Предлагается способ получения антибиотика, ранее в патентной литературе не описанный, заключающийся в том, что культуру Streptomyces micarofaciens SF-837 (АТСС 21454) выращивают в аэробных условиях на среде, содержащей источники углерода, азота и минеральные соли, в течение 2-5 дней при температуре 15-38°С до накоиления антиб.нотика в культуральной жидкостн. Затем культуру отф.ильтровывают и прп рН 7-9 экстрагируют органическим растворителем, не смещивающимся с водой, например этилацетатом, этиловым эфиром, метилизобутилкетоном, бутилацетатом, хлороформом, бутиловым спиртом. Получеиный экстракт повторно экстрагируют ири рН 2-3 водным раствором кислоты, например соляной, серной, устанавливают рН 7-9 с помощью щелочи. Полученный экстракт вновь экстрагируют не смешиваюидимся с водой органическим растворителе.м. Экстракт упаривают в вакууме досуха, растворяют сырой продукт в этилацетате или бутилацетате, пропускают раствор через колонку с активированным углем и элюируют активные фракции этилацетатом или бутилацетатом. Активные фракции очищают новториой экстракцией и хроматографическпмн методами. Полученное предложенным способом антибиотическое вещество обладает низкой токсичностью. При испытаннц на .мышах
установлено. что аО%-ная смертность через оцределен 1ые промежуткн временн достигалась соответственно при дозе, 3200, 3100, 2900 и 2850. Штамм, используемый в предложенном способе, выделен из образца иочвы, относится к Streptomyces micarofaciens иод номером 21454 в соответствии с нормами АТСС и имеет след ющие особенное
на разлнчны.х питательных средах.
Сахарозный агар Чапека. Субстратный мицелий от бесцветного до кремового, рост плохой. Воздушны л 1щелнй скудный хлопкоиоНОДоб Ь 1 беловато-серый. РасТВОр МОГО .мента нет.
Глицериновый агар . Субстратный мицелий темно-коричневый. Воздушный м 1целий от белого до кремового, частично с образованием окрашеиного в серый оттенок, хлопко юдобный. Растворимого Н 1гмента нет или очень .мало слабо-розового цвета.
Глюкозо-аспарагпновый агар Чапека. Субстратны светло-коричневый до красновато-коричневого. Воздушный .мицел Й белый бело-розовый. Растворимого пигмента нет 1ли слабо-коричневато-и елтый.
Глюкозо-аспарагиновый агар УЩННСКОГО. Субстратный мицелий коричневый с красноватым оттенком. Воздушный м щелий розоватый. Растворимый 1игмент светло-кор;1чневыи.
Кальци11-малатный агар. Субстратный л ицелий кремовый, рост слабый. Воздушного мицелия нет. Растворимого пигмента нет.
Глицерин-кальциевый агар. Субстратный мицелий светло-коричневый. Воздушный мицелий розовый, переходящий в сероватый. Растворимого ппгмента нет.
Крахмально-си1птетический агар. Субстратный мицелий тем-но-корич«евый с пуроурным оттепком. Воздушный мицелий розовый со значительным красноватым оттенком, постепенно переходящим ъ сероватый. Растворимого пигмента нет.
Бульонный агар. Субстратный мицелИЙ желто-коричневый. Воздушный мицелий очень скудный, белый. Растворимого пигмента нет.
Глю козо-бульоппый агар. Субстратный мицелий от коричневого до темно-коричневого, рост хороший. Растворимого пигмента нет.
Глюкозо-пептонный агар. Субстратный мицелий коричневый. Воздушный мицелий скудный, белый.
Тирозиновый агар. Субстратный мицелий белый.
На картофельных дольках субстратный мицелий от коричневого до темно-коричневого, рост хороший; воздушный мицелий обильный, серовато-коричневый, растворимый пигмеит темновато-коричневый.
На дольках моркови субстратный мицелий коричневый, воздушный мицелий от белого до кремового; растворимый пигмент коричневый но периферии колонии.
Сиятое молоко. Субстратиый мицелий светло-коричневый, рост поверхностный. Воздушный мицелий скудный, белый.
Яйцо. Субстратный мицелий бледно-коричневый, воздушного мицелия пет.
Коагулированная сыворотка Лсффлера. Субстратный мицелий бледно-коричневый. Воздушного мицелия иет.
