I
Изобретение относится к улучшенному способу пбследовательного замещения атомов хлора хлористого цианура аминами с образованием 2-алкиламино-4,6-дихлор- или 2,4-бис-Диалкиламино-6-хлор-с«жл -триазинов, которые находят применение в качестве гербицидов и исходных продуктов для их синтеза.
Известен способ получения 2,4-ди-(алкиламин)-6-хлор-сыл лг-триазинов путем последовательного замещения хлора в хлористом циануре двумя одинаковыми или различными аминами в органическом растворителе, таком как толуол, бензол, четыреххлористый углерод, в присутствии акцептора кислоты, такого как щелочи, добавляемого периодически 1.
Недостатками этого известного способа является недостаточно высокий выход целевого продукта (не более 95%) и загрязненность его больщими количествами побочных продуктов.
Известен способ последовательного замещения хлора в хлористом циануре двумя одинаковыми или различными аминами в ацетоне или смеси ацетона с водой при О-20°С при регулировании рН путем периодичеЬкого добавления щелочи 2.
Недостатками такого способа является недостаточно высокий выход целевого продукта и их загрязненность побочными продуктами. Так, выход 2-изопропиламино-4-этиламино-6-хлор-сижлг-триазина составляет 97,5%; в случае синтеза цианалкиламинопроизводных выход не выще 93%.
Известентакже наиболее близкий по достигаемому эффекту к предлагаемому способ последовательного замещения атомов хлора хлористого цианура амином или двумя одинаковыми или различными аминами в кетонах, ограниченно смещивающи.хся с водой, например, метилэтилкетоне, при температуре О-20°С и регулировании рН путем периодического добавления щелочи 3.
Выход 2,4-диалкиламино-6-хлор-с«лгл -триазинов при различных аминах достигает 97,5% при частоте 99,6%.
Недостатки этого способа заключаются в недостаточно высоких выходах и загрязненности целевых продуктов побочными про20дуктами.
Цель изобретения - повыщение выхода целевого продукта и повышение его чистоты.
Поставленная цель достигается способо.м последовательного замещения атомов хлора
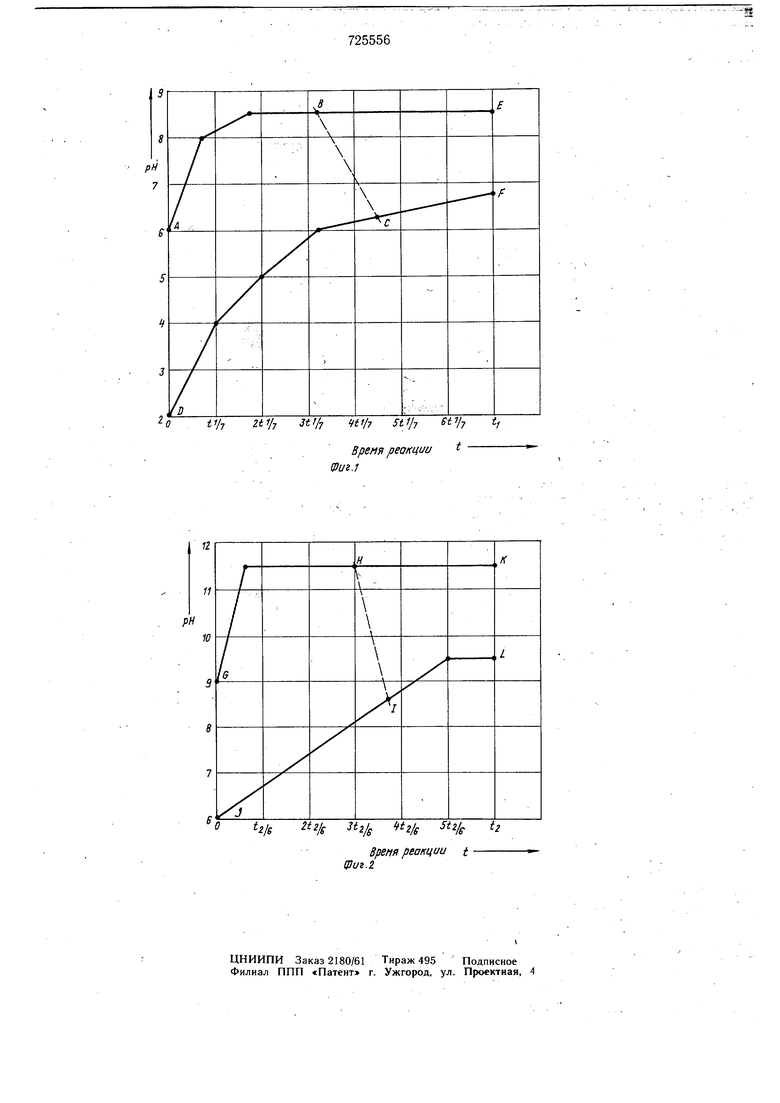
хлористого цианура амином или двумя одинаковыми или различными аминами в смеси 65-85 вес. % ксилола, этилбензола, бензола и/или алифатического, и/или циклоалифатического углеводорода с 5-ТО атомами углерода и 35-15 вес. % кетона с 3-8 атомами углерода и рН реакционной смеси на первой стадйй айинйрования в зависимости Ш врёмени реакции поддерживают непрерывно путем добавления щелочи таким образом, чтобы оно соответствовало точке внутри области, ограниченной линиями ABCD (фиг. 1) и рН реакционной смеси проходило область, начинающуюся с времени реакции t О до значений в области, ограниченной линиями BCEF, И по достижении рН 7,0-7,2 температура реакционной смеси возрастает до 10-60°С, на второй стадии рН реакционной смеси путем добавлений щелочМ устанавливают таким образом, чтобы оно соответствовало точке внутри области, которая ограничена линиями GHIJ (фиг. 2), при температуре 40-70°С и рН реакционной смеси проходят область, начинающуюся с времени реакции второй стадий t О до значений в облати, ограниченной линиями HIKL при уело-, ВИИ, что i 4-10 ч и tj 2-8 ч, и линия ВС. соответствует уравнению рН -(12,64i) t + 14,35 и линия HI соответствует уравнению рН -(24,857 t г) t + + 23,9285.
Предлагаемь1й способ предпочтительно осуществляют следующим образом. Процесс на первой стадии начинают при 10-18°С и на 1 моль хлористого цианура добавляют 1,00-1,02 моль первого амина и при 25:-40°С в конце первой стадии и по достижению рН 7,2 температура возрастает до 25-40°С и на 1 моль хлористого цианурата добавляют .0,98-1,02 экв. щелочи, на второй стадии добавляют 1,,02 моль второго амина на 1 моль используемого хлорйСтбТо цианура, при этом темЪературу повыщают до 45-55°С и при этом периодическими добавками щелочи регулируют указанное выше значение рН реакционной смеси, причем Ч, 7 4,t а 6 ч.
Способ предпочтительно осуществляют таким образом, что после добавления первого реакционную смесь выдерживают в следующих условиях при температуре 10- 18°С путем ддбавления щелочи и при выдержке воды в известных условиях.
рНВремя реакции, мин
1а) 3,5-5,03-43
16) 4,5-6,250-56
1 в) 5,5-7,017-189
1г) 7,0-8,0.30-493
и температуре 10-50°С, причем стадии 1а и 16, 16 и 1в могут бь1ть oбъe инeны в одну, а после добавки второго амина реакционную смесь при 40-70°С путем добавления щелочи выдерживают в условиях:
рНВремя реакции, мин 2а) 6,5-8,02-60 26) 7,25-9,0О-Я2
2в) 8,0-10,00-172
2г) 10,0-11,2515-408
причем стадии 2а и 26, 26 и 2в могут быть объединень в одну.
По предлагаемому способу предпочтительно смесь выдерживают в условиях:
рНВремя реакции, мин
1 а) 4,25-4,75
9-21 16) 5,0-5,5
3-18 51-93 1в)5,75-6,25
при температуре от15до35°С 135-330 1г) 7,25-7,9 10-30 2а) 6,75-7,25 0-36 26) 7,5-8,0 2в) 8,25-8,75 9-66
при температуре
2г) 10,25-10,75 45-55°С135-306
При непрерывном осуществлении способа которое выгодно из-за достигаемых при этом выходов по объему и во времени, целесообразно использовать растворы или суспензии хЛо рйстого ци а ну р а в кон це нтр а ци и, которая определяется содержанием кетона в используемой органической среде и устанавливается следующим образом.
