(5) СПОСОБ ПРОВЕДЕНИЯ ФЕРМЕНТАТИВНЫХ
1
Изобретение относится к способам проведения ферментативных реакций с помощью препаратов ферментов, которые могут быть регенерированы из реакционной среды для повторного использования, и может найти широкое применение в химической и фармацевтической промышленности, а также для очистки промышленных и сточных вод от многих токсичныхпримесей органической природы.
До настоящего времени npofeauленное применение ферментов несколько ограничено из-за высокой их стоимости и невозможности повторного использования, поскольку фермент растворен в реакционной среде и часто бывает трудно после окончания реакции отделить фермент от полученного продукта. Как правило, процесс такого отделения вызывает значительное снижение активности фермента или полную его инактивацию.
Известны способы отделения фермента от субстратов и продуктов ферментативных реакций и многократного использования ферментов в реакторах, когда широко используРЕАКЦИЙ
ется иммобилизация ферментов на нерастворикых в воде носителях посредством адсорбции или ковалентного связывания Ij.
В этих способах часто удается осуществлять получение продуктов ферментативных реакций в непрерывном режиме.
Однако иммобилизованные препа10раты фервюнтов обладают существенных недостатков.
Во-первых, такие препараты подвержены механическому разрушению. Во-вторых, эти препараты часто име15ют иизкую удельную активность изза низкой степени обогащения ферментом поверхности твердого носителя. В-третьих, доступ субстратов к активному центру иммобилизованных
20 ферментов затруднен, особенно в случае высокомолекулярных субстратов.
Попытки повысить Сдельную активность таких нерастворимых препаратов путем увеличения внешней поверхности твердого носителя приводят к уменьшению размеров частиц твердого препарата, затрудняет обработ30 У частности, отделение фильтрованиеМ, и вызывает увеличение внутренней поверхности вследствие повышения пористости частиц, и к образованию частиц с меньшей механической прочностью.
Известен способ, проведения ферментативных реакций с помощью водорастворимых фермент-полимерных комплексов, в которы с фермент связан с водорастворимым полимерным носителем или непосредственно, или при помощи мостика, путем контактирования в водной фазе указанного препарата фермейта с субстратом с последующим отделением препарата фермента ультрафильтраций и выделением продуктов реакции 2}.
Однако осуществление ультрафильтрования для вьзделения препарата фермента из реакционной среды сложный и дорогостоящий процесс, особенно, если проводить его в широком масштабе, поэтому применение его в про14ьтшенном процессе нежелательно.
Цель изобретения - упрощение процесса.
Указанная цель достигается тем, что в контакт с препаратом фермента, находящимся в водной фазе, вводят инертную не смешивающуюся с водой жидкость, а в качестве препарата фермента используют препарат, включакхций фермент, обработаннный соединением формулы R - X, где X - функциональная группа, вступающая во взаимодействие с ферментом, а R неполярная группа, представляющая собой: алкил C,-C.jo, алкинил , , циклоалкил С, -Сjo циклоалкеиил ,o арил, С,-С(,-алкиларил, аралкил, обеспечивающая ассоциирование препарата фермента с не смешивающейся с водой жидкостью.
В качестве не смешивакнцейся с водой жидкости используют алканы, ароматические углеводороды, высшие сшифатические слозхные эфяра или QJ алифатические спирты.
Контакт между препарате фермента и не смешивающейся с водой жидкостью осуществляют-обычно до проведения ферментативной реакции, путем перемешивания.
Предпочтительно используют пре- . парат фермента на основе пенициллинацилазы, а в качестве субстрата; ферментативной реакции - бензилпенициллин или феноксиметилпенициллин.
Вырг1жение инертная не смешивгиощаяся с водой жидкость означает жидкость, не реагирующую с препаратом фермента и не вызывающую суще|ственного изменения активности фермента.
Термин ассоциирует с ... означает любую форму взаимодействия неполярных групп препарата фермента с молекулами не смешивающейся с водой жидкости, которое приводи к образованию достаточно устойчивой формы для обеспечения возможности отделения препарата фермента от водной среды.
Для некоторых ферментов связывание неполярных групп непосредственно с молекулой фермента вызывает изменение структуры и изменение активности фе жента. Из-за этого бы|вает иногда невозможно привязать достаточное количество иеполярных групп к ферменту, чтобы придать препарату способность отделяться йт водной среды без неприемлемого уменьшения активности.
Предпочтительно использовать препарат фермента с ферментом, связанным с полимерным носителем или адсорбированным на полимерном но-, сителе, который неполярные группы(вместо или в дополнение неполярных групп, содержащихся на самом ферменте. Это позволяет также регулировать соотношение количества неполярных групп и фермента в препарате. Можно также привязывать фермент, имеющий достаточное количество неполярных групп для обеспечения возможности отделения его от водной среды, к полимерному носителю, который может содержать дополнительные неполярные группы.
Привязка фермента к полимеру в 1 принятом значении включает как ковалентную связь с полимерным носителем, так и адсорбцию на пол11мерно носителе, поскольку адсорбция достаточно прочна для удержания фермента на носителе, когда препарат ассоциирован с не смешивающейся с водой жидкостью.
Пригодный для описанных препаратов носитель может быть водорастворимьлм или водонерастворимым.
Если в препаратах по предлагаемому изобретению применяется водорастворимое полимерное вещество, оно должно быть тонко измельчено, чтобы обеспечить большую поверхность для привязки фермента и таким образом придать препарату большую удельную активность.
Пригодными водонерастворимыми веществами являются порошок целлюлозы и производные целлюлозы, например, ионообменные смолы на основ карбоксиметилцеллюлозы, нейлон, высокомол кулярные полисахариды, например, агароза и сшитые декстраны, полисахариды, модифицированные модифицирукхцими агентами, например эпихлоргидрином, или модифицированные для введения карбоксиметильной или аминометильной групп, полиакрилаты и полиме1гакрилаты.
Предпочтительными являются агароза и макроретикулярно сшитые полиакриловая и полиметакриловая смолы.
Пригодными водорастворимьп14и веществами являются полимеры природного происхождения, например полисахариды, например декстран, декстрины или крахмал, и модифицированные полимеры природного происхождения, например частично расщепленные крахмал или целлюлоза, полисахариды, олигосахариды и сахариды, модифицированные модифицирующими агентс1ми, например эпихлоргидрином и целлюлозой, модифицированная введением карбоксиметильных или с1Миноэтильных групп.
Из упомянутых модифицированных схаридов и олигосахаридов наиболее пригодны эпихлоргидрин и лактоза, либо декстроза, либо сахароза. Предпочтительным полимером является полимер сахарозы-эпихлоргидрина фикол (Ficoll) с молекулярным весом около 400000.
Кроме полимеров природного происхождения можно также применять синтетические водорастворимые полимеры, например полимеры поливинилового спирта и сополимеры малеинового или акрилового ангидридов с этиленом, стиролом, метилвиниловьал эфиром, дивиниловым эфиром или винилацетатом.
