Для защиты о -аминогрупп применяют карбобензокси или трвт,-бути-локсикарбонил или такие родственные им группы, как/ например, параметокси, паранитрб- или параметоксифенилаэокарбобензокси.
Для защиты (-гуанидиновой функции аргинильной группы используют протонизацию группы НО2Или паратолуолсульфонил; -аминогруппы в лизине защищаиот карбобензокси-, трет 1-бутилоксикарбонил- или пара-толуол сульфонильной группой; оС карбоксигруппу защищают метиловый этиловый или -бензиловый эфир,
В описании использованы следующие сокращения:
Аминокислоты {в левовращающей форма, кроме особо отмеченных случаев) г Apr аргинин;Азо - 2-азе,тидинкарбоновая кислота; Ала - алаНИН Гли - глицин Иле - изолейцин; Лей -- лейцин; Лиз - лизин; Фе - феНилаланин; Пип - пипеколиновая кислота; Про пролин; Вал - валик; АцОН - уксусная кислота; Бз - бенЗоил; КБо - карбобензокси; ДЦКИ дицикло;е ексилкарбОдиимид; ДМФ димвтилформамид; ТЭА - триэтиламин; ЭА этилацетат; ГФХ - хроматография с фильтрованием через гель; ОБТ - Й-оксибензотриазол; Осу 4/-ОКСИСУ1СЦИНИМИД.; МеОН - метанол; ПЫФ - паранитрофенокси; ПНА - паранитроанилид; ТБК - трет.-бутилоксикарбонил; ТФУК - трифторуксусная кислота ТСХ - хроматография в тонком слое
Типы реакций,, применяемых для синтеза,
