1
Изобретение цредсназначено для использования в пищевой промышлвННосчти и отиасится к способам определения содержания в жирах :пестиЦИ1а,О|В, преимущественно проиавоаных ка|рбаМ-иновой кислоты.
Известеи способ олределения содержания пести-ц-идав в лшрах путе.ч Э1кст1ракци 1 исходной навеоки оргаиИче|С1К№м (раюпзскрптелем, воздействия на пестицид реагентаМ и, вызывающи.мн образование а рашещиого соединения и колори-метриро1ваи 1Я с послед ующим расчетом искомого 31иачения. Этот способ сложен, так как предлс-матривает очи1СТ1ку зюст ракта от жи1ра, а точнооть анализа недостаточна, поскольку знаЧ|11телыное кол1гчест)во пестицидов теряется iB троцессе очистки.
Для }1П;рощения способа опроделе)1ия содержа«ия пестицидов и павыше1ния точности оп раделеиия лред-лагается воздействие «а пестицид реагентами ос)Щвствляют непосредстванйЮ в экстракте (, а очфа-шенное соединение .перед колориметрИ(роваИИе|М выделять И.З ipacT(30ipa дополн-ительной экстракцией.
Для осущаствлемия способа «авеску растительного масла Э1К|С7)раги|ру-ют органическим растворителем, цреимуществеино в ацетоне, обрабатывают ipacTSQp реагентами, вызывающими оЕ разоваяие акрашеинаго соединения с пести ц идом, ок(рашеинь й продукт выделяют из экстракта реакционной среды одноюратной
дополнительной экстракцией, налрнмер, хлорофор.мом, эасстракт колориметрируют с помощью фотоэлектроколориметра и рассчитывают искомое 31наче1ние оаде|ржания пести1Цйда по «ал1ибр0 воч но.му графику, |Построенно.му для .испытуемого и станда ртного растворов (пестицид без жира).
Реагенты, воздействующие па лестицид, выби1рают в зависимости от вида пестицида.
Таким способом оп ределяют содвржаияе в жи;рах пестицидов-икропзводных к арб а.ми новой кислоты, например тетраметилтиура-ндисульфида, 4-.xлopбyтин-2-J л-N-м-xлo pфвн.илкapбoмата («арбина) и т. д.
колориметрического определения экстрагированных из жи.рав пестиадгдор на основе карбамино1вой к ислоты.
Пример 1. Одределеппе тетраметилтиурамдисульфида (ТМТД).
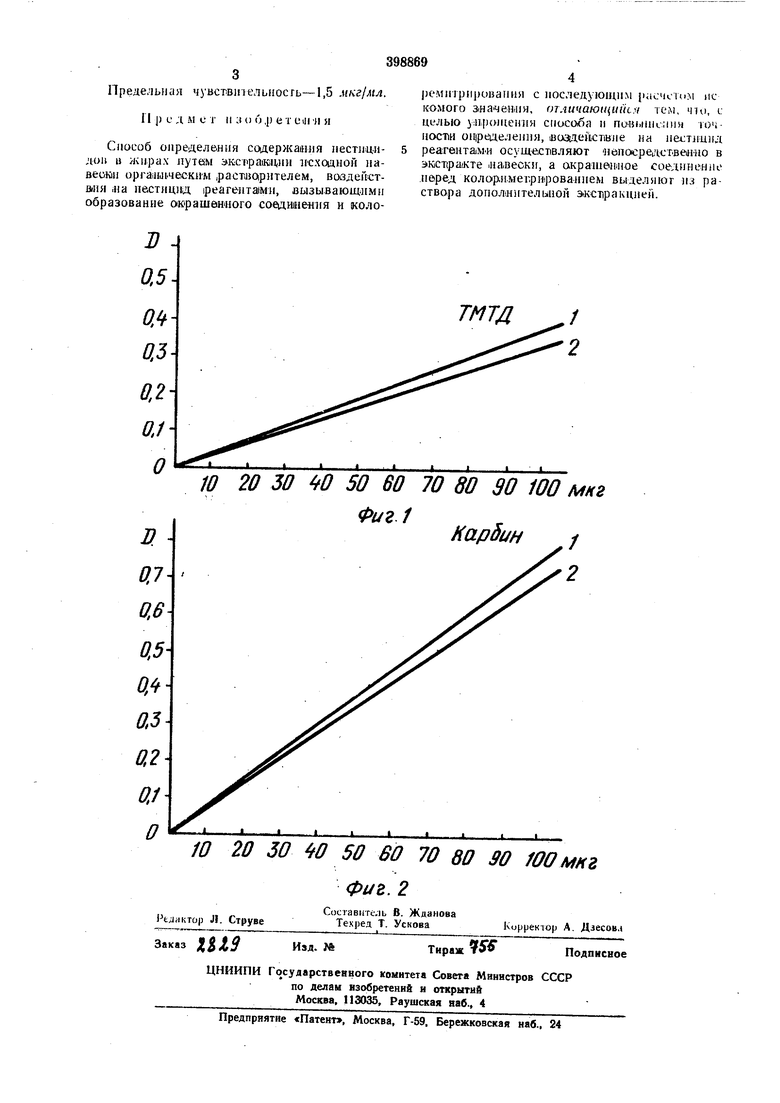
ТМТД определяют в лро,бах растительного (в данном п.ри.мере - олдавкового) масла следующим образом. Реактивы: ацетон «хч, дистиллированная вода, 0,1 и. водный раствор серной кислоты «хч, 0,1%нный водный раство,р гидрох/инона марки «А, 0,1%-ный водный, растюор ацетата меди «хч, .медиц гаекий хлороформ и се|рноиислый безводный «атрий. Аппаратура: калориметр тила ФЭК-56, оветоф«льт)р № 4 с MaiKJCHMyiMOM пропусканпя 434 нм -1В диапазоне 400-450 нм. ll-риоы ОЛИВКОВОГО масла объемом, например, 1 мл, вносят в проби р:ки со сгеклявными пришлифоваиньшп пробками и приливают в каждую двой-ное «оли-чество зюсхрагента ацегана. Смесь вптеноивно ;вс11ряХИ1вают 30- Ш сек, шюсят дистиллшрованную воду в количеспве 1/5 объема ацетона и вновь встряхивают. Пасле Д1вухмииут1ного отстаивания и расслоения смеси пробирки охлаждают до -20°С, шатрлшер в сосуде Дьюара, используя для этой цели смесь 200 г льда со 100 лы «онцентрэрованиой азотнюй кислоты. Охлаждение проводят в течение 20 мин. Экстракт сливают с за;стЫ|Вшего .