Пригодным классом водорастворимых сополимеров являются сополимеры метилвиниловый эфир/малеиновый ангидрид, известные под названием гантреэ AN (Gantrez AN)
Фермент может быть, привязан непосредственно к полимеру, содержащему неполярные группы, или же он может быть связан с мостиками полимерного носителя. Пригодными мостиками являются алифатический о6,а) -диамин, н априме р, 1,3-диаминпропан или 1,6-диамингексан. Одна аминогруппа мостика привязана к полимеру, а другая свободна и может быть присоединена- к ферменту, например, посредством водорастворимого альдегида, например, глиоксаля или глутарового альдегида. Применение таких бифункциональных мостиков приводит к сшиванию фермент/полимер. Мостики могут быть образованы между молекулами фермент/носитель, фермент/неполярная группа (с носителем или без нвг носитель/неполяриая группа.
Количество неполярных групп, вводиколх в препараты по предлагаемому изовретению, зависит от ряда факторов. Привязка неполярных групп к полимеру и/или ферменту создает гидрофобное окружение вблизи фермента. Слишком высокая степень гидрофобиости приводит к дезактивации фермеита вследствие сопутствующих измеиений структуры.
Важно найти оптимальное количество неполярных групп, с одной стороны - минимальное, обеспечивающее возможность отделения фермента от водной , а с другой стороны - максимальное, обеспечивающее стабильность фермента. Нахождение такого оптимального количества может оказаться причиной ошибок. Критериями для установления такого равновесия являются: молекулярный
0 вес полимерного носителя молекулярный вес мономера; содержание в полимере ячеек мономера, замещенных неполярными группами; размер неполярных групп; индивидуальные осо5бенности данного фермента; степень замещения полимера ферментом.
Используемый в предлагаемом изобретении препарат фермента получают посредством контактирования соеди0нения формулы I R - X, где R - неполярная группа, а X - функциональная группа, с ферментом, привязанньм к активной группе, способной реагировать с группой X.
Группа X может представлять соSбой, например, аминогруппу, карбоксил, сшьдегид, гидроксил, тиол или азогруппу, или реакционноспособные связывающие группы, являющиеся, например, производными диальдеОгИдов, например глиоксашя или глицеральдегида, и/или диаминов, например 1,6-гексаметилендиамина или этилендиамина, и/или oc,u} -аминоалифатических карбоиовых кислот, на5пример глициновой или 6-амингексг1миновой кислоты; или полимерные носителем, содержащим любую такую фуикционсшьную группу. Предпочтительно X является аминогруппой или же час0тью, содержащей аминогруппу.
Активная группа, привязанная к ферменту и способная реагировать с группой X, может быть группой, содержащейся на ферменте либо по природе самого фермента, либо выве5денной посредством модификации, как, например, каЕ боксил, аминогруппа, тиол или аксифенольная группа, либо ангидридные связи, или полимерный носитель, содержсидий такую
0 активную группу; или реакционноспособная связывающая группа, например, из тех, что приведены для групп X..
Если используемый препарат фер5ме.нта состоит в основном только из фермента и неполярных групп, предпочтительно, чтобы группа X была производным диальг.вгидом и/или ot,u) диамина, т.е., чтобы неполярные,
0 группы были привязаны к ферменту через такие мостики.
Если иcпoльзye «Iй препарат фер мента содержит также полимерный носитель, предпочтительно сначала до привязки к ферменту модифициро5вать но ситель для введения в него неполярной группы и, если необходимо, мостика. Группа X в формуле R - X обозначает полимер, связанны с неполярной группой R и с функциональной группой. Предлагаемый способ проведения ферментативных реакций осуществляют следующим образом. Препарат фермента приводят в контакт с субстратом фермента, наход5пцимся в водной фазе с последую щим отделением указанного препарата фермента от водной реакционной смеси посредством контактирования с не смешивакндейся с водой жидкостью. Затем производят отделение водной фазы и извлечение из нее продукта реакции при помощи известных приемов. Возможно повторное использование отделенного от водной фазы препарата фермента. Для отделения препарата фермент от водной среды могут быть примене ны различные не смешивающиеся с во дой жидкости. Пригодными жидкостям являются алканы, например, гептан октан, нонак, гексадекан, аромати ческие углеводороды, высшиэ алифа тические сложные эфиры, например, триолеиновый глицерид, и фатические спирты, например, п-де канол. При контактировании препарата фермента с такой жидкостью на пов ности контакта образуется поверх(ностный слой. В целях максимально го увеличения этой поверхности ко такта стадия контактирования вклю чает обычное перемешивание, дости гаемое либо перемешиванием, либо встряхиванием, чтобы разбить не смешивающуюся с водой жидкость на мельчайшие капельки. При этом каж дая капелька окружается препарато фермента, прилипая к его поверхно сти. Такое контактирование /препарата фермента с не смешивающейся с водой жидкостью может быть осущест лено до, во время или после проведения ферментативной реакции. Пред почтительно осуществлять его до проведения ферментативной реакции путем диспергирования (посредством перемешивания) препарата фермента не смешиваемой с водой жидкости в водной среде, содержа11ей субстрат. При этом в ходе ферментативной реакции препарат ассоциирует с капельками не смешивающейся с водой жидкости, которая в виде эмульсии присутствует в водной среде, и дисперсное состояние поддерживается во время реакции путем перемешивания. Когда требуется отделит препарат фермента, перемешивание ,прекращается. Возможен и другой вариант, по которому препарат фермента присутствует в водной реакционной смеси в виде раствора или суспензии, а после проведения реакции производят контактирование реакционной среды с не смешивающейся с водой жидкостью для отделения препарата фермента от продукта реакции; оста(ощегося в водной фазе. Механизм ассоциации препарата фермента с не смешивающейся с водой жидкостью меняется, он зависит от полимера и/или неполярных групп, введенных в препарат фермента. Например, препаратами фермент/полимер содержащими в основном сшитые молекулы, образуются твердые агрегаты и эти частицы прилипают к капелькам не смешивающейся с водой жидкости. Однако у препаратов, не содержащих сшитых солекул, типичными из которых ЯВЛЯЮТС5 описанные носители гантрез (и не содержащие полимеров препараты), обычно не наблюдается большого прилипания твердых частиц к капелькам, и на капельках образуется мономолекулярный слой препарата фермента. Это особенно выгодно, когда ферментативную реакцию осуществляют после первоначального контактирования препарата с не смешивающейся с водой жидкостью. каков бы ни был механизм ассоциации, контакт прерапата фермента с не смешивающейся с водой жидкостью (обычно в форме капель) позволяет отделить препарат фермента от водной среды. Капельки, покрытые препаратом фермента, имеют обычно такую же плотность, как и не смешивгиощаяся с водой жидкость. После прекращения перемешивания капельки либо плава эт на поверхности водной среды (если плотность жидкости меньше плотности воды), либо опускаиотся на дно (если хидкость более плотная). Так образуется разделительный слой, содержащий не смешивающуюся с водой Жидкость, ассоциированную с препаратом фермента, которую легко отделить от водной среды. Жидкости, которые не всплывают ли не оседают достаточно быстро з водной среды, могут быть отделеы другими способами, например ценрифугированием или путем пропускаия смеси через водоотталкивающий ильтр, через который проходит водая среда, а не смешивающаяся с одой жидкость (и вместе с ней преарат фермента) остается на фильте и таким образом отделяется от одной среды. Предпочтительным ферентативным процессом является олучение б-аминопенициллиновой ислоты из пенициллина с применением репарата на основе фep :eнтa пенииллин ацил азы. В этом случае фермент пенициллинацилаэу предпочтительно получают из бактерий, например, штаммов Eseherichia соП для расцепления бенэилпенициллина, или грибков actinomycetes для расщепления феноксиметилпенициллина. Эти ферменты хорошо известны, они применяются для получения б-аминопенициллиновой кислоты в интервале рН 6,0-9,0, предпочтительно 7,7-8,5 Так как деацилирование пенициллина приводит к выделению свободной кислоты из пенициллиновой части цепи, необходимо поддерживать рН в указанном пределе в течение процесса получения 6-аминопенициллиновой кислоты путем добавления, если необходимо, щелочи, например расвора гидроокиси натрия или аммония или триэтиламина.
