1
Изобретение относится к опосо-бу получения дезацетоксидефалоопоринов, которые находят применение в фармацевтической промышленности.
Известен снособ получения сложных эфиров 7;в-зцилами:Но-3-метил-1цеф-3-ем-4-карбо 10вой КИСЛОТЫ, заключающийся s том, что сложный эфир 1-оксида бр-ациламинопен-ициллановой кислоты ннгревают л.ри 80-150° С в в среде инертного органического растворителя в присутствии кислоты, такой как /г-толуолсульфокислота, и третичного эмида карйоновой .кислоты, с одновременным удалением или инактивацией образующейся в результате реакции воды и:31вест1НЫ|МИ дриемами. Продукты выделяют известным способом. Выход продуктов не превышает 77,7%. При этом продукт, полученный с наибольшим выходом, имеет т. пл. 184-196,5° С, что указывает на значительное содержание примесей в нем (температура плавления чистого вещества 189-191°С).
С целью получения продуктов лучшего каче(ст1ва и с большим выходом, предлагается способ полумения сложных эфиров 7 у9-ацилаадино-3-метил-цеф-3-е1М-4-1кар(боновой кислоты, заключающийся в том, что сложный эфир 1-оксида 6 -ациламинопвнициллановой кислоты Нагревают в диоксане или кетоне, и, т. кип. 75-1,20°С, или в сложном эфире, имеющем т. кип. 75-140° С, или в простом диметилоБом эфире диэтиленгликоля в присутствии соли азотистого оснавания, .щего значение рКв не менее 4, и .кислоты, или смеси соединений, образующих такую соль непосредственйо в реа.кщионной смеси. Продукты выделяют илвестны.м способом. По предлагаемому способу выход некоторых продуктов повыщается до 89% и качество (судя по темлературе плавления) несколько выше.
Кислота, образующая соль, представляет собой, наетример, органическую сульфокислоту, фосфорную или трифторуксусную -кислоту. Органическая сульфокислота представляет собой алкил-, арал;кил-, или арилсульфокислоту (иапри1мер, метансульфокислоту, толуолпара-сульфокислоту, лара-1ксилолсульфОкислоту или нафталин-2-сульфокислоту), либо пиридилсульфокислоту. Фосфорная кислота представляет собой ортофосфорную, полифоофориую, пирофосфорную или фосфористую кислоты, либо фосфиновую кис.тоту. Фосфиновая кислота представляет собой алифатич-ескую, аралифатическую, или арилфосфиновую кислоты. Алифатическая, ар.алифатическая или ариловая прулпа такой фосфиновой кислоты является углеводородной группой (налример, грЗПпой ппзщего avткил.a, фенила низшего алкила или фенила), либо углеводородиой группой, за;мещенной, налример атомом галогена
или л-штрогру:ппой. Примерами алифатических фосфиновых .кислот являются низшие алкилфосфиновые кислоты и замещенные (на.пример, галогенза/мбщенные низшие алкилфосфино;вые кислоты, такие как .метанофосфнкоеая, этайфосфиновая, дихлормета:Нфосфиновая, трихлорлштанфосфшювая и иодометаифосфи:Иовая кислоты. Примерами арилфосфшювых кислот являются бензолфосфиновые кислоты и замещенные (например, галотен- или нитрозалгещенные бензолфосфиновые кислоты, такие как бромббизолфосфиновые и нитробензолфосфиновые).
Азотистое основание может представлять собой как органическое, так и неорганическое соединение. Термин «-азотистое основание иснользуется здесь как понятие, с помощью которого обычно определяется основание, включающее азот, хотя это основание может ва лю1чать и другие гетероато1мы, на иример кислород. Одна-ко, предпочтительно использовать органические амины. Основа тия, которьг° можно использовать, имеют знамение рКв лля лротони.рования не менее 4 (измеренное в воде при 25°С). Данное основание может представлять СО-бой многофункциональное (включающее много фушщиональных г.рулн) основание, включающее фунициональную группу азота с указанной величиной рКв для первой стадии протаии.рования. Эти основания имеют величину рКв (в воде) преимущественно не менее 7.
Органргческое основание может быть первИ(Чным, вторичным или третичным, однако предпочтительно использовать слабые третичные органические основания. Примерами таких третичдых Орг аличеоких оснований являются непредельные гетероциклические основания, такие как лиридин, хииолин, изохинолин, бензиашдазол, и их гомологи, например ал«илзамещенные пиридины и хинолины, такие как а-, Р- и -а1иколииы, а также 2- и 4-метилхинолины. Другие замещенные гетероциклические основания, которые могут использоваться в соответствии с предлагаемым способом, включают основания, замещенные атомам галогена (например, хлором или бромам), ацилом (наиришер, формилО|М или ацетилом), ащиламидо- (например, ацетамидо-), циано-, карбоксил-, альдоксиминогрупной и т. д. Другие органические основания, которые могут ис/пользоваться в соответствии с предлагаемым способом, включают анилин и замещенные в ядре анилины, такие как галогеноаиилины (например, орто-хлорл-нилин, метахлоранилин и пара-хлоранилин), низшие алкиланилины (например, ортометиланилин и метаметил.анилин), окси- и низшие алкоксианилины (например, ортометаксианилин и мета-оксианилии), нитроанилины (например, 1мета-нитроанили«) и карбокси.аиилины (например, мета-1карбоксиа.нилин), а также низшие N-алкилаНилины (например, N-метиланилин).
Наиболее предпочтительными солями азотистых оснований являются соли, получаемые в результате химической фосфорной кислоты или сульфокислоты с азотистым основанием, например в эквимолярных соотнощениях. Положительные результаты были получены в тех случаях, когда в качестве катализатора использовали соли ортофосфорной кислоты или фосфиновой кислоты. Примерами
фосфИНовых кислот являются алифатические фосфиновые КИСЛОТЫ и арилфосфиновые кислоты, упомянутые выше. Наиболее предпочтительны соли, полученные в результате химической реакции, в основном, взятых в эквимолярных соотношениях .кислоты с ароматически гетероциклически м третичиым органичес ки.м азотистым основанием.
Положительные рез льтаты лолучены в том случае, когда в качестве катализатора использовали соли пиридина, хинолина, изохинолина или их производные, замещенные низшим алкилом, галогеном, ацилом, ациламлдо-, циано- , карбоксил- или альдо1кси1миногруплой.
Наиболее предпочтительными солями азотистых оснований являются соли, полученные в результате химической реакции фосфорной кислоты с арО|Матически1М гетероциклическим третичным органически-м азотистым основанием, в особенности в результате указанной химической реакции в основном при эгавимолярном соотношении реагентов. Положительные результаты были получены в случаях, когда использовались соли ортофосфорной
или фосфиновой кислоты с пиридином, хинолинам, изохинолином, или такие основания, замещенные например, низшим алкилом, атомом галогена, ацилом, ациламидом, цианом, карбоксилом, или альдаксимином. Так, например, используемые в лредлагаемО М способе катализаторы включают; пиридин, 2-,метил4:(метил-п,иридин; хинолиновые и изохинолиновые соли ортофосфорной, метанфосфиловой, этанфосфиновой, иодометанфосфиновой, дихлорметанфосфиновой, трихлорметанфосфиновой, бромбензолфосфиновой и нитробензолфосфиновой кислот.
Соли, используемые в способе, могут быть лолучены из кислоты и основания, взятых в
таких соотнащениях, что одна или несколько кислотных функциональных групп полностью нейтрализуются основанием. Ка лравило, в данном способе используются раюнамолярные соотиощения основания и кислоты. Однако,
если это необходимо, могут июпользаваться иные молярные соотношения, чем тем, которые определены выше. Например, может использаваться менее, чем молярное количество азотистого основания, так что, наряду с солью,
катализатор включает некоторое количество свободной кислоты. Кроме того, можно использовать более чем молярное количество азотистого основания, в результате чего лолучается соль, средний состав которой соответствует соединению, занимающему промажуточное положение между солью люно- и ля(азотистаго основания). Оонование может использоваться в колияесшве, ,п,ре)вышаю:це.м общее необходимое моляр-ное количество для нейтрализации кислотных функцнональных групп, одЕаКО оно ие должно лревы Ш.ать указанный избыток, например, оно не должно использоваться в количестве, превышающем 5-ти моляриый объем.
