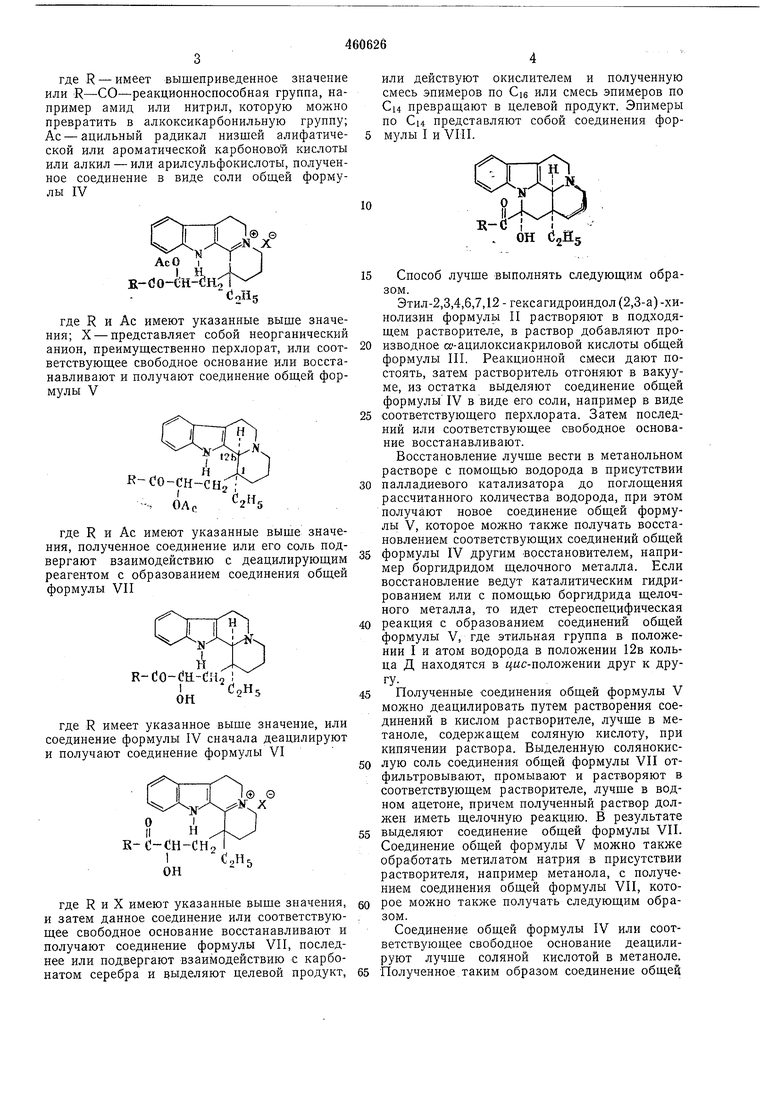
где R - имеет вышеприведенное значение или R-СО-реакционноспособная группа, например амид или нитрил, которую можно превратить в алкоксикарбонильную группу; Ас - ацильный радикал низшей алифатической или ароматической карбоновой кислоты или алкил - или арилсульфокислоты, полученное соединение в виде соли обш,ей формулы IV Yf Н k J К-ЙО-СН-ЙНЛ где R и Ас имеют указанные выше значения; X - представляет собой неорганический анион, преимущественно перхлорат, или соответствующее свободное основание или восстанавливают и получают соединение общей формулы V к-Со-сн-сы где R и Ас имеют указанные выше значения, полученное соединение или его соль подвергают взаимодействию с деацилирующим реагентом с образованием соединения общей формулы VII R-dO-OE-Ciio , I С ОН где R имеет указанное выше значение, или соединение формулы IV сначала деацилируют и получают соединение формулы VI S-0-CH-CH где R и X имеют указанные выще значения, и затем данное соединение или соответствующее свободное основание восстанавливают и получают соединение формулы VII, последнее или подвергают взаимодействию с карбонатом серебра и выделяют целевой продукт. или действуют окислителем и полученную смесь эпимеров по Cie или смесь эпимеров по Си превращают в целевой продукт. Эпимеры по Си представляют собой соединения формулы I и VIII. Способ лучше выполнять следующим образом. Этил-2,3,4,6,7,12 - гексагидроиндол(2,3-а)-хинолизин формуль П растворяют в подходящем растворителе, в раствор добавляют производное то-ацилоксиакриловой кислоты общей формулы III. Реакционной смеси дают постоять, затем растворитель отгоняют в вакууме, из остатка выделяют соединение общей формулы IV в виде его соли, например в виде соответствующего перхлората. Затем последний или соответствующее свободное основание восстанавливают. Восстановление лучше вести в метанольном растворе с помощью водорода в присутствии палладиевого катализатора до поглощения рассчитанного количества водорода, при этом получают новое соединение общей формулы V, которое можно также получать восстановлением соответствующих соединений общей формулы IV другим восстановителем, например боргидридом щелочного металла. Если восстановление ведут каталитическим гидрированием или с помощью боргидрида щелочного металла, то идет стереоспецифическая реакция с образованием соединений общей формулы V, где этильная группа в положении I и атом водорода в положении 12в кольца Д находятся в цис-положеник друг к друПолученные соединения общей формулы V можно деацилировать путем растворения соединений в кислом растворителе, лучше в метаноле, содержащем соляную кислоту, при кипячении раствора. Выделенную солянокислую соль соединения общей формулы VII отфильтровывают, промывают и растворяют в соответствующем растворителе, лучше в водном ацетоне, причем полученный раствор должен иметь щелочную реакцию. В результате выделяют соединение общей формулы VII. Соединение общей формулы V можно также обработать метилатом натрия в присутствии растворителя, например метанола, с получением соединения общей формулы VII, которое можно также получать следующим образом. Соединение общей формулы IV или соответствующее свободное основание деацилируют лучше соляной кислотой в метаноле. Полученное таким образом соединение общей