Глюкозный раствор Чапека. Субстратный мицелий бледно-коричиевый. Воздушиый мицелий скудный, белый.
Желати} а. Субстратный мицелий от кремового до бледио-коричневого. Воздушно1о мицелия нет.
Среда Беинета. Субстратный мицелий от коричиевого до темно-коричневого. Воздушиый М1шелий розовый с красноватым оттенком.
Предложенным способом иолучено веш,ество, обладающее свойствами антибиотика, которое ингибирует рост граммиоложительных бактерий. Оио включает четыре активные фракцни. Антибиотик растворяется в метиловом спирте, этиловом снирте, ацетоне, хлороформе, этилацетате, бутилацетате, подкисленной воде, бензоле, этнловом эфире и четыреххлористом углероде, плохо растворяется в петролейном эфире, нормальном гексане и нейтральной воде. Имеет осиовиую реакцию, образует с кислотами соли.
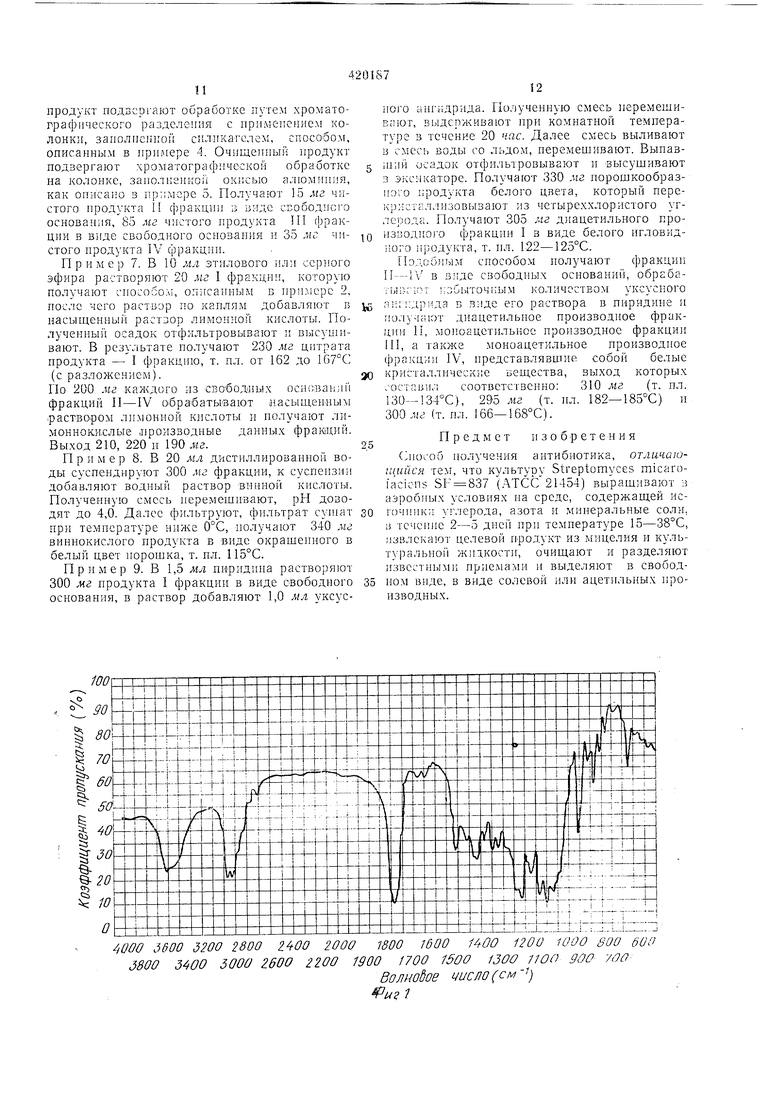
Антнбиотик содержит в молекуле углерод, водород, азот и кислород и вращает плоскость поляризации влево в этиловом спирте, причем это веиюство проявляет свойства макролпдных антнбиотиков. I фракция в свободной основной форме образует белый иорошок с т. нл. 122-124°С, проявляющий степень рКа 6,9 в 50 %-ном растворе этилового сцир5 та в воде. Элементарный состав, %: С 60,38, Н 8,35, N-1,65 и остальное О, мол. вес 813, что оиределено путем масс-анализа, эмпирическая формула C4iH67O 5N, при концентрации в этиловом спирте 1 % оптическое враще0 ние «1 - 67°. Максимальная абсорбционная снособность лучей ультрафиолетового участка спектра раствора в этиловом спирте при длине .вол.ны 232 нм раВна(Е}Д 325). Вещество в виде пилюль, т. е. в виде свободного основания в бромистом калии, обладает сиособностью поглощать лучи в инфракрасном участке спектра при следующих волиовых числах, 3500, 2970, 2930, 1731460, 1408, 1376, 1360, 1330, 1295, 1275, 1190,
0 1165, 1120, 1082, 1050, 1015, 910, 860, 840, 805, 780, 735 и 680. Диацетилпроизводпое продукта образует окрашенные в белый цвет иглы, т. ил. 122-125°С при перекристаллизации из четыреххлористого углерода.