Оптимальное соотношение кислоты к основанию будет зависеть от различных факторов, в том числе и от природы кислоты и основания, а т,аК1же от природы окиси пенициллина. Предпочтительной является такая соль, которая получается непосредсшенно в реакционной смеси в результате |ХИ1мической реакции в основном между равномолярными количествами Пиридина и ортофосфорной кислоты, или ,соль, образующаяся из пиридина п дихлор.метанфосфииавой кислоты. Эту соль получают в результате химической реакции между в основном равномолярными коли1чествами пиридина и дихло1р|МетаН|фо1сфиновой кислоты; она рассм.атривается здесь как вдонопиридиндихлорметанфосфинат. Другую соль получают в результате хим:ической реакции в основном 2 моль пиридии.а с 1 моль фосфинощой кислоты; она рассматривается здесь как диеириндихлорметанфасфинат.
Соли, образующиеся из азотисто.го основания, вмеющето значение рКв не менее 4, и фосфиновой кислоты, являются новыми соединения.ми. Фос|фи 0|вая кислота представляет собой преимущественно низший алка« или галогено-иизший алкан фосфиновой кислоты. К таки1М новым соединениям относятся; пиридинтрихлорметанфосфинат, М-метилан,илинтрихлорметанфосфинат, бис- (бензиламмоний)три1хлор,метанфосфинат, а-1пиколинтрихлорметаифосфинат, пиридин-орто-,бр01м;бе1нзолфосфинат, монолиридиндихларметанфосфинат моноизохи1нолинди1хлорметанфосфин.ат и мо:)ш-3-1метилизохинолиндихлорметанфосфинат. Монопиридиндихлорметанфссфинат может быть легко Получен путем постоянного введения пиридина в раствор дихларметанфосфиновой кислоты, находящейся в полярном раст1ворителе (например, кетоне, таком как ацетон, или низшем ал,каноле, та-ком как метанол, этанол, н-пропанол, или изопропанол). Монолиридихлор,мета«фосфонат является стойким кристаллическим веществам белого цвета, имеющим т. пл. 142-145° С. Примерами кетонов и сложных эфиров, используемых в данном способе, являются алифатические кетоны и сложные эфиры, имеющие соответствующие температуры кипения, в TOiM числе этилметиловый кетон, изобутилметиловый кетоН, метиЛН- пропиловый кетон, н-пропилацетат, «-бутилацетат, изобутилацетат, вторичный бутилацетат и диэтилкарбонат.
Время реакции, необходимое для достижения оптимальных выходов продуктов реакции по предлагаемому способу, изменяется в зависимости от тила используемого растворителя. Реакция ;аТомной церетруппировки, как пра..
вило, протекает гфи температуре кипения выбранных для да.нной цели растворителей, и, в случае использования та1ких растворителей, предел температуры кипения которых на.ходится в нижней части ужазанного интервала температуры киления, может потребоваться, соответственно, большее время реакции (например, 48 час), чем при иопользовании растворителей, предел температуры ки.пения которьгх выше. Время, необходимое на осуществление реа1К1Ции перегруппировки в среде диокт сана, при котором достигаются олтим.альные результаты, составляет обычно 7-15 час, в то время лри протекании реакции в метилизобутиловом кетоне обычно требуется 1-8 tac. Выходы продуктов реакции перегрушпировки зависят (|но уже в менъщей степени) от концентрации катализатора, находящегося в растворителе, причем большее время реакции
требуется соответственно для более низких концентраций катализатора.
В предлагаемом способе отдается предпочтение использованию в качестве органического растворителя диоксана. ОкисЛы пенициллина могут растворяться в данном растворителе до получения высокой концентрации. Обычно с увеличением концентрации (до величины порядка 35%) выход продукта не. уменьшается. Количество используемой соли,
как , не должно превышать 1 моль на 1 моль окиси пенициллина, однако предпочтительно использовать 0,01-0,2 моль соли на 1 моль окиси пенициллина. Соли, ислользуемые в соответствии с предлагаемым способом,
оказывают относительно незначительное окрашивающее действие в процессе реакции перегруп1пировки по сравнению с аналогичными реакциями перегруппировки, протакающи ми Б присутст1вии кислотного катализатора. Побочные продукты реакции, (которые обычно образуются при использовании таких кислот-, ных катализаторов, в данном случае образуются в значительно меньшей степени, и использов,анне солей имеет важное практическое
значение в том отношении, что перед удалением растворителя реакционной среды в данных условиях нет необходимости удалять, обесцвечивающие агенты и связующие кислоту а.генты.
Окончание химической реа.кции может быть определено при .помощи одного или нескольких следующих .видов испытания реакционного раствора.
1. С помощью жидкостной хроматографии
с использованием, например силикагелевого наполнителя хроматографической колонки, и с использоваиием смеси бензола и этилацетата в отношении 2: 1, и при идентификации видимых точек путем обра ботки иодом (раствором азида). В случае, когда, напри.мер, исходных продуктом является сложный 2,2,2-трихл-орэтиловый эфир 1 |б-окиси. 6 уб-феноилацетаминоиенициллановой кислоты, то продукт реакции (/0,65) придает реакционной массе ный продкут (,5) - темно-желтый цвет.
о.ранжево-.бурый цвет, в то время как исход7
2.С помощью ротациомного спектра лосле соответствующего разбавления реа,иционной смеси, налрилтер хлороформом. При использовании указанного выше исходного продукта .ротадионный опектр снижается до 1/3-1/4 iqacTH исходной велвчи.иы.
3.С 1ПО(М01Щью ультрафиолетового спектра образца .реаюциоННОЙ смеси, разбавленного соот)ветствующим образ1ом этиловым спиртом. Используя указанный вьвше исходный п.роГ-|1 й
дукт, рассчитанная вел-ичИна см при ддине волны 64 нм (на.но.метра) повышается примерно до 100 при успешно протекающей реакции. Максимумы поглощения дри более высоких энамениял длин волн имеют преимущ-еспвенно нвбольщие згначения или отсутствуют. Эт1И определения не могут быть Осуществлены в случаях, кагда в качестве реакционной среды используются кетонные раст(ворители.
Следует отметить, что хотя удовлетворительные выходы продуктов (реакции могут быть получены при протекании реакции в условиях абьмного .на грева с обратным холодиль-ни1ком, выходы продуктов реакции можно у1величить с помощью осу) вещества (HainpHMep, акиси алюминия, окиси кальция, гидрата окиси натрия или молекулярных сит), которое вводится в растворитель пю линии обратного стока конденсирующегося пара этого растворителя для удаления воды, образующейся в процессе химической реакции. Кро.ме того, вода, образующаяся при химической реакции, может быть удалена при помощи ректификационной колонны, в результате фракционной перегонки. |По завершении химической реа1кции соль может быть удалена либо перед, либо после выпаривания реакционной смеси. Если растворитель несмешигваем с водой, то соль можно удалить простой операцией промывания. Если же реакционная среда смешиваема с водой, то приемлемьш способом очиски является удаление этого раство.рителя (на1ПрИ мер, отгонкой при пониженном давлении) и последующая очистка полученного остатка лшбым известным способом, например, путем хроматографии с использованием силикагелевого наполнителя для хроматографической колонки.
