Изобретение относится к медицине, микробиологической промышленности, биотехнологии и может быть использовано для мониторинга окружающей среды, а также в пищевой, фармацевтической и химической промышленности.
Применение ракетного топлива при запусках и заправке ракет, розлив и хранение этого топлива и выбросы остатков несгоревшего горючего в районах сброса первых ступеней ракет создают существенную опасность для проживающего в этих районах населения, животному и растительному миру. Важной задачей является оценка мутагенного воздействия различных синтезируемых химических соединений, разрабатываемых лекарственных препаратов и продуктов их метаболизма в организме, экотоксикантов из окружающей среды.
Известно использование репортерных генов, кодирующих биолюминисцентные белки, для обнаружения агентов, повреждающих генетический аппарат клетки.
Известен патентный документ WO 9844149 (А1-1998-10-08) «Detection of DNA damaging agents». Предложены рекомбинантные молекулы ДНК, содержащие регуляторный элемент для активации генной экспрессии в ответ на повреждение ДНК, оперативно связанный с последовательностью ДНК, которая кодирует испускающий свет репортерный белок; рекомбинантные векторы, содержащие такие молекулы ДНК.
Известно семейство патентных документов, связанное с данным патентным документом: 1) AU 747297 B2 - 2002-05-16; 2) AU 6841998 А - 1998-10-22; 3) СА 2285703 А1 - 1998-10-08; 4) ЕР 0970247 A1 - 2000-01-12; 5) GB 9706414D DO - 1997-05-14; 6) JP 2001517083T Т - 2001-10-02; 7) US 6489099 B1 - 2002-12-03; 8) US 2003049690 A1 - 2003-03-13; 9) ЕР 1561822 А1.
В патентных документах (1-7, 9) предложены рекомбинантная молекула ДНК, содержащая регуляторный элемент RAD54 дрожжей, состыкованный с последовательностью ДНК, которая кодирует флуоресцирующий в зеленой области спектра белок (Green Fluorescent Protein - GFP) или его излучающие свет производные; рекомбинантный вектор, содержащий вышеуказанную молекулу ДНК, и ДНК вектор.
В патентном документе (8) предложены рекомбинантная молекула ДНК, содержащая регуляторный элемент RNR, выбранный из группы, состоящей из регуляторных элементов из генов RNR1, RNR2, RNR3, где указанный регуляторный элемент оперативно связан с последовательностью ДНК, которая кодирует GFP или его флуоресцирующие производные; рекомбинантный вектор, содержащий данную молекулу ДНК, и ДНК вектор.
Данные плазмиды обладают двумя ограничениями. Определение мутагенов с использованием конструкций ДНК, описанных в вышеуказанных патентах, где используются рекомбинантные штаммы дрожжей Saccharomyces cerevisiae, занимает существенно большее время, чем в аналогичных системах, в которых применяют рекомбинантные штаммы E.coli, поскольку скорость деления клеток E.coli в 3-5 раз выше таковой у Saccharomyces cerevisia.
У дрожжевой клетки сравнительно низкая проницаемость клеточной стенки, поэтому использование E.coli позволяет проводить тестирование более экспрессно.
В вышеуказанных изобретениях в опытах с метилметансульфонатом (MMS), вызывающим метилирование ДНК, MMS используется в сравнительно высокой концентрации 0,01-0,002%, которая для клеток Е.coli является субтоксичной и существенно подавляет метаболизм самой клетки. Многие экотоксиканты, присутствующие в окружающей среде, имеют значительно более низкие концентрации и не могут быть определены с помощью вышеописанных систем.
Кроме того, метилметансульфонат является метилирующим агентом и в строгом смысле не является повреждающим ДНК агентом. Метилирование ДНК происходит при нормальной жизнедеятельности клетки, и оно репарируется ada-системой репарации, а в патентном документе (8) использована система sos-репарации.
Известна работа авторов Kostrzynska M, Leung KT, Lee H, Trevors JT. "Green fluorescent protein-based biosensor for detecting SOS-inducing activity of genotoxic compounds» /J.Microbiol. Methods, 2002 Jan; 48 (1); 43-51 (Department of Environmental Biology, University of Guelph, N1G 2W1, Guelph, ON, Canada) (прототип).
Авторами получены флуоресцирующие индикаторные штаммы Escherichia coli, содержащие плазмиду, в состав которой входит recA промотор штамма JM 101, сшитый с геном белка GFP, флуоресцирующего в зеленой части спектра, или с геном, кодирующим мутантный вариант GFP со сдвинутой в красную область более интенсивной флуоресценцией (мутант 3). Биосенсоры, основанные на GFP, позволяют определять дозозависимые ответы на действие таких генотоксических агентов как митомицин С, N-methyl-N'-nitro-N-nitrosoguanidine и налидиксовой кислоты.
Полученные авторами данной работы плазмиды по отношению к ряду мутагенов проявляют меньшую чувствительность по сравнению с плазмидами, описываемыми в настоящем изобретении. Кроме того, в работе Kostrzynska M. и др. не проводились исследования с повреждающими воздействиями физической природы и чувствительность к ним данных плазмид не известна.
Раскрытие изобретения
Задачей изобретения является создание рекомбинантных плазмидных конструкций с большей чувствительностью по отношению к повреждающим ДНК воздействиям химической природы, а также чувствительных по отношению к повреждающим ДНК факторам физической природы.
Рекомбинантные плазмидные ДНК pRTGFP и pRTGFP2 обеспечивают синтез флуоресцирующего мутантного белка GFP из Aequorea victoria в Rec+ штаммах E.coli под воздействием индукторов, активирующих систему SOS-репарации.
Рекомбинантная плазмидная ДНК pRTGFP содержит:
- EcoRI-BstEII фрагмент ДНК размером 0.72 тысяч пар оснований (т.п.о.), содержащий фрагмент мутантного гена gfp Aequorea victoria размером 0,71 т.п.о.;
- BstEII-BamHI фрагмент ДНК размером 0,024 т.п.о., кодирующий глицин, шесть гистидиновых аминокислотных остатков и содержащий стоп кодон трансляции;
- BamHI-EcoRI. фрагмент ДНК плазмиды pIL-2/21 (SU 1761805 A1) размером 3,42 т.п.о. с внутренним уникальным сайтом рестрикции AatII, содержащий ген bla β-лактамазы в качестве генетического маркера, определяющего устойчивость трансформированных плазмидой клеток E.coli к ампициллину, и регуляторную область гена recA Proteus mirabilis;
- уникальные сайты рестрикции AatII, EcoRI, BstEII, BamHI.
Ниже приведено описание рекомбинантной плазмиды pRTGFP, которая представлена в перечне последовательностей SEQ ID NO 1. Указанная плазмида имеет размер 4170 п.о., молекулярную массу 2,71 мегадальтон и состоит из следующих элементов (все позиции нуклеотидных остатков указаны относительно плазмиды pRTGFP).
