1. РОДСТВЕННЫЕ ЗАЯВКИ
Данная заявка основана на заявках на патент США №60/589058, поданной 19 июля 2004 г., №60/619153, поданной 15 октября 2004 г., №60/632578, поданной 2 декабря 2004 г., и №60/655838, поданной 24 февраля 2005 г., и №60/655803, поданной 24 февраля 2005 г., и включает полные описания данных изобретений посредством ссылки. Эта заявка также включает посредством ссылки следующие заявки, поданные 19 июля 2005 г. Radhakrishnan с соавт.: заявку на патент США №11/184668 под названием "Cation complexes of insulin compound conjugates, formulations and uses thereof; заявку на патент США №11/184594 под названием "Insulin-oligomer compound conjugates, formulations and uses thereof"; заявку на патент США №11/184528 под названием "Fatty acid formulations for oral delivery of proteins and peptides, and uses thereof".
2. ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к новым конъюгатам соединений инсулинов, в которых инсулин или аналог инсулина связан с модифицирующей группировкой. Изобретение также относится к катионным комплексам таких конъюгатов соединений инсулинов и к фармацевтическим препаратам, содержащим такие конъюгаты соединений инсулинов и/или модифицирующие группировки.
3. ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Цинковый комплекс соединения инсулина имеется в продаже, например, под торговыми названиями HUMULIN® и HUMALOG®. Цинковый комплекс инсулина обычно существует в гексамерной форме.
Описаны различные способы применения цинка в кристаллизации ацилированного инсулина. Например, в патентной публикации США №20010041786, опубликованной 15 ноября 2001 г., Mark L. Brader с соавт., под названием "Stabilized acylated insulin formulations", описан препарат с водным раствором для парентеральной доставки, в частности, в виде препарата для инъекций, имеющий рН от 7,1 до 7,6, содержащий инсулин, ацилированный жирной кислотой, или аналог инсулина, ацилированный жирной кислотой, и стабилизированный с помощью цинка и предпочтительно фенольного соединения. В патенте США №6451970 под названием "Peptide derivatives", опубликованном 17 сентября 2002 г. Schaffer с соавт., права на который переданы Novo Nordisk A/S, описаны производные соединения инсулина и аналогов инсулина, где N-концевая аминогруппа В-цепи и/или е-аминогруппа Lys в положении В28, В29 или В30 ацилирована с использованием длинноцепочечной углеводородной группы, содержащей от 12 до 22 атомов углерода, и их цинковые комплексы.
Описали применение протаминов и фенольных соединений для кристаллизации ацилированного инсулина. В патентах США №№6268335 (31 июля 2001 г.) и 6465426 (10 октября 2002 г.), опубликованных Brader, которые оба озаглавлены "Insoluble insulin compositions", описаны нерастворимые композиции, содержащие ацилированный инсулин, комплексообразующее соединение протамина, гексамер-стабилизирующее фенольное соединение и двухвалентный катион металла.
Существующие подходы специально предназначены для кристаллизации нативного соединения инсулина, или аналогов соединений инсулинов, или для ацилированных соединений инсулинов, имеющих повышенную липофильность по сравнению с неацилированными соединениями инсулинами. В данной области техники существует необходимость в фармацевтически приемлемых комплексах, включающих производные соединений инсулинов, отличные от ацилированного соединения инсулина, такие как гидрофильные и/или амфифильные производные соединений инсулинов, и в стабилизации неацилированных липофильных аналогов соединений инсулинов. В данной области техники существует необходимость также в новых белковых конъюгатах, имеющих повышенную биодоступность или другие улучшенные фармацевтические свойства по сравнению с существующими конъюгатами. В данной области техники существует необходимость в новых препаратах, которые облегчают пероральную доставку белков и белковых конъюгатов. Наконец, существует необходимость в комбинированном подходе для улучшения пероральной биодоступности белка, такого как соединение инсулин, который включает улучшенный пероральный белковый конъюгат, предложенный в виде твердого вещества в улучшенном препарате для максимизации преимуществ пероральной доставки белков.
4. КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В общем, в изобретении предложен комплекс, включающий конъюгат соединения инсулина, в котором соединение инсулин конъюгировано с модифицирующей группировкой, и катион, где конъюгат соединения инсулина образует комплекс с катионом. Соединение инсулин может представлять собой, например, нативный инсулин или аналоги инсулина. Примеры соединений инсулинов включают в себя инсулин человека, лизпроинсулин, des30 инсулин, нативный проинсулин, искусственный проинсулин и так далее. Катионный компонент может представлять собой, например, двухвалентный катион металла, выбранный из группы, состоящей из Zn++, Mn++, Са++, Fe++, Ni++, Cu++, Co++ и Mg++.
Модифицирующая группировка может быть выбрана так, чтобы сделать конъюгат соединения инсулина более, менее или таким же растворимым, как соответствующее неконъюгированное соединение инсулин. Предпочтительно, модифицирующая группировка выбрана так, чтобы сделать конъюгат соединения инсулина по меньшей мере в 1,05; 1,25; 1,5; 1,75; 2; 2,5; 3; 3,5; 4; 4,5; 5; 5,5; 6; 6,5; 7; 7,5; 8; 8,5; 9; 9,5; 10; 10,5; 11; 11,5; 12; 12,5; 13; 13,5; 14; 14,5 или 15 раз более растворимым, чем соответствующее неконъюгированное соединение инсулин, в водном растворе при рН приблизительно 7,4. Предпочтительно, модифицирующая группировка выбрана так, чтобы конъюгат соединения инсулина имел растворимость в воде, которая превышает приблизительно 1 г/л, 2 г/л, 3 г/л, 4 г/л, 5 г/л, 10 г/л, 15 г/л, 20 г/л, 25 г/л, 50 г/л, 75 г/л, 100 г/л, 125 г/л или 150 г/л при рН приблизительно 7,4. Кроме того, модифицирующая группировка выбрана так, чтобы сделать конъюгат соединения инсулина таким же или более растворимым, чем соответствующее неконъюгированное соединение инсулин, и растворимость в воде конъюгата соединения инсулина уменьшена путем добавления цинка. В другом воплощении модифицирующая группировка выбрана так, чтобы сделать конъюгат соединения инсулина таким же или более растворимым, чем соответствующее неконъюгированное соединение инсулин; растворимость в воде конъюгата соединения инсулина уменьшена путем добавления цинка; и растворимость в воде комплекса выше растворимости в воде соединения инсулина. В еще одном воплощении относительная липофильность конъюгата соединения инсулина по сравнению с соответствующим исходным соединением инсулином (kотн) равна 1 или меньше 1.
В изобретении также предложены новые конъюгаты соединений инсулинов, содержащие соединение инсулин, конъюгированное с модифицирующей группировкой. Например, в изобретении предложены соединения инсулины, связанные с модифицирующей группировкой, имеющей формулу:
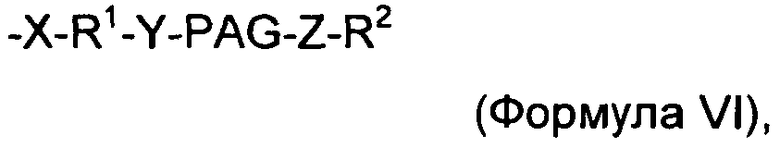
где:
X, Y и Z представляют собой независимо выбранные связывающие группы, и каждая из них возможно присутствует, и X, когда присутствует, связан с соединением инсулином ковалентной связью,
по меньшей мере один из R1 и R2 присутствует и представляет собой низший алкил и возможно может включать карбонильную группу,
R2 представляет собой блокирующую группу, такую как -CH3, -Н, тозилат, или активирующую группу, и
PAG представляет собой линейную или разветвленную углеродную цепь, включающую одну или более алкаленгликолевых группировок (т.е. оксиалкаленовых группировок) и возможно включающую одну или более дополнительных группировок, выбранных из группы, состоящей из -S-, -O-, -NH- и -С(O)-, и
где наибольшее число тяжелых атомов, имеющихся в модифицирующей группировке, составляет 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 или 25.
В воплощениях изобретения любой один или более из X, Y и Z могут отсутствовать. Кроме того, X, Y и/или Z, когда присутствуют, независимо могут быть выбраны из -С(O)-, -O-, -S-, -С- и -NH-. В одном воплощении Z представляет собой -С(O)-. В другом воплощении Z не присутствует.
В некоторых воплощениях R1 представляет собой низший алкил, и R2 не присутствует. В других воплощениях R2 представляет собой низший алкил, и R1 не присутствует.
В другом воплощении модифицирующая группировка может включать линейную или разветвленную группировку, представляющую собой замещенную углеродную цепь, имеющую основную цепь из 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 19, 19, 20, 21, 22, 23, 24 или 25 атомов, выбранных из группы, состоящей из -C, -C-, -O-, =O, -S-, -NH-, -Si-. Тяжелые атомы обычно включают в себя один или более атомов углерода и один или более неуглеродных тяжелых атомов, выбранных из группы, состоящей из -O-, -S-, -NH- и =O. Атомы углерода и неуглеродные тяжелые атомы обычно присутствуют в соотношении по меньшей мере 1 атом углерода на каждый неуглеродный тяжелый атом, предпочтительно по меньшей мере 2 атома углерода на каждый неуглеродный тяжелый атом, более предпочтительно по меньшей мере 3 атома углерода на каждый неуглеродный тяжелый атом. Атомы углерода и атомы кислорода обычно присутствуют в соотношении по меньшей мере 1 атом углерода на каждый атом кислорода, предпочтительно по меньшей мере 2 атома углерода на каждый атом кислорода, более предпочтительно по меньшей мере 3 атома углерода на каждый атом кислорода. Модифицирующая группировка может включать одну или более блокирующих групп, таких как разветвленный или линейный C1-6, разветвленный или линейный или карбонил. Модифицирующая группировка обычно включает атомы водорода, и один или более из данных атомов водорода может быть замещен фтором (который является тяжелым атомом, но не следует его рассматривать в качестве тяжелого атома в вышеприведенной формуле). В некоторых случаях модифицирующая группировка, в частности, может исключать незамещенные алкильные группировки. Модифицирующая группировка может быть связана, например, с подходящей группой на аминокислоте, такой как аминогруппа, гидроксильная группа или свободная карбоновокислотная группа полипептида, например, посредством связывающей группы, такой как карбаматная, карбонатная, простая эфирная, сложноэфирная, амидная группа или вторичная аминная группа, или посредством дисульфидной связи. Молекулы в связывающей группе считаются частью модифицирующей группировки. В предпочтительном воплощении молекулярная масса модифицирующей группировки меньше молекулярной массы модифицирующей группировки HIM2.
Изобретение включает конъюгаты соединений инсулинов, имеющие модифицирующие группировки формулы:
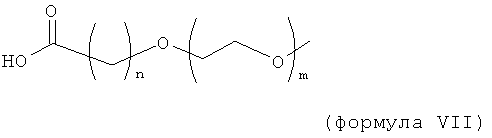 ,
,
где n равно 1, 2, 3 или 4, и m равно 1, 2, 3, 4 или 5; и/или
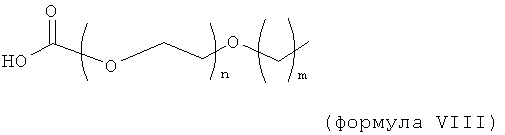 ,
,
где n равно 1, 2, 3, 4 или 5, и m равно 1, 2, 3 или 4.
Следует принимать во внимание, что новые модифицирующие группировки, а также применение таких группировок для модификации инсулина и других полипептидов сами являются аспектами данного изобретения.
В изобретении также предложены новые препараты, содержащие конъюгаты соединений инсулинов и/или конъюгаты катиона и соединения инсулина по изобретению. Авторы изобретения неожиданно обнаружили, что некоторые композиции жирных кислот являются особенно полезными, в частности, для пероральной доставки полипептидов и полипептидных конъюгатов, таких как инсулин и конъюгаты соединений инсулинов, и/или для пероральной доставки комплексов конъюгатов соединений инсулинов с катионами по изобретению. В одном аспекте в изобретении предложены композиции жирных кислот с одной или более насыщенными или ненасыщенными С4, С5, С6, С7, С8, С9 или С10 жирными кислотами и/или солями таких жирных кислот. Предпочтительными жирными кислотами являются каприловая, каприновая, миристиновая и лауриновая. Предпочтительными солями жирных кислот являются натриевые соли каприловой, каприновой, миристиновой и лауриновой кислот. Содержание жирных кислот в композиции обычно находится в диапазоне, имеющем в качестве нижнего предела приблизительно 0,1; 0,2; 0,3; 0,4; 0,5; 0,6; 0,7; 0,8; 0,9; 1,0; 1,1; 1,2; 1,3; 1,4; 1,5; 1,6; 1,7; 1,8; 1,9; 2,0; 2,1; 2,2; 2,3; 2,4; 2,5; 2,6; 2,7; 2,8; 2,9 или 3,0% масс./масс., и имеющем в качестве верхнего предела приблизительно 3,0; 3,1; 3,2; 3,3; 3,4; 3,5; 3,6; 3,7; 3,8; 3,9; 4,0; 4,1; 4,2; 4,3; 4,4; 4,5; 4,6; 4,7; 4,8; 4,9; 5,0; 5,1; 5,2; 5,3; 5,4; 5,5; 5,6; 5,7; 5,8; 5,9; 6,0; 6,1; 6,2; 6,3; 6,4; 6,5; 6,6; 6,7; 6,8; 6,9; 7,0; 7,1; 7,2; 7,3; 7,4; 7,5; 7,6; 7,7; 7,8; 7,9; 8,0; 8,1; 8,2; 8,3; 8,4; 8,5; 8,6; 8,7; 8,8; 8,9; 9,0; 9,1; 9,2; 9,3; 9,4; 9,5; 9,6; 9,7; 9,8; 9,9; 10,0; 10,1; 10,2; 10,3; 10,4; 10,5; 10,6; 10,7; 10,8; 10,9; 11,0; 11,1; 11,2; 11,3; 11,4; 11,5; 11,6; 11,7; 11,8; 11,9 и 12,0% масс./масс. В еще одном воплощении содержание жирных кислот в композиции обычно находится в диапазоне, имеющем в качестве нижнего предела приблизительно 0,1; 0,2; 0,3; 0,4; 0,5; 0,6; 0,7; 0,8; 0,9; 1,0; 1,1; 1,2; 1,3; 1,4; 1,5; 1,6; 1,7; 1,8; 1,9; 2,0; 2,1; 2,2; 2,3; 2,4; 2,5; 2,6; 2,7; 2,8; 2,9 или 3,0% масс./масс., и имеющем в качестве верхнего предела приблизительно 3,0; 3,1; 3,2; 3,3; 3,4; 3,5; 3,6; 3,7; 3,8; 3,9; 4,0; 4,1; 4,2; 4,3; 4,4; 4,5; 4,6; 4,7; 4,8; 4,9; 5,0; 5,1; 5,2; 5,3; 5,4; 5,5; 5,6; 5,7; 5,8; 5,9; 6,0; 6,1; 6,2; 6,3; 6,4; 6,5; 6,6; 6,7; 6,8; 6,9; 7,0; 7,1; 7,2; 7,3; 7,4; 7,5; 7,6; 7,7; 7,8; 7,9; 8,0; 8,1; 8,2; 8,3; 8,4; 8,5; 8,6; 8,7; 8,8; 8,9; 9,0; 9,1; 9,2; 9,3; 9,4; 9,5; 9,6; 9,7; 9,8; 9,9; 10,0; 10,1; 10,2; 10,3; 10,4; 10,5; 10,6; 10,7; 10,8; 10,9; 11,0; 11,1; 11,2; 11,3; 11,4; 11,5; 11,6; 11,7; 11,8; 11,9 или 12,0% масс./масс., и содержание одной жирной кислоты, предпочтительно каприловой, каприновой, миристиновой или лауриновой, или ее соли в содержании жирных кислот в композиции обычно выше приблизительно 90; 91; 92; 93; 94; 95; 96; 97; 98; 99; 99,5; 99,6; 99,7; 99,8 или 99,9% масс./масс.
В изобретении также предложен способ лечения недостаточности инсулина или иного пополнения инсулина у субъекта с использованием конъюгатов соединений инсулинов, комплексов конъюгатов соединений инсулинов с катионами и/или препаратов по изобретению. Данные способы обычно включают введение терапевтически эффективного количества одного или более конъюгатов соединений инсулинов, комплексов конъюгатов соединений инсулинов с катионами и/или препаратов по изобретению субъекту, нуждающемуся в этом.
5. КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На фиг.1-15Б представлены микрофотографии различных кристаллических твердых веществ по изобретению. На фиг.1 и 2 представлены микрофотографии, полученные с использованием микроскопа Zeiss Axiovert, показывающие Zn-комплекс Т-типа для HIM2 в концентрации 30 г/л, кристаллы выращивали в течение 24 ч. На фиг.3 представлена микрофотография, полученная с использованием микроскопа Zeiss Axiovert, показывающая Zn-комплекс Т-типа для HIM2 в концентрации 30 г/л, кристаллы выращивали в течение 5 суток. На фиг.4 представлена микрофотография, полученная с использованием микроскопа Zeiss Axiovert, показывающая Zn-комплекс R-типа для HIM2 в концентрации 30 г/л, кристаллы выращивали в течение 4 суток. На фиг.5 представлена микрофотография кристаллического Zn-комплекса R-типа для IN105, содержащего 30% органического вещества. На фиг.6А-10Б представлены микрофотографии различных Zn-комплексов R-типа для HIM2, полученных с использованием органического растворителя. На фиг.11А-14Б представлены микрофотографии кристаллов различных Zn-комплексов R-типа, полученных в результате сокристаллизации HIM2 и IN105. На фиг.15А-15Б представлены микрофотографии кристаллов различных Zn-комплексов R-типа, полученных в результате сокристаллизации HIM2 и инсулина человека. Данное изобретение включает кристаллы, имеющие морфологию, показанную на любой из фиг.1-15Б.
На фиг.16-20 представлены результаты анализа глюкозы в крови у мышей (MBGA от англ. Mouse Blood Glucose Assay) для HIM2 и различных комплексов Zn-HIM2. На фиг.16 показаны MBGA-профили биологической активности для HIM2. На фиг.17 показаны MBGA-профили биологической активности для продукта Zn HIM2 соединения инсулина R-типа. На фиг.18 показаны MBGA-профили биологической активности для продукта Zn HIM2 соединения инсулина Т-типа. На фиг.19 показаны MBGA-профили биологической активности для продукта Zn HIM2 соединения инсулина с протамином. На фиг.20 показан эффект протаминового комплекса R-типа на снижение уровня глюкозы через 30 и 90 минут после введения дозы.
На фиг.21-24 показаны MBGA-профили биологической активности для IN-186, IN-192, IN-190, IN-191, IN-189, IN-178, IN-193, IN-194, IN-185, IN-196 и IN-197.
На фиг.25 и 26 приведены результаты исследований с фиксированным уровнем глюкозы у собак для комплексов Zn-HIM2 по изобретению.
На фиг.27 и 28 приведены результаты исследований с фиксированным уровнем глюкозы у собак для комплексов Zn-IN105 по изобретению.
На фиг.29 и 30 приведены результаты исследований с фиксированным уровнем глюкозы у собак, которым вводили IN105 в дозе 0,25 мг/кг в пероральном жидком препарате [820], содержащем 3% (масс./об.) натриевой соли каприновой кислоты в фосфатном буфере без дополнительных эксципиентов.
На фиг.31-33 приведены результаты исследований с фиксированным уровнем глюкозы у собак, которым давали таблетки, содержащие 6 мг IN105 и 150 мг маннита, 30 мг Exlotab с 143 мг капрата с или без 143 мг лаурата (пероральная доза составляла 0,25 мг/кг).
На фиг.34-37 приведены результаты исследований с фиксированным уровнем глюкозы у собак, которым давали таблетки-прототипы, содержащие 150 мг и 280 мг капрата, и таблетки, содержащие 140 мг/140 мг капрата/лаурата (доза ZN IN-105 составляла 6 мг или 0,25 мг/кг) и сравнение с подкожным* и ингаляционным* введением стандартного инсулина.
На фиг.38-42 приведены результаты исследований с фиксированным уровнем глюкозы у собак, которым давали таблетки, содержащие 150 мг и 286 мг капрата, и таблетки, содержащие 140 мг/140 мг капрата/лаурата (доза ZN IN-105 составляла 0,25 мг/кг).
6. ОПРЕДЕЛЕНИЯ
Ниже приведены определения терминов, используемых в данном описании изобретения и в формуле изобретения. Предложенные определения, если не оговорено особо, использованы везде в описании настоящего изобретения. Термины, не определенные в данном описании, имеют значение, которое обычно подразумевается в той области техники, к которой относится данный термин.
Термин "вставка", когда его используют в отношении аминокислотной последовательности, включает добавления одной или более аминокислот с одного или обоих концов последовательности, а также инсерции в пределах данной последовательности.
Термин "комплекс" относится к ассоциации молекул, в которой одно или более соединений инсулинов или конъюгатов соединений инсулинов образуют координационные связи с одним или более атомами или ионами металла. Комплексы могут существовать в растворе или в виде твердого вещества, такого как кристалл, микрокристалл, или аморфного твердого вещества. Термин "смесь комплексов" означает смесь, содержащую два или более различных комплексов, либо в растворе, либо в твердой форме. Смеси комплексов могут включать в себя, например, комплексы с разными соединениями инсулинами, разными конъюгатами соединений инсулинов, разными гибридными комплексами, разными катионами, комбинации вышеупомянутого и тому подобное. Термин "гибридный комплекс" означает комплекс конъюгата соединения инсулина с катионом, содержащий два или более разных соединений инсулинов и/или конъюгатов соединений инсулинов.
Термин "комплексообразующий агент" означает молекулу, которая имеет множество зарядов и которая связывается или образует комплекс с конъюгатами соединений инсулинов. Примеры комплексообразующих агентов, подходящих для применения в настоящем изобретении, включают в себя протамины, сурфен, глобиновые белки, спермин, спермидин, альбумин, аминокислоты, карбоновые кислоты, поликатионные полимерные соединения, катионные полипептиды, анионные полипептиды, нуклеотиды и антисенс. Смотри Brange, J., Galenics of Insulin compound, Springer-Verlag, Berlin Heidelberg (1987), полное описание которого включено в данное изобретение посредством ссылки.
Термин "консервативный", используемый в отношении вставки, делеции или замены аминокислоты, означает вставку, делецию или замену в аминокислотной цепи, которая не уменьшает полностью терапевтическую эффективность соединения инсулина, т.е. данная эффективность может уменьшаться, оставаться такой же или увеличиваться относительно терапевтической эффективности приемлемого с научной точки зрения контроля, такого как соответствующее соединение нативного инсулина.
Термин "гидрофильный" означает проявление свойств растворимости в воде, и термин "гидрофильная группировка" относится к группировке, которая является гидрофильной, и/или которая, при присоединении к другой химической структурной единице, увеличивает гидрофильность такой химической структурной единицы. Примеры включают в себя сахара и полиалкиленовые группировки, такие как полиэтиленгликоль, но не ограничиваются этим. Термин "липофильный" означает проявление свойств растворимости в жирах, таких как накопление в жире или жировых тканях, способность растворяться в липидах и/или способность проникать в биологические мембраны, взаимодействовать с ними и/или проходить через них, и термин "липофильная группировка" означает группировку, которая является липофильной, и/или которая, при присоединении к другой химической структурной единице, увеличивает липофильность такой химической структурной единицы. Термин "амфифильный" означает проявление свойств гидрофильности и липофильности, и термин "амфифильная группировка" означает группировку, которая является амфифильной, и/или которая, при присоединении к полипептидному или неполипептидному лекарственному препарату, увеличивает амфифильность (т.е. увеличивает как гидрофильность, так и амфифильность) полученного конъюгата, например некоторые PEG-жирнокислотные модифицирующие группировки и сахар-жирнокислотные модифицирующие группировки.
Термин "низший алкил" означает замещенные или незамещенные, линейные или разветвленные алкильные группировки, содержащие от одного до шести атомов углерода, т.е. C1, С2, С3, С4, С5 или С6. Термин "высший алкил" означает замещенные или незамещенные, линейные или разветвленные алкильные группировки, содержащие шесть или более атомов углерода, например С7, C8, С9, С10, С11, С12, С13, С14, С15, С16, С17, C18, С19, С20 и так далее.
Термин "монодисперсный" описывает смесь соединений, где приблизительно 100 процентов соединений в смеси имеют одинаковую молекулярную массу. Термин "по существу монодисперсный" описывает смесь соединений, где по меньшей мере приблизительно 95 процентов соединений в смеси имеют одинаковую молекулярную массу. Термин "чисто монодисперсный" описывает смесь соединений, где приблизительно 100 процентов соединений в смеси имеют одинаковую молекулярную массу и имеют одинаковую молекулярную структуру. Соответственно, чисто монодисперсная смесь является монодисперсной смесью, но монодисперсная смесь не обязательно является чисто монодисперсной смесью. Термин "по существу чисто монодисперсный" описывает смесь соединений, где по меньшей мере приблизительно 95 процентов соединений в смеси имеют одинаковую молекулярную массу и одинаковую молекулярную структуру. Соответственно, по существу чисто монодисперсная смесь является по существу монодисперсной смесью, но по существу монодисперсная смесь не обязательно является по существу чисто монодисперсной смесью. В композициях конъюгатов соединений инсулинов с катионами компоненты конъюгатов соединений инсулинов предпочтительно являются монодисперсными, по существу монодисперсными, чисто монодисперсными или по существу чисто монодисперсными, но могут быть также полидисперсными. Термин "полидисперсный" означает имеющий дисперсность, которая не является монодисперсной, по существу монодисперсной, чисто монодисперсной или по существу чисто монодисперсной.
Термин "соединение нативный инсулин", специфически используемый в данном описании, означает соединение инсулин млекопитающего (например инсулин человека, соединение бычий инсулин, соединение инсулин свиньи или соединение инсулин кита), полученное из природного, искусственного или генно-инженерного источника. Инсулин человека состоит из А-цепи, имеющей двадцать одну аминокислоту, и В-цепи, имеющей тридцать аминокислот, которые сшиты дисульфидными связями. Правильно сшитый инсулин человека содержит три дисульфидных мостика: один - между А7 и В7, второй- между А20 и В19, и третий - между А6 и А11. Инсулин человека имеет три свободные аминогруппы: В1-фенилаланин, А1-глицин и В29-ЛИЗИН. Свободные аминогруппы в положениях А1 и В1 представляют собой α-аминогруппы. Свободная аминогруппа в положении В29 представляет собой е-аминогруппу. Термин "аналог инсулина" означает полипептид, проявляющий частичную, полную или повышенную активность относительно соответствующего нативного инсулина, или полипептид, который превращается in vivo или in vitro в полипептид, проявляющий частичную, полную или повышенную активность относительно соответствующего нативного инсулина, например полипептид, имеющий структуру инсулина человека со вставками, делециями и/или заменами одной или более консервативных аминокислот. Аналоги инсулина могут быть идентифицированы с использованием известных методов, таких как методы, описанные в патентной публикации США №20030049654, "Protein design automation for protein libraries", поданного 18 марта 2002 г. на имя Dahiyat с соавт. Проинсулины, препроинсулины, предшественники инсулина, одноцепочечные предшественники инсулина человека и животных, не относящихся к человеку, и аналоги любого из вышеперечисленного в данном описании также называются аналогами инсулина, как и инсулины немлекопитающих. В данной области техники известно много аналогов инсулина (смотри обсуждение ниже). Если в контексте не оговорено особо (например, кроме тех случаев, где указан конкретный инсулин, такой как "инсулин человека" или тому подобное), термин "соединение инсулин" используют в широком смысле для включения нативных инсулинов и аналогов инсулинов.
Термин "полиалкиленгликоль" или PAG относится к замещенным или незамещенным, линейным или разветвленным полиалкиленгликолевым полимерам, таким как полиэтиленгликоль (PEG), полипропиленгликоль (PPG) и полибутиленгликоль (PBG), и их комбинациям (например линейным или разветвленным полимерам, включающим комбинации двух или более различных PAG-субъединиц, такие как комбинации двух или более различных PAG-субъединиц, выбранных из PEG-, PPG-, PPG- и PBG-субъединиц) и включает простые моноалкилэфиры полиалкиленгликоля. Термин PAG-субъединица означает одну структурную единицу PAG, например термин "PEG-субъединица" относится к одной полиэтиленгликолевой структурной единице, например к -(СН2СН2О)-, термин "PPG-субъединица" относится к одной полипроленгликолевой структурной единице, например к -(СН2СН2СН2О)-, и термин "PBG-субъединица" относится к одной полибутиленгликолевой структурной единице, например к -(СН2СН2СН2СН2О)-. PAG и/или PAG-субъединицы также включают в себя замещенные PAG и/или PAG-субъединицы, например PAG, содержащие алкильные боковые цепи, такие как метильные, этильные или пропильные боковые цепи, или карбонильные боковые цепи, а также PAG, содержащие одну или более разветвленных PAG-субъединиц, таких как изо-PPG или изо-PBG.