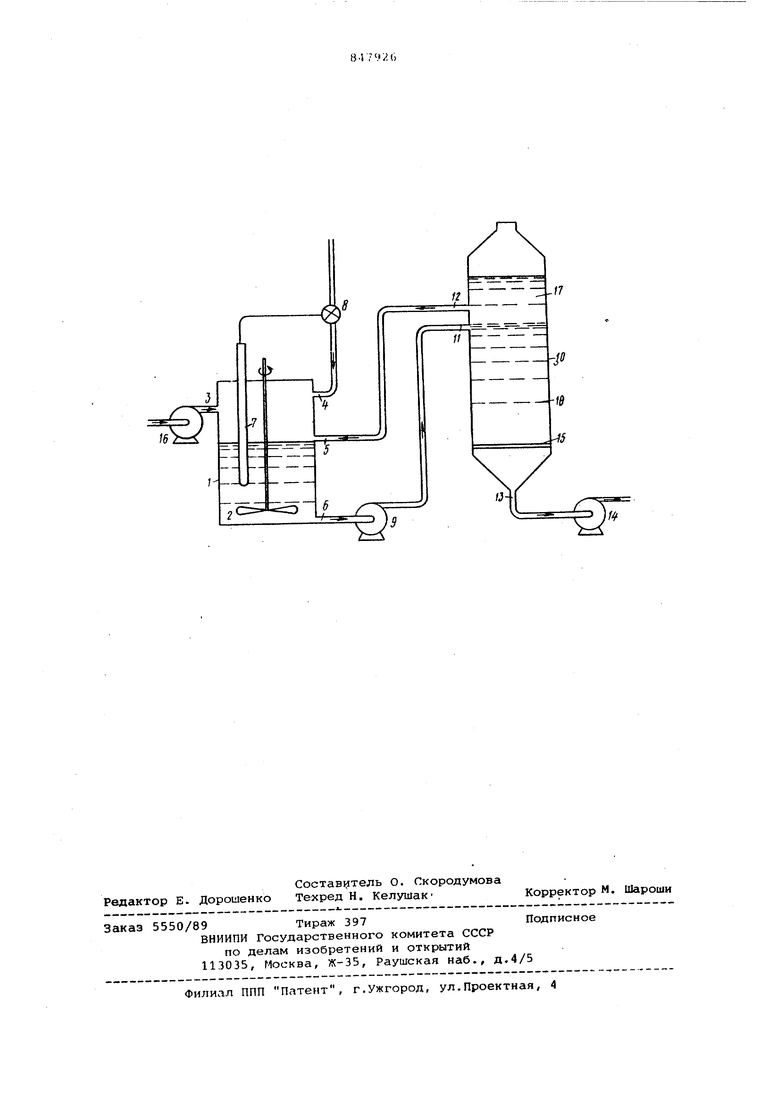
Особенным преимуществом применения препаратов ферментов по изобретению является возможность их применения для непрерывного способа осуществления ферментативных реакций в реакторе, схема которого представлена на чертеже.
Схема содержит реакционный сосуд 1, оборудованный перемешивающим устройством в виде мешалки .2, впускными отверстиями 3, 4 и 5 для субстрата, щелочи и фермента соответственно и выходным отверстием 6. Сосуд 1 имеет также пробник 7 для определения рН, который управляет клапаном 8, соединенным с впускным отверстием 4 для щелочи. Из выходного .отверстия 6 жидкость посредством насоса 9 поступает в делительный сосуд 10 через впускное отверстие 11. Делительный сосуд имеет верхнее выходное отверстие 12, связанное с впускным отверстием 5 реакционного сосуда,.нижнее выпускное отверстие 13 для удаления продукта реакции посредством насоса 14 и фильтр в виде стеклянного агломератора 15, расположенный над нижним выпускным отверстием 13. Во впускное отверстие 3 субстрат подается насосом 16. Насосы 14 и 16 спарены и работают с одинаковой производительностью.
Во время работы реакционный сосу 1 загружают .через впускное отверстие 3 субстратом в водной среде вместе с препаратом фермента по изобретению, и не смешивакяцейся в воде жидкостью. Смесь диспергируется мешалкой 2 для получения эмульсии. Сосуд 1 может поддерживаться при постоянной температуре при помощи, например, водяной рубашки (не показана), окружающей еосуд. Щелочь добавляют в сосуд к эмульсии через впускное отверстие 4, и скорость добавления регулируется клапаном 8, управляемым пробником 7, так, что рН реакционной среды
поддерживается в оптимальном для реакции интервале. В ходе реакции работает насос 9 и смесь удаляется через выпускное отверстие 6 и поступает в .делительный сосуд 10 через входное отверстие 11. В сосуде 10 не смешивающаяся с водой жидкость (в данном случае менее плотная, чем вода) образует отдельный верхний слой 17, содержащий пренарат фермента. Нижний водный
0 слой 1В, содержащий продукт реакции, удаляется через фильтр 15 и выпускное отверстие 13 насосом 14.
Верхний слой 17, содержащий препарат фермента, ассоциированный с
5 не смешивающейся с водой жидкостью вытекает через выпускное отверстие 12 и возвращается в реакционный сосуд 1 через впускное отверютне 5. По мере удаления продукта через выпускное отверстие 13, в сосуд 1
0 через впускное отверстие 3 при помощи насоса 16 подеиот дополнительное количество раствора субстрата. Работа насосов 14 и 16 скоординирована так, что скорюсть подачи раст5iBopa субстрата равна скорости вы1текания раствора, содержащего продукт реакции.
Скорость потоков, продолжительность нахождения в реакционном со0суде и другие параметры реакции зависят от данной системы препарат фермента/субстрат. По предварительным результатам можно заключить, что в реакторе емкостью 10000 л с приме5нением 1000 кг препарата фермента на основе пенициллинацилазы, привя- . занной к носителю гё1нтрез AN 149, можно достигнуть производительности около 2000 кг/сут пенициллина G с 5%-ной концентрацией (вес/объем)
0 при 27Рс и рН 7,8, хотя эти данные могут быть улучшены, если применять препарат фермента большой удельной активности. ,
ПолН чение некоторых препаратов
5 ферментов и их использование для получения продуктов ферментатив- . ных реакций, получение и свойства некоторых комплексов фермент/полимер рассматривается в примерах.