Было обнаружено, что степень хИ|МНческого превращения может быть такой, что можно отказаться от процессов сложной очистки, и получаемый продукт реакции может быть выделен ,в основном в чисто.м виде после простой перекристаллизации. Однако желательно, что(бы продукт реакции был выделен путем сливания реакционной смеси с водой, фильтрования продукта реакции и, если необходимо, дальнейшей пере К1ристаллизацией его из соответствующего растворителя или суспендирова1ВИЯ его с соответствующим растворителем. Лри ислолвзовании наиболее предпочтительных в данном способе катализаторов, например фосфата пиридина или монопиридиндихлорметанфосфината в растворе диоксанового растворителя, для получения высокого вы8
хода продукта реакции довольно высокой степени чистоты может оказаться необходимым лишь выпаривание растворителя и перекристаллизация продукта реакции из соотоетствующего растворителя. Может быть введена также стадия обесцвечивания с использованием, например, древесного угля; однако, как правило, нет необходимости вводить эту стадию лри выполнении наиболее предпочтительных условий процесса.
Группы ацила, находящиеся в б/З-аминоположении ОКИСИ пенициллина, могут представлять собой любую требуемую ацильную групщ, но должны преимущественно находиться
в довольно устойчивом состоянии в условиях перегруппировки. Обычно, ацильная группа, находящаяся в 6/5-положении, представляет собой группу пенициллина, получаемого путе.м фе1рментации, например группу фенилацетила
или феноксиацетила. Такая группа может быть нежелателыной в целевом продукте-цефалоопорине, но она может быть аннулирована при последующих превращениях, о которых будет говориться ниже. Другой группой, которая может быть использована, является группа формила. Наряду с этим группа ацила, находящаяся в б/3-1положении окиси пенициллина, может представлять собой желаемую группу в цефалоспориновом соединении, например
пруппу тиенилацетила или фенилогликоксилили, либо может представлять собой группу ацила, содержащую защищенную функциональную группу, , как защищенную амиHorpyniny. ПрИмером такой ацильной группы
является защищенная а-аминофенилацетильная группа.
Аминозащитная группа представляет собой обычно такую группу, которая может быть впоследствии удалена восстановлением
или гидролизом, не воздействуя при этом iia остаток молекулы, особенно лактамовые- или 7/5-а мидо-связи полученного в результате данного процесса цефалоспоринового соединения. Такая же защитная rpynna может
использоваться также в качестве этерифицирующей группы в 3-СООНнположении, и обе группы могут быть одновременно удалены, как будет показано ниже. Наиболее успешным является способ удаления обеих групп
в последней стадии последовательно осуществляемых процессов. Защитные группы включают уретановую, аридметиловую (например, тритиламиновую), арилметиленаминовую, сульфениламиновую и енаминовую группы. Такие грулпы, как правило, могут быть удалены посредством одного или нескольких реагентов, такжх как разбавленные неорганические кислоты, напри .мер разбавленная соляная кислота, концентрированные органические кислоты, например концентрированная уксусная кислота, трифторуксусная кислота и жидкий бромистый водород при очень визких температурах, например при -80° С. Обычно используемая защитная группа -
третичная бутоксикарбонильная группа, которая легко удаляется путем гидрол за разбавленной соляной нислотой 1илл (предпочтительно) СИЛЬНОЙ органической 1кислогой (такой,, как муравьиная или трифторуксусная кислоты) -при 0-40° С, преи-мущественно при комлатной температтуре (15-25° С). Другая обычно используемая защитная группа -2,2,2-трихлорэтоксика|рбоН(ИЛЬная группа, которая может ..расщепляться пссредстшом реагента, такого как динк в уксусной и муравьиной кислотах, НИЗШИХ опиртах или пиридине.
Сложный эфир пенИЦйЛланОВОЙ кислоты образуется нреи.муществвнно при взаимодействии со спиртом или фенолом, который может легко расщепляться 1путем, например, пь дролиза или восстановления на последней стадии, ериводя в результате к ооразова-шю цеф-3-емо-вого соединения в виде свободной кислоты. Стиртовые или фенольные остатк::. которые могут легко расщепляться, представляют собой такие группы, которые содержат притягивающие электроны за-местители, iiaпри-мер сульфогруипы и этерйфицировапные карбоксильные группы, (пржчем эти группы могут далее расщепляться под действием щелочных реагентов. Бензильные и ортобензилоксифеноксиэфирные группы могут быть удалены путем восстановления сложных эфиров в спирты дейст1вием водорода, хотя в результате этого М01жет иметь место отравление катализаторным ядом. Наи более предпачтителен способ удаления этих групп - расщепление кислоты. Группы, которые могут быть удалены путем расщепления кислоты, включают: .нтиловый, третичный бутиловый, бензиловый остатки, напри-мер, анизил, и остат1ки алканолов, содержащие элехТ1ронные доноры в «-положении, например ацилокси-, алкокси-, бензоилоксигруппы, замещенный бензилоксигалоген, тиоалкил, фенил, алкаксифенил, или ароматический гетероциклический радикал. Эти радикалы могут быть получены из бензиловых спиртов, например пара-метоксибензиловаго спирта, д;.1пара-метоксвфенилметанола, тр фенил метанола, дифенилметанола, бензо- и локсчметанола, пара-н итробенз1илового спирта и фурфурИлового спирта. Спиртовыми остатка1м-и, которые затем могут быть легко расщеплены с помощью восстанавливающего агента, ЯВлЯЮтся 2,2,2-триталогенэтанол, например 2,2,2-трихлорэтанол, нара-нитробеизиловый спирт или 4-Н:Иридилметанол, 2,2,2тригалогенэтиловые группы могут быть легко удалены посредством реагентов: цинк (уксусная кислота, цинк) муравьиная кислота, цинк (низший спирт или цинк) пиридин, либо посредством хромистых .реагентов. Паранитробензиловые группы могут быть легко Здалены восстановлением сложных эфиров в спирты действием -водорода, и 4-пиридилметилОвые группы .могут быть легко удалены электролитическим восстановлением. В случае,, когда группа сложного эфира может
оыть удалена в результате реакции катализированной кислотой, процесс удаления этой группы ускоряется при использова1Еи« муравьиной или трифторуксусной КИСЛОТ (преимущественно в сочетании с остатком анисо.вой кислоты), лИ|бо при использовании соляной кислоты в смеси, например с уксусной кислотой.
Наиболее предпочтительны окислы пенициллина, имеющие группу дифенилметоксикар;бонила, 2,2,-трП|Хлорэтоксикарбонила, третичного бутоксикарбонила, пара-нитробензилоксикарбонила, бензил.метоксикарбонила или пара-метоксибензилокси1карбонила, бензоилметоксикарбонила или пара-метоксибензнлоксикарбонила, находящуюся в 3-положении, поскольку соединения цеф-3-ема, полученные из сложных эфиров да))ного типа, не претерпевают заметной изомеризации в процессе деэтерификации.
В приведенных примерах, в случае, если нет специальных оговорок, процесс жидкостной хроматографии (или хроматография в тонком слое) осуществляется на силижагеле
с использованием смеси бензола с этилацетато,м (в соотнощении бензол: этилацетат 2:1), служащей ,в качестве растворителя с проявлением видимых точек раствором иод-азид.
Пример 1. 2,2,2-Трихлорэтил-6Д-фенилацетамидоненнциллат - 1/8 - оксид (19, 28 г, 40 ммоль) растворяют в нагретом диаксане (400 мл). Затем добавляют пиридинфосфат (С5Н52НзРО4, 0,0704 г, 2,56 ммоль) и раствор
нагревают с обратным холодильником в течение 16 час в аппаратуре, сконструированной таким o.6pa3QN, что конденсирующийся растворитель, стекая, проходит через слой окиси кальция (размером частиц 6-12 меш, 40 г),
после чего возвращается -в реакционный сосуд. Охлажденную смесь выпаривают при по)П1жениом давлении, получая смолистый продукт, который обрабатывают нагретым этиловым спиртом (техническим метилированным
спиртом, 20 мл). Твердый остаток кристаллизуют, смесь охлаждают в течение 3 час п фильтруют. Извлеченный твердый продукт промывают техническим .метилированным спиртом (ГО .1/л) и диэтиловым эфиром
(20 Л1л) и высущивают, в результате получают 2,2,2-трихлэрэтил-3-1метил-7Д-фенилацетамидоцеф-3-ем-4-карбаксилат (12,4 г, 66,9% от теоретического выхода) + 51,9° (с 1,0 в СНС1з); т. пл. 162-164° С. Вторую дополни.
