Изобретение относится к способам получения новых химических соединеНИИ, конкретнее к способам получени спирооксазолидиндионов формулы где X - атом кислорода или серы; R., RJ, R - независимо друг от друга атом водорода, лора, брома или фтора. Эти соединения обладают способностью снижать активность фермента альдозоредуктазь и подавлять накопление сорбита в седалищном нерве кр и могут быть использованы при лечеНИИ некоторых хронических осложнени диабета таких, как диабетические катаракты и неврастении. Известен способ получения N-{3,5 дигалоидфенил)-6ксазолидинонали N-(3,5-Дигалоидфенил)-тиазолидиOHa-t формулы атом водорода, низший алкил; галоида; кислорода или серы, не могут одновременно означать атом кислорода, если оба Х - атом хлора, который заключается в том, что производное формулы Хг где В - атом водорода или низший алВ, В, У. и X. имеют указан ные значения подвергают циклизации в присутствии основания при SO-ZOO C в среде растворителя р J. N-(3,5-Дигалоидфенил)-оксазолидинон-4 и N-(3,5-Дигaлoидфeнил)-тиaзoлидинон-4 обладают биологической активностью. Цель изобретения - получение новы соединений, расширяющих арсенал средств воздействия на живой организ Эта цель достигается способом получения спирооксазолидиндионов общей формулы 1, который заключается в том что соединение формулы т в OCigHs где R - R-iH X имеют указанные значения, подвергают взаимодействию или с фосгеном в присутствии основания при температуре от -10 до в течени мин с дальнейшим перемешиванием реакционной смеси при 20-50 С в течение 12-48 ч с образованием промежуточного соединения формулы NH HOv JL где R. - R--, и X имеют указанные значения , и дальнейшим взаимодействием соедине ния формулы Ш непосредственно в реак ционной смеси с фосгеном при температуре от -10 до +10 С в течение 15-75 мин с последующим перемешиванием при комнатной температуре в те чение ч для превращения промежуточного продукта формулы Ш в целевой продукт формулы .1; или с галоидалкилформиатом с I- атомами углеро да в алкиле при температуре от -10 до в течение 30 мин - 2 ч с последующей выдержкой раствора при 50-150°С в течение 1 ч и дальнейшей выдержкой образовавшегося промежуточного продукта формулы Ш при указанной температуре 2-6 ч с конверсией промежуточного продукта формулы Ш в целевой продукт формулы 1; или с 1,1-карбонилдиимидазолом при 50150С в течение 30-90 мин и дальнейшей выдержкой промежуточного продукта- формулы Ш при указанной температуре 12-36 ч с конверсией промежуточного продукта формулы Ш в целевой продукт формулы 1; или с карбонатом щелочного металла или карбонатом аммония при 15-50с в течение 5-24 ч. При реакции с фосгеном в качестве основания используют триэтиламин или другой триалкиламин с 1-4 атомами углерода в алкиле. Реакцию проводят в инертном органическом растворителе: диэтиловом эфире, тетрагидрофуране, диметоксиэтане или диоксане. В качестве галоидалкилформиата предпочтительно использовать этилхлорформиат и реакцию проводить в пиридине. Взаимодействие с 1,1 -карбонилдиимидазолом проводят или без растворителя, или в инертном органическом растворителе таком, как диоксан, тетрагидрофуран, диметоксиэтан или диметиловый простой эфир. Пример 1. 6-Хлор-4-циано-4-триметил.