Термин "соединение проинсулин" означает соединение инсулин, в котором С-конец В-цепи связан с N-концом А-цепи посредством природного или искусственного С-пептида, имеющего 5 или более аминокислот. Термин "соединение препроинсулин" означает соединение проинсулин, дополнительно содержащее лидерную последовательность, соединенную с N-концом В-цепи, такую как последовательность, выбранную для стимуляции экскреции в виде растворимого белка, или последовательность, выбранную для предотвращения конъюгирования N-конца, или последовательность, выбранную для улучшения очистки (например последовательность со сродством связывания к колонке для очистки). Термин "одноцепочечный предшественник соединения инсулина" или "соединение минипроинсулин" означает соединение инсулин, в котором С-конец В-цепи (или укороченной В-цепи, у которой на С-конце удалены 1, 2, 3 или 4 аминокислоты) связан с N-концом А-цепи или укороченной А-цепи, которая укорочена на N-конце на 1, 2, 3 или 4 аминокислоты, без промежуточного С-пептида или через укороченный С-пептид, имеющий 1, 2, 3 или 4 аминокислоты.
Термин "протамин" относится к смеси сильно основных белков, полученных из природных (например из спермы рыб) или рекомбинантных источников. Смотри Hoffmann, J. A., et al., Protein Expression and Purification, 1:127-133 (1990). Композиция протамина может быть представлена в виде препарата данных белков, относительно свободного от солей, часто называемого как "протаминовое основание" или в виде препарата, содержащего соли данных белков.
Термины "белок", "пептид" и "полипептид" используются в данном описании взаимозаменяемо и относятся к соединениям, имеющим аминокислотные последовательности любой длины, но по меньшей мере из двух аминокислот.
Термин "R-тип" означает конформацию комплекса, образованного в присутствии конъюгата соединения инсулина, катиона и стабилизирующего соединения, такого как фенол. Термин "Т-тип" означает конформацию комплекса, образованного в присутствии конъюгата соединения инсулина и катиона без стабилизирующего соединения, такого как фенол. Комплекс Т-типа или R-типа может включать или исключать протамин.
Термин "приемлемый с научной точки зрения контроль" означает экспериментальный контроль, который является приемлемым для среднего специалиста в той области техники, к которой относится предмет эксперимента.
Термин "твердое" относится к состоянию вещества, у которого существует трехмерная регулярная структура; в данном описании этот термин широко используется для упоминания как кристаллических твердых веществ, аморфных твердых веществ, так и комбинаций кристаллических твердых веществ и аморфных твердых веществ. Термин "твердый конъюгат соединения инсулина с катионом" относится к твердому веществу, которое включает конъюгат соединения инсулина с катионом, предпочтительно координированный одновалентным или поливалентным катионом. Термин "кристалл" означает твердое вещество в форме правильного многогранника. Термин "кристаллический" относится к твердым веществам, имеющим свойства кристаллов. Термин "микрокристалл" означает твердое вещество, которое образовано, главным образом, из вещества в кристаллическом состоянии, которое является микроскопическим по размерам, наибольший размер которого обычно находится в диапазоне от 1 микрона до 100 микрон. В некоторых случаях отдельные кристаллы, имеющие микрокристаллическую структуру, представляют собой кристаллы, имеющие преимущественно одну кристаллографическую структуру. В некоторых воплощениях кристаллы по изобретению не являются микрокристаллами. Термин "микрокристаллический" относится к состоянию вещества, являющегося микрокристаллом. Термин "аморфный" относится к твердому веществу, которое не находятся в кристаллической форме. Средний специалист в данной области может отличить кристаллы от аморфных веществ, используя стандартные методы, например, используя рентгеновские кристаллографические методы, сканирующую электронную микроскопию или оптическую микроскопию. Термин "смесь твердых веществ" означает смесь двух разных твердых веществ Термин "смесь кристаллов" означает смесь двух разных кристаллов. Термин "сокристалл" означает кристалл, содержащий два или более различных соединений инсулинов и/или конъюгатов соединений инсулинов. Комплексы конъюгатов соединений инсулинов с катионами по изобретению могут быть представлены в любой из вышеупомянутых форм или в виде смесей двух или более таких форм.
Термин "замена" означает замену одного или более аминокислотных остатков в последовательности соединения инсулина другой аминокислотой. В некоторых случаях аминокислотная замена действует как функциональный эквивалент, что приводит к "молчащему" изменению. Замены могут быть консервативными; например, консервативные замены могут быть выбраны из других представителей того же класса, к которому принадлежит замещенная аминокислота. Примеры неполярных (гидрофобных) аминокислот включают в себя аланин, лейцин, изолейцин, валин, пролин, фенилаланин, триптофан и метионин. Примеры полярных нейтральных аминокислот включают в себя глицин, серии, треонин, цистеин, тирозин, аспарагин и глутамин. Примеры положительно заряженных (основных) аминокислот включают в себя аргинин, лизин и гистидин. Примеры отрицательно заряженных (кислых) аминокислот включают в себя аспарагиновую кислоту и глутаминовую кислоту.
"Растворимость в воде" или "водная растворимость", если не оговорено особо, определяют в водном буферном растворе при рН 7,4.
7. ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В изобретении предложены комплексы конъюгатов соединений инсулинов с катионами и различные композиции, содержащие такие комплексы, а также способы получения и применения таких комплексов и композиций. Данные комплексы являются полезными для введения соединения инсулина для лечения различных медицинских состояний, таких как состояния, которые характеризуются дефицитом соединений инсулинов. Комплексы обычно включают в себя катионный компонент и компонент, представляющий собой конъюгат соединения инсулина. Компонент, представляющий собой конъюгат соединения инсулина, обычно включает соединение инсулин, связанное с модифицирующей группировкой. Примеры других подходящих компонентов данных комплексов и/или композиций включают в себя стабилизирующие агенты, комплексообразующие агенты и другие известные в данной области компоненты для применения в получении катион-белковых комплексов. В изобретении также предложены новые конъюгаты соединений инсулинов и препараты жирных кислот, содержащие такие конъюгаты соединений инсулинов и/или комплексы конъюгатов соединений инсулинов с катионами.
7.1. Соединение инсулин
Конъюгат соединения инсулина с катионом включает компонент, представляющий собой соединение инсулин. Соединение инсулин может представлять собой, например, соединение инсулин млекопитающего, такое как инсулин человека, или аналог соединения инсулина.
В данной области техники известно большое количество различных аналогов соединений инсулинов. Предпочтительными аналогами соединений инсулинов являются те аналоги, которые содержат лизин, предпочтительно лизин в пределах 5 аминокислот С-конца В-цепи, например в положении В26, В27, В28, В29 и/или В30. Группа подходящих аналогов, имеющих последовательность соединения инсулина, за исключением того, что аминокислотный остаток в положении В28 представляет собой Asp, Lys, Leu, Val или Ala; аминокислотный остаток в положении В29 представляет собой Lys или Pro; аминокислотный остаток в положении В10 представляет собой His или Asp; аминокислотный остаток в положении В1 представляет собой Phe, Asp или делетирован один или в комбинации с делецией остатка в положении В2; аминокислотный остаток в положении В30 представляет собой Thr, Ala или делетирован; и аминокислотный остаток в положении В9 представляет собой Ser или Asp; при условии, что в положении либо В28, либо В29 находится Lys, описана в ЕР-А 1227000107 (полное описание которого включено в данное изобретение посредством ссылки).
Другие примеры подходящих аналогов соединений инсулинов включают в себя AspВ28 инсулин человека, LysB28 инсулин человека, LeuB28 инсулин человека, ValВ28 инсулин человека, AlaВ28 инсулин человека, AspB28ProВ29 инсулин человека, LysВ28ProB29 инсулин человека, LeuВ28ProВ29 инсулин человека, ValB28ProВ29 инсулин человека, AlaВ28ProВ29 инсулин человека, а также аналоги, полученные с использованием описанных выше принципов замещения. Фрагменты соединений инсулинов включают в себя В22-В30 инсулин человека, В23-В30 инсулин человека, В25-В30 инсулин человека, В26-В30 инсулин человека, В27-В30 инсулин человека, В29-В30 инсулин человека, В1-В2 инсулин человека, В1-В3 инсулин человека, В1-В4 инсулин человека, В1-В5 инсулин человека, А-цепь инсулина человека и В-цепь инсулина человека, но не ограничиваются этим.
Другие дополнительные примеры подходящих аналогов соединений инсулинов можно найти в патентной публикации США №20030144181 А1 под названием "Insoluble compositions for controlling blood glucose", 31 июля 2003 г.; патентной публикации США №20030104983 А1 под названием "Stable insulin formulations", 5 июня 2003 г.; патентной публикации США №20030040601 А1 под названием "Method for making insulin precursors and insulin analog precursors", 27 февраля 2003 г.; патентной публикации США №20030004096 А1 под названием "Zinc-free and low-zinc insulin preparations having improved stability", 2 января 2003 г.; патенте США №6551992 В1 под названием "Stable insulin formulations", 22 апреля 2003 г.; патенте США №6534288 В1 под названием "С peptide for improved preparation of insulin and insulin analogs", 18 марта 2003 г.; патенте США №6531448 В1 под названием "Insoluble compositions for controlling blood glucose", 11 марта 2003 г.; патенте США № RE 37971 E под названием "Selective acylation of epsilon-amino groups", 28 января 2003 г.; патентной публикации США №20020198140 А1 под названием "Pulmonary insulin crystals", 26 декабря 2002 г.; патенте США №6465426 В2 под названием "Insoluble insulin compositions", 15 октября 2002 г.; патенте США №6444641 В1 под названием "Fatty acid-acylated insulin analogs", 3 сентября 2002 г.; патентной публикации США №20020137144 А1 под названием "Method for making insulin precursors and insulin precursor analogues having improved fermentation yield in yeast", 26 сентября 2002 г.; патентной публикации США №20020132760 А1 под названием "Stabilized insulin formulations", 19 сентября 2002 г.; патентной публикации США №20020082199 А1 под названием "Insoluble insulin compositions", 27 июня 2002 г.; патенте США №6335316 В1 под названием "Method for administering acylated insulin", 1 января 2002 г.; патенте США №6268335 В1 под названием "Insoluble insulin compositions", 31 июля 2001 г.; патентной публикации США №20010041787 А1 под названием "Method for making insulin precursors and insulin precursor analogues having improved fermentation yield in yeast", 15 ноября 2001 г.; патентной публикации США №20010041786 А1 под названием "Stabilized acylated insulin formulations", 15 ноября 2001 г.; патентной публикации США №20010039260 А1 под названием "Pulmonary insulin crystals", 8 ноября 2001 г.; патентной публикации США №20010036916 А1 под названием "Insoluble insulin compositions", 1 ноября 2001 г.; патентной публикации США №20010007853 А1 под названием "Method for administering monomeric insulin analogs", 12 июля 2001 г.; патенте США №6051551А под названием "Method for administering acylated insulin", 18 апреля 2000 г.; патенте США №6034054 А под названием "Stable insulin formulations", 7 марта 2000 г.; патенте США №5970973 А под названием "Method of delivering insulin lispro", 26 октября 1999; патенте США №5952297 А под названием "Monomeric insulin analog formulations", 14 сентября 1999; патенте США №5922675 А под названием "Acylated Insulin Analogs", 13 июля 1999; патенте США №5888477 А под названием "Use of monomeric insulin as a means for improving the bioavailability of inhaled insulin", 30 марта 1999; патенте США №5873358 A под названием "Method of maintaining a diabetic patient's blood glucose level in a desired range", 23 февраля 1999; патенте США №5747642 A под названием "Monomeric insulin analog formulations", 5 мая 1998; патенте США №5693609 А под названием "Acylated insulin compound analogs", 2 декабря 1997; патенте США №5650486 А под названием "Monomeric insulin analog formulations", 22 июля 1997; патенте США №5646242 А под названием "Selective acylation of epsilon-amino groups", 8 июля 1997; патенте США №5597893 А под названием "Preparation of stable insulin analog crystals", 28 января 1997; патенте США №5547929 A под названием "Insulin analog formulations", 20 августа 1996; патенте США №5504188 А под названием "Preparation of stable zinc insulin compound analog crystals", 2 апреля 1996; патенте США №5474978 A под названием "Insulin analog formulations", 12 декабря 1995; патенте США №5461031 А под названием "Monomeric insulin analog formulations", 24 октября 1995; патенте США №4421685 А под названием "Process for producing an insulin", 20 декабря 1983; патенте США №6221837 под названием "Insulin derivatives with increased zinc binding" 24 апреля 2001 г.; патенте США №5177058 под названием "Pharmaceutical formulation for the treatment of diabetes mellitus" 5 января 1993 (описывает фармацевтические препараты, содержащие производное соединения инсулина, модифицированное основанием в положении В31 и имеющее изоэлектрическую точку в диапазоне от 5,8 до 8,5, и/или по меньшей мере одну из его физиологически допустимых солей в фармацевтически приемлемом эксципиенте, и относительно высокое содержание ионов цинка в диапазоне от приблизительно 1 мкг до приблизительно 200 мкг цинка/МЕ, включая соединение инсулин-В31-Arg-ОН и инсулин-В31-Arg-В32-Arg-ОН человека). Полное описание каждого из вышеупомянутых патентных документов включено в данное описание посредством ссылки, особенно в части, относящейся к получению, применению и композициям различных аналогов соединений инсулинов.
Соединение инсулин, используемое для получения конъюгатов соединения инсулина с катионом, может быть получено с использованием любой из множества общепризнанных методов синтеза пептидов, например классических методов (синтеза в растворе), методов твердофазного синтеза, полусинтетических методов и методов рекомбинантной ДНК. Например, Chance с соавт. в заявке на патент США №07/388201, ЕР 0383472; Brange с соавт. ЕР 0214826 и Belagaje с соавт. в патенте США №5304473, включенных в данное описание посредством ссылки, раскрывают получение различных соединений проинсулинов и аналогов соединений инсулинов. А- и В-цепи аналогов соединений инсулинов могут быть получены также с помощью молекулы предшественника проинсулин-подобного соединения или молекулы одноцепочечного предшественника соединения инсулина с использованием методов рекомбинантной ДНК. Смотри Frank at al., "Peptides: Synthesis-Structure-Function", Proc. Seventh Am. Pept. Symp., Eds. D. Rich and E. Gross (1981); Bernd Gutte, Peptides: Synthesis, Structures, and Applications, Academic Press (October 19, 1995); Chan, Weng and White, Peter (Eds.), Fmoc Solid Phase Peptide Synthesis: A Practical Approach, Oxford University Press (March 2000 г.); полные описания которых в части, относящейся к пептидному синтезу, рекомбинантной продукции и производству, включены в данное описание посредством ссылки.
7.2. Модифицирующая группировка
Комплексы конъюгатов соединений инсулинов с катионами включают в себя модифицирующую группировку, связанную (например посредством ковалентной или ионной связи) с соединением инсулином с получением конъюгата соединения инсулина. Модифицирующие группировки представляют собой связанные с соединением инсулином группировки, которые дают соединение инсулин с желательными свойствами, как описано в данном изобретении. Например, модифицирующая группировка может уменьшать скорость распада соединения инсулина в различном окружении (таком как желудочно-кишечный тракт и/или кровоток), так что меньше соединения инсулина распадается в модифицированной форме, чем распадалось бы в таком окружении в отсутствие модифицирующей группировки. Предпочтительные модифицирующие группировки представляют собой группировки, которые позволяют конъюгату соединения инсулина сохранить терапевтически значимый процент биологической активности исходного соединения инсулина. Кроме того, предпочтительные модифицирующие группировки представляют собой группировки, которые являются амфифильными или гидрофильными и/или которые делают конъюгат соединения инсулина амфифильным, или гидрофильным, или менее липофильным, чем приемлемый с научной точки зрения контроль, такой как соответствующее соединение инсулин или соответствующее неконъюгированное соединение инсулин.
Примеры подходящих модифицирующих группировок и конъюгатов соединений инсулинов, полезных в композициях конъюгатов соединений инсулинов с катионами, можно найти в следующих патентах, полные описания которых включены в данное описание посредством ссылки: патенте США №6303569 под названием "Trialkyl-lock-facilitated polymeric prodrugs of amino-containing bioactive agents", 16 октября 2001 г.; патенте США №6214330, "Coumarin and related aromatic-based polymeric prodrugs", 10 апреля 2001 г.; патенте США №6113906 под названием "Water-soluble non-antigenic polymer linkable to biologically active material", 5 сентября 2000 г.; патенте США №5985263 под названием "Substantially pure histidine-linked protein polymer conjugates", 16 ноября 1999; патенте США №5900402 под названием "Method of reducing side effects associated with administration of oxygen-carrying proteins", 4 мая 1999; патенте США №5681811, "Conjugation-stabilized therapeutic agent compositions, delivery and diagnostic formulations comprising same, and method of making and using the same" 28 октября 1997; патенте США №5637749 под названием "Aryl imidate activated polyalkylene oxides", 10 июня 1997; патенте США №5612460 под названием "Active carbonates of polyalkylene oxides for modification of polypeptides", 18 марта 1997; патенте США №5567422 под названием "Azlactone activated polyalkylene oxides conjugated to biologically active nucleophiles", 22 октября 1996; патенте США №5405877 под названием "Cyclic imide thione activated polyalkylene oxides", 11 апреля 1995; и патенте США №5359030 под названием "Conjugation-stabilized polypeptide compositions, therapeutic delivery and diagnostic formulations comprising same, and method of making and using the same", 25 октября 1994. Дополнительные примеры конъюгированных пептидов, полезных в препаратах по настоящему изобретению, можно найти в следующих заявках на патент США, полные описания которых включены в данное описание посредством ссылки: в заявке на патент США №09/134803, поданной 14 августа 1998; №10/018879, поданной 19 декабря 2001 г.; №10/235381, поданной 5 сентября 2002 г.; №10/235284, поданной 5 сентября 2002 г.; и №09/873797, поданной 4 июня 2001 г. Полное описание каждого из вышеупомянутых патентов и каждой из вышеупомянутых заявок на патент в части, относящейся к группировкам, используемым для модификации полипептидов, включено в данное описание посредством ссылки.
Основные цепи модифицирующих группировок могут включать слабые или способные к распаду связи. Например, PAG могут включать гидролитически нестабильные связи, такие как лактидная, гликолидная, карбонатная, сложноэфирная, карбаматная и тому подобное, которые чувствительны к гидролизу. Данный подход позволяет расщеплять такие полимеры на фрагменты с меньшей молекулярной массой. Примеры таких полимеров описаны, например, Hubbell с соавт. в патенте США №6153211, полное описание которого включено в данное описание посредством ссылки. Смотри также патент США №6309633, Ekwuribe с соавт., полное описание которого включено в данное описание посредством ссылки.
Модифицирующая группировка может включать в себя любые гидрофильные группировки, липофильные группировки, амфифильные группировки, солеобразующие группировки и их комбинации. Типичные гидрофильные, амфифильные и липофильные полимеры и модифицирующие группировки описаны более подробно ниже.
7.2.1. Гидрофильные группировки
Примеры подходящих гидрофильных группировок включают в себя PAG-группировки, другие гидрофильные полимеры, сахарные группировки, полисорбатные группировки и их комбинации.
7.2.2. Полиалкиленгликолевые группировки
PAG представляют собой соединения с повторяющимися алкиленгликолевыми структурными единицами. В некоторых воплощениях все структурные единицы являются идентичными (например, PEG или PPG). В других воплощениях алкиленовые структурные единицы являются разными (например, полиэтилен-со-пропиленгликоль или PLURONICS®). Данные полимеры могут быть статистическими сополимерами (например, когда сополимеризуются этиленоксид и пропиленоксид) или разветвленными или привитыми сополимерами.
PEG представляет собой предпочтительный PAG и является полезным в биологических применениях, так как он имеет весьма желательные свойства и признан FDA (Управлением США по санитарному надзору за качеством пищевых продуктов и медикаментов) безопасным (GRAS). PEG обычно имеет формулу Н-(СН2СН2О)n-Н, где n может варьировать от приблизительно 2 до приблизительно 4000 или более, однако блокирующие группировки могут варьировать, например, представлять собой монометокси или дигидрокси. Обычно PEG не имеет цвета, запаха, является водорастворимым или смешивается с водой (в зависимости от молекулярной массы), термостойким, химически инертным, гидролитически стабильным и, как правило, нетоксичным. PEG является также биосовместимым и обычно не вызывает в организме иммунной реакции. Предпочтительные PEG-группировки включают в себя 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50 или более PEG-субъединиц.
PEG может быть монодисперсным, по существу монодисперсным, чисто монодисперсным или по существу чисто монодисперсным (например таким, как описано ранее авторами данной заявки в патенте США №09/873731 и патенте США №09/873797, которые оба поданы 4 июня 2001 г. и полные описания которых включены в данное описание посредством ссылки) или полидисперсным. Одно из преимуществ использования относительно низкомолекулярных монодисперсных полимеров заключается в том, что они образуют легко определяемые молекулы конъюгатов, что может облегчить как воспроизводимый синтез, так и одобрение FDA).
PEG может быть линейным с гидроксильной группой на каждом конце (до его конъюгирования с остатком соединения инсулина). PEG также может представлять собой алкокси-PEG, такой как метокси-PEG (или mPEG), где один конец представляет собой относительно инертную алкокси-группу (например линейную или разветвленную OC1-6), тогда как другой конец представляет собой гидроксильную группу (которая связана с соединением инсулином).
PEG также может быть разветвленным, который в одном из воплощений может быть представлен как R(-PEG-nOH)m, где R представляет собой центральный (обычно многоатомный) коровый агент, такой как пентаэритрит, сахар, лизин или глицерин, n представляет собой число PEG-субъединиц и может варьировать для каждой ветви, и обычно равно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49 или 50, и m представляет собой число ветвей и варьирует от 2 до максимального числа сайтов присоединения на коровом агенте. Все эти ветви могут быть одинаковыми или разными и могут быть терминированы, например, простыми эфирами и/или сложными эфирами. Число ветвей m может варьировать от трех до ста или более, и одна или более концевых гидроксильных групп могут быть связаны с остаточным соединением инсулином или иным образом подвергнуты химической модификации.
Другие разветвленные PEG включают в себя PEG, представленные формулой (CH3O-PEG-)pR-Z, где р равно 2 или 3, R представляет собой центральный кор, такой как лизин или глицерин, и Z представляет собой группу, такую как карбоксил, которая легко подвергается химической активации. Еще одна разветвленная форма, PEG с боковыми подвешенными группами, имеет химически активные группы, такие как карбоксилы, вдоль основной цепи PEG, а не на конце, или не только на конце, PEG-цепей. Разветвленный PEG может быть представлен формулой PEG(-LCHX2)n, где L представляет собой связывающую группу, и X представляет собой активированную концевую группу.
7.2.3. Сахарные группировки
Модифицирующие группировки, описанные в данном изобретении, могут включать в себя сахарные группировки. Обычно сахарная группировка представляет собой углеводный продукт из по меньшей мере одной сахарозной группы. Типичные сахарные группировки включают в себя, но не ограничиваются этим, глицериновые группировки, моно-, ди-, три- и олигосахариды, и полисахариды, такие как крахмалы, гликоген, целлюлоза и полисахаридные камеди. Конкретные моносахариды включают в себя С6 и выше (предпочтительно от С6 до С8) сахара, такие как глюкоза, фруктоза, манноза, галактоза, рибоза и седогептулоза; ди- и трисахариды включают в себя группировки, имеющие две или три моносахаридные структурные единицы (предпочтительно С5-С8), такие как сахароза, целлобиоза, мальтоза, лактоза и раффиноза. Конъюгирование с использованием сахарных группировок описано в патентах США №№5681811, 5438040 и 5359030, полные описания которых включены в данное описание посредством ссылки.
7.2.4. Полисорбатные группировки
Модифицирующие группировки могут включать в себя одну или более полисорбатных группировок. Примеры включают в себя сложные эфиры сорбитана и полиоксиэтиленовые производные полисорбата. Конъюгирование с использованием полисорбатных группировок описано в патентах США №№5681811, 5438040 и 5359030, полные описания которых включены в данное описание посредством ссылки.
7.2.5. Биосовместимые водорастворимые поликатионные группировки
В некоторых воплощениях могут быть использованы биосовместимые водорастворимые поликатионные полимеры. Биосовместимые водорастворимые поликатионные полимеры включают в себя, например, любую модифицирующую группировку, содержащую протонированные гетероциклы, присоединенные в виде концевых групп."Водорастворимый" в данном контексте означает, что вся модифицирующая группировка растворяется в водных растворах, таких как забуференный физиологический раствор или забуференный физиологический раствор с небольшими количествами добавленных органических растворителей в качестве сорастворителей, при температуре от 20 до 37°С. В некоторых воплощениях модифицирующая группировка сама по себе не является достаточно растворимой в водных растворителях, но становится растворимой в результате прививки водорастворимыми полимерами, такими как PEG-цепи. Примеры включают в себя полиамины, имеющие аминогруппы либо на основной цепи модифицирующей группировки, либо на боковых цепях модифицирующей группировки, такие как поли-L-Lys и другие положительно заряженные полиаминокислоты из природных или синтетических аминокислот или смесей аминокислот, включая поли(D-Lys), поли(орнитин), поли(Arg) и поли(гистидин), и непептидные полиамины, такие как поли(аминостирол), поли(аминоакрилат), поли(N-метиламиноакрилат), поли(N-этиламиноакрилат), поли(N,N-диметиламиноакрилат), поли(N,N-диэтиламиноакрилат), поли(аминометакрилат), поли(N-метиламинометакрилат), поли(N-этиламинометакрилат), поли(N,N-диметиламинометакрилат), поли(N,N-диэтиламинометакрилат), поли(этиленимин), полимеры четвертичных аминов, такие как поли(N,N,N-триметиламиноакрилатхлорид), поли(метилакриламидопропилтриметиламмонийхлорид), и природные или синтетические полисахариды, такие как хитозан.
7.2.6. Другие гидрофильные группировки
Модифицирующие группировки могут включать в себя также другие гидрофильные полимеры. Примеры включают в себя поли(оксиэтилированные полиолы), такие как поли(оксиэтилированный глицерин), поли(оксиэтилированный сорбит) и поли(оксиэтилированная глюкоза); поливиниловый спирт ("PVA"); декстран; полимеры на основе углеводов и тому подобное. Данные полимеры могут представлять собой гомополимеры, или статистические или блок-сополимеры и тройные сополимеры на основе мономеров вышеуказанных полимеров, с линейной цепью или разветвленные.
Конкретные примеры подходящих дополнительных полимеров включают в себя поли(оксазолин), бифункциональный поли(акрилоилморфолин) ("РАсМ") и поли(винилпирролидон) (PVP), но не ограничиваются этим. PVP и поли(оксазолин) представляют собой хорошо известные в данной области техники полимеры, и их получение будет очевидным для специалиста. РАсМ и его синтез и применение описаны в патенте США №5629384 и патенте США №5631322, описания которых во все своей полноте включены в данное описание посредством ссылки.
7.2.7. Биоадгезивные полианионные группировки
Некоторые гидрофильные полимеры, по-видимому, имеют потенциально полезные биоадгезивные свойства. Примеры таких полимеров можно найти, например, в патенте США №6197346 (Mathiowitz с соавт.). Данные полимеры, содержащие карбоксильные группы (например, полиакриловую кислоту) проявляют биоадгезивные свойства и также могут быть легко конъюгированы с соединениями инсулинами, описанными в данном изобретении. Быстро биодеградируемые полимеры, которые экспонируют карбоновокислотные группы для деградации, такие как поли(лактид-со-гликолид), полиангидриды и полиортоэфиры, также являются биоадгезивными полимерами. Эти полимеры можно использовать для доставки соединений инсулинов в желудочно-кишечный тракт. Так как эти полимеры деградируют, они могут экспонировать карбоновокислотные группы, что дает им возможность прочно прилипать к желудочно-кишечному тракту, и могут содействовать доставке конъюгатов соединений инсулинов.
