(54) СПОСОБ ПОЛУЧЕНИЯ ОПТИЧЕСКИ АКТИВНЫХ ПРОИЗбОДНЫХ ПРОСТАГЛАНДИНОВ ИЛИ ИХ ОПТИЧЕСКИХ АНТИПОДОВ, ИЛИ ИХ РАЦЕМАТОВ
В качестве исходного соединения формулы I используют предпочтительно соединение формулы у .0
О-С-СНз
eNH$02 iHj
,.-Ч
с но
где R - диметил-трет-бутилсилилркси,
Для осуществления процесса обычно вначале приготовляют илид в атмосфере азота в таком растворителе, как диметоксиэтан, эфир или тетрагидрофуран, при медленном добавлении такого основания, как гидрид натрия или бутиллитий, к раствору подходящего фосфоната в том же рас1ворителе при температуре реакции 0-25°С, обычно при . Образование илида обычно заканчивается в течение 1ч. К реакционной смеси добавляют альдегид формулы П растворенный в том же растворителе, и реакдионную смесь перемешивают при 0-80°С обычно при комнатной температуре, до окончания реакции, обычно в течение 1 ч. После установления рН-б-7 реакционной смеси выделяют целевой продукт. Обычно продукт очищают хроматографией на силикагеле.
«.Выбор фосфоната, из которого полуЧс1ют илид, зависит от структуры желаемого конечного продукта. Например, когда желательно, чтобы конечный продукт имел боковую цепь у С, , содержащую 5 атомов углерода, т.е. А CgH , применяют диметил 2-оксогептилфосфонат, а если желательна метиленфенокси боковая цепь у (т.е. А CgHg-О-СН2-), то применяют диметил-2-оксо-3-феноксипропилфосфонат.
Следукядие примеры иллюстрируют приготовление промежуточных соединений и конечных простагландиновых продуктов в соответствии со способом данного изобретения. Температуры плав ления и кипения указаны в С без поправок. Данные ИК-анализа указаны в .микронах, а данные ЯМР указаны в ч. на млн.
Пример 1. (парадифенил карбОкси)-5оС-окси-2р|-(2-тиоксаланил) -циклопент-1а(.-ил уксусная кислота, J лактон.
К раствору 2- 3bt- (парадифенилкарбокси) -5оС-окси-2 -формилциклопент-1 ;4.-ил уксусной кислоты, ЯГ-лактона (21 г, 0,06 мЬль) в безводном хлористом метилене (500 мл) и 2-меркаптоэтаноле (4,68 г, 0,06 моль), охлажденному до в атмосфере азота, добавляют эфират трехфтористого бора (4 мл, 0,03 моль) в течение 15 мин. Полученный раствор после нагревания до комнатной температурят перемешиваю 2 ч. Затем реакционную смесь разбавляют 300 мл хлористого метилена и промывают водой (2 раза по 100 мл). Слой хлористого метилена высушивашзт над безводным сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Получают масло, которое затвердевает при обработке гексаном. После фильтрования, промывки гексаном и сушки в вакууме получают целевое соединение (23,9 г, выход 97%, т.пл. 14б°С).
При замене 2-меркаптоэтанола 3-меркапто-1-пропанолом описанной выше процедурой получают соответствующий гомологический защищенный альдегид.
Пример 2, , 5оС.-Диокси-2р1- (2-тиоксаланил) циклопент-1-ил уксусная кислота, j -лактон.
Гетерогенную смесь сырой (парадифенилкарбокси) -5с с-окси-2(Ь- (2-тиоксаланил) циклопент-1-ил уксусной кислоты, J -лактона (23,5 г, 0,057-моль абсолютного метанола (230 мл) ,и тонко измельченного безводного;карбоната калия (3,93 г, 0,028 моль) перемешивают при комнатной температуре в течение 10-15 ч. Осажденное твердое вещество отфильтровывают и промывают метанолом Фильтрат выпаривают примерно до 100 мл И охлаждают на льду. К охлажденному IpacTBOpy добавляют по каплям 0,1 н. соляную кислоту до рН-3 раствора и выпавшее восадок твердое вещество отфильтровывают. Водный слой насыщают хлористым натрием и экстрагируют этилацетатом (3 раза по 50 мл). После .сушки объединенного органического слоя безводным сульфатом натрия и выпаривания получают целевое соединение (12,6 г, выход 96%). ИК (СНСЕз, ) 1770 (карбонил лактон) , 3420 широкая абсорбция (-ОН)..