силилокси-4Н-2 ,3-Дигидробензпиран. Смесь 6-хлор-4Н-2,ЗДигидробензпиран-4-она (.20,0 г, 0,11 моль), триметилсилилцианида И 3,0 г, 0,13 моль) и йодида цинка (0,2 г в 50 мл простого эфира перемешивают в течение 18 ч. Раствор обесцвечивают при помощи древесного угля, фильтруют и выпаривают под вакуумом с образованием оранжевого масла, которое при добавлении пентана дает кристаллический 6-хлор-4-циано-4-триметилсилилокси-4Н-2,3-дигидробензпиран, 26,4 г т.пл. б7-б9°С. Пример 2. Этил-6-хлор-4-окси-4Н-2,3-дигидробензпиран-4-карбоксимидат. Насыщенный раствор 6-хлор-4-ц1 1ано-4-триметилсилилокси-4Н-2, 3 дигидробензпирана ( 273,0 г, 0,97 моль) в 2,0 л этанола охлаждают до и в него в течение 40 мин подают сухой хлористый водород. При слабой экзотермии смесь становится однородной. Спустя 16 ч при А°С летучие вещества удаляют под вакуумом, при этом образуется полутвердый остаток. Растирание в порошок в 800 мл диэтилового простого эфира с последующей фильтрацией дает твердый материал, который разделяют между 3,8 л хлороформа И 500 мл насыщенного бикар боната натрия. Органический слой про мывают дополнительным количеством (500 мл) насыщенного бикарбоната нат рия, сушат над сульфатом магния, фильтруют и выпаривают под вакуумом, в результате чего образуется твердый этил-6-хлор- -окси-4Н-2 ,З-ДИГИДРОбензпиран- -карбоксимидат, 193,0 г С . Растирание в порошок с простым эфиром дает материал с т.пл. 12 -12б°С. Пример 3. 6-Хлор-спиро CtH-2, 3-дигидробензпиран ( j.S) оксазолидинЗ -2 , ( - дион. Смесь этил-6-хлор- -окси- Н-2,3-дигидробензпиранкарбоксимидатаЦОО г, 1,95 моль) и. триэтиламина ЦОО г, 3,96 моль) в 13 л сухого тетрагидрофурана при продувают фосгеном (1818 г, 18,+ моль) с такой скоростью, чтобы поддержать температуру ниже 27 С. Перемешивание продолжают при подаче фосгена, температуре дают возможность достичь 20С и поддерживают ее такой в течение +8 ч. Анализ при помощи тонкослойной хроматографии показывает точку в R 0,57 и отсутствие точки в Rr 0,29(1:1, хлороформ:этилацетат, на силикагеле). (Материал с R 0,29 - это исходный имидат, тогда как материал с Rr 0,57 - это промежуточный этоксиоксазолин - 2 -он из примера б). Смесь затем сливают в 13 л колотого льда при перемешивании, фосген и двуокись углерода пр этом высвобождаются. Двухфазную смес нейтрализуют 1,7 л 50 -ного раствора гидрата окиси натрия до рН 7. Далее добавляют карбонат натрия ( г 2,0 моль) и смесь перемешивают в течение 16 ч при 20 С. Продукт выделяют при помощи следующей процедуры экстрагирования: в смесь добавляют 12 л этилацетата и после встряхивания водный слой отбирают. Органическую фазу дважды промывают при помощи 12 л 7 -ного водного раствора бикарбоната натрия. Объединенные водные слои подкисляют до рН 1 с охлаждение до 10-15°С при добавлении концентри g 6 рованной соляной кислоты. Водный слой экстрагируют три раза 12 л этилацетата. Объединенную органическую фазу промывают 12 л насыщенного раствора хлорида натрия,,сушат над сульфатом магния, фильтруют и выпаривают под вакуумом до вёрдого остатка; 392 г (79%), т.пл. 191-195°С. Перекристаллизация из толуола дает 6-хлор-спиро-Е4Н-2,3-дигидробензпиран (t.SоксазолидинЗ-2 , t-дион с т. пл. 19б-198°С; Rp О, (1:1, СНСЦ : :EtOAc на салициловой кислоте). Пример 4, 6-Хлор-спиро 4Н-2,3-дигидробензпиран (+,5 ) оксазолидинЗ-2, 4 -дион. Смесь этил-6-хлор-4-окси-2Н-2 ,3-дигидpoбeнзпиpaн-+-кapбoкcимидaтa(5,0 г, 0,019 моль ) и 1,1 -карбонилдиимидазола (3,7 г 0,023 моль) нагревают в 5 мл диоксана до температуры 90 С, которую поддерживают в течение 16 ч. Анализ реакционной смеси при помощи тонкослойной хроматографии спустя 1 ч показывает точку Rr 0,57 (1:1, хлороформ: этилацетат, на салициловой кислоте, которая соответствует этоксиоксазолин-2 - ону из примера 6. После охлаждения смесь разбавляют 100 мл этилацетата и дважды промывают . 