Количество хлористого цианура равно 0,2-кратному количеству кетона в смеси раст-ворителей плюс 1,5, но не больше К, причем
К равно 15, предпочтительно 5.
В качестве смеси растворителей используют из группы алифатических углеводородов: пентан,гексан, гептан, октан, нонан, декан и/или их изомеры, а также цнклогексан и/или, ИЗгруппы ароматических углеводородов: бензол, толуол, этилбензол или ксилолы и одного или нескольких кетонов таких, как ацетон, метилэтилкетон, диэтилкетон, мётил-н-пропилкетон, метилизопропилкетон, метил-н-бутилкетон, метилизобутилкетон, этил-н-амилкетон, этилизоамилкетон, или циклогексанон.
При синтезе цианоалкиламинохлортриазинов особенно благоприятно использовать в качестве растворителя смеси из 70вес. % толуола и 30 вес. % кетона, который соответствует применяемому цианоалкиламину, т.- е. давал бы его при взаимодействии с синильной кислотой и аммиаком.
Желательно, если перед или сразу после начала добавки акцептора кислоты при введении первого амина, например,цианоалкиламина, к реакционной смеси добавляют 0,5- 25 вес. % воды по отношению к используемому количеству растворителя, т. е. устанавливают формальный состав системы углеводород (кетон): вода-99,5-80 вес. /о смеси углеводород - кетон и 0,5-:20 вес. % воды. Особенно благоприятно до начала добавления акцептора кислоты в первой стадии взаимодействия добавл;|ть к реакционной смеси столько воды, чтобы образовалась формальная смесь растворителей углеводород (кетон): вода состава 99,5-98,0 вес. /о смеси углеводород - кетон и 0,5-2 вес. % воды, т. е. с учетом водной щелочи, до формального состава сМеси углеводород(кетон)вода: 95-80 вес. %; предпочтительно 88-
84 вес. °/о, смеси углеводород(кетон) и 5- 20 вес. °/о, предпочтительно 12-16 вес., %,-, воды; добавляется вода только тогда,когда в реакционной смеси достигнуто значение рН по крайней мере, 4,5, предпочтительно по крайней мере 5,0, не:зависимо от указанной добавки воды до или в начале дозировки акцептора кислоты для способа по изобретению в качестве связующего кислоту средства можно использовать водные растворы неорганических оснований, т. е. окиси, гидроокиси, карбонаты и гидрокарбонаты щелочных ю и щелочноземельных металлов; Предпочтительно применять гидроокиси, в особенности гидроокиси щелочных металлов. Их используют предпочтительно в форме водных растворов, которые содержат акцептор в концентрйциях 10-50 вес. %, в особенности 20-40 вес. %, предпочтительно 20-30 вес.%.
При осуществлении способа по изобретению хлористый цианур растворяют в смеси растворителей или суспендируют в ней и затем растворы или суспензии последова- jo тельно вводят во взаимодействие с одинаковыми или различными аминами сначала для получения 2-алкиламино-4,б-дихлор-сыуиж-триазинов и затем до 2,4-ди-(алкила-мино)-6-хлор-сижж-триазинов. Так как взаимодействие происходит в две отдельные ста- 21
дии, предлагаемый способ также выгоден для получения 2-алкиламино-4,6-дихлорсимм-триазиноВ.
В качестве аминов для предлагаемого способа пригодны 1-цианоалкиламины-{Г), такие Kai а-аминоизобутиронитрил (1-циано-1-метилэтиламина-(1)), 1-циано-Т-метилпропиламин,1-циано-1,2-димеТилпропиламин, ц1 ано-1-аминоциклогексан, а также простые алкиламины такие, как метиламин, диметиламин, этиламин, н- или изопропи ламин, цик- 35 лопропиламин, этиленимин или диэтиламин. Если хлористый цианур должен превращаться в 2-алкиламин6-4,6-дихлор-с«.л ж-триазины или в 2,4-ди-(алкиламино)-6-хлор-с«л«л -триазины с различными алкиламиновыми заместителями в 4- или 6-положении, то из названных аминов предпочтительны Г-цианалкйламины. Особенно предпочтителен во всех случаях а-аминоизобутиронитрил для введения первого аминозаместителя в сие-, тему триазина. В качестве аминов для вто- j рой стадии синтеза используют простые первичные и вторичные алкиламины, такие, как метиламин, дим.етиламин, -этиламин, н- или изо- или циклопропиламин, этиленамин или диэтиламин. Предпочтительны этиламин и, циклопропиламин, особенно предпочтите- 50 лен этиламин.
По способу изобретения аминь добавляют в молярных количествах, по отнощению к взятому хлористому циануру; незначительный избыток амина, максимально 5%, пред- . почтительно максимально 3%, допускается для первой стадии взаимодействия, особенно, если используется цианалкиламцн,. Такой избыток также допускается во второй стадии превращения независимо от рода используемого . Менее, чем молярные количества амина на 1 моль используемого хлористого цианура, всегда приводят к уменьщению выхода и чистоты продукта.
При этом амины могут применяться как в чистом виде, так и в виде растворов, в инертных растворителях, предпочтительно таких, которые должны присутстйовать в реакции как компоненты системы углеводород-кетон, или в случае, когда речь идет не об цианоалкиламине - также в воде.
Цианоалкиламины можно использовать в форме их равновесной смеси из кетонциангидрина и аммиака, растворенной в стехиометрическ(Л1 избытке кетона.
Добавка аминов или растворов аминов должна производиться так, чтобы за счет выделяющегося при этом тепла реакции или сольватации не происходило повыщения температуры реакционной смеси вь1ще температурных границ, указанных для предлагаемого способа. Поэтому добавку амина нужно производить соответственно медленнее и/или при соответствующем охлаждении. Однако непосредственно для установления соответствующей температуры реакции можно полностью или частично использовать теплоту реакции или сольватации при добавке амина. В особенности это возможно в начале второй стадии синтеза при синтезе 2,4-ди- (алкиламино) -6-xлop-cuжJИ-тpиaзинoв, т.е. при добавке второго амина.
Способ по изобретению позволяет достичь выходов свыше 99% от теории при чистоте продукта свыще 98 или 99%. Полученные непосредственно по этому способу после обычной обработки продукты содержат менее, чем 0,7 вес. /о непрореагировавщего хлористого цианура и/или продуктов его гидролиза.
В качестве реакционных продуктов по способу изобретения в зависимости от выбранной исходной концентрации хлористого цианура и в зависимости от рода, т. е. растворимости, полученного аминохлортриазина получают двух- или трехфазные смеси, причем третьей фазой является твердое вещество.
При обработке этих продуктов руководствуются этими фазовыми свойствами. Например, при получении 2-(1 -циано-1 -метилэтиламино)-4-этиламино-6-хлор-сы ж-триазинов, если исходят примерно из 10%-ных растворов хлористого цианура в смеси толуол/ацетон, при температуре выще 40°С, образуются прозрачные двухфазные растворы, которые можно легко разделить на нижнюю водно-ацетоновую, содержащую хлорид натрия фазу, и верхнюю толуольно-ацетоновую, содержащую целевой продукт. Предпочтительно -обработкой из нее можно получить продукт путем упаривания растворителя, например, путем отгонки или после частачного упаривания путем распылительной сушки или вальцовой сушки.
Следующими целесообразным методом является осаждение продукта из «верхней фазы путем частичного испарения растворителя путем концентрирования, например, примерно до 50°/о-ной концентрации твердого вещества и таким образом отделение основной части с помощью обычнь1х способов отделения твердого вещества, как центрифугирование или фильтрование, в то время как маточный раствор возвращают для концентрирования. Полученный таким образом не содержащий растворителя продукт можно затем сущить: электросущкой, сушкой в псевдоожиженном слое, сущкой с использованием тарельчатой сушилки, или просто в сушильном шкафу. Аналогично эти способы можно использовать также: для образующихся по предлагаемому способу трехфазных реакционных смесей, с отделением твердого продукта до и/или после концентрирования после отделения преобладающей водной фазы.