I. EcoRI-BstEII фрагмент ДНК (позиции 190-910 п.о), содержащий фрагмент мутантного гена gfp Aequorea victoria (позиции 196-909 п.о.), который кодирует аминокислотную последовательность белка GFP. Мутантный ген gfp включает в себя кодон инициации трансляции - ATG (позиция 196-198 п.о.). Нуклеотидная последовательность мутантного гена gfp Aequorea victoria и кодируемая им аминокислотная последовательность белка GFP представлены в перечне последовательностей: SEQ ID NO 2; аминокислотная последовательность отдельно представлена в SEQ ID NO 3;
II. BstEII-BamHI фрагмент ДНК (позиции 911-934), содержащий два нуклеотида из трех, кодирующих глицин (позиции 911-912), шесть кодонов гистидина (позиции 913-930) и стоп кодон трансляции (позиции 931-933);
III. BamHI-EcoRI фрагмент ДНК плазмиды pIL-2/21 (SU 1761805 A1) (935-189 п.о.), который содержит:
- фрагмент ДНК плазмиды pBR322 (позиции 1872-4168 п.о.) длиной 2297 п.о. (соответствующий фрагменту 2065-4361 п.о. оригинальной последовательности pBR322), который содержит:
а. участок начала репликации (позиция 2342 п.о.);
b. ген bla, кодирующий (3-лактамазу в качестве генетического маркера, определяющего устойчивость трансформированных плазмидой клеток E.coli к ампициллину (позиции 3100-3963 п.о.);
с. регуляторную область гена recA Proteus mirabilis, обеспечивающую под воздействием индукторов (налидиксовая кислота, митомицин С, УФ-воздействие и другие мутагенные факторы) инициацию транскрипции матричной РНК, кодирующей флуоресцирующий в зеленой области спектра белок GFP (позиция 45-189 п.о.);
- фрагмент генома бактериофага лямбда (позиции 1771-1865 п.о.).
Уникальные сайты узнавания эндонуклеазами рестрикции имеют следующие координаты: AatII (GACGTC) 4091-4096 п.о., EcoRI (GAATTC) 189-194 п.о., BstEII (GGTCACC) 910-916 п.о., BamHI (GGATCC) 934-939 п.о.
Рекомбинантная плазмидная ДНК pRTGFP2 содержит:
- EcoRI-BamHI фрагмент ДНК размером 0,74 т.п.о., содержащий фрагмент мутантного гена gfp Aequorea victoria размером 0,71 т.п.о., кодоны глицина, шести гистидинов и стоп кодон трансляции;
- BamHI-XbaI фрагмент ДНК размером 0,10 т.п.о. (включающий XbaI-сайт полностью) и содержащий фрагмент генома бактериофага лямбда;
- фрагмент плазмидной ДНК pBR322 размером 2,22 т.п.о.; содержащий ген bla β-лактамазы в качестве генетического маркера, определяющего устойчивость трансформированных плазмидой клеток E.coli к ампициллину; 5'-конец фрагмента содержит часть сайта AatII-рестриктазы;
- AatII-EcoRI фрагмент ДНК размером 0,24 т.п.о., содержащий фрагмент гена recA Proteus mirabilis, энхансер трансляции фага Т7 (ген 10) (Cheng X, Patterson ТА. Construction and use of lambda PL promoter vectors for direct cloning and high level expression of PCR amplified DNA coding sequences// Nucleic Acids Res. 1992. Sep 11; 20 (17):4591-8.) и последовательность Шайна-Дальгарно (De Boer N.A., Comstock L.J., Vasser M. The tac promoter: A functional hybrid derived from the trp and lac promoters // Proc. Nat. Acad. Sci. USA, Biol. Sci. - 1983b. - V.80, N 1. - P.21-25);
- уникальные сайты рестрикции AatII, EcoRI, MfeI, BstEII, BamHI, XbaI.
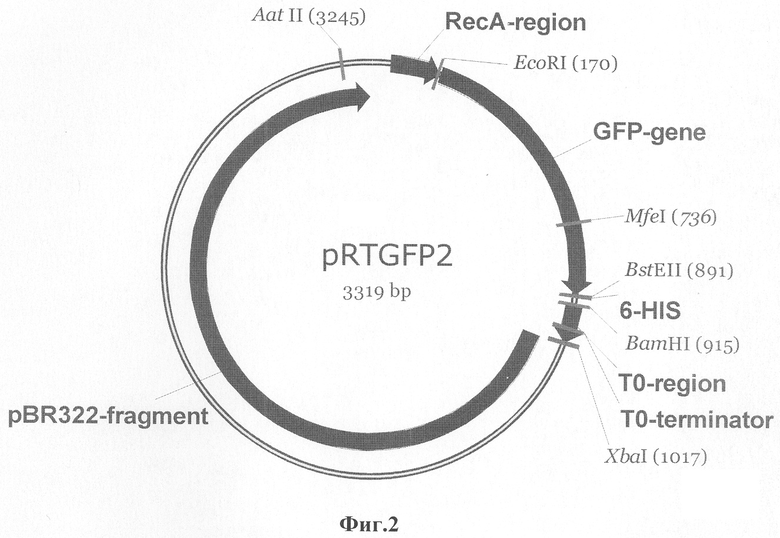
Ниже приведено описание рекомбинантной плазмиды pRTGFP2, которая представлена в перечне последовательностей SEQ ID NO 4. Плазмида pRTGFP2 имеет размер 3319 п.о., молекулярную массу 2,16 мегадальтон и состоит из следующих элементов (все позиции нуклеотидных остатков указаны относительно плазмиды pRTGFP2).
I. EcoRI-BamHI фрагмент плазмидной ДНК (позиции 170-914 п.о.), который содержит:
a) фрагмент мутантного гена gfp Aequorea victoria (позиции 176-889 п.о., который кодирует аминокислотную последовательность белка GFP. Мутантный ген gfp включает в себя кодон инициации трансляции - ATG (позиция 176-178 п.о.);
b) фрагмент ДИК, кодирующий шесть гистидиновых аминокислотных остатков (893-910 п.о.);
c) стоп кодон трансляции (911-913 п.о.);
II. BamHI-XbaI фрагмент ДНК (915-1021 п.о.), (включая JM-сайт полностью), содержащий фрагмент генома бактериофага лямбда (920-1014 п.о.);
III. фрагмент ДНК плазмиды pBR322 (позиции 1022-3317 п.о.) длиной 2296 п.о., соответствующий фрагменту (позиции 2065-4361 п.о.) оригинальной последовательности pBR322, который содержит:
a) участок начала репликации (позиция 1491 п.о.);
b) ген bla, кодирующий β-лактамазу в качестве генетического маркера, определяющего устойчивость трансформированных плазмидой клеток Е.coli к ампициллину (позиция 2249-3112 п.о.);
c) 5'-конец фрагмента содержит часть сайта AatII-рестриктазы;
IV. AatII-EcoRI фрагмент ДНК (3245-169 п.о.), который содержит:
a) фрагмент регуляторной области гена recA Proteus mirabilis, обеспечивающий под воздействием индукторов (мутагенные факторы) инициацию транскрипции матричной РНК, кодирующей белок GFP (позиция 45-141 п.о.);
b) энхансер трансляции фага Т7 (ген 10) - ТТААСТТТА (позиция 149-157 п.о.);
c) последовательность Шайна-Дальгарно из плазмидной ДНК рКК223-3 - AGGA (позиция 161-164 п.о.)