100 мл 1 н. раствора соляной кислоты. Органический слой экстрагируют дважды 100 мл насыщенного раствора бикарбоната натрия. Основной слой подкисляют 6н.раствором соляной кислоты до рН 1 и экстрагируют трижды 100 мл этилацетата. Этот последний органический слой промывают солянокислым раствором, сушат над сульфатом магния, фильтруют и выпаривают под вакуумом до твердого остатка; 1,20 г (25%), т.пл. 189-191 0. Перекристаллизация из толуола дает 6-хлор-спиро С+Н-2,3дигидробензпиран (,) оксазолидинЗ-2 , 4 дион с т.пл. 192-193С. Пример 5-6- Хлор-спироt4H-2,3-Дигидробензпиран (4,5) оксазолидин -2, Ц-дион. Смесь этилхлорформиата (2,00 г 2,82 моль), этил-6-хлоо-4-окси-4Н-2,3 дигидробензпиран-4-карбоксимидата (1,69 г, 1,5б ммоль) и пиридина (5 мл) подвергают взаимодействию при 0°С в течение 1 ч, затем нагре- вают до комнатной температуры и дефлегмируют в течение k ч. Концентрирование под вакуумом и экстрап
гирование, описанное в примере 3, дает 6-хлор-спиро ftH-ZjS-flHrHflpo-бензопиран (5,Оксазолияин -2 , 4 -дион с выходом 10%, т.пл. 195198°С.
Пример 6, б-Хлор- -ЭТОКСИ-, спиро 4Н-2,3-дигидробензпиран С,5) оксазолинЗ-2-он.
Раствор этил-6-хлор- -этокси-|Н-2,3-Дигидробензпиран-«-кар6оксимидата и,15 г 4,00 ммоль) в 60 мл тетрагидрофурана охлаждают до 0°С и перемешивают. При этом в течение 5 мин подают фосген. Спустя 30 мин тонкослойная хроматография реакции показы вает новую точку с Rr 0,57 (1:1, хлороформ: этилацетат, на салицилово кислоте) и отсутствие исходного материала с RP 0,29. Смесь слибают в 90 мл смеси лед-вода и экстрагируют дважды при помощи 50 мл этилацетата. Органический слой промывают дважды 30 мл 5%-ного водного раствора бикарбоната натрия, сушат над сульфатом магния, фильтруют и выпаривают под вакуумом до маслянистой жидкости (0,б51 г), которая кристаллизуется при низкой температуре из смеси простой эфир - гексан 0,350 г (3U), т.пл. ЮВ-ПО С. Этот этоксиоксазолин также получают из с использованием этилхлорформиата в пиридине с выходом 62. с использованием этилхлорформиата в пиридине с выходом 62I.
Пример 7. 6-Хлор-спиро Ь4Н-2,3-дигидробензпиран (4,5) оксазолидинЗ-2 , 4 -дион.
Смесь 6-хлор-4-этоксиспиро ,3-Дигидробензпиран (4,5) оксазолин -2-она (100 мг, 0,355 ммоль) и карбоната натрия (88 мг, 0,710 ммоль в 2 мл смеси (1:1) тетрагидрофурана и воды перемешивают при в течение 16 ч..После добавления 10 мл этилацетата и 10 мл воды осуществляют экстрагирование, как описано в примере 3, которое дает 6-хлор-спиро 4Н-2,3-дигидробензпиран ( 4,5Л)ксазолидин -2, 4-дион, 63 мг (70), т.пл. 192-195с.
Примеры 8-15.Способом, аналогич ым приведенному в примере 1, получают соединения с использованием соответствующих кетонов вместе 6-хло-4Н-2,3-Дигидробензпиран-4-она,представленные в таблЛ. Все соединения выделяют в виде масел после промывки раствором бикарбоната натрия и соляным раствором, сушки над сульфатом магния с последующим выпариванием под вакуумом. Структуру соединений определяют при помощи ЯМРспектра и/или тонкослойной хроматографии на салициловой кислоте.
N(i. 0$1((Нэ)5
Сохраняется продукт при комнатной температуре (20°С), охлаждается до 4С.
П р и м е р 22. Спиро 4Н-2,3-дигидробензпиран (4,7 оксазолидин -2, 4-дион.