Элемеитариый состав, %: С 60, 35, Н 8,02, К 1,58, а остальное О - 30,05, что соответствует эмпирической формуле C45H7iOi7N,причем максимальная абсорбционная способность при 232 нм равна (Ej-.; 295) в ультрафиолетовой области спе(тра в растворе в этиловом снирте. Это нроизводиое характеризуется способностью иоглощать излучение в инфракрасной области спектра граиул в бромистом калии при следующих волновых числах, с.н. 3450, 2970, 2930, 1728, 1454, 1368, 1232, 1167, 1122, 1082, 1054, 1024, 1002, 957, 907, 868, 837, 788, 762. И фракция в виде свободиого основания иредставляет собой окрашенный в белый цвет порошкообразный продукт, т. пл. 125-128°С; показатель рКа в 50%-ном растворе этилового спирта в воде 6,8. Элементарный состав, %; С 60,58, Н 8,85, N 1,72, остальное О - 28,85; мол. вес 827; эмпирическая формула C42H69Oi5N; оптическое вращение а 1 -68° нрн концентрацин 1% в этнловом спирте. Максимальная абсорбционная способпость при длине волны 232 нм (Е 320) в ультрафиолетовом участке
0 снектра в растворе этилового спирта. Характеризуется абсорбционной способностью излучения в инфракрасной области снектра и в виде свободного основания в форме гранул в бромистом калии при следующих волновых
5 числах, 3500, 2970, 2985, 1737, 1460, 1410, 1377, 1360, 1300, 1275, 1190, 1170, 1123, 1082, 1052, 1017, 990, 920, 910, 860, 840, 805, 780, 740 Н 700. Диацетилпроизводиое II фракции образует окрашенные в белый цвет иглы с
0 т. нл. 130-134°С. Элемеитариый состав, %; С 60,68, Н 8,28, N 1,49, а остальное ,60. Эмнирическая формула С4бН7з017К.
III фракция в виде свободного основания иредставляет собой окрашенный в белый цвет
)5 норошок с т. ил. 122-125°С, в 50%-лом водно-сп 1ртовом растворе рКа равен /,0. Элементарный состав, %; С 60,58, Н 8,23, N 1.87, а остальное 0-29,32; мол. вес 811; эмпирическая формула C4iH65Oi5N. Оптическое вращение равно а JJ -42° при 1%-ной концентрации в этиловом спирте. Максимальная абсорбционная способность при длине волны 280 им равна (Е ,;, 295) при излучении в ультрафиолетовой области спектра в растворе этилового спирта. Поглощает излучение в ипфракраспой области спектра в форме гралул в бромистом калии в виде свободного оснозалия при следующих волновых числах, С.1Г: 3500, 2970, 2930, 1738, 1680, 1640, 1600, 1460, 1378, 1360, 1300, 1275, 1252, 1190, 1168, 1121, 1088, 1052, 1015, 980, 910, 863, 840, 805, 780. Моноацетильное производное III фракции образует кристаллы, подойные кристаллам речного песка, т. пл. 182-185°С. Элементарный состав, %: С 60,5, Н 7,92, N 1,68, а остальное О-29,84. Эмпнрическая формула С4зНб7М.