Однако также можно осаждать часть продукта путем сильного разбавления реакционной смеси водой, осадивщееся твердое вещество отделять и лишь потом обрабатывать оставшуюся «органическую фазу путем испарения растворителя. При этом способе переработки возникает опасность снижения выхода путем определенной раст-. воримости продукта в (кетонсодержащей) воде, что играет существенную отрицательную роль в обычных ранее известных способах, в которых для достижения достаточной чистоты продукта нужно еще промывать целевой продукт.
2,4-Ди-алкиламино-6-хлор-с«л{ж-триазины, которые можно получать по предлагаемому способу, соответствуют общей формуле
,,
VA
где Ri и Ri одинаковые или разные и означают заМещенные при известных условиях ОН-, ORs-, SRs-, CN-группами или атомами галогенов, линейные или разветвленные алкильные с 1-4 атомами углерода, алкенильные, циклоалкильные или метилциклопропильные, предпочтительно незамещенные этил- или циклопропильные группы и где RS - алкильная группа с 1-4 атомами углерода и причем предпочтительно Ri или RI означает атом водорода; Rj и R4 могут иметь значения, как R, и Ra, однако, предпочтительно Rs означает атом водорода и R4-rpynny,B которой Re и R/ - одинаковые или разные и означают линейную или разветвленную алкильную или алкенильную группу с 1-8 атомами углерода, которые также могут быть замкнуты в 5-7-членное кольцо или циклоалкильный, предпочтительно метильный, метилциклопропильный или циклопропильный остаток, в особенности метильный остаток, причем также Re или R7 могут являться атомом водорода.
Предпочтительно получают такие соединения, в которых Ri-R4 означают метильный, этильный, изопропильный и циклопропйльный остаток.
Пример 1. В пятигорлую колбу емкостью 2 л, снабженную механической мещалкой, обратным холодильником, термометром, стеклянным электродом (одностёржневая измерительная цепь фирмы Schott и Gen., Jena тип Н., нулевая точка рН 7, платиновая диафрагма; калиброванная по водным буферным растворам: рН 7 (фосфат) и рН 9 (борная кислота - раствор едкого натра - (КС), капельной воронкой и охлаждающей баней, помещают 10%-ный раствор
5 92,2 г (0,5 моль) хлористого цианура в 829,8 г смеси, содержащей 70 толуола и 30 вес. °/о ацетона (650,9 г толуола и 278,9 г ацетона). Раствор при перемешивании охлаждают до , затем в течение 20 мин прибавляют по каплям 44,6 г 98/о-ного
(0,52 моль) а-аминоизобутиронитрила. После этого добавляют 15 мл воды и начинают,, прибавлять по каплям 25%-ный водный раствор едкого натра. Добавку едкого натра проводят непрерывно и именно таким образом,
S чтобы поддерживать следующим ход зависимости рН - время:
В начале добавления едкого натра рН 3,5;
Спустя 26 мин после начала добавления едкого натра рН 5,5;
Спустя 51 мин после начала добавления едкого натра добавляют 140 мл воды и устанавливают рН 6,5 путем прибавления едкого натра.
Спустя 77 мин после начала добавления
J едкого натра рН 7,0;
103 мин - рН 7,5; 129 мин температура реакционной смеси повышается до S5°C и путем дозировки едкого натра устанавливают рН 7,5; Спустя 154 мин рН ,7;
о180 мин рН 7,7.
В целом добавляют 79 г 25/о-ного раствора едкого натра. Затем в течение следующих 20 мин прикапывают 57,1 г 50%-ного водного раствора циклопропиламина и после этого реакционную смесь нагревают до 50°. По достижении этой температлры (спустя 15 мин) прибавляют дополнительное количество 25%-ного едкого натра, и опять непрерывно, по следующему ходу зависимости рН - время; в начале второго добавления едкого натра рН 8,5. Спустя 17 мин после начала второго добавления едкого натра рН 9,5; спустя 34 мин - рН 10,5; спустя 51 мин рН 10,5; спустя 69 мин - рН 10,5; спустя 86 мин - рН 10,5; спустя 103 мин - рН 10,5; спустя
120 мин - рН 10,5;
Всего добавляют 81 г едкого натра (25°/о-ного) при втором добавлении едкого натра. Полученную реакционную смесь разбавляют 200 мл ацетона и затем при 45°С осуществляют разделение фаз. Водную фазу отбрасывают, органическую фазу вместе с выкристаллизовавшимися из нее частями выпаривают в вакууме досуха и затем остаток сушат в вакууме при 60°С до постоянного веса. Получают 120,1 г продукта, который по анализу содержит 99,8% 2-(1-циано-1-метилэтиламино)-4-циклопропиламино-6-хлорсылж-триазина и что соответствует 99,6% теоретического выхода. Пример 2. В непрерывно работающей аппаратуре к току 10%-ного раствора хлористого цианура в смеси 65 вес. % толуола и 35 вес. % ацетона непрерывно добавляют 1,02 моль а-аминойзобутиронитрила на 1 моль хлористого цианура. Смесь выдерживают при температуре + 10°С с по1у1ощью охлаждения и тотчас после получения этой смеси, т. е. после добаЬления всего х-аминоизобутиронитрила, ее подают в каскад реакторов. Объем каскада, состоящего из 4 реакторов, соразмеряется таким образом, чтобы среднее время пребывания в нем составляли 20, затем 10, затем 70, и наконец, 175 мин. , Температуры реакционной смеси в отдельных реакторах выдерживаются равные 10,10, 10 соответственно Зр°С. В первый реактор каскада, кроме того, непрерывно вводят в6ду, и с такой скоростью, чтобы при скорости прохождения 1 моль/ч триазина вводилось 27 г 1ч воды. Кроме того, путем непрерывной подачи 25%-ногб раствора едкого натра и в первом каскаде реактора поддерживается рН 4,6 (измерение с помощью стеклянного электрода, как описано в примере 1). К продукту во втором каскаде реактора также добавляют воду и 25%-ный раствор едкого натра, а именно добавляют воду на 1 моль расходуемого триазина со скоростью 240 г/ч; едкий натр добавляют таким образом, чтобы постоянно поддерживалось рН 5,0. В третьем и четнГертом реакторах путем подачи 25%-ного водного раствора едкого натра и именно таким образом, чтобы в третьем каскаде реактора поддерживалось рН 6,0 и в четвертом каскад- ном реакторе - рН 7,8. Реакционную смесь, выходящую из четвертого каскадного реактора, в смесительном сопле смещивают с 50%-ным водным этиламином и при соотношении скоростей 1 моль/ч этиламина с 1 моль/ч расходуемого триазина. Реакционную смесь при использовании теплоты смещения обоих компонентов и легкого подогрева доводят до температуры 50°С, тотчас вводят во второй каскад реакторов, опять состоящий из четырех реакторов. В первом реакторе этого второго каскада поддерживают рН 7,1 путем непрерывного добавления 25%-ного раствора едкого натра, во вто.ром реакторе этого второго каскада путем
725556
