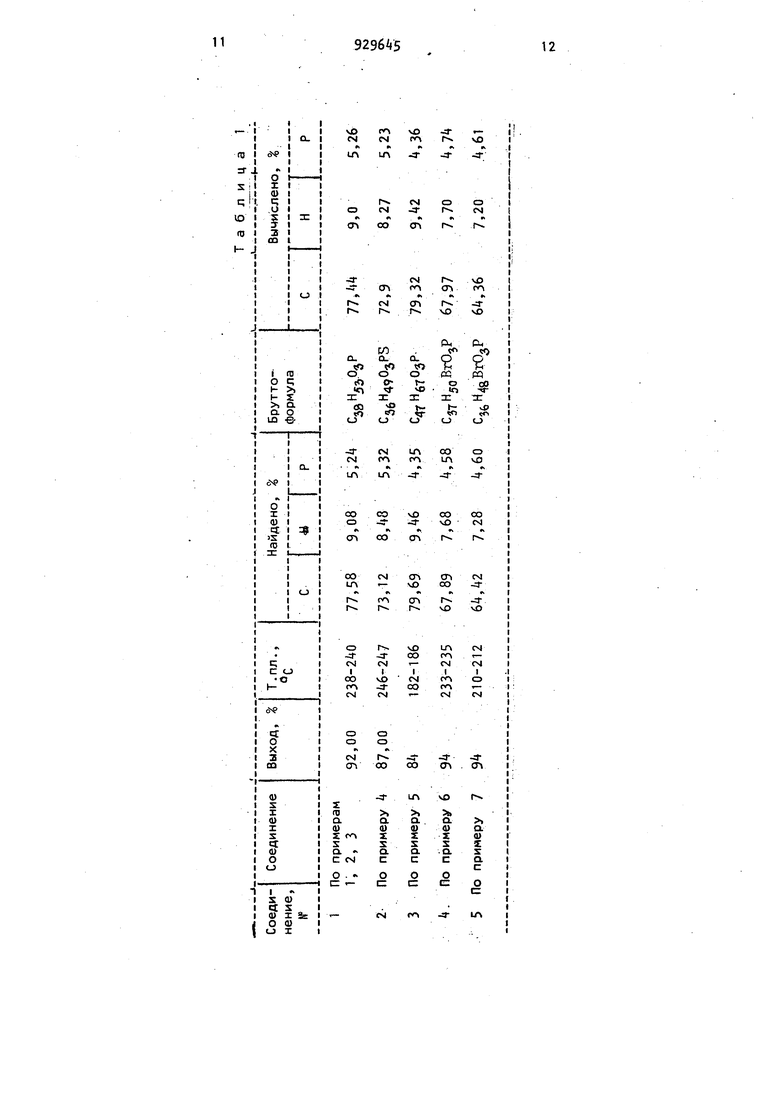
3 ми формулы I, являющимися ингибиторами крксообразования, которые позвЪляют значительно снизить коксообразовакие при пиролизе бензина. . Известен способ получения циклич ких фосфитов общей формулы где R и алкил или алкарил; алкил, арил или алкарил;X - метилен или сера, путем взаимодействия циклического хлорфосфита с гидроксилсодержащим соединением К2Ш, где Rj имеет указанные значения, в присутствии о , нования в среде органического растворителя, например бензола, толуола iгептана 33. Однако этот способ не пригоден для получения восьмичленных цикличе ких фосфитов формулы Т, так как их выход составляет лишь . Целью изобретения является разра ботка простого и доступного способа получения в.осьмичленных циклических фосфитов формулы I. , Поставленная цель достигается те что согласно способу получения вось мичленных циклических фосфитов форм лы I циклический хлорфосфит общей формулы где U и R,имеют указанные значения, подвергают взаимодействию с фенолом общей формулы (W) где R2И Rj имеют указанные значения в среде полярного апротонного раст.ворителя в присутствии основания при 80-100°С. В качестве полярного апротонного растворителя используют диметилформамид СДМФА) или ацетонитрил. ,4 , Предлагаемый способ позволяет получать новые вбсьмичленные циклические фосфиты с выходом до . , Способ отличается простотой, а использованные в реакции полярные апротонные растворители доступны. Для исключения образования побочнык продуктов и сдвига реакции в желаемом направлении органическое основание вводят в процесс в количестве на 1-5 более стехиометрически необходимого. Ввод компонентов на синтез лучше осуществлять в следующем порядке: к фенольному компоненту добавлять хлорфосфит, затем растворитель и, используя понижение температуры смеси на за счет теплоты растворения, вводить по каплям органическое основание за 15-20 мин. При этом температура реакционной массы повышается на и смесь можно сразу подогревать. В случае применения ацетонитрила, например, до 82°С (флегма),при использовании ДМФА нагрев кипящей водяной баней обеспечивает температуру реакционной массы . Возможно также вводить основание с растворителем-. Во всех случаях синтез ведут при интенсивном перемешивании в отсутствии контакта с влагой воздуха и s iдостаточно сухих растворителях.. Для выделения продуктов реакции опробовано несколько вариантов,дающих практически одинаковые вы.сокие выходы фосфитов. Так,при использовании ацетонитрила после окончания синтеза реакционную массу отпаривают в колбе роторного испарителя в техническом вакууме до (баня) от растворителя досуха. К остатку добавляют воду и при нагреве до 100°С в техническом вакууме хлоргидрат основания, растворяют, с спензию охлаждают, переносят на вакуум-фильтр, чтобы отделить кристаллический продукт от жидкой фазы фосфита, промывают водой до отсутствия хлор-иона, измельчают и сушат в вакуум-сушильном шкафу при 80-100°С (200-tOO мм рт. ст.) до постоянного веса. В случае применения ДМФА, После окончания выдержки реакционную массу охлаждают водой до или рассолом до , фосфит в виде осадка отфильтровывают на вакуум-фильтре, отмывают водой до отсутствия хлор-иона и сушат анологично. Маточник используют при последую щих операциях синтеза или отпаривают от растворителя. В последнем случае остаток, содержащий целевого продукта, растворяют в подходящем низкокипящем растворителе, не растворяющем хлор-гидрат основания, таком как петролейный эфир, гексан, гептан, хлорметаны, метиловый или этиловый спирты и т.