тельную порцию продукта (1,65 г, 8,9/о от теоретического выхода) получают из жидкого щелока. Он имеет следующие данные: + 52,6°С (с 1,0 в CHCls), т. пл. 160- 1В1°С.
При .м е р 2. Смесь 2,2,2-трихлорэтил-6Дфенилацетамидопениц:иллат-1 -оксида (4,82 г, 10 ммоль), пиридина (0,078 г, 1 ммоль) ортофосфорной 1КИСЛОТЫ (уд. вес 1,75; 0,112 г, 1 ммоль) и изобутилового «етона (200 мл)
кипятят с обратным холодильником в течение
3 час. После этого раствор охлаждают и промывают водой (первый раз - 100 мл, второй - 50 мл). Органическую фазу выпа,ривают при пониженном давлении. Полу-ченный в .результате см-олистый продукт растворяют в этиловом спирте (технический метилироваи.ный спирт, 20 мл). Раствор выпаривают досу-ха, ивердый осадок суспендируют этиловы1М спи,ртом (техническим метилированным спиртом, 15 мл). Полученную суспензию выдерживают 16 час 1П:р.и 0° С, затем фильтруют. Извлеченный твердый продукт про.мывают простым Эф:И1ро-м и высушивают, в результате получают 2,2,2-трихлорэтил-3-метил-7;б-фвнилацетам1идо-1Цбф-3-ё1м-4нка1р|бо1К1силат (3,20 г, 69,0% от теоретического выхода), т. пл. 161 - 162° С, аЬ+52,1 (с 1,0 в СНС1з).
Пример 3. 2,2,2-т1рихло1рэтил-6 -фенилацетамидопениц.иллат- -оксид (4,82 г, IQ ммоль) и пиридияметансульфонат (0,175 г, 1 ммоль) растворяют iB го,ря1чем изобутилметиловом кетоне (200 мл) и нагревают с о.братныим холодильником в течение 1,5 час. Затем раствор охлаждают и промывают водой (первый раа- 50 мл, второй - 25 мл), после чего выпаривают досуха 1П.ри пониженном давлении. Полученный полукристаллический смолистый .осадок суспендируют этиловым спиртом (техническим метилированным спиртом, 20 мл и охлаждают в течение 4 час. В результате извлечения путем фильтрации твердого продукта, прочмытого этиловым спиртом (20 мл) и высушенного в вакууме, получают 2,2,2-трихлорэтил-3-метил-7 -фенилацетаМидо - цеф-3ем-4-карбоксилат (2,79 г, 60,1%- от теоретичеокого выхода), т. пл. 160-16ГС, аЬн-55,5° (с 1,0 в НС1з). Из щелока дополнительно получают продукт (0,1 г, 2,2% от теоретического выхода), т. пл. 160° С.
ПрИ1Мер 4. Пиридин (0,4 мл; 0,05 молярный эквивалент) и 88 вес. % (от общего объема) ортафоофарной кислоты (0,032 мл, 0,05 молярный эквивалент) добавляют к н-юропилпрапионату. Омесь кипятят с обратным холодильником при тажих условиях, что конденсат, стекая, проходит через коловку, наполненную окисью кальция (10 г), после чего возвращается в реакционный сосуд. После нагревания с обратным холодильником в течение 15 мин смесь немного охлаждают и добавляют -к ней 2,2,2-трИ1Хлорэтил- буЗ-1фенила1цетамидопенициллинат - 1 - оксид (4,82 г, 0,01 моль). Эту смесь иагревают с обратным холодильником в течение 2 час, выпаривают до получения небольшого объема. Полученный осадок перекристаллибовьгоают из этанола (12 мл), в результате получают 2,2,2-трихлорзтил-31метил-7/в-фенилацетам1ИДо - цеф - 3 - ем1кар|боксилат, 3,04 г (65,5% от теоретического (выхода), т. пл. 161-162° С, аЬ + 52,2° (с 08 :в СНС1з); А„,кс (этанол) при 264 нм (ff. 130).
Пример 5. а) Пара-метоксибензил-3-метил-7|б-:фенилацетамидо .. цеф-З-ем-4-карбоксилат получают в результате перегруппировки
пара - мето:кси:бензил-6/3-фенилацетамидопенициллат-1 -оксида. Пара-метоксибензил-6/9-фенилацетаыидопенициллат -IP- оксид превращают в пара - метоксибензил - 3 - метил7/3 - фенилацетамидо - цеф - 1уЗ - оксида (9,41 г, 20 ммоль), пиридина (0,16 г, 2 ммоль) и 89 /о-1Ной фосфорной кислоты (0,22 г, 2 ммоль) в высушенном, не содержащем перекиси диоксане (200 мл), нагревают
с обратным холодильничком в течение 15,25 час, причем конденсирующиеся .пары, стекая, проходят через молекулярные сита (сита Лино
де, 4А, 40 г), после чего они снова возвращаются в реакционный сосуд. Результаты
анализа методом жидкостной хроматографии, в тонком слое (с использованием смеси бензол - зтилацетат в отношении 2:1), показали лишь следы оставшегося исходного 1/3-оксида, при идентифичкации с использованием
иодоазидного реагента. Диоксан удаляют при 30°С (15 мл), в результате чего получают бурое желеобразное вещество, которое суспендируют с техническим метилированным спиртом (50 мл). .Полученное бледно-бурое желеобразное вещество промывают техническим метилированным спиртом и простым эфиром и высушивают. В результате получают параметокси - бен.зил-3-метил-7/3-|фенилацетамидоцеф-3-ем-4-«ар|боксилат, 6,025 г (66,5% от теоретического выхода) « jD+44° с 1,24в Хмакс (этанол) 228 нм (, 355) и 264- 274 нм ( 134) Я.М.Р. (СДС1з) 7,92 (Сз-СНз), определенный для 2,64 протона по
отношению к 6,22 (СбН40СНз). В результате выпаривания соединенных фильтратов и промывных щелоков и К1ристаллизации из технического метилированного спирта получается дополнительно менее чистый сложный эфир
цеф-3-ема (0,69 г, 7,6% от теоретического выхода). Я. М. Р. (СОС1з) 7,92, определенный для 2,25 протона по отношению к т 6,22.
б) Пиридин (320 мг, 4 ммоль) и 89%-ную фосфорную кислоту (220 мг, 2 ммоль) добавляют к раствору сложного эфира 1 -оксида (9,41 г, 20 ммоль) в высушенном, не содержащем перекиси дноксане (200 мл). Омесь нагревают с обратным холодильником в течение 16 час таким образом, что конденсирующиеся пары, стекая, проходят через слой молекуля.рных сит (сит Линде, 4А, 1/16, 40 г), после чего они возвра.щаются в реакционный сосуд. Результаты анализа методом жидкостной хроматографии (с использованием смеси бензол - этилацетат в отношении 1 : 1) с последующей обработкой иодо-азидным реагентом, показали присутствие в реакционной смеси следов исходного 1/3-окси.да, в связи с чем эту
реак|Ц|ИОНную смесь нагревают с обратным холодильниКО м еще 4 час. Диоксан удаляют при 30°С (15мм). При этом получают твердое вещество оранжевого цвета, которое перекристаллизовывают из кипящего метанола (300 мл), в результате получают иглообразный пара-|метоксйбензил-3-1метил-7/вчфенилацетамидо-цеф-3-ем-4-кар1боксилат. Этот продукт отфильтровывают, промывают простым эфиром (20 мл) и высушивают. В результате по.лучают продукт, 5,81 г, что составляет 64% от теаретического выхода, т. .пл. 151 -152° С, +38,5 (с 1,00 в СНС1з), Я„,, (этанол) 227 нм, (ЕЦ 382) ,и 269 нм (f« 176). В результате вы1пари вания фильтрата и промывных щелоков до объема 100 мл получают дополнительно сложный эфир цеф-З-ема (0,98 г, lil% ОТ теоретического выхода), т. пл. 150-152° С, aPi, + 38° (с 0,98 в СНС1з), ыакс (этанол) 226 нм ( 361,5) и 269 нм
162).