7.2.8. Липофильные группировки
В некоторых воплощениях модифицирующие группировки включают в себя одну или более липофильных группировок. Липофильная группировка может представлять собой различные липофильные группировки, которые, как известно специалистам в данной области, включают в себя алкильные группировки, алкенильные группировки, алкинильные группировки, арильные группировки, арилалкильные группировки, алкиларильные группировки, жирнокислотные группировки, адамантантил и холестерил, а также липофильные полимеры и/или олигомеры, но не ограничиваются этим.
Алкильная группировка может представлять собой насыщенную или ненасыщенную, линейную, разветвленную или циклическую углеводородную цепь. В некоторых воплощениях алкильная группировка имеет 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50 или более атомов углерода. Примеры включают в себя насыщенные линейные алкильные группировки, такие как метил, этил, пропил, бутил, пентил, гексил, гептил, октил, нонил, децил, ундецил, додецил, тридецил, тетрадецил, пентадецил, гексадецил, октадецил, нондецил и эйкозил; насыщенные разветвленные алкильные группировки, такие как изопропил, втор-бутил, трет-бутил, 2-метилбутил, трет-пентил, 2-метилпентил, 3-метилпентил, 2-этилгексил, 2-пропилпентил; и ненасыщенные алкильные группировки, происходящие из вышеуказанных насыщенных алкильных группировок, включая, но не ограничиваясь этим, винил, аллил, 1-бутенил, 2-бутенил, этинил, 1-пропинил и 2-пропинил. В других воплощениях алкильная группировка представляет собой низшую алкильную группировку. В других воплощениях алкильная группировка представляет собой С1-С3 низшую алкильную группировку. В некоторых воплощениях модифицирующая группировка конкретно не состоит из алкильной группировки, или конкретно не состоит из низшей алкильной группировки, или конкретно не состоит из алкановой группировки, или конкретно не состоит из низшей алкановой группировки.
Алкильные группы могут быть либо незамещенными, либо замещенными одним или более заместителями, и предпочтительно, чтобы такие заместители не препятствовали способам синтеза конъюгатов и не уничтожали биологическую активность конъюгатов. Потенциально мешающая функциональная группа может быть подходящим образом блокирована защитной группой для того, чтобы сделать данную функциональную группу немешающей. Каждый заместитель возможно может быть замещен дополнительными немешающими заместителями. Термин "немешающий" характеризует заместители, которые не уничтожают возможности осуществления любых реакций в соответствии со способом по данному изобретению.
Липофильная группировка может представлять собой жирнокислотную группировку, такую как природная или синтетическая, насыщенная или ненасыщенная, линейная или разветвленная жирнокислотная группировка. В некоторых воплощениях жирнокислотная группировка содержит 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 или более атомов углерода. В некоторых воплощениях модифицирующая группировка конкретно не состоит из жирнокислотной группировки; или конкретно не состоит из жирнокислотной группировки, содержащей 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 или более атомов углерода.
Когда модифицирующая группировка включает в себя арильное кольцо, тогда данное кольцо может быть функционализировано нуклеофильной функциональной группой (такой как ОН или SH), которая расположена так, что она может вступать во внутримолекулярное взаимодействие с карбаматной группировкой и способствовать ее гидролизу. В некоторых воплощениях данная нуклеофильная группа защищена защитной группой, способной гидролизоваться или иным образом деградировать in vivo, в результате, когда защитная группа удалена, тогда гидролиз конъюгата и последующее высвобождение исходного соединения инсулина облегчается.
Другие примеры подходящих модифицирующих группировок включают в себя -С(СН2ОН)3; -СН(СН2ОН)2; -С(СН3)3; -СН(СН3)2.
7.2.9. Амфифильные группировки
В некоторых воплощениях модифицирующая группировка включает в себя амфифильную группировку. Многие полимеры и олигомеры являются амфифильными. Они часто представляют собой блок-сополимеры, разветвленные сополимеры или привитые сополимеры, которые включают в себя гидрофильные и липофильные группировки, которые могут находиться в форме олигомеров и/или полимеров, таких как линейные, разветвленные или привитые полимеры или сополимеры.
Амфифильные модифицирующие группировки могут включать в себя комбинации любых липофильных и гидрофильных группировок, описанных в данном изобретении. Такие модифицирующие группировки обычно включают в себя по меньшей мере одну реакционноспособную функциональную группу, например галогено, гидроксил, амин, тиол, сульфоновую кислоту, карбоновую кислоту, изоцианат, эпокси, сложный эфир и тому подобное, которые часто находятся на терминальном конце модифицирующей группировки. Эти реакционноспособные функциональные группы могут быть использованы для присоединения липофильной алкильной, алкенильной, алкинильной, арилалкильной или алкиларильной группы с линейной или разветвленной цепью или липофильного полимера или олигомера, что увеличивает липофильность модифицирующей группировки (и тем самым делает ее обычно амфифильной). Липофильные группы могут быть производными, например, моно- или дикарбоновых кислот или, когда это целесообразно, реакционноспособными эквивалентами карбоновых кислот, такими как ангидриды или хлорангидриды. Примерами подходящих предшественников липофильных групп являются уксусная кислота, пропионовая кислота, масляная кислота, валериановая кислота, изомасляная кислота, триметилуксусная кислота, капроновая кислота, каприловая кислота, гептановая кислота, каприновая кислота, пеларгоновая кислота, лауриновая кислота, миристиновая кислота, пальмитиновая кислота, стеариновая кислота, бегеновая кислота, лигноцериновая кислота, цератиновая кислота, монтановая кислота, изостеариновая кислота, изононановая кислота, 2-этилгексановая кислота, олеиновая кислота, рицинолеиновая кислота, линолевая кислота, линоленовая кислота, эруковая кислота, жирная кислота сои, жирная кислота льна, жирная кислота дегидратированного касторового масла, жирная кислота таллового масла, жирная кислота тунгового масла, жирная кислота подсолнечника, жирная кислота сафлора, акриловая кислота, метакриловая кислота, малеиновый ангидрид ортофталевый ангидрид, терефталевая кислота, изофталевая кислота, адипиновая кислота, азелаиновая кислота, себациновая кислота, тетрагидрофталевый ангидрид, гексагидрофталевый ангидрид, янтарная кислота и полиолефиновые карбоновые кислоты.
Концевые липофильные группы не обязательно должны быть эквивалентными, т.е. полученные сополимеры могут включать концевые липофильные группы, которые являются одинаковыми или разными. Липофильные группы могут быть производными более чем одной моно- или бифункциональной алкильной, алкенильной, алкинильной, циклоалкильной, арилалкильной или алкиларильной группы, как определено выше.
7.2.10. PAG-алкильные модифицирующие группировки
Модифицирующая группировка может быть линейной или разветвленной полимерной группировкой, содержащей одну или более линейных или разветвленных PAG-группировок и/или одну или более линейных или разветвленных, замещенных или незамещенных алкильных группировок. В некоторых случаях такие группировки считаются амфифильными; однако PAG и алкильные группировки можно изменять таким образом, чтобы сделать такие группировки более липофильными или более гидрофильными. В некоторых воплощениях модифицирующая группировка конкретно не состоит из алкильной группировки, а в других воплощениях модифицирующая группировка конкретно не состоит из алкановой группировки.
PAG-группировки в некоторых воплощениях включают в себя 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 или 25 PAG-субъединиц, организованных в линейную или разветвленную форму. PAG-группировки в некоторых воплощениях включают в себя PEG-, PPG- и/или PBG-субъединицы. Алкильные группировки в некоторых воплощениях предпочтительно имеют 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20 атомов углерода. Предпочтительно, алкильные группировки представляют собой алкановые группировки. Модифицирующая группировка может включать в себя блокирующую группировку, такую как -ОСН3. Кроме того, модифицирующая группировка может включать в себя гидрофобную группу, такую как пивалоильная группа.
В одном воплощении модифицирующая группировка имеет формулу:
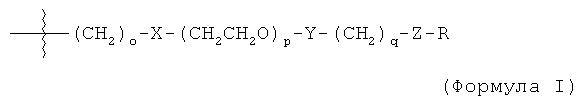 ,
,
где о, р и q независимо равны 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49 или 50, и по меньшей мере одно из о, р и q равно по меньшей мере 2. X, Y и Z независимо выбраны из -С-, -О-, -С(О)-, -С(O)O-, -ОС(О)-, -NH-, -NHC(O)- и -C(O)NH-, и R представляет собой Н или алкил, предпочтительно низший алкил, более предпочтительно метил. Переменные о, р и q предпочтительно выбраны таким образом, чтобы получить гидрофильную или амфифильную модифицирующую группировку, и предпочтительно выбраны относительно соединения инсулина таким образом, чтобы получить гидрофильный или амфифильный конъюгат соединения инсулина, предпочтительно моноконъюгат, диконъюгат или триконъюгат. В одном предпочтительном воплощении конъюгата соединения инсулина, который следует использовать для базального поддержания соединения инсулина, о, р и q выбраны таким образом, чтобы получить PAG, который является проксимальным относительно соединения инсулина, и алкильную группировку, которая является дистальной относительно соединения инсулина. Альтернативно, О, Р и Q могут быть выбраны таким образом, чтобы получить PAG, который является дистальным относительно соединения инсулина, и алкил, который является проксимальным относительно инсулина. В альтернативном воплощении R представляет собой пивалоильную группу или алкил-пивалоильную группу.
В родственном воплощении модифицирующая группировка имеет формулу:
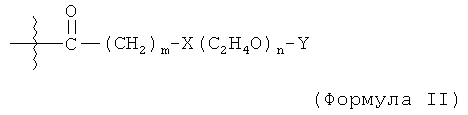 ,
,
где m равно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 или 25, и n равно от 2 до 100, предпочтительно от 2 до 50, более предпочтительно 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 или 25, X представляет собой -С-, -О-, -С(О)-, -NH-, -NHC(O)- или -C(O)NH-, и Y представляет собой низший алкил или -Н. X представляет собой предпочтительно О, и Y представляет собой предпочтительно -СН3. В некоторых случаях карбонильная группа (-С(О)-) может отсутствовать и группировка -(СН2)- может быть связана с подходящей группой на аминокислоте, такой как гидроксильная группа или свободная карбоновокислотная группа.
В предпочтительном воплощении модифицирующая группировка имеет структуру, выбранную из следующих:
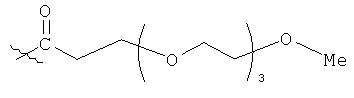 и
и
(когда непосредственно предшествующая модифицирующая группировка связана с инсулином человека в положении В29, полученный моноконъюгат называется IN105).
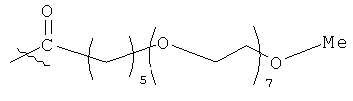 ,
,
(когда непосредственно предшествующая модифицирующая группировка связана с инсулином человека в положении В29, полученный моноконъюгат называется HIM2). Любая из вышеупомянутых группировок может быть связана, например, с инсулином человека по нуклеофильному остатку, например А1, В1 или В29. В некоторых случаях карбонильная группа (-С(О)-) может отсутствовать или может быть заменена алкильной группировкой, предпочтительно низшей алкильной группировкой, и группировка -(СН2)- может быть связана с подходящей группой на аминокислоте, такой как гидроксильная группа или свободная карбоновокислотная группа.
В другом воплощении модифицирующая группировка имеет формулу:
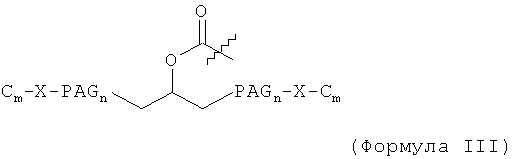 ,
,
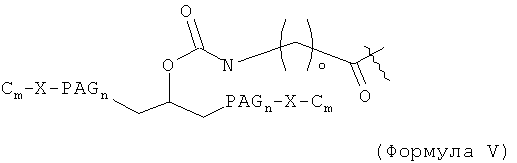
где каждый С независимо выбран и представляет собой алкильную группировку, имеющую m атомов углерода, и m равно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20; и каждый PAG независимо выбран и представляет собой PAG-группировку, имеющую n субъединиц, и n равно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 или 25; каждый X независимо выбран и представляет собой связывающую группировку, присоединяющую PAG к С, и представляет собой предпочтительно -С-, -О-, -С(О)-, -NH-, -NHC(O)- или -C(O)NH-. В некоторых воплощениях группировка Cm-X отсутствует, и группировка PAGn заканчивается группировкой -ОН или группировкой -ОСН3. Например, PAG может представлять собой PAG с концевой метоксигруппой или концевой гидроксигруппой, содержащий 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20 PAG-субъединиц, включая PEG-, PPG- и/или PBG-субъединицы. В некоторых случаях карбонильная группа (-С(О)-) может быть заменена алкильной группировкой, предпочтительно низшей алкильной группировкой, которая может быть связана с подходящей группой на аминокислоте, такой как гидроксильная группа или свободная карбоновокислотная группа.
Модифицирующая группировка может, например, иметь формулу:
 ,
,
где каждый С независимо выбран и представляет собой алкильную группировку, имеющую m атомов углерода, и гл равно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20; и каждый PAG независимо выбран и представляет собой PAG-группировку, имеющую n субъединиц, и n равно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 или 25; X представляет собой -О- или -NH-; каждое о независимо выбрано и равно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 или 15. Например, PAG может иметь 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20 PAG-субъединиц, включая PEG-, PPG- и/или PBG-субъединицы. В некоторых случаях карбонильная группа (-С(О)-), проксимальная к точке присоединения, может отсутствовать или может быть заменена алкильной группировкой, предпочтительно низшей алкильной группировкой, и группировка -(СН2)- может быть связана с подходящей группой на аминокислоте, такой как гидроксильная группа или свободная карбоновокислотная группа.
Модифицирующая группировка может, например, иметь формулу:
 ,
,
где каждый С независимо выбран и представляет собой алкильную группировку, имеющую m атомов углерода, и m равно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20; и каждый PAG независимо выбран и представляет собой PAG-группировку, имеющую n субъединиц, и n равно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 или 25; каждый X независимо выбран и представляет собой связывающую группировку, присоединяющую PAG к С, и предпочтительно представляет собой -С-, -О-, -С(О)-, -NH-, -NHC(O)- или -C(O)NH-; каждое о независимо выбрано и равно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 или 15. В некоторых воплощениях группировка Cm-X отсутствует и группировка PAGn содержит концевую группировку -OH или концевую группировку -OCH3. Например, PAG может представлять собой PAG с концевой метоксильной группой или концевой гидроксигруппой, содержащий 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20 PAG-субъединиц, включая PEG-, PPG- и/или PBG-субъединицы. В некоторых случаях карбонильная группа (-C(O)-), проксимальная к точке присоединения, может отсутствовать, и группировка -(CH2)- может быть связана с подходящей группой на аминокислоте, такой как гидроксильная группа или свободная карбоновокислотная группа.
В другом воплощении модифицирующая группировка может иметь формулу:
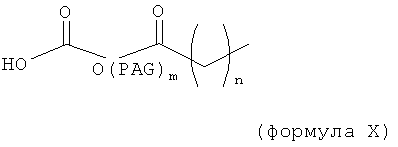
-X-R1-Y-PAG-Z-R2 (формула VI),
где X, Y и Z представляют собой независимо выбранные связывающие группы, и каждая из них возможно присутствует, и X, когда присутствует, связан с соединением инсулином ковалентной связью, а когда X не присутствует, тогда R1 связан с группой аминокислоты соединения инсулина,
по меньшей мере один из R1 и R2 присутствует и представляет собой низший алкил, и возможно может включать карбонильную группу,
R2 представляет собой блокирующую группу, такую как -CH3, -H, тозилат, или активирующую группу, и
PAG представляет собой линейную или разветвленную углеродную цепь, включающую одну или более алкаленгликолевых группировок (т.е. оксиалкаленовых группировок) и возможно включающую одну или более дополнительных группировок, выбранных из группы, состоящей из -S-, -O-, -NH- и -C(O)-, и
где максимальное число тяжелых атомов в модифицирующей группировке составляет 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 или 25.
В воплощениях изобретения любой один или более из X, Y и Z может отсутствовать. Кроме того, X, Y и/или Z, когда присутствуют, могут быть независимо выбраны из -C(O)-, -O-, -S-, -C- и -NH-. В одном воплощении Z представляет собой -C(O)-. В другом воплощении Z не присутствует.
В некоторых воплощениях R1 представляет собой низший алкил, и R2 не присутствует. В других воплощениях R2 представляет собой низший алкил, и R1 не присутствует.
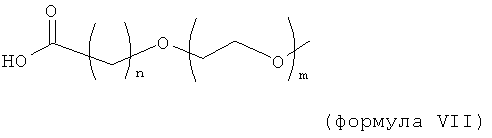
В другом воплощении модифицирующая группировка может включать в себя группировку линейной или разветвленной замещенной углеродной цепи, имеющей основную цепь из 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 19, 19, 20, 21, 22, 23, 24 или 25 атомов, выбранных из группы, состоящей из -C, -C-, -O-, =O, -S-, -NH-, -Si-. Тяжелые атомы обычно включают в себя один или более атомов углерода и один или более неуглеродных тяжелых атомов, выбранных из группы, состоящей из -O-, -S-, -NH- и =O. Атомы углерода и неуглеродные тяжелые атомы обычно присутствуют в соотношении по меньшей мере 1 атом углерода на каждый неуглеродный тяжелый атом, предпочтительно по меньшей мере 2 атома углерода на каждый неуглеродный тяжелый атом, более предпочтительно по меньшей мере 3 атома углерода на каждый неуглеродный тяжелый атом. Атомы углерода и атомы кислорода обычно присутствуют в соотношении по меньшей мере 1 атом углерода на каждый атом кислорода, предпочтительно по меньшей мере 2 атома углерода на каждый атом кислорода, более предпочтительно по меньшей мере 3 атома углерода на каждый атом кислорода. Модифицирующая группировка может включать в себя одну или более блокирующих групп, таких как разветвленный или линейный C1-6, разветвленный или линейный, или карбонил. Модифицирующая группировка обычно включает атомы водорода, и один или более из данных атомов водорода могут быть замещены фтором (который представляет собой тяжелый атом, но не должен рассматриваться тяжелым атомом в вышеупомянутой формуле). Модифицирующая группировка в некоторых случаях может конкретно исключать незамещенные алкильные группировки. Модифицирующая группировка может быть связана, например, с подходящей группой на аминокислоте, такой как аминогруппа, гидроксильная группа или свободная карбоновокислотная группа полипептида, например, с помощью связывающей группы, такой как карбаматная, карбонатная, простая эфирная, сложноэфирная, амидная или вторичная аминная группа, или посредством дисульфидной связи. Молекулы в связывающей группе рассматриваются как часть модифицирующей группировки. В предпочтительном воплощении молекулярная масса модифицирующей группировки меньше молекулярной массы модифицирующей группировки HIM2.
Изобретение включает модифицирующие группировки, имеющие формулу:
 ,
,
где n равно 1, 2, 3 или 4, и m равно 1, 2, 3, 4 или 5.
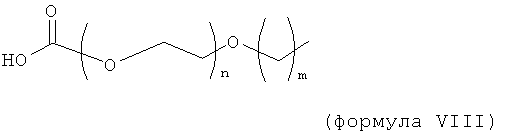
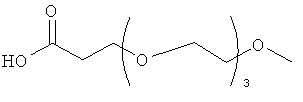
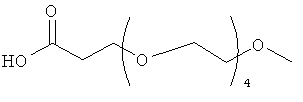
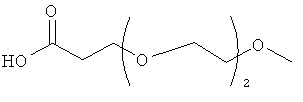
Изобретение включает модифицирующие группировки, имеющие формулу:
 ,
,
где n равно 1, 2, 3, 4 или бит равно 1, 2, 3 или 4.
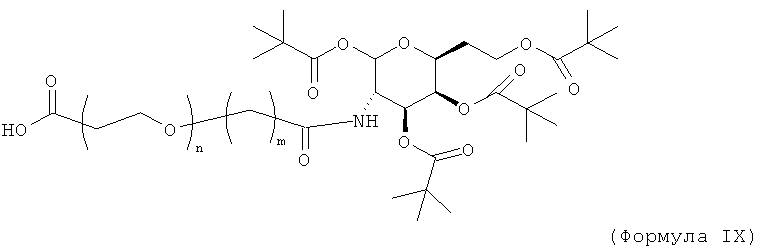
Изобретение включает модифицирующие группировки, имеющие формулу:
 ,
,
где m равно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20, и n равно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20.
Изобретение также включает модифицирующие группировки, имеющие формулу:
 ,
,
где PAG представляет собой PAG-группировку, имеющую m субъединиц, и m равно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20, и n равно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 или 20.
Другие предпочтительные модифицирующие группировки включают в себя:
 ,
,
 ,
,
 ,
,
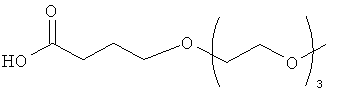 ,
,
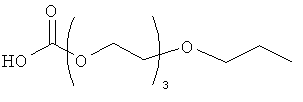 ,
,
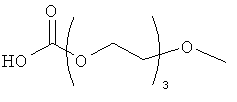 ,
,
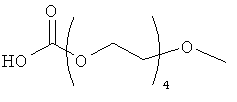 и
и
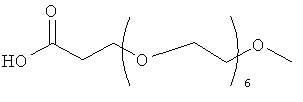 .
.
Следующие модифицирующие группировки могут быть особенно предпочтительными для применения в схеме замещения базального соединения инсулина.
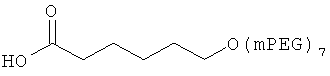 ,
,
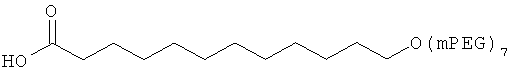 ,
,
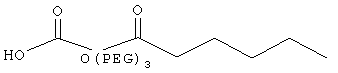 ,
,
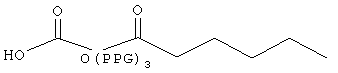 ,
,
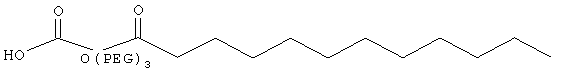 ,
,
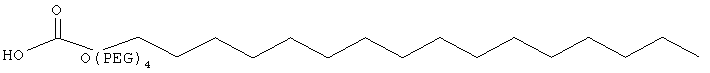 ,
,
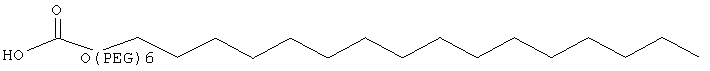 ,
,
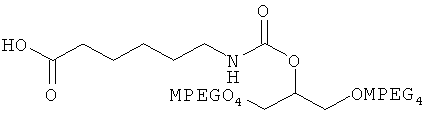 и
и
 .
.
Другие модифицирующие группировки включают в себя следующие группировки:
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-GH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2 _CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2 _CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2 _CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH3,
R-CH2 _CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2 _CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2 _O-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-O-,
R-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-CH2 _CH2-CH2-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-CH2 _CH2-O-CH2-CH2-CH2 _O-CH3,
R-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-О-CH2-CH2-О-CH2-CH2-CH2-CH2CH2-СН3,
R-CH2-CH2-CH2-CH2-CH2-О-CH2-СН2-О-CH2-CH2-CH2-CH2-СН3,
R-CH2-CH2-CH2-CH2-CH2-О-CH2-CH2-О-CH2-СН2-CH2-СН3,
R-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-CH2-CH2-O-CH3,
R-CH2 _CH2-CH2-CH2-O-CH2-CH2-,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2 _O-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-СН2-СН2-СН2-O-СН2-СН2-СН2-СН2-СН2-СН3,
R-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-,
R-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-,
R-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-,
R-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2 _CH3,
R-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-О-CH2-CH2-О-CH2-CH2-CH2-О-CH2-CH2-CH2-СН3,
R-СН2-СН2-СН2-O-СН2-СН2-O-СН2-СН2-СН2-O-СН2-СН2-СН3,
R-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-CH2-O-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-H3,
R-CH2-CH2-O-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-CH2-O-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CHZ-O-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-O-CH3,
R-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH3,
Р-CH2-CH2-О-CH2-CH2-О-CH2-CH2-О-CH2-CH2-О-CH2-СН3,
R-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-CH2-O-CH2 _CH3,
R-CH2-CH2-O-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2 _CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-СН2-О-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-СН3,
R-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-O-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-O-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-O-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-O-CH2-CH2-CH2-O-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-CH2-O-CH3,
R-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2 _CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-O-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-,
R-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-CH2-O-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-CH2-O-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-O-CH2-CH3,
R-CH2-O-CH2-CH2-O-CH2-CH2-O-CH3,
R-CH2-O-CH2-CH2-O-CH3,
R-CH2-O-CH2-CH3,
R-CH2-O-CH3,
где R представляет собой -H, -ОН, -СН2ОН, -СН(ОН)2, -С(O)ОН, -СН2С(O)ОН, или активирующую группировку, такую как карбодиимид, смешанный ангидрид или N-гидроксисукцинимид, или блокирующую группу. Изобретение также включает такие группировки, присоединенные к белку или пептиду, предпочтительно к соединению инсулину. Конкретные стратегии коъюгации обсуждаются более подробно ниже. Из этих модифицирующих группировок предпочтительными группировками являются такие группировки, которые делают соединение инсулин менее липофильным и/или более гидрофильным, чем соответствующее неконъюгированное соединение инсулин. Изобретение включает такие модифицирующие группировки, дополнительно включающие одну или более карбонильных групп, предпочтительно 1, 2, 3, 4 или 5 карбонильных групп; данные карбонильные группы могут быть встроены в модифицирующую группировку, или -О- или -СН2- могут быть заменены карбонилом. Дополнительно, любая из группировок -СН2- или -СН3 может быть замещена, например, низшим алкилом, или -ОН, или PAG-цепью, имеющей 1, 2, 3, 4 или 5 PAG-субъединиц, которые могут быть одинаковыми или разными. Предпочтительно, R выбран таким образом, что каждый -О- отделен от ближайшего -О- по меньшей мере 2 атомами углерода. В изобретение также включены разветвленные модифицирующие группировки, в которых две или более из группировок присоединены к разветвленной группировке, такой как лизин.
Фармацевтические характеристики, такие как гидрофильность/липофильность конъюгатов согласно воплощениям изобретения, можно изменять, например, путем регулирования липофильной и гидрофильной частей модифицирующей группировки, например, путем увеличения или уменьшения числа PAG-мономеров, типа и длины алкильной цепи, природы PAG-пептидной связи и числа сайтов конъюгирования. Точная природа связи модифицирующей группировки и пептида может быть изменена таким образом, чтобы она стала стабильной и/или чувствительной к гидролизу при физиологическом рН или в плазме. Изобретение также включает любую из вышеупомянутых модифицирующих группировок, связанных с полипептидом, предпочтительно с соединением инсулином. Предпочтительно, модифицирующая группировка делает данный полипептид более растворимым, чем соответствующий неконъюгированный полипептид, например, по меньшей мере в 1,05; 1,25, 1,5; 1,75; 2; 2,5; 3; 3,5; 4; 4,5; 5; 5,5; 6; 6,5; 7; 7,5; 8; 8,5; 9; 9,5; 10; 10,5; 11; 11,5; 12; 12,5; 13; 13,5; 14; 14,5 или 15 раз. Модифицирующая группировка по изобретению может быть связана, например, с соединением инсулином, таким как инсулин человека, в любой подходящей точке присоединения. Предпочтительная точка присоединения представляет собой нуклеофильный остаток, например А1, В1 и/или В29.
Кроме того, следует принимать во внимание, что один из аспектов изобретения включает новые модифицирующие группировки, такие как, но не ограничиваясь этим, группировки Формул VII и VIII, в карбоновокислотной форме. Кроме того, когда модифицирующая группировка включает карбоксильную группу, ее можно превратить в смешанный ангидрид и подвергнуть взаимодействию с аминогруппой пептида с образованием конъюгата, содержащего амидную связь. В другом методе карбоксильная группа может быть обработана водорастворимым карбодиимидом и подвергнута взаимодействию с пептидом с образованием конъюгатов, содержащих амидные связи. Следовательно, изобретение включает активированные формы новых группировок, представленных в данном описании, таких как активированные формы модифицирующих группировок Формул VII и VIII, и другие новые олигомеры по изобретению, такие как карбодиимиды, смешанные ангидриды или N-гидроксисукцинимиды.
В некоторых случаях модифицирующая группировка может быть связана с полипептидом с помощью аминокислоты или последовательности из 2 или более аминокислот, связанных с С-концом или боковой цепью полипептида. Например, в одном воплощении модифицирующая группировка связана с -ОН или -С(O)ОН Thr, и mm-модифицированный Thr (mm, модифицирующая группировка) связан с полипептидом на карбоксильном конце. Например, в одном воплощении модифицирующая группировка связана с -ОН или -С(O)ОН Thr, и данный модифицированный Thr связан с аминокислотой В29 (например В29 Lys в случае инсулина человека) соединения des-Thr-инсулина. В другом примере mm связана с -ОН или -С(O)ОН Thr концевого октапептида из В-цепи соединения инсулина, и mm-модифицированный октапептид связан с аминокислотой В22 соединения des-окта-инсулина. Другие вариации станут очевидны для специалиста в данной области в свете данного описания изобретения.
