измерения составляет в среднем 1,96%.
5
0
5
0
Q ней регистрируемые в эксперименте
5
0
5
величины экстраполируют на калибровочную кривую, а затем на ось абсцисс с известными концентрациями ТДФ.
Пример 4. Интервал концентраций ТДФ, в котором сохраняется линейность сопряженного процесса с использованием комплекса.
Н№ фиг. 1-3 представлены зависимости суммарной скорости сопряженного ферментативного процесса от концентрации ТДФ при различных полярных концентрациях предлагаемого комплекса в отсутствие (фиг. 1-2 а) и при добавочном внесении (фиг. 1 и 2 б) коммерческой алкогольдегидрогеназы. Как видно из графиков, в исследуемом интервале концентраций кофермента, при содержании белка 0,18-0,60 мг, удовлетворительная линейность суммарной скорости процесса сохраняется до 0,3 мкг ТДФ. При более низком чем 0,18 мг содержании комплекса в силу недостаточной разности экстинкции между контрольной и опытной пробами в случае малых концентраций ТДФ снижается точность ферментативного определения (см. таблицу 3). Концентрация комплекса 0,6 мг практически обеспечивает белковое насыщение и ее увеличение способствует непроизводительным затратам комплекса. Дополнительное внесение коммерческой алкогольдегидрогеназы (50 мкг на пробу) к комплексу не ведет к увеличению скорости (см. фиг. 1-2) сопряженного процесса, свидетельствуя тем самым о достаточности ферментативной активности алкогольдегидрогеназы дрожжей в составе комплекса.
и
Пример 5. Чувствительность точность определения.
Согласно данным табл. 3 ферментативный метод количественного определения общего ТДФ с использованием комплекса позволяет фиксировать концентрации кофермента в дозе 0,005 мкг, а точность измерения составляет в среднем 1,96%.
Формула изобретения Способ получения ферментного препарата для количественного определения тиаминдифосфата. в биологических объектах, предусматривающий выделени пируватдекарбоксилазы из экстракта пивных дрожжей путем фракционирования ацетоном, фракционирования сульфатом аммония в интервале концентраций 44-53% насыщения и хроматогра- фической очистки, отличающийся тем, что, с целью расширения технологических возможностей способа, ускорения процесса и удешев ления конечного продукта, совместно
с пируватдекарбоксилазой выделяют алкогольдегидрогеназу, при этом фракционирование экстракта ацетоном проводят в интервале концентраций 35,5- 41,1% и хроматографическую очистку проводят методом адсорбционной хроматографии на гидроксиапатите в 0,12 М фосфатном буфере до соотношения пиру- ватдекар&оксилазной и алкогольдегид- рогеназной активностей в ферментном препарате 1:(0,15-0,5) и до величины пируватдекарбоксилазной активности не менее 14,0 ед/мг белка, а пиру- ватдекарбоксилазу в составе препарата получают в виде апофермента.
Таблица t
Продолжение табл.2
| Ullrih.I | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Accay of Thiamine Pyro- phosphate, Methods in Enzymology | |||
| Кинематографический аппарат | 1923 |
|
SU1970A1 |
| Шкив для канатной передачи | 1920 |
|
SU109A1 |
| Способ получения пируватдекарбоксилазы из пивных дрожжей | 1988 |
|
SU1541255A1 |
| Способ гальванического снятия позолоты с серебряных изделий без заметного изменения их формы | 1923 |
|
SU12A1 |