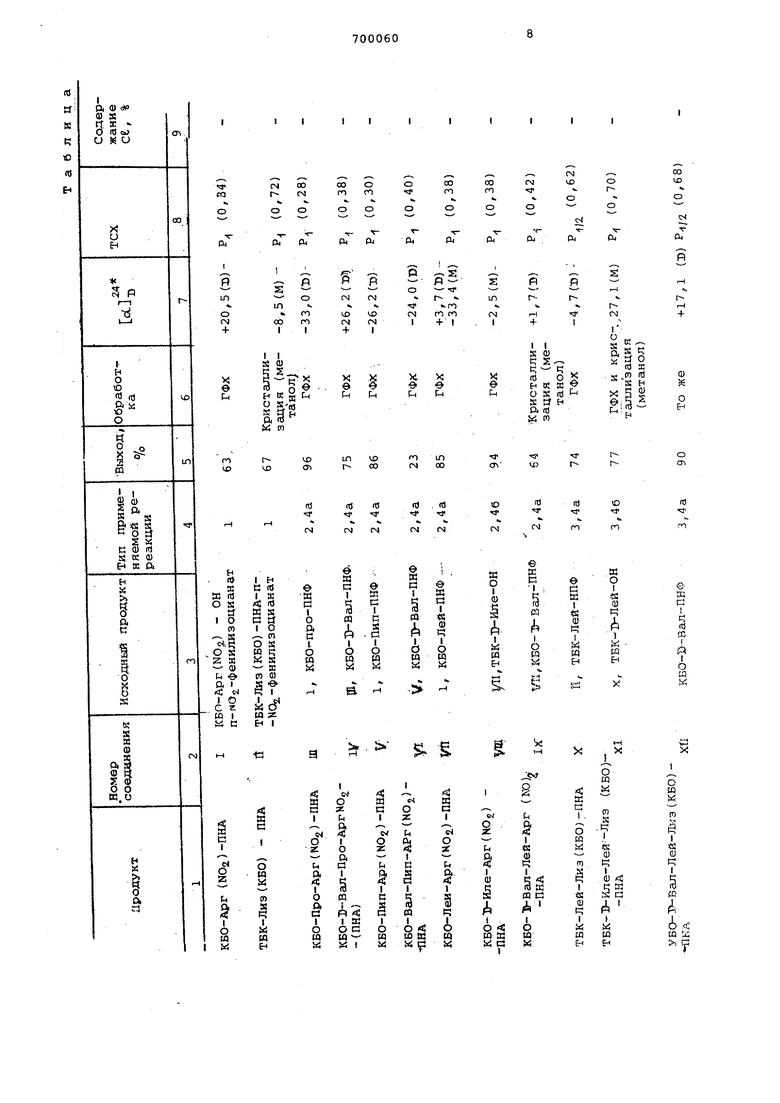
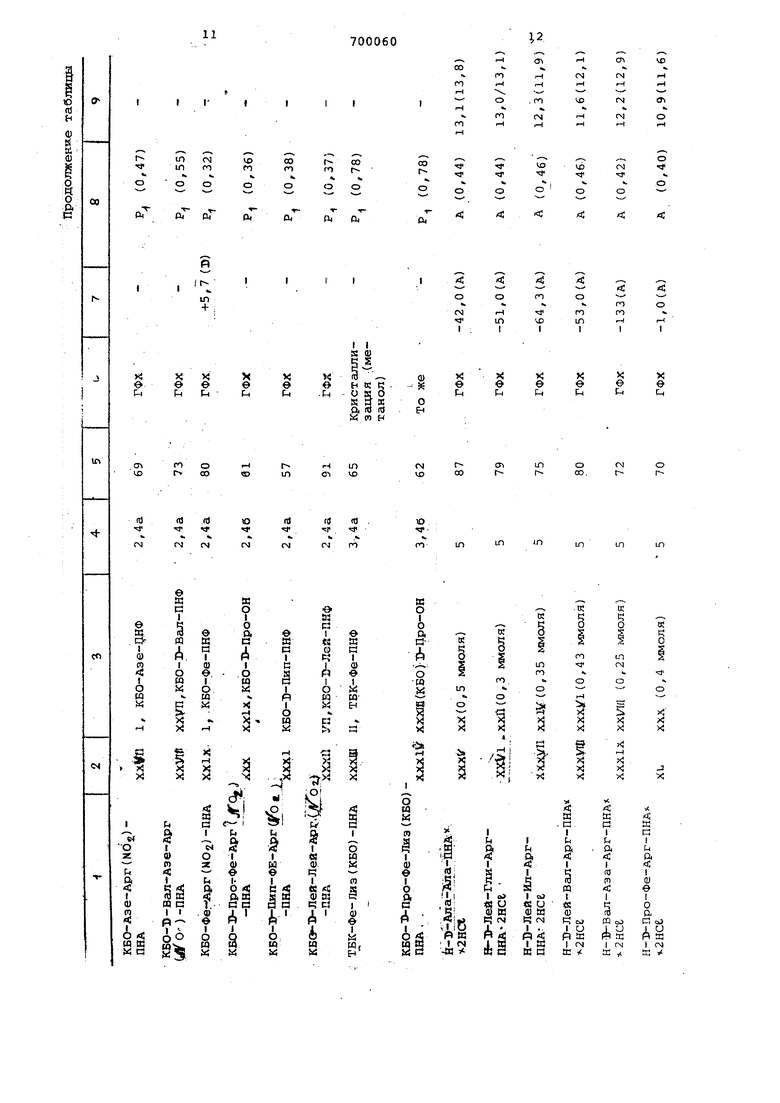
Для синтеза пептидов (см, таблицу, № XXV - XLffl) разные стадии проводят одинаковым способом, поэтому дано описание реакций разных типов и в таблице указаны промежуточные и тсонечные продукты, методы, примененные ,тшя реакций разных типов, и некоторые физические данные. Реакция типа I, Сочетание хромофорной груапк (R),
20 ммолей Nf N-защищенного аргинина или W-f м защищенного орнитина или: лизина или соответствующим образом за1у1ещенкого производного пептида измельчают, тщательно высушивают и растворяют в 50 мл свежеперегнанной N,N,N,N,N,N -reCclмвтиJIфocфopнoй кислоты при комнатной гёглиоратуре. Затем при перемешивании в отсутствии влаги добавляю 20 ммолей триртиламина и 30 ммолей амина, содержащего хромофорную группу в виде его изоцианатного производного. Смесь вьтдерживают сутки, реакционный раствор вливают в 0,5 л 2%-ного раствора бикарбоната натрия при перемешивании. Образовавшийся осадок отфильтровывают и тщательно
промывают раствором бикарбоната, водой, 0,5 н„ соляной кислотой и снова водой. Из осеуцка продукт извлекают метанолом, Метанольный экстракт упаривают, остаток пере кристаллизовывают или чистят с помой;ью хроматографии в тонком слое.