IV фракция представляет собой свободное оспова.ние, которое образует порошкообразный продукт, окрашенный в белый цвет, т. лл. 120-122°С, показатель рКа з 50%-ном водном ра-створе этилового спирта равен 7.0. Элементарный состав, %; С 60,82, И 8,52, N 1,73, а остальное О-28,93, молекулярньи: вес 825. Эмпи рическая фсрмула C.i2H67Oi5N. Оптическое вращение -40° при концентрации 1% в этиловом -спирте. Максимальная абсорбционная способиость :1злуче1 ия г ультрафиолетовой области спектра при длине волны 280 им (Е 1 285) в растворе в этиловом спирте. В виде гранул в форме свободного основаиия в бромистом калии характеризуется иоглопиющей способностью в инфраKpacHoii области излучения ири следуюидих волновых числах, слг: 3500, 2970, 2930, 1738, 1680, 1640, 1600, 1460, 1378, 1,360, 1300, 1276. 1252, 1190, 1170, 1120. 1082, 1052, 1017, 980, 920, 910, 863, 840 и 780.
Моноацстилпроизводное IV фракции образует кристаллы, имеюидае форму круииц речного песка, т. ил. 166-168°С. Элементарный состав, о/с: С 60,85, Н 8,06, N 1,65, а остальное О-29,44. Эмп;1рическая формула C4.iHG90i6N. Все перечисленные фракции могут быть получены одновременно в ходе проведения процесса кyльтивиpoвaiпия П1тамма и характер:.1зу1отся аналогичными физико-хим-ическими свойствами.
Па ф:ИГ. 1-4 иредставлепы характеристики I-IV фракций.
Пример 1. В 60 л жидкой иитательной среды, содержащей 2,5% осахаренного крахмала, 4% растворимого растительного протеина, 0,3% хлористого калия и 0,3% углекислого кальция, при рП 7,0 инокулируют штаммом II при постоянном перемешивании культивируют ири 28°С в течение 35 час з условиях непрерывной аэрации. Полученную культуру отфнльтровывают, мицелий промываю:
ВОДНЫМ растворо.м соляной кислоты. Фильтрат объединяют с промывной жидкостью, в результате оби1.ее количество полученной жидкой фракции составляет 50 л (потенция или сила равна 150 мкг/мл). Далее из фильтрата при рП 8, с помощью 25 л этилацетата экстрагируют активное вещество, далее 22 л этилацетатксй фракции концентрируют до конечного объема, равного приблнз 1тельно 3 л, 3 условиях пониженного давления. Полученный конпентрат разбавляют 1,5 л воды, доводят рП среды до 2,0 добавлением 5 н. раствора соляной кислоты ii тщательно перемеапизают вcтp ixивaниe i. Затем водную фазу отделяют от орган 1чеСкой фазы, доводят рН ;гэдного раствора до 8,0 добавлением 3 н. раствора гидрата о-киси натрия. Далее экстрагируют 800 мл этилацетатов. Полученный этилацетатный экстракт перемешивают встряхиванием с 5GO мл водного раствора соляной югслоты. в результате чего активные фракции переходят в этот раствор, который затем повторно экстраг; руют с 400 .ил серного эфира при рП 8. Далее эфирный экстракт сушат безводным сульфатом натрия с последующим лонцентрмрованием ири пониженном давлении, в результате получают 16,5 г светло-желт;)го порошК.;образного продукта.
После это;-,) 12 г продукта растворяют в 200 мл. эти.-ишетата и раствор ироиускают через колонку с 600 мл тонкодисиерсного угля, иредварггельно пропитанного этилацетатом. Элюирхют этилацетатом. после чего акт; виые фра-кцли элюата объединяют, обп1,ее количество конечной фракции 2500 мл. Затем ее выпаривают досхха в условпях пониженного давления, получают 5 г отвращенного з белый цвет и.ороикообразного продукта. Далее этот продукт растворяют в 0 мл бензола и отф1:льтровывают нерастнсри:5И1уюся чпсть. После этого фильтрат иропускают через хромг:тсграф :чсскую колонку, в KOTOpoii содерЖ1 тс)1 700 мл силикагеля, предварительно иропитанного бензолом, с иоследуюп1ей элюпией с помоицпо бензола
и ацетона в соотнашен1Ч 4;1. Полученный
элюат собирают в виде фракгип. солпчество
каждой из ,которых составляет 20 .i/.г. После
этого активные
фракции Ло 90-380, характеризующиеся только одним пятном в ходе проведения хроматографнческого анализа в тонком слое окиси алюминия, что соответствует содержанию чистой I фракции, в соответствии с величиной Rf для одного пятна, объедиияют, o6niee ко3 .личестБо составляет 4000 мл. После этого продукт концентрирмот в условиях поииженного давления, получая 1.5 t окрашенного в белый цвет иорош.кообразного иродукта, т. ил. 122-124°С. В результате а 1ализа устаповле0 но, что он представляет собой ч 1стый продукт в виде свободного основания.