,в) При протекании химической .реакции, аналогичной описанной вьвше (б), но при ис.пользовании пиридина (320 мг, 4 м-моль) и монопИрИ|Д1ИЛдихлОгр|Мета1Нфосфи1ната (488 мг, 2 ммоль), после удаления дио ксана получают желатинообразное твердое вещество ора нжевого цвета, которое переюристаллизовывают из кипящего метанола (250 мл), в результате получают иглоабразяый слож;ныйэфир цеф-З-ема, который отфильтровывают, промывают простым эфиром (20 мл) и высущивают, получая при этом ерощукт (6,08 г), что составляет67Vo от теорети}4еско1го выхода, т. пл. 151 -152,5° С, a)%-f390° (с 1,08 IB СНС1з); «акс (этанол) 227 нл (f, 392) и 269 нм (ЕЧ 175).
ф|ильтрат и промывные щелоки выпаривают .до остаточлого абъема 75 мл, до начала выделения желеобразного вещества. Это вещество повторно растворяют при нагревании с обратным холодильничком. В результате охлаждения получают дополнительно иглообразный продукт сложного эфира цеф-З-ема (1,28 г, 14% от теоретического выхода), т. пл. 149-150,5° С, аРд+,39° (с 0,98 в СНСЬ); .Амакс (этанол) 227 нм (fif 375) и 269 нм (Г°, 164).
Пример 6. а) Раствор 1 -оксида бД-фепилацетаминопеницИллаиовой кислоты (7,02 г, 20 ммоль), фенацилбромида (4,02г,20.«лголь), и триетилагмина (2,02 г, 20 ммоль) в высущенном N, Ы-диметилформамиде (100 .д) перемешивают три комнатной температуре в течение 1 час, выливаютиа воду (700 мл) и экстрагируруют метиленхлоридом (200 лгл). Соединенные экстракты органических фаз промывают водой (четыре раза по 350 мл), сушат над безводным сулыфатом магния и выпаривают. В ре-зультате получают желтое пенистое вещество (8,81 г), которое очищают методам хроматографии с использованием силйкагелевого наполнителя колонки (340 г), используя в качестве элюента смесь бензол-этилацетат. С помощью смеси бензол-этила.детат (вотнащении 1 : 1) происходит элюирование из адсорбента сложного афира 1 -оксида фенацила в виде «е совсем .белого пенистого вещества (6,17 г, 66%), « z +18:i° (с 1 в СНС1з), Х„.кс (этанол) 243 «ж ( 11,850).
б) Раствор фенаадил-б/З-фенилацетамидопенициллат-1 -оксида (1,18 г, 2,5 ммоль), пиркдина (21 мг, 0,25 ммоль) и 89% фосфорной кислоты (28 мг, 0,25 ммоль) в высушенном, не содержащем перекиси диоксале (50 мл),нагревают с обратным холодильником 27 час, причем конденсирующиеся пары, стекая вниз, проходят через молекулярные сита (сита Линде, 4А, /1б, 40 г), после чего возвращаются в реакционный сосуд. Результаты анализа методом жидкостной хроматографии (с использовайием смеси бензол - этилацетат в отношении 1:1) при обработке иодоазидным реагентом показали лишь следы исходного 1 оксида, оставшегося в реа1кционной смеси. Диоюсан удаляют при 30° (15 мл). Остаточный твердый продукт желтого цвета растворяют при нагревании в смеси этилацетата (100 мл) и 2 н серной кислоты (100 мл). Слой органической фазы промывают водой (100 мл), соединяют с этилацетато-м повторной промывки водных слоев, высушивают и выпаривают до образования кристаллического твердого продукта. Этот продужт перекристаллизовывают
из технического метилированного спирта, в
результате получают фена(цил-3-метил-7 фенилацетамидо - цвф-3-ем-4 - жарбоксияат
(0,56 г, 50%); т. пл. 184-190° С, а о+12°
(с 1,0 в СНС1з); Амакс (этанол) 244 нм
... 270 нм (ЕЦ 162). В результате последующей перекр|И1Сталл;иаа.ции из технического адетилированного спирта полу(чаегся следующий продукт; т. лл. 190-193° С, (а1о + 7° (с 1,ОвСНС1з); А„акс (этанол) 244 нл
( 19,800) и 27D нм (I 7,480). Найдено экспери.ментально: С 64,2; Н 5,0; N 6,2; S 7,0. C24H2 N2O5S (450,5). Требуется: С 64,0; Н 4,9; ,N 6,2; S 7,1.
/Пример 7. а) Пара-бромфенацилбромид
(3.2 г, 1 молярный эквивалент) в течение 5 мин добавляют по частял к перемещиваемому раствору 1 (в-оксида 6 /8-фенилацетамидопенициллановой кислоты (4,0 г) и триэтиламина (1,56 мл, 1 ,МОЛЯ|рный эквивалент) в
NN-димeтилфopмalмидe (45 лгл). Реакционную смесь пере.мещивают при комнатной температуре в течение 2,5 час, смещивают с водой (150 мл) и экстра1лируют этилацетом (два раза по 75 мл). Соединенные этетракты органических фаз повторно промывают водой, высушивают и выпаривают, осадок перекристаллизовывают из этилацетата простого эфира, в результате получают сложный эфир 1 /З-оксид парабромфенацила (5,32 г, 86,5%), т. пл.
150 153°С, аЬ+1б1° (с 2,03, тетрагидрофуран), XM.KC (СНВгз) 1800 /8-лактам) и 1766 см- 1 (-CO2R).
б) Распвор пара-|бромфенацил-6/3-(фенилацетамидопеницилланат - Ift -оксида (1,095 г,
2 ммоль), пиридина (16 мг, 0,2 ммоль) и 89% фосфорной кислоты (22 ме, 0,2 ммоль) в высушенном, не содержащем перекиси диоксане (50 мл) нагревают с обратным холодильникО|М в течение 30 час, причем сконденсированные пары, стекая, проходят через
15
молекулярные сита (сита Линде, 4А, 1/16, 40 г), п0сле чего возвращаются в реакционный сосуд. Результаты анализа методом жидкостной хроматографии (с использованием смеси бензол-этилацетата в отношении 3:1) цри аб,ра|ботке иодоазидным реаге«том паказали, что в реакционной смеси совершен.но отсутствует исходный 1-оксид. Диоксаи удаляют при 30° С, 15 мм, и осадок растворяют в нагретой смеси этилацетата (125 мл) и 2 Н-ной серией кислоты (125 мл). Слой органической фазы промывают водой, соединяют с этилацетатом обратной промЫ)ВК) водных слоев, высушивают и обесцвечивают путем ,перембши1ван,ия со смесью безводного сульфата магиия и древесного угля, фильтруют и вьша.ривают. Кристаллический осадок перек,ристаллизовы.вают из горячего технического метилированного спирта, в результате Получают -пара - б:ром фенацил - 3 - метил -7фенилацетамидо - деф - 3 - ем-4-карбо1ксилат (0,50 г, 47%), т. пл. 194-198° С, + 8°, (с 1,05 в СНС1з); макс (этанол) (258 нм I 25,900). В результате последующей нерекристаллизации из технического метиллирова:нного спирта получают следующий продукт: т. пл. 196-199° С + 9° (с 1,0 в СНС1з). Найдено экопариментально: С 54,3; Н 4,Г; Вг 14,9; N 5,0; S 5,8%. C24H2iBrN2O5S (мол. в. 529,4). Требуемый состав: С 54,4; Н 4,0; Вг 15,1; N5,3; S 6,1%. .