формулы VI восстанавливают до соединения общей формулы V. Восстановителем может служить боргидрид щелочного металла или другие восстанавливающие реагенты. Соединения общей формулы VI можно также восстанавливать водородом в присутствии катализатора. Если восстановление ведут каталитическим гидрированием или боргидридом щелочного металла, то идет стереоспецифическая реакция.
Соединения общей формулы VII можно окислить в целевой продукт, если эти соединения растворить в соответствующем растворителе, например диметилсульфоксиде, и к нему добавить раствор фосфорной кислоты в диметилсульфоксиде. Затем по каплям добавляют свежеперегнанный дициклогексилкарбодиимид и смеси дают постоять. Через несколько часов смесь выливают на лед (раствор должен иметь щелочную реакцию) и смесь экстрагируют соответствующим растворителем, лучше дихлорометаном. Дихлорметановый раствор сушат, растворитель отгоняют в вакууме и получают смесь (±)-винкамина и (±)-эпивинкамина или смесь других соединений формулы I. Остаток кристаллизуют из соответствующего растворителя, с получением (±)-винкамина или относящихся к нему соединений общей формулы I. Согласно другому методу остаток не кристаллизуют, а подвергают эпимеризации и получают (±)-винкамин или относящиеся к нему соединения общей фоомулы I.
Соединения общей формулы VII можно также окислять в бензольной среде карбонатом серебра. Реакцию ведут при температуре кипения реакционной смеси. Растворитель выпаривают в вакууме и получают остаток, представляющий собой смесь {±)-винкамина и С±)-эпивинкамина или смесь соединений общей формулы I, и полученную смесь обрабатывают как указано выше.
Окисление соединений общей формулы VII и последующую эпимеризацию продуктов можно проводить в одну стадию. В этом случае реакцию ведут с карбонатом серебра, а бензол заменяют другим инертным растворителем или смесью растворителей. В качестве растворителя можно применять углеводороды бензольного типа или их смеси, предночтительно применяют ксилол.
Ксилольный раствор выпаривают приблизительно до 20% от начального объема в вакууме в атмосфере азота, оставляют стоять, выпавшие кристаллы отфильтровывают,- промывают и сушат. Таким образом получают (±)-винкамин или относящиеся к нему соединения общей формулы I.
(±)-Винкамин или относящиеся к нему соединения формулы I можно разделять с помощью 0,0-дибензоилвинной кислоты, й-винной кислоты, й-камфарсульфокислоты или других оптически активных кислот. Оптически активные конечные продукты можно так-. же получать разделением любых рацемических промежуточных- продуктов в процессе синтеза и проведением последующих стадий с оптически активными соединениями.