10 добавления 25%-ного раствора едкого натра поддерживают рН 7,6, в третьем реакторе этого второго каскада также путем добавки 25%-ного раствора едкого натра поддерживак)т рН 7,8 и в четвертом (последнем) реакторе этого второго каскада поддерживают рН 10,4 (опять же путем непрерывного добавления 25%-ного раствора едкого натра). Объемы отдельных реакторов каскада соразмеряются таким образом, чтобы они соответствовали среднему времени пребывания непрерывного тока продукта - 20, затем 10, 35, 175 мин. Все реакторы первого и также второго каскада снабжены хорощо действующей системой перемешивания, так Чторасслоение содержащихся в них реакционных смесей невозможно, и гомогенные суспензии Или растворы из реактора поступают в ближайщ1/е. Выходящую из последнего (4-го) реактора второго каскада реакционную смесь подают в автоматический разделитель фаз, в котором непрерывно через дифференциальный регулятор плотности отделяется верхняя фаза и подается в аппарат для упаривания раствЬрителя. Из последнего путем непрерывного выделения получают 98,5%-ный 2-(1 -циано- 1 -метилэтиламино) -4-этиламино-6-хлор-сиж и-триазин с выходом, 99% от теории, (в расчете на хлористьт цианур, вводимый в начале в реакцию). В продукте с помощью тонкослойной хроматографии не обнаруживается 2-(1 ;Циано-1-метилэтиламино)-4,6-дихлор-сылж-триазин и 2,4-диэтиламино-6-хлор-смл.«-триазин. Сточная вода, выходящая из разделителя фаз, после отделения перегонкой содержащегося в ней ацетона содержит практически поваренную соль наряду со следами циануровой кислоты. Пример 3. В прибор, аналогичньш примеру 1, помещают 10%-ный раствор 92,2 г (0,5 моль) хлористого цианура и 829,8 г смеси, состоящей из 70 вес. % «-ксилола и 30% ацетона (650,9 г «-ксилола и 278,9 г ацетона). Реактор при перемешивании охлаждают до 10°С, затем в течение 20 мин прибавляют по каплям 44,6 г 98%--ного раствора (0,52 моль) (х-аминоизобутиронитрила. После этого добавляют 15 мл воды и начинают прибавление по каплям 25%-ного водного раствора едкого натра. Добавление едкого натра происходит непрерывно, а именно таким образом, что поддерживается следующий режим изменения рН во времени. К моменту начала добавления едкого натра рН 3,3. Через 26 мин после начала добавления едкого натра рН 5,5. Через 51 мин после начала добавления едкого натра прибавляют очередные 140 мл водь и устанавли- . вают рН 6,5 путем изменения скорости доба1вления едкого натра. Через 77 мин после начала добавления едкого натра рН 7,0. ерез 103 мин после начала добавления едкого натра рН 7,5. Через 129 мин после начала добавления едкого, натра температуру реакционной .смеси повышают до 35°С и рН 7,6 устанавливают рн /,ь путем изменения скорости добавления едкого натра. Через 154 мин после начала добавления едкого натра рН 7,7. Через 180 мин после начала добавления едкого натра рН 7,7. Всего добавляют 79 г 25°/о-ного раствора едкого натра. После этого в течение следующих 20 мин прибавляют по каплям 57,1 г БО /О-НОГО раствора циклопропиламина в воде, а затем нагревают реакционную смесь до 50°С. По достижении этой температуры (через 15 мин) опять начинают непрерывно добавлять 25°/о-ный раствор едкого натра, соблюдая следующий режим изменения величин рН во времени. К моменту начала второй очереди добавления едкого натра рН 8,3; через 7 мин после начала второй очереди добавления едкого натра рН 9,5; через 34 мин рН 10,5; через 51 мин рН 10,5; через 69 мин рН 10,5; через 86 мин рН 10,5; через 103 мин рН 10,5; через 120 мин рН 10,5. Всего во время второй очереди добавления едкого натра прибавляют 81 г 25%-ного раствора едкого натра. Полученную реакционную смесь разбавляют 200 мл ацетона и после этого проводят разделение фаз при 45°С. Водную фазу выбрасывают, органическую фазу вместе с выкристаллизовавщейся частью упаривают в вакууме досуха, а затем остаток высушивают в вакууме при 60°С до постоянного веса. Таким образом, получают 119,86 г продукта, содержащего пО результатам анализа 99,5% 2- (1 -циано-1 -метилэтиламино) -4-цик лопропиламино-6-хлор-сыл{.л(-триазина, что соответствует 99,5% от теоретического выхода чистого продукта. Пример 4. Помещают 10%-ный раствор 92,2 г (0,5 моль) хлористого цианура в 829,8 г смеси, состоящей из 80 % и-ксилола и 20% метилэтилкетона (663,8 г ж-ксилола и 166,0 г метилэтилкетона). Раствор охлаждают до 10°С при перемещивании, затем в течение 20 мин прибавляют по каплям 44,6 г 98%-ного раствора (0,52 моль) а-аминоизобутиронитрила. После этого добавляют 15 мл воды и начинают ирибавление по каплям 25%-ного водного раствора едкого натра. Добавление едкого натра происходит непрерывно, а именно таким образом, что поддерживается следующий режим изменения рН во времени. К моменту начала добавления едкого натра рН 3,6; через 26 мин после начала добавления едкого натра рН 5,5; через 51 мин прибавляют следующие 140 мл воды и устанавливают рн 6,5 путем изменения скорости добавления едкого натра; через 77 мин рн 7,0; через 103 мин рН 7,5; через 129 мин температуру реакционной смеси повыщают до 35°С и устанавливают рН 7,6 путем изменения скорости добавления едкого натра; через 154 мин рН 7,7; через 180 мин рН 7,7.Всего добавляют 79 г 25%-ного раствора. 7 6 После этого в течение следующих 20 мин прибавляют по каплям 57,1 г 50%-ного водного раствора циклопропиламина и затем нагревают реакционную смесьдо 50°С. По достижении этой температуры (через 15 мин) опять начинают непрерьшно добавлять 25%-ный раствор едкого натра, соблюдая следующий режим изменения ве тичин рН во времени. . К моменту начала второй очереди добавления едкого натра рН 8,4 через 17 мин после начала второй очереди добавления едкого натра рн 9,5; через 34 мин рН 10,5; через 51 мин рН 10,5; через 69 мин рН 10,5; через 86 мин рН 10,5; через 103 мин рН 10,5; через 120 мин рН 10,5. Всего во время второй очереди добавления едкого натра прибавляют 81 г 25%-ного раствора едкого натра. Полученную реакционную смесь разбавляют 200 мл ацетона и после этого проводят разделение фаз при.45°С. Водную фазу выбрасывают; органическую фазу вместе с выкристаллизовавщейся частью упаривают в вакууме досуха, затем остаток высущивают в вакууме при 60°С до постоянного веса. Таким образом, получают 119,61 г продукта, состоящего по данным анализа, из 99,7% 2-(1 -циано-1 -метилэтиламино)-4-циклопропилаМино-6-хлор-сил(.д -триазина, что соответствует 99,1% от теоретического выхода чистого продукта. Пример 5. В прибор, аналогичный примеру 1, помещают 10%-ный раствор 92,2 г (0,5 моль) хлористого цианура в 829,8 г смеси, состоящей из 75 вес. % целлозольва и 25-вес. % ацетона (622,4 г целлозольва и 207,5 г ацетона). Раствор охлаждают до 10°С при перемещивании, затем в течение 20 мин прибавляют по каплям 44,6 г 98%-ного раствора (0,52 моль) а-аминоизобутиронитрила. После этого добавляют 15 мл воды и начинают прибавление по каплям 25%-ного водного раствора едкого натра. Добавление едкого натра происходит непрерывно, а именно таким образом, что поддерживается сле.дующий режим изменения рН во времени. К моменту начала добавления едкого натра рН 3,4; через 26 мин после начала добавления рН 5,5; через 51 мин прибавляют следующие 140 мл воды и устанавливают рН 6,5 путем изменения скорости добавления едкого натра; через 77 мин рН 7,0; через 129 мин температуру реакционной смеси повыщают до 35°С и устанавливают рН 7,6 путем изменения скорости добавления едкого натра; через 154 мин рН 7,7; через 180 мин рН 7,7; Всего добавляют 79 г 25%-ного раствора едкого натра. После этого в течение следующих 20 мин прибавляют по каплям 57,1 г 50%-ного водного раствора циклопропиламина и затем нагревают реакционную смесь до 50°С. По достижении- этой температуры (через 15 мин) опять начинают непрерывно добавлять 25%-ный раствор едкого натра,
