Изобретение относится к способу получения дипептидов или их приемлемых для пищевых продуктов солей - новых биологически активных соединений, которые могут найти применение в пищевой промышленности. .
Цель изобретения - получение новых производных дипептидов - малотоксичных и обладающих более высоким подслащивающим эффектом.
Пример 1. Д+)Фенхиловый сложный эфир, а -1 -аспартил-2-метилаланина.
Аминоизомасляную кислоту, защищенную группой Ы-СВ2,фастворяют в 1,2-ди- хлорэтане (50 мл) в атмосфере аргона при 0°С. В 1,2-дихлорэтан (10 мл) добавляют раствор N.N-диметиламинопиридина (0,5 эквивалента) и/3(+)фенхилового спирта эквивалент). Затем добавляют дицикло- гексилкарбодиимид (1,1 эквивалента) в твердом виде. После пяти дней перемешивания при комнатной температуре из раствора удаляют мочевину фильтрованием, а полученный фильтрат разбавляют петро- лейным эфиром (50 мл). Этот раствор затем снова осветляют фильтрованием, а полученный фильтрат выпаривают до образования пасты в высоком вакууме в роторном испарителе. Колоночной хроматографией на си- ликагеле с использованием петролейного эфира и этилацетата при соотношении 15;1 получают чистый продукт с выходом в 75-79% в виде белого кристаллического вещества.
ЯМР (CDCIa). $ 0,90 (с. ЗН): 1,05 (с, ЗН); 1,10 (с, ЗН); 1,20-1,80 (м, 7Н); 1,60 (с. 6Н); 1,20 (с, 1Н); 5,10 (с, 2Н); 5,55 (с. 1Н);7,40 (с, 5Н).
af5D -11,65°(MeOH).
Т. пл. 83-85°С.
Защитную группу удаляют гидрогенизацией в присутствии палладия на угле (10%) в метаноле с количественным выходом свободного аминоэфира.
Амин тут же растворяют в N.N-диме- тилформамиде и связывают с предшественником аспаргиновой кислоты с использованием хлорида меди (II) с получением 90%-ного выхода сложного бензиловоео эфира - а 2-метилаланин Д+)фенхилового эфира N-CBZ or-L-аспарги- новой кислоты.
ЯМР (CDCIs): 5 0,90 (С, ЗН); 1,05 (с. ЗН); 1.10 (с. ЗН); 1,20-1,80 (м, 7Н); 1,6 (д, 6Н); 2,70-3,15(м,2Н); 4,1-4,2 (м, 1Н);4,20(с, 1Н); 4,60 (с, 1Н); 5,10 (с, 4Н); 5,60 (д, 1Н); 5,80 (д, 1Н);5,90(д, 1Н);7,40(с, ЮН).
Защитную группу продукта удаляют гидрогенизацией с последующей очисткой колоночной хроматографией, Rp Сш, с использованием элюанта метанол - вода при соотношении 85:15.
-3,30°(МеОН).Т. пл. 121-123°С.
Определение сладости этих соединений дало следующие результаты (табл. 1).
Пример 2. а-Ь-Аспартил-О-аланин Д+)фенхиловый сложный эфир.
А. Экзо-Д- (+) - фенхол.
В кипящую суспензию, состоящую из 72,65 г изопропилата алюминия в 300 мл только что перегнанного изопропилового спирта, добавляют по каплям 27,1 г R-(-)- фенхона в 50 мл изопропанола. Реакцию прекращают через шесть дней, когда с помощью газовой хроматографии (Карбовакс 20 М) определяют, что содержание кетона уменьшилось более чем на 50%. С помощью капиллярной хроматографии (Су- пелковакс 10) также определяют, что отношение экзо/эндоизомеров для фенхо- ла равно 3/1. После охлаждения эту смесь фильтруют и тщательно промывают дихлор- метаном. Полученный осадок растворяют в 5%-ной HCI (100 мл) и экстрагируют дих- лорметаном. Затем объединенные растворы дихлорметана промывают 50%Гной НС (50 мл), насыщенным раствором NaHCOa (50 мл) и водой (50 мл) и высушивают над MgSCXi. Фильтрованием и удалением рас- творителя получают 23,44 г масла, которое на 40% состоит из непрореагировавшего фенхона и на 60% из изомеров а- и/ -фен- хола.
0
5
0
5 0 5
5
0
5
0
В течение 24 ч кипятят смесь из 12 г (0,78 моль) /3 - и а -фенхола, 11,9 мл (1,1 эквивалента) триэтиламина и 15,9 г пара-нит- робензоилхлорида (1,1 г) в 500 мл сухого дихлорметана. Затем разделяют смесь /8 la сложных эфиров с помощью вытеснитель- ной хроматографии на силикагеле с использованием гексан/этилацетат при их соотношении 40/1. Выделяют 6,0 г экзо- фенхил-пара-нитробензата, ,1 ° (в бензоле). После гидролиза в щелочной среде нитробензатного эфира (кипячение в избытке NaOH в метаноле) получают 3 г фенхола (9/1:/ /а). ft - (+ фенхол; ар5о +23,4° (чистый).