масла в друпне ПробнрКн с пришлифова.цны.ми пробками, а масляную фазу повторно подвергают вышеон.исан1ной обработке, увелашивая л.ишь время экс-прак;ции (встряхивания смеои) до 10- 12 мин. Оба экстракта для (Каждой И1робы объедц1ияют (общий объем 4-5 мл) н подвергают х.И1Ми.чеокои o6pai6oT.K€ для перевода в окрашенное соединение. Для этого в каждую пробу послвдов ательно вносят: 0,2 мл 0,1 н. |)асшора сернай ,Й11С.лоть1: (в качестпзе регулягара рИ), 0,3 .«л О,l%-iioro.раствора пидрох)нона (в качест.ве восстановителя) ; л 0,5 мл 0,1%-лого ipacTBopa ацетата даухвалентиой меди (в качестве (ком плексообразователя). Смесь тщательно встряхивают, а затем отстаивают 25 мин. Протекающий при этом процесс обобщенно онисывается уравнением: (НзС)2МС(:8)- S-S-C(:S)--N(CH3)2-t-Cu (MsCjsNC S2C4. ПолЗченное окрашениюе соединение ак сцрагнруют 0,6 мл хлороформа 1ФИ .вст1рЯ:ХИ1ва1№ии ,в течеч.яе 10 мин, экстракт (нижний слой) отби-рают лялеткой в пробирки. Г1впосрадспве111но перед .вынесением хларофо|). aiccTipaiKTa в кювету колориметра его обеэволуивают, щюся 0,3-0,4 г безводного су.-)ьфата напрня. Пробы носледователыю переносят в кювету фотоэлект|роколо1риметра и намеряют оптическую плотность при 436 нм против хлороформа или воды лри толщине с.юм 3 мм. О.краска остается стабильной в течанпе 30 мин. Количество ТМТД в н-робе ( мкг) определяют то каЛ|И.бро;воч Ному графику см. фиг. 1, где кривая / относится к случаю, .корда ТМТД взято в виде cTawaaipTиых растворов; кривая 2 показывает количесгво ТМТД в пробе, находя его относительпое содержание в масле расчетом по обп1еизвестпой -методиасе. Мезнанительная (разница в оптической плотности растворов для одной и той же исходшой «онцентра-ции свидетельстиует о точHOiCTiH определения. Предельная чуясивительность метода 4,1 или 1 мкг/мл в анализируемой пробе. П рнме,р 2. Определение 4-хло1рбуги1И-2-ил-Ы-,м-хлорфен1ИЛка|рба.мата (жарбила). РеактИвыгацетон «хч, длстилли-рованная вода, 20 %-ный раствор едкого натра, 10%-ный (20%-ныГ|) раствор серной кислоты, 409й-ный ргитвор уксусной кислоты, смешанный растБОр нитрита натрия (6%) и бромида натрия (10%), 10-о-ный раствор аммиака, 0,05%-иый раствор ос нафтола в этаноле, 50% NaOH и бутанол «хч. Аппаратура:колориметр ФЭК-56, светофильтр № 6 с максимумом пропускания 540 нм в диапазоне 500-560 нм. Пробы загрязненного ка|рби1иом оливкоозого .масла объемом, 1наДриме(р, 1 мл, вносят s дроби1рш1 с пришлифованными стекляины-ми Н робка.ми. Ка)рбИН iipoeKipaTHO -эоистрагируют двойным Количествам ацетона с доба1влением после встряхиваиия анистИлли-раваиной войы в объеме /5 количе|ства адетона и повторнььм встряхиванием, отстаиваиием ,в течение 2 мин и охлаждением смеси до -20°С, как описаяо в лримаре 1. 