13 соблюдая следующий режим изменения величин рН во времени: К моменту начала второй очереди добавления едкого натра рН 8,6; через 17 мин после начала второй очереди добавления едкого натра рН 9,5; через 34 мин после начала второй очереди добавления едкого натра рН 10,5; через 51 мин рН 10,5; через 69 мин рН 10,5; через 86 мин рН 10,5; через 103 мин рН 10,5; через 120 мин рН 10,5. Всего во время второй очереди добавления едкого натра прибавляют 81 г 25%-ного раствора едкого натра. Полученную реакционную смесь разбавляют 200 мл ацетона и после этого проводят разделение фаз при 45°С. Водную фазу выбрасывают; органическую фазу вместе с выкристаллизовавшейся частью упаривают досуха в вакууме, а остаток высушивают в вакууме при 60°С до постоянного веса. Таким образом получают 119,61 г продукта, содержащего по данным анализа 99,6% 2-(1 -циано-1 -метилэтиламино)-4-циклопропиламино-б-хлор-сыжж-триазина, что соответствует 99, от теоретического выхода чистого продукта. Пример 6. В прибор, аналогичный примеру 1, помещают Ю /о-ный раствор 92,2 г (0,5 моль) хлористого цианура в 829,8 г смеси, состоящей из 70 вес. % толуола и 30 вес. % ацетона (650 г толуола и 278,9 г ацетона). Раствор охлаждают до 10°С при перемешивании, затем в течение 20 мин прибавляют по каплям 43,8 г свежеперегнанного 98%-ного (остальное вода) а-аминоизобутиронитрила. После этого добавляют 15 мл воды и начинают прибавление по каплям 25/о-ного BojiHoro раствора едкого натра. Добавление едкого натра происходит непрерывно, а именно таким образом, что поддерживается следующий режим измененеия рН во времени. К моменту начала добавления едкого натра рН 3,5; через 26 мин рН 5,5; через 51 мин прибавляют следующие 140 мл воды и уС: танавлйвают рН 6,5 путем изменения скорости добавления едкого натра; через 77 мин рН 7,0; через 103 мин рН 7,5; через 129 мин температуру реакционной смеси повыщают до 30°С и устанавливают рН 7,6 путем изменения скорости добавления едкого натра; через 154 мин рН 7,7; через 180 мин рН 7,7: Всего добавляют 79 г 25%-ного раствора едкого натра. После этого в течение следующих 20 мин прибавляют по каплям 31,3 г 50°/о-ного водного раствора метиламина и затем нагревают реакционную смесь до 50°С. При достижении этой температуры (через 15 мин) опять начинают непрерывно добавлять 25°/о-ный раствор едкого натра, соблюдая следующий режим изменения величины рН во времени. К моменту начала второй очереди добавления едкого натра рН 8,5;- через 7 мин
725556
14 после начала второй очереди добавления едкого натра рН 9,5; через 34 мин после начала второй очереди добавления едкого натра рН 10,5; через 51 мин после начала второй очереди добавления едкого натра рН 10,5, через 69 мин после начала второй очереди добавления едкого натра рН 10,5; через 86 мин после начала второй очереди добавления едкого натра рН 10,5; через 103 мин после начала второй очереди добавления едкого натра рН 10,5; через 120 мин после начала второй очереди добавления едкого натра рН 10,5. Всего во время второй очереди добавления едкого натра прибавляют 81 г 25%-ijoro раствора едкого натра. Полученную ргакционную смесь разбавляют 200 мл ацетона и после этого проводят разделение фаз при 45°С. Водную фазу выбрасывают, органическую фазу вместе с выкристаллизовавшейся частью упаривают в вакууме досуха, а затем остаток высушивают в вакууме при 60°С до постоянного веса. Таким образом, получают 113,22 г продукта, содержапдего по результатам анализа 99,8% 2-(1 циано-1 -метилэтиламино)-4-метиламино-6-хлор-с«лж-триазина, что соответствует 99,7% от теоретического выхода чистого продукта. Пример 7. В прибор по примеру 1 помещают 10%-ный раствор 92,2 г (0,5 моль) хлористого цианура в 829,8 г смеси, состоящей из 70 вес. % толуола и 30 вес. % ацетона (650,9 г толуола и 278,9 г ацетона). Раствор охлаждают до 10°С при перемешивании, затем в течение 20 мин прибавляют по каплям 43,8 г свежеперегнанного 98%-но-. го (остальное вода) а-амин6изобутиронитрила. После этого добавляют 15 мл воды и начинают прибавление по каплям 25%-ного водного раствора едкого натра. Добавление едкого натра происходит непрерывно, а именно таким образом, что поддерживается следующий режим изменения рН во времени: к моменту начала добавления едкого натра рН 3,5; через 26 мин после.начала добавления едкого натра рН 5,5; через 51 мин после начала добавления едкого натра прибавляют следующие 140 мл воды и устанавливают рН 6,5 путем изменения скорости добавления едкого натра; через 77 мин после начала добавления едкого натра рН 7,0; через 103 мин рН 7,5; через 129 мин после начала добавления едкого натра температуру реакционной смеси повышают до 30°С и устанавливают рН 7,6 путем изменения скорости добавления едкого натра; через 154 мин после начала добавления едкого натра рН 7,7; через 180 мин рН 7,7. Всего добавляют 79 г 25%-ного раствора едкого натра. После этого в течение следующих 20 .мин прибавляют по каплям 59,7 г смеси, состоящей из 50:50 вес. ч. изопропиламина и воды, и затем нагревают реакционную смесь до 50°С. По достижении этой температуры (через 15 мин) 8Ш|Ё ;нэТинают непрерывно добавлять 25°/о-ный .раствор едкого натра, соблюдая следующий режим изменения величин рН во времени: к моменту начала второй очереди добавления едкого натра рН 8,5; через 17 мин после начала второй очереди добавления едкого натра рН 9,5; через 34 мин после начала второй очереди добавления едкого натра рН 10,5; через 51 мин после начала второй очереди добавления едкого натра рН 10,5; через 69 мин после начала второй очереди добавления едкого натра рН 10,5; через 86 мин после начала второй очереди добавления едкого натра рН 10,5; через 103 мин после начала второй очереди добавления едкого натра рН 10,5; через 120 мин после начала второй очереди добавления едкого натра рН 10,5. Всего во время второй очереди добавления едкого натра прибавляют 81 г 25/о-ного раствора едкого натра. Полученную реакционную смесь разбавляют 200 мл ацетона и после этого проводят разделение фаз при 45°С. Водную фазу выбрасывают, органическую фазу вместе с выкристаллизовавшейся частью упаривают в вакууме досуха, а затем остаток высушивают в вакууме до постоянного веса при 60°С. Таким образом, получают 126,98 г продукта, содержаш,его по результатам анализа 99,8% 2-(1 -циано-1 -метилэтиламина)-4-изоп.ропиламино-6-хлор-с«л«лг-триазйна,что соответствует 99,5% от теоретического выхода чистого продукта. Пример 8. В прибор, аналогичный примеру 1, помеш.ают 10 /о-ный раствор 92,2 (0,5 моль) хлористого цианура в 829,8 г смеси, состоящей из 70 вес. % толуола .и 30 вес. % ацетона (650,9 г толуола и 278,9 г ацетона). Раствор охлаждают до 10°С при перемешивании, затем в течение 20 мин прибавляют по каплям 43,8 г свежеперегнанного 98%-ного (остальное вода) а-аминоизобутйронитрила. После этого добавляют 15 мл воды и начинают прибавление по каплям 25%-ного раств ора едкого натра. Добавление едкого jaTpaпроисходит непрерывно, аименно таким образом, что поддерживается следующий режим изменения рН во временигк моменту начала добавления едкого натра рН 3,5; через 26 мин после начала добавления ёдкого натра рН 5,5; через 51 мин после начала добавления едкого натра при-, бавляют следующие 140 мл,воды и устанав: ливают рН 6,5 путем изменения скорости добавления едкого натра; через 77 мин после начала доб..оления едкого натра рН 7,0; через 103 мин после начала добавления едкого натра рН 7,5; через 129 мин после начала добавления едкого натра температуру реакционной смеси повь1щают до 30°С и устанавливают рН 7,6 путем изменения скорости добавления едкого натра;. через 154 мин после начала добавления едкого натра рН 7,7; через 180 мин после начала добавления едкого натра рН 7,7. Всего добавляют 79 г 25%-ного раствора едкого натра После этого в течение следующих 20 мин прибавляют по каплям 57,7 г смеси, состоящей из равнь1х весовых частей циклопропиламина и воды, и затем нагревают реакционную смесь до 50°С. По достижении этой температуры (через 15 мин) опять начинают непрерывно добавлять 25°/о-ный раствор едкого натра, соблюдая следующий режим изменения величин рН во времени. К моменту начала второй очереди добавления едкого натра рН 8,5; через 17 мин после начала второй очереди добавления едкого натра рН 9,5; через 34 мин после начала второй очереди добавления едкого натра рН 10,5; через 51 мин после начала второй очереди добавления едкого натра рН 10,5; через 69 мин после начала второй очереди добавления едкого натра рН 10,5; через 86 мин после начала второй очереди добавления едкого натра рН 10,5; через 103 мин после начала добавления едкого натра рН 10,5; через 120 мин после начала второй очереди добавления едкого натра рН 10,5. Всего во время второй очереди добавления едкого натра прибавляют 81 г 25%-ного раствора едкого натра. Полученную реакционную смесь разбавляют 200 мл ацетона и после этого проводят разделение фаз при . Водную фазу выбрасывают, ограническую фазу вместе с выкристаллизовавшейся частью упаривают в вакууме досуха, а затем остаток высущивйют в вакууме при 60°С до постоянного веса. Таким образом получают 126,10 г продук- . та, содержащего по результатам анализа 99,8%2-(1 -циано-1 -метилэтиламино)-4-циклопропиламино-6-хлор-сил{.