Пример 1. Получение {Н-)-винкамина. А. Получение 1-(3-ацетокси-(3-метоксикарбонилэтил)-1-этил-1,2,3,4,6,7- гексагидро-12Ниндол (2,3-а)-хинолизин перхлората (формула IV; R - метоксильная группа; Ас - ацетильная группа; Х-перхлорат ион).
7,09 г (2,8 ммоль) 1-Этил-2,3,4,6,7,12-гексагидро-12Н-индол(2,3-а)-хинолнзина (соединение формулы II) растворяют в 100 мл сухого дихлорметана. К раствору добавляют 10 мл метилового эфира го-ацетоксиакриловой кислоты и реакционной смеси дают постоять при комнатной температуре в течение 2 дней. Дихлорметан отгоняют в вакууме, темный маслянистый остаток растирают -с 3x50 мл петролейного эфира, растворяют в 20 мл метанола, раствор подкисляют до рН-5 с помощью 70%-ной надхлорной кислоты. Последнюю операцию проводят при охлаждении. Выделившийся перхлорат 1-(р-ацетокси-(3-метоксикарбонилэтил) - 1-этил-l,2,3,4,6,7-гeкcaгидpo12Н-ИНДОЛ) 2,3-а-хинолизина отфильтровывают и промывают холодным метанолом и эЛиром. Получают 7,00 г продукта (выход 50%), т. пл. 152-154°С. Найдено, %: С 55,40; Н 5.60; N 5,90.
C23H2oN2O8Cl (мол. вес. 496.93).
Вычислено, %: С 55,56; Н 5.88; N 5,54. ИК спектр; СОН), 3500 (NH). 1730 (СО2СНз)т, 1760 (СООСНз), 1630, 1538 (C N) см-1.
Б. Получение 1-(6-ацетокси-р-метоксикарбонилэтил) - 1 -этил-1,2,3,4,6.7,12,12в-октагидроиндол (2,3-а)-хинолизина (формула V; R-метоксильная группа; Ас-ацетильная группа).
бь 7,50 г Перхлората 1-((5-ацетокси-р-метоксикяпбонилэтил - 1-этил-1,2,3,4,6,7-гексагидро-12Н-индол (2,3-а)-хинолизина растворяют в 350 мл метанола и к раствору добавляют 4,5 г 10%-ного палладия на угле. Реакционную смесь гидрируют. Смесь поглощает рассчитанное количество водорода в течение 2 ч. Смесь отфильтровывают, фильтрат упаривают, получают 7 г перхлората 1-(6-ацетокси-р-метоксикарбонилэтил) -1 -этил-1,2,3,4,6.7,
12,12в-октагидроиндол(2,3-а)-хинолизнна, который можно использовать в следующей стадии без очистки.
1 г перхлората 1-(В-ацетокси-6-метоксикарбонилэтил) -1 -этил-1.2.3,4,6,7,12,12в-октагидроиндол (2,3-а)-хинолизина растворяют в 5 мл 80%-ного водного ацетона, раствор подщелачивают гидроокисью аммония до рН-8. Выпавший кристаллический 1-(6-ацетокси-6-метоксикарбонилэтил) - 1 - этил-1,2,3,4,6,7,12,12воктагидроиндол(2,3-а) -хинолизин плавится при после перекристаллизации из метанола. Выход 0,67 г.
Найдено, %: С 69,58; Н 7.77; N 7,06. С2зНзоМ2О4 (мол. вес 398,49).
Вычислено, %: С 69,32; Н 7,58; N 7,03.
ИК спектр: (NH), 1720 (СО2СНз), 1760 (ОСОСНз) см-Ч
62- 1 г перхлората 1-(р-ацетокси-р-метоксикарбонилэтил)-1-этил - 1,2,3,4,6,7 - гексагидро12Н-ИНДОЛ (2,3-а)-хинолизина суспендируют в 100 мл метанола, в суспензию добавляют 0,7 г боргидрида натрия при охлаждении ледяной водой. Во время добавления реакционную смесь сильно перемешивают. Через 10 мин смесь подкисляют до рН-б ледяной уксусной кислотой, смесь упаривают в вакууме досуха. Остаток распределяют между 5%-ным раствором бикарбоната натрия в воде и эфиром. Эфирный слой сушат и упаривают досуха. Остаток перекристаллизовывают из метанола. Получают 0,5 г 1-(р-ацетокси- |3-метоксикарбонилэтил)-1-этил-1,2,3,4,6,)7, 12,12в-октагидроиндол (2,3-а)-хинолизина.