7.2.11. Солеобразующие группировки
В некоторых воплощениях модифицирующая группировка содержит солеобразующую группировку. Солеобразующая группировка может представлять собой различные подходящие солеобразующие группировки, которые, как известно специалистам в данной области, включают в себя карбоксилат и аммоний, но не ограничиваются ими. В некоторых воплощениях, где модифицирующая группировка включает солеобразующую группировку, конъюгат соединения инсулина предложен в форме соли. В этих воплощениях конъюгат соединения инсулина ассоциирован с подходящим фармацевтически приемлемым противоионом, который, как известно специалистам в данной области, включает в себя, но не ограничивается этим, отрицательные ионы, такие как хлоро, бромо, иодо, фосфат, ацетат, карбонат, сульфат, тозилат и мезилат, или положительные ионы, такие как натрий, калий, кальций, литий и аммоний.
Подразумевается, что вышеупомянутые примеры модифицирующих группировок являются иллюстративными и не должны рассматриваться в качестве ограничивающих каким-либо образом. Специалисту в данной области будет понятно, что подходящие группировки для конъюгирования с целью достижения конкретной функциональности, возможно будут ограничены связями химических механизмов конъюгирования, раскрытыми и заявленными в данном описании. Соответственно, дополнительные группировки могут быть выбраны и использованы в соответствии с принципами, раскрытыми в данном описании.
7.3. Стратегии конъюгирования
Факторы, такие как степень конъюгирования с модифицирующими группировками, выбор сайтов конъюгирования на молекуле и выбор модифицирующих группировок, можно варьировать для того, чтобы получить конъюгат, который, например, является менее чувствительным к деградации in vivo и, соответственно, имеет увеличенный период полураспада в плазме. Например, соединения инсулины могут быть модифицированы таким образом, чтобы включать модифицирующую группировку в одном, двух, трех, четырех, пяти или более сайтах в структуре соединения инсулина в соответствующих сайтах присоединения (т.е. конъюгирования модифицирующей группировки), подходящих для облегчения ассоциации модифицирующей группировки. В качестве примера, такие подходящие сайты конъюгирования могут содержать такие аминокислотные остатки, как аминокислотный остаток лизина.
В некоторых воплощениях конъюгаты соединений инсулинов представляют собой моноконъюгаты. В других воплощениях конъюгаты соединений инсулинов представляют собой поликонъюгаты, такие как диконъюгаты, триконъюгаты, тетраконъюгаты, пентаконъюгаты и тому подобное. Количество модифицирующих группировок в соединении инсулине ограничено только количеством сайтов конъюгирования в соединении инсулине. В других воплощениях конъюгаты соединений инсулинов представляют собой смеси моноконъюгатов, диконъюгатов, триконъюгатов, тетраконъюгатов и/или пентаконъюгатов. Предпочтительные стратегии конъюгирования представляют собой такие стратегии, которые дают конъюгат, проявляющий некоторую или полную биологическую активность исходного соединения инсулина.
Предпочтительные сайты присоединения включают в себя А1 N-конца, В1 N-конца и лизин В29 боковой цепи. Весьма предпочтительными являются В29 моноконъюгат и В1, В29 диконъюгаты. Другой предпочтительной точкой присоединения является функциональная аминогруппа на С-пептидном компоненте или лидерном пептидном компоненте соединения инсулина.
С соединением инсулином могут быть связаны одна или более модифицирующих группировок (т.е. одна или множество структур модифицирующих группировок). Модифицирующие группировки в данном множестве предпочтительно являются одинаковыми. Однако необходимо понимать, что модифицирующие группировки в данном множестве могут отличаться друг от друга, или, альтернативно, некоторые из модифицирующих группировок в данном множестве могут быть одинаковыми, а некоторые могут быть разными. Когда с соединением инсулином связано множество модифицирующих группировок, может быть предпочтительно, чтобы одна или более модифицирующих группировок были связаны с соединением инсулином гидролизуемыми связями и одна или более модифицирующих группировок были связаны с соединением инсулином негидролизуемыми связями. Альтернативно, все связи, соединяющие множество модифицирующих группировок с соединением инсулином, могут быть гидролизуемыми, но имеют разную степень гидролизуемости, так что, например, одна или более модифицирующих группировок могут быть относительно быстро удалены из соединения инсулина путем гидролиза в организме, и одна или более модифицирующих группировок удаляются более медленно из соединения инсулина путем гидролиза в организме.
7.3.1. Связывание модифицирующей группировки с соединением инсулином
Модифицирующая группировка предпочтительно ковалентно связана с соединением инсулином. С соединением инсулином может быть ковалентно связано более одной группировки на модифицирующей группировке. В связывании могут быть использованы гидролизуемые или негидролизуемые связи или комбинации этих двух связей (т.е. разные связи в разных сайтах конъюгирования).
В некоторых воплощениях соединение инсулин связано с модифицирующей группировкой с использованием гидролизуемой связи (например сложноэфирной, карбонатной или гидролизуемой карбаматной связи). Использование гидролизуемого связывания дает конъюгат соединения инсулина, который действует как пролекарство. Подход, предполагающий использование пролекарства, может быть желательным в том случае, когда конъюгат соединения инсулина и модифицирующей группировки является неактивным (т.е. конъюгат теряет способность воздействовать на организм через основной механизм действия соединения инсулина), например, когда сайт конъюгирования модифицирующей группировки находится в связывающей области соединения инсулина. Использование гидролизуемого связывания также может обеспечить замедленное или контролируемое высвобождение, введение соединения инсулина в течение заданного периода времени по мере того, как одна или более модифицирующих группировок отщепляются от соответствующих конъюгатов соединения инсулина и модифицирующей группировки с получением активного лекарства.
В других воплощениях соединение инсулин связано с модифицирующей группировкой с помощью негидролизуемой связи (например негидролизуемой карбаматной, амидной или простой эфирной связи). Использование негидролизуемой связи может быть предпочтительным в том случае, когда желательно, чтобы терапевтически значимые количества конъюгата соединения инсулина циркулировали в кровяном русле в течение продолжительного периода времени, например в течение по меньшей мере 2 часов после введения. Связи, используемые для ковалентного связывания соединения инсулина с модифицирующей группировкой в негидролизуемой форме, обычно выбраны из группы, состоящей из ковалентной(ых) связи(ей), сложноэфирных группировок, карбонатных группировок, карбаматных группировок, амидных группировок и вторичных аминных группировок.
Модифицирующая группировка может быть связана с соединением инсулином по разным нуклеофильным остаткам, которые включают в себя нуклеофильные гидроксильные функциональные группы и/или функциональные аминогруппы, но не ограничиваются этим. Нуклеофильные гидроксильные функциональные группы можно найти, например, в остатках серина и/или тирозина, и нуклеофильные функциональные аминогруппы можно найти, например, в остатках гистидина и/или Lys, и/или на одном или более из N-концов А- или В-цепей соединения инсулина. Когда модифицирующая группировка связана с N-концом натрийуретического пептида, тогда при связывании предпочтительно образуется вторичный амин.
Модифицирующая группировка может быть связана с соединением инсулином по свободной группе -SH, например, путем образования сложной тиоэфирной, простой тиоэфирной или сульфонатной связи.
Модифицирующая группировка может быть связана с соединением инсулином через одну или более аминогрупп. В случае инсулина человека примеры включают в себя аминогруппы в А1, В1 и В29. В одном воплощении одна модифицирующая группировка связана с одиночной аминогруппой в соединении инсулине. В другом воплощении две модифицирующие группировки связаны с разными аминогруппами в соединении инсулине. Когда имеются две модифицирующие группировки, связанные с двумя аминогруппами, предпочтительным расположением является связывание в В1 и В29. Когда имеется несколько полимеров, все данные полимеры могут быть одинаковыми или один или более полимеров могут отличаться от остальных. Различные способы и типы связывания полимеров с соединениями инсулинами описаны в заявке на патент США №09/873899 под названием "Mixtures of insulin compound conjugates comprising polyalkylene glycol, uses thereof, and methods of making same", поданной 4 июня 2001 г., полное описание которой включено в данное описание посредством ссылки.
В других воплощениях может быть использован подход, предполагающий использование частичного пролекарства, когда гидролизируется часть модифицирующей группировки. Например, смотри патент США №6309633, Ekwuribe с соавт.(полное описание которого включено в данное описание посредством ссылки), где описаны модифицирующие группировки, имеющие гидрофильные и липофильные компоненты, в которых липофильные компоненты гидролизируются in vivo с образованием микропэгилированного конъюгата.
7.3.2. Выбор модифицирующей группировки и свойства конъюгата соединения инсулина и его комплексов
Модифицирующая группировка может быть выбрана таким образом, чтобы получить конъюгат соединения инсулина и его комплексы с желательными свойствами. Предпочтительные модифицирующие группировки выбраны таким образом, чтобы сделать соединение инсулин более растворимым в водном растворе, чем растворимость в воде соединения инсулина в отсутствие модифицирующей группировки, предпочтительно, чтобы оно стало по меньшей мере в 1,05; 1,25; 1,5; 1,75; 2; 2,5; 3; 3,5; 4; 4,5; 5; 5,5; 6; 6,5; 7; 7,5; 8; 8,5; 9; 9,5; 10; 10,5; 11; 11,5; 12; 12,5; 13; 13,5; 14; 14,5 или 15 раз более растворимым в водном растворе, чем исходное соединение инсулин (т.е. соответствующее неконъгированное соединение инсулин). Например, не находящийся в комплексе нативный инсулин человека имеет растворимость примерно 18 мг/мл при рН приблизительно 7,4. Авторы изобретения неожиданно обнаружили способ получения комплексов конъюгатов инсулина человека, которые являются по меньшей мере в 1,05; 1,25; 1,5; 1,75; 2; 2,5; 3; 3,5; 4; 4,5; 5; 5,5; 6; 6,5; 7; 7,5; 8; 8,5; 9; 9,5; 10; 10,5; 11; 11,5; 12; 12,5; 13; 13,5; 14; 14,5 или 15 раз более растворимыми, чем инсулин человека.
В некоторых воплощениях модифицирующая группировка выбрана таким образом, чтобы растворимость в воде конъюгата соединения инсулина превышала 1 г/л, 2 г/л, 3 г/л, 4 г/л, 5 г/л, 20 г/л, 50 г/л, 100 г/л или даже 150 г/л при рН, варьирующем от приблизительно 4 до приблизительно 8, предпочтительно при рН, варьирующем от приблизительно 5 до приблизительно 7,5, в идеале при рН приблизительно 7,4.
Конъюгат соединения инсулина может иметь большую пероральную биодоступность в организме млекопитающего, чем приемлемый с научной точки зрения контроль, такой как соответствующее неконъгированное соединение инсулин. В других воплощениях конъюгат соединения инсулина имеет большую пероральную биодоступность в организме человека, чем приемлемый с научной точки зрения контроль, такой как соответствующее неконъгированное соединение инсулин. В некоторых воплощениях абсорбция конъюгата соединения инсулина, например, определенная путем измерения уровней конъюгата в плазме, по меньшей мере в 1,5; 2; 2,5; 3; 3,5 или 4 раза больше абсорбции контрольного неконъюгированного соединения инсулина.
Следует принимать во внимание, что хотя в некоторых аспектах изобретения модифицирующая группировка выбрана таким образом, чтобы сделать конъюгат соединения инсулина более растворимым, чем соответствующее неконъгированное соединение инсулин, в других аспектах модифицирующая группировка также, или альтернативно, может быть выбрана таким образом, чтобы сделать конъюгат соединения инсулина таким же или более гидрофильным, чем соответствующее неконъгированное соединение инсулин. Кроме того, модифицирующая группировка может быть выбрана таким образом, чтобы сделать конъюгат соединения инсулина более амфифильным, чем соответствующее неконъгированное соединение инсулин.
В некоторых воплощениях комплекс конъюгата соединения инсулина с катионом является таким же или более водорастворимым, чем (а) соответствующий не находящийся в комплексе конъюгат соединения инсулина, (б) соответствующее не находящееся в комплексе и неконъюгированное соединение инсулин и/или (в) соответствующее находящееся в комплексе, но неконъюгированное соединение инсулин.
В предпочтительном воплощении растворимость в воде конъюгата соединения инсулина уменьшена путем добавления Zn++. В некоторых воплощениях модифицирующая группировка выбрана таким образом, чтобы сделать конъюгат соединения инсулина таким же или более растворимым, чем соответствующее неконъюгированное соединение инсулин, и растворимость в воде конъюгата соединения инсулина уменьшена путем добавления цинка. В других воплощениях модифицирующая группировка выбрана таким образом, чтобы сделать конъюгат соединения инсулина таким же или более растворимым, чем соответствующее неконъюгированное соединение инсулин, растворимость в воде конъюгата соединения инсулина уменьшена путем добавления цинка, и растворимость в воде катионного комплекса выше растворимости в воде соединения инсулина. В другом аспекте конъюгат соединения инсулина представляет собой соединение инсулин, ацилированное жирной кислотой, катион представляет собой цинк, и растворимость в воде конъюгата соединения инсулина уменьшена путем добавления цинка. В еще одном воплощении конъюгат соединения инсулина представляет собой соединение инсулин, ацилированное жирной кислотой, которое является таким же или более водорастворимым, чем соответствующее неконъюгированное соединение инсулин, катион представляет собой цинк, и растворимость в воде конъюгата соединения инсулина уменьшена путем добавления цинка.
В некоторых предпочтительных воплощениях липофильность конъюгата соединения инсулина относительно соответствующего исходного соединения инсулина равна 1 или меньше 1. Относительная липофильность конъюгата соединения инсулина (котн) по сравнению с соответствующим исходным соединением инсулином может быть определена, например, следующим образом: котн=(tконъюгат-t0)/(tчеловек-t0), где относительную липофильность измеряют на колонке LiChroSorb RP18 (5 мкм, 250×4 мм) для высокоэффективной жидкостной хроматографии путем изократической элюции при 40°С. В качестве элюентов могут быть использованы следующие смеси: 0,1 М натрий-фосфатный буфер, рН 7,3, содержащий 10% ацетонитрила и 50% ацетонитрила в воде. "Мертвый" объем (t0) определяют путем введения 0,1 мМ нитрата натрия. Время удерживания для инсулина человека доводили до по меньшей мере 2t0 путем варьирования соотношения между смесями (в)(1) и (b)(2). Предпочтительно, в этих воплощениях относительная липофильность приблизительно равна 1 или меньше 1, или по существу меньше 1. В предпочтительном воплощении соединение инсулин представляет собой инсулин человека, и относительная липофильность меньше 1. Предпочтительно относительная липофильность меньше приблизительно 0,99; 0,98; 0,97; 0,96; 0,95; 0,94; 0,93; 0,92; 0,91 или 0,90. Обсуждение методов определения растворимости и/или липофильности инсулина и конъюгатов инсулина приведено в патенте США №5750499 под названием "Acylated insulin", опубликованном Harelund с соавт.12 мая 1998, полное описание которого включено в данное описание посредством ссылки.
В одном воплощении относительная липофильность является такой, как описано выше, и модифицирующая группировка представляет собой углеродную цепь, имеющую 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17 или 18 атомов углерода, причем данная углеродная цепь содержит 2, 3, 4, 5, 6, 7, 8, 9 или 10 встроенных в нее оксигрупп. В другом воплощении относительная липофильность является такой, как описано выше, и модифицирующая группировка представляет собой углеродную цепь, имеющую 5, 6, 7, 8, 9 или 10 атомов углерода, причем данная углеродная цепь содержит 2, 3 или 4 встроенных в нее оксигруппы. В родственном воплощении относительная липофильность является такой, как описано выше, и модифицирующая группировка содержит 2, 3, 4, 5, 6, 7, 8, 9 или 10 полиалкаленгликолевых структурных единиц. В другом родственном воплощении относительная липофильность является такой, как описано выше, и модифицирующая группировка содержит 1, 2 или 3 полиэтиленгликолевые структурные единицы и 1, 2 или 3 полипропиленгликолевые структурные единицы.
7.4. Компонент, представляющий собой катион металла, и характеристики комплексов
Комплексы конъюгатов соединений инсулинов с катионами включают катион металла. Подходящие катионы металлов для использования в качестве катионного компонента включают в себя любой катион металла, способный к комплексообразованию, агрегированию или кристаллизации с конъюгатом соединения инсулина. Предпочтительно, чтобы катион металла образовывал комплекс с конъюгатом соединения инсулина. Может быть использован один или несколько катионов. Предпочтительно, катион не является существенным окислителем для конъюгата соединения инсулина, т.е. не является окислителем в такой степени, чтобы сделать комплексы непригодными для заданной цели.
В некоторых воплощениях катион металла является биосовместимым. Катион металла является биосовместимым, если данный катион не оказывает слишком значительных вредных воздействий на организм реципиента, таких как значительная иммунологическая реакция в месте инъекции. Однако следует принимать во внимание, что в некоторых обстоятельствах риск токсичности и другие вредные воздействия могут быть компенсированы общей пользой от композиции конъюгата соединения инсулина с катионом и поэтому могут быть приемлемыми при таких обстоятельствах.
Пригодность катионов металлов для стабилизации биологически активных агентов и отношение катиона металла к необходимому биологически активному агенту могут быть определены средним специалистом в данной области путем выполнения ряда тестов на стабильность, таких как электрофорез в полиакриламидном геле, изоэлектрофокусирование, обращенно-фазная хроматография и ВЭЖХ-анализ (анализ с использованием высокоэффективной жидкостной хроматографии) на частицы биологически активных агентов, стабилизированных катионом металла, до и после уменьшения размера частиц и/или инкапсулирования.
Компонент, представляющий собой катион металла, включает соответственно один или более одновалентных, двухвалентных или трехвалентных катионов металлов или их комбинации. В предпочтительном воплощении катион металла представляет собой катион металла группы II или катион переходного металла. Примеры подходящих двухвалентных катионов включают в себя Zn++, Mn++, Са++, Fe++, Ni++, Cu++, Со++ и/или Mg++. Когда включен одновалентный катион, тогда он представляет собой предпочтительно Na+, Li+или К+. Катион добавляют предпочтительно в виде соли, такой как хлоридная или ацетатная соль, наиболее предпочтительными являются ZnCl2 и ZnAc.
Молярное отношение конъюгата соединения инсулина к катиону обычно составляет от приблизительно 1:1 до приблизительно 1:100, предпочтительно от приблизительно 1:2 до приблизительно 1:12, и более предпочтительно от приблизительно 1:2 до приблизительно 1:7, или составляет приблизительно 1:2, 1:3, 1:4, 1:5, 1:6 или 1:7. В конкретном воплощении в качестве катионного компонента используют Zn++, он присутствует в молярном отношении компонента, представляющего собой катион цинк, к конъюгату соединения инсулина, составляющем от приблизительно 1:1 до приблизительно 1:100, предпочтительно составляющем от приблизительно 1:2 до приблизительно 1:12, и более предпочтительно от приблизительно 1:2 до приблизительно 1:7, или составляющем приблизительно 1:2, 1:3, 1:4, 1:5, 1:6 или 1:7.
Предпочтительно, катионный компонент состоит более чем на приблизительно 90% из одного катиона, такого как Zn++. Предпочтительно, данный катион представляет собой более чем приблизительно 95%, 99% или 99,9% Zn++.
Предпочтительно, устойчивость к расщеплению химотрипсином находящегося в комплексе конъюгата соединения инсулина выше, чем устойчивость к расщеплению химотрипсином соответствующего не находящегося в комплексе конъюгата соединения инсулина. Предпочтительно, устойчивость к расщеплению химотрипсином находящегося в комплексе конъюгата соединения инсулина выше, чем устойчивость к расщеплению химотрипсином соответствующего находящегося в комплексе, но неконъюгированного соединения инсулина,.
Находящийся в комплексе конъюгат соединения инсулина может иметь большую пероральную биодоступность в организме млекопитающего, чем приемлемый с научной точки зрения контроль, такой как соответствующий не находящийся в комплексе конъюгат соединения инсулина. В других воплощениях находящийся в комплексе конъюгат соединения инсулина имеет большую пероральную биодоступность в организме человека, чем приемлемый с научной точки зрения контроль, такой как соответствующий не находящийся в комплексе конъюгат соединения инсулина. В некоторых воплощениях абсорбция находящегося в комплексе конъюгата соединения инсулина, например, как определено уровнями данного конъюгата в плазме, по меньшей мере в 1,5; 2; 2,5; 3; 3,5 или 4 раза больше абсорбции не находящегося в комплексе конъюгата соединения инсулина.
Находящийся в комплексе конъюгат соединения инсулина может иметь большую пероральную биодоступность в организме млекопитающего, чем приемлемый с научной точки зрения контроль, такой как соответствующее находящееся в комплексе, но неконъюгированное соединение инсулин. В других воплощениях находящийся в комплексе конъюгат соединения инсулина имеет большую пероральную биодоступность в организме человека, чем приемлемый с научной точки зрения контроль, такой как соответствующее находящееся в комплексе, но неконъюгированное соединение инсулин. В некоторых воплощениях абсорбция находящегося в комплексе соединения инсулина, например, как определено уровнями данного конъюгата в плазме, по меньшей мере в 1,5; 2; 2,5; 3; 3,5 или 4 раза больше абсорбции находящегося в комплексе, но неконъюгированного соединения инсулина.
7.5. Комплексообразующие агенты
В некоторых воплощениях композиции конъюгата соединения инсулина с катионом включают один или более комплексообразующих агентов. Примеры комплексообразующих агентов, подходящих для применения в настоящем изобретении, включают в себя протамины, сурфен, глобиновые белки, спермин, спермидин, альбумин, аминокислоты, карбоновые кислоты, соединения поликатионных полимеров, катионные полипептиды, анионные полипептиды, нуклеотиды и антисмысловые последовательности. Смотри Brange, J., Galenics of Insulin compound, Springer-Verlag, Berlin Heidelberg (1987), полное описание которого включено в данное описание посредством ссылки. В свете описания настоящего изобретения пригодность комплексообразующих агентов для стабилизации данных композиций может быть определена средним специалистом в данной области. В некоторых воплощениях композиции конъюгата соединения инсулина с катионом конкретно исключают комплексообразующий агент или по существу лишены комплексообразующего агента.
Предпочтительным комплексообразующим агентом является протамин. В твердой форме протамин обычно присутствует в молярном отношении соединения инсулина к протамину, предпочтительно составляющем от приблизительно 3:1 до приблизительно 1:3, более предпочтительно от приблизительно 2:1 до приблизительно 1:2, в идеале в молярном отношении приблизительно 1:1. В некоторых воплощениях композиции конъюгата соединения инсулина с катионом конкретно исключают протамин или по существу лишены протамина.
В качестве комплексообразующих агентов могут быть использованы также аминокислоты, например глицин, аланин, валин, лейцин, изолейцин, серии, треонин, фенил аланин, пролин, триптофан, аспарагин, глутаминовая кислота и гистидин, и олигопептиды, такие как диглицин.
Карбоновые кислоты также подходят для применения в качестве комплексообразующих агентов; примеры включают в себя уксусную кислоту и оксикарбоновые кислоты, так как лимонная кислота, 3-гидроксимасляная кислота и молочная кислота.
7.6. Стабилизирующие агенты
В некоторых воплощениях композиции конъюгатов соединений инсулинов с катионами содержат один или более стабилизирующих агентов. Предпочтительные стабилизирующие агенты включают в себя фенольные соединения и ароматические соединения. Предпочтительными фенольными соединениями являются фенол, мета-крезол и мета-парабен или их смеси. Стабилизирующий агент может присутствовать в любом количестве, которое улучшает стабильность композиций конъюгатов соединений инсулинов с катионами относительно приемлемого с научной точки зрения контроля, такого как соответствующая композиция конъюгата соединения инсулина с катионом в отсутствие стабилизирующего агента.
7.7. Представление комплексов
Комплексы могут быть предложены в виде сухого твердого вещества, такого как по существу чистый порошок конъюгата соединения инсулина с катионом или порошок, содержащий твердый конъюгат соединения инсулина с катионом вместе с другими фармацевтически приемлемыми компонентами. Комплексы могут быть также предложены в растворенном состоянии в водной или органической среде и/или в виде нерастворимых твердых веществ в таких средах.
7.7.1. Твердые композиции
Комплекс конъюгата соединения инсулина с катионом может быть предложен в виде твердого вещества. Данное твердое вещество может находиться, например, в высушенном состоянии или в нерастворенном состоянии в водном растворе, органическом растворителе, эмульсии, микроэмульсии или в другой невысушенной форме.
В одном воплощении комплекс конъюгата соединения инсулина с катионом предложен в виде чистой обработанной твердой композиции. В чистой обработанной твердой композиции молярное отношение конъюгата соединения инсулина к катиону обычно составляет от приблизительно 3:4 до приблизительно 3:0,5 (конъюгат соединение инсулин катион), от приблизительно 3:3,5 до приблизительно 3:1 или в идеале приблизительно 3:1.
В обработанной чистой твердой композиции Т-типа (с катионом, конъюгатом соединения инсулина и без протамина) молярное отношение конъюгата соединения инсулина к катиону обычно составляет от приблизительно 3:4 до приблизительно 3:0,5 (конъюгат соединение инсулин: катион), от приблизительно 3:3,5 до приблизительно 3:1 или в идеале приблизительно 3:1. В обработанной чистой твердой композиции Т-типа с протамином (с катионом, конъюгатом соединения инсулина и протамином) молярное отношение конъюгата соединения инсулина к катиону обычно составляет от приблизительно 3:6 до приблизительно 3:0,5 (конъюгат соединение инсулин: катион), от приблизительно 3:5 до приблизительно 3:1 или в идеале приблизительно 3:2.
В обработанной чистой твердой композиции R-типа (lente) (с катионом, соединением инсулином и стабилизирующим соединением (например с фенольным соединением) и без протамина) молярное отношение конъюгата соединения инсулина к катиону обычно может варьировать от приблизительно 3:4,5 до приблизительно 3:0,9, предпочтительно от приблизительно 3:3,9 до приблизительно 3:2,4. В обработанной чистой твердой композиции R-типа (ultralente) (с катионом, соединением инсулином и стабилизирующим соединением (например с фенольным соединением) и без протамина), молярное отношение конъюгата соединения инсулина к катиону обычно может варьировать от приблизительно 3:12 до больше чем приблизительно 3:4,5, предпочтительно от приблизительно 3:9 до приблизительно 3:4,8, более предпочтительно от приблизительно 3:6 до приблизительно 3:5,4. В обработанной чистой твердой композиции R-типа с протамином (с катионом, соединением инсулином и стабилизирующим соединением (например с фенольным соединением) и протамином) молярное отношение конъюгата соединения инсулина к катиону обычно может варьировать от приблизительно 3:12 до приблизительно 3:3, предпочтительно от приблизительно 3:9 до приблизительно 3:4,5, более предпочтительно от приблизительно 3:6,9 до приблизительно 3:5,4.
В случае одновалентного катиона, такого как Na+, как следовало ожидать, отношение конъюгата соединения инсулина к катиону в твердом веществе составляет от приблизительно 3:6 до приблизительно 3:3.
Твердые композиции по изобретению могут включать в себя, например, такие композиции, как порошки, содержащие конъюгаты соединений инсулинов и/или комплексы конъюгатов соединений инсулинов с катионами по изобретению. Предпочтительно, данные твердые композиции имеют фармацевтически приемлемый уровень чистоты, т.е. свободны от примесей, которые неприемлемо снижали бы пригодность композиций для использования людьми.
В некоторых воплощениях предложены композиции, в которых компонент, представляющий собой конъюгат соединения инсулина с катионом, является более чем на приблизительно 90% кристаллическим, предпочтительно более чем на приблизительно 95% кристаллическим, более предпочтительно более чем на приблизительно 99% кристаллическим. В других воплощениях предложены композиции, в которых компонент, представляющий собой конъюгат соединения инсулина с катионом, представляет собой более чем на приблизительно 90% аморфные твердые вещества, предпочтительно более чем на приблизительно 95% аморфные твердые вещества, более предпочтительно более чем на приблизительно 99% аморфные твердые вещества.