Пример 2. 20 л питательной среды, сндержаще (%) 3 глюкозы, 1 пептона, 0,5 мясного экстракта, 0,2 хлористого иатрия, 0,3 уг5 лекислого кальция и 0,3 соевого acлa, рП 7.0,
помещают в вибрационный бродильный чан. емкостью 30,0 л, стерилизуют при 120°С 20 мин. Стерилизованную питательную среду .ино,кулируют Streptomyces micarofaciens с последующим культивирова-нием .при иоетоян«ом перемешл-вании при 28°С в течение 48 час с одновреме;нной аэрацией. Далее жидкую ферментативную среду фильтруют при рН 6 н получают 18 л фильтрата (сила 200 .икгЛил). рН фильтрата доводят до 7,5, добавляя 3 н. раствор гидрата окиси натрия, и экстрагируют 8 л бутилацетата. Полученный бутилацетатный экстракт подвергают обра|бот:ке, оиисанной ,в 1примере 1, т. пл. окрашенного в белый цвет конечного порошкообразного продукта 122- 124°С. Выход антибиотика 800 мг.
Пример 3. 4,0 г сырого порошкообразного вещества, окрашенного в желтый цвет, полученного оии;санным в примере 1 способом, суспендируют в 1000 мл воды. Величину рН суспензии доводят до 3,5 добавлением 5 н. раствора соляной ки-слоты. Раствор пропускают через колонку, содержащую 100 мл продукта Амберлит СС-50 (кокаино-активного типа). Смолу промывают 800 мл дистиллировап.ной воды с последующей элюцией смесью 0,4 н. раствора соляной кислоты с этиловым спиртом в соотношении 1:2. Объединяют активные фра;кции элюата, общ,ий объем их составляет 180 мл. Величину рП элюата доводят до 6,0 добавлением 3 н. раствора гидрата окиси натрия и концентрируют до объема 40 мл в условиях пониженного да1Вле И1Я. Далее величину рП сконцеитр.ированного продукта доводят до 8,0 добавлеиием 3 н. раствора гидрата окиси натрия с иоследующим двойным экстрагированием двумя порция-М-: по 50 мл каждая этилового эфира. Полученные фракции объединяют и концентрируют в условиях иониженного давления. В результате получают 2,1 г окра1щенного в белый цвет порошкообразного продукта, который растворяют в 10 мл этилацетата. Полученный раствор пропускают через колон.ку, заполненную 200 мл окиси алюминия, предвар1 тельно проиитанной этилапетатом, с последующей элЕоцией этилацетатом. Активные фракции элюата объединяют. Об1цее количество этого элюата 200 мл. Далее в элюат добавляют 40 мл дистиллированной воды и доводят рН до 1,8 добавлением 5 н. раствора соляной кислоты с иоследующим тщательным перемещением путем встряхивания. Далее водную фазу отделяют, доводят рП до 8,5, добавляя I н. раствор гидрата окиси натрия, в результате твердый продукт выпадает в осадок. Осадок отфильтровывают, растворяют в бензоле, а затем раствор иропускают через колонку с еиликагелем (как описано в нрнмере 1), причем (разделение производят в соответствии с вариантом осуществления иредлагаемого способа. Получают норошок белого цвета. Выход антибиотика 300 мг.
П р и м е р 4. Штамм АТСС 21454 Streptomyces micarofaciens инокулируют в 300 л жидкой
питательиой среды, содержащей (%) 2,0 глюкозы, 1 пептона, 0,5 мясного экстракта, 0,4 кукурузного экстракта, 0,2 хлористого натрия, 0,3 углекислого кальция и 0,2 лярда, при рН 7,0, и культивируют при 28°С в течение 70 час в условиях аэрации.