В. Получение перхлората 1-(р-окси-р-метокарбонилэтил)- -этил -1,2,3,4,6,7-гексагидро12Н-ИНДОЛ (2,3-а)-хинолизина (формула VI; JR - метоксильная группа; - перхлорат ион).
0,3 г перхлората 1-(|р-ацетокси-р-метоксикарбонилэтил)-1-этил - 1,2,3,4,6,7-гексагидро12Н-ИНДОЛ (2,3-а) -хинолизина (полученного, как описано в п. А) растворяют в 10 мл метанола, содержаш,его 0,165 г соляной кислоты на 1 мл. Раствор кипятят 2 ч и упаривают досуха. Остаток растворяют в воде и к раствору добавляют 70%-ную водную надхлорную кислоту. Получают 0,2 г перхлората 1-(рокси-;р-метоксикарбонилэтил) - 1-этил-1,2,3,4,6, 7-гексагидро- 12Н-ИНДОЛ (2,3-а) - хинолизина с т. пл. 180-18ГС.
Найдено, %: С 55,49; Н 5,98; N 6,08.
C2iH27N207C (мол. вес 454,90).
Вычислено, %: С 55,44; Н 5,81; N 6,15.
ИК спектр (NH), 3360 (ОН), 1719 (СООСНз) 1620, 1535 (C N), 1442, lIOOcM-i.
Г. Получение 1 - (р - окси - р-метокарбонилэтил)-1-этил-1,2,3,4,6,12,12в - октагидроиндол (2,3-а)-хинолизииа (формула VH; R - метоксильная группа).
РЬ 7 г перхлората 1-(р-ацетокси-;р-метоксикарбонилэтил) - 1 -этил-2,2,3,4,6,7,12,12в-октагидроиндол (2,3-а)-хинолизина растворяют в 150 мл метанола, содержаш;его 0,165 г соляной кислоты на 1 мл, раствор нагревают с обратным холодильником в течение 2 ч. Затем раствор упаривают в вакууме до 30 мл. Выпавшую соль отфильтровывают и промывают холодным метанолом н эфиром. Полученные 5,5 г соли растворяют в смеси 80 мл ацетона и 80 мл воды, раствор подшелачивают 5%-ным водным раствором карбоната натрия. Выпавший белый кристаллический 1-(р-окси-р-метоксикарбонилэтил) - 1-этил-1,2,3,4,6,7,12,12в-октагидроиндол (2,3-а) - хинолизин отделяют фильтрованием и промывают водой. Получают 8,9 г (80%) целевого продукта с т. пл. 234°С
Найдено, %: С 70,47; Н 7,92; N 8,14.
С9чН28Ы2Оз (мол. вес 356,45).
Вычислено, %: С 70,75; Н 7,91; N 7,86. ИК спектр: (ОН, NH), 1742 (С02СНз), 1200, ИЗО, 745 см-.
Гг. 1 г перхлората 1-(р-ацетокси-р-метоксикарбонилэтил) - 1 -этил-1,2,3,4,6,7,12,12в-октагидроиндол (2,3-а)-хинолизина смешивают с 30 мл метанольного раствора метилата натрия (0,18 г натрия на 100 мл метанола), смесь кипятят 40 мин. Метилат натрия разлагают
ледяной уксусной кислотой и раствор упаривают досуха в вакууме. Остаток суспендируют в 5%-ном водном растворе бикарбоната натрия и суспензию экстрагируют дихлорметаном. Дихлорметановый раствор сушат над
сульфатом магния и отгоняют растворитель.
Получают 0,73 г 1-(р-окси-р-метоксикарбонилэтил) - 1 -этил-1,2,3,4,6,7,12,12в-октагидроиндол
(2,3-а)-хинолизина с т. пл. 234°С.