0
Ферменты обозначены: L - липаза, РА - пенициллинацилаза, Т - трипсин.
Носители: Г - фикол, GAN - гантрез, SR - сефароза (Sepharose), Т2000 - декстран Т.2000.
5
Связывакяцая группа фермент/носитель: ИД - 1,6 диаминогексан.
Неполярные группы: Д - п-дециламин, ДД - п-додег,иламин, ОД -. п-окТсщециламин.
Связьргоощие агенты, активирующие
О агенты и- активированные группы: CMC - 1-циклогексил-З-(2-мopфoлинэтил)-кapбoдиимид-мeтaI:-n-тoлyoлcyльфoнaт, G - глутаровый альдегид, А ги; разид. Цифра, стоящая после условного обозначения, указывает номер партии .Например, препарат фермента в примере 1 обозначен РЛРНДДС. Следовате прно, фермент - пенициллинацилаза (РА).Носитель - фикол (F), содержащий группы гексаметилендиамина (НД) для привязки к ферменту, а так же дециловые группы (д) в качестве неполярных групп. Привязка свободной аминогруппы гексаметилендиамина к феЕ менту осуществляется при п мсхци глутаровогр альдегида. Точно так же , GAN 149 НДОД обоз начает модифицированный носитель, полученный из гантрез 149, модифицированного 1,6-диаминогексаном и октадециламином, а PAGAN 149 НДОД - G обозначает вышеприведенны модифицированный носитель, к котор му привязана пенициллинацилаза в присутствии глутарового альдегида. Пример 1.а п-Дециламин, (6-аминогексиламин)-фикол(РНДЦ-1). Фикол (5 г) растворяют в воде (300м и доводят рН до 11,0. Добавляют бро циан (1 г) и во время реакции рН поддерживают постоянным путем добав ления 2 н. NaOH. Реакция продолжается 20 мин при комнатной температу после чего ее прекрацгиот и добавля ют раствор ч1-дециламина (4 г) и 1,6-диаминогексана (6,5 г) в метаноле (100 мл). Доводят рН до 9,5 при помо1ци 2н. НС1 и смесь перемеши вают в течение 16 ч при . Образовавшийся коллоидный раствор отцентрифуговывают и получают белую гранулу (вес во влажном состоянии 35 г). б Пенициллинацилаза п-дециламин, (б-аминогексиламин)-фикол ,(РАРНДДа-). РНДД-1 (вес во влажном состоянии 21,6 г) смешивают с буферным раствором 0,1 М фосфата натрия рН 6,0 (20 мл) и рН доводят до 6,2 при помощи 2 н. НС1. Раствор пенициллинацилазы Е. coli (50 мл, 340 мг частично очшденного белка) доводят до рН 6,2 при-О С и добавляют глутаровый альдегид (5 мл, 25% (вес/ объем) водный раствор). После перемешивания при 0°С в течение 30 мин добавляют суспензии РНДД-1. Смесь перемешивают при в течение 3 ч и при в.течение 4 ч, затем охлаждают (до - 20°С) и выдерживают в течение 16 ч. После оттаивания суспензию центрифугируют при 20000 g (4°С) 45 мин и получают коричневые гелеобразные гранулы. Гель повторно суспендируют в буферном 0,1 М растворе фосфата натрия рН 7,0 (30 мл) и повторно центрифугируют при 20000 g ( ) 30 мин. Всплывшие верхние слои соединяют, и коричневые гранулы (вес во влажном состоянии приблизительно 6,3 г) суспендируют в буферном 0,1 М растворе фосфата натрия рН 7,0 и хранят при 4с. При смешивании суспензии гранул по меньшей мере с таким же весовым количеством п-деканола они всплывают с деканолом на поверхность водного раствора, и остается нижний слой, не содержащий частиц фермента. Ферментативная активность гранул, верхнего слоя и исходного фермента приведена в табл.1. Регенерировано приблизительно 43% начальной активности Фермента, и фактически вся - в конъюгате. Активность комплекса может быть существенно регенерирована из растворов пенициллина либо путем флотации либо обычным центрифугированием. Таблица












| название | год | авторы | номер документа |
|---|---|---|---|
| Препарат фермента | 1977 |
|
SU687080A1 |
| Способ получения антибиотика, обладающего -лактамазной ингибирующей активностью | 1975 |
|
SU576965A3 |
| Способ получения 6-аминопенициллановой кислоты | 1973 |
|
SU578834A3 |
| Способ получения антибиотиков | 1975 |
|
SU528038A3 |
| Способ получения антибактериального вещества | 1975 |
|
SU648117A3 |
| Способ получения водонерастворимого ферментного препарата | 1973 |
|
SU499813A3 |
| Способ получения антибиотика, проявляющего активность в отношении -лактамазы | 1976 |
|
SU581881A3 |
| Способ получения 6-аминопенициллановой кислоты | 1974 |
|
SU654170A3 |
| Способ получения 6-метокси- -карбоксипенициллиной или их солей | 1976 |
|
SU623519A3 |
| Способ получения аминов клавулановой кислоты или их солей или сложных эфиров | 1976 |
|
SU639454A3 |
Повторные анализы
Методика 1 Активность пенидиллинацилазы в РЛРНДЦО - 1. Методика анализа: пенициллин G (натриевая соль) растворяют в соот ветствуюцей концентрации в 20 мл буферного раствора 0,02 М фосфата натрия рН 7,8 при и добавля бт пробу фермента. рН поддерживают постоянным путем добавления 0,1 ил 0,5 H.NaOH и активность выражают как начальную скорость образования 6-аминпенициллиновой кислоты (б-ЛРА При повторном анализе 1,2 млп- де канола добавляют к перемешиваемой реакционной смеси и после определе ,ния суспензию оставляют для разделения на два слоя. Верхний слой удаляют шприцем и используют повторно. В методике 2 конечную суспенз центрифугируют при 20000 д/15 мин и гранулы используют повторно. Пример 2.а п-Октадециламин, (б-амингексиламин)-фикол (РОДН Д-1). Фикол (5 г) растворяют в воде (300 мл) и раствор обрабатывают бромцианом (1 г) при рН 11,0 как описано. Через 20 мин рН раствора доводят до 10,0 с помощью 2 н НС1 и добавляют раствор п-октаде.циламина (5 г) и 1,б-диамингексана (0,5 г) в метаноле (20 мл). рН раст вора снова доводят до 10,0 с помощью 2 н.НС1, смесь перемешивают при в течение 72 ч. Продукт центрифугируют при 20000 д(4с)45 мин.