ЯМР: д 0,95-1,8 (16Н, м, СН2, СНз); 3,0 ч на млн. (1Н, с, СН-0).
B.N-Cbz-D-Аланин, (З-(+)-фенхиловый сложный эфир.
В тщательно перемешанный раствор 1,3 (+}-фенхола в 20 мл сухого дихлорметана добавляют 1, 9 г (0,0084 моль) N- Cbz-D-аланина и полученный раствор охлаждают до 0°С. Затем добавляют сюда 0,113 г пара-диметиламинопиридина и 1,91 г дициклогексилкарбодиимида. После 24 ч реакцию прекращают и раствор фильтруют. Растворитель испаряют, а масляный остаток растворяют в диэтиловом эфире, затем тщательно промывают5%-ной HCI (25 мл), насыщенным ЫаНСОз (25 мл), водой (25 мл) и высушивают над МдЗОд. После фильтрации и испарения растворителя полученный продукт очищают хроматографией на силикагеле с конечным выходом 1,86 г N-Cbz-D-аланин З -(+)-фен- хилового эфира.
af5D +3,86°ЯМР:(5 0,8-1,8 ч. на млн. (19Н, м, СНа, СНз); 4,2 ч. на млн. (1Н, с, СН-0); 4,4 ч. на млн. (1Н, м, СН-С); 5,1 ч. на млн. (2Н, с, CH2-Ph); 5,4 ч. н/млн. (1Н, д, NH); 7.4 ч. на млн. (5Н, с, Ph).
C.0-Аланин, / -фенхиловый сложный эфир.
N-Cbz-D-аланин, ft -(+)-фенхиловый сложный эфир растворяют в 50 мл метанола и гидрируют 2 ч над 0,1 г 5% Pd/C в трясучке Паара. Затем раствор фильтруют через целит, промывают метанолом, концентрируют и кристаллизованный остаток растворяют в дихлорметане.
D.М-СЬг-}8-Бензил-1--аспартил-0-ала- нин,(+) фенхиловый сложный эфир.
В дихлорметановый раствор, содержащий сложный эфир D-аланина (0.00355 моль), добавляют эквимолярное количество/3 -бензил N-Cbz-L-аспаргиновой кислоты (1,27 г) и 0,526 г Си (II) CI2. После растворения CuCIa
добавляют ДЦК (0,81 г). После 24 ч реакцию заканчивают, отфильтровывают мочевину и испаряют растворитель. Желтое масло растворяют в диэтиловом эфире (25 мл) и промывают 5%-ной HCI (25 мл), затем насыщенным раствором ЫаНСОз(25 мл) и НаО (25 мл). Слой простого эфира высушивают над МдЗСм и испаряют с получением 0,95 г конечного продукта.
ЯМР: д 0,85-1,80 (19Н, м, СН2, СНз); 4,2 ч. на млн. (1Н, с, СН-0); 4,5-4,7 ч. на млн. (2Н, м, М-СН-р); 5,1 ч. на млн. (4Н, с, ОСН2- Ph); 5,95 ч. на млн. (1 Н, д. NH); 7,05 ч. на млн. (1 Н. д, NH); 7,4 ч. на млн. (ЮН, с, Ph).
Е. a-L-Аспартил-О-аланин, $ -(+)-фенхи- ловый сложный эфир.
0,95 г защищенного дипептида растворяют в 50 мл метанола, в который добавляют 0,1 г 10% Pd/C. Полученный раствор гидрогенизируют в течение 24 ч в трясучке Паара. Полученный таким образом раствор фильтруют и испаряют досуха с получением 0,194 г твердого вещества: а о - -0,867°С,
Полученный продукт очищают на ВЭМХ с обращением фаз (85% метанол/вода) с получением 75 мл 1--аспартил-О-аланина,/3 - (+) фенхилового сложного эфира.
ЯМР: 0.8-1,8 (19Н, м,0СН2. СНз); 2,3 - 2,4 ч. на млн. (2Н, м, СНа-С) ; 4,2 ч. на млн. (1Н, с, ОСН); 4,5 ч. на млн. (2Н, м, N-CH); 8.8 ч. на млн. (1Н, с. Н-С).
Определение сладости этого соединения дало следующие результаты (табл. 2).