Реакция типа 2. Отщепление карбобензокси защитной группы (КБО)
Q 10 ммолей тщательно высушенного производного карбобензокси (КВО) суспендируют в 25 мл уксусной кислоты и при комнатной температуре в отсутствии влаги добавляют 15 мл 5,6 н.раствора НВГ в уксусной кис- лоте. Через 45-60 мин реакционный
раствор по каплям и при энергичном перемешивании вливают в 800 мл безводного эфира. Выпавший осадок отделяют и промывают 2-3 раза эфиром
0 порциями по 100 м/ Полученный бромгидрат N -деблокированного соединения сушат над таблетками гидроокиси натрия в вакууме при 40°С в течение ч,
5 Реакция типа 3, Отщепление трет,-бутилоксикарбонильной защитной группы (ТБК).
10 ммолей тщательно высущенного ТБК - производного растворяют в
0 200 мл 25%-ного раствора трифторуксусной кислоты в , в отсутст зии влаги при комнатной температу-. ре. Спустя 20 мин раствор по каплям вливают в 500 мл безводно1О эфира. Выпавший осадок отфильтровывает и тщательно промывают эфиром. Полученный трифторацетат Н --деблокированного соединения сушат над таблетками гидроокиси натрия з вакууме
0 при 30°С 2-3 ч.
Реакци типа 4 .Деблокирование с -аминогруппы.
При ацилировании производного,полученного при реакции типа 2 или 3,
5 должна,, присутствовать свободная oi,-аминогруппа. Деблокирование этой группы осуществляют разными путяглИ; например добавляют один эквивалент- безводного третичного амина (триэ0 или К-зтилморфолина) к раствору производного НВг- или трифторуксусной кислоты в диметилформамиде, охлажденному до . При применении триэтиламина и НВгс производных,выпавший в осадок триэтиламин , гидр обр .E( удаляют фильтрованием.
Производные с КВг или трифторуксусной кислотой можно также растворить в растворе бикарбоната натрия, из которого выделяемое производное экстрагируют, например этилацетатоМр а затем органическую фазу сушат и упаривают. Реакция сочетания: , А. С производным Н-эащищенного эфира. К раствору 10 ммолей производного пептида или аминокислоты, вы деленного в 20-50 мл свежеперегна ного диметилформамида, добавляют 11 ммолей ,№-защииенного паранитрофенил- или N-оксисукцинимидозфи производного аминокислоты при --lO Смесь выдерживают 1 ч при , затем добавляют к ней в качестве буфера 5 ммолей третичного амина. Охлаждение убирают, температура реакционной массы самопроизвольно повышается до комнатной. За ходом реакции наблюдают с помощью TCDC. В случае необходимости реакционную массу охлаждают вновь и добав ляют 5 ммолей основания. По окончании реакции раствор упаривают пр пониженном давлении, полученный маслообразный остаток смешивают с двумя порциями воды и чистят перекристаллизацией или гельфильтрацией. При хроматографии фильтрованием через гель, примененной для очистки продукта, объем элюента полностью или частично совпадает с объемом производного активного эфира сочетаемой аминокислоты. По окончании реакции (перед выпариваниём) неизрасходованное производное активного эфира заменяют в течение 30 мин при комнатной темпе ратуре избытком (3-5 ммолей) первичного , например бутиламина, Б. С N -защищенной аминокислотой или пептидом. Образование акти ного эфира по ходу реакции. К раствору 10 ммолей указанного производного пептида или аминокисл в 20-50 мл свежеперегнанного диметилформами а, 11 ммоле и М -защищенной аминокислоты или соответстЕую-иим образом защищенного произйодного пептида со свободной С-кон цевой карбоксильной группой добавляют 11 ммолей N-оксибензотриазола или N -оксисукцинимида при В течение ,1-3 ч температуру реакционного раствора (-10°С) повышают до комнатной. За,ходом реакции следят с помощью хроматографии в jOHKOM слое. По окончании реакци раствор вливают при перемешивании в 100-300 мл 5%-ного водного раствора бикарбоната натрия. Полученный осадок отфильтровывают и промывают водой. Затем чистят методом гель-фильтрации или перекристаллизацией. Реакция типа 5. Отщепление всех защитных групп, очистка и ионный обмен. 