Уникальные сайты узнавания эндонуклеазами рестрикции имеют следующие координаты: AatII (GACGTC) 3240-3245 п.о., EcoRI (GAATTC) 169-174 п.о, Mfel (CAATTG) 735-740, BstEII (GGTCACC) 890-896 п.о., Ватт (GGATCC) 914-919 п.о., XbaI (TCTAGA) 1016-1021 п.о.
Рекомбинантная плазмида pRTGFP2 является усовершенствованным вариантом плазмиды pRTGFP, обладающим более высокой чувствительностью к воздействию индуктора, активирующего систему SOS-репарации, что выражается в увеличении количества белка GFP, синтезируемого плазмидой pRTGFP2 по сравнению с плазмидой pRTGFP, в ответ на воздействие одной и той же дозы мутагена.
Перечень фигур иллюстративного материала
Фиг.1. Физико-генетическая карта рекомбинантной плазмидной ДНК pRTGFP. GFP-gene: фрагмент ДНК (позиции 190-910 п.о.), содержащий кодирующую область мутантного гена gfp Aequorea victoria; 6-HIS: фрагмент ДНК (позиции 913-930 п.о.), кодирующий 6 аминокислотных остатков (а.о.) гистидина; pBR322-fragment: фрагмент оригинальной последовательности ДНК плазмиды pBR322 (позиции 1872-4168 п.о.); RecA-region: регуляторная область гена recA Proteus mirabilis (Позиции 45-189 п.о.); ТО-terminator: терминатор транскрипции ТО бактериофага лямбда (позиции 1771-1865 п.о.).
Фиг.2. GFP-gene: фрагмент ДНК (позиции 176-889 п.о.), содержащий кодирующую область мутантного гена gfp Aequorea victoria; 6-HIS: фрагмент ДНК (позиции 893-910 п.о.), кодирующий 6 аминокислотных остатков (а.о.) гистидина; pBR322-fragment: фрагмент оригинальной последовательности ДНК плазмиды pBR322 (позиции 1022-3317 п.о.); RecA-region: регуляторная область гена recA Proteus mirabilis (позиция 45-141 п.о.); ТО-terminator: терминатор транскрипции ТО бактериофага лямбда (позиции 977-1003 п.о.).
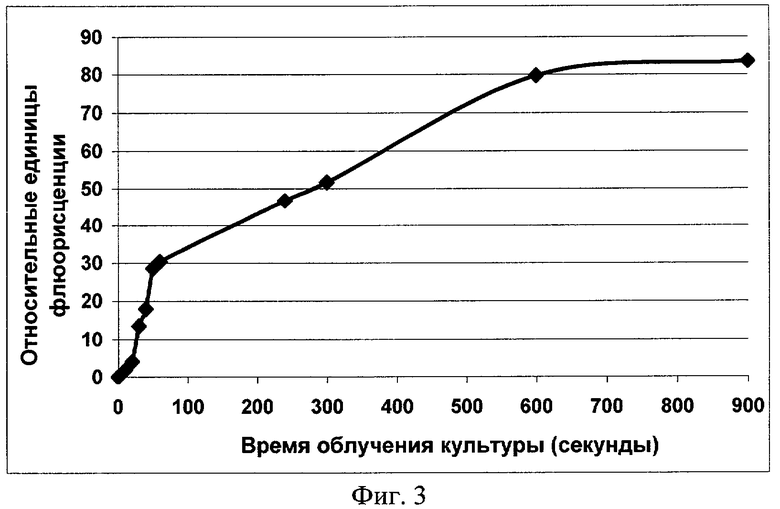
Фиг.3. Возрастание интенсивности флуоресценции культуры клеток E.coli BL21 (DE3), содержащих плазмиду pRTGFP, с увеличением времени (дозы) облучения клеток ультрафиолетовым светом (λ 260 нм, мощность 200 Вт).
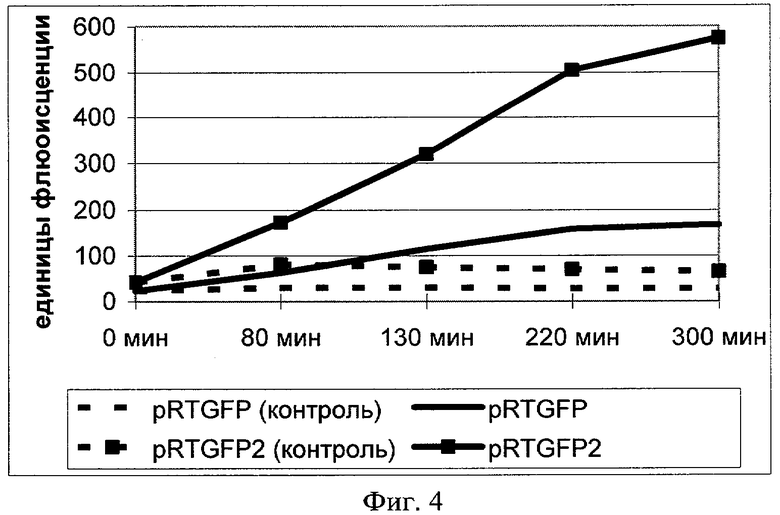
Фиг.4. Флуоресценция клеток культуры E.coli штамма BL21 (DE3) в контроле и в опыте (в опыте те же клетки содержат плазмиды pRTGFP или pRTGFP2 в присутствии налидиксовой кислоты).
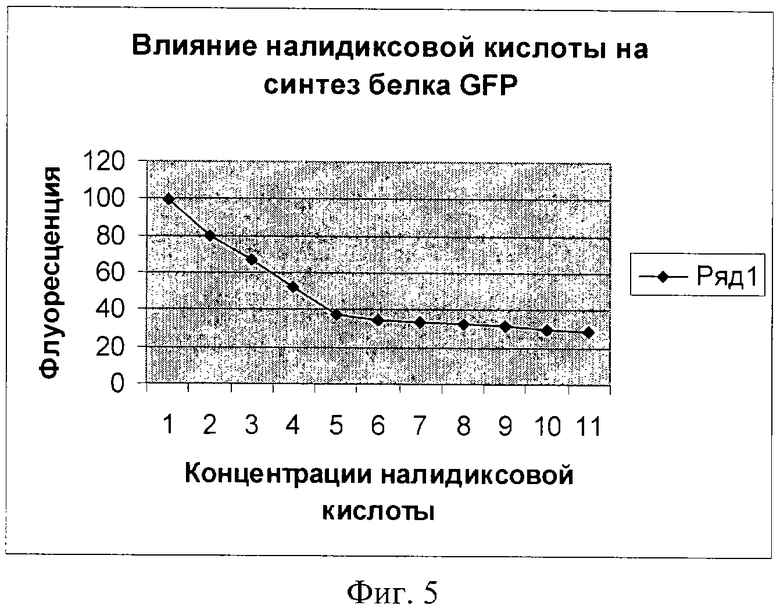
Фиг.5. Влияние налидиксовой кислоты на синтез белка GFP (плазмида pRTGFP). По оси абсцисс указаны значения концентрации в мкМ (1 деление соответствует 1,25 мкМ). По оси ординат указано значение фактора индукции.