Полученные дипептиды получают или в виде свободной кислоты, или в виде в пищевом отношении приемлемых солей, например в виде соответствующих амино-. солей, т.е. в виде следующих соединений гидрохлорида, сульфата, гидросульфата, нитрата, гидробромида, гидройодида, фосфата или гидрофосфата; или в виде солей щелочного металла, например натрия, калия, лития, или же в виде таких солей щелочноземельных металлов, как кальций или магний, а также солей алюминия, цинка и других подобных солей.
Превращение производных свободного пептида в их физиологически приемлемые соли осуществляется обычными хорошо известными способами, например непосредственным контактированием соединений формулы I с минеральной кислотой, гидроокисью щелочного металла, окисью щелочного металла или карбонатом, либо гидроокисью щелочноземельного металла,
его окисью, карбонатом, либо его прочими более сложными соединениями.
Перечисленные выше и физиологически приемлемые соли можно также использо- вать в качестве подслащивающих агентов, которые обычно имеют повышенную растворимость и стабильность по сравнению со своими свободными формами.
Пример 3. Изучена стабильность оН -аспартил-2-метилаланин Г/(+)фенхило- вого сложного эфира (пример 1), a ,L-ac- партил-0-аланин-|Д+)фенхилового сложно- го эфира (пример 2) и аспартам (С-аспар- тил-1 -фенилаланинметилового эфира) в буферных растворах при рН 3,5 и 7 при 50, 75 и 100°С, были получены следующие результаты:
Период вращения, при
100°СрНЗ рН5 рН7
Пример 1 8,4 33 67
Пример 2 3,9 14 10
Аспартам 5,3 5,3 « 1
Период полупревращения, дни, при
75°СрН 3 рН5
Пример 1 3,0 15,0
Пример 2 1.3 5,1
Аспартам 0,9 1,1
Период полупревращения, дни, при
50°СрНЗ рН5
Пример 1 64 150
Пример 2 22 131
Подслащивающие вещества по примерам 1 и 2 имеют исключительно высокую стабильность в буферных растворах при рН 3, 5 и 7. Соединения примеров 1 и 2 обладают лучшей стабильностью в буферных растворах по сравнению с аспартамом, за исключением случая с рН 3 при температуре 100°С, когда аспартам имел промежуточную стабильность между соединениями примеров 1 и 2. Соединение примера 1 более стабильно, чем соединение примера 2 Период полупревращения соединения примера 1 был в два- и три раза продолжительнее в буферных растворах при 75 и 100°С и рН 3 и 5, чем период полупревращения соединения примера 2. Период полупревращения соединения примера 1 в буферном растворе при 50°С и рН 5 был в 1,1-2,9 раза продолжительнее, чем у соединения примера 2.
Проведенные испытания показали, что производные дипептидов, полученные а условиях предлагаемого способа. - соедине- ния практически нетоксичные и слаще сахарозы в 900-5900 раз и превосходят по сладости аспартам в 5-23 раза.
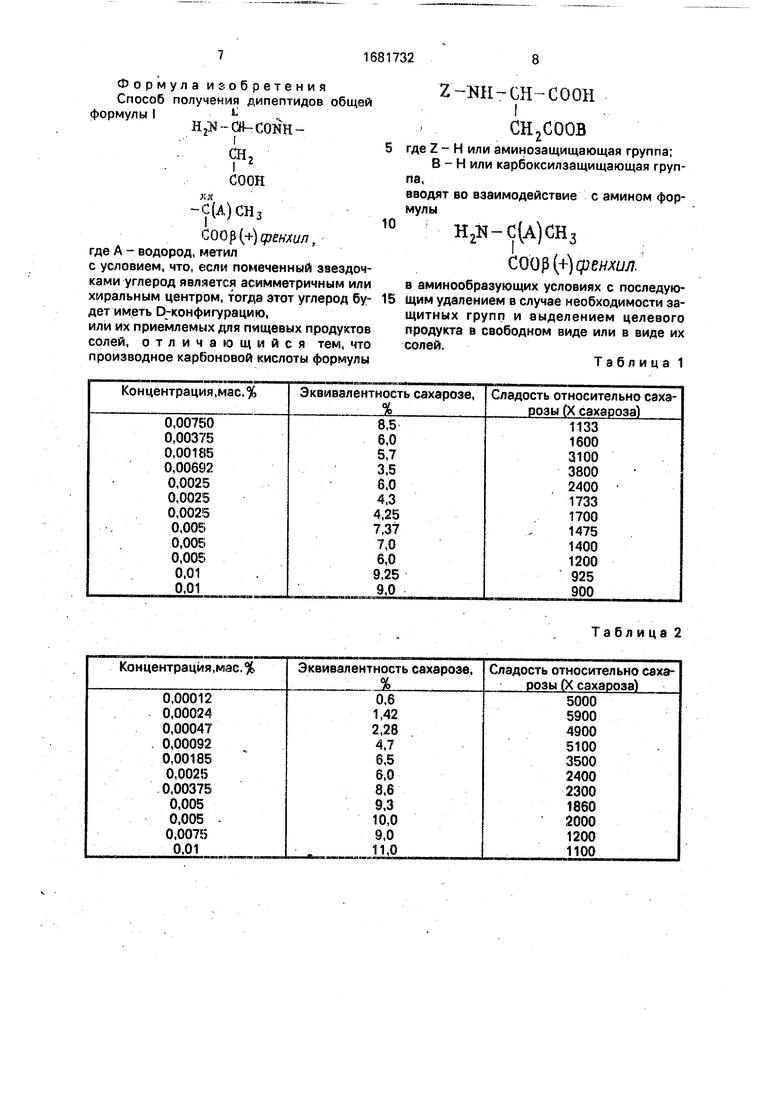
Формула изобретения Способ получения дипептидов общей формулы
Н2Я-СН-СОШсн,
СООН -С(А) СН3
XX