0,2 - 1,0 ммоль защищенного производного пептида с хромофорной группой деблокируют обработкой 5-20 мл безводного HF- в присутствии 0,21,0 мл анизола в аппарате Сакакибара, в течение 60 мин при 0°С. После окончания реакции и отгонки всего Нр,- сырой продукт растворяют в 33%ном водном растворе уксусной кислоты и раствор чистят с помощью гель-фильтрации. Продукт сушат лиофилизацией, затем подвергают ионному обмену в колонке, содержащей слабо основную ионообменную смолу. Сефадекс (R)Q АЕ - 25 в виде хлорида выдерживают до набухания в смеси метанола и воды (95:5), Для растворения и элюирования используют эту же смесь. Чистый продукт сушат при температуре ниже и пониженном давлении. Полученный продукт растворяют в метаноле и раствор вносят в колонку подходящего размера (объем 0,5 - 7,5 л, длина 100 см), заполненную сефадексом (К)Н - 20, набухшем в метаноле. Для элюирования используют этот же растворитель. Элюат разделяют на фракции подходящих объемов и непрерывно определяют их УФ-абсорбцию (254 нм.). Фракции, содержащие продукт, проверяют на чистоту методомТСХ, чистые фракции объединяют и упаривают. Производные пептида, полученные после удаления защитных групп с помощью HF (аналогично реакции типа 5), в виде 30%-ного раствора в водной уксуснойкислоте вносят в колонку (объем 0,5 - 2,0 л, длина 30см), заполненную сефадекром (R)G-15, набухшим предварительно в водной уксусной кислоте, продукт элюируют тем же растворителем. Фракции, содержащие продукт (контроль УФ абсорбция при 254 нм), объединяют, концентрируют в вакууме на роторном испарителе при 25°С, затем сушат при температуре ниже . Хроматография в тонком слое. Стеклянные пластинки покрывают тонким слоем кизельгеля F (Мерк).. Для проявления используют следующие системы растворителей (объемное соотношение): А - бутанол-уксусная кислота - вода (3:2:1) ; Р - хлороформ - метанол (9:1); P-j/a- хлороформ-метанол (19:1). По окончании хроматографии пластинку исследуют в УФ-свете (254 нм) и обрызгивают реактивом - хлорортотолуидином. ff--i J Tlxf Формула изобретения Способ получения цептидов об1Ц€1й формулы l ,-A,2. NHR, где А4 аланил-, валил-,.лейцил-, пропил-, пипеколиновая -кислота А аланил валил-, глицил-, лейцил-, изолейцил-, фенилаланил-, азетилинкарбоновая кислота; А - аргинил-, лизил; R - нитрофенил; или их солей, . f личающийся тем, что соединение общей формулы , где R имеет указанные значения, псдверггиот взаимодействию с соответствующей аминокислотой и далее требуемую пептидную структуру постепенно наращивают путем сочетания остальных аминокислот, причем R используют в качестве защитной группы для С-концевого карбоксила ,п первой аминокислоты. Источники информации, принятые во внимание при экспертизе . Шредер Э., Любке К. Пептиды, ч« 1 - Москва; Мир, 1967, с. 116,





| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения пептидов или их солей | 1975 |
|
SU957762A3 |
| Способ получения трипептидов | 1980 |
|
SU1277904A3 |
| Способ определения активности протеаз серина | 1977 |
|
SU736889A3 |
| Способ получения трипептидов или их солей | 1973 |
|
SU671721A3 |
| Способ получения производных трипептидов | 1980 |
|
SU1034603A3 |
| Способ получения производных тетрапептидов или их солей | 1978 |
|
SU908246A3 |
| Способ получения высокоочищенного тетрадекапептида | 2020 |
|
RU2759377C1 |
| ЗАЩИЩЕННЫЕ ПРОИЗВОДНЫЕ ВАЗОПРЕССИНА | 1997 |
|
RU2123498C1 |
| Способ получения пептидов | 1970 |
|
SU439967A1 |
| ЗАЩИЩЕННЫЕ ПРОИЗВОДНЫЕ ОКСИТОЦИНА | 1997 |
|
RU2125062C1 |