Смесь ;этил-4-окси-4Н-2,3-дигидробен пиран-4-карбоксимидата (5,50 г, 24,9 ммоль) и триэтиламина (5,03 г,49,7 ммоль) в 50 мл тетрагидрофурана при насыщают газообразным фосгеном в течение 30 мин. Сразу же после начала подачи фосгена образуется твердый осадок. Смесь перемешивают 16 ч при . При этих условиях i оксазолидиндион получают непосредственно без выделения и гидролиза промежуточного оксазолина. Реакционную смесь сливают на 200 мл колотого льда и экстрагируют этилацетатом. Спиро Э ЛН-2,3-дигидробензпиран (А,5 оксазолидин1-2, 4 -дион выделяют из это органической фазы описано в примере 3, с выходом 5б, т.пл. 168-170°С. Пример 23. 6-Фтор-спиро ,3-дигидробензпиран { k,S)окса золидин -2j -дион. 6-Фтор-спиро , 3-дигидробензпиран (4,5 о сазолидинЗ-2, -дион получают способом, аналогичным описанному в примере , из этил-6-фтор-4-окси- Н-2,3-ДИГИдробензпиран- -карбоксимидата с выходом 3%. Исклю чение составляет то, что смесь нагревают до температуры 100°С, которую поддерживают 1 ч; т.пл. 177,5180 С после перекристаллизации из толуола. Пример 2. 6-Бром-спиро ин-2,3-дигидробензпиран ( ,S) окса золидинЗ-2, 4-дион 6-Бром-спиро 4Н-2,3-.дигидробенз пиран (4,5)оксазолидин -2 ,-дион получают способом, аналогичным описанному в примере , из этил-6-бром -4-окси- Н-2,3-дигидробензпиран-4-карбоксимидата с выходом 38. Исклю чение составляет то, что смесь нагре вают до темпер атуры 100 С, которую поддерживают в течение 0,5 ч; т.пл. IBS-ISI C после перекристаллизации из толуола. Пример 25. Спиро ,3-дигидробезтиопиран С,5 оксазолидин 1-2 4-дион. Спиро kH-2,3-дигидробензтиопира (J,5) оксазолидин -2 , -дион при готавливается способом, аналогичным описанному в примере , из этил- -окси- Н-2,З-Дигидробензтиопиран-4-карбоксимидата с выходом 5. Исключение составляет то, что смесь выдерживается 7,0 ч при 100 С; т.пл 165-167 С после перекристаллизации толуола. Пример 26. 6-Фтор-спиро tH-,3-дигидробензтиопиран С,5) оксазолидинЗ-2, -дион. 6-Фтор-спиро L Н-2,3-дигидроб нз тиопиран (,5)оксазолидинЗ-2, -дион получают в соответствии со способом, описанным в примере , из этил-6-фтор- -окси-4Н-2,3-дигидробензтиопиран-4-карбоксимидата с выходом Ц}%. Отличие в том, ццо смесь выдерживают в течение 48 ч при ЮО т.пл. 193-19, после перекристал лизации из толуола. 7 Пример 27. (- -$-хлор-спиро Н-2,3-дигидробензпиран (,S-) оксазолидин -2, Л-дион. Рацемический 6-хлор-спиро L4H-2,3 -дигидробензпиран С,) - oкcaзoлидинJ -2, t-дион (32,0 г, 0,126 моль) и цинхонидин (37,1 г, 0,126 моль) растворяют при нагревании в 290 мл безводного этанола. После охлаждения твердого материала аддукт С оксазолидиндиона и цинхонидина выделяют при помощи фильтрации, 28,0 г (81). Фильтрат концентрируют под вакуумом до твердого остатка, который разделяют между 500 мл этилацетата и tOO мл 1н,раствора соляной кислоты. Органический слой промывают дополнительно tOO мл 1н.раствора соляной кислоты, 200 мл соляного раствора, сушат над сульфатом магния, фильтруют и концентрируют до 150 мл. К этому органическому раствору добавляют жидкий L-амфетамин (10, 13 г, 0,075 моль). Образующийся обильный осадок выделяют фильтрованием, промывают простым эфиром и сушат под вакуумом,в результате чего образуется аддукт (-) - оксазолидиндион-L-амфетамина, 1,85 г (60%) ;Co(l 36, т.пл. 171-17 с.15,80 г этого аддукта разделяют между 300 мл этилацетата и 200 мл Зн.раствора соляной кислоты. Органический слой промывают дополнительно 200 мл Зн. раствора соляной кислоты, 100 мл соляного раствора, сушат над сульфзтоь магния, фильтруют и выпаривают под вакуумом до бесцветного твердого остатка; 9,5 г С выход rg7;8S); Cotafto - 60,58°; т.пл. 201-202,5с. Перекристаллизация из толуола дает С-) -6-хлор-спиро 1.,3-дигидробензпиран (,5) оксазолидинЗ-2 , -дион, 8,203 г; т.пл. 200-200, 161,. Пример 28. Расщепление 6-хлор-спиро ЦН-2,3-Дигидробензпиран (,,5) оксазолидин -2, Ц -диона. 