В других воплощениях предложены композиции, в которых компонент, представляющий собой конъюгат соединения инсулина с катионом, присутствует в смеси аморфных твердых веществ и кристаллических твердых веществ. Например, отношение аморфного твердого вещества к кристаллическому твердому веществу может составлять от приблизительно 1:10 до приблизительно 10:1, или от приблизительно 1:9 до приблизительно 9:1, или от приблизительно 1:8 до приблизительно 8:1, или от приблизительно 1:7 до приблизительно 7:1, или от приблизительно 1:6 до приблизительно 6:1, или от приблизительно 1:5 до приблизительно 5:1, или от приблизительно 1:4 до приблизительно 4:1, или от приблизительно 1:3 до приблизительно 3:1, или от приблизительно 1:2 до приблизительно 2:1, или приблизительно 1:1.
Кроме того, могут быть предложены композиции, в которых использованы смеси твердых соединений инсулинов с катионами, содержащих разные соединения инсулина, такие как твердое вещество, включающее соединение нативного инсулина, с твердым веществом, включающим конъюгаты соединений инсулинов, или твердые вещества, включающие один конъюгат соединения инсулина с твердым веществом, включающим другой конъюгат соединения инсулина.
Кроме того, тип твердого вещества и компонент, представляющий собой соединение инсулин/конъюгат соединения инсулина, все могут варьировать. Например, могут быть предложены композиции, которые содержат кристаллы соединений Zn-инсулина, где использовано соединение нативного инсулина, и аморфные конъюгаты соединений инсулинов, или могут быть предложены композиции, которые содержат аморфные твердые соединения Zn-инсулина, где использовано соединение нативного инсулина, и кристаллические конъюгаты соединений Zn-инсулина. Такие смеси могут быть использованы для достижения вариаций в физических характеристиках, таких как профиль растворения, и/или вариаций в фармакокинетическом профиле.
Средний размер частиц данных твердых веществ предпочтительно составляет от приблизительно 0,1 до приблизительно 100 микрон, более предпочтительно 1-50 микрон, еще более предпочтительно 1-25 микрон, в идеале 1-15 микрон. Частицы маленького размера могут быть получены в условиях микрокристаллизации, посредством распылительной сушки, измельчения, сушки в вакууме, сублимационной сушки и тому подобного.
В одном воплощении композиция, когда она высушена, содержит более приблизительно 96% (масс./масс.) конъюгата соединения инсулина и от приблизительно 0,05; 0,1; 0,15 или 0,2 до приблизительно 4% (масс./масс.) цинка. В другом воплощении композиция, когда она высушена, содержит более приблизительно 91% (масс./масс.) конъюгата соединения инсулина, от приблизительно 0,05; 0,1; 0,15 или 0,2 до приблизительно 4% (масс./масс.) цинка и от приблизительно 0,2 до приблизительно 5% (масс./масс.) фенола. В еще одном воплощении композиция, когда она высушена, содержит более приблизительно 82% (масс./масс.) конъюгата соединения инсулина, от приблизительно 0,05; 0,1; 0,15 или 0,2 до приблизительно 4% (масс./масс.) цинка, от приблизительно 0,2 до приблизительно 14% (масс./масс.) протамина. В еще одном воплощении композиция, когда она высушена, содержит более приблизительно 71% (масс./масс.) конъюгата соединения инсулина, от приблизительно 0,05; 0,1; 0,15 или 0,2 до приблизительно 4% (масс./масс.) цинка, от приблизительно 0,2 до приблизительно 14% (масс./масс.) протамина и от приблизительно 0,2 до приблизительно 15% (масс./масс.) фенола.
В другом воплощении композиция, когда она высушена, содержит от приблизительно 0,1 до приблизительно 2% (масс./масс.) Zn++ и от приблизительно 0,08 до приблизительно 1% (масс./масс.) фенола, предпочтительно от приблизительно 0,5 до приблизительно 1,3% (масс./масс.) Zn++ и от приблизительно 0,1 до приблизительно 0,7% (масс./масс.) фенола, более предпочтительно от более или равно 1 до приблизительно 3,5% (масс./масс.) Zn++ и от приблизительно 0,1 до приблизительно 3% (масс./масс.) фенола, и еще более предпочтительно от более или равно 1,3 до приблизительно 2,2% (масс./масс.) Zn++ и от приблизительно 0,4 до приблизительно 2% (масс./масс.) фенола.
Данные комплексы могут быть предложены в препарате lente-типа. Например, в предпочтительном высушенном препарате lente-типа Zn присутствует в количестве, варьирующем от приблизительно 0,1 до приблизительно 2% (масс./масс.), и фенол присутствует в количестве, варьирующем от приблизительно 0,08 до приблизительно 1% (масс./масс.), остальные % (масс./масс.) приходятся на долю конъюгата соединения инсулина. В идеале, в случае высушенного препарата lente-типа, Zn присутствует в количестве, варьирующем от приблизительно 0,5 до приблизительно 1,3% (масс./масс.), и фенол присутствует в количестве, варьирующем от приблизительно 0,1 до приблизительно 0,7% (масс./масс.), остальные % (масс./масс.) приходятся на долю конъюгата соединения инсулина.
Комплексы могут быть предложены в препарате ultralente-типа. Например, в предпочтительном высушенном препарате ultralente-типа Zn присутствует в количестве, варьирующем от более или равно 1 до приблизительно 3,5% (масс./масс.), и фенол присутствует в количестве, варьирующем от приблизительно 0,1 до приблизительно 3% (масс./масс.), остальные % (масс./масс.) приходятся на долю конъюгата соединения инсулина. В идеале, в случае высушенного препарата ultralente-типа, Zn присутствует в количестве, варьирующем от более или равно 1,3 до приблизительно 2,2% (масс./масс.), и фенол присутствует в количестве, варьирующем от приблизительно 0,4 до приблизительно 2% (масс./масс.), остальные % (масс./масс.) приходятся на долю конъюгата соединения инсулина.
7.7.2. Жидкие композиции
Комплексы конъюгатов соединений инсулинов с катионами могут быть предложены в виде нерастворимых компонентов жидкости. Например, жидкость может представлять собой водный раствор, содержащий конъюгат соединения инсулина с катионом в виде осадка, или конъюгат соединения инсулина с катионом может быть предложен в виде компонента суспензии, эмульсии или микроэмульсии. Вместе с нерастворимыми компонентами жидкость может содержать также растворимые компоненты или комплексы.
7.7.3. Смеси и сокристаллы
Композиции по изобретению могут содержать, например, смеси комплексов, смеси твердых веществ, гибридные комплексы и сокристаллы.
Соответственно, в изобретении предложены композиции, которые содержат, например, два или более конъюгатов соединений инсулинов и/или неконъюгированных соединений инсулинов. Кроме того, когда композиции содержат твердые вещества, тогда данные твердые вещества могут иметь различные формы. Так, например, одно твердое вещество может быть кристаллическим, а другое твердое вещество может представлять собой аморфное твердое вещество. Как указано в другом месте, твердые вещества могут быть предложены в высушенной форме или могут быть предложены в виде твердых компонентов жидкой смеси. В предпочтительном воплощении смесь по изобретению содержит два или более разных конъюгатов соединений инсулинов, и эти разные конъюгаты соединений инсулинов имеют разные растворимости. В одном воплощении один из комплексов включает липофильный конъюгат соединения инсулина, а другой включает гидрофильный конъюгат соединения инсулина. В еще одном воплощении комплексы могут включать разные конъюгаты соединений инсулинов, где один или более комплексов имеют период полувыведения из кровотока от приблизительно 1 до приблизительно 4 часов, и один или более комплексов имеют период полувыведения из кровотока, который значительно больше периода полувыведения из кровотока первого комплекса. В родственном воплощении один из комплексов имеет профиль быстрого действия, а другой из комплексов имеет профиль от среднего до длительного действия. Кроме того, один из комплексов может иметь профиль, подходящий для базального контроля соединения инсулина, в то время как другой имеет профиль, подходящий для постпрандиального контроля глюкозы. Предпочтительными смесями являются смеси HIM2 и инсулина, смеси HIM2 и IN105, смеси IN105 и соединения инсулина, смеси IN105 и инсулина, ацилированного жирной кислотой, смеси HIM2 и инсулина, ацилированного жирной кислотой. Подходящие инсулины, ацилированные жирными кислотами, описаны в следующих патентах США, полные описания которых включены в данное описание посредством ссылки: в патенте США №6531448 под названием "Insoluble compositions for controlling blood glucose", опубликованном 11 марта 2003 г.; патенте США № RE 37971 под названием "Selective acylation of epsilon-amino groups", опубликованном 28 января 2003 г.; патенте США №6465426 под названием "Insoluble insulin compositions", опубликованном 15 октября 2002 г.; патенте США №6444641 под названием "Fatty acid-acylated insulin analogs", опубликованном 3 сентября 2002 г.; патенте США №6335316 под названием "Method for administering acylated insulin", опубликованном 1 января 2002 г.; патенте США №6268335 под названием "Insoluble insulin compositions", опубликованном 31 июля 2001 г.; патенте США №6051551 под названием "Method for administering acylated insulin", опубликованном 18 апреля 2000 г.; патенте США №5922675 под названием "Acylated Insulin Analogs", опубликованном 13 июля 1999; патенте США №5700904 под названием "Preparation of an acylated protein powder", опубликованном 23 декабря 1997; патенте США №5693609 под названием "Acylated insulin analogs Granted", опубликованном 2 декабря 1997; патенте США №5646242 под названием "Selective acylation of epsilon-amino groups", опубликованном 08 июля 1997; патенте США №5631347 под названием "Reducing gelation of a fatty acid-acylated protein", опубликованном 20 мая 1997; патенте США №6451974 под названием "Method of acylating peptides and novel acylating agents", опубликованном 17 сентября 2002 г.; патенте США №6011007 под названием "Acylated insulin", опубликованном 4 января 2000 г.; патенте США №5750497 под названием "Acylated insulin Granted: 12 мая 1998; патенте США №5905140 под названием "Selective acylation method", опубликованном 18 мая 1999; патенте США №6620780 под названием "Insulin derivatives", опубликованном 16 сентября 2003 г.; патенте США №6251856 под названием "Insulin derivatives", опубликованном 26 июня 2001 г.; патенте США №6211144 под названием "Stable concentrated insulin preparations for pulmonary delivery", опубликованном 3 апреля 2001 г.; патенте США №6310038 под названием "Pulmonary insulin crystals", опубликованном 30 октября 2001 г. и патенте США №6174856 под названием "Stabilized insulin compositions", опубликованном 16 января 2001 г.Особенно предпочтительными являются инсулины, моноацилированные жирными кислотами, содержащие 12-, 13-, 14-, 15- или 16-углеродные жирные кислоты, ковалентно связанные с Lys(B29) инсулина человека.
В одном воплощении в изобретении предложен сокристалл, имеющий два разных соединения инсулина и/или конъюгата соединения инсулина. Предпочтительно, данный сокристалл имеет одну или более следующих характеристик: по существу гомогенное растворение, одну кривую растворения in vivo и/или фармакодинамический профиль с одним пиком. Предпочтительными сокристаллами являются сокристаллы HIM2 и инсулина, сокристаллы HIM2 и IN105, сокристаллы IN105 и соединения инсулина.
В одном воплощении сокристалл включает инсулин человека, и сокристаллизация с инсулином человека уменьшает растворимость кристалла относительно растворимости соответствующего кристалла конъюгата соединения инсулина. В другом воплощении сокристалл включает инсулин человека, и сокристаллизация с инсулином человека уменьшает растворимость кристалла относительно растворимости соответствующего кристалла конъюгата соединения инсулина.
В другом воплощении сокристалл включает быстродействующий, быстро выводящийся и/или высокоэффективный конъюгат соединения инсулина и длительно действующий, медленно выводящийся и/или низкоэффективный конъюгат соединения инсулина. Предпочтительно, данный сокристалл имеет PK/PD (фармакокинетический/фармакодинамический) профиль, подходящий для постпрандиального контроля глюкозы или для ночного базального контроля соединения инсулина.
В другом воплощении в изобретении предложены смесь или сокристалл, которые содержат конъюгат соединения инсулина вместе с инсулином человека или лизпроинсулином. Смеси по изобретению могут включать два разных конъюгата соединения инсулина. Смеси могут включать конъюгат соединения инсулина и неконъгированное соединение инсулин. Смеси могут включать разные конъюгаты соединений инсулинов вместе с разными соединениями инсулинами.
Кроме того, в изобретении предложены комплексы, включающие два разных конъюгата соединения инсулина и/или конъюгат соединения инсулина и неконъюгированное соединение инсулин. В изобретении предложены гибридные сокристаллы из двух, трех или более разных конъюгатов соединений инсулинов. В изобретении предложен комплекс, включающий конъюгат соединения инсулина вместе с неконъюгированным соединением инсулином. В изобретении предложен сокристалл с двумя или более разными гидрофильными конъюгатами соединений инсулинов; с двумя или более разными гидрофобными конъюгатами соединений инсулинов; с двумя или более разными амфифильными конъюгатами соединений инсулинов; с гидрофильным конъюгатом соединения инсулина и липофильным конъюгатом соединения инсулина; с гидрофильным конъюгатом соединения инсулина и неконъюгированным соединением инсулином; HIM2 вместе с неконъюгированным соединением инсулином; IN105 вместе с неконъюгированным соединением инсулином; HIM2 вместе с IN105; HIM2 вместе с соединением инсулином и IN105; и другие комбинации вышеупомянутых элементов. Как указано в другом месте, данные комплексы могут быть предложены в виде сухих твердых веществ, в виде растворимых комплексов в растворе и/или в виде нерастворимых комплексов в растворе. Различные комбинации могут быть использованы, например, для получения комплекса или сокристалла, имеющего пролонгированный профиль.
7.8. Растворимость комплексов по изобретению
Предпочтительно, растворимость в воде комплекса конъюгата соединения инсулина с катионом при рН приблизительно 7,4 составляет от приблизительно 1/15, 1/14, 1/13, 1/12, 1/11, 1/10, 1/9, 1/8, 1/7, 1/6, 1/5 вплоть до приблизительно 0, 1, 2, 3, 4, 5, 6, 7, 8, 9 или менее 10 раз растворимости в воде растворимости в воде не находящегося в комплексе конъюгата соединения инсулина. Любые комбинации вышеупомянутых верхних или нижних пределов входят в объем изобретения. Однако предпочтительным диапазоном является диапазон от приблизительно 1/15 до менее 5, более предпочтительным - от приблизительно 1/10 до приблизительно 2, идеальным - от приблизительно 1/10 до менее 0. В особенно неожиданном аспекте изобретения растворимость в воде конъюгата соединения инсулина с катионом в растворе при рН приблизительно 7,4 зачастую по существу меньше растворимости в воде конъюгата соединения инсулина в растворе при рН приблизительно 7,4. Однако следует принимать во внимание, что в некоторых воплощениях растворимость в воде конъюгата соединения инсулина с катионом в растворе при рН приблизительно 7,4 может быть такой же, выше или существенно выше растворимости в воде конъюгата соединения инсулина в растворе при рН приблизительно 7,4.
В одном неожиданном воплощении растворимость в воде комплекса конъюгата соединения инсулина с катионом при рН приблизительно 7,4 по существу меньше растворимости соответствующего не находящегося в комплексе конъюгата соединения инсулина в растворе при рН приблизительно 7,4, и комплекс конъюгата соединения инсулина с катионом остается растворимым в концентрации более чем приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 30, 40, 50, 60, 70, 80, 90, 100, 110, 120 или 130 г/л в водном растворе в диапазоне рН, начинающемся при приблизительно 5,5; 5,6; 5,7; 5,8; 5,9; 6; 6,1; 6,2; 6,3; 6,4; 6,5; 6,6; 6,7; 6,1 или 6,9 и заканчивающемся при приблизительно 7,5; 7,6; 7,7; 7,8; 7,9; 8; 8,1; 8,2; 8,3; 8,4; 8,5; 8,6; 8,7; 8,8 или 8,9. В еще одном воплощении растворимость в воде комплекса конъюгата соединения инсулина с катионом при рН приблизительно 7,4 по существу меньше растворимости соответствующего конъюгата соединения инсулина в растворе при рН приблизительно 7,4, и комплекс конъюгата соединения инсулина с катионом остается растворимым в концентрации более чем приблизительно 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 30, 40, 50, 60, 70, 80, 90, 100, 110, 120 или 130 г/л в водном растворе в диапазоне рН от приблизительно 5,8 до приблизительно 8,5, предпочтительно в диапазоне рН от приблизительно 6,5 до приблизительно 8, более предпочтительно в диапазоне рН от приблизительно 6,9 до приблизительно 7,8. [ВОПРОС: Растворимость, выраженную в г/л, измеряли с использованием массы комплекса или массы конъюгата соединения инсулина?]
Предпочтительно, конъюгаты соединений инсулинов по изобретению выбраны таким образом, чтобы получать кристаллы в водном растворе при значении рН, равном pI +/- приблизительно 2,5, где концентрация конъюгата соединения инсулина составляет от приблизительно 0,5 мг/мл до приблизительно 50 мг/мл, предпочтительно от приблизительно 5 мг/мл до приблизительно 30 мг/мл, более предпочтительно от приблизительно 15 мг/мл до приблизительно 30 мг/мл, и образование кристаллов начинается при приблизительно 3, 4 или 5% (масс./масс.) катиона относительно конъюгата соединения инсулина, где катион предпочтительно представляет собой Z++. Предпочтительно, кристаллы представляют собой моноконъюгат без протамина в водном растворе при рН, варьирующем от приблизительно 4; 4,1; 4,2; 4,3 или 4,4 до приблизительно 5,2; 5,3; 5,4; 5,5; 5,6; 5,7 или 5,8; предпочтительно при рН от приблизительно 4 до менее 6,5, предпочтительно от приблизительно 4 до менее 5,8, предпочтительно от приблизительно 4,2 до приблизительно 5,5, более предпочтительно от приблизительно 4,4 до приблизительно 5,2. Предпочтительно, кристаллы присутствуют для диконъюгата без протамина при рН от приблизительно 3,5 до менее 5.8, предпочтительно от приблизительно 3,8 до приблизительно 5,5, более предпочтительно от приблизительно 4,0 до приблизительно 5,2. Предпочтительно, кристаллы присутствуют для триконъюгата без протамина при рН от приблизительно 3 до менее 5,5, предпочтительно от приблизительно 3,3 до приблизительно 5, более предпочтительно от приблизительно 3,8 до приблизительно 4,8.
7.8.1. Комплексы R-типа
Предпочтительно, растворимость в воде Zn-комплекса конъюгата соединения инсулина R-типа при рН приблизительно 7,4 составляет от приблизительно 10 до приблизительно 150 г/л, более предпочтительно от приблизительно 20 до приблизительно 130 г/л, более предпочтительно от приблизительно 30 до приблизительно 110 г/л, более предпочтительно от приблизительно 35 до приблизительно 60 г/л.
Предпочтительно, растворимость в воде Zn-комплекса конъюгата соединения инсулина R-типа с протамином при рН приблизительно 7,4 составляет от приблизительно 10 до приблизительно 110 г/л, более предпочтительно от приблизительно 20 до приблизительно 85 г/л, более предпочтительно от приблизительно 30 до приблизительно 70 г/л.
7.8.2. Комплексы Т-типа
Предпочтительно, растворимость в воде Zn-комплекса конъюгата соединения инсулина Т-типа при рН приблизительно 7,4 составляет от приблизительно 30 до приблизительно 175 г/л, более предпочтительно от приблизительно 50 до приблизительно 160 г/л, более предпочтительно от приблизительно 70 до приблизительно 150 г/л.
Предпочтительно, растворимость в воде Zn-комплекса конъюгата соединения инсулина Т-типа с протамином при рН приблизительно 7,4 составляет от приблизительно 10 до приблизительно 150 г/л, более предпочтительно от приблизительно 20 до приблизительно 130 г/л, более предпочтительно от приблизительно 30 до приблизительно 110 г/л, более предпочтительно от приблизительно 35 до приблизительно 60 г/л.
7.8.3. Комплексы NPH-типа
Предпочтительно, растворимость в воде комплекса NPH-типа конъюгата соединения инсулина при рН приблизительно 7,4 составляет от приблизительно 1 до приблизительно 150 г/л, более предпочтительно от приблизительно 5 до приблизительно 120 г/л, еще более предпочтительно от приблизительно 10 до приблизительно 90 г/л.
7.9. Фармацевтические свойства
Образование комплекса конъюгата соединения инсулина с катионом обычно приводит к улучшению фармацевтических свойств конъюгата соединения инсулина относительно приемлемого с научной точки зрения контроля, такого как соответствующий не находящийся в комплексе конъюгат соединения инсулина.
В некоторых случаях находящийся в комплексе конъюгат соединения инсулина будет проявлять пролонгированный или иным образом измененный профиль рК относительно приемлемого с научной точки зрения контроля, такого как соответствующий не находящийся в комплексе конъюгат соединения инсулина. В конкретных случаях профиль рК будет демонстрировать лизпро-подобный профиль. Профиль рК может быть определен с помощью стандартных экспериментов in vivo, например, на мышах, крысах, собаках и людях. Приведенные в данном описании анализы по определению характеристик комплексов конъюгатов соединений инсулинов с катионами представляют собой аспект данного изобретения.
Комплексы могут проявлять улучшенную химическую стабильность. Различные характеристики стабильности могут быть определены, если подвергнуть комплекс воздействию различных условий анализа, таких как присутствие плазмы, присутствие протеаз, присутствие гомогената печени, присутствие кислотных условий и присутствие щелочных условий. Стабильность улучшается относительно не находящегося в комплексе конъюгата соединения инсулина, когда стабильность находящегося в комплексе конъюгата соединения инсулина при любом одном или более, чем одном из данных условий анализа выше стабильности не находящегося в комплексе конъюгата соединения инсулина в тех же условиях. Предпочтительный анализ для определения стабильности в кислой среде включает воздействие на находящийся в комплексе конъюгат соединения инсулина раствором, имеющим рН 2, в течение по меньшей мере 2 часов, где уменьшенная деградация находящегося в комплексе конъюгата соединения инсулина относительно приемлемого с научной точки зрения контроля, такого как соответствующий не находящийся в комплексе конъюгат соединения инсулина, указывает на улучшение стабильности. Для тестирования стабильности могут быть использованы также анализы in vivo. Например, стабильность находящегося в комплексе конъюгата соединения инсулина может быть протестирована путем воздействия на него желудочно-кишечного тракта субъекта и сравнения с подходящим контролем.
7.10. Способ приготовления
В изобретении также предложен способ приготовления композиций конъюгатов соединений инсулинов с катионами, приведенных в данном описании. Этот способ обычно включает приведение в контакт одного или более конъюгатов соединений инсулинов, описанных в данном изобретении, с одним или более катионами, как описано в данном изобретении, с образованием твердого вещества.
В случае двухвалентного катиона, такого как Zn++, молярное отношение конъюгата соединения инсулина к катиону, используемое для приготовления композиции в водном растворе с концентрацией соединения инсулина, варьирующей от приблизительно 2 мг/мл до приблизительно 50 мг/мл, обычно может варьировать от приблизительно 1:15 (конъюгат соединение инсулин: катион) до приблизительно 1:0,4, предпочтительно от приблизительно 1:9 до приблизительно 1:2.
Для приготовления твердого вещества Т-типа (с катионом и конъюгатом соединения инсулина и без протамина) в условиях водного раствора, описанных выше, молярное отношение конъюгата соединения инсулина к катиону предпочтительно составляет от приблизительно 1:1,5 до 1:3, в идеале приблизительно 1:2. Для приготовления твердого вещества R-типа (с катионом, соединением инсулином и стабилизирующим соединением (например фенольным соединением) и без протамина) в условиях водного раствора, описанных выше, молярное отношение конъюгата соединения инсулина к катиону предпочтительно составляет от приблизительно 1:4 до 1:9, предпочтительно от приблизительно 1:7 до приблизительно 1:9, в идеале приблизительно 1:8.
Для приготовления твердого вещества Т-типа с протамином (с катионом и конъюгатом соединения инсулина и с протамином) в условиях водного раствора, описанных выше, молярное отношение конъюгата соединения инсулина к катиону предпочтительно составляет от приблизительно 1:1,5 до 1:9, в идеале приблизительно 1:2. Для приготовления твердого вещества R-типа с протамином (с катионом, соединением инсулином и стабилизирующим соединением (например фенольным соединением) и с протамином) в условиях водного раствора, описанных выше, молярное отношение конъюгата соединения инсулина к катиону предпочтительно составляет от приблизительно 1:2 до 1:15, предпочтительно от приблизительно 1:7 до приблизительно 1:9, в идеале приблизительно 1:8.
Конъюгат соединения инсулина предпочтительно добавляют к буферу в количестве, которое, как рассчитано, позволяет достичь концентрации в диапазоне от более 2 до приблизительно 100 г/л, предпочтительно от приблизительно 3, 4, 5, 6, 7, 8, 9 или 10 до приблизительно 40 г/л, более предпочтительно от приблизительно 10 до приблизительно 30 г/л.
Когда катион является двухвалентным (например Zn++, Са++), его предпочтительно добавляют в количестве, которое, как рассчитано, позволяет достичь концентрации в диапазоне от приблизительно 0,04 до приблизительно 10 г/л, предпочтительно от приблизительно 0,1 до приблизительно 5 г/л, более предпочтительно от приблизительно 0,2 до приблизительно 4 г/л. В случае кристаллов Т-типа или кристаллов Т-типа с протамином, концентрация катиона предпочтительно находится в диапазоне от приблизительно 0,04 до приблизительно 1 г/л, более предпочтительно от приблизительно 0,1 до приблизительно 0,3 г/л. В случае кристаллов R-типа или кристаллов R-типа с протамином, концентрация катиона предпочтительно находится в диапазоне от приблизительно 1 до приблизительно 5 г/л, более предпочтительно от приблизительно 1,5 до приблизительно 4 г/л.
Когда катион является одновалентным, его предпочтительно добавляют в количестве, которое, как рассчитано, позволяет достичь концентрации в диапазоне от приблизительно 0,08 до приблизительно 40 г/л, предпочтительно от приблизительно 0,4 до приблизительно 20 г/л, более предпочтительно от приблизительно 0,8 до приблизительно 16 г/л.
Способ может дополнительно включать объединение стабилизирующего агента с катионом и конъюгатом соединения инсулина. Предпочтительные стабилизирующие агенты описаны выше. При использовании стабилизирующий агент добавляют в количестве, достаточном для получения большей степени образования твердого вещества по сравнению с той, которая достигается при использовании тех же самых реагентов и условий реакции в отсутствие стабилизирующего агента. Там, где стабилизирующий агент представляет собой фенольное соединение (например фенол, мета-крезол, мета-парабен), его можно добавлять в количестве, варьирующем от приблизительно 10 до приблизительно 50% масс./масс., более предпочтительно от приблизительно 20 до приблизительно 40% масс./масс., еще более предпочтительно от приблизительно 25 до приблизительно 35% масс./масс. В более предпочтительном воплощении стабилизирующий агент представляет собой фенольное соединение (например фенол, мета-крезол, мета-парабен), его можно добавлять в количестве, варьирующем от приблизительно 0,01 до приблизительно 10% масс./масс., более предпочтительно от 0,01 до приблизительно 5% масс./масс., еще более предпочтительно от 0,01 до приблизительно 1% масс./масс. Соответственно, в одном воплощении способ включает объединение конъюгата соединения инсулина, катиона и стабилизирующего агента в водном растворе с получением композиции конъюгата соединения инсулина с катионом, где данная комбинация может давать растворимые комплексы и/или кристаллические или некристаллические твердые вещества.
Способ может дополнительно включать использование комплексообразующего агента, такого как протамин, который объединен с катионом и конъюгатом соединения инсулина, и возможно также включает стабилизирующий агент.
Для приготовления твердого вещества в водном растворе, имеющем рН в диапазоне от приблизительно 5 до приблизительно 8, протамин предпочтительно используют в количестве от приблизительно 4 до приблизительно 45% масс./масс., относительно конъюгата соединения инсулина (протамин/соединение инсулин), предпочтительно от приблизительно 8 до приблизительно 25% масс./масс., более предпочтительно от приблизительно 9 до приблизительно 20% масс./масс., в идеале от приблизительно 10 до приблизительно 12% масс./масс. Для твердых веществ Т-типа предпочтительный диапазон рН составляет от приблизительно 5 до приблизительно 6, более предпочтительно от приблизительно 5 до приблизительно 5,5, еще более предпочтительно от приблизительно 5,1 до приблизительно 5,3, в идеале приблизительно 5,2. Для твердых веществ R-типа предпочтительный диапазон рН составляет от приблизительно 6 до приблизительно 7, более предпочтительно от приблизительно 6,2 до приблизительно 6,8, еще более предпочтительно от приблизительно 6,4 до приблизительно 6,6, в идеале приблизительно 6,5.