С00р(+)фенхил, где А - водород, метил с условием, что, если помеченный звездочками углерод является асимметричным или хиральным центром, тогда этот углерод будет иметь D-конфигурацию, или их приемлемых для пищевых продуктов солей, отличающийся тем, что производное карбоновой кислоты формулы
0
2-Ш1-СН-СООН
I
СН2СООВ
где Z - Н или аминозащищающая группа;
В - Н или карбоксилзащищающая группа,
вводят во взаимодействие с амином формулы
Н2К
С(А)СН3 С00р(+)фекхш7.
в аминообразующих условиях с последую- щим удалением в случае необходимости защитных групп и выделением целевого продукта в свободном виде или в виде их солей.
Таблица 1


| название | год | авторы | номер документа |
|---|---|---|---|
| Состав пищевого продукта | 1988 |
|
SU1834647A3 |
| Способ получения производных цефалоспорина или их солей | 1981 |
|
SU1190987A3 |
| НОВЫЕ ПРОИЗВОДНЫЕ АСПАРТИЛОВОГО ДИПЕПТИДНОГО ЭФИРА И ПОДСЛАСТИТЕЛИ | 1999 |
|
RU2192430C2 |
| Способ получения производных бензиламина или их солей | 1973 |
|
SU571188A3 |
| Способ получения сложных эфиров 6-амидинопенициллановых кислот или их аддитивных солей с кислотами и его вариант | 1980 |
|
SU1015830A3 |
| Способ получения производных аминокислоты | 1985 |
|
SU1468411A3 |
| Способ получения 6-замещенных метиленпенициллановых кислот,или их сложных эфиров,или их солей с щелочными металлами | 1985 |
|
SU1395144A3 |
| Способ получения имидазохинолиновых соединений или их фармацевтически приемлемых солей | 1987 |
|
SU1470192A3 |
| КОМПОЗИЦИИ ВЫСОКОИНТЕНСИВНЫХ ПОДСЛАСТИТЕЛЕЙ, ИМЕЮЩИЕ УСОВЕРШЕНСТВОВАННУЮ СЛАДОСТЬ, МОДИФИКАТОР ВКУСА И ИХ ПРИМЕНЕНИЕ | 2000 |
|
RU2238945C2 |
| Способ получения дипептидныхэфиРОВ | 1978 |
|
SU841583A3 |
Изобретение касается пептидов, в частности получения дипептидов общей ф-лы НО-С(ОЬСН2-СН(МН2)-С(0)-МН-С (АХСНз) - С(0}-ОД+)фенхил, где А Н или СНз с условием, что, если С является асимметричным или хиральным центром, то этот углерод будет иметь D-конфигурацию, или их приемлемых для пищевых продуктов солей, что может быть использовано для обеспечения подслащивающего эффекта. Цель изобретения - повышение сладости и снижение токсичности нового соединения указанного класса. Синтез ведут реакцией производного карбоновой кислоты ф-лы Z-NH-CH(C/0/OB)-CH2-C(0)-OH. где Z Н, аминная защита, с амином ф-лы СНз- C(AXNH)2-C(0}-0 Д+)фенхил, в аминообразу- ющих условиях с последующим удалением, в случае необходимости, защитных групп и выделением целевого продукта в свободном виде или в виде их солей. Новые вещества имеют высокую стабильность в буферных растворах при рН 3 или 5, или 7, практически не токсичны и превосходят по сладости аспартам 5-23 раза и сахарозу в 900-5900 раз. 2 табл. fe
Таблица 2
| Шредер Э., Любке К | |||
| Пептиды, М.: Мир, 1967, ч | |||
| I, с | |||
| Способ получения бензидиновых оснований | 1921 |
|
SU116A1 |