Пол чают белые рыхлые пванулы(25 мл ост лены) и прозрачный верхний слой с по верхностной пленкой(октадециламин). б Пенициллинацилаза fn-октаде циламин, (6-амингексиламин)-фиколЗ (РАРОДНДй-). рН пенициллинацилазы (25 мл, 170 мг белка) доводят до 6,2 при комнатной температуре, затем добавляют глутаровыП альдегид 2,5 мл, 25% (вес/объем). Смесь перемешивают при комнатной температуре в течение 15 мин, добавляют РОДНД(10 мл) и доводят рН суспензии до 6,2 с помощью 2 н.. НС1 . После перемешивания при 4с в течение 3 ч материал центрифугируют при 20000 g (.С) 30 мин. Розовые гранулы повторно суспендируют в буферном 0,1 Н растворе фос.фата натрия рН 7,0 и хранят при 4С. Суспензия .заметно всплывает при смешивании с. п-декано лом, ее активность приведена в табл.2. Таблица 2 Исходный 1 мл 0,885 фермент 0,5 г 0,940 Продолжение таблицы Повторные анализы (ме одика 2) -Восстановление активности составляет приблизительно 60%. Пример З.а п-Дециламин, (б-амингексиламин)-декстран Т2000 (Т 2000 НОО-1). Декстран (средний мол.вес 2000000) (10 г) растворяют в воде (800 мл) и активируют бромциачом (2,5 г), как описано. Через 20 мин добавляют п-дециламин (5 г) и 1,6-диамиигексан (1,0 г) и доводят рН до 10,0. Раствор перемешивают при 40с в течение 16 ч и затем центрифугируют при 20000 g (40с) 1-1/2 ч. Образовавшиеся гранулы повторно суспендируют в 0,1 н. НСJ (100 мл) и снова центрифугируют. Гранулы (вес во влажном состоянии 50 г) оставляют. б Пенициллинацилаза п-дециламин, (6-амингексиламин)-декстран Т 2000 РАТ 2000 НДЦС-1. Т 2000 НДЦ-1 (10 Г ВО влажном состоянии)- гомогенизируют в буферном 0,1 М растворе фосфата натрия рН 6,0 и рН суспензии до 6,2, рН пейициллинацилазы (50 мл, 340 мг белка) доводят до 6,2 и добавляют глутаровый альдегид 5 мл, 25% (вес/ объем). Этот раствор перемешивают в течение 45 мин при 4с и затем дoбaвJfe ют т 2000 НДД-1. Смесь перемешивают при 4Ос в течение 30 мин и затем оставляют при -20°с в течение 16 ч. После оттаивания суспензию перемешивают при комнатной температуре в течение 1 ч и затем центрифугируют (6000 g (220С) ЗОмин) Всплывший слой оставляют, и гранулы повторно суспендируют в 0,1 М фосфате натрия рН 7,0 (75 мл) и гомогенизируют. После центрифугирования, как прежде, последующего повторного суспендирования и следующего центрифугирования получа от коичневые гранулы ( во влажном остоянии 8,0 г). Они хорошо сплывают при смешиваиии с п-деанолом и хуже при смешивании с -гептаном. Активность препарата приведена табл.3.
Таблица
PAT 2000 НДДа-1
Всплывший слой PAT 2000 НДЦС-1
Повторные анализы (методика 2) ,
Приблизительно 10% первоначальной активности фермента сохраняет.ся в конъюгате.
Пример 4. а п-Дециламин, (6-амингексиламин) сефароза 4В (SRHOD-1).
Активированная бромцианом сефароза 4В 2 г Б сухом состоянии набухает в 50 мл н. HCt при в течение 15 мин и затем промывается на стеклянном фильтре мл 10 н. нее.. Гель добавляют к 0,1 М раствору бикарбоната натрия (20 мл), этанола (5 мл), п -дециламина 0,5г и 1,6-диаминогексана С0,1 г). Смесь медленно вращают при 4С в течение 16 ч и
Приблизительно 72% первоначальной активности сохраняется в конъюгате .
П р и м е р 5. п-Дециламинглу тараль-пенициллинацилаза (РАДДС-). рН пенициллинацилазы (40 мл,. 272 мг) доводят до 6,2 добавляют глутаровый альдегид 4 мл, 25% (вес/ объем). После перемешивания в течение 20 мин при добавляют п-деканол (6 мл)и п-дециламин (0,6 мл) и продолжают энергичное перемешивание при в течение 6 ч и при комнатной температуре в течение 1 ч. Смесь центрифугируют при 2000 д(22 С 10 мин, образуется коричневый верхний слой, который отделяется и хранится суспендированным в буферпод вакуумом 4x20 . HCt , .200 мл этанола и мл воды, затем повторно суспендируют в воде и хранят при .
б Пенициллинацилаза п-дециламин, (б-аминогексиламин)-сефароза 5 ,4В РА5«НДДС-1.
Высушенный под вакуумом гель SRHOD (1,5 г) суспендируют в буферном 0,1 М растворе фосфата натрия рН 6,0 (5 мл), затем добавляют глу0 таровыП альдегид (0,5 мл, 25% (вес/ объем) . Смесь перемешивают в течение 2 ч при комнатной температуре затем высушивают под вакуумом на стеклянном фильтре. Затем гель добавляют к смеси пенициллинацилазы (2 мл, 13,6 мг) и буферного 0,1 М раствора фосфата натрия рН 6,0 (З мл). Эту смесь перемешивают в течение 24 ч при 4°С, высушивают и
0 промывают на стеклянном фильтре 5X20 мл буферного (f, 1 М раствора фосфата натрия рН 7,0. Продукт суспендируют в 5 мл того же буферного раствора. Образовавшиеся шарики всплывают с п-деканолом (в противоположность сефарозе, которая осаждается из водных суспензий, содержащих п-деканол).
ном ОД М растворе фосфата натрия Н 7,0 (30 мл). Материал спонтанно
всплывает на поверхность водных суспензий, но он в меньшей степени диспергируется, чем полимерные конъюгаты, и проявляет тенденцию к образованию крупных агрегатов. Один мл суспензии, проанализированной как
в примере1, показывает активность 0, моль/мин, что соответствует 42% сохранения активности ацялазы в конъюгате.
Пример 6. Трипсин- п-октадециламин, (6-г1миногексиамин)-фикол (ТГОДНД -1).
Трипсин (50 мг,не содержащий солей) растворяют в буферном 0,1 М
растворе фосфата натрия рН 6,0
(20 мл) при . Добавляют глутаровый альдегид (1 мл, 25% (вес/объе и смесь перемешивают в течение 15 мин при . Добавляют РОДНД(10 мл) (пример 2 а) и рН раствора доводят до 6,2 при помощи 2 н. НС1 Смесь перемешивают в течение 16 ч при 4°С и затем центрифугируют при 20000 g (4°С) 30 мин. Гранулы повторно суспендируют в 20 мл воды и фильтруют с применением фаэоразделительной фильтровальной бумаги Ватман ИПС (alman IPS), Остаток на фильтре повторно суспендируют в 10 М НС1 (20 мл) и снова фильтруют. Затем два)Ц}ь1 проводят суспендирование (10 М ПС1) и центрифугирование (10000 g (14°С) 20 мин) и получают 2,65 г. коричневого геля Этот материал всплывает и с п-деканолом, и с п-гептаном.
Активность полученного материала измеряют следующим образом.