Авторы изобретения неожиданно обнаружили, что комплексы Т-типа могут быть превращены в протаминовые комплексы Т-типа в отсутствие стабилизирующего агента, такого как фенол. Комплекс Т-типа получают в результате образования комплекса Zn с молекулой соединения инсулина в водном растворе в отсутствие фенола. Затем добавляют протамин для того, чтобы превратить комплекс Т-типа в протаминовый комплекс Т-типа. Количества и диапазоны являются такими, как описано выше.
Соответственно, в одном воплощении способ включает объединение конъюгата соединения инсулина, катиона и комплексообразующего агента в водном растворе с получением композиции конъюгата соединения инсулина с катионом, где данная комбинация может давать растворимые комплексы и/или кристаллические или аморфные твердые вещества. В другом воплощении способ включает объединение конъюгата соединения инсулина, катиона, комплексообразующего агента и стабилизирующего агента в водном растворе с получением композиции конъюгата соединения инсулина с катионом, где данная комбинация может давать растворимые комплексы и/или кристаллические или аморфные твердые вещества.
В некоторых воплощениях композиции могут содержать консерванты. Примеры подходящих консервантов включают в себя бензиловый спирт, сложные эфиры лара-гидроксибензойной кислоты, глицерин. Стабилизирующие агенты, такие как фенол, мета-крезол и мета-парабен, также могут быть использованы в качестве консервантов. Соответственно, глицерин и фенол добавляют вместе для повышения противомикробной эффективности.
Другие компоненты, полезные в приготовлении твердых веществ, включают в себя изотонические агенты, такие как NaCl, глицерин и моносахариды.
Твердые конъюгаты соединений инсулинов с катионами типично могут образовываться относительно быстро. Например, образование твердого вещества обычно завершается в пределах трех суток, часто в пределах 24 часов. В некоторых случаях может оказаться желательным замедлить данную реакцию для того, чтобы улучшить образование кристаллов.
В одном воплощении изобретения твердые вещества образуются при комнатной температуре (25°С) без необходимости снижения температуры для индукции осаждения твердых веществ. Например, комнатная температура является эффективной для кристаллов R-типа и Т-типа. Температура для образования твердых веществ находится предпочтительно в диапазоне от приблизительно 0 до приблизительно 40°С, предпочтительно от приблизительно 17 до приблизительно 30°С, и более предпочтительно от приблизительно 22 до приблизительно 27°С, в идеале приблизительно 25°С.
В одном воплощении способ включает объединение в водном растворе конъюгата соединения инсулина и катиона металла с получением кристаллического или аморфного твердого вещества. Водный раствор, содержащий конъюгат соединения инсулина, к которому будет добавлен катион, представляет собой предпочтительно забуференный раствор, имеющий рН в диапазоне pI конъюгата соединения инсулина +/- приблизительно 1,5, предпочтительно pI +/- приблизительно 1, более предпочтительно pI +/-приблизительно 0,75. Данные диапазоны также применимы к комплексам Т-типа, R-типа и протаминовым комплексам. Однако в случае нейтральных протаминовых комплексов (NPH-типа), предпочтительный рН составляет от приблизительно 7 до приблизительно 8,5, более предпочтительно от приблизительно 7,5 до приблизительно 8. При добавлении катиона металла, рН может слегка измениться, и рН можно довести до заданных диапазонов рН, описанных выше. При использовании фенольных соединений, может иметь место незначительное изменение рН, и для доведения рН до предпочтительных диапазонов можно использовать кислоту или основание.
Значения pi для конъюгатов соединений инсулинов обычно требуют рН менее приблизительно 7, предпочтительно менее приблизительно 6, более предпочтительно менее приблизительно 5,5. Моноконъюгаты инсулина человека с нейтральными модифицирующими группировками обычно имеют pI в диапазоне приблизительно 4,75+/-0,25. Для диконъюгатов инсулина человека диапазон pi обычно составляет 4,25+/-0,25. Для триконъюгатов инсулина человека диапазон pi обычно составляет 3,5+/-0,25.
Примеры подходящих буферных систем включают в себя аммоний-ацетатный буфер, натрий-фосфатный буфер, трис-буфер, смесь фосфата натрия и ацетата аммония, натрий-ацетатный буфер, смесь ацетата натрия и ацетата аммония и цитратный буфер, и любую из вышеупомянутых буферных систем [А], также содержащую этанол и/или ацетонитрил [Б] (например в процентном соотношении А:Б от приблизительно 1:1 до приблизительно 10:1). Неожиданным аспектом изобретения является то, что твердый конъюгат соединения инсулина с катионом может быть образован в водной смеси, содержащей органический растворитель, такой как этанол или ацетонитрил.
Одним из уникальных признаков изобретения является то, что дополнительно к предложенным полезным комплексам конъюгатов соединений инсулинов с катионами в изобретении также предложен способ отделения конъюгатов соединений инсулинов с катионами от неконъюгированного соединения инсулина в процессе производства. В этом способе конъюгаты соединений инсулинов с катионами можно осаждать из раствора, и растворенное неконъюгированное соединение инсулин можно удалять, например, фильтрацией. Данный признак исключает 2 стадии в получении конъюгатов соединений инсулинов: стадию концентрирования и стадию лиофилизации.
Обработанная чистая твердая композиция может быть получена с помощью стандартных методов, таких как центрифугирование и/или фильтрация с последующими промывкой (например смесью этанол/вода) и лиофилизацией или вакуумной сушкой. Многократные промывки могут быть использованы для регуляции содержания фенола и/или катионов.
7.11. Препарат
Комплексы могут быть приготовлены в виде препарата для введения в фармацевтический носитель в соответствии с известными методами. Смотри, например, Alfonso R. Gennaro, Remington: The Science and Practice of Pharmacy, Lippincott Williams & Wilkins Publishers (июнь 2003 г.) и Howard С.Ansel, Pharmaceutical Dosage Forms and Drug Delivery Systems, Lippincott Williams & Wilkins Publishers, 7th ed. (октябрь 1999), полные описания которых в части, относящейся к выбору, приготовлению и использованию фармацевтических лекарственных форм, включены в данное описание посредством ссылки
Комплексы, обычно в форме аморфного или кристаллического твердого вещества, могут быть объединены с фармацевтически приемлемым носителем. Носитель должен быть приемлемым в смысле его совместимости с любыми другими ингредиентами в фармацевтической композиции и не должен быть чрезмерно вредным для субъекта, что касается пользы, полученной от активного(ых) ингредиента(ов). Носитель может представлять собой твердое вещество или жидкость, или и то и другое. Его приготавливают предпочтительно в виде препарата в стандартной лекарственной форме, например, в виде таблетки. Данная стандартная лекарственная форма может содержать, например, от приблизительно 0,01 или 0,5% до приблизительно 95% или 99% по массе комплекса соединения инсулина с катионом. Фармацевтические композиции могут быть приготовлены с помощью любых хорошо известных методов фармации, включая, но не ограничиваясь этим, смешивание компонентов, возможно включение одного или более дополнительных ингредиентов.
Примеры подходящих фармацевтических композиций включают в себя композиции, приготовленные для перорального, ректального, ингаляционного (например, с помощью аэрозоля), трансбуккального (например сублингвального), вагинального, парентерального (например подкожного, внутримышечного, интрадермального, внутрисуставного, внутриплеврального, внутрибрюшинного, интрацеребрального, внутриартериального или внутривенного), местного введения, для введения через поверхности слизистых оболочек (включая поверхности дыхательных путей), носовые поверхности и для трансдермального введения. Наиболее подходящий путь в любом случае будет зависеть от природы и тяжести состояния, которое лечат, и от природы конкретных комплексов соединений инсулинов с катионами, которые используют. Предпочтительные пероральные композиции представляют собой композиции, приготовленные для проглатывания внутрь субъектом. В идеале пероральные композиции приготавливают таким образом, чтобы они выдерживали или по существу выдерживали прохождение через желудок и полностью или по существу полностью растворялись в кишечнике для доставки активного ингредиента. Примеры подходящих трансдермальных систем включают в себя ультразвуковые, ионтофоретические системы доставки и системы доставки с использованием пластыря.
В одном аспекте в изобретения предложены композиции жирных кислот, содержащие одну или более насыщенных или ненасыщенных С4, С5, С6, С7, С8, С9 или С10 и/или солей таких жирных кислот. Предпочтительными жирными кислотами являются каприловая, каприновая, миристиновая и лауриновая. Предпочтительными солями жирных кислот являются натриевые соли каприловой, каприновой, миристиновой и лауриновой кислоты.
Предпочтительные композиции жирных кислот содержат одну жирную кислоту или одну соль жирной кислоты и не содержат значительные количества других жирных кислот или солей жирных кислот. В одном аспекте изобретения жирнокислотное содержание композиции более чем на приблизительно 90; 91; 92; 93; 94; 95; 96; 97; 98; 99; 99,5; 99,6; 99,7; 99,8 или 99,9% (масс./масс.) представлено одной жирной кислотой. В другом воплощении жирнокислотное содержание композиции находится в диапазоне, имеющем в качестве нижнего предела приблизительно 0,1; 0,2; 0,3; 0,4; 0,5; 0,6; 0,7; 0,8; 0,9; 1,0; 1,1; 1,2; 1,3; 1,4; 1,5; 1,6; 1,7; 1,8; 1,9; 2,0; 2,1; 2,2; 2,3; 2,4; 2,5; 2,6; 2,7; 2,8; 2,9 или 3,0% масс./масс., и имеющем в качестве верхнего предела приблизительно 3,0; 3,1; 3,2; 3,3; 3,4; 3,5; 3,6; 3,7; 3,8; 3,9; 4,0; 4,1; 4,2; 4,3; 4,4; 4,5; 4,6; 4,7; 4,8; 4,9; 5,0; 5,1; 5,2; 5,3; 5,4; 5,5; 5,6; 5,7; 5,8; 5,9; 6,0; 6,1; 6,2; 6,3; 6,4; 6,5; 6,6; 6,7; 6,8; 6,9; 7,0; 7,1; 7,2; 7,3; 7,4; 7,5; 7,6; 7,7; 7,8; 7,9; 8,0; 8,1; 8,2; 8,3; 8,4; 8,5; 8,6; 8,7; 8,8; 8,9; 9,0; 9,1; 9,2; 9,3; 9,4; 9,5; 9,6; 9,7; 9,8; 9,9; 10,0; 10,1; 10,2; 10,3; 10,4; 10,5; 10,6; 10,7; 10,8; 10,9; 11,0; 11,1; 11,2; 11,3; 11,4; 11,5; 11,6; 11,7; 11,8; 11,9 или 12,0% масс./масс. В еще одном воплощении жирнокислотное содержание композиции находится в диапазоне, имеющем в качестве нижнего предела приблизительно 0,1; 0,2; 0,3; 0,4; 0,5; 0,6; 0,7; 0,8; 0,9; 1,0; 1,1; 1,2; 1,3; 1,4; 1,5; 1,6; 1,7; 1,8; 1,9; 2,0; 2,1; 2,2; 2,3; 2,4; 2,5; 2,6; 2,7; 2,8; 2,9 или 3,0% масс./масс., и имеющем в качестве верхнего предела приблизительно 3,0; 3,1; 3,2; 3,3; 3,4; 3,5; 3,6; 3,7; 3,8; 3,9; 4,0; 4,1; 4,2; 4,3; 4,4; 4,5; 4,6; 4,7; 4,8; 4,9; 5,0; 5,1; 5,2; 5,3; 5,4; 5,5; 5,6; 5,7; 5,8; 5,9; 6,0; 6,1; 6,2; 6,3; 6,4; 6,5; 6,6; 6,7; 6,8; 6,9; 7,0; 7,1; 7,2; 7,3; 7,4; 7,5; 7,6; 7,7; 7,8; 7,9; 8,0; 8,1; 8,2; 8,3; 8,4; 8,5; 8,6; 8,7; 8,8; 8,9; 9,0; 9,1; 9,2; 9,3; 9,4; 9,5; 9,6; 9,7; 9,8; 9,9; 10,0; 10,1; 10,2; 10,3; 10,4; 10,5; 10,6; 10,7; 10,8; 10,9; 11,0; 11,1; 11,2; 11,3; 11,4; 11,5; 11,6; 11,7; 11,8; 11,9 или 12,0% масс./масс., и более приблизительно 90; 91; 92; 93; 94; 95; 96; 97; 98; 99; 99,5; 99,6; 99,7; 99,8 или 99,9% (масс./масс.) жирнокислотного содержания композиции представлено одной жирной кислотой.
Активные компоненты данных препаратов могут включать в себя конъюгированные или неконъюгированные, находящиеся в комплексе или не находящиеся в комплексе белки и/или пептиды. Предпочтительные белки и/или пептиды представляют собой белки и/или пептиды, описанные в данном изобретении. Предпочтительные конъюгаты представляют собой конъюгаты, описанные в данном изобретении. Предпочтительные комплексы представляют собой комплексы, описанные в данном изобретении. Предпочтительные пероральные композиции представляют собой композиции, приготовленные для проглатывания внутрь субъектом. В идеале, данные пероральные композиции приготавливают таким образом, чтобы они выдерживали или по существу выдерживали прохождение через желудок и полностью или по существу полностью растворялись в кишечнике для доставки активного ингредиента. В некоторых случаях препарат может включать энтеросолюбильное покрытие, и в некоторых случаях препарат будет конкретно исключать энтеросолюбильное покрытие. Данная композиция предложена предпочтительно в виде таблетки, порошка, твердой желатиновой капсулы или мягкой желатиновой капсулы, хотя другие формы, описанные в данном изобретении, также являются подходящими.
Композиции жирных кислот по изобретению могут содержать инсулины, ацилированные жирными кислотами. Примеры подходящих инсулинов, ацилированных жирными кислотами, описаны в следующих патентах США, полные описания которых включены в данное описание посредством ссылки: в патенте США №6531448 под названием "Insoluble compositions for controlling blood glucose", опубликованном 11 марта 2003 г.; патенте США № RE 37971 под названием "Selective acylation of epsilon-amino groups", опубликованном 28 января 2003 г.; патенте США №6465426 под названием "Insoluble insulin compositions", опубликованном 15 октября 2002; патенте США №6444641 под названием "Fatty acid-acylated insulin analogs", опубликованном 3 сентября 2002; патенте США №6335316 под названием "Method for administering acylated insulin", опубликованном 1 января 2002; патенте США №6268335 под названием "Insoluble insulin compositions", опубликованном 31 июля 2001 г.; патенте США №6051551 под названием "Method for administering acylated insulin", опубликованном 18 апреля 2000 г.; патенте США №5922675 под названием "Acylated Insulin Analogs", опубликованном 13 июля 1999; патенте США №5700904 под названием "Preparation of an acylated protein powder", опубликованном 23 декабря 1997; патенте США №5693609 под названием "Acylated insulin analogs Granted", опубликованном 2 декабря 1997; патенте США №5646242 под названием "Selective acylation of epsilon-amino groups", опубликованном 08 июля 1997; патенте США №5631347 под названием "Reducing gelation of a fatty acid-acylated protein", опубликованном 20 мая 1997; патенте США №6451974 под названием "Method of acylating peptides and novel acylating agents", опубликованном 17 сентября 2002; патенте США №6011007 под названием "Acylated insulin", опубликованном 4 января 2000 г.; патенте США №5750497 под названием "Acylated insulin Granted: 12 мая 1998; патенте США №5905140 под названием "Selective acylation method", опубликованном 18 мая 1999; патенте США №6620780 под названием "Insulin derivatives", опубликованном 16 сентября 2003 г.; патенте США №6251856 под названием "Insulin derivatives", опубликованном 26 июня 2001 г.; патенте США №6211144 под названием "Stable concentrated insulin preparations for pulmonary delivery", опубликованном 3 апреля 2001 г.; патенте США №6310038 под названием "Pulmonary insulin crystals", опубликованном 30 октября 2001 г.; и патенте США №6174856 под названием "Stabilized insulin compositions", опубликованном 16 января 2001 г. Особенно предпочтительными являются инсулины, моноацилированные жирными кислотами, содержащие 12, 13, 14, 15 или 16-углеродные жирные кислоты, ковалентно связанные с Lys(B29) инсулина человека.
Фармацевтические композиции, подходящие для перорального введения, могут быть представлены в виде дискретных единиц, таких как капсулы, облатки, лепешки или таблетки, каждая из которых содержит заданное количество смеси конъюгатов соединений инсулинов; в виде порошка или гранул; в виде раствора или суспензии в водной или неводной жидкости или в виде эмульсии типа масло в воде или вода в масле. Такие препараты могут быть приготовлены с помощью любого подходящего метода фармации, который включает стадию объединения смеси конъюгатов соединений инсулинов и подходящего носителя (который может содержать один или более дополнительных ингредиентов, как отмечено выше). Препараты могут включать суспензии твердых веществ, находящиеся в комплексе конъюгаты соединений инсулинов с катионами, не находящийся в комплексе активный ингредиент (например соединение нативного инсулина, конъюгаты соединений инсулинов) и смеси вышеупомянутого.
Обычно фармацевтические композиции по изобретению приготавливают путем равномерного и тщательного перемешивания комплексов с жидким или твердым носителем, или и с тем и с другим, и последующего, если необходимо, формования полученной смеси. Например, таблетка может быть приготовлена путем прессования или формования порошка или гранул, содержащих смесь конъюгатов соединений инсулинов, возможно с одним или более дополнительными ингредиентами. Прессованные таблетки могут быть приготовлены путем прессования смеси, находящейся в сыпучей форме, такой как порошок или гранулы, возможно смешанной со связующим веществом, смазывающим веществом, инертным разбавителем и/или поверхностно-активным(и)/диспергирующим(и) агентом(ами), в подходящем устройстве. Формованные таблетки могут быть изготовлены путем формования порошкообразной композиции, увлажненной инертным жидким связующим веществом, в подходящем устройстве.
Фармацевтические композиции, подходящие для трансбуккального (сублингвального) введения, включают в себя лепешки, содержащие смесь конъюгатов соединений инсулинов в ароматной основе, обычно сахарозе и гуммиарабике или трагаканте; и пастилки, содержащие смесь конъюгатов соединений инсулинов в инертной основе, такой как желатин и глицерин или сахараза и гуммиарабик. Примеры подходящих препаратов можно найти в патентных публикациях США №20030229022 ("Pharmaceutical formulation"); №20030236192 ("Method of modifying the release profile of sustained release compositions"); №20030096011 ("Method of producing submicron particles of a labile agent and use thereof); №20020037309 ("Process for the preparation of polymer-based sustained release compositions"); №20030118660 ("Residual solvent extraction method and microparticles produced thereby"); а также в патентах США №№6180141 ("Composite gel microparticles as active principle carriers"); 6737045 ("Methods and compositions for the pulmonary delivery insulin compound"); 6730334 ("Multi-arm block copolymers as drug delivery vehicles"); 6685967 ("Methods and compositions for pulmonary delivery of insulin compound"); 6630169 ("Particulate delivery systems and methods of use"); 6589560 ("Stable glassy state powder formulations; 6592904 ("Dispersible macromolecule compositions and methods for their preparation and use"); 6582728 ("Spray drying of macromolecules to produce inhaleable dry powders"); 6565885 ("Methods of spray drying pharmaceutical compositions"); 6546929 ("Dry powder dispersing apparatus and methods for their use"); 6543448 ("Apparatus and methods for dispersing dry powder medicaments"); 6518239 ("Dry powder compositions having improved dispersivity"); 6514496 ("Dispersible antibody compositions and methods for their preparation and use"); 6509006 ("Devices compositions and methods for the pulmonary delivery of aerosolized medicaments"); 6433040 ("Stabilized bioactive preparations and methods of use"); 6423344 ("Dispersible macromolecule compositions and methods for their preparation and use"); 6372258 ("Methods of spray-drying a drug and a hydrophobic amino acid"); 6309671 ("Stable glassy state powder formulations"); 6309623 ("Stabilized preparations for use in metered dose inhalers"); 6294204 ("Method of producing morphologically uniform microcapsules and microcapsules produced by this method"); 6267155 ("Powder filling systems, apparatus and methods"); 6258341 ("Stable glassy state powder formulations"); 6182712 ("Power filling apparatus and methods for their use"); 6165463 ("Dispersible antibody compositions and methods for their preparation and use"); 6138668 ("Method and device for delivering aerosolized medicaments"); 6103270 ("Methods and system for processing dispersible fine powders"); 6089228 ("Apparatus and methods for dispersing dry powder medicaments"); 6080721 ("Pulmonary delivery of active fragments of parathyroid hormone"); 6051256 ("Dispersible macromolecule compositions and methods for their preparation and use"); 6019968 ("Dispersible antibody compositions and methods for their preparation and use"); 5997848 ("Methods and compositions for pulmonary delivery of insulin compound"); 5993783 ("Method and apparatus for pulmonary administration of dry powder, alpha. 1-antitrypsin"); 5922354 ("Methods and system for processing dispersible fine powders"); 5826633 ("Powder filling systems, apparatus and methods"); 5814607 ("Pulmonary delivery of active fragments of parathyroid hormone"); 5785049 ("Method and apparatus for dispersion of dry powder medicaments"); 5780014 ("Method and apparatus for pulmonary administration of dry powder alpha 1-antitrypsin"); 5775320 ("Method and device for delivering aerosolized medicaments"); 5740794 ("Apparatus and methods for dispersing dry powder medicaments"); 5654007 ("Methods and system for processing dispersible fine powders"); 5607915 ("Pulmonary delivery of active fragments of parathyroid hormone"); 5458135 ("Method and device for delivering aerosolized medicaments"); 6602952 ("Hydrogels derived from chitosan and poly(ethylene glycol) or related polymers") и 5932462 ("Multiarmed, monofunctional, polymer for coupling to molecules and surfaces"). Кроме того, подходящие препараты замедленного высвобождения описаны в патенте США №5968554 (Cardinal Health) под названием "A sustained release pharmaceutical preparation", опубликованном 19 октября 1999, полное описание которого включено в данное описание посредством ссылки. Подходящие препараты микрочастиц описаны в международной патентной публикации WO/2003-049701 (Spherics, Inc.) под названием "Methods and products useful in the formation and isolation of microparticles", опубликованной 30 октября 2003 г. Подходящие биоадгезивные препараты описаны в международной патентной публикации WO/2003-051304 (Spherics, Inc.) под названием "Bioadhesive drug delivery system with enhanced gastric retention", опубликованной 6 мая 2004 г.
Фармацевтические композиции согласно воплощениям изобретения, подходящие для парентерального введения, содержат стерильные водные и неводные инъекционные растворы комплексов, препараты которых являются предпочтительно изотоническими относительно крови предполагаемого реципиента. Эти препараты могут содержать антиоксиданты, буферы, бактериостатики и растворенные вещества, которые делают композицию изотонической относительно крови предполагаемого реципиента. Водные и неводные стерильные суспензии могут содержать суспендирующие агенты и загустители. Композиции могут быть представлены в однодозовых или многодозовых контейнерах, например в герметичных ампулах или флаконах, и могут храниться в сублимированном (лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя, например физиологического раствора или воды для инъекций непосредственно перед применением. Инъекционные растворы и суспензии для немедленного приема могут быть приготовлены из стерильных порошков, гранул и таблеток, виды которых описаны ранее. Например, может быть предложена инъекцируемая стабильная стерильная композиция со смесью комплексов в стандартной лекарственной форме в герметичном контейнере. Смесь комплексов может быть предложена в форме лиофилизата, который может быть растворен в подходящем фармацевтически приемлемом носителе с образованием жидкой композиции, подходящей для инъекции субъекту. Парентеральная стандартная лекарственная форма обычно содержит от приблизительно 1 микрограмма до приблизительно 10 мг смеси комплексов. Когда комплексы по существу не растворимы в воде, эмульгирующий агент, который является физиологически приемлемым, может быть использован в количестве, достаточном для эмульгирования данных комплексов в водном носителе. Одним из таких полезных эмульгирующих агентов является фосфатидилхолин.
Твердая лекарственная форма для перорального введения обычно содержит от приблизительно 2 мг до приблизительно 500 мг, предпочтительно от приблизительно 10 мг до приблизительно 250 мг, в идеале от приблизительно 20 мг до приблизительно 110 мг комплексов.
Фармацевтические композиции, подходящие для ректального введения, предпочтительно представлены в виде суппозиториев, содержащих стандартную дозу. Данные фармацевтические композиции могут быть приготовлены путем смешивания комплексов с одним или более традиционными твердыми носителями, например с маслом какао, и последующего формования полученной смеси.
Фармацевтические композиции, подходящие для местного нанесения на кожу, предпочтительно имеют форму мази, крема, лосьона, пасты, геля, спрея, аэрозоля или масла. Носители, которые могут быть использованы, включают в себя вазелин, ланолин, PEG, спирты, трансдермальные усилители и комбинации двух или более таких носителей.
Фармацевтические композиции, подходящие для трансдермального введения, могут быть представлены в виде отдельных пластырей, приспособленных для того, чтобы оставаться в тесном контакте с эпидермисом реципиета в течение длительного периода времени. Композиции, подходящие для трансдермального введения, могут быть доставлены также путем ионтофореза (смотри, например, Pharmaceutical Research 3 (6):318 (1986)) и обычно имеют форму возможно забуференного водного раствора смеси конъюгатов соединений инсулинов. Подходящие препараты содержат цитратный или бис/трис-буфер (рН 6) или смесь этанол/вода и содержат от 0,1 до 0,2 М активного ингредиента.
В предпочтительном воплощении комплексы вводят в виде компонентов препаратов твердых жирных кислот, таких, как описано в заявке на патент США №60/494821, поданной 13 августа 2003 г., Opawale с соавт., полное описание которой включено в данное описании посредством ссылки.
В некоторых воплощениях конъюгат соединения инсулина может быть предложен отдельно от катионных и/или других компонентов, необходимых для образования твердых веществ. Например, конъюгат соединения инсулина может быть предложен в виде сухого твердого вещества, а буферный раствор, содержащий катион, стабилизирующий агент, консервант и/или другой компонент, может быть предложен отдельно, так что потребитель может комбинировать данные отдельные компоненты для получения комплексов конъюгатов соединений инсулинов с катионами.
7.12. Способы лечения
Композиции конъюгатов соединений инсулинов с катионами и их препараты являются полезными в лечении состояний, при которых увеличение количества циркулирующего соединения инсулина (относительно количества, имеющегося у субъекта в отсутствие введения соединения инсулина из экзогенного источника) дает желаемый терапевтический или физиологический эффект. Например, состояние, которое лечат, может представлять собой диабет типа I или типа II, преддиабет и/или метаболический синдром. В одном воплощении данные композиции вводят для облегчения симптомов диабета. В другом воплощении композиции вводят субъекту, находящемуся в преддиабетическом состоянии, для предупреждения или замедления развития диабета.
Эффективное количество композиции конъюгата соединения инсулина с катионом для введения согласно способам по изобретению будет несколько различаться от смеси к смеси и от субъекта к субъекту и будет зависеть от таких факторов, как возраст и состояние субъекта, путь доставки и состояние, которое лечат. Такие дозировки могут быть определены в соответствии с общепринятыми фармакологическими методами, известными специалистам в данной области.
В качестве общего предложения, терапевтическую эффективность будет иметь пероральная лекарственная форма, содержащая от приблизительно 0,025 до приблизительно 10 мг/кг активного ингредиента (т.е. конъюгата), причем все массы рассчитаны на основе массы смеси конъюгатов соединений инсулинов. Более предпочтительный диапазон составляет от приблизительно 0,06 до приблизительно 1 мг/кг, и еще более предпочтительный диапазон составляет от приблизительно 0,125 до приблизительно 0,5 мг/кг.
Парентеральная дозировка обычно варьирует от приблизительно 0,5 мкг/кг до приблизительно 0,5 мг/кг, причем все массы рассчитаны на основе массы смеси конъюгатов соединений инсулинов. Более предпочтительный диапазон составляет от приблизительно 1 мкг/кг до приблизительно 100 мкг/кг.
Частота введения обычно составляет один, два или три раза в сутки или является такой, как требуется для контроля состояния. Альтернативно, композиции конъюгатов соединений инсулинов с катионами можно вводить путем длительной инфузии. Продолжительность лечения зависит от типа соединения инсулина, недостаточности, которую лечат, и лечение может продолжаться в течение всей жизни субъекта. Комплексы можно вводить, например, за 0-30 минут до еды. Комплексы можно вводить, например, за 0-2 часа до сна.
8. Примеры синтеза
Следующие примеры представлены для иллюстрации и объяснения изобретения.