При м ер 8. 2,2,2-Т1рихлорэтил-7,8-(N-Tperбутокси1кар:бонил - D - а-аминофенилацетамидо) -3-метил-|цеф-3-е.м-4-карбоксилат получают в результате химической реакции, протекающий ло следую1щей схеме: 2,2,2-трихлорэтил - 6(3 - фенилацетамидопеиищиллат (Кислый .пара-толуолсульфонат-2,2,2 - трихлорэтил - 6/5аминопенициллат2,2,2-трИХЛОрэтил - 6Д(М-трет.-1бутоксикар1бонил - D - а-а,минофенилацетамидо) -пенициллат2,2,2-трихлоэтил6/3-(Ы-трет.- бутоксика|рбонил - D - а-аминофенилацетамидо) - пенициллат-1Д - оксид 2,2,2трихлор1Этил-7|б-(Н-трет.- бутакси.карбонил - Da-aминoфeнйлaцeтaLMИдo)-3-мeтил-lцeф-3 - ем4-кар1бож:силат.
Пример 9. PacTiBQp 2,2,2-трихлорэтил-бу5(Ы-трихлоретилоксикарбойил - D -а-аминофенилацетамидо)-(пенициллат-1/5-оксида (328 мг, 0,45 ммоль) и лиридинфосфата 9 мг, 0,033 ммоль) в высушеннам диоксане (3 мл) нагревают с обратным холодильником в течение 16 час. Растворитель выпаривают, осадок растворяют IB этилацетате (20 мл), раствор промывают водой, высушивают, этилацетат выпаривают, оставляя стекловидный продукт, который суспендируют нетролейным эфиром, т. К|ИЛ. 40-60° С. В результате получают 2,2,2-трихлорвтил- 7/5 -трихлор1этилокси«арбонил-Г-ю-а-минофенилацетамидо (-3-метил-цеф3-ем-4-1кар|бок1силат в виде не совсем белого порощкообразного продукта (294 мг). Хотя результаты жидкостной ;хроматографии (система, описанная выше) показывают наличие
16
синглетной тоЧ1ки 0,72, спектр протонного магнитного резонанса показывает чистоту 30%, остальные составляют смесь продуктов.
П р и м е р 10. 2,2,2-трихлорэтил-6 3-фенилацетамидоненициллат-Ггв-оксид (4,82 г, 10 ммоль), Ы-|Метиланилин (0,107 г, 1 ммоль) и ортафосфорную кислоту (уд. вес 1,75; 0,112 г,
1 ммоль) добавляют к изобутилметиловому кетону мл). Смесь нагревают с обратным холодильником в течение 1,25 час. ДобавляютК смеси Ы-метиланилин (0,107 г, 1 ммоль) и ортофосфорную кислоту (уд. вес 1,75; 0,112 г,
1 ммоль) и кипятят еще 2,75 час. Раствор охлаждают до комнатной температуры и лромывают водой (один раз - 100 мл, второй - 50 мл). Слой органической фазы досуха выпаривают, полученный полукристаллический
осадок растворяют в этило вом спирте (техническом метилированном спирте, 20 лы). Спиртовый расивор аналогичным образом выпаривают, в результате суспендирования осадка с этиловым спиртом (технИ)Ческим метилированным спиртом), 15 мл, получают кристаллический продукт. Смесь выдержи1вают при 0° С в течение 16 час. Продукт извлекают фильтрацией, промывают диэтиловым эфиром и высущивают, в результате получают 2,2,2трИхлорэт1ил-3-метил-7-фенилацета.мидо - цеф3-ем-4нкарбоксилат (1,90 г, 41,0% от теоретичеокдго выхода), т. пл. 161-162° С, 51° (с 1,0 в СНС1з).
Пример 11. PacTiBOip 2,2,2-трихлорэтил6/в - фенилацетамидопенициллат - Ift - оксид (14,46 г, 0,03 ммоль) в диоксане (300 мл) обра1баты1вают мета-аитробензосульфокислотой (0,609 г) и пиридином (0,24 мл) и полученный в результате раствор нагревают с обратным холодильником в течение 16 час. В процессе нагревания с обратным холодильником конденсат проходит через слой нейтральной окиси алюминия, после чего вновь возаращается в реакционный сосуд. Далее реакционную смесь выпаривают в вакууме, осадок суспендируют с горячим техническим метилированным спиртом (30 мл), охлаждают до комнатной температуры и продолжают это охлаждение в течение 2 час. Твердый продукт извлекают фильтрацией, промывают техническим метилированным спиртом (10 мл) и простым дизтиловым эфиром (20 мл), высушивают .в вакууме при 40° С, в результате получают 2,2,2-трихлорэтил-3-метил-7 -фенилацетамидо - цеф-3-вм-4-1кар.боксилат (11,32 г, 81, от теоретического выхода) в виде белого кристаллического твердого вещества, т. пл. 160-162° С, аЬ + 52,8° (с 0,9 в СНСЬ); Х,,,,кс (этанол) 264 нм 136,5). В результате выпаривания маточного раствора и последующей кристаллизации при 0° С получают дополнительный вьгход продукта (0,6 г, 4,3%),. т. 1ПЛ. 160-182° С, «Ь 52° (с 0,8 в.
СНС1з).
17
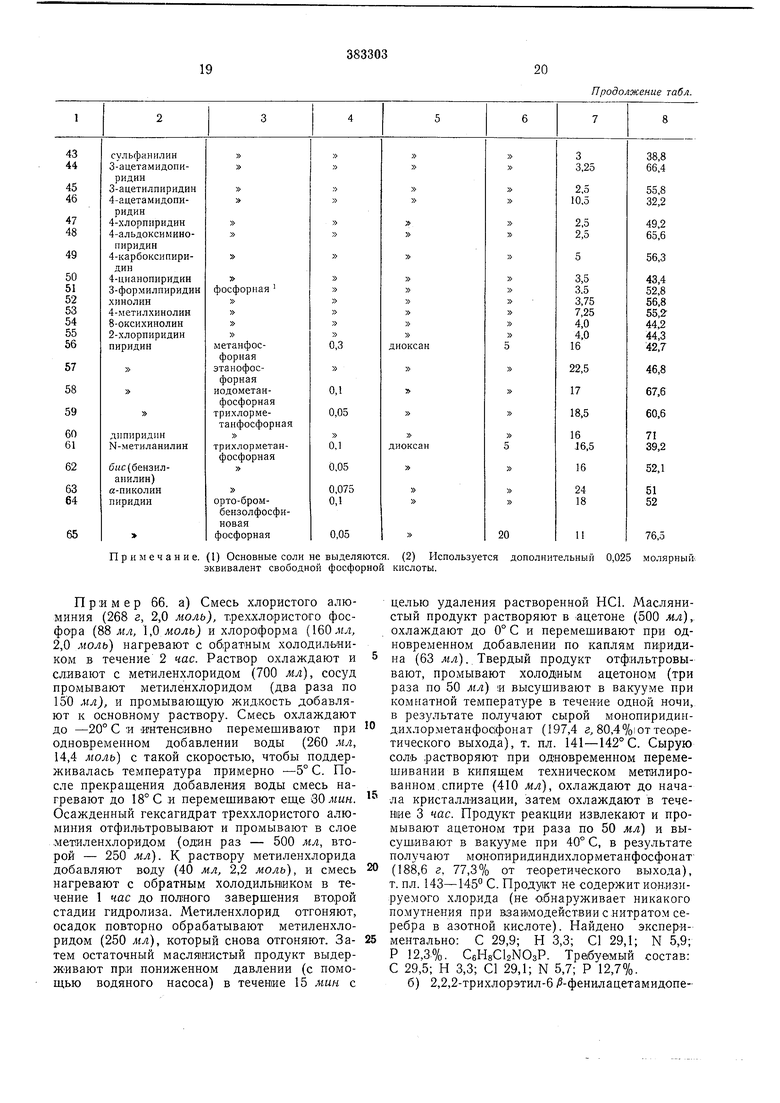
Пр.иМеры 12-65. Иопользуя различные растворители и катализаторы, 2,2,2-трихлорэтил- 6/3 -фенилацетамидопенициллат- оксид превращают в 2,2,2-трихлорэтил-З-ме18
тил-7|й-фенила цетамидо - цеф-3-ем-4-ка,рбоксилат. Условия превращения и выходы продукта реакции предст iвлeньI в табл.