л -триазина, что соответствует 99,6% от теоретического выхода чистого продукта. Пример 9. В прибор, аналогичный примеру 1, помещают 10%-ный раствор 92,2 г (0,5 моля) хлористого цианура в 829,8 г смеси, состоящей из 70 вес. % толуола и 30 вес. % ацетона (650,9 г толуола и 278,8 г ацетона). Раствор охлаждают до 0°С при перемешивании, затем в течение 20 прибавляют по каплям 29,7 г изопропиламина. После этого добавляют 15 мл воды и начинают прибавление по каплям 25%-ного водного раствора едкого натра. Добавление едкого натра происходит непрерывно, а именно таким образом, что поддерживается следующий режим изменения рН во времени: к моменту начала добавления едкого натра рН 3,5; через26 мин после начала добавления едкого натра рН 5,5; через 51 мин после начала добавления едкого натра прибавлякэт следующие 140 мл воды и,устанавливают рН 6,5 путем изменения скорости добавления едкого натра; через 77 мин после начала добавления едкого натра рН 7,0; через 103 мин после начала добавления едкого натра 7,5; через 129 мин после начала добавления едкого натра температуру реакционной смеси повышают до 20°С и устанавливают рН 7,6 путем изменения скорости добавления едкого натра; через 154 мин после начала добавления едкого натра рН 7,7; через 180 мин после начала добавления едкого натра рН 7,7.
Всего добавляют 79 г 25%-ного раствора едкого натра: После этого в течение следующих 20 мин прибавляют по каплям 45,5 г 50%-ного водного раствора этиламина и затем нагревают реакционную смесь до 50°С. По достижении этой температуры (через 15 мин) оцять начинают непрерывно добавлять 25%-ный раствор едкого натра, соблюдая следующий изменения величины рН во времени:
к моменту начала второй очереди добавления едкого натра рН 8,5; через 17 мин после начала второй очереди добавления едкого натра рН 9,5; через34 мин после начала второй очереди добавления едкого натра рН 10,5, через 51 мин после начала второй очереди добавления едкого натра рН 10,5; через 69 мин после начала второй очереди добавления едкого натра рН 10,5; через 86 мин после начала второй очереди добавления едкого натра рН 10,5; через 103 мин после начала второй очереди добавления едкого натра рН 10,5; через 120 мин после начала второй очереди добавления едкого натра рН 10,5.
Всего во время второй очереди добавления едкого натра прибавляют 81 г 25%-ного раствора едкого натра.
Полученную реакционную смесь разбавляют 200 мл ацетона и после этого прово.дят разделение фаз при 45°С. Водную фазу вместе с выпавщим продуктом тщательно отделяют, делительную воронку промывают ацетоном от следов осадка и полученную суспензию в смеси ацетона и толуола после добавления еще около 50 мл толуола промывают водой (без уноса твердой фазы) до отсутствия в промывных водах ионов хлора. Полученную (в основном) толуольную суспензию триазина вместе с другой органической фазой, и выкристаллизовавшейся частью упаривают в вакууме досуха, а затем остаток высушивают в вакууме при 60°С до постоянного веса. Таким образом, получают 107 г продукта, содержащего по результатам анализа 99,8% 2-изопропиламино-4-этил-б-хлор-сцлж-триазина («атразин), что соответствует 99,0% от теоретического выхода чистого продукта.
Пример 10. В прибор, аналогичный примеру 1, помещают Швее. °/о-ный раствор 92,2г (0,5 моля) хлористого цианура в 829,8 г смеси, состоящей из 70 вес. % толуола и 30% ацетона (650,9 г толуола и 278,9 г ацетона).
Раствор охлаждают до 10°С при перемешивании, затем в течение 20 мин прибавляют по каплям 29,9 г изопропиламина. После этого добавляют 15 мл воды и начинают прибавление по каплям 25%-ного водного раствора едкого натра. Добавление едкого натра происходит непрерывно, а именно таким образом, что поддерживается следующий режим изменения рН во времени:
к моменту начала добавления едкого натра рН 3,5; через 26 мин после начала добавления едкого натра рН 5,5; через 51 мин после начала добавления едкого натра прибавляют следующие 140 мл воды и устанавливают рН 6,5 путем изменения скорости добавления едкого натра; через 77 мин после начала добавления едкого натра рН 7,0; через 103 мин после начала добавления едкого натра рН 7,5; через 129 мин после начала добавления едкого натра температуру реакционной смеси повышают до 30°С и устанавливают рН 7,6 путем изменения скорости добавления едкого натра; через 154 мин посленачала добавления едкого натра рН 7,7; через 180 мин после начала добавления едкого натра рН 7,7.
Всего добавляют 79 г 25%-ного раствора едкого натра. После этого в течение следующих 20 мин прибавляют по каплям 30,1 г изопропиламина и затем нагревают реакционную смесь до 50°С. По достижении этой температуры (через 15 мин) опять начинают непрерывно добавлять 25%-ный раствор едкого натра, соблюдая следующий режим изменения велиЧ:йны рП во времени:
к моменту начала второй очереди добавления едкого натра рН 8,5; через 17 мин после начала второй очереди добавления едкого натра рН 9,5; через 34 мин после начала второй очереди добавления едкого натра рН 10,5; через 51 мин после начала второй очереди добавления едкого натра рН 10,5; через 69 мин после начала второй очереди добавления едкого натра рН 10,5; через 86 мин после начала втор.ой очереди добавления едкого натра рН 10,5; через 103 мин после начала второй очереди добавления едкого натра рН 10,5; через 120 мин после начала второйочереди добавления едкого натра рН 10,5.
Всего во время второй очереди добавления едкого натра прибавляют 81 г 25%-ного раствора едкого натра.
Полученную реакционную смесь разбавляют 200 мл ацетона и после этого проводят разделение фаз при 45°С. Водную фазу вместе с выпавщим продуктом тщательно отделяют, делительную воронку промывают ацетоном от следов осадка и полученную суспензию в смеси ацетона и толуола после добавления еще около 500 мл толуола промывают водой (без уноса твердой фазы.) до отсутствия в промывных водах ионов хлбра. Полученную в основном толуольную суспензию триазина вместе с другой органической фазой и выкристаллизовавщейся
частью упаривают в вакууме досуха, а за ёйостаток высушивают в вакууме при 60°С до постоянного веса. Таким образом, получают 114,7 г продукта, содержащего по результатам анализа 99,8% 2,4-бис-изопропиламино-6-хлор-симм-1гриазина (пропазин), что соответствует 99,7% от теоретического
5й,
1ШШШ ЭДстЪго г1рб1укта: .,-..
Пример 11. В прибор, аналогичный примеру 1, помещают Ю /о-ный раствор 92,2 г (0,5 моль) хлористого цианура в 829,8 гсмеси, состоящей из. 70 вес. % толуола и 30 вес. % ацетона (650,9 г толуола и 278,9 г ацетона).