8.1. Синтез защищенного MPEG6C3-олигомера (трет-бутилового эфира 3-{2-[2-(2-{2-[2-(2-метоксиэтокси)этокси]этокси}этокси)этокси]этокси}-пропионовой кислоты)
Метилгексаэтиленгликоль (1,0 г, 3,37 ммоль) и трет-бутилакрилат (0,216 г, 1,69 ммоль) растворяли в сухом THF (тетрагидрофуране) (10 мл). К раствору добавляли металлический натрий (0,4 мг, 0,016 ммоль). После перемешивания в течение 4 ч при комнатной температуре реакционную смесь гасили путем добавления 1 М HCl (15 мл). Затем погашенную реакционную смесь экстрагировали CH2Cl2 (1×50 мл, 1×25 мл). Органический слой сушили (MgSO4) и концентрировали. После очистки посредством хроматографии на силикагеле (этилацетат в качестве элюента) получали продукт в виде масла (0,832 г, 58%).
8.2. Синтез MPEG6C3-олигомерной кислоты (3-{2-[2-(2-{2-[2-(2-метоксиэтокси)этокси]этокси}этокси)этокси]этокси}пропионовой кислоты)
С трет-бутилового эфира (0,165 г, 0,0389 ммоль) удаляли защитную группу путем перемешивания при комнатной температуре в трифторуксусной кислоте (2,0 мл). Затем содержимое концентрировали до постоянной массы (0,125 г, 87%).
8.3. Синтез активированного MPEG6C3-олигомера (2,5-диоксопирролидин-1-илового эфира 3-{2-[2-(2-{2-[2-(2-метоксиэтокси)этокси]этокси}этокси)этокси]этокси}пропионовой кислоты)
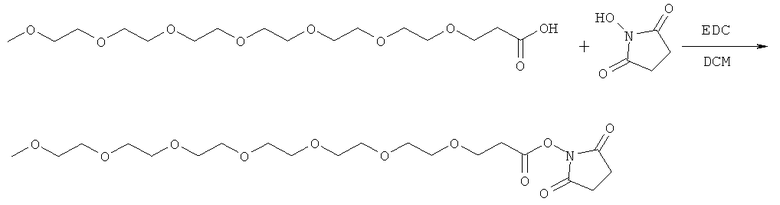
Кислоту (0,660 г, 1,79 ммоль) и N-гидроксисукцинимид (0,2278 г, 1,97 ммоль) растворяли в сухом CH2Cl2 (15 мл). Добавляли гидрохлорид этилдиметиламинопропилкарбодиимида (EDC, 0,343 г, 1,79 ммоль). После перемешивания при комнатной температуре в течение ночи реакционную смесь разбавляли CH2Cl2 и промывали водой (2×45 мл). Органический слой сушили (MgSO4) и концентрировали до постоянной массы. Продукт представлял собой масло (0,441 г, 53%).
8.4. Синтез защищенного MPEG4C3-олигомера (трет-бутилового эфира 3-(2-{2-[2-(2-метоксиэтокси)этокси]этокси}этокси)пропионовой кислоты)
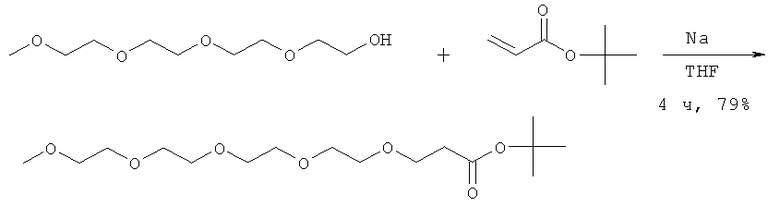
Метилтетраэтиленгликоль (1,0 г, 4,80 ммоль) и трет-бутилакрилат (0,308 г, 2,40 ммоль) растворяли в сухом THF (10 мл). К данному раствору добавляли металлический натрий (0,6 мг, 0,024 ммоль). После перемешивания в течение 4 ч при комнатной температуре реакционную смесь гасили путем добавления 1 М HCl (15 мл). Затем погашенную реакционную смесь экстрагировали CH2Cl2 (1×50 мл, 1×25 мл). Органический слой сушили (MgSO4) и концентрировали. После очистки посредством хроматографии на силикагеле (этилацетат в качестве элюента) получали продукт в виде масла (1,28 г, 79%).
8.5. Синтез MPEG6C3-олигомерной кислоты (3-(2-{2-[2-(2-метоксиэтокси)этокси]этокси}этокси)пропионовой кислоты)
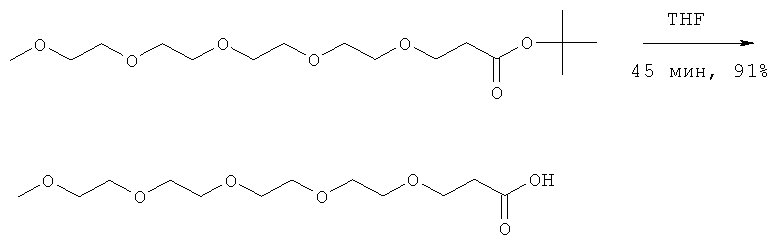
С трет-бутилового эфира (1 г, 3,42 ммоль) удаляли защитную группу путем перемешивания при комнатной температуре в трифторуксусной кислоте (6,0 мл). Затем содержимое концентрировали до постоянной массы (0,87 г, 91%).
8.6. Синтез активированного MPEG4C3-олигомера (2,5-диоксопирролидин-1-илового эфира 3-(2-{2-[2-(2-метоксиэтокси)этокси]этокси}этокси)пропионовой кислоты)
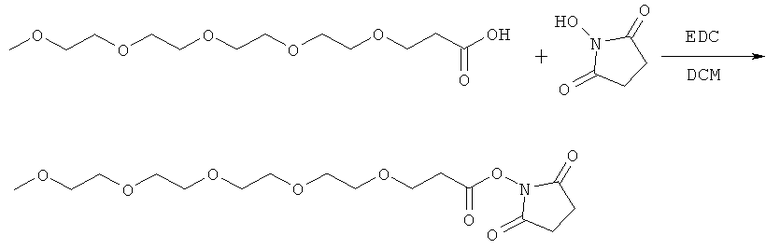
Кислоту (0,6 г, 2,14 ммоль) и N-гидроксисукцинимид (0,271 г, 2,35 ммоль) растворяли в сухом CH2Cl2 (20 мл). Добавляли гидрохлорид этилдиметиламинопропилкарбодиимида (EDC, 0,409 г, 2,14 ммоль). После перемешивания при комнатной температуре в течение ночи реакционную смесь разбавляли CH2Cl2 и промывали водой (2×45 мл). Органический слой сушили (MgSO4) и концентрировали до постоянной массы. Продукт представлял собой масло (0,563 г, 69%).
8.7. Синтез защищенного MPEG4C3-олигомера (трет-бутилового эфира 3-(2-метоксиэтокси)пропионовой кислоты)
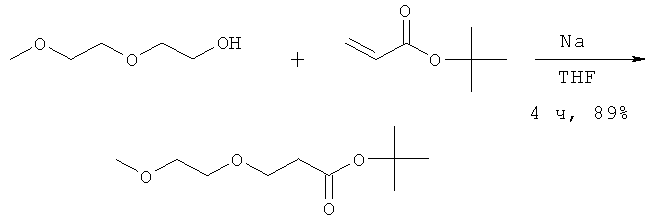
Метилтетраэтиленгликоль (5,0 г, 41,6 ммоль) и трет-бутилакрилат (2,66 г, 20,8 ммоль) растворяли в сухом THF (20 мл). К данному раствору добавляли металлический натрий (0,47 мг, 20,8 ммоль). После перемешивания в течение 4 ч при комнатной температуре реакционную смесь гасили путем добавления 1 М HCl (30 мл). Затем погашенную реакционную смесь экстрагировали CH2Cl2 (1×100 мл, 1×50 мл). Органический слой сушили (MgSO4) и концентрировали. После очистки посредством хроматографии на силикагеле (этилацетат в качестве элюента) получали продукт в виде масла (7,5 г, 89%).
8.8. Синтез MPEG6C3-олигомерной кислоты (3-[2-(2-метоксиэтокси)этокси]пропионовой кислоты)
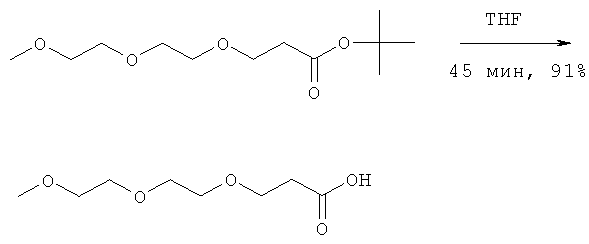
С трет-бутилового эфира (1 г, 4,90 ммоль) удаляли защитную группу путем перемешивания при комнатной температуре в трифторуксусной кислоте (6,0 мл). Затем содержимое концентрировали до постоянной массы (0,652 г, 89%).
8.9. Синтез 2-[2-(2-пропоксиэтокси)этокси]этанола (1)
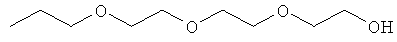
Триэтиленгликоль (19,5 г, 0,13 моль) растворяли в тетрагидрофуране (150 мл) и добавляли порциями гидрид натрия (2,60 г, 0,065 моль) в течение 0,5 ч, и реакцию перемешивали в течение еще 1 ч. Затем через воронку для вливания добавляли по каплям 1-бромпропанол (8,0 г, 0,065 моль), растворенный в тетрагидрофуране (30 мл), и реакцию перемешивали в течение ночи при комнатной температуре. Неочищенную реакционную смесь фильтровали через целит, промывали CH2Cl2 и упаривали досуха. Полученное масло растворяли в CH2Cl2 (250 мл), промывали насыщенным NaCl (250 мл), H2O (250 мл), сушили MgSO4 и упаривали досуха. После колоночной хроматографии (силикагель, этилацетат) получали 1 в виде желтоватого масла (2,24 г, выход 18%).
8.10. Синтез 2-[2-(2-пропоксиэтокси)этокси]этилового эфира 4-нитрофенилового эфира угольной кислоты
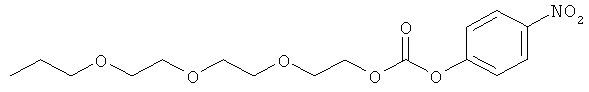
4-Нитрохлорформиат (3,45 г, 17,1 ммоль) и 1 (2,2 г, 11,4 ммоль) растворяли в CH2Cl2 (20 мл). После перемешивания в течение 10 минут добавляли TEA (2,1 мл, 15 ммоль), и реакцию перемешивали в течение ночи при комнатной температуре. Неочищенную реакцию разбавляли CH2Cl2 (50 мл), промывали 1 М HCl (50 мл), H2O (50 мл), сушили MgSO4 и упаривали досуха. После колоночной хроматографии (силикагель, этилацетат/гексаны, 3:2) получали 2 в виде желтоватого масла (2,57 г, выход 63%).
8.11. Синтез 2-[2-(2-метоксиэтокси)этокси]этилового эфира 2,5-диоксопирролидин-1-илового эфира угольной кислоты
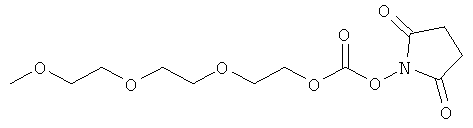
Триэтиленгликоля монометиловый эфир (1,0 г, 6,1 ммоль) и N,N'-дисукцинимидилкарбонат (1,87 г, 7,3 ммоль) растворяли в ацетонитриле (10 мл). Затем добавляли триэтиламин (1,3 мл, 9,15 ммоль), и реакцию перемешивали в течение ночи при комнатной температуре. Неочищенную реакцию упаривали досуха, растворяли в насыщенном NaHCO3 (50 мл), промывали этилацетатом (2×50 мл), сушили MgSO4 и упаривали досуха. После колоночной хроматографии (силикагель, этилацетат) получали 1 в виде прозрачного масла (0,367 г, выход 20%).
8.12. Синтез 2-{2-[2-(2-метоксиэтокси)этокси]этокси}этилового эфира 2,5-диоксопирролидин-1-илового эфира угольной кислоты (1).
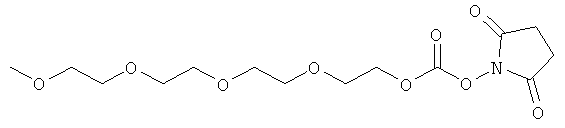
Тетраэтиленгликоля монометиловый эфир (1,0 г, 4,8 ммоль) и N,N-дисукцинимидилкарбонат (1,48 г, 5,8 ммоль) растворяли в ацетонитриле (10 мл). Затем добавляли триэтиламин (1,0 мл, 7,2 ммоль), и реакцию перемешивали в течение ночи при комнатной температуре. Неочищенную реакцию упаривали досуха, растворяли в насыщенном NaHCO3 (30 мл), промывали этилацетатом (2×30 мл), сушили MgSO4 и упаривали досуха. После колоночной хроматографии (силикагель, этилацетат/МеОН, 20:1) получали 1 в виде прозрачного масла (0,462 г, выход 28%).
8.13. Синтез этилового эфира бут-3-еновой кислоты
Винилуксусную кислоту (10,0 г, 0,12 моль) растворяли в этаноле (200 мл) и добавляли концентрированную серную кислоту (0,75 мл, 0,014 моль). Реакцию нагревали до температуры дефлегмации в течение 4 ч. Неочищенную реакцию разбавляли этилацетатом (200 мл), промывали Н2О (200 мл), насыщенным NaHCO3 (200 мл), сушили MgSO4 и упаривали досуха с получением 1 в виде прозрачного масла (3,17 г, 23%).
8.14. Синтез этилового эфира 4-{2-[2-(2-метоксиэтокси)этокси]этокси}-масляной кислоты
Триэтиленгликоля монометиловый эфир (4,27 г, 0,026 моль) и этиловый эфир бут-3-еновой кислоты (1,5 г, 0,013 моль) растворяли в тетрагидрофуране (10 мл). Затем добавляли кусковой Na0 (0,030 г, 0,013 моль), и реакцию перемешивали в течение 4 ч. Неочищенную реакцию гасили 1 М HCl (20 мл), промывали этилацетатом (3×20 мл). Органические слои объединяли и промывали Н2О (2×10 мл), сушили MgSO4 и упаривали досуха с получением 2 в виде желтоватого масла (1,07 г, выход 30%).
8.15. Синтез 4-{2-[2-(2-метоксиэтокси)этокси]этокси}масляной кислоты
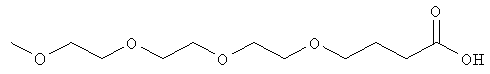
Этиловый эфир 4-{2-[2-(2-метоксиэтокси)этокси]этокси}масляной кислоты (1,07 г, 4,0 ммоль) растворяли в 1 М NaOH (10 мл), и реакцию перемешивали в течение 2 ч. Неочищенную реакцию разбавляли насыщенным NaCl (40 мл), подкисляли до рН примерно 2 концентрированной HCl, промывали CH2Cl2 (2×50 мл), сушили MgSO4 и упаривали досуха с получением 3 в виде прозрачного масла (0,945 г, выход 94%).
8.16. Синтез 2,5-диоксопирролидин-1-илового эфира 4-{2-[2-(2-метоксиэтокси)этокси]этокси}масляной кислоты
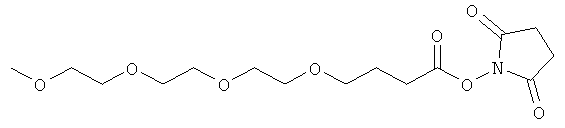
N-гидроксисукцинимид (0,55 г, 4,8 ммоль) и EDCI (1,15 г, 6,0 ммоль) растворяли в CH2Cl2 (7 мл). Затем добавляли 4-{2-[2-(2-метоксиэтокси)этокси]этокси}масляную кислоту (0,940 г, 3,8 ммоль), растворенную в CH2Cl2 (2 мл). Реакцию перемешивали в течение ночи при комнатной температуре. Неочищенную реакцию разбавляли CH2Cl2 (21 мл), промывали 1 М HCl (30 мл), H2O (30 мл), сушили MgSO4 и упаривали досуха. После колоночной хроматографии (силикагель, этилацетат) получали 4 в виде прозрачного масла (0,556 г, выход 43%).
9. Получение комплексов
Изучали способы получения цинковых комплексов конъюгатов соединений инсулинов. Для получения цинкового комплекса HIM2 разработали новые способы, необычные относительно опубликованных способов, используемых для комплексообразования/кристаллизации соединения инсулина и аналогов соединения инсулина. HIM2 представляет собой моноконъюгат инсулина человека с модифицирующей группировкой, связанной в положении В29, где модифицирующая группировка имеет следующую структуру:
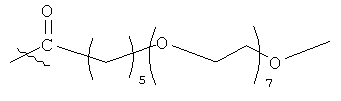 .
.
Другие комплексы получали с использованием IN105, моноконъюгата инсулина человека с модифицирующей группировкой, связанной в положении В29, где модифицирующая группировка имеет следующую структуру:
.
Данные способы давали три основных типа катионных комплексов твердых конъюгатов соединений инсулинов: комплекс "Т-типа" и "R-типа" и "протаминовый" комплекс.
9.1. Получение и анализ твердых веществ Т-типа
9.1.1. Попытка получения Zn-комплекса HIM2 Т-типа (2 г/л)
Раствор HIM2 в концентрации приблизительно 2 г/л доводили 10% HCl до конечного рН примерно 3. Ледяную уксусную кислоту добавляли к аликвоте вышеуказанного раствора объемом 10 мл (20 мг белка) до конечной концентрации 0,25 М. К данному образцу добавляли двадцать (или сорок) мкл 2% (масс./масс.) раствора ZnCl2. рН доводили до 5,1 (или 5,5) концентрированным гидроксидом аммония. Раствор перемешивали в течение 15 минут при комнатной температуре (или +5°С) и затем оставляли стоять в течение одних суток при комнатной температуре (или +5°С), чтобы дать возможность образоваться твердому веществу. После того, как реакции позволили стоять одни сутки при комнатной температуре (или при +5°С), не образовалось никаких кристаллов или осадка. Смотри Пример 2 патента США №5504188 под названием "Preparation of stable zinc insulin compound analog crystals".
9.1.2. Zn-комплекс HIM2 Т-типа (концентрация 10 г/л)
Раствор HIM2 в концентрации приблизительно 10 г/л доводили 10% HCl до получения конечного рН примерно 3. Ледяную уксусную кислоту добавляли к аликвоте вышеуказанного раствора объемом 10 мл (100 мг белка) до конечной концентрации 0,25 М. К данному образцу добавляли сорок мкл 10% (масс./масс.) раствора ZnCl2. рН доводили до 5,2 концентрированным гидроксидом аммония. Раствор перемешивали в течение 15 минут при +5°С и затем оставляли стоять в течение пять суток при +5°С, чтобы дать возможность образоваться твердому веществу.
Реакционную смесь переносили в центрифужную пробирку и центрифугировали при 2000 об/мин в течение 10 минут. Раствор декантировали, и твердое вещество промывали 5 мл холодной DI (деионизированной) воды. Этот раствор центрифугировали при 2000 об/мин в течение 10 минут перед тем, как декантировать воду, и твердые вещества промывали дополнительно 5 мл холодной DI воды. Снова образец центрифугировали приблизительно при 2000 об/мин в течение приблизительно 10 минут перед тем, как декантировать Н2О. Образец промывали 5 мл стандартного холодного EtOH 200 и центрифугировали при 2000 об/мин в течение 10 минут перед тем, как декантировать EtOH. Образец сушили в лиофилизаторе с получением белого твердого вещества.
9.1.3. Zn-комплекс HIM2 Т-типа (концентрация 20 г/л)
Раствор HIM2 в концентрации приблизительно 20 г/л доводили 10% HCl до получения конечного рН примерно 3. Ледяную уксусную кислоту добавляли к аликвоте вышеуказанного раствора объемом 10 мл (200 мг белка) до конечной концентрации 0,25 М. К данному образцу добавляли восемьдесят мкл 10% (масс./масс.) раствора ZnCl2. рН доводили до 5,37 концентрированным гидроксидом аммония. Раствор перемешивали в течение 15 минут при +5°С и затем оставляли стоять в течение четырех суток при +5°С, чтобы дать возможность образоваться твердому веществу.
Реакционную смесь переносили в центрифужную пробирку и центрифугировали при 2600 об/мин в течение 20 минут. Раствор декантировали, и твердое вещество промывали 5 мл холодной DI воды. Раствор центрифугировали при 2600 об/мин в течение 20 минут перед тем, как декантировать воду, и твердые вещества промывали дополнительно 5 мл холодной DI воды. Снова образец центрифугировали приблизительно при 2600 об/мин в течение приблизительно 20 минут перед тем, как декантировать H2O. Образец промывали 5 мл стандартного холодного EtOH 200 и центрифугировали при 2600 об/мин в течение 20 минут перед тем, как декантировать EtOH. Образец сушили в лиофилизаторе с получением белого твердого вещества.
9.1.4. Zn-комплекс HIM2 Т-типа (концентрация 30 г/л)
Раствор HIM2 в концентрации приблизительно 30 г/л доводили 10% HCl до получения конечного рН примерно 3. Ледяную уксусную кислоту добавляли к аликвоте вышеуказанного раствора объемом 50 мл (1,5 г белка) до конечной концентрации 0,25 М. К данному образцу добавляли шестьсот мкл 10% (масс./масс.) раствора ZnCl2. рН доводили до 5,34 концентрированным гидроксидом аммония. Раствор оставляли стоять при +5°С в течение пяти суток, чтобы дать возможность образоваться твердому веществу.
Реакционную смесь переносили в центрифужную пробирку и центрифугировали при 2800 об/мин в течение 15 минут. Раствор декантировали, и твердое вещество промывали три раза 10 мл холодной DI воды, центрифугируя и декантируя Н2О при каждой промывке. Образец затем промывали три раза 10 мл, стандартного холодного EtOH 200. Его центрифугировали при 2800 об/мин в течение 15 минут и декантировали после каждой промывки образец. Образец сушили в лиофилизаторе с получением белого твердого вещества.
На фиг.1 и 2 представлены микрофотографии, полученные с использованием микроскопа Zeiss Axiovert, показывающие кристаллы, выросшие в течение 24 часов. На фиг.1 размер кристалла составляет приблизительно 11,3 мкм в длину и приблизительно 5,3 мкм в диаметре. На фиг.2 размер кристалла слева составляет приблизительно 15,1 мкм в длину и приблизительно 5,9 мкм в диаметре, а размер кристалла справа составляет приблизительно 9,1 мкм в длину и приблизительно 5,3 мкм в диаметре. На фиг.3 представлена микрофотография, полученная с использованием микроскопа Zeiss Axiovert, показывающая кристаллы, выросшие в течение 5 суток. В одном аспекте изобретение включает кристаллы, имеющие такую морфологию, как показано на фиг.1, 2 или 3.
9.1.5. Zn-комплекс HIM2 Т-типа (концентрация 50 г/л)
Раствор HIM2 в концентрации приблизительно 50 г/л доводили 10% HCl до конечного рН примерно 3. Ледяную уксусную кислоту добавляли к аликвоте вышеуказанного раствора объемом 10 мл до конечной концентрации 0,25 М. К данному образцу добавляли двести мкл 10%-ного раствора ZnCl2. рН доводили до 5,23 концентрированным гидроксидом аммония. Раствор перемешивали при +5°С в течение 15 минут и затем оставляли стоять в течение четырех суток при +5°С, чтобы дать возможность образоваться твердому веществу.
Реакционную смесь переносили в центрифужную пробирку и центрифугировали при 2600 об/мин в течение 20 минут. Раствор декантировали, и твердое вещество промывали 5 мл холодной DI Н2О. Раствор центрифугировали при 2600 об/мин в течение 20 минут перед тем, как декантировать H2O, и твердое вещество промывали дополнительно 5 мл холодной DI H2O. Снова образец центрифугировали при 2600 об/мин в течение 20 минут перед тем, как декантировать Н2О. Образец промывали 5 мл стандартного холодного EtOH 200 и центрифугировали при 2600 об/мин в течение 20 минут перед тем, как декантировать EtOH. Образец сушили в лиофилизаторе в течение трех суток.
9.1.6. Zn-комплекс HIM2 Т-типа (масштаб 1 г)
Раствор HIM2 в концентрации приблизительно 10 г/л доводили 10% HCl до конечного рН примерно 3. Ледяную уксусную кислоту добавляли к аликвоте вышеуказанного раствора объемом 50 мл (500 мг белка) до конечной концентрации 0,25 М. К данному образцу добавляли двести мкл 10%-ного раствора ZnCl2. рН доводили до 5,49 концентрированным гидроксидом аммония. Раствор перемешивали при +5°С в течение 15 минут и затем оставляли стоять при +5°С в течение семи суток, чтобы дать возможность образоваться твердому веществу.
Реакционную смесь переносили в центрифужную пробирку и центрифугировали при 2600 об/мин в течение 20 минут. Раствор декантировали, и твердое вещество промывали 10 мл холодной DI Н2О. Раствор центрифугировали при 2600 об/мин в течение 20 минут перед тем, как декантировать H2O. Промывки водой повторяли еще два раза. Образец затем промывали 10 мл стандартного холодного EtOH 200 и центрифугировали при 2600 об/мин в течение 20 минут перед тем, как декантировать EtOH. Таким же образом осуществляли еще две промывки EtOH перед тем, как образец помещали сушиться в лиофилизатор на четверо суток.
9.1.7. Zn-комплекс HIM2 Т-типа при нейтральном рН (масштаб 5 г)
Раствор HIM2 в концентрации приблизительно 10 г/л доводили 10% HCl до конечного рН примерно 3. К данному образцу добавляли два миллилитра 10%-ного раствора ZnCl2. рН доводили до 7,05 концентрированным гидроксидом аммония. Раствор перемешивали при комнатной температуре в течение ночи, чтобы дать возможность образоваться твердому веществу.
Молочную реакционную смесь Zn-HIM2 (500 мл) добавляли частями в воронку объемом 350 мл с пористым (4,5-5 мкм) диском (ChemGlass CG1402-28, диаметр 90 мм). Фильтрат собирали в колбу с боковым отводом в течение приблизительно 4-6 ч, создавая в ней вакуум. В качестве одного из вариантов, осадок можно промыть 100 мл холодного 1% ZnCl2, и фильтрат можно собрать отдельно. Осадок промывали 100 мл ледяной воды, и фильтрат снова собирали. Осадок также дополнительно промывали 100 мл ледяного 100% этанола, и фильтрат собирали еще раз. Конечную промывку осадка осуществляли 100 мл свежей ледяной воды и собирали конечный фильтрат. Осадок сушили под вакуумом и/или сушили на воздухе в течение 12-18 ч. После сушки осадок соскребали с воронки, взвешивали и измеряли содержания влажности/белка посредством ВЭЖХ. Фильтраты, собранные на разных стадиях промывки, также анализировали с помощью ВЭЖХ для того, чтобы определить концентрацию потерянного Zn-HIM2 во время процесса. В результате фильтрации получали 2,5% (масс./масс.) содержание Zn с общим выходом 98%.
9.1.8. Zn-комплекс HIM2 Т-типа при нейтральном рН (масштаб 500 мг)
Раствор HIM2 в концентрации приблизительно 10 г/л доводили 10% HCl до конечного рН примерно 3. К данному образцу добавляли двести мкл 10%-ного раствора ZnCl2. рН доводили до 7,06 концентрированным гидроксидом аммония. Раствор перемешивали при +5°С в течение 15 минут и затем оставляли стоять при +5°С в течение двух суток, чтобы дать возможность образоваться твердому веществу.
Реакционную смесь переносили в центрифужную пробирку и центрифугировали при 2800 об/мин в течение 15 минут. Раствор декантировали, и твердое вещество промывали 10 мл холодной DI H2O. Раствор центрифугировали при 2800 об/мин в течение 15 минут перед тем, как декантировать H2O. Промывки водой повторяли еще два раза. Образец затем промывали 10 мл стандартного холодного EtOH 200 и центрифугировали при 2600 об/мин в течение 20 минут перед тем, как декантировать EtOH. Таким же образом осуществляли еще две промывки EtOH перед тем, как образец помещали сушиться в лиофилизатор на двое суток.