6-Хлор-спиро 4Н-2,3-дигидробензпиран C.SV оксазолидинЗ-2 , -дион (1,00 г, 3,93 ммоль) и цинхонидин (1,16 г, ммоль) растворяют в орячем этаноле. После выпадения в осадок аддукт собирают и перекристаллизовывают из этанола, т.пл. 206-. 207°С. Этот твердый материал разделяют между 50 мл этилацетата и -50 мл 1н.раствора соляной кислоты. Кислую 119 фазу экстрагируют при помощи 50 мл этилацетата. Объединенные органичес кие слои промывают при помощи 50 мл соляного раствора, сушат над сульфа том магния, фильтруют и выпаривают под вакуумом до твердого остатка, к торый перекристаллизовывают из толу ола; 288 мг (57); т.пл. таЗ-ТЭУ С, Перекристаллизация его из толуола дает ( + ) -6-хлор-спиро 4Н-2,3-дигидробензпиран ( k,S) оксазолидинЗ -2, -дион, 200 мг (0); т.пл. 2QO-201,5°C;Cc jE ° -. +60,55°. Из первог этанольного маточного раствора получаюттакже наибольшое количество )-аддукта в виде кристаллов после того, как он отстоится в течение ночи. После фильтрации это маточный раствор разделяют между 50 мл 1н.раствора соляной кислоты и 50 мл этилацетата. Оксазолидиндион полученный таким образом, дважды перекристаллизовывают из толуола, в результате чего образуется 6-хлорспиро 4Н-2,3-дигидробензпиран (1,5) оксазолидинЗ-2, - дион, 143 мг С29); т.пл. 199-200°С;Со; Е4-° - 61,. Пример 29. Расщепление 6-хлор-спиро 4Н-2 ,3-Дигидробензпиран (4,5 оксазолидин 3-2 ,- 4-диЪна . 6-Хлор-спиро +N-2,3-Дигидробензпиран (,) оксазолидин - 2, V-ди он (1,00 г 3,95 ммоль) и L -амфетами (26k мг, 2,00 ммоль) растворяют в горячем этилацетате. Высушенные крис таллы, собранные после охлаждения (Э мг, т.пл. 1б5-1б8 с), перекристаллизовывают из этилацетата; 221 мг, т.пл. 171-173 0 ;W3|tOH . - 32,7°. Расщепление аддукта и выделение (-)-оксазолидиндиона осуществляют в соответствии с примерном соответствии с 198-20ос;Ш О 27; 53 мг, т.пл. - 60,83°С,. Пример 30. Спирооксазолидин дионы, полученные в соответствии с предыдущими примерами, проверяют на способность снижать или подавлять активность фермента альдозоредуктазы При этом используют материал, который является частично очищенным ферментом альдозоредуктазы, полученным из хрусталика глаза теленка. Результаты, полученные для каждого соединения при концентрации 10м, выражают в процентах подавления активности фермента. 7 Подавление активности фермента соединениями формулы 1 представлены в табл.3. Пример 31. Спирооксазолидиндионы, полученные в соответствии с приведенными примерами, проверяют на способность снижать или подавлять накапливание сорбита в седалищном нерве у крыс, которым был введен стрептозотоцин (т.е. больных диабетом). В этом исследовании количество сорбита, которое накапливается в седалищном нерве, измеряют спустя 27 ч после поражения крысы диабетом. Соединрния применяют стоматически в дозах, которые указаны спустя 4,8 и 2k ч после применения стрептозотоцина. Полученные результаты приведены в процентах подавления для каждого соединения по сравнению со случаем, когда соединение не применялось (т.е. для необработанных животных, где в общем случае количество сорбитола возрастало с 50-100 до/ 00 мм/г ткани спустя 27 ч) В этом эксперименте значения ниже 20 не всегда имеют экспериментальный и статистический смысл. Подавление накапливания сорбита в седалищном нерве крыс соединениями формулы 1 дано в табл.. Активность новых соединений формулы 1 в качестве агентов для регулирования осложнений при хроническом диабете может быть определена при помощи нескольких стандартных биологических или фармакологических экспериментов, к которым относятся измерение способности: подавлять активность фермента выделенной альдозоредуктазы; снижать или полностью исключать накапливание сорбита в седалищном нерве крысы, которой введена сильная доза стрептозотоцина (т.е. пораженной диабетом);снижать уже высокий уровень содержания сорбита в седалищном нерве и хрусталике глаза у крыс с хроническим диабетом, вызванным введением стрептозотоцина; предотвращать или снижать образование галактита в хрусталике глаза у крыс с острым нарушением синтеза галактита; тормозить образование катаракты и снижать степень замутненности хруста; лика глаза у крыс с хроническим нарушением процесса синтеза галактита . Новые спироок-сазолидин-2, 4-дионы используются в качестве ингибиторов