Культуральную жидкость фильтруют и мицелиальную массу промывают соляной кислотой. Фильтрат объединяют с промывными жидкостями, в реззльтате чего общий объем конечной массысоставляет 280 л (сила 320 мкг1мл). Далее фильтрат экстрагируют 70 л этилацетата и 71 л полученной этилацетатной фазы концентрируют до 20 л в условиях пониженного давления. Концентрат разбавляют 10 л воды, доводят рН до 2,0 добавлением 5 н раствора соляной кислоты с последующим тщательным перемещнванием путем встряхивания. Далее водную фазу отделяют, доводят рН до 9,0 добавлением 3 н. раствора гидрата окиси натрия и экстрагируют 4 л этилацетата. Полученный экстракт перемешивают встряхиванием с применением 2 л водного раствора соляной кислоты. Затем вновь экстрагируют 1 л этилацетата при рН 8,0. После этого экстракт сушат безводным сульфатом натрия с последующим концентрированием в условиях пониженного давления. Получают 92 г сырого порошкообразного продукта, окрашенного в желтый цвет.
90 г этого продукта растворяют в 1 л этнлацетата, раствор пропускают через колонку, заполненную 1,5 л угля, пропитанного этилацетатом е последующей элюцией этнлацетатом, после чего активные фракции элюата объединяют. Получают 5,5 л. Далее объедипеппую фракцию выпаривают до сухого соетояиия в условиях попижеппого давлеиия. Получают 55 г порошкообразного продукта белого цвета (сила коиечпого продукта 700 мкг/мг).
Порощкообразный продукт подвергают обработке методом распределения по принципу противотока, применяя бензол и 0,ЗМ раствор фосфорного буффера (рП 4,40), используют 2,5 л каждого растворителя и нижний подвижHbni слой подвергают десятикратной обработке методом распределения. Антибиотик распределяется но пробиркам или трубкам с первой по третью и сосредотачивается преимущественно во второй пробирке, тогда как большая часть, т. е. приблизительно 80% остальных фракций - П-IV сосредотачиваются в первой пробирке.
После этого содержимое первой пробирки или трубки (2,4 л) концентрируют, получают 7,7 г продукта, окрашенного в белый цвет (сила 720 мкг/мг). После этого продукт растворяют в 15 мл бензола, нерастворившийся остаток отфильтровывают, фильтрат пропускают через колонку, заполненную 800 мл силикагеля, который предварительно пропитывают бензолом. Активные вещества элюируют смесью бензола с ацетоном в соотношении 4:1. Элюаты объединяют. Далее те фракции элюата (№ 28-30), которые характеризовались только одним пятном, объединяют в вакууме. Получают 140 мг порошкообразного продукта, окрашенного в белый цвет, т. пл. 120-122°С. Это IV активная .фракция в чистом виде в форме свободного основания.
Далее фракции № 38-57, которые характеризовались образованием трех пятен в ходе проведения хроматографического анализа в тонком слое окиси алюминия, объединяют и концентрируют в условиях пониженного дав.ления. Получают 1,1 г порошкообразного продукта белого цвета, который представляет фракции II и III и незначительное количество I фракции. Фракции № 70-95, которые характеризовались образованием только одного пятна в ходе проведения хроматографического анализа в тонком слое окиси алюминия содержали только I фракцию. Затем концентрируют в условиях пониженного давления и получают 0,8 г порошкообразного продукта, окрашенного в белый цвет, т. пл. 122-124°С. Он представляет собой I фракцию в виде свободного основания.
П|ример 5. В 4 мл бензола раствсряют 1 г продукта, окрашенного в белый цвет и содержаш.еео II и 1П активные фракции и значительное количество I фракции. После этого раствор пропускают через колонну, заполненную 100 мл окиси алюминия, пропитанной бензолом, и элюируют смесью этнлацетата с .бензолом в соотношении 1:1. Элюат собирают в виде отдельных фракций, количество каждой из которых составляет 20 мл. Каждую фракцию подвергают испытанию хроматографическим путем в тонком слое окиси алюминия с применением в качестве элюата смеси этилацетата с бензолом в соотношении 2:1. После этого те фракции, номера которых находятся Б пределах от 30 до 48 и которые характеризуются только одним пятном в ходе ироведени : хроматографического анализа в тонком слое окиси алюминия, объединяют и концентрируют в условиях пониженного давления до порошкообразного продукта, окрашенного в белый цвет с т. пл. 122-125°С. Получают IV активпую фракцию в виде свободного основанрт.