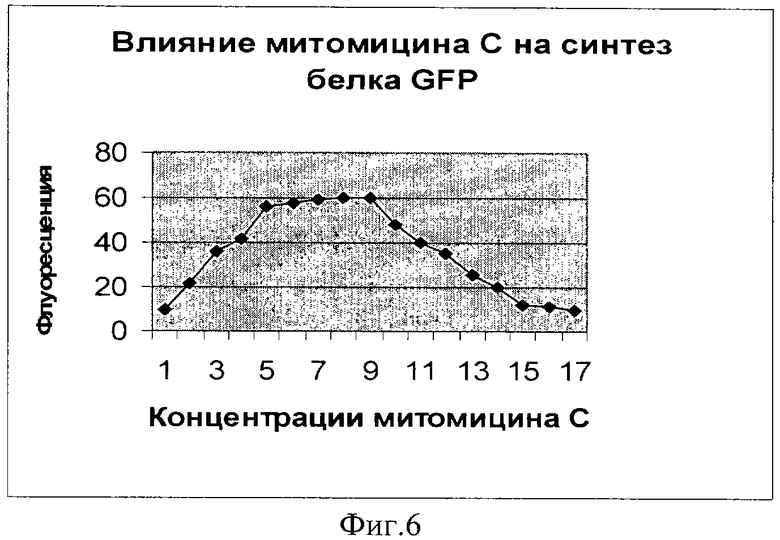
Фиг.6. Влияние митомицина С на синтез белка GFP (плазмида pRTGFP). По оси абсцисс указаны значения концентрации в наномолях (1 деление соответствует 1 наномолю). По оси ординат указано значение фактора индукции.
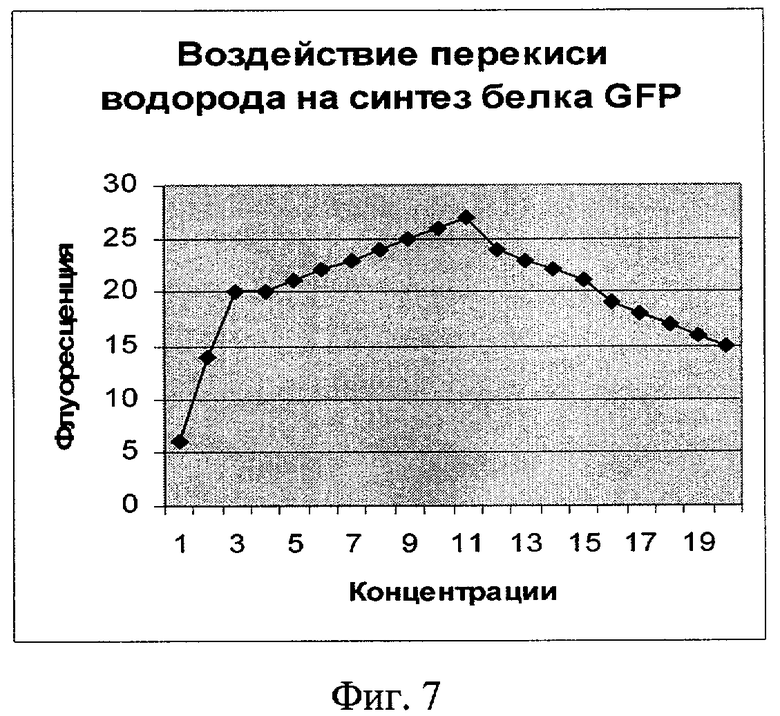
Фиг.7. Влияние перекиси водорода на синтез белка GFP (плазмида pRTGFP). По оси абсцисс указаны значения концентрации в мкМ (1 деление соответствует 100 мкМ). По оси ординат указано значение фактора индукции.
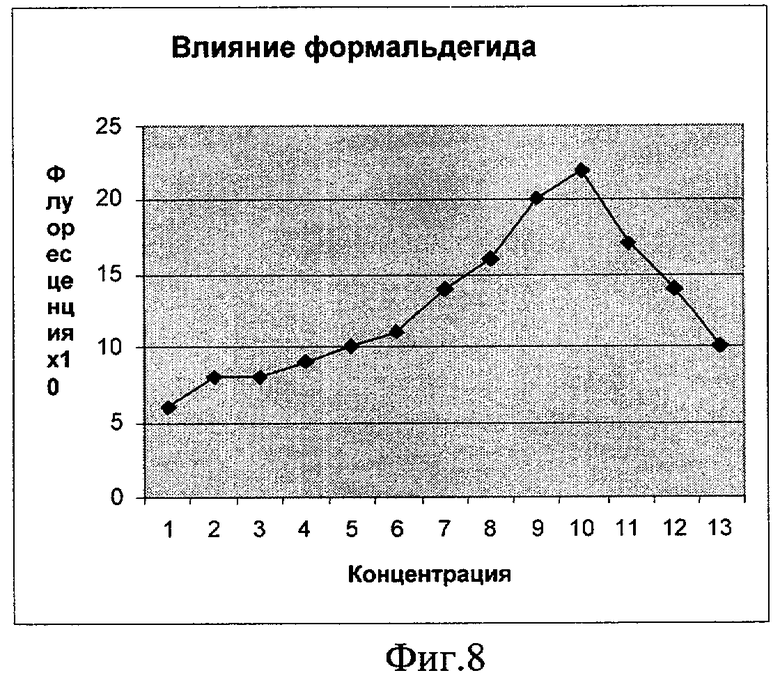
Фиг.8. Влияние формальдегида на синтез белка GFP (плазмида pRTGFP). По оси абсцисс указаны значения концентрации в мкМ (1 деление соответствует 50 мкМ). По оси ординат указано значение фактора индукции.
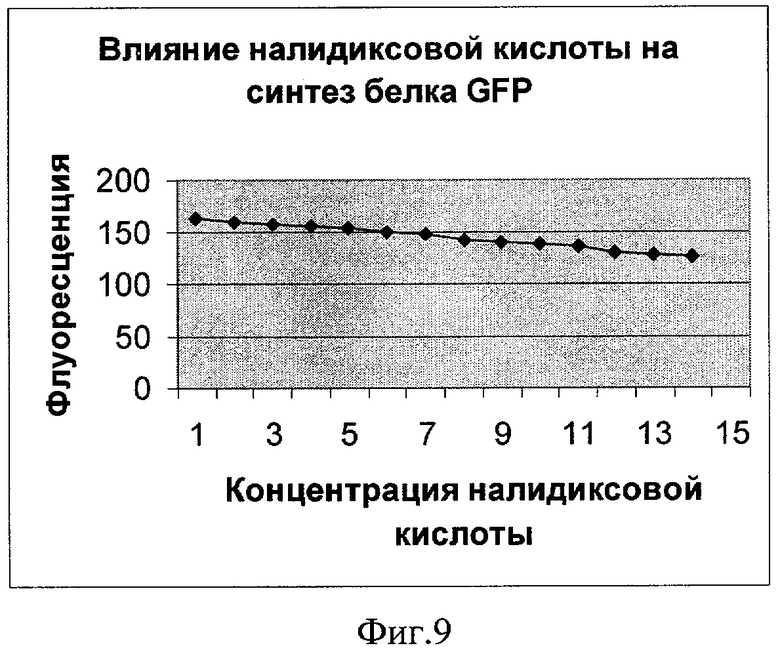
Фиг.9. Влияние налидиксовой кислоты на синтез белка GFP (плазмида pRTGFP2). По оси абсцисс указаны значения концентрации в мкМ (1 деление соответствует 1,25 мкМ). По оси ординат указано значение фактора индукции.