альдозоредуктазы, являясь при этом терапевтическими агентами при лечени осложнений хронического диабета, таких как катаракта, неврастения и заболевание сетчатки ( ретинит). Лечение состоит в предотвращении или облегчении упомянутых осложнений. Соединения формулы 1 могут быть при-. менены различными способами, включая стоматический и парентеральный. В общем случае эти соединения применяются в дозах 1-500 мг/кг веса тела больного в день, предпочтительно 125 мг/кг в день. Однако могут быть необходимы некоторые изменения доз в зависимости от состояния больного и конкретного соединения, которое применяется для лечения. Соответствующая доза определяется в каждом конкретном случае.
Соединения могут быть применены как таковые или в смеси с приемлемыми с фармацевтической точки зрения носителями как в виде одной дозы так и в виде нескольких. Соответству-25 кол;
ющими приемлемыми с фармацевтическо1 1 точки зрения носителями являются инертные твердые разбавители или наполнители, стерильные водные растворы и различные нетоксичные органические растворители.
Фармацевтические композиции, образованные в результате смешивания спирооксазолидин-2,4-диона и приемлемого с фармацевтической точки зрения носителя, применяются в различных формах таких, как таблетки, порошки, пилюли, сиропы и растворы для инъекций. Эти фармацевтические композиции, если это необходимо, могут содержать дополнительные ингредиенты такие, как душистые вещества носители или наполнители. Таким образом, для стоматического применения таблетки, содержащие различные наполнители, такие как цитрат натрия, карбонат кальция и фосфат кальция, могут применяться вместе с различными диспергирующими материалами, таким как крахмал, предпочтительно картофельный, или крахмал тапиоки,
альгиновая кислота и некоторые ком- плексы силикатов.вместе со связы-. вающим агентом, таким как поливинилпирролидон, глюкоза, желатин и акация. Кроме того, при изготовлении таблеток часто используются смазывающие агенты, такие как стеарат магния, лаурил, сульфат натрия и тальк. Твердые композиции того же типа можно
также использовать в качестве наполнителей легких и тяжелых капсул из желатина. Предпочтительными материалами для этого являются лактоза или молочный сахар, а также полиэтилен-.
гликоли с высоким молекулярным весом. Если для стоматического применения используются эликсиры или суспензии, активный ингредиент в них смешивается с различными дезодорирующими или ароматизирующими агентами, окрашивающими материалами и, если это необходимо, эмульгирующими или суспендирующими агентами вместе с разбавителями, такими как вода, этанол, пропиленглиПри парентеральном применении можно использовать растворы сгтирооксазолидин-2,-дионов в кунжутном или арахисовом масле или в водном
растворе пропиленгликоля, а также стерильные водные растворы соответствующих растворимых в воде солей щелочных или щелочно-земельных металлов. Если это необходимо, такие
водные растворы дополняются буферным раствором или жидким разбавителем, который перед добавлением превращается в изотонический раствор при помощи солевого раствора или раствора глюкозы. Упомянутые водные растворы, в частности, пригодны при внутривенном, внутримышечном, подкожном и внутрибрюшинном введении. Стерильные водные растворы, которые применяются при упомянутом использовании предлагаемых соединений, можно легко получить известными способами. Кроме того, спирооксазолидин-2,-дион можно применять местно, используя соответствующий оптальмовый раствор и применяя его в виде капель в глаза. глицерин или их смеси.