После окончания вымывания фракции N° 80 с хроматографической колонки смесью этилацетата с бензолом в соотношении 2:1, фракции № 90-125 концентрируют в условиях пониженного давления. В результате получают 280 мг порошкообразного продукта белого цвета, представляющего собой 11 активную фракцию. Далее 250 мг этого продукта растворяют в 3 мл бензола, раствор пропускают через колонку, заполненную 50 Л1л окиси алюминия, предварительно пропитанной бензолом, затем элюируют 300 мл смеси этилацетата с бензолом в соотношении 1:3, после чего заменяют эту смесь смесью этилацетата с бенлом в соотношении 2:1. Элюат собирают в виде отдельных фракций, количество каждой из которых составляет 5 мл, после чего каждую фракцию испытывают путем хроматографического анализа в тонком слое окиси алюмин::я. Далее ()ракции Ло 3G-47. образуюш,ие только одио пятно в ходе хроматографического анализа в тонком слое, объединяют и концентрируют в условиях пониженного давления. Получают 80 мг порошкообразного вещества белого цвета, представляющего II активную фракцию.
Пример 6. Штамм , который получил наименование Streptomyces mkarofaciens, инокулпруют в 30 л жидкой питательной среды, содержащей (%) 3,0 глюкозы, 5 растворимого растительного белка, 0,2 хлористого калия и 0,3 углекислого кальция, при рН 7,0. Затем культивируют при 30°С в течение 85 час при постоянной аэрации. Далее ферментативную среду фильтруют, м щелиальную массу нромывают разбавленным раствором соляной кислоты. Фильтрат объединяют с промывной жидкостью до объема 28,1 л (сила 300 мкг/мл). Полученный фильтрат при рП 8,4 экстрагируют с 7 л бутилацетата, а полученный экстракт, как описано в примере 4. Получают 15,2 г. После этого в 200 мл бутилацетата растворяют 15 г сырого иорошкообразного же.ттого продукта, раствор пропускают через колонку, заполненную 400 мл угля, предварительно нропитан«ого бутилацетатом, с последующей элюцией бутплацетатом. Активные фракции элюата объединяют. Общее количество объединенной ()ракци11 составило 1,8 л. Общую фракцию концентрируют в условиях пониженного давления до мл. Далее концентрат разбавляют 50 мл дистиллированной воды, рП раствора доводят до 2,0 добавлением 6 п. раствора соляной кислоты, после чего конечную массу перемен ивают встряхиван11См. Затем водную фазу отделяют, величину рП этого водного раствора доводят до 9,0 добавлением 1 н. раствора гидрата окиси натрия, в результате чего выпадает в осадок конечньп продукт. Затем осадок отфильтровывают и высушивают в эксикаторе, в результате получают 6,3 г окрашенного в белый цвет порошкообразного продукта, спла которого 720 мкг/мг.
Порошкообразный белый продукт растворяют в 12 мл бензола, а нерастворнвшуюся часть отфильтровывают. Фильтрат пропускают через колопку, заполненную 600 мл силикагсля, нропитапного бензолом с последующей элюписй смесью бензола с ацетоном в соотношении 6:1. Элюат получают в виде отдельных фракций по 20 мл. Активные фракции П-IV вымывают ранее I фракции. После этого каждую из полученных фракций подвергают испытанию путем хроматографического анализа в тонком слое снликагеля с применением в качестве элюата смеси бензола с ацетоном в количественном соотношении 2:1. Фракции 64-82 концентрируют в условиях пониженного давления, в результате получают 800 мг окрашенного в белый цвет порошкообразного продукта, который включает фракци; II-IV. с незначительным количеством 1 фракц. После этого порошкообразный
И
продукт подвергают обработке путем хроматографического разделения с iipsiMeneni-ieM колонки, заполненной еиликагслем, способом, описанным в лрпмере 4. Очищенный нродзкт подвергают хроматографической обработке на колонке, занолнеикой окясью алюминия, как описано з примере 5. Получают 15 мг чпетого продукта 1 фракции з виде ссободпсго основания, 85 мг чистого продукта 1П фракции в виде свободного осиозанпя и 35 мг чистого нродукта IV фракции.