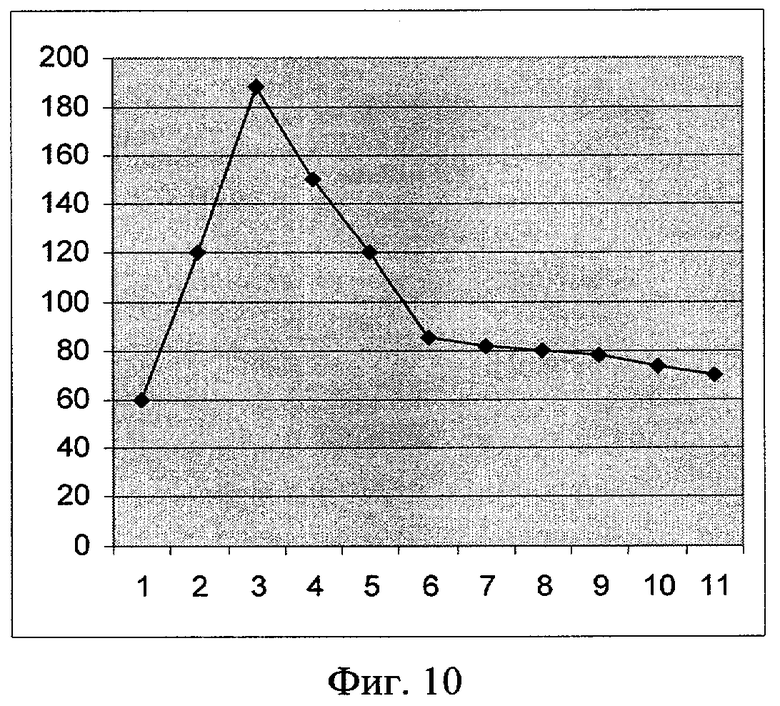
Фиг.10. Влияние митомицина С на синтез белка GFP (плазмида pRTGFP2). По оси абсцисс указаны значения концентрации в наномолях (1 деление соответствует 1 наномолю). По оси ординат указано значение фактора индукции.
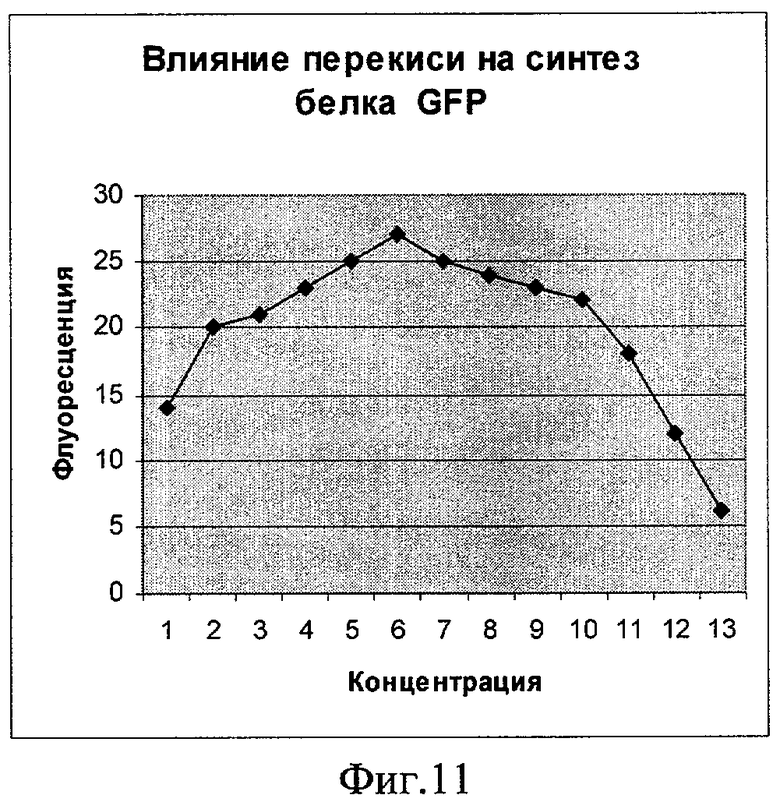
Фиг.11. Влияние перекиси водорода на синтез белка GFP (плазмида pRTGFP2). По оси абсцисс указаны значения концентрации в мкМ (1 деление соответствует 100 мкМ). По оси ординат указано значение фактора индукции.
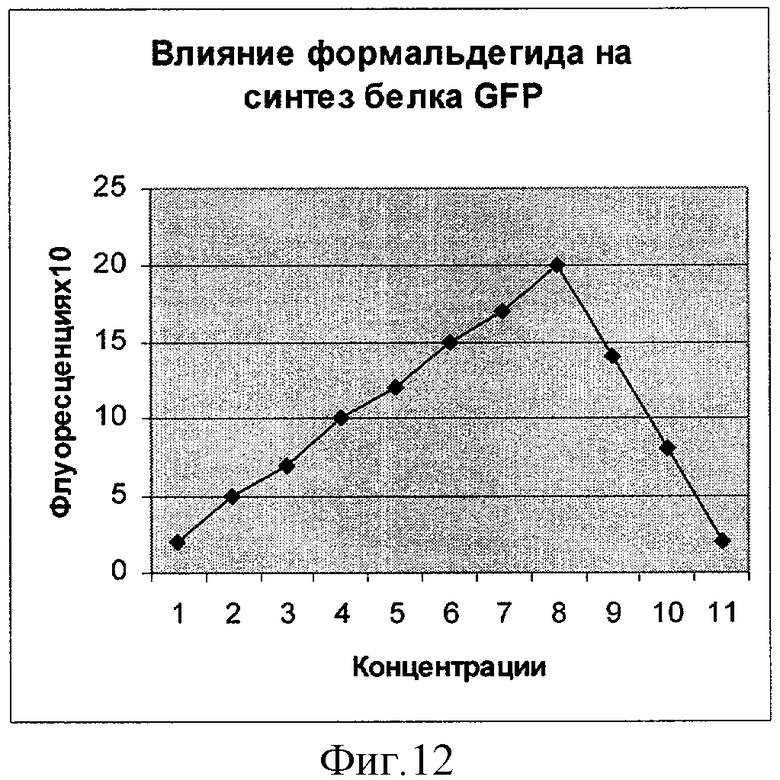
Фиг.12. Влияние формальдегида на синтез белка GFP (плазмида pRTGFP2). По оси абсцисс указаны значения концентрации в мкМ (1 деление соответствует 50 мкМ). По оси ординат указано значение фактора индукции.
Осуществление изобретения
Пример конструирования плазмиды pRTGFP и использования ее для оценки мутагенного воздействия различных факторов химической и физической природы.