Пример 3. (тетрагидропиран-2-илокси)-5о1.-окси -2р-( 2-тиоксаланил) -циклопент-1о.-ил уксусная кислота, 3 лактон.
К охлажденному раствору (0-5°С) сырой 2- 3otf 5с6-диокси-2 - (2-тиоксаланил)циклопент-1оС-ил уксусной кислоты,
-лактона (2,5 г, 10,9 ммоль) и свежеперегнаного-дигидропирана (1,47 мл, 16,3 ммоль) в безводном хлористом метилене (24 мл) добавляют моногидрат паратолуолсульфокислоты(250 мг 1,31 ммоль). Полученную смесь перемешивают 1,5 ч при 0°С и разбавляют эфиром (60 мл). Органический слой промывают насыщенным раствором бикарбоната натрия (10 мл), насыщенным раствором хлористого натрия (10 мл) и высушивают над безводным сульфатом натрия. При концентрировании в вакууме получают целевое соединение (3,4 г, выход 100%) в виде масла. ИК (CHCfj ) 1770 см (карбонил лактон).
Пример 4. .-(Тетрагидропиран- 2-илокси- 5с --окси- 2 &-) 2-тио кс алинилциклопент-1 ;Л-ил ацетальдегид, у-гемиацеталь.
В охлажденный до (-75)С раствор в атмосфере азота сырой .-(тетрагидропиран-2-илокси) -5с -окси 21i- { 2-тиоксалинил )-циклопент-1о1-ил уксусной кислоты, J -лактона (3,24 г, 10,3 ммоль) в безводном толуоле (50 (50 мл) в течение 25 мин добавляют 20.%-ный раствор гидрида диизобутилал миния в гексане (14,9 мл, 12,0 ммоль Через 30 мин реакцию прекращают при добавлении по каплям метанола. После нагревания до комнатной температуры выпаривают толуол при пониженном дав лении и остаток разбавляют эфиром (200 мл). Органический раствор промы BcuoT 50%-ным раствором виннокислого калия (3 раза) и насыщенным растворо хлористого натрии и высушивгшзт над безводным сульфатом натрия. После ко центрирования раствора получают целе вое соединение (3,1 г, выход 95%) в виде масла. Этот продукт очищают на 90 г окиси кремния Бэкера (0,0740,177 мм)-, применяя бензол-этил ацета в качестве растворителя для элюирова ния, и получают чистое целевое соеди нение (2,9) г, ИККСПа 3350 см , ОН-группа , Пример 5. 7- 2ji-(2-Тиоксала нил) -ЗоС- (тетрагидропиран-2-илоксп) -5о.-оксициклопент-1с -ил -цис-5-гепте новая кислота. 1К раствору 5-трифенилфосфониопент новой кислоты (23,04 г, 52 ммоль) в безводном диметилсульфоксиде (46 мл) по каплям добавляют примерно 2,0 н. раствор метилсульфинилметила натрия (49,3 мл, 98,6 ммоль) в диметилсульфоксиде. К полученному красному раствору в течение 1 ч добавляют раствор 2 ( 2тиоксапанил) -ЗоС- (тетрагидропиран-2-илокси) -5о -оксициклопент-1оС-ил ацетальдегида, -т -гемиацеталя (6,6 г, 20,8 ммоль) в безводном диметилсульфоксиде (63 мл). После перемешивания в течение 0,5 ч реакционную смесь выпивают на смесь льда с водой (600 мл). Основной водный раствор экстрагируют смесью этилацетата и эф ра (2:1, 2 раза по 300 мл). Холодный водный слой покрывает этилацетатом и подкисляют 10%-ной соляной кислотой до рН-3. Затем водный слой дополнительно экстрагируют этилацетатом (2 раза по 200 мл) и объединенные органи чески.е экстракты промывают водой и раствором хлористого натрия. При суш ке- органического слоя над безводным сульфатом натрия и концентрировании получают масло желтого цвета (20 г). При добавлении 150 мл смеси этиладетата и эфира (2:1) в осадок выпадает твердое вещество, которое отфйльтровывсцот, промывают эфиром и фильтрат выпаривают. Получают сырое целевое соединение (10,2 г, 120%), которое используют непосредственно на следующей стадии. , 3400 см (связанный гидроксил) .и абсорбции между 2400-2800 см , для карбоксильных групп. 