гз. 1 г Перхлората 1-(р-окси-р-метоксикарбонилэтил) - 1-ЭТИЛ-1,2,3,4,6,7- гексагидро-12Ниндол (2,3-а)-хинолизина растворяют в 50 мл метанола, смесь гидрируют в присутствии 0,6 г 10%-ного палладия на угле. Катализатор отфильтровывают, фильтрат упаривают
досуха. Остаток обрабатывают карбонатом натрия как описано в п. Г. Получают 0,63 г 1-(р-окси-р-метоксикарбонилэтил)-1-этил-1,2,3, 4,6,7,12,12в - октагидроиндол (2,3-а) - хинолизина.
Д. Получение винкамина и 16-эпивинкамина (формула I и VHI; -R - метоксильная группа).
Д. 1,00 г (2,3 ммоль) 1-(р-окси-р-метоксикарбонилэтил)- - 1,2,3,4,6,7,12,12в - октагидроиндол (2,3-а)-хинолизина растворяют в смеси 13 мл, сухого диметилсульфоксида и 4 мл 1 М раствора фосфорной кислоты в диметилсульфоксиде. Когда все твердое вещество переходит в раствор, в него добавляют 1,66 г
свежеперегнанного дициклогексилкарбодиимида, реакционную смесь оставляют на 7 ч при комнатной температуре. Затем смесь выливают в 130 мл ледяной воды, подщелачивают 5%-ным водным раствором карбоната натрия
и экстрагируют дихлорметаном. Дихлорметановый раствор объединяют, сушат над сульфатом магния, растворитель отгоняют в вакууме. Получают 0,25 г остатка, представляющего собой смесь (±)-винкамина и (±)эпивинкамина.
Остаток перекристаллизовывают из смеси дихлорметана, метанола и диэтилового эфира 1:1:1. Получают 0,095 г, (±)-винкамина с т. пл. 224-225°С.
Маточный раствор из этого процесса упаривают и получают 0,098 г смеси эпивинкамина и винкамина. ту смесь перекристаллизовывают из метанола и получают 0,072 г чистого (±)-эпивинкамина с т. пл. 210°С.
ИК спектр: (С02СНз), 1462, 1370, 1330, 1310, 1250, 1258, 1240, 1188, 1088, 1020, 810, 756 см-1.
Д2. 0,1 Г 1-(р-окси-р-метоксикарбонилэтил)1 -этил-1,2,3,4,6,7,12,12в-октагидроиндол (2,3-а) хинолизина растворяют в 10 мл бензола, добавляют 1,5 г реактива - карбонат серебра- целит (50% карбоната серебра), смесь кипятят в течение 48 ч. Смесь фильтруют, бензольный фильтрат упаривают в вакууме. Остаток в количестве 0,095 г представляет собой смесь (±)-винкамина и (±)-эпивинкамина. Смесь подвергают фракционной кристаллизации (растворитель 1 : 1 смесь дихлорметана и метанола) и получают 0,015 г (±)эпивинкамина.
Е. Получение (±)-винкамина (формула I; R - метоксильная группа).
61.1 г смеси (±)-винкамина и (±)-эпивинкамина, полученной в п.д, растворяют в 100 мл ксилола и к раствору добавляют 7,5 г реактива - карбонат серебра - целит (50% карбоната серебра). Реакционную смесь кипятят 3-4 ч. Степень конверсии проверяют методом тонкослойной хроматографии. В конце кипячения смесь фильтруют, осадок промывают горячим ксилолом, ксилольный раствор объединяют и упаривают в вакууме до 20% начального объема. Эту операцию ведут в атмосфере азота. Концентрату дают постоять и на следующий день выпавшие кристаллы отфильтровывают, промывают и сушат. Получают 0,8 г (±)-вицкамина с т. пл. 227-228°С.
62.2,0 г 1-(р-Окси-р-метоксикарбонилэтил)1-этил-1,2,3,4,6,7,12,12в-октагидроиндол(2,3-а) хинолизина (полученного как в п.д) растворяют в 100 мл ксилола, раствор нагревают до кипения и к горячему раствору при перемешивании добавляют 10,0 г реактива карбонат серебра - целит (50% карбоната серебра). Реакционную смесь затем кипятят в течение 5 ч. Степень конверсии определяют с помош.ью тонкослойной хроматографии. Затем ксилольную смесь фильтруют в горячем состоянии, фильтрат охлал{дают смесью лед - соль, через несколько часов отфильтровывают выпавшие кристаллы (+)-винкамина. Выход составляет 1,44 г (75%), т. пл. 225°С.