М-о /-бензил-01-аргинин-пара-нитроанилид растворяют в диметилформамиде (5 мл) и добавляют к 0,05 М буферному раствору рН 8,3 (содержащему 10 моль СаС) и получают раствор хромогенного субстрата. Добавляют г.-гептан (5 мл) и смесь энергично перемешиваиот при 220С. Добавляют пробу фермента и через равные интервалы отбирают 5 мл аликвоту. При недолгом стоянии или осторожном центрифугировании отделяется гептановый слой (вместе ТРОДНДа-). Нижний водный слой отделяют и измеряют ОД определения остаточного паранитроанилида. В некоторых случаях для измерения поглощения нижний слой разбавляют. После процедуры измерения гептановый и водный слои добавляют к пере 1ешиваемоЯ реакционной смеси. Общая активность, сохраняемая конъюгатом, составляет 17% от первоначальной активности.
Пример 7. Липаза- п-дециламин, (б-амингексиламин)-декстран Т 2000 (Т 2000 НДЦО-1).
Липазу из Candida Cykindracia (200 мг) растворяют в ОД М растворе фосфата натрия рН 6,0 (20 мл) и рН доводят до 6,2. Добавляют глутаровый альдегид (2 мл, 23% (вес ) и смесь перемешивают в течение 30 мин при комнатной температуре. Т 200 НДЦ-1 (пример 3) осаждают из суспензии этанолом и осадок оставляют повторно набухать в воде. 10 г образовавшегося геля гомогенизируют в 0,1 М растворе фосфата натрия рН 6,0 (20 мп) и добавляют к смеси фермент-глутаровый альдегид. рН доводят до 6,2 и смесь перемешивают при в течение 16 ч. Суспензию центрифугируют при 20000 g (4°С) 30 мин, гранулы повторно суспендируют в
0,1 и растфоре фосфата натрия рН 7,0 (50 мл) и повторно центрифугируют . Эту операцию повторяют еще два раза. Полученные гранулы (вес во влажном состоянии 8,4 г) суспендируют в 20 мл 0,1 М раствора фосфата натрия рН 7,0. Суспензия всплывает и с п-деканолом, и с п-гептаном.
Методика активности конъюгата состоит в следующем.
0
Пара-нитрофениллаурат (10 М в п-гептане), 5 мл, перемешивают с 0,1 М раствором фосфата натрия рН 8,0 (20 мл) при комнатной температуре. При этих условиях неЛерментный
5 гидролиз сложного эфира не наблюдается в течение 1 ч или более. После добавления фермента равные промежутки времени отбиргцот 5 мл аликвоты, центрифугируют при 4000 g 1-1/2 мин и нижний (подный) слой
0 отделяпт.
О Д -1A-S,.;. определяют точно чеHVJvl Г1 М
3 мин после отделения аликвот. -Затем водный и гептановыП слои добавляют к перемешиваемой смеси. (Так
5 как в данном эксперименте субстрат содержится в неводной фазе, скорость перемешивания должна оставаться постоянной в т&чение всего эксперимента) .
0
рриблизительно 1% начальной активности липазы обнаружено в конъюгате.
8,
а GAN 149
Приме НДОД-1.
5
п-Октадециламин (1 г) и 1,6 диамингексан (0,2 г) суспендируют в метаноле 10 мл и добавляют к буферному 0,2 М раствору фосфата натрия рН 8,0 (200 мл), содержаще0му метанол (40 мл), Добавляют гантреэ 149 (2 г) маленькими порциями при энергичном перемешивании. Смесь перемешивают в течение 16 ч при комнатной температуре и затем центрифугируют при 12000 g в течение 1ч
5 при комнатной температуре. Поверхностную пленку и верхний слой отбрасывают, а гранулы повторно суспендируют в буферном 0,1 И растворе фосфата натрия рН 7,0 (100 мл) и
0 повторно центрифугируют, как указано, Гелеобразные гранулы (80. г) хранят при 4°С.
б PAGAN 149 НДОД-1-G.
рН раствора пенициллинацилазы
5 (20 мл, 1, единиц, 136 мг белка) доводят до 6,2 и добавляют глутаровый альдегид (2 мл 25% (вес/ объем) в воде. Смесь перемешивают при комнатной температуре и добавляют суспензию GAN 149 НДОД-1 (10 г)
0 в буферном 0,1 М растворе фосфата натрия рН 6,0 (12 мл). Поддерживают рН 6,2 и продолжгцот перемешивание при комнатной температуре в течение 2 ч. Суспензию центрифугяру5
ют в течение 1 ч при 20000 g и и гранулы (14 г) сохраняют. Гранулы повторно суспендируют в 0,1 М растворе фосфата натрия рН 7,0 (10 мл) и повторно отделяют центрифугированием. Гранулы хранят при
дОр
--б
Удельная активность: 3x10 молей
б-АРА мнн (5% пенициллин G, рН 7,8, 22С).
Активность после регенерации: 21 Всплывает с п-деканолом или с п-деканом.
П р и м е р 9. а GAN 149 НДОД-2
п-Октадециламин (5 г) растворяют в этаноле (50 мл) и добавляют к буферному 0,1 М раствору фосфата натрия рН 9,0 (1 л), содержащему этанол (200 мл). Добавляют гантрез 149 -(5 г) маленькими порциями в течение 45 мин при перемешивании. рН поддерживают в интервале между 8 и 9 припомощи 2 н. МаОН. После добавления гантреза добавляют 1,б-диаминогексан (5 г) и смесь перемешивают при комнатной температуре в течение 6ч. рН понижают до 3,0 при помощи концентрированной HCI и суспензию центрифугируют при 12000 g в течение 1 ч при комнатной температуре. Верхний слой отбрасывают и гранулы повторно суспендируют в воде (550 мл) и повторно центрифугируют, получают белые гранулы (150 г), которые хранят при 4°С.
б PAGAN 149 НДОД-2-G.
Раствор пенициллинацилазы (100 м 9,8x10 единиц, 88 мг белка смешивают с GAN 149 НДОД-2 (15 г), буферны 0,1 М раствором фосфата натрия рН 6,0 (20 мл) и 2-% (вес/объем) азидом, натрия (1 мл) и быстро гомогенизируют при 0°С. рН смеси доводят до 6,8 и оставляют при в течение 4,5 ч, затем центрифугируют при 20000 д(4°С) 1 ч.
Гоанулы повторно суспендируют в 0,1 М растворе фосфата натрия рН 7,0 и повторно центрифуг1 руют, Эти операции повторяют, получают конечный продукт в виде гранул, вес 11,2 хранение при 4С.
Удельная активность: 1,8x10 молей 6-АРА мин г .
Активность после регенерации: 55%. Всплывает с п-деканолом или п-деканом.