Раствор охлаждают до 20°С при перемешивании, затем в течение 20 мин прибавля-,: ют по каплям 46 г 50/о-ногов6дйбгб раствора этиламина. После этого добавляют 15 мл воды и начинают прибавление по каплям 25°/о-ного водного раствора еДкого натра. Добавление едкого натра происходит непрерывно, 9 именно таким образом,что поддерживается следующий режим изменения рН во времени.
К моменту начала добавления едкого натра рН 3,5; через 26 мин после начала добавления едкого натра рН 5,5; через 51 мин после начала добавления едкого натра прибавляют следующие 140 мл воды и устанавливают рН 6,5 путем изменения скорости добавления едкого натра; через 77 мин после начала добавления едкого натра рН 7,0; через 103 мин после начала добавления едкого натра рН 7,5; через 129 мин после начала добавления едкого натра температуру реакционной смеси повышают до 30°С и устанавливают рН 7,6 путем изменения скорости добавления едкого натра; через J54 мин после начала добавления едкого натрз рН 7,7; через 180 мин после начала добавления едкого натра рН 7,7.
Всего добавляют 79 г 25 /о-ного раствора едкого натра. После, этого в течение следующих 20 мин прибавляют по каплям 47 г 50°/о-ного водного раствора этиламина и затем нагревают реакционную смесь до 50°С. По достижении этой температуры (через 15 мин) опять начилают неггрерывно добавлять 25%-ный раствор едкого натра,соблюдая следующий режим изменений величины рН во времени;..
к моменту начала второй очереди добавления едкого натра рН 8,5; через 17 мин после начала второй очереди добавления едкого натра рН 9,5; через 34 мин после начала второй очереди добавления рН 10,5; через 51 мин после начала второй очереди добавления едкого натра рН 10,5; через 69 мин после начала второй очереди добавления едкого натра рН 10,5; через 86 мин после начала второй очереди добавления едкого натра рН 10,5; через 103 мин после начала второй очереди добавления едкого натра рН .10,5; через 120 мин после начала второй очереди добавления едкого натра рН
10,5....;,;.::.:,,. ,;..д,..; , - ,.
Всего во время второй очереди добавления едкого натра прибавляют 81 г 25% ного раствора едкого натра.
Получёнйую реакционную смесь ра.збавляют 20 мл ацетона и лосле этого проводят,. разделение фаз при 45°С. Водную фазу вместе с выпавшим продуктон тщательно отделяют, делительную воронку промывают ацетоном от следов осадка и полученную суспензию в смеси ацетона и толуола после добавления еще около 500мл, толуола промывают водой (без:уноса твердой фазы) до отсутствия в промывных водах ионов хлора. Полученную (в основном) толуольную суспензию триазина вместе с другой органической фазой и выкристаллизовавшейся частью упаривают в вакууме досуха, а затем остаток высушивают в вакууме при 60°С до постоянного веса. Таким образом, получают 100,9 г Продукта, содержащего по результатам анализа 99,8% 2,4-б«с-этиламино-6-хлор-смлл -триазина, что соответствует 99,9% от теоретического выхода чистого продукта.
Пример 12. В прибор, аналогичный при меру 1, помешают 10%-ный раствор 92,2 г (0,5 моль) хлористого цианура в 829,8 г смеси, состоящей из 70 вес. % толуола и 30 вес. % ацетона (650,9-г толуола и 278,9 г ацетона).
Раствор охлаждают до 0°С при перемешивании, затем в течение 20 мин прибавляют по каплям 57,5 г смеси, состоящей из равных весовых частей циклопропиламина и воды.-После этого добавляют 15 мл воды и на.Минают прибавление по каплям 25%-ного водного раствора едкого натра. Добавление едкого натра происходит непрерывно, а именно таким образом что поддерживается следующий режим изменения рН во времени: - -. - --: . .
к моменту начала добавления едкого.йатра рН 3,5. Через 26 мин после начала добавления едкого натра рН 5,5; через 51 мин после начала добавления едкого натра при ..г.. бавляют следующие 140 млводы и устанавливают рН 6,.5 путем изменения скорости добавления едкого натра; через 77 мин после начала добавления едкого натра рН 7,0; через 103 мин после начала добавления едкого натра рН 7,5; через 129 мин после начала добавления едкого натра температуру реакционной смеси повышают до 10°С и устанавливают рН 7,6 путем изменения скорости добавления еДкого натра; через 154 мин после начала добавления едкого натра рН 7,7; через 180 мин после начала добавления едкого натра рН 7,7.
Всего добавляют 79 г 25%-ного раствора едкого натра. После этого в течение следующих 20 мин прибавляют по каплям 45,5 г 55 50%-ного водного раствора этиламина и затем нагревают реакционную смесь до 50°С. По достижении этой температуры (через 15 мин) опять начинаю непрерывно добавлять 25%-ный раствор едкого натра, соблюдая следующий режим изменения рН во времени:., к моменту начала второй очереди добавления едкого натра рН 8,5; через 17 мин после начала второй очереди добавления едкого натра рН 9,5; через 34 мин после начала второй очереди добавления едкого натра рН 10,5; через 51 мин после начала второй очереди добавления едкого натра рН 10,5; через 69 мин после начала второй очереди добавления едкого натра рН 10,5; через 86 мин после: начала второй очереди добавления едкого натра рН 10,5; через 103 мин ггосле начала второй очереди добавления едкого натра рН 10,5; через 120 мин после начала второй очереди добавления едкого натра ,рН 10,5. , . Всего во время второй очереди добавления едкого натра прибавляют 81 г 25%-ного раствора едкого натра. Полученную реакционную смесь разбавляют 200 мл ацетона и после этого проводят разделение фаз при 45°С. Водную фазу вместе с выпавшим продуктом тщательно отделяют, делите;|ьную воронку промывают ацетоном от следов осадка и полученную суспензию в смеси ацетона и толуола после добавления еще около 500 мл толуола, промывают водой (без уноса твердой фазы) до отсутствия в промывных водах ионов хлора. Полученную, в основном толуольную суспензию триазина вместе с другой органической фазой и выкристаллизовавшейся частью упаривают в вакууме досуха, а затем остаток высущивают в вакууме при 60°С до постоянного веса. Таким образом, получают 106,5г продукта, содержащего по результатам анализа 99,8% 2-циклопропиламино-4-этиламино-6-хлор-с«л ж-триазина, что соответствует 99,5% от теоретического выхода чистого продукта. Пример 13. В прибор, аналогичный примеру 1, помещают 10%-ный раствор 92,2 г (0,5 моль) хлористого цианура и 829,8 г смеси, состоящей из 70 вес. % толуола и 30 вес. % ацетона (650,9 г толуола и 278,9 г ацетона). Раствор охлаждают до 0°С, при перемешивании, затем в течение 20 мин прибавляют по -каплям 57,2 г смеси, состоящей из равных весовых частей аллиламина и воды. После этого добавляют 15 мл воды и начинают прибавление по каплям 25%-ного водного раствора едкого натра. Добавление едкого натра происходит непрерывно, а именно таким образом, что поддерживается следующий режим изменения рН во времени: к моменту начала добавления едкого натра рН 3,5; через 26 мин после начала добавления едкого натра рН 5,5; через 51 мин после начала добавления