Получение гена gfp Aequorea victoria
Источник мутантного гена gfp Aequorea victoria
В качестве источника мутантного гена gfp Aequorea victoria служила плазмида pGreenTIR (William G. Miller, Steven E. Lindow. An improved GFP cloning cassette designed for prokaryotic transcriptional fusions // Gene, V.191, 1997. Р.149-153), ген амплифицировали с помощью полимеразной цепной реакции. Для амплификации структурной области мутантного гена gfp, использовали пару праймеров PR149F+PR150R.
Нуклеотидные последовательности данных праймеров:
прямой праймер PR149F
5'-ATGAATTCTATGAGTAAAGGAGAAGAACTT-3';
обратный праймер PR150R
5'-TTGGTGACCTTTGTATAGTTCATCCATGC-3'.
На 5'-конце праймера PR149F был предусмотрен сайт узнавания рестриктазой EcoRI, а на 5'-конце праймера PR150R был предусмотрен сайт узнавания рестриктазой BstEII (сайты подчеркнуты). Ампликоны гидролизовали соответствующими рестриктазами, очищали фенол хлороформной экстракцией. Гидролиз ДНК эндонуклеазами рестрикции и лигирование Т4 ДНК-лигазой проводили согласно инструкциям фирмы-изготовителя ферментов (СибЭнзим, Новосибирск, Россия).
Полимеразная цепная реакция
Для проведения ПНР использовали амплификатор МС-2 («ДНК-технология», Москва). Реакцию проводили в пластиковых пробирках объемом 0,5 мл ('QSP'', USA). Объем реакционной смеси в одной пробирке составлял 50 мкл. Использовали следующие концентрации компонентов реакционной смеси: 67 мМ Трис-HCl, рН 8,3 при 25°С; 16 мМ (NH4)2SO4; 1,5 mM MgCl2; 1-2 е.а./мкл Pfu ДНК-полимеразы; 0,03% (об/об) Tween 20; 0,2 мкМ каждого праймера; 100 мкМ каждого дНТФ, ˜200 pmol плазмидной ДНК pGreenTIR. ПНР проводили в режиме активного регулирования температуры реакционной смеси в следующих температурно-временных условиях каждого цикла реакции.
1 этап: денатурация ДНК при 94°С 30 сек; отжиг праймеров с ДНК при 52°С 30 сек; синтез цепи ДНК при 72°С 90 сек; количество циклов - 5.
2 этап: денатурация ДНК при 94°С 30 сек; отжиг праймеров с ДНК при 62°С 30 сек; синтез цепи ДНК при 72°C 90 сек; количество циклов - 30.
Получение BstEII-EcoRI фрагмента ДНК плазмиды pREB9-H6-F7
Плазмидную ДНК pREB9-H6-F7 (патент РФ № 2260047 на изобретение «Рекомбинантная плазмидная ДНК, обеспечивающая синтез иммунодоминантного белка Borrelia garinii, используемого для диагностики Лайм-боррелиоза») гидролизовали рестриктазами BstEII и EcoRI. Линейную форму плазмидной ДНК размером 3449 п.о. (фрагмент BstEII-EcoRI) отделяли от продуктов гидролиза в 0,8% агарозном геле. Необходимый фрагмент плазмидной ДНК вырезали из агарозного геля и растворяли в равном объеме раствора, содержащего NaClO4 и ЭДТА в следующих концентрациях 8 М NaClO4, 2мМ ЭДТА, рН раствора 7.0. Раствор остужали до комнатной температуры и вносили в него «силику» (мелкодисперсный оксид кремния) в количестве, достаточном для связывания 3-кратного количества ДНК по отношению к тому ее количеству, которое находится в вырезанной полоске геля. Содержимое пробирки инкубировали в течение 15 мин. После сорбции ДНК силику осаждали центрифугированием в течение 15-20 сек при 10000g. Осадок силики с сорбированной ДНК дважды промывали раствором, содержащим NaClO4 и ЭДТА (4 М NaClO4, 1мМ ЭДТА, рН 7,0). Для освобождения осадка силики от NaClO4 осадок дважды промывали 70% этанолом, затем обезвоживали 96% этанолом и высушивали. Сорбированную на силике ДНК элюировали 50 мкл бидистиллированной воды, инкубируя суспензию в термостате в течение 10 мин при 50°С. После осаждения силики центрифугированием при 10000g в течение 2 мин в растворе оказывалось около 70% исходной ДНК, не содержащей примесей в виде моно - или олигонуклеотидов.
Получение плазмиды pRTGFP
Очищенный фрагмент плазмидной ДНК pREB9-H6-F7 (BstEII-EcoRI) лигировали с предварительно гидролизованным мутантным геном gfp. Лигирование проводили с помощью Т4 ДНК-лигазы. На завершающем этапе лигазной смесью трансформировали клетки Е.coli шт. BL21 (DE3) (ГНЦ вирусологии и биотехнологии, НИИ «Коллекция культур микроорганизмов», п.Кольцове, Новосибирская область, Россия).
Трансформация клеток E.coli рекомбинантной плазмидной ДНК pRTGFP
Трансформацию клеток E.coli рекомбинантной плазмидной ДНК проводили с помощью электропоратора в соответствии с руководством фирмы-производителя прибора - «PeqLab, Biotechnologie GmbH».
После электропорации трансформированные клетки быстро переносили в 2 мл среды ЛБ (Луриа-Бертани), не содержащей антибиотики, и инкубировали в течение 1 час при 37°С. Подращенные трансформированные клетки концентрировали центрифугированием при 10000g в течение 1 мин на настольной центрифуге Beckman и рассевали на агаризованной среде ЛБ с ампициллином в концентрации 100 мкг/мл. Чашки инкубировали при 37°С до образования колоний размером 0,5-1 мм в диаметре.
Часть выросших клонов клеток E.coli была проанализирована с помощью ПЦР с праймерами PR149 и PR150 на наличие в составе рекомбинантной плазмиды pRTGFP мутантного гена gfp. Клоны, давшие положительный ответ, были исследованы на способность синтезировать белок GFP под действием индуктора экспрессии - налидиксовой кислоты. Один из клонов, давших положительный результат во всех анализах, использовался далее для оценки мутагенного воздействия различных веществ.
Детекция экспрессии гена белка GFP в клетках E.coli
Для детекции использовали культуры клеток E.coli, трансформированных рекомбинантной плазмидой pRTGFP. Детекцию флуоресценции синтезируемого белка GFP осуществляли с помощью флуориметра «F-3000» (фирма «Hitachi», Япония).
Индукция экспрессии гена белка GFP в клетках E.coli при действии на клетки различных химических мутагенов
Стадия подготовки культур
Клетки E.coli, штамм BL21 (DE3), трансформированные плазмидой pRTGFP, выращивали в течение ночи при 37°С в среде LB с добавлением ампициллина в концентрации 100 мкг/мл. Пересев ночной культуры E.coli проводили на свежую среду LB с добавлением ампициллина до 100 мкг/мл в отношении 1:50 (к 50 мл среды добавляли 1 мл культуры). Культуру выращивали в течение 4-5 часов до плотности 0,6-0,7 (λ=600 нм).