Пример 10. Трипсин (п-дециламино гантрез АН 119) (TGAN 119Д
Гантрез AN 119 (1 г) растворяют диметилформамиде (ДМФ) (5 мл) и энергично перемешивают. Добавляют ri-дециламин (0,3 г) в ДМФ (1 мл), и раствор становится более вязким. После перемешивания в течение 10 ми раствор по каплям добавляют к раствОру бычьего трипсина (300 мл) в буферном 0,1м растворе фосфата натрия, рН 8,0 (60 мл), при 22°С. рН
поддерживают постоянным при помощи 2 и. NaOH, и после того как расход титранта прекращается, добавляют хлорид натрия (4 г). Образовавшуюся суспензию выливают в буферный „ 0,1 М раствор фосфата натрия, рН 7,6 (175 мл) и охлаждают до 4с. в течение 20 мин. Смесь центрифугируют при 25000 д(4°С) 1 ч и гелеобразные гранулы повторно гомогенизируют в буферном 0,1 М растворе фосфата натрия. рН 7,6 (30 мл) Повторяют центрифугирование и затем цикл гомогенизации (центрифугирование повторяют еще дважды, получают конечный рыхлый гель (7,5 г). Активность определяется спектрофотометрически. В качестве субстрата берут N-бензил-L-аргинии-паранитроанилид (BANA), 2 моль в буферном 0,1 М растфоре веронала, рН
0 8,3, при 22с. Одна единица соответствует повышению оптической плотности на 0,001 в мин при 400 нм. Так как гель вызывает значительное рассеяние света, необходимо периодически встряхивать кювету (длина кюветы 1 см).
Активность геля: 168 единиц/мг во влажном состоянии, 2000 единиц/мг |В сухом состоянии (лиофилизованный гель).
/активность после регенераций: 24,2%.
Иммобилизация ферментного конъювгата на капельках растворителя.
2 Гель (1 г во влажном состоянии) гомогенизируют в небольшом дефибре с п-деканом (25 мл) и буферным 0,1 М растворе фосфата натрия, рН 8,0 (10 Nin) при 4000 об/мин и в течение- 5 мин. Образовавшуюся молочную сг спензию после трехчасового
стояния при разделяют на верхний слой, состоящий из плотно упакованны : капелек декана, и нижний водный слой. Оба слоя анализируют
5 описанным спектрофотометрическим методом. Кювету встряхивают через равные промежутки времени при анализе верхнего слоя и измеряют среднюю скорость увеличения оптической
0 плотности.
Активность верхнё й фазы: 2280 единиц/мл. Активность нижней фазы: 57 единиц/мл.
Так как при измерении предлагаемым методом кажущаяся активность верхней фазы, по-видимому, гораздо меньше ее истинной активности, получаемый результат показывает, что по меньшей мере 90% активности трипсина с капельками растворителя.
0 Пример 11. Пенициллинацилаза (п-октадециламин гантрез AN 119) (PAGAN 119 ОД). .
Гантраз AN 119 (0,5 г) растворяют в диметилформамиде (2,5 мл) и
5 подогревают до 80С на водяной бане., Октадециламин (0,28 г) также растворяют в диметилформамиде (2,5 мл) при и быстро добавляют к еще горячему раствору гантреза. Смесь перемешивают и добавляют пиридин (0,1 мл). После охлаждения до комнатной температуры в течение 1 ч весь раствор добавляют к пенициллинацилаэе (175 мг) в воде (30 мл) при рН 7,0 и быстро гомогенизируют при 0°С. Затем смесь розового цвета перемешивают при в течение 3 ч, рН поддерживают постоянным при 4 С в течение 16 ч, растворяют хлорид натрия (5,8 г) в суспензии и смесь центрифугируют при 25000 g () 1 ч. Образовавшиеся гранулы малиновой окраски весят 4,О г и повторно суспендируются в буферном 0,1 М растворе фосфата натрия, рН 7,0 (20 мл). Удельная активность этой суспензии, определенная с помощью 5% (вес объем) пенициллина G в буферном 0,02 М растворе фосфата натрий, рН 7,8 (20 мл), при 25°С, составляет 4,0x10 моль/мин/МП. Это соответствует приблизительно 70 % регенерированной активности в конъюгате Описанную суспензию (4,0 мл) сме шивают с буферным О,02 М раствором фосфата натрия (20 мл) и п-деканолом (5 мл) и гомогенизируют при 4000 об/мин в течение 5 мин при О С Эмульсию центрифугируют при 100 g и комнатной температуре в течение 20 мин и разделяют на верхнюю органическую эмульсию и никний вод.ный слой. Нижний слой оставляют, а верхний слой повторно суспендируют в буферном 0,02 Н растворе фосфата натрия (20 мл) и повторно центрифугируют . Первоначальный нижний слой имеет общую активность 1, мол мин, а промытый верхний слой - 9,5 ЛО моль/мин. Отношение удельной активности (на основе объемов) составляет 21,6 (верхний слой/нижний слой). После определения активности описанньвя методом органический слой обрабатывают повторным путем осторожного центрифугирования и отделения от водного сЛоя. Активность показана в табл.5. Таблица 5
Во втором эксперименте 2 мл растff вора конъюгата фермента гомогенизируют при тех Же условиях с п-деканолом (2,5 мл) и буферным 0,02 М раствором фосфата натрия (10 мл). После центрифугирования и проьывки тем же буферньм раствором верхний 60 слой имеет активность 1,8x10 моль/ мкн, а первоначальный нижний слой имеет активность 8,0«10 моль/мин. Отнесение удельной активности (верх-,иийслой/нижний слой, на основе 65объемов) составляеЬ 7,5. Пример 12. Пенициллинацилаза (п-додециламин-гантрез AN 119) (PAGAH 119 ДД). п-ДодецилаМингантрез AN 119 (0,25 г) растворяют в диметилформамиде (1 мл) и добавляют к раствору пенициллинацилазы ( мг в 15 мл буферного 0,1 11 раствора фосфата натрия, рН 7,0). После недолгой гомогенизации прИО с суспензию перемешивают в течение 16 ч при 4°С. К гомогенному раствору добавляют при 4Ос.сульфат .аммония (7,8 г), поддерживая постоянный рН 7,0 при помощи 1 М раствора карбоната натрия. После растворения сульфата аммония образуется мутная суспензия. Этот материал центрифугируют при 25000 g () 1 Ч. Гранулы повторно суспендируют в растворе сульфата аммония (7,8 г)в воде (30 мл), рН 7,О,и повторно центрифугируют. Продукт в виде гранул весит 1,36 г и повторно растворен в воде (10 мл) с образованием вязкого раствора. Активность гранул (определяют описаинЕЛ методом) составляет 1,62 10моль/мин/г веса во влаэхном состоянии, что составляет 80% регенерированной активности. ИммобилизащЛ водорастворимого конъюгата на капельках п-деканола. 1 мл указанного раствора растворяпт в 0,02 М буферном растворе фосфата натрия (20 мл) и гомогенизируют с п-деканолом (10 мл) при 4000 об/ мин в течение 3 мин при . Эмульсию разделяют на слои путем центрифугирования при 300g .в течение 10 мин. Верхний слой имеет общую активность 1,14х10 моль/мин, и его активность после повторения последовательных циклов показана в табл.6. Таблица 6
Прлмер 13. Трипсин (гидролизoвaнн ;Jй п-октадециламин гантреЭОМ 149) (ТСЛМ 149 ДСИС).