Таблица. 1






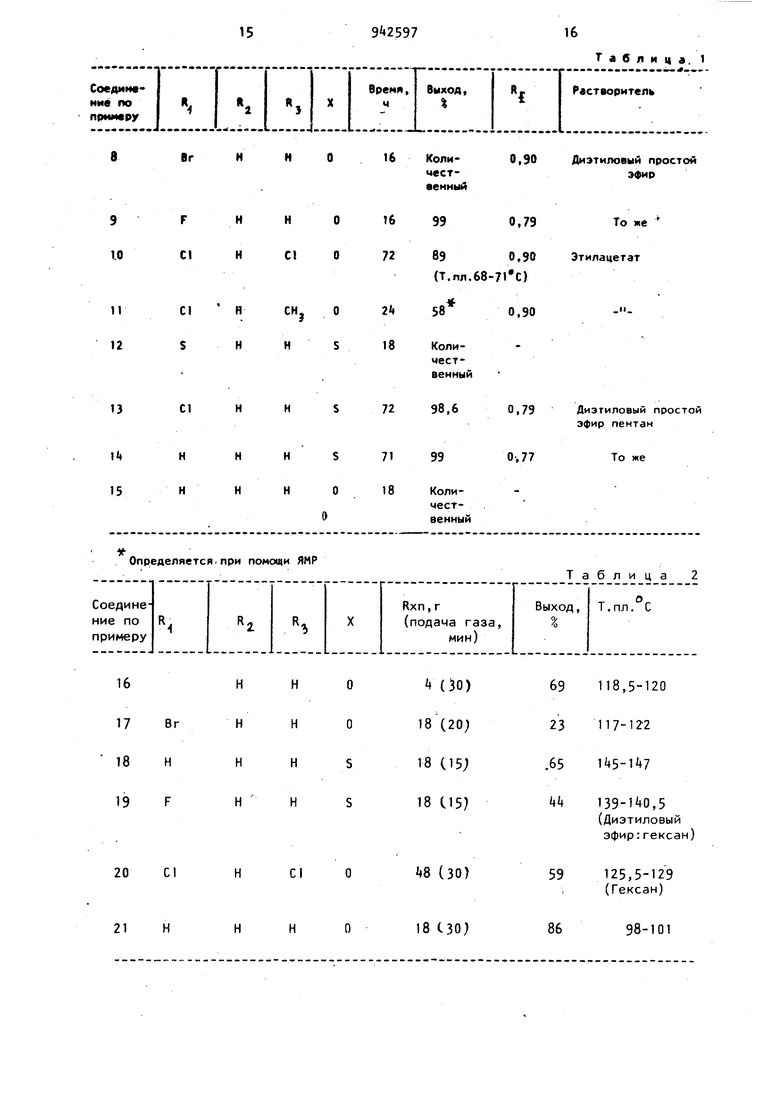

8r
Cl
Cl
HCHj 0
Cl
21Н
0.90
КолиДиэтиловый простой
эфир чественный
99
0,79
То же
890,90 Этилацетат
(Т.пя.68-71 С)
58
0,90
18 СЗО)
98-101
86
Примечание. Через косую даны результаты повторного эксперимента.