Пример 7. В 10 мл зтилового или серного эфира растворяют 20 мг I фракции, которую получают сиособом, описаниым в нри.лиере 2, после чего раствор по каплям добавляют в насыщенный растзор лимонной кислоты. Полученный осадок отфильтровывают и высушивают. В результате получают 230 мг цитрата продукта - I фракцию, т. пл. от 162 до 167°С (с разложением).
По 200 мг каждого из свободиых основаh;iи фракций И-IV Обрабатывают насыщенным .раствором лимонной кислоть и получают лимоннокЕслые лроизводные дапных фракций. Выход 210, 220 и 190 лгг.
Пример 8. В 20 мл диетнллироваппой воды еуспепдируют 300 м.г фракции, к суспензии добавляют водный раствор винной кислоты. Полученную смесь иеремегпивают, рП доводят до 4,0. Далее фильтруют, фильтрат сушат при температуре ниже 0°С, получают 340 мг виннокислого продукта в виде окрашенного в белый цвет иорошка, т. пл. 115°С.
Пример 9. В 1,5 мл пиридина растворяют 300 мг продукта I фракции в виде свободного основания, в раствор добавляют 1,0 мл уксус12
ного ангидрида. Полученную смесь перемешивают, выдерживают при комнатной температуре в течение 20 гас. Далее смесь выливают 13 смесь воды со льдом, перемешивают. ВьшавИ1ИЙ осадок отфильтровывают и высушивают 3 эксикаторе. Получают 330 мг порошкообраз1 ого продукта белого цвета, который перекрисгаллизовывают из четыреххлориетого углерода. Получают 305 мг диацетильного производиоо фракции I з виде белого игловидiioro продукта, т. пл. 122-125°С. Подобным способом получают фракции в виде свободпых оснований, обраба: ;зСьггоч ;ым количеством уксусного ангидрида в виде его раствора в пиридине и ;л,иацетильиое производное фрак)Ц и и 1 i, моноацетнльное пронзводиое фракции
II, а также моноацетильное производпое фракц 1 IV, представлявшие собой белые кристаллические вещества, выход которых составил соответственно; 310 мг (т. пл. 130--134°С), 295 мг (т. пл. 182-185°С) и 300 лс (т. пл. 166-168°С).
Предмет изобретения
(йюсоб нолучения антибиотика, отличающийся тем, что культуру Streptomyces micarolacic-ns (.-VrCC21454) выращивают ; аэроб1гых условиях па среде, содержащей исгочпшчи углерода, азота и минеральные соли, и течение 2-5 дней прн температуре 15-38°С, извлекают целевой продукт из мицелия и культуральноГ жидкости, очищают и разделяют известными нриемами и выделяют в свободном виде, в виде солевой или ацетильных производных.




| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения антибиотиков | 1967 |
|
SU528883A3 |
| Способ получения антибиотического комплекса а-35512 | 1977 |
|
SU751332A3 |
| Способ получения антибиотика | 1977 |
|
SU741804A3 |
| Способ получения антибиотика | 1965 |
|
SU556732A3 |
| Способ получения алкалоидов спорыньи | 1976 |
|
SU845827A1 |
| СПОСОБ ПОЛУЧЕНИЯ АНТИБИОТИЧЕСКОГО КОМПЛЕКСА | 1972 |
|
SU352469A1 |
| Способ получения антибиотического комплекса | 1967 |
|
SU884575A3 |
| Способ получения антибиотика @ -15003 @ -3 | 1978 |
|
SU1036251A3 |
| Способ получения антибиотика, обладающего -лактамазной ингибирующей активностью | 1975 |
|
SU576965A3 |
| Способ получения антибиотика казугамицина | 1964 |
|
SU454749A3 |


4000 ддОО 5200 2800 2.00 2000 1800 1600 1200 1000 800 600 дЗОО 5000 260О 2200 1900 t700 15ОО 130О 1WO ЭОО 7ОО
Волновое Ljucno(CM)
О
000 3000 WOO 1800 1600
000 3000 2000 1800
1200 1000 80п
f-no 00
Волнобое число (см} 1риг.2
бОП 0
1600 то 1200 1000 800 Волнодое число (см }
Во Л И О бое /исло (см )