д.,раствор фосфита отделяют от кристаллических остатков хлоргидрата основания фильтро ванием, охлаждают до температуры кристаллизации и фильтруют для выделения целевого- продукта. Второй вариант использован Также и при применении ацетонитрила. Способ не исключает применения и других возможных вариантов выделения целе.вых продуктов. Например, после синтеза реакционную массу охлаждают до комнатной тем пературы, фильтрованием отделяют от основного количества хлоргидрата основания, отпаривают от растворителя Для отделения от остатков хлоргидрата основания реакционную массу растворяют в петролейном эфире, а затем из охлаждаемого фильтрата выделяют целевой продукт и т.д. Новые фосфиты - кристаллические вещества с высокими температурами плавления, бесцветны или .слабожелтого цвета, не имеют запаха, по данным дифференциально-термического анализа их распад начинается при 2 0-320 0. Идентификация проведена определением молекулярных весов элементным анализом, ИК- и ЯМР-спектроскопией, тонкосл(;йной хроматографией. В ИК :пектрах полученных соединений отсутствуют полосы поглощения в области 3350-3640 см соответствующие валентным колебаниям ОН-групп фенолов, отсутствует также полоса поглощения в области +50 , характерная для связи Р-СГ. Вспектрах ЯМР на яд рах 31Р наблюдается сигнал с химическим- сдвигом 137-1+0 М.Д., характерный для фосфитов. Найденные значе ния элементного анализа и определения молекулярных весов соответствуют вычисленным значением. В табл. 1 представлены свойства восьмичленных циклических фосфитов. При тонкослойной хроматографии в системе гексан-ацетон-хлороформ (4:2:) на пластинках Silufol, проявитель перманганат калия или йод, значения получаемых фосфитов на 0,5-1 см больше исходных фенолов и хлорфосфитов. Пример. Получение -метил-2,6-дитретбутилфенилового эфира 2,2 -метиленбис(4-метил-6-третбутилфенил) фосфористой кислоты. К 107 г (0, моль) 2,2-метиленбис{Л-метил-6-третбутил-фенил)хлорфосфита в 150 мл диметилформамида добавляют 58,2 г (0,26 моль) I-MBтил-2,6-дитретбутилфенола (ионола) и при интенсивном перемешивании прикапывают 38 мл (0,275 моль) триэтиламина при 2б-Зб С. Реакционную массу выдерживают при 95-97С 4 ч, проверяя периодически рН среды (рН должен быть больше 7). Суспензию охлаждают до минус 10 С, отделяют осадок на вакуум-фильтре, получают 180 г белого кристаллического порошка.Последний переносят в 300 мл смеси воды со льдом, хорснио перемешивают. Осадок, остающийся после обработки водой, отделяют на вакуум-фильтре, промывают дистиллятом до отсутствия хлор-иона.После сушки получают 126 г кристаллического порошка це левого продукта. Органический маточник отпаривают до ДМФА до 150°С/5 мм рт. ст., к кубу добавляют 60-75 мл метанола, нагревают до кипения, охлаждают до минус 10 С и фильтрацией с последующей отмывкой от остатков хлор-иона и сушкой дополнительно выделяют 16 г кристаллического порошка. Общий выход фосфита 91, 23б-240 С иа октана. Свойства фосфита представлены в табл. 1, соединение 1 . П р и м е р 2. Получение А-метил-2,6-дитрет-бутйлового эфира 2,2 метилен-бис{ -метил-6-трет-бутилфенил) фосфористой кислоты. К раствору 0,15 моль ионала и 0,15 моль хлорфосфмта, указанного в примере 1, в 100 мл