Соединение по примеру
Таблица 3
Таблица
Процент подавления при дозе, мг/кг
15
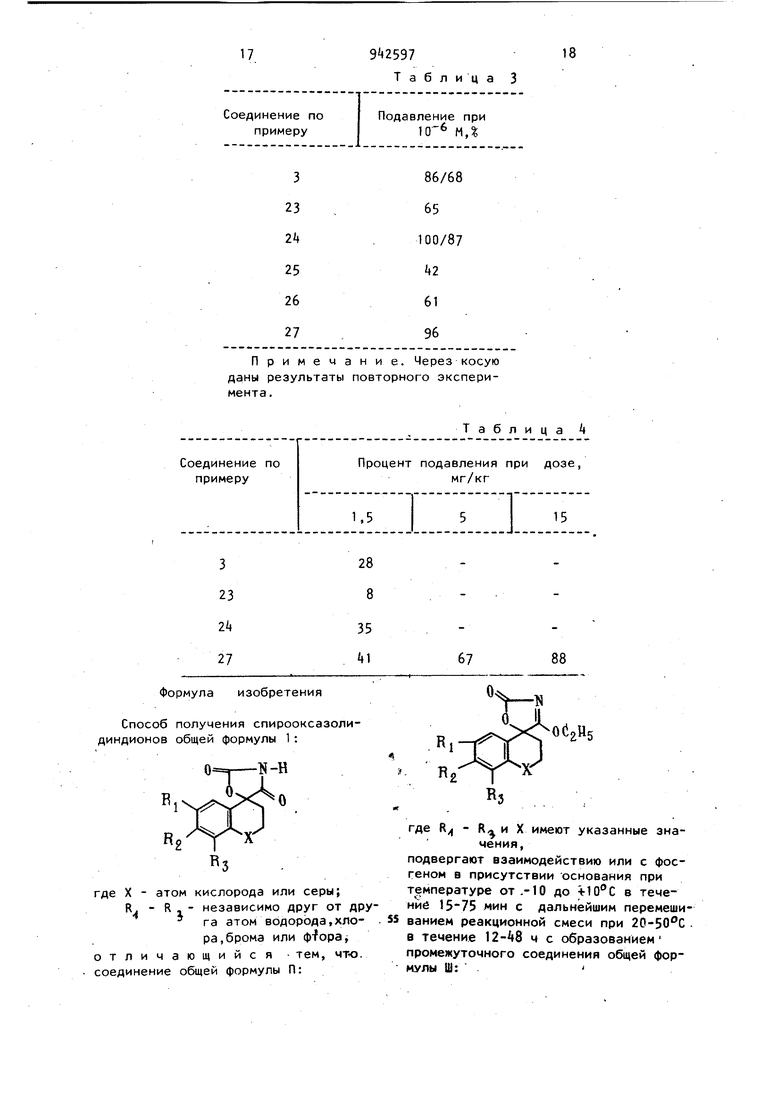
диндионов общей формулы 1:
«-Н
где X - атом кислорода или серы;
R. - R - - независимо друг от дру га атом водорода,хлора, брома или ф-fopaj отличающийся тем, что. соединение общей формулы П:
где - R и X имеют указанные значения,
подвергают взаимодействию или с фосгеном в присутствии основания при температуре от.-10 до в течение 15-75 мин с дальнейшим перемеши ванием реакционной смеси при 20-50 0 в течение 12-А8 ч с образованием промежуточного соединения общей формулы Ш: где Rj и X имеют указанные значения, и дальнейшим взаимодействием соедине ния формулы Ш непосредственно в реак ционной смеси с фосгеном при температуре от -10 до в течение 15-75 мин с последующим перемешиванием при комнатной температуре в течение ч для превращения промежуточного продукта формулы Ш в целевой продукт формулы 1; или с галоидалкилформиатом с I- атймами углерода в алкиле при температуре от -10 до в течение 30 мин - 2 ч последующей выдержкой раствора при 50-Т50 С в течение 1 ч и дальнейшей выдержкой образовавшегося промежуточ-. ного продукта формулы Ш при указанной температуре 2-6 ч с конверсией промежуточного продукта формулы Ш в целевой продукт формулы 1; или с 1,1-карбонилдиимидазолом при 50 150с в течение 3090 мин и дальнейшей выдержкой промежуточного продукта формулы Ш при указанной температуре 12-36 ч с конверсией промежуточного продукта форг мулы Ш в целевой продукт формулы 1; или с карбонатом щелочного метал- - ла или карбонатом аммония при 1550 С в течение 5-24 ч. Источники информации,, принятые во внимание при экспертизе 1 . Патент СССР № 23300, кл. С 07 О-абЗ/ , 1969.