986 Пример 6. 7- 2 Ь-Формил-Зо.- (тетрагидропиран-2-илокси) -5огоксициклопент-1с1 -ил -цис-5-гептановая кислота. К раствору (2-тиоксалинил)-3ot- (тетрагидропиран-2-илокси) -5ot-оксициклопент-ЬзС-ил -ци -5-гептеновой кислоты (2,0 г, 0,005 моль) в смеси ацетонитрила и воды, (4:1, 85 мл) последовательно добавляют безводный карбонат кальция (2,87 г, 0,029 моль) и двуххлористую ртуть (5,4 г, 0,020 моль). Смесь перемешивают и нагревают при 50С в атмосфере азота 1/2 ч, а затем фильтруют через Целит и промывают эфиром (250 мл). Фильтрат обрабатывают при перемешивании 1н. соляной кислотой (3 мл), эфирный слой отделяют и промывают рассолом (3 раза по 15 мл). После сушки над безводным .сульфатом натрия и концентрирования при пониженном давлении получают 1,7 г (100%) целевого соединения в виде масла. ИК (четкий) 1725 см (карбонил альдегид) . Пример 7. 9о1.-окси-11оСг-(тетрагидропиран-2-илокси)-15-оксо-цис-5-транс-13-простадиеновая кислота. К раствору в атмосфере азота диметил (2-оксогептил)-фосфоната (2 г, 0,009 моль) в диметоксиэтане (30 мл), охлажденному до , по каплям до:бавляют 2,2 М бутиллития (3,96 мл, 0,0087 моль). После перемешивания в течение 1 ч быстро добавляют 7- 2р -формил-ЗоС:- (тетрагидропиран-2-илокси) -5оС.-оксициклопент-1 -ил -цис-5-гептеновую кислоту (1,02 г, 0,003 моль), растворенную в диметоксиэтане (6 мл), и смесь перемешивают при комнатной температуре 1/2 ч, добавляя ледяную уксусную кислоту до рН-7. Нейтрализованный раствор концентрируют при ротационном выпаривании и полученный продукт суспендируют в бензоле и фильтруют. При концентрировании фильтрата получают сырое целевое соединение, которое очищают хроматографически силикагелем, применяя смесь бензола и этилацетата в качестве растворителя для элюирования. Получают чистое целевое соединечие (10 мг). ИК (четкий) 3450 см (ОН) 1720 (карбоксил) 1670 (нез.амещенный карбонил) 970 (слабый/ транс-двойная связь). Пример 8. N-метансульфонил (1,3-оксатиалан-2-ил)Зс1С- (тетрагидропиран-2-илокси) -5с -оксициклопент-loi-ил -цис-5-гептенамид, К раствору 27,0 г (52,0 ммоль) бромистого (4-метансульфониламинокарбонилбутил)трифенилфосфония в 46 мл диметилсульфоксида по каплям добавляют 49,3 мл (98,6 ммоль) 2,0 М раствора метилсульфинилметила натрия в диметилсульфоксиде. К полученному красному раствору в течение 15 мин добавляют 6,6 г (20,8 моль) целевого продукта примера 4 в 63 мл, диметилсульфоксида. После перемешивания в течение 2 ч реакционную смесь выливают в 60 мл смеси льда и воды. Водный охлажденный слой покрывают этилацета-ом и подкисляют 10%-ной соляной кислотой до рН-3 Подкисленный водный слой экстрагируют этилацетатом (2 раза по 200 мл) и объединенные органические экстракты промывают водой и рассолом. ГЬсле сушки их над безводным сульфатом натрия и концентрирования получают сырой продукт, который обрабатывают при растирании эфиром. После выпаривания эфира получают N-метансульфонил 7- (1, З-оксатиалан-2-ил) -ЗоС- (тетрагидропиран-2-илокси)-5Ы-оксициклопент-1оС-ил -циc-5-гeптeнa 1ИД.
ИК (СНСЕэ ) 1720 (сульфонамид) Продукт этого примера можно анилиО
ровать,применяя( о -ufu СГ
С1
где к - С -Сд-алкил, нафтил, фенил, парадифенил, или д-фенил ал кил. Этот продукт можно превращать в Е2-простагландины.
При мер 9, N-метансульфонил 7- 2/)- (1 , 3--оксатиолан-2-ил) -3«- (тетра1идропира,н-2-илокси) -SX-ацетоксициклопент-1,х1-ил -цис-5- гептенамид.