ацетонитрила вводят 20 мл триэтиламина при 2035°С, выдерживают реакционную массу k ч на кипящей водяной бане (82 С, легма) и отфильтровывают. Полученный кристаллический осадок промыват водой до отсутствия хлор-иона и сушат. Получают технический целевой продукт с т. пл. 188 С. Маточник тпаривают от ацетонитрила в техни79меском вакууме, остаток растворяют в петролейном эфире, отделяют от остатков хлоргидрата триэтиламина и кристаллизацией выделяют 25,5 г крис таллического порошка с т. пл. 186 С. Суммарный выход 92. После перекристаллизации из ДМФА получают продукт с т.пл, 238-2 0 С (табл. 1, соединение 1) . П р и м е р 3. Получение -метил-2,6-дитрет-бутилового эфира 2,2 -метилен-бис(-метил-6-трет-бутилфенил) фосфористой кислоты. К 0,2 моль ионола, 0,2 моль хлорфосфита, указанного в примере 1, приливают 3t,5 мл (0,25 моль) триэти амина в 150 мл ацетонитрила и выдерживают k.ч при флегмировании растворителя. После окончания выдержки сус пензию переносят в колбу роторного испарителя и отпаривают в техническом вакууме до 100°С (баня), возвращая 98 введенного ацетонитрила. К остатку добавляют 200 мл воды и нагревают до начала отгонки воды в техниМеском вакууме (температура бани 100-110 С). Суспензию охлаждают, отфильтровывают, промывают осадок водой от остатков хлор-иона, отжимают от. воды, сушат до постоянного веса, получают 117,9.г белого порошка. Пос ле перекристаллизации из октана получают целевой продукт ст. пл, 238240°С. Выход 92. П р и м е р 4. Получение 2,6-дитретбутилфенилового эфира 2,2-тиоби -/ -метил-6-трет-бутилфенил/фосфористой кислоты. В условиях примера 1 из 21,15 г (0,05 моль) 2,2-тиобис(-метил-6 трет-бутилфенил)хлорфосфита, 10,3 г (0,05 моль) 2,6-дитрет-бутил-фенола 9 мл (0,0б5 моль) триэтиламина в 50 мл ацетонитрила получают .25,8 г (87) кристаллического порошка.После промывки ацетоном получают целе,вой продукте т.пл. (табл.1, «соединение 2) . Пример 5. Получение 2,-6-тритрет-бутилфенилового эфира 2,2 -метилен-бис-(4-метил-6-в6-метилциклогексилфенил)фосфористой кислоты. К 31,53 г (0,0б5 моль)2,2 -мети-, ленбис (t-метил-б-оС-метил-циклогек.силфвнил)хлорфосфита и 17,06 г (0,065 моль) 2,4,6-тритрет-бутилфено ла в 75 мл ДМФА прикапывают 9 мл (0,0б5 моль) триэтиламина при 25 8. .Реакционную массу выдерживают ,5 чпри 95-97 С, охлаждают до отфильтровывают осадок, промывают его на фильтре 50 мл ДМФА. Плав маточника растирают в порошок, промывают 200 мл метанола, сушат, получают 5 г (92,7%) белого кристаллического порошка с т.пл. 127С. При перекристаллизации из ацетонитрила целевой продукт имеет т. пл. 182-186 C. Выход 84 (табл. 1, соединение 3). П р и м е р 6. Получение 4-бром-2,6-дитрет-бутилфенилового эфира 2,2 -метиленбис(-метил-б-третбутилфенил фосфористой кислоты. К суспензии 81 г (0,2 моль) 2,2 -метиленбис{4-метил-6-трет-бутилфенил)хлорфосфита и 57 г (0,20 моль) 4-бром-2,6-дитрет-бутилфенола в 100 мл ацетонитрила прикапывают раствор 30 мл (0,217 моль) триэтиламина в 35 мл ацетонитрила от 28 до . После б-ти часовой выдержки на кипящей водяной бане осадок отфильтровывают, промывают ацетонитрилом, отжимают, сушат на фильтре, получают бе-лый кристаллический порошок в количестве 130 г. Осадок, как указано в примере 1,. промывают водой, сушат, получают 111,2 г (86) целевого продукта с т. пл. . Органический маточник отпаривают от растворителей до 130 с/мм рт. ст., куб растворяют в петролейном эфире (80 мл), отделяют от осадка фильтрованием, петролейный эфир о1-паривают из маточника и из метанола кристаллизацией выделяют дополнительно 11,8 г порошка с т.. пл. 233-235°С. Суммарный выход (табл. 1, сое„ ..