Ксилольный маточный раствор упаривают до 1/3 начального объема и концентрат охлаждают. Получают еш,е 0,16 г кристаллического веш,ества (общий выход 80%), однако, эта вторая часть может быть смесью (±)-винкамина и (±)-эпивинкамина. Эту последнюю фракцию либо перекристаллизовывают, или используют в первой стадии окисления в качестве исходного вещества.
Ж. Получение (±)-винкамина.
Ж. 0,3 г (±)-винкамина и 0,3 г О,О-дибензоилвинной кислоты растворяют в 6 мл горячего метанола. При необходимости смесь фильтруют в горячем состоянии. Раствору дают охладиться и затравливают кристаллами (±)-винкамин О,0-дибензоилтартрата (т. пл. 205°С). Смеси дают постоять при комнатной температуре в течение 2 дней, затем выпавшие кристаллы фильтруют и промывают холодным метанолом. Выход 0,15 г (50%), т. пл. 205°С.
Полученную таким образом соль обрабатывают 5%-ным водным раствором карбоната натрия. Свободное основание экстрагируют дихлорметаном.. Дихлорметановый раствор сушат и упаривают. Получают 0,08 г ()винкамина с т. пл. 228°С, (а) о +41,32 (с 1, пиридин).
Ж2. Повторяют процесс, описанный в п.Жь, с той разницей, что в качестве агента для разделения применяют d-винную кислоту.. (+)-Винкаминтартрат имеет т. пл. 218°С. Выход и оптическая чистота ( + )-винкамина, полученного этим способом, такая же, как
в п.Ж).
жз. Процесс, описанный в п.Жь повторяют
с той разницей, что в качестве агента для
разделения применяют rf-камфарсульфокислоту. ( + )-Винкамин-й-камфарсульфонат имеет
т. пл. 130°С (вещество кристаллизуется с метанолом). Выход оптически чистого (+)-винкамина по этому способу такой же, как в п.Жь
Пример 2. Этиловый аналог (-|-)-винкамина (формула I; R - этоксильная группа).
Повторяют опыт по примеру 1, но с той разницей, что вместо метилового эфира а-ацетоксиакриловой кислоты применяют этиловый
эфир а-ацетоксиакриловой кислоты.
Этиловый аналог (+)-винкамина (формула I; R - этоксильная группа) имеет т. пл. 245°С (а) ,1 (, пиридин).
Пример 3. Получение перхлората 1-(рокси-|3-метоксикарбонилэтил) - 1-этил-1,2,3,4,6,
7-гексагидро - 12Н - индол (2,3-а)-хинолизина
(формула VI; R - метоксильная группа;
- перхлорат ион).
Повторяют способ из примера 1А, но с той разницей, что вместо а-ацетоксиакриловой кислоты применяют а-ацетоксиакрилонитрил. Маслянистый остаток, полученный после упаривания дихлорметана, очищают через перхлоратную соль. Очищенное вещество кипятят в метаноле, содержащем соляную кислоту, до тех пор, пока нитрильные и амидные полосы исчезают в инфракрасном спектре смеси. Затем раствор обрабатывают как в примере 1 В. Получают перхлорат 1-(р-окси-|3-метокарбонилэтил)-1-этил - 1,2,3,4,6,7-гексагидро-12Пиндол (2,3-а) -хинолизина.
Этот продукт идентичен соединению, полученному в примере 1 В.