Стадия индукции синтеза белка
После достижения вышеуказанной плотности к среде LB добавляли мутагены. Синтез белка GFP оценивали при длине волны возбуждения флуоресценции 480 нм и длине волны эмиссии 520 нм. В качестве отрицательного контроля были взяты параллельные культуры клеток E.coli, в которые мутагены не добавляли. В качестве мутагенов использовали налидиксовую кислоту, митомицин С, перекись водорода, формальдегид. Налидиксовая кислота и митомицин С являются стандартными веществами для индукции мутаций в геноме клеток E.coli. Налидиксовая кислота воздействует на ДНК-гиразу и создает протяженные неспаренные участки в ДНК. Митомицин С вызывает сшивки двойной цепи ДНК. Перекись водорода окисляет нуклеотиды ДНК, формальдегид вызывает образование сшивок ДНК.
Интенсивность флуоресценции нормировали на оптическую плотность клеток, равную 1.0, при 600 нм. Интенсивность флуоресценции оценивали с помощью фактора индукции (условные, или относительные единицы).
Фактор индукции (F инд.) определяется как отношение интенсивности флуоресценции в опыте (нормированной по плотности клеток культуры в опыте) к интенсивности флуоресценции в контроле (нормированной по плотности клеток культуры в контроле):
F инд.=(F1 оп. × D к.) / ( F1 к. × D оп.),
F1 оп. - интенсивность флюоресценции в опыте,
F1 к. - интенсивность флюоресценции в контроле,
Dоп. - плотность клеток в опыте,
Dk. - плотность клеток в контроле.
Как видно из фигур 5-8, плазмида pRTGFP не уступает лучшей из двух плазмид в прототипе (вышеуказанная работа Kostrzynska M, Leung KT, Lee Н, Trevors JT.) по чувствительности к перекиси водорода и формальдегиду, а по чувствительности к налидиксовой кислоте и митомицину С значительно ее превосходит (при использовании плазмиды pRTGFP значение фактора индукции равно 65 для концентрации налидиксовой кислоты, равной 4 мкМ; в прототипе значение фактора индукции для указанной концентрации налидиксовой кислоты равно 2. При использовании плазмиды pRTGFP значение фактора индукции равно 35 для концентрации митомицина С, равной 12 наномолей, в прототипе значение фактора индукции для указанной концентрации митомицина С равно 2).
Индукция экспрессии гена белка GFP в клетках E.coli, при действии на клетки физического мутагена
Кроме воздействия химических мутагенов на геном E.coli, нами было исследовано воздействие на геном мутагена физической природы, а именно - электромагнитного излучения ультрафиолетовой области спектра (УФ). УФ вызывает образование димеров тимина в структуре ДНК.
Стадия подготовки культуры была аналогична той, что описана выше. В качестве источника УФ был использован LKB 2011 MacroVue Transilluminator (мощность 8m W/cm2). Культуры облучали и подращивали в пластиковых культуральных планшетах. Объем пробы составлял 2 мл. Время облучения культур составило: 10, 20, 30, 40, 50, 60, 240, 300, 600 и 900 секунд. После облучения культуры инкубировали при 37°С и постоянном покачивании на орбитальном шэйкере в течение 3 часов, после чего осуществляли замер флуоресценции. В качестве отрицательного контроля использовали культуры, не подвергавшиеся облучению. Результаты эксперимента представлены на фиг.3. Из полученных данных видно, что возрастание времени облучения ультрафиолетовым светом (и, соответственно, дозы УФ) приводит к росту количества синтезируемого белка GFP и увеличению интенсивности флуоресценции.
Таким образом, разработанная генетическая конструкция может быть использована для обнаружения генетических повреждений клетки при воздействии на нее мутагенов химической и физической природы.
Пример конструирования плазмиды pRTGFP2 и использование ее для оценки мутагенного воздействия различных веществ
За основу конструкции плазмидной ДНК pRTGFP2 была взята плазмидная ДНК pRTGFP.
Удаление фрагмента ДНК BamHI-XbaI из состава плазмиды pRTGFP и получение плазмидной ДНК pRT
Плазмиду pRTGFP гидролизовали рестриктазами BamHI и XbaI, вектор отделяли от продуктов неполного гидролиза в 0,8% агарозном геле, полосу геля, соответствующую вектору, вырезали, элюировали из нее вектор, очищали его с помощью «силики» и лигировали с ампликонами ТО-региона (методика описана выше в примере конструирования плазмиды pRTGFP). Ампликоны предварительно гидролизовали ВатHI и XbaI рестриктазами и очищали фенол-хлороформной экстракцией. Ампликоны ТО-региона получали в препаративных количествах с помощью ПЦР, в качестве матрицы использовали pRTGFP. Праймеры подбирали так, чтобы после амплификации ТО-регион на 5'-конце содержал сайт BamHI рестриктазы -PR145F, а на 3'-конце сайт AM - PR146R:
Прямой праймер PR145F:
5'-AGGGATCCGACTCCTGTTCATAGATCCAG-3'
(Tm при 200000 pMol = 72,4°С);
Обратный праймер PR146R:
5'-CGTCTAGAGATTCTCACCAATAA-3'
(Tm при 200000 pMol - 57,5°С).
Лигирование проводили с помощью Т4 ДНК-лигазы (методика описана выше в примере конструирования плазмиды pRTGFP), лигазной смесью трансформировали клетки E.coli шт. BL21 (DE3) с использованием электропорации (методика описана выше в примере конструирования плазмиды pRTGFP). Часть выросших клонов клеток E.coli была проанализирована с помощью ПЦР с праймерами PR149F и PR146R. По размерам амплифицируемого фрагмента - 847 п.о. (ген gfp + TO-регион) судили о выбросе фрагмента ДНК из состава плазмиды pRTGFP и получении плазмидной ДНК pRT. Клоны, давшие положительный ответ, были дополнительно исследованы на способность синтезировать белок GFP под действием индуктора экспрессии - налидиксовой кислоты. Один из клонов, давших положительный результат во всех анализах, был использован для дальнейшей работы.
Изменение промотора плазмидной ДНК pRT - получение плазмиды pRTGFP2.
С целью усиления экспрессирующей силы промотора в область промотора плазмидной ДНК pRT был введен энхансер трансляции из фага Т7, ген 10 (Cheng X, Patterson ТА. Construction and use of lambda PL promoter vectors for direct cloning and high level expression of PCR amplified DNA coding sequences. // Nucleic Acids Res. 1992. Sep 11; 20(17):4591-8.). Кроме того, был заменен рибосомосвязывающий сайт - последовательность Шайна-Дальгарно (SD-сайт), SD-сайт позаимствован из синтетического taq-промотора (De Boer N.A., Comstock L.J., Vasser M. The tac promoter: A functional hybrid derived from the trp and lac promoters // Proc. Nat. Acad. Sci. USA, Biol. Sci. - 1983 b. - V.80, N 1. - P.21-25).
Сборка pRTGFP2 происходила из трех фрагментов. В качестве акцептора использовали плазмиду pRT, предварительно гидролизованную AatII и EcoRl рестриктазами. В результате гидролиза вырезали фрагмент ДНК, содержащий регуляторную область гена recA Proteus mirabilis. Фрагмент ДНК (EcoRI-AatII), полученный гидролизом плазмиды pRT, очищали и подготавливали для лигирования с помощью стандартных методик, описанных в примере конструирования плазмиды pRTGFP.