Набухший гель полимера (15 г) смшивают с буферным 0,1 М раствором фосфата натрия, рН 7,0 и рН доводят до 4,7 при помощи 2 н. HCI, Добавляют 1-циклогексил-3-(2-морфолинэтил) карбодиимид-мета-пара-толуолсульфонат (CMC, 0,5 г) и поддерживают рН 4,7 при помощи 2 н. НС1, .после пятиминутной обработки при комнатной температуре добавляют бычий трипсин (-75 мг в буферном 0,1 М растворе фосфата натрия, рН 7,0, 2 мл), доводят рН смеси до 5,9 и поддерживают это значение рН в течение 1 ч при помощи 2 н. NaOH. После трехчасовой обработки при комнатной температуре гель центрифугиру1от при 25000 g () 30 мин. Гранулы повторно суспендируют в буферном 0,1 М растворе фосфата натрия, рН 7,0 (40 мл), и повторно центрифугируют. Затем гранулы смешивают с описанным буферным раствором (15 мл) и п-деканом (10 мл) и быстро гомогенизируют. В результате этой процедуры происходит неполная дисперсия, и смесь подвергают действию ультразвука при 20 кГ и 5х 5 мин импульсов с периодическим охлалсдением во льду. После этого эмульсию отцентрифуговывапт при 100 д/15 мин. Верхний слой трижды промывают фосфатлым буфером (25 мл). Конечный объем верхнего слоя 11 мл, а активность (определяется, как в примере 3) 1050 SANA единиц/мл. Кажущаяся регенерированная активность 50%.
Пример 14. а Гидролизованный п-октадециламин гантрез AN 119 гидразид (GAN 1190ДН).
Гантрез AN 119 (5 г) растворяют в диметилформамиде (20 мл) и нагревают до 6О®С на водяной бане. До.бавляют раствор п-октадециламина (2,8 г) в горячем (),диметилформамиде (30 мл) и смесь поддерживают при температуре приблизительно в течение 1 ч. После охлаждения до комнатной температуры добавляют к смеси гидразингидрата (10 мл) и диметилформамида (50 мл) при комнатной температуре. Образовывается незначительный коричневый осадок, смесь перемешивают в течение 10 мин и добавляют воду (500 мл Получёиот мыльный зеленый раствор и после перемешивания в течение 10 мин рН доводят до 7,0 при помощи концентрированной НС1, затем добавляют хлорид натрия (50 г). Образуются хлопья и суспензию центрифугируют при 10000 g (4°С) 30 мин. Гранулы повторно суспендируют в воде (400 мл), перемешивают в течение 16 ч при 4Ос и затем отцентрифуговывают, как указано выше. Образуются серо-голубые гранулы (вес во влажном состоянии 39 г) б Пенициллинацилаза (п-октадециламин гантрез AN 119)-гидразнд, связанный PAGAN 119ОДН.
5 GAN 119ОДН (5 г во влажном состоянии) суспендируют в 2 н. HCI (50 мл) и перемешивают в течение 15 мин при 0°С. Добавляют раствор нитрата натрия (0,5 г) в воде (5 мл) и смесь перемешивают при (Яс в течение 10 мин, за-. тем центрифугируют при 25000 g () 30 мин и гранулы повторно суспендируют в буферном 0,1 Н растворе Лосфата натрия, рН 8,0 (50 мл), при .
5 Эту суспензию добавляют к раствору пенициллинацилазы (100 мл, 55 мг белка) и доводят рН до 8,0. Суспензию перемешивают при в течение 72 ч и затем центрифугируют при
0 25000 g () ч, повторно суспендируют в буферном б, 1 М растворе фосфата натрия, рН 7,0 и повторно центрифугируют. Вес гранул 3,74 г. Удельная активность (анализ выпол, няют по описанной методике): 5,2х 10 моль/мин/г. Восстановленная активность: 32,5%.
Суспензию указанного конъюгата в буферном 0,1 М растворе фосфата натрия, рН 7,0 (0,92 г/мл, 2,5 мл)
0 смешивают с буферным 0,02 М раствором фосфата натрия, рН 7,8 (20 мл) и гомогенизируют с п-деканолом (7,5 мл) при в течение 2 мин, затем отцентрифуговывают при скорости 5000 об/мин (100 g, 10 мин), при этом образуется 2 слоя. Общая активность нижнего слоя 1, моль/ мин, а активность верхнего слоя после промывки указанным буферным
0 раствором (20 мл) 1,9)10 моль/мин. Отношение удельной активности (верхний слой/нижний слой, на основе объемов) составляет 30.
Результаты повторного цикла обработки верхней Фазы приведены в
табл.7.
Таблица 7
Активность, %
Цикл, №
90 84 74
П.ример 15.. Ферментативное расщепление бензилпенициллина для получения 6-аминопенициллиновой 60 кислоты.
1,0 г гантреза AN 119 растворя1ЭТ в 10 мл диметилформамида и добавляют 0,56 г октадециламина. Смесь перемешивают в течение нсчи при комнатной температуре.
48 wi частично очшчсиного препар.та пеинииллннацилази лонодпт до рН 9,0 фн ПОМО1ЧИ 1 М растфора карбоната натрия. Затем добавляют раствор смолы гантрез двумя порциями при гомогенизации и поддержании постоянного рН в интервалах между последовательными добавлениями смолы. Затем смесь перемешивают в течение 3 ч, поддерживая рН 9,0 путем добавления 1 М раствора NaOH.
Затем систему фермент-смола регенерируют путем центрифугирования, повторно суспендируют в 120 мл буферного 0,1 М раствора фосфата натрия, рН 7,0 и гомогенизируют в течение 30 с. Систему фермент-смола регенерируют путем-центрифугировани и повторно промывают.
20 г геля Лермента гомогенизируют с 80 мл п-декана и 20 мл буферного 0,02 М фосфата, рН 7,8, в течение 1 мин. Гомогенат доЪавляют к 25 мл дистиллированной РОДЫ. Смесь подогревают до , рН доводят до 7, и добавляют бензилпенициллин калия (21,3 г). Смесь перемешивают в течение 5 ч и поддерукивают рН 7,8 путем добавления 4% (вес/объем) раствора NaOH. К концу реакции смес выливают в делительную воронку, и фермент отделяется от водной фазы Это продолжается 30 мин. Затем воднып слой удаляют и извлекают аминопенициллиновую кислоту следующим способом.
Лидкость упаривают до 1/4 объема охлахадают до и при перемешивании добавляют равный объем метйлизобутилокетона. рН поних ают до 4,3 путем добавления концентрированной азотной кислоты, при этом выпадает в осадок 6-аминопенициллиновая кислота. Твейдое вещество отделяют фильтрованием, прюмывают водой и ацетоном и сушат в течение ночи при 40°С.
Получают 7,65 г 6-аминопенициллиновой кислоты.
Формула изобретения
Cff-C Q , ЦИКЛОПЛКИЛ , ЦИКЛОалкенил С -С , арил, ,-алкил5арил, аралкил, обеспечиваю1-1ая ассоциирование препарата фермента с не смешивающейся с водой жидкостью,
5
5
Источники информации, принятые во внимание при экспертизе
J
n
w
/7
12
-(-
n
«-.
;5