Смесь 1,69 г (3,54 ммоль) сырого целевого соединения примера 8 5,9 мл пиридина и 0,368 мл (3,89 ммоль) уксусного ангидрида перемешивают в атмосфере азота в течение 10-15 ч при 50 С. После охлгикдения до комнатной температуры смесь разбавляют эфиром (75 мл). Эфирный раствор промывают во яой (1 раз) и насыщенным раствором сульфата меди (3 раза), высушивают над безводным сульфатом магния и концентрируют до получения целевого N-метансульфонил )-(1, 3-оксатиалан-2-ил) ЗоС-(тетрагидропиран-2-илокси) -5сзСгацетоксициклопент-1 Я.-ил -цис-5-гептенамида.
ИК (CHCEj) 1740 см (ацетаткарбонил)
1725 см (сульфонамид) Пример 10. N-Метансульфонил 7- 2|5-формил-Зо(.- (тетрагидропиран-2-ил-окси) .-ацетоксициклопент-1с.-ил -цис-5-гептенамид.
К раствору 2,9 г (5 ммоль) геметио ацеталя, полученного в примере 9, в 85 мл смеси ацетонитрила и воды (4:1 последовательно добавляют 2,87 г (0,029 ммоль) безводного карбоната кальция и 5,4 г (0,020 ммоль) двухлористой ртути. Смесь перемешивают при в атмосфере азота 1/2 ч, а затем фильтруют через Целит и промывают 250 мл эфира. Фильтрат перемешивают и обрабатывают 3 мл 1 н. соляной кислоты. Эфирный слой отделяют и промывают рассолом (3 раза по 5 мин). После сушки над безводным сульфатом натрия и концентрирования при пониженном давлении получают целевой N-метансульфонил 7- 2|6-Формил-3.чС-(тeтpaгидpoпиpaн-2-илoкcи) -5о1.-ацетоксициклопент- с(-ил цис-5-гептенамид .
ИК (СНсе,, ) 1740 см (ацетат карбонил).
1725-1720 см (альдегид карбонил и сульфонамид) ,
Пример 11. N-Метансульфонил -9с(-ацетокси-11о(.- (тетрагидропиран-2-илокси)-15 оксо-5-цис-1З-транс-16-фенокси-СО -тетранопростадиенамид.
К суспензии 220 мг (5,22 ммоль) 57,0%-ной дисперсии гидрида натрия в минеральном масле в 20 мл тетрагидрофурана добавляют 1,34 (5,22 ммоль) диметил-2-оксо-З-феноксипропилфосфоната. Смесь перемешивают в течение 1 ч в атмосфере азота, а затем к ней добавляют раствор 1,23 г (2,37 ммоль) сырого целевого продукта примера 10 в 4 мл тетрагидрофурана. Эту смесь перемешивают при комнатной температуре в атмосфере азота 2 ч. Затем реакцию прекращают при добавлении ледяной уксусной кислоты до рН-6 и реакционную смесь концентрируют ротационным выпариванием.
Полученную смесь растворяют в этилацетате, -органический слой промывают 0,1 н. соляной кислотой, водой и рассолом, высушивают над безводным сульфатом магния и концентрируют. При очистке сырого продукта на хроматографической колонке получают целевой Н-метансульфонил 9(Л-ацетокси-11оС- (тетрагидропиран-2-илокси)-15-оксо-5-цис-13-транс-16-фенокси- (О-тетранорпростадиенамид.
ИК (CHCEj) 1840 см- (ацетат карбонил) 1720 см (сульфонамид)
1670 см
(енон карбовил) .
Формула изобретения
1. Способ получения оптически ак тивных производных простагландинов формулы Г
OR
или их оптических антиподов или их
рацематов
С где R - водород или группа - С.