Таблица
19
20
Продолжение табл.










| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения сложных эфиров 7 -ациламидо-3-метилцеф-3-ем-4-карбоновой кислоты | 1970 |
|
SU735170A3 |
| СПОСОБ ПОЛУЧЕНИЯ З-ОКСИ-З-МЕТИЛ-7- АМИНОЦЕФАЛОСПОРИНОВ | 1973 |
|
SU361571A1 |
| Способ получения соединений 7- аминоцефем-3-ол-4-карбоновой кислоты или их солей в виде смеси изомеров 2-и 3-цефем или отдельных изомеров | 1975 |
|
SU655316A3 |
| Способ получения производных 3-оксииминометилцефалоспорина или их солей | 1972 |
|
SU525429A3 |
| Способ получения производных 7-ациламино-цеф-3-ем-4-карбоновой кислоты | 1971 |
|
SU446969A1 |
| Способ получения производных 7- -аминоцефам-3-он-4- карбоновой кислоты или 3-кетальпроизводных или 1-окисей или их солей | 1973 |
|
SU583760A3 |
| Способ получения 3-ацилоксиметил- цефемов | 1977 |
|
SU703023A3 |
| Способ получения 7-ациламидо-7метокса-3-р-3-цефем-4-карбоновой кислоты | 1972 |
|
SU454742A3 |
| Способ получения производных 7-аминоцеф-3-ем-4-карбоновой кислоты | 1971 |
|
SU468428A3 |
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНОГО ЭФИРА ДЕЗАЦЕТОКСИЦЕФАЛОСПОРИНА | 1972 |
|
SU352464A1 |
Примечание. (1) Основные соли не выделяются. (2) Используется дополнительный 0,025 молярный-, эквивалент свободной фосфорной кислоты. Пример 66. а) Смесь хлористого алюминия (268 г, 2,0 моль), т,рех.хло1ристого фосфора (88 ж л, ,моль) и хлороформа (160 .«л 2,0 моль) нагревают с обратным холодильником в течение 2 час. Раствор охлаждают и сливают с метиленхлоридом (700 мл), сосуд промывают метиленхлоридом (два раза по 150 мл), и промывающую жидкость добавляют к основному раствору. Смесь охлаждают до -20° С и интенсивно перемешивают при одновременном добавлении воды (260 мл, 14,4 моль) с такой скоростью, чтобы поддерживалась температура примерно -5° С. После прекращения добавления воды смесь нагревают до 18° С и перемешивают еще ЗОлшн. Осажденный гексагидрат треххлористого алюминия отфильтровывают и промывают в слое мет иленхлоридом (один раз - 500 мл, второй - 250 мл). К раствору метиленхлорида добавляют воду (40 мл, 2,2 моль), и смесь нагревают с обратным холодильником в течение 1 час до полгного заверщения второй стадии гидролиза. Метиленхлорид отгоняют, осадок повторно обрабатывают метиленхлоридом (250 мл), который снова отгоняют. Затем остаточный масляиистый продукт выдерживают при пониженном давлении (с помощью водяного насоса) в течение 15 мин с целью удаления растворенной НС1. Маслянистый продукт растворяют в ацетоне (500 мл), охлаждают до 0° С и перемещивают при одновременном добавлении по каплям пиридина (63 мл). Твердый продукт отфильтровывают, промывают холодным ацетоном (три раза по 50 мл) и высушивают в вакууме при комнатной температуре в течение одной ночи, в результате получают сырой монопиридиндихлорметанфос|фонат (197,4 г, 80,4 %i от теоретического выхода), т. пл. 141-142°С. Сырую солЬ растворяют при одновременном перемешивании в кипящем техническом метилированном спирте (410 мл), охлаждают до начала кристаллизации, затем охлаждают в течение 3 час. Продукт реакции извлекают и промывают ацетоном три раза по 50 мл) и высушивают в вакууме при 40° С, в результате получают монопиридиндихлорметанфосфонат(188,6 г, 77,3% от теоретического выхода), т. пл. 143-145 С. Проду1кт не содержит ио«изируемого хлорида (не обнаруживает никакого помутнения при нзавмодействиис.нитратом серебра в азотной кислоте). Найдено экспериментально: С 29,9; Н 3,3; С1 29,1; N 5,9; Р 12,3-%. CsngCbNOsP. Требуемый состав: С 29,5; Н 3,3; С1 29,1; N 5,7; Р 12,7%. б) 2,2,2-трихлорэтил-6 /3-фенилацетамидопеииц:илат-1-/3-оксг1д (96,4 г, 0,2 Л;(,ъм) и могэпиридипди.хлорметанфосфонат (,Э5 г, О.ООВлоль) прибавляют к диоксану, 482 мл, высушенному основной окисью алюминия), который находится в трехгорлой колбе, снабженной мешалкой и холодильником. Смесь перемешивают и нагревают с обратным холоднЛЬником, причем конденсирую нанеся па.ры, стекая в реакционный сосуд, .проходят предварительно через гранулы гидрата окиси натрия. Раствор неремешивают при нагревании с обратным ХОЛОДИЛЬНИКОМ в течение 8 час до тех пор, пока в реакционной смеси совершенно не останется исходного продукта (как показывают результаты жидкостной хроматографии). Диоксан удаляют при пониженном давлении, оставляя влажный твердый осадок. К нему добавляют технический метилированный спирт (200 мл. Этот твердый продукт суспензируют в течение нескольких минут, в результате нолучают однородный кристаллический продукт. Смесь выдерживают при 0° С в течение i3 час, твердый продукт извлекают путем фильтрации, промывают техническим метилированным спиртом (100 мл), простым эфиром (100 мл) и высушивают в вакууме при ко.мнатной температуре, в результате получают 2,2,2-т.рихлорэтил-3-метил-7 -фенилацетамидо-цеф-З-ем-4-карбоксилат (75,1 г, 80,9% от теоретического выхода), в виде белого кристаллического вещества, т. пл. 162-165° С, «7, 53° (с 0,91 в CHCls), Хмакс (этанол) 264 им (,1 130). Результаты жидкостной хроматографии (с использованием смеаи бензол-этилацетат в отношении 2:1) показали синглетную точку Rf 0,65. В результате выпаривания промывных щелоков получают дополнительно продукт (2,3 г, 2,5% от теоретического выхода), т. пл. 159-162° С, WB 53,1 (с 0,97 в СНС1з),Х„акс (этанол) 264 нл (fi-, 131). Результаты жидкостной хроматографии (с использованием смеси бензол-этилацетат п отношении 2:1) показали синглетную точку Rf 0,65.