Предмет изобретения
Способ получения винкамина или его производных общей формулы I
.N
но ,
60 j{( lie
С2Н5
О
где R - алкоксильная группа, содержащая 65 1-5 атомов углерода, или R-СО- реакци11
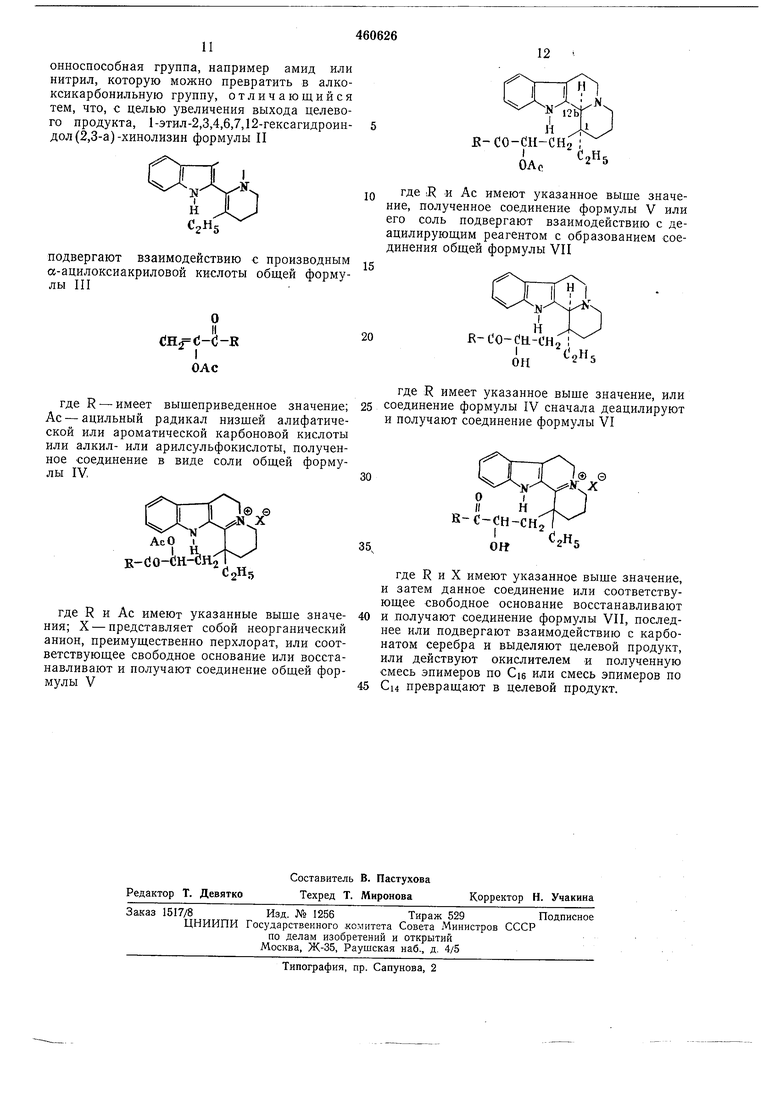
онноспособная группа, например амид или нитрил, которую можно превратить в алкоксикарбонильную группу, отличающийся тем, что, с целью увеличения выхода целевого продукта, 1-этил-2,3,4,6,7,12-гексагидроиндол(2,3-а)-хинолизин формулы II
подвергают взаимодействию а-ацилоксиакриловой кислоты лы III
О
II
( I ОАС
где R - имеет вышеприведенное значение; Ас - ацильный радикал низщей алифатической или ароматической карбоновой кислоты или алкил- или арилсульфокислоты, полученное соедкнение в виде соли общей формулы IV,
к-(3о-с1н- с нф;
CgHs
где R и Ас имеют указанные выше значения; X - представляет собой неорганический анион, преимущественно перхлорат, или соответствующее свободное основание или восстанавливают и получают соединение общей формулы V
460626
12
И
-ьN,
Jl il
СО-СН-СНг I
СгН
ОАо
где R и Ас имеют указанное выще значение, полученное соединение формулы V или его соль подвергают взаимодействию с деацилирующим реагентом с образованием соединения общей формулы VII
к-оо-сц-он
С,11, I он
где R имеет указанное выше значение, или 25 соединение формулы IV сначала деацилируют и получают соединение формулы VI
ОI
/IН
с-сн-сн
2«5 он
где R и X имеют указанное выше значение, и затем данное соединение или соответствующее свободное основание восстанавливают
и получают соединение формулы VII, последнее или подвергают взаимодействию с карбонатом серебра и выделяют целевой продукт, или действуют окислителем и полученную смесь эпимеров по Cig или смесь эпимеров по
Си превращают в целевой продукт.