31 стра1кты собирают вместе в пробирКи с притертыми пробками и проводят их .Х)имическ ую обработку. Сначала, добавив в Э1К|СТ1ракт из каждой лробы по 1 мл 20%-iHoro раствора едкого натра, карбпн гидролизуют, нагревая на кипящей водяяой ба«е в течеИИе 45 мин. Схема 1реаКЦии гидролиза: VNiicoeH.(..a Y L/II IT В охлаиадепНый гищролизат- доба.вл:яют ло 10 .ил во:дь1 и .иейт1рализук)Т;.избыток едкого натра 10 11ли-20%-н.рй cepiioh кислотой, доводя ipH до 4-Ч0 ;с кбнтроле.м по у11жве|рсальной индикаторной 6yiMaire. Затем ina основе содерж.ан1егося в гшдролизате З-хлораадилина получают окрашенное соединение, внося в каждую пробу: 0,4 .нл 40%-,ной уюсуоной кислоты; 0,6 мл cMeujaiHiHoro раствора нитрита « бромида наррня, а после 1выде|рж1ки 5 .иын - 0,4 .«л 10%-ного а.м.миака, 1 .«л 0,05%-ного спиртового раствора аннафтола « 0,4 мл едкого натра. После внесения каждого реактива смесь тщательно взбалтывают. Пригот.о влепные о.меси выдерживают 20- 25 лмн, чтобы 1кол1ичест1вен1но прошла реакция аао1сочета1ния З- хлораиилина с а-нафтоло.м до получения мрачного К|расителя, который эжстра;гируют бутаполам. Бутаиол даносят ло 5 мл ,в (Каждую inpotdy, -встряхивают, отсталвают до разделения и Эгкстракт отб|ирают пипеткой в пробирки, в которые иносят по 0,5 мл алетона. Измерен1ие оптической плот1ности должно быть проведено в течение 20 мин, лооколыоу позд1нее воз можио помут1нение раствора. Количество карбина в пробе (пределяют по кал1и6ровоч1ному (Прафику (см. фиг. 2, кривая 2), а его относительное содержание в масле Пахадят расчетом ino иэвестиой методике. Каж и в Примере /, кр.ивая / на фиг. 2 отноюптся « случаю, копда карбин использован для1Пост1роениЯ графика IB ввде стандаръных paCTiBopoB (без масла). И в этом случае точность метода, иллклстрИруемая расхождением кали1бровачных прямых, оказывается згилчительной и пра1Ктическ1И достаточ-ной.

| название | год | авторы | номер документа |
|---|---|---|---|
| Способ определения остаточных количеств феноксикарба в почве методом высокоэффективной жидкостной хроматографии | 2020 |
|
RU2760530C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ ЦИНЕБА В КОРМОВЫХ КОРНЕПЛОДАХ | 1991 |
|
RU2013771C1 |
| Способ определения неионогенных поверхностно-активных веществ | 1979 |
|
SU900173A1 |
| Способ определения 2,4-дихлорфеноксиуксусной кислоты | 1980 |
|
SU974261A1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ОСТАТОЧНЫХ КОЛИЧЕСТВ ПЕСТИЦИДА В МОЛОКЕ И МЯСЕ | 1991 |
|
RU2034295C1 |
| Способ определения 17-кетостероидов в моче | 1985 |
|
SU1328758A1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ВИТАМИНА C | 2002 |
|
RU2229132C2 |
| Способ определения кватерона в фармацевтических препаратах | 1980 |
|
SU938150A1 |
| Способ определения фосфорорганических нестицидов в воде | 1985 |
|
SU1359741A1 |
| Способ определения катапина-бактерицида в сточных водах | 1986 |
|
SU1418606A1 |