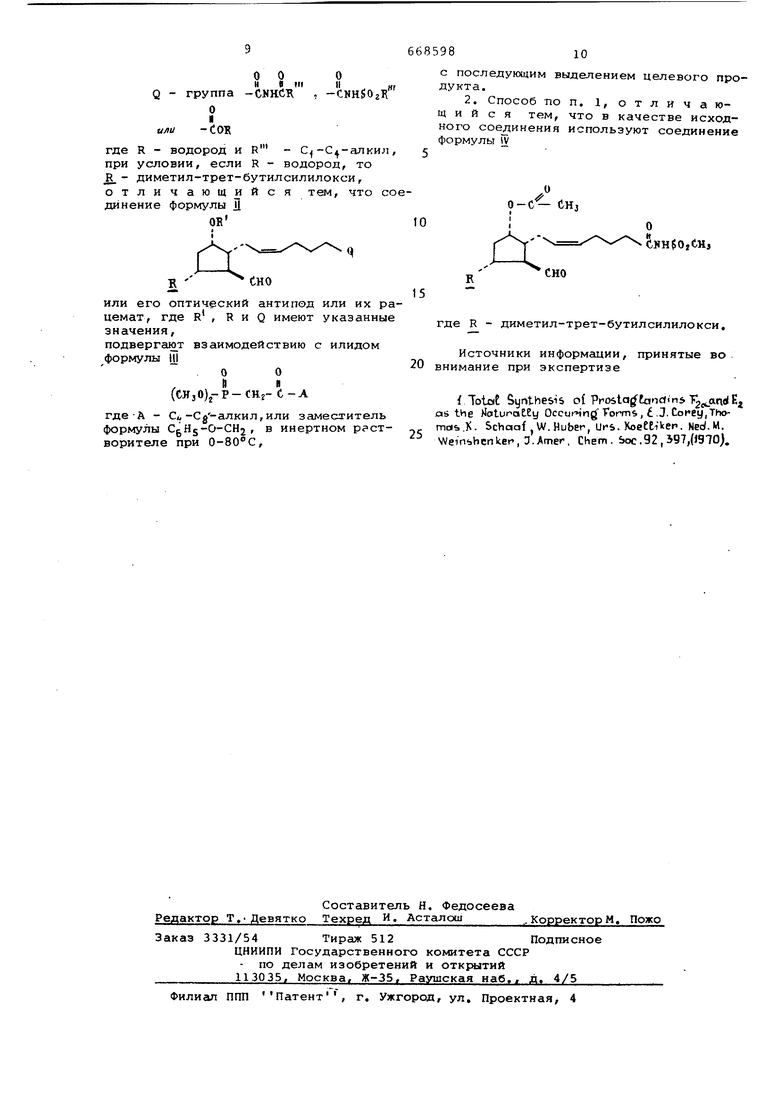
R - С| -С4-алкил, R - водород, тетрагилропиран-2-йлокси или диметил-трет-бутилсилилокси, О о н а (м -СИН$Ог/ Q - группа -СЛНСЖ где R - водород и - С -С -алкил при условии, если R - водород, то R - диметил-трет-бутилсилилокси, отличающийся тем, что с динение формулы Д Ой или его оптический антипод или их р цемат, где R , R и Q имеют указанны значения, подвергают взаимодействию с илидом формулы Ш О О И И (CHjOjj-P-CHj-C-A где-А - Си-Cg-алкил,или заместитель формулы CgHg-O-CHj, в инертном р створителе при 0-80°С, с последующим выделением целевого продукта. 2. Способ по п. 1, отличающийся тем, что в качестве исходного соединения используют соединение формулы W CHj - - cuH$o,tH, .. где R - диметил-трет-бутилсилилокси. Источники информации, принятые во внимание при экспертизе f Totat Synthesis of Prosta anc((n5 T25jin Ej as the fetunatEy Occut-Hng Forms, tJ. Corey,Thomoib K. Schoaf W.Huber, Urs. KoeEEiVen. Ned.M. Wem hcnker, D.Amer, Chem. boc.92,b97,(JgiO).



| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения 11-дезокси-16арилокси- -тетранорпростагландинов или их солей | 1976 |
|
SU679134A3 |
| Способ получения аналогов природных простагландинов | 1973 |
|
SU665799A3 |
| Способ получения промежуточных соединений для синтеза простагландинов и их -эпимеров | 1976 |
|
SU640660A3 |
| Способ получения оптически активныхпРОСТАглАНдиНОВ или иХ ОпТичЕСКиХАНТипОдОВ, или иХ РАцЕМАТОВ | 1978 |
|
SU843736A3 |
| Способ получения промежуточных соединений для получения простагландинов | 1975 |
|
SU645563A3 |
| Способ получения производных простагландина | 1977 |
|
SU900806A3 |
| Способ получения аналогов природных простагландинов | 1974 |
|
SU522789A3 |
| Способ получения простагландинов или их эпимеров с15 или с9 и с15 эпимеров | 1975 |
|
SU893130A3 |
| Способ получения аналогов природных простагландинов | 1974 |
|
SU515438A3 |
| Способ получения 15-замещенных простановых производных или их солей | 1973 |
|
SU644384A3 |