Пример 67. 2,2,2-Трихлорэтил-6/3-феНилацетамидопенициллат - 1 - оксид (96,4 г, 0,2 люль),монопи.р.идиндйхлорметанфосфонат
(1,708 г, 0,007 моль) и пиридин (0,56 мл, 0,007 люль добавляют к диоксану (4,82 мл, высушенному основной окисью алюминия), находящемуся в трехгорлой колбе, снабженной мешалкой и конденсатором. Химическая реакция и процесс выделения продукта реакции осуществляются аналогично примеру 66, б, в результате получают 2,2,2-трихлорэтил - 3- метил-7-/ -фенилацетамидо-цеф-3-ем-4,карбо.ксилат (73,5 г, 79,2%: от теоретического выхода), в виде белого кристаллического твердого продукта, т. пл. 161,5-164,5° С, Ив+ 53,9 (с 0,02 в СНС1з), я„акс (этанол) 264 нм ( 131). Результаты жидкостной хроматографии (с использованием смеси бензол-этилацетата в отношении 2:1) показали синглетную точку Rf 0,65. Из маточного
раствора извлекают дополнительно продукт (2,74 г, 2,95/о от теоретического выхода), т. пл. 60-162°С + 53,6° (с 0,85 в СНС1з), -м,а,с (этанол) 264 («, 131). П р и м е р 68. 2,2.2-Трихлорэтил-6 -фенилацетамндопенициллат - 1/5 - оксид (96,4 г, 0,2 -иоль) и монопиридиндихлорметанфосфинат (1,95 г, 0,008 моль) нагревают с обратным холодильником в высушенном, не содержащем
° перекиси дноксане (482 мл). Конденсат проходит через колонку, наполненную осущающнм веществам - гра1нулированным гидратом окнсп натрия (200 г), после чего возвращается в реакционный сосуд. После химической реакции проводят аналнз методом жидкостной хроматографии. После нагревания с обратным холодильником в течение 7,5 час в реакционной смеси совершенно не остается исходного продукта. Раствор охлаждают до
0 комнатной температуры и сливают с водой (2,5 л) прн перемешивании. Твердый продукт отделяют фильтрованием, промывают водой (два раза по 100 мл) и высушивают в вакууме при комнатной температуре, в результате получают 2,2,2-тр,ихлорэтил-3-метил-7Д-фенилацетампдо-цеф-З-ем-4-карбоксилат (89,6 г, 96,6Vo от теоретического выхода), т. пл. 156° С), :«,) 58° (СПС1з), Я„акс (этанол) 264 нм
0 2 Пример 69. 2,2,2-Трихлорэтил-буЗ-фенилацетамндопенициллат - 1 jS - оксид-(48,2 г, 0,1 моль) и моноизохинолиндихлорметанфосфонат (1,471 г, 0,005 моль) подвергают реакции в диоксане (482 мл) в течение 8 час. Получен5ный продукт отделяют аналогично примеру 66, б, в результате получают 2,2,2-трихлорэтил-3-метнл - 7 - фенилацетамидо-цеф-З-ем-4карбоксилат (37.02 г, 79, от теоретического выхода), т. пл. 162-164°С, аЬ + 52,1° (с 0,8 в ), макс (этанол) 264 нлг (, 140). В результате выпар:ивания щелоков получают дополнительно такой же продукт (0,08 г, 2,6я/о от теоретического выхода), т. пл. 159- 5 161°С, :а:в-52,4°.
П р и мер 70. 2,2,2-Трнхлорэтнл-6,5-фенилацетамидопенициллат - 1 - оксид (48,2 г, 0,1 моль) и моно-3-метилизохинолиндихлорметанфосфонат (1,54 г, 0,005 моль) подвергают peo акции в диоксане (482 мл) в течение 81,25 час. Продукт выделяют аналогично примеру 66, б, в результате ползчают 2,2,2трихлорэтил-З-метил-73-фенилацетамидо - цефЗ-ем-4-карбоксилат (30,42 г, 65,5в/о от теоретического выхода), т. пл. 162-164° С, MB + 52° (с 0,8 СНС1з), -макс (этанол) 264 нм ( 138).
Пример 71. 2,2,2-Трихлорэтил-3-1Метил7/3-феноксиацетамидо-цеф-ё-ем-4-карбокснлат. Раствор 2,2,2-трихларэтил-6/ -феноксиацетамидопенициллат-1/ -оксида (2,45 г, 5,0 моль) в диоксане, высушенном при прохождении через слой основного окисла алюминия (50 мл), нагревают с обратным холодильником таким образом, что конденсированные пары, стекая.
проходят через молекулярные сита (сита
о
Линде, 4А, 30 г), после чего возвращаются в реакционный сосуд. Смесь нагревают с обратньим холодильником в течение 20 мин, затем добавляют к ней монопиридиновую соль дихлорметанфосфиновой кислоты (0,1345 г, 0,52 ммоль, 0,104 эквивалента) и пи.ридин 0,0391 г, 0,50 ммоль, 0,100 экнивалента), -нагревают с обратным холодильником 5,5 час. За это время исходный продукт полностью реагируют (на что указывают результаты анализа методом жидкостной хроматографии с использованием в качестве элюента 2 /о-ного раствора ацетона в метиленхлориде с использованием детектирующего реагента раствора иодид-иод-аз;ид калия). Диоксан удаляют при пониженном давлении, полученный маслянистый остаток желтого цвета растворяют в метаноле (5 мл и простом эфире (5 мл). В результате выпаривания этого раствора досуха остается бледно-желтый твердый продукт, который суспензируют с простым эфиром (10 мл), содержащим метаиол (0,5 мл), в результате получают требуемое соединение в виде темно-желтого твердого продукта (1,45 г, 60,5Vo от теоретического выхода), т. пл. 112-115° С, а1в + 57,2 (с 1,19 в СНСЬ). В результате выпаривания маточных растворов и повторных процессов суспензирования с простым эфиром (2 мл), содержащим метанол (0,1 мл), получают дополнительно продукт, окращенный в темно-желтый цвет (0,436 3, 18,6%), т. пл. 105-114° С, |а 53,4° (с 1,20 в СНС1з).
Третичный бутил-3-метил-7;8-фенилацетамидо-це,ф-3-ем-4-карбоксилат. Раствор третичяого бутил-6/5-фенилацетамидопенициллат-1,б-оксида (9,28 г, 22,9 ммоль) и дипиридиндихло.рметилфосфината (0,729 г, 2,65 ммоль) в высущенном диоксане (180 мл) -нагревают с обратным холодильником в течение 7 час, причем конденсирующийся диоксан, стекая, проходит через слой молекулярных сит (сита Лииде, 4А). Расивор выпаривают досуха при пониженном давлении, остаток растворяют в дихлорметане (200 мл), промывают насыщенным водным раствором бикарбоната натрия (50 мл), водой (50 мл), высущивают MgS04
и выпаривают досуха при повиженном давлении до получения желатинообразного твердого продукта (Э,83 г). Указанный твердый продукт перемещивают с простым эфиром
5 (60 мл) в течение 6 час при 25° С, охлаждают при 4° С в течение 62 час и отфильтровывают, осадок промывают простым эфиром (30 мл) и высушивают. В результате получают третичный бутил-3-метил-7р-фенилацетамидо-цеф-310 ем-4-карбоксилат (5,80 г, 15,0 ммоль, 65,5«/о), т. пл. 120-123° С, MB+ 62°.
Предмет изобретения
5 1. Способ получения сложных эфиров 7/3ациламино-З-метил-цеф-З-ем-4-карбоновой кислоты нагреванием сложного эфира 1-оксида .илами;нопенициллановой кислоты в орга.ническом растворителе, с последующим выделением продуктов известным способом, отличающийся тем, что, с целью повыщения выхода и качества продуктов, процесс проводят в диоксапе или кетоне, имеющем температуру кипения 75-120°, или в сложном эфире, имеющем температуру кипения 75-140° С, или в простом диметилавом эфире диэтиленглпколя в присутствии соли азотистого осяованая, жмеющего рКв не менее 4, и кислоты или смеси соединений, образующих такую соль непосредственно в реакционной смеси.