„ динение 4). П р и м е р 7. Получение -бром-2,6-дитрет-бутилфенилового эфира 2,2-тио-бис-(-метил-б-трет-бутилфенил)фосфористой кислоты. К смеси 42,3 г(0,1 моль)2,2-тиобис ( -метил-6-третбутилфенил)-хлорфосфита, 28,5 г (0,1 моль) 4-бром-2,6-дитрет-бутилфенола в 75 мл ацетонитрила при 20-36 С прикапывают 8,57 мл (1,1 моль) пиридина в 25 мл ацетонитрила. Реакционную массу выдерживают k ч при флегмировании (8082 С), охлаадают до , отфильтровывают осадок, промывают его охлажденным ацетонитрилом, сушат на фильтре до постоянного веса, промывают водой до отсутствия хлор-иона. Получают белый кристаллический порошок в количестве 52 г с т. пл. 210212 0. Из маточника, как указано в . примере 1, дополнительно выделяют 11 г продукта. Суммарный выход - 9t (табл. 1, соединение 5). Новые циклические фосфиты испытаны в качестве ингибиторов коксообразо вания при пиролизе прямогонного бензина. (Т.н.к. - Л5°С, Т.к.-к. - 160®с) содержащего изопарафиновых углеводородов 36%, нормально-парафиновых 42, нафтеновых 19. Пиролизуемое сырье, в котором пред варительно растворяли заданное количество используемого ингибитора, подавали в реакционную печь в смеси с водяным паром (массовое соотношение водяной пар:бензин равно 0,5:1)« Пиролиз проводили при 828®С и времени контактирования 0,08 с на лабораторной установке а металлическом реакторе с подачей углеводорода и воды дозировочными насосами с точностью дозировки 1-2 мл/ч при подаче 0,5 л/ч. Реактор помещали в электри929to ческую печь с точностью регулировки температуры . Продукты пиролиза после прохождения печи поступали пос ледовательно в водяной конде11сатор, холодильник (), а газообразные продукты - в газометр. Газы пиролиза анализировали хроматографически. Выход продуктов пиролиза:этилена - дивинила - 2,8 %. Количёство сырьяj разложившегося до кокса определяли по содержанию ;СОй в газах регенерации по привесу аскаритной трубки. Результаты испытаний представлены в табл. 2. Как видно из табл. 2, по сравнению с известным ингибитором трифенилфосфитом новые циклические фосфиты позволяют значительно сни-г зить коксообразование при пиролизе . бензина. Предлагаемый способ получения фосфитов позволяет повысить их выход по сравнению с известным ог 5-10% до .





| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения циклических фосфитов | 1980 |
|
SU910644A1 |
| Способ получения циклическихфОСфиТОВ | 1979 |
|
SU794016A1 |
| Способ получения циклических арилхлорфосфитов | 1979 |
|
SU787412A1 |
| Циклические амидофосфиты в качестве антиокислительной присадки к сложным эфирам карбоновых кислот | 1981 |
|
SU958425A1 |
| Способ получения бисфениловых эфиров фосфористой кислоты | 1977 |
|
SU732269A1 |
| Способ получения циклических арилхлорфосфитов | 1980 |
|
SU859369A1 |
| Способ получения комплексов триарилфосфита с галоидами | 1980 |
|
SU982545A3 |
| Способ получения алкиловых эфиров @ -(4-окси-3,5-ди-трет-бутилфенил)-пропионовой кислоты | 1980 |
|
SU1001649A1 |
| СПОСОБ ПОЛУЧЕНИЯ 1,3-ДИГИДРО-7-НИТРО-5-ФЕНИЛ-2H-1,4-БЕНЗОДИАЗЕПИН-2-ОНА (НИТРАЗЕПАМА) (ВАРИАНТЫ) | 1998 |
|
RU2150467C1 |
| Способ получения кислых циклических фосфитов | 1976 |
|
SU585169A1 |