едкого, натра прибавляют следующие 140 мл воды и устанавливают рН 6,5 путем изменения скорости добавления едкого натра; через 77 мин после начала добавления едкого натра рН 7,0; через 103 мин после начала добавления едкого натра рН 7,5; через 129 Аин после начала добавления едкого натра темпера1туру реакционной смеси повь1шают до 10°С и устанавливают рН 7,6 изменения скорости добавления едкого натра; через 154 мин после начала добавления едкого натра рН 7,7 через 180 мин после начала добавления едкого натра рН 7,7. Всего добавляют 79 г 25%-ного раствора едкого натра. После этого в течение следующих 20 мин прибавляют по каплям 59,5 г смеси изопропиламина и воды при их весовом сботношёнии 1:1 и затем нагревают реакционную смесь до 50°С. По достижении этой температуры (через 15 мин) опять начинают непрерывно добавлять 25%-ный раствор едкого натра,соблюдая следующий режим изменения величины рН во времени. К моменту начала второй очереди добавления едкого натра рН 8,5; через 17 мин после начала второй очереди добавления едкого натра рН 9,5; через 34 мин после начала второй очереди добавления едкого натра рН 10,5; через 5Гмин после начала второй очереди добавления едкого натра рН 10,5; через 69 мин после начала второй очереди добавления едкого натра рН 10,5; через 86 мин после начала второй очереди добавления едкого натра рН 10,5; через 103 мин после начала второй очереди добавления едкого натра рН 10,5; через 120 мин после начала второй очереди добавления едкого натра рН 10,5. Всего во время второй очереди добавления едкого натра прибавляют 81 г 25%-ного раствора едкого натра. Полученную реакционную смесь разбавляют 200 мл ацетона и после этого проводят разделение фаз при 45°С. Водную фазу вместе с выпавшим продуктом тщательно отделяют, делительную воронку промывают ацетоном от следов осадка и получедную суспензию в смеси .ацетона и толуола после добавления еще около 500 мл толуола промывают водой (без уноса твердой фазы) до отсутствия в промывных водах ионов хлора. Полученную (в основном) толуольную суспензию триазина вместе с другой органической фазой и выкристаллизовавщейся частью упаривают в вакууме досуха, а затем остаток высущивают в вакууме при 60°С до постоян- . ного веса. Таким образом получают 113,0 г продукта, содержащего по результатам анализа 99,8% 2-аллиламино-4-изопропИламино-6-хЛор-с«уИл-триазина, что -соответствует 99,1% от теоретического выхода чистого продукта. Формула изобретения Способ последовательного замещения атомов хлора хлористого цианура амином или двумя одинаковыми или различными
аминами в органичес1(ом растворителе при регулировании рН путем добавления щелочи отличающийся тем, что, с цеяЬю повышения выхода целевого продукта и повышения его чистоты, процесс проводят в смеси 65- 85 вес. °/о ксилола, этилбензола, бензола и/или алифатического, и/илл ци клоалифатического углеводорода с5-10 атомами углерода и 35-15 вес. /о кетона с 3-8 атомами углерода и рН реакционной смеси на первой стадии аминированИя в зависимости от времени реакции поддерживают непрерывно путем добавления щелочи таким образом, чтобы оно соответствовало точке внутри области, ограниченной линиями АВСД (фиг. 1) и рН реакционной смеси проходило область, начинающуюся с времени реакции t О до значений в области, ограниченной линиями BCEF и по достижении рН 7,0-7,2 температура реакционной смеси возрастает до 10-60°С, на второй стадии рН реакционной смеси путем добавления щелочи устанавливают таким образом чтобы оно соответствовало точке внутри области, которая ограничена линиями GHIJ (фиг. 2) при 40-70°С и рН ре-акционной смеси проходит область, начинающуюся с времени реакции второй стадии t О до значений в области, ограниченной линиями HIKL при условий, что ti 4-104nta 2-8 ч и линия ВС соответствует уравнению рН -(12,6 t,) t 4 14,35 и линия HI соответствует уравнению рН - (24,857 t г) t + -4- 23,9285.
2. Способ по п. 1, отличающийся тем, что процесс на первой стадии начинают при 10-18°С и на 1 моль хлористого цианура добавляют 1,00-1,02 моль первого амина и при 25-40 С в конце первой стадии и по достижении рН 7,2 температура возрастает до 25-40°С, а на 1 моль хлористого цианура добавляют 0,98-1,02 экв. щелочи, на второй стадии добавляют- 1,00- 1,02 моль второго амина на 1 моль используемого хлористого цианура, при этом температуру повыщают до 45-55°С, причем ti 7 ч, ta - 6 ч.
3.Способ по пп. 1 и 2, отличающийся тем, что после добавления первого амина реакционную смесь выдерживают в следующих условиях при 10-18°С путем добавления щелочи и вьщержке воды в известных условиях
рНВремя реакции,мин
1 а) 3,5-5,03-43
16)4,5-6:250-56
1в) 5,5-7,0.17-189
1г) 7,0-8,030-493
и температуре 10-50°С, причем стадии 1а и 16, 16 и 1в могут быть объединены в одну, а после добавки второго амина реакционную смесь при 40-70°С путем добавления щелочи выдерживают в условиях:
рНВремя реакции, мин
2а) 6,5-8,02-60
26) 7,25-9,00-92
2в) 8-10,0 и при температуре О-172 2г) 10,0-11,2545-55°С15-408
причем стадии 2а и 26, 26 и 2в могут быть объединены в одну.4.Способ по пп. , отличающийся тем, что смесь выдерживают в условиях
5рНВремя реакции, мин
1а) 4,25-4,75.9--21
16) 5,0-5,5 3-18
IB) 5,75-6,25 и при температуре 51-93 1г) 7,25-7,90 15-35°С135-330
2а) 6,75-7,2510-30
26) 7,5-8,00-36
2в) 8,25-8,75 и при температуре 9-66 2г) 10,25-10,7545-55°С135-306
Источники информации, принятые во внимание при экспертизе 5 1. Патент США № 3590040, кл. 260-249.5, опублик. 1971.
2.Патент ФРГ № 1670541, кл. 12 р 10/05, опублик. 1974.
3.Заявка № 1695117,
кл. 12 р 10/05, опублик. 1974 (прототип).
рН 7











| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения производных триазина | 1976 |
|
SU635869A3 |
| СПОСОБ ПОЛУЧЕНИЯ ХЛОРАМИНО-5-ТРИАЗИНОВ | 1972 |
|
SU348003A1 |
| Способ получения стильбеновых соединений | 1974 |
|
SU633479A3 |
| Способ получения производных 2,4,-бис/азиридинил-1/-6-амино-симмтризина | 1974 |
|
SU521272A1 |
| ДИНАТРИЕВЫЕ СОЛИ 4-СУЛЬФОКСИЭТИЛСУЛЬФОНИЛ- 2-X-2' -Y-4' -АМИНО-N-МЕТИЛСУЛЬФО-N- N-2" -( 4" -ХЛОР- 6" -АМИНО-ДИ-( β -ГИДРОКСИЭТИЛ)- 1", 3", 5" -ТРИАЗИНИЛ)-АЗОБЕНЗОЛА ДЛЯ КРАШЕНИЯ ИЛИ ПЕЧАТИ МАТЕРИАЛОВ ИЗ НАТУРАЛЬНЫХ И/ИЛИ СИНТЕТИЧЕСКИХ ВОЛОКОН | 1993 |
|
RU2064949C1 |
| СПОСОБ ПОЛУЧЕНИЯ S-ЗАМЕЩЕННЫХ 2-МЕРКАПТО-4,6- ДИХЛОР-8-ТРИАЗИПА | 1971 |
|
SU304746A1 |
| ТЕТРАНАТРИЕВАЯ СОЛЬ 4,4'-БИС-4-МОРФОЛИНО- 6-(N-СУЛЬФОАНИЛИНО)- СИМ-ТРИАЗИНИЛ-2-ИЛ- АМИНОСТИЛЬБЕН-2,2'- ДИСУЛЬФОКИСЛОТЫ В КАЧЕСТВЕ ОПТИЧЕСКИ ОТБЕЛИВАЮЩЕГО ВЕЩЕСТВА | 1989 |
|
SU1646260A1 |
| Способ получения 2-имино-3-(2-окси -2-фенилэтил)тиазолидина или его солей | 1973 |
|
SU559651A3 |
| Соли N,N @ -дихлор-N,N @ -ди(3-хлор-2-оксо-4-окси-1,3,5-триазин-6-ил)-1,2-этилендиамина в качестве отбеливателей текстильных материалов | 1990 |
|
SU1747445A1 |
| Способ получения производных триазина | 1974 |
|
SU565631A3 |
,{D
f/j ZHh ft 1/7 Л//7 «/7 tf Vui.l
i2/e 3i2/g ftz/e stij tz фиг. 2
Время реакции
Время реакции t