Донорами являлись два фрагмента: первый - фрагмент оригинальной регуляторной области гена recA Proteus mirabilis (231 п.о.), содержащий SOS-ВОХ; второй - синтетический фрагмент: два олигонуклеотида, гибридизованные вместе и образующие дуплекс с липкими концами для Mfel и EcoRI рестриктаз. Ниже приведена нуклеотидная последовательность синтетического дуплекса, содержащего энхансер (ENH) и рибосомосвязывающий сайт (SD):
Фрагмент оригинальной регуляторной области гена recA Proteus mirabilis, содержащий SOS-BOX, нарабатывали в препаративных количествах с помощью ПЦР, где в качестве матрицы использовали pRT. Праймеры подбирали так, чтобы амплифицируемый фрагмент на 5'-конце содержал сайт AatII рестриктазы - PRAatF, а на 3'-конце сайт MfeI - PR141R:
Прямой праймер PRAatIIF: 5'-AGTGCCACCTGACGTCTAAG-3'
(Tm при 200000 pMol = 60,3°С).
Обратный праймер PR141R:
5'-GCCAATTGTATATGATTTTGCTTGTTGAAA-3'
(Tm при 200000 pMol - 68,0°С).
Подготовленные фрагменты лигировали Т4 ДНК-лигазой, лигазной смесью трансформировали клетки Е.coli шт. BL21 (DE3) с использованием электропорации (методы описаны выше). Часть выросших клонов клеток E.coli была проанализирована с помощью ПЦР с праймерами PRAatIIF и PR144 на наличие в составе рекомбинантной плазмиды pRTGFP2 встроенного синтетического дуплекса, содержащего энхансер и SD-сайт. Клоны, давшие положительный ответ, были дополнительно исследованы на способность синтезировать белок GFP под действием индуктора экспрессии - налидиксовой кислоты. Один из клонов давших положительный результат во всех анализах был использован далее для оценки мутагенного воздействия различных веществ.
Индукция экспрессии гена белка GFP в клетках E.coli при действии на клетки различных химических мутагенов
Эксперименты проводили со следующими мутагенами: налидиксовой кислотой, митомицином С, перекисью водорода, формальдегидом.
Как показывают фигуры 9-12, плазмида pRTGFP2 не уступает лучшей из двух плазмид в прототипе по чувствительности к перекиси водорода и формальдегиду, а по чувствительности к налидиксовой кислоте и митомицину С значительно ее превосходит. При использовании плазмиды pRTGFP2 для концентрации налидиксовой кислоты, равной 4 мкМ, значение фактора индукции равно 155; в прототипе значение фактора индукции для указанной концентрации налидиксовой кислоты равно 2. При использовании плазмиды pRTGFP2 для концентрации митомицина С, равной 12 наномолей, значение фактора индукции равно 20; в прототипе значение фактора индукции для указанной концентрации митомицина С равно 2.
Сравнение экспрессии GFP в клетках E.coli. штамм BL21 (DE 3), несущих плазмидные ДНК pRTGFP и pRTGFP2.
Культуры выращивались в дублированном варианте, один вариант индуцировался налидиксовой кислотой, а второй вариант культур оставался не индуцированным как контроль. После достижения ОП600 культур 0.8-1.2 о.е., все культуры разбавляли до одинаковой оптической плотности, равной 0,8 о.е. Затем осуществляли индукцию экспрессии белка GFP налидиксовой кислотой (50 мкг/мл) в течение 5 часов, предварительно отобрав нулевую точку во всех культурах. По мере инкубации культур и накопления белка GFP через определенный промежуток времени из всех культур отбирали аликвоту (3 мл). Аликвоты хранили при 20°С. По окончании индукции измеряли значение флюоресценции всех аликвот. По данным флюоресценции строили графики накопления белка GFP в индуцируемых и контрольных культурах.
Из данных фиг.4 видно, что уровень экспрессии белка GFP в штамме E.coli, содержащем плазмиду pRTGFP2 примерно в 3 раза больше, чем в исходной конструкции pRTGFP. Таким образом, изменения привели к увеличению силы промотора, т.е. к увеличению экспрессии целевого белка - GFP и, следовательно, к увеличению чувствительности плазмидной ДНК pRTGFP2 к мутагенному воздействию индукторов, активирующих систему SOS-репарации.






Изобретение относится к области биотехнологии и может быть использовано в пищевой, фармацевтической и химической промышленности. Рекомбинантная плазмидная ДНК обеспечивает синтез флуоресцирующего мутантного белка GFP из Aequorea victoria в Rec + штаммах E.coli под контролем регуляторной области гена recA Proteus mirabilis. Применение изобретения обеспечивает повышенную чувствительность обнаружения ряда повреждающих ДНК факторов химической природы, а также повреждающих воздействий физической природы. 2 н.п. ф-лы, 12 ил.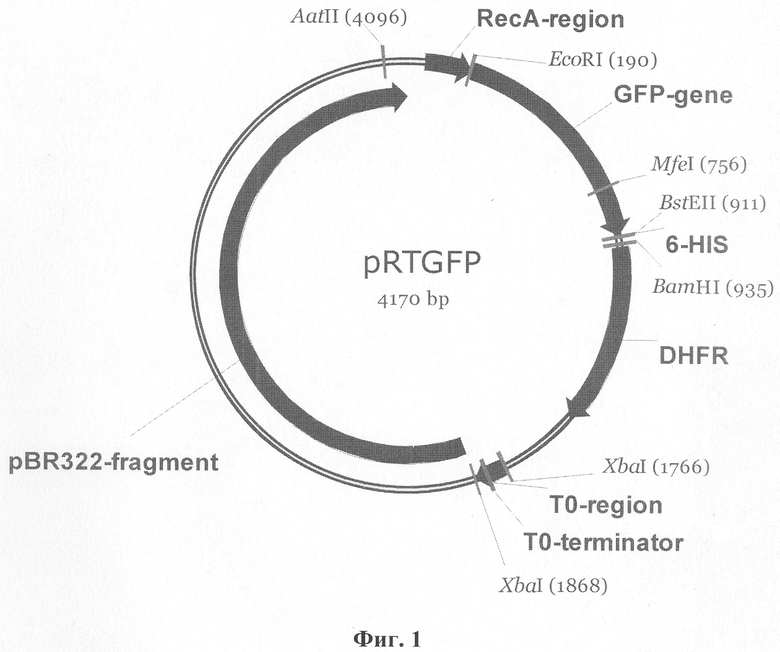
| J Microbiol Methods | |||
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| KR 1020040026253, 31.03.2004 | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Appl Environ Microbiol | |||
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Mol Gen Genet | |||
| Устройство для видения на расстоянии | 1915 |
|
SU1982A1 |
| Рекомбинантная плазмидная ДНК pIL-2/21, кодирующая человеческий интерлейкин-2, способ ее конструирования и штамм бактерий ЕSснеRIснIа coLI - продуцент человеческого интерлейкина-2 | 1990 |
|
SU1761805A1 |