9.1.9 Результаты для твердых композиций Т-типа
NEM = недостаточно вещества
ND = нет данных
9.1. Получение и анализ твердых веществ R-типа
9.2.1. Zn-комплекс HIM2 R-типа с фенолом при концентрации 2 г/л
Раствор HIM2 в концентрации приблизительно 2 г/л доводили ледяной уксусной кислотой до конечного рН примерно 3. Тридцать три мкл жидкого фенола добавляли к аликвоте вышеуказанного раствора объемом 10 мл. рН доводили до 5,89 концентрированным гидроксидом аммония. К данному образцу добавляли сто шестьдесят мкл 10% (масс./масс.) раствора ZnCl2. Раствор перемешивали при комнатной температуре в течение 15 минут и затем оставляли стоять при комнатной температуре в течение трех суток, чтобы дать возможность сформироваться большему количеству осадка.
Реакционную смесь переносили в центрифужную пробирку и центрифугировали при 3400 об/мин в течение 15 минут. Супернатант декантировали, и твердое вещество промывали 5 мл холодной DI воды. Образец затем промывали 5 мл стандартного холодного EtOH 200 и центрифугировали при 3200 об/мин в течение 15 минут перед тем, как декантировать EtOH. Образец снова промывали 5 мл холодного EtOH, однако его не центрифугировали. Твердому веществу давали возможность осесть на дно пробирки и затем помещали в быструю вакуумную сушилку для сушки.
9.2.2. Получение Zn-комплекса HIM2 R-типа при концентрации 20 г/л
Раствор HIM2 в концентрации приблизительно 20 г/л доводили 10% HCl до конечного рН примерно 3. Шестьдесят шесть мкл жидкого фенола добавляли к аликвоте вышеуказанного раствора объемом 10 мл. рН доводили до 6,43 концентрированным гидроксидом аммония. К данному образцу добавляли триста двадцать мкл 10%-ного раствора ZnCl2. Раствор перемешивали при комнатной температуре в течение 15 минут и затем оставляли стоять при комнатной температуре в течение четырех суток, чтобы дать возможность сформироваться большему количеству осадка.
Реакционную смесь переносили в центрифужную пробирку и центрифугировали при 2600 об/мин в течение 20 минут. Раствор декантировали, и твердое вещество промывали 5 мл холодной DI H2O. Раствор центрифугировали при 2600 об/мин в течение 20 минут перед тем, как декантировать H2O, и твердое вещество промывали дополнительно 5 мл холодной DI H2O. Образец снова центрифугировали при 2600 об/мин в течение 20 минут перед тем, как декантировать H2O. Образец промывали 5 мл стандартного холодного EtOH 200 и центрифугировали при 2600 об/мин в течение 20 минут перед тем, как декантировать EtOH. Образец подвергали лиофилизации в течение трех суток.
9.2.3. Получение Zn-комплекса HIM2 R-типа при концентрации 30 г/л
Раствор HIM2 в концентрации приблизительно 30 г/л доводили 10% HCl до конечного рН примерно 3. Девяносто девять мкл жидкого фенола добавляли к аликвоте вышеуказанного раствора объемом 10 мл. рН доводили до 6,47 концентрированным гидроксидом аммония. Затем к данному образцу добавляли 480 мкл 10%-ного раствора ZnCl2. Раствор перемешивали при комнатной температуре в течение 15 минут и затем оставляли стоять при комнатной температуре в течение четырех суток, чтобы дать возможность сформироваться большему количеству осадка.
Реакционную смесь переносили в центрифужную пробирку и центрифугировали при 2600 об/мин в течение 20 минут. Раствор декантировали, и твердое вещество промывали 5 мл холодной DI H2O. Раствор центрифугировали при 2600 об/мин в течение 20 минут перед тем, как декантировать H2O, и твердое вещество промывали дополнительно 5 мл холодной DI Н2О. Образец снова центрифугировали при 2600 об/мин в течение 20 минут перед тем, как декантировать H2O. Образец промывали 5 мл стандартного холодного EtOH 200 и центрифугировали при 2600 об/мин в течение 20 минут перед тем, как декантировать EtOH. Образец подвергали лиофилизации в течение трех суток.
На фиг.4 показано твердое вещество, образовавшееся в течение 4 суток. Фотография получена с использованием микроскопа Zeiss Axiovert. Средняя длина кристаллов составляет приблизительно 9,7 мкм.
9.2.4. Получение Zn-комплекса HIM2 R-типа при концентрации 50 г/л
Раствор HIM2 в концентрации приблизительно 50 г/л доводили 10% HCl до конечного рН примерно 3. Сто шестьдесят пять мкл жидкого фенола добавляли к аликвоте вышеуказанного раствора объемом 10 мл. рН доводили до 6,82 концентрированным гидроксидом аммония. К данному образцу добавляли восемьсот мкл 10%-ного раствора ZnCl2. Раствор перемешивали при комнатной температуре в течение 15 минут и затем оставляли стоять при комнатной температуре в течение четырех суток, чтобы дать возможность сформироваться большему количеству осадка.
Реакционную смесь переносили в центрифужную пробирку и центрифугировали при 2600 об/мин в течение 20 минут. Раствор декантировали, и твердое вещество промывали 5 мл холодной DI Н2О. Раствор центрифугировали при 2600 об/мин в течение 20 минут перед тем, как декантировать H2O, и твердое вещество промывали дополнительно 5 мл холодной DI Н2О. Образец снова центрифугировали при 2600 об/мин в течение 20 минут перед тем, как декантировать H2O. Образец промывали 5 мл стандартного холодного EtOH 200 и центрифугировали при 2600 об/мин в течение 20 минут перед тем, как декантировать EtOH. Образец подвергали лиофилизации в течение трех суток.
9.2.5. Получение Zn-комплекса HIM2 R-типа в масштабе 1 г
Раствор HIM2 в концентрации приблизительно 10 г/л доводили 10% HCl до конечного рН примерно 3. Сто шестьдесят пять мкл жидкого фенола добавляли к аликвоте вышеуказанного раствора объемом 50 мл. рН доводили до 6,42 концентрированным гидроксидом аммония. К данному образцу добавляли восемьсот мкл 10%-ного раствора ZnCl2. Раствор перемешивали при комнатной температуре в течение 15 минут и затем оставляли стоять при комнатной температуре в течение семи суток, чтобы дать возможность сформироваться большему количеству осадка.
Реакционную смесь переносили в центрифужную пробирку и центрифугировали при 2600 об/мин в течение 20 минут. Раствор декантировали, и твердое вещество промывали 10 мл холодной DI Н2О. Раствор центрифугировали при 2600 об/мин в течение 20 минут перед тем, как декантировать Н2О. Таким же образом осуществляли еще две промывки водой. Образец затем промывали 10 мл стандартного холодного EtOH 200 и центрифугировали при 2600 об/мин в течение 20 минут перед тем, как декантировать EtOH. Таким же образом осуществляли еще две промывки EtOH перед тем, как поместить образец в лиофилизатор на четверо суток.
9.2.6. Zn-комплекс HIM2 R-типа в масштабе 5 г
Раствор HIM2 в концентрации приблизительно 10 г/л доводили 10% HCl до конечного рН примерно 3. Одну тысячу пятьсот мкл жидкого фенола добавляли к 450 мкл вышеуказанного раствора. рН доводили до 7,1 концентрированным гидроксидом аммония. К данному образцу добавляли одну тысячу восемьсот мкл 10%-ного раствора ZnCl2. Раствор перемешивали при комнатной температуре в течение 15 минут и затем оставляли стоять при комнатной температуре в течение ночи, чтобы дать возможность сформироваться большему количеству осадка.
Полученную выше реакционную смесь разделяли на три пробы для фильтрации. Реакционную смесь из первой пробы фильтровали через мелкопористую воронку и затем промывали 1%-ным раствором ZnCl2. Вещество сушили в течение ночи путем вакуумной фильтрации. Вторую пробу фильтровали через среднепористый фильтр, который также содержал фильтровальную бумагу. Вещество затем промывали этанолом и водой и сушили в течение ночи путем вакуумной фильтрации. Наконец, третью пробу фильтровали через мелкопористую воронку, промывали 1%-ным раствором ZnCl2 и также промывали этанолом и водой. Вещество также сушили в течение ночи путем вакуумной фильтрации.
9.2.7. Zn-комплекс HIM2 R-типа при нейтральном рН
Раствор HIM2 в концентрации приблизительно 10 г/л доводили 10% HCl до конечного рН примерно 3. Сто шестьдесят пять мкл жидкого фенола добавляли к 50 мл вышеуказанного раствора. Затем к данному образцу добавляли двести мкл 10%-ного раствора ZnCl2. рН доводили до 7,18 концентрированным гидроксидом аммония. Раствор находился при комнатной температуре в течение двух суток, чтобы дать возможность сформироваться осадку.
Реакционную смесь переносили в центрифужную пробирку и центрифугировали при 2800 об/мин в течение 20 минут. Однако сначала вещество не оседало на дно пробирки, и поэтому его центрифугировали в течение приблизительно 2 ч. Раствор декантировали, и твердое вещество промывали 5 мл холодной DI Н2О. Раствор центрифугировали при 2800 об/мин в течение 60 минут перед тем, как декантировать Н2О. Промывку водой повторяли еще два раза. Образец затем промывали 5 мл стандартного холодного EtOH 200 и центрифугировали при 2800 об/мин в течение 60 минут перед тем, как декантировать EtOH. Таким же образом осуществляли еще две промывки EtOH. После третьей промывки EtOH реакция оставалась мутной, и ее помещали на ночь в холодильник, чтобы дать возможность продуктам реакции осесть более полно. Растворитель декантировали, и вещество помещали в лиофилизатор на 2 суток.
9.2.8. Результаты для твердых композиций R-типа
ND = нет данных
9.3. Получение и анализ твердых веществ с протамином
9.3.1. Получение Zn-комплекса HIM2 Т-типа с протамином при кислом рН
Протамин добавляли к исходному раствору HIM2 с концентрацией 10 г/л, который имел конечный рН примерно 3, подведенный 10% HCl. Ледяную уксусную кислоту добавляли к аликвоте вышеуказанного раствора объемом 10 мл (100 мг белка) до конечной концентрации 0,25 М. К данному образцу добавляли двести мкл 10%-ного раствора ZnCl2. рН доводили до рН приблизительно 5 концентрированным гидроксидом аммония. Раствор перемешивали при +5°С в течение 15 минут и затем оставляли стоять при +5°С в течение двух суток, чтобы дать возможность образоваться твердому веществу.
Реакционную смесь переносили в центрифужную пробирку и центрифугировали при 2600 об/мин в течение 20 минут. Раствор декантировали, и твердое вещество промывали 10 мл холодной DI H2O. Раствор центрифугировали при 2600 об/мин в течение 20 минут перед тем, как декантировать H2O. Таким же образом осуществляли еще две промывки H2O. Образец затем промывали 10 мл стандартного холодного EtOH 200 и центрифугировали при 2600 об/мин в течение 20 минут перед тем, как декантировать EtOH. Таким же образом осуществляли еще две промывки EtOH перед тем, как поместить образец в лиофилизатор на двое суток.
9.3.2. Получение Zn-комплекса HIM2 Т-типа с протамином при нейтральном рН
Раствор HIM2 в концентрации приблизительно 30 г/л доводили 10% HCl до конечного рН примерно 3. Один миллилитр ледяной уксусной кислоты добавляли к аликвоте вышеуказанного раствора объемом 50 мл (1,5 г белка). К данной реакции добавляли шестьсот мкл 10%-ного раствора ZnCl2 с последующим добавлением 225 миллиграммов протамина. рН доводили до 6,95 концентрированным гидроксидом аммония, и реакцию оставляли стоять в течение двух суток при +5°С, чтобы дать возможность образоваться твердому веществу.
Реакционную смесь переносили в центрифужную пробирку и центрифугировали при 2600 об/мин в течение 20 минут. Раствор декантировали, и твердое вещество промывали 10 мл холодной DI H2O. Раствор центрифугировали при 2600 об/мин в течение 20 минут перед тем, как декантировать Н2О. Таким же образом осуществляли еще две промывки H2O. Образец затем промывали 10 мл стандартного холодного EtOH 200 и центрифугировали при 2600 об/мин в течение 20 минут перед тем, как декантировать EtOH. Таким же образом осуществляли еще две промывки EtOH перед тем, как поместить образец в лиофилизатор на трое суток.
9.3.3. Получение Zn-комплекса HIM2 R-типа с протамином при кислом рН
Раствор HIM2 в концентрации приблизительно 10 г/л доводили 10% HCl до конечного рН примерно 3. Жидкий фенол (2,48 мл) добавляли к аликвоте вышеуказанного раствора объемом 150 мл (1,5 г белка). рН реакции доводили концентрированным гидроксидом аммония до рН примерно 6,57. К этой реакции добавляли двенадцать мкл 10%-ного раствора ZnCl2 с последующим добавлением 225 миллиграммов протамина. Реакционную смесь перемешивали при комнатной температуре в течение 15 минут перед тем, как ее оставляли стоять в течение двух суток при комнатной температуре, чтобы дать возможность образоваться твердому веществу.
Реакционную смесь переносили в центрифужную пробирку и центрифугировали при 2800 об/мин в течение 15 минут. Раствор декантировали, и твердое вещество промывали 50 мл холодной DI H2O. Раствор центрифугировали при 2800 об/мин в течение 15 минут перед тем, как декантировать H2O. Таким же образом осуществляли еще две промывки H2O. Образец затем промывали 10 мл стандартного холодного EtOH 200 и центрифугировали при 2800 об/мин в течение 15 минут перед тем, как декантировать EtOH. Таким же образом осуществляли еще две промывки EtOH перед тем, как поместить образец в лиофилизатор на двое суток.
9.3.4. Получение Zn-комплекса HIM2 R-типа с протамином при нейтральном рН
Раствор HIM2 в концентрации приблизительно 10 г/л доводили 10% HCl до конечного рН примерно 3. Жидкий фенол (495 мл) добавляли к 150 мл реакции. Затем к реакции добавляли 600 мкл 10%-ного раствора ZnCl2 с последующим добавлением 75 мг протамина. рН доводили концентрированным гидроксидом аммония до рН 7,01. Реакцию оставляли стоять в течение трех суток при комнатной температуре, чтобы дать возможность образоваться твердому веществу.
Реакционную смесь переносили в центрифужную пробирку и центрифугировали при 2800 об/мин в течение 15 минут. Раствор декантировали, и твердое вещество промывали 50 мл холодной DI H2O. Раствор центрифугировали при 2800 об/мин в течение 15 минут перед тем, как декантировать H2O. Таким же образом осуществляли еще две промывки H2O. Образец затем промывали 50 мл стандартного холодного EtOH 200 и центрифугировали при 2800 об/мин в течение 15 минут перед тем, как декантировать EtOH. Таким же образом осуществляли еще две промывки EtOH перед тем, как поместить образец в лиофилизатор на двое суток.
9.3.5. Результаты для твердых композиций с протамином
ND = нет данных
9.4. Получение и анализ комплексов диконъюгатов соединений инсулинов
9.4.1. Zn-комплекс Т-типа (А1 и В29)-диконъюгата соединения инсулина
Диконъюгат соединения инсулина, содержащий модифицирующую группировку -С(O)(CH2)5(OCH2CH2)7ОСН3, связанную в положениях В29 и А1 инсулина человека (DICON-1), добавляли к раствору в концентрации приблизительно 10 г/л и доводили 10% HCl до конечного рН 3,15. Ледяную уксусную кислоту добавляли к аликвоте вышеуказанного раствора объемом 3,75 мл до конечной концентрации 0,25 М. Затем к данному образцу добавляли 15 мкл 10%-ного раствора ZnCl2. рН доводили до рН 4,90 концентрированным гидроксидом аммония. Раствор перемешивали в течение 15 минут при +5°С и затем оставляли стоять в течение шести суток при +5°С, чтобы дать возможность образоваться твердому веществу (с выходом белого твердого вещества).
9.4.2. Zn-комплекс R-типа (А1 и В29)-диконъюгата соединения инсулина
DICON-1 добавляли к раствору в концентрации приблизительно 10 г/л и доводили 10% HCl до конечного рН 3,15. Приблизительно 12 мкл жидкого фенола добавляли к аликвоте вышеуказанного раствора объемом 3,75 мл. рН доводили до 5,75 концентрированным гидроксидом аммония. К данному образцу добавляли 60 мкл 10%-ного раствора ZnCl2. Раствор перемешивали при комнатной температуре в течение 15 минут и затем оставляли стоять при комнатной температуре в течение шести суток, чтобы дать возможность сформироваться большему количеству осадка (с выходом белого твердого вещества).
Реакционную смесь переносили в центрифужную пробирку и центрифугировали при 2600 об/мин в течение 20 минут. Раствор декантировали, и твердое вещество промывали 5 мл холодной DI H2O. Раствор центрифугировали при 2600 об/мин в течение 20 минут перед тем, как декантировать H2O, и твердое вещество промывали дополнительно 5 мл холодной DI H2O. Образец снова центрифугировали при 2600 об/мин в течение 20 минут перед тем, как декантировать H2O. Образец промывали 5 мл стандартного холодного EtOH 200 и центрифугировали при 2600 об/мин в течение 20 минут перед тем, как декантировать EtOH. Образец подвергали лиофилизации в течение шести суток.
9.4.3. Диконъюгат В1, В29 (10 мг/мл)
DICON-1 добавляли к раствору в концентрации приблизительно 10 г/л и добавляли 33 мкл жидкого фенола. рН доводили до 5,34 концентрированным гидроксидом аммония. Затем к данному образцу добавляли 160 мкл 10%-ного раствора ZnCl2. Раствор оставляли стоять при комнатной температуре в течение двух недель, чтобы дать возможность образоваться твердому веществу (с выходом белого твердого вещества).
Реакционную смесь переносили в центрифужную пробирку и центрифугировали при 2800 об/мин в течение 15 минут. Раствор декантировали, и твердое вещество промывали три раза 5 мл холодной DI Н2О. Раствор центрифугировали в течение 15 минут при 2800 об/мин и декантировали после каждой промывки. Образец затем промывали три раза 5 мл стандартного холодного EtOH 200. Образец снова центрифугировали при 2600 об/мин в течение 15 минут и декантировали после каждой промывки. Образец подвергали лиофилизации в течение двух суток.
9.4.4. Диконъюгат В1, В29 (20 мг/мл)
DICON-1 добавляли к раствору в концентрации приблизительно 20 г/л и добавляли 66 мкл жидкого фенола. рН доводили до 7,65 концентрированным гидроксидом аммония. Затем к данному образцу добавляли 320 мкл 10%-ного раствора ZnCl2. Раствор оставляли стоять при комнатной температуре в течение двух недель, чтобы дать возможность образоваться твердому веществу (с выходом белого твердого вещества).
Реакционную смесь переносили в центрифужную пробирку и центрифугировали при 2800 об/мин в течение 15 минут. Раствор декантировали, и твердое вещество промывали три раза 5 мл холодной DI Н2О. Раствор центрифугировали в течение 15 минут при 2800 об/мин и декантировали после каждой промывки. Образец затем промывали три раза 5 мл стандартного холодного EtOH 200. Образец снова центрифугировали при 2600 об/мин в течение 15 минут и декантировали после каждой промывки. Образец подвергали лиофилизации в течение двух суток.
9.5. Получение и анализ твердых веществ IN105 Т-типа
9.5.1 Zn-комплекс Т-типа моноконъюгата IN105 (концентрация 10 г/л)
Раствор IN105 в концентрации приблизительно 10 г/л (100 мг) доводили 10% HCl до получения конечного рН примерно 3. К данному образцу добавляли 50 мкл 10%-ного раствора (масс./масс.) ZnCl2. рН доводили до 7,52 концентрированным гидроксидом аммония. Мутный раствор перемешивали и затем оставляли стоять в течение пяти суток при комнатной температуре, чтобы дать возможность образоваться твердому веществу.
Реакционную смесь переносили в центрифужную пробирку и центрифугировали при 2900 об/мин в течение 15 минут. Раствор декантировали, и твердое вещество промывали 3×10 мл холодной DI воды. Раствор центрифугировали при 2900 об/мин в течение 10 минут перед тем, как декантировать воду, и твердые вещества промывали новой порцией холодной DI воды. Образец затем промывали 3×10 мл стандартного холодного EtOH 200 и центрифугировали при 2900 об/мин в течение 10 минут перед тем, как декантировать EtOH. Образец сушили под вакуумом с получением белого твердого вещества (90 мг).
9.5.2. Zn-комплекс Т-типа моноконъюгата IN105 (масштаб 1 г)
Раствор IN105 в концентрации приблизительно 10 г/л (1 г) доводили 10% HCl до конечного рН примерно 3. К данному образцу добавляли пятьсот мкл 10%-ного раствора ZnCl2. рН доводили до примерно 7,4 концентрированным гидроксидом аммония. Мутный раствор перемешивали в течение 15 минут и затем оставляли стоять при комнатной температуре в течение примерно 2 суток перед фильтрацией.
Реакционную смесь фильтровали через воронку с пористым стеклом (мелкопористым) под вакуумом. Воронку с пористым стеклом с отфильтрованным веществом выдерживали в стеклянном эксикаторе под вакуумом в течение ночи с получением белого тонкого порошка (900 мг).
9.5.3. Zn-комплекс Т-типа моноконъюгата IN105 при нейтральном рН (масштаб 5 г)
Раствор IN105 в концентрации приблизительно 10 г/л (5 г, lot#Nobex040706L) доводили 10% HCl до конечного рН примерно 3. К данному образцу добавляли два миллилитра 10%-ного раствора ZnCl2. рН доводили до примерно 7,4 концентрированным гидроксидом аммония. Затем мутный раствор оставляли стоять при комнатной температуре в течение ночи, чтобы дать возможность образоваться твердому веществу перед фильтрацией.
Полученную выше реакцию распределяли в 4×50 мл центрифужные пробирки и сначала центрифугировали при 3200 об/мин в целом в течение 2 ч. Затем вещество центрифугировали при 9000 об/мин в течение 20 минут и хранили при 5°С в течение ночи. Супернатант декантировали, и твердое вещество в каждой пробирке промывали 10 мл холодной DI H2O. Пробирки переворачивали и центрифугировали при 3200 об/мин в течение примерно 1 ч перед тем, как декантировать Н2О, и твердые вещества промывали дополнительно 10 мл холодной DI H2O. Образец снова центрифугировали при 3200 об/мин в течение примерно 1 ч перед тем, как декантировать H2O. Образец промывали 2×10 мл стандартного холодного EtOH 200 и центрифугировали при 3200 об/мин в течение 1 ч перед тем, как декантировать EtOH. Образец сушили под вакуумом в течение двух суток с получением 1,64 г (lot#Nobex040730L-A) белого порошка.
9.5.4. Результаты для твердых композиций IN105 Т-типа
9.6. Получение и анализ твердых веществ IN105 R-типа
9.6.1. Zn-комплекс R-типа конъюгата IN105 с фенолом при нейтральном рН
Раствор IN105 в концентрации приблизительно 10 г/л (500 мг) доводили 10% HCl до конечного рН примерно 3. К вышеуказанному раствору добавляли двести мкл 10%-ного раствора ZnCl2 и 165 мкл жидкого фенола. рН доводили до примерно 7,37 концентрированным гидроксидом аммония. Мутный раствор находился при комнатной температуре в течение 2 суток для того, чтобы дать возможность образоваться твердому веществу перед фильтрацией.
Реакционную смесь фильтровали через воронку с пористым стеклом (мелкопористым) под вакуумом. Воронку с пористым стеклом с отфильтрованным веществом выдерживали в стеклянном эксикаторе под вакуумом в течение ночи с получением белого тонкого порошка (440 мг).
9.6.2. Zn-комплекс R-типа конъюгата IN105 в масштабе 5 г
Раствор IN105 в концентрации приблизительно 10 г/л (4,2 г, lot#Nobex040706L) доводили 10% HCl до конечного рН примерно 3. К вышеуказанному раствору добавляли 1,5 жидкого фенола и 1,8 мл 10%-ного раствора ZnCl2 и 165 мкл. рН доводили до примерно 7,4 концентрированным гидроксидом аммония. Данный очень мутный раствор оставляли стоять при комнатной температуре в течение ночи, чтобы дать возможность сформироваться большему количеству осадка.
Полученную выше реакцию распределяли в 4×50 мл центрифужные пробирки и сначала центрифугировали при 3200 об/мин в целом в течение 2 ч. Затем вещество центрифугировали при 9000 об/мин в течение 20 минут и хранили при 5°С в течение ночи. Супернатант декантировали, и твердое вещество в каждой пробирке промывали 10 мл холодной DI H2O. Пробирки переворачивали и центрифугировали при 3200 об/мин в течение примерно 1 ч перед тем, как декантировать H2O, и твердые вещества промывали дополнительно 10 мл холодной DI H2O. Образец снова центрифугировали при 3200 об/мин в течение примерно 1 ч перед тем, как декантировать H2O. Образец промывали 2×10 мл стандартного холодного EtOH 200 и центрифугировали при 3200 об/мин в течение 1 ч перед тем, как декантировать EtOH. Образец сушили под вакуумом в течение 2 суток с получением 2,34 г белого порошка.
9.6.3. Результаты для твердых композиций IN105 R-типа
* В фосфатном буфере, рН приблизительно 7,4
9.7. Получение и анализ твердых веществ IN105 с протамином
9.7.1. Получение Zn-комплекса R-типа моноконъюгата IN105 с протамином при кислом рН
Раствор IN105 в концентрации приблизительно 10 г/л доводили 10% HCl до конечного рН примерно 3. Жидкий фенол (248 мкл) добавляли к аликвоте вышеуказанного раствора объемом 15 мл (150 мг белка). рН реакции доводили концентрированным гидроксидом аммония до рН примерно 6,50. К этой реакции добавляли один мкл 10%-ного раствора ZnCl2 с последующим добавлением 22,5 миллиграмма протамина. Реакционную смесь перемешивали при комнатной температуре в течение 15 минут перед тем, как ее оставляли стоять в течение двух суток при комнатной температуре, чтобы дать возможность образоваться твердому веществу.
Реакционную смесь переносили в центрифужную пробирку и центрифугировали при 2800 об/мин в течение 15 минут. Раствор декантировали, и твердое вещество промывали 5 мл холодной DI H2O. Раствор центрифугировали при 2800 об/мин в течение 15 минут перед тем, как декантировать H2O. Таким же образом осуществляли еще две промывки H2O. Образец затем промывали 10 мл стандартного холодного EtOH 200 и центрифугировали при 2800 об/мин в течение 15 минут перед тем, как декантировать EtOH. Таким же образом осуществляли еще две промывки EtOH перед тем, как образец сушили под вакуумом в течение двух суток.
9.7.2. Получение Zn-комплекса R-типа конъюгата IN105 с протамином при нейтральном рН
Раствор IN105 в концентрации приблизительно 10 г/л доводили 10% HCl до конечного рН примерно 3. Жидкий фенол (49,5 мкл) добавляли к реакции объемом 15 мл. Затем к реакции добавляли 60 мкл 10%-ного раствора ZnCl2 с последующим добавлением 7,5 мг протамина. рН доводили концентрированным гидроксидом аммония до рН 7,00. Реакцию оставляли стоять в течение трех суток при комнатной температуре, чтобы дать возможность образоваться твердому веществу.
Реакционную смесь переносили в центрифужную пробирку и центрифугировали при 2800 об/мин в течение 15 минут. Раствор декантировали, и твердое вещество промывали 5,0 мл холодной DI H2O.
Раствор центрифугировали при 2800 об/мин в течение 15 минут перед тем, как декантировать H2O. Таким же образом осуществляли еще две промывки H2O. Образец затем промывали 50 мл стандартного холодного EtOH 200 и центрифугировали при 2800 об/мин в течение 15 минут перед тем, как декантировать EtOH. Таким же образом осуществляли еще две промывки EtOH перед тем, как образец сушили под вакуумом в течение двух суток.
9.7.3. Получение кристаллического Zn-комплекса IN105 R-типа
Неочищенный раствор IN105 в концентрации 15 мг/мл, содержащий 25% органического вещества, доводили до рН 3,47 с помощью 1 М HCl. Твердый фенол плавили на водяной бане при 40-60°С, и 0,218 мл добавляли в реакционную колбу. Затем к реакции добавляли 0,4 мл 4%-ного подкисленного водного раствора ZnCl2. рН раствора доводили 1 М NaOH до конечного рН 6,6. Во время подведения рН отбирали аликвоты объемом 10 мл при следующих значениях рН: 4,8; 5,0; 5,2; 5,4; 5,6; 5,8; 6,0; 6,2; 6,4 и 6,6. Данные образцы оставляли стоять без перемешивания в течение 24 ч. Под микроскопом наблюдали иглоподобные кристаллы.
9.7.4. Получение кристаллического Zn-комплекса IN105 R-типа, содержащего 30% органического вещества
Свежий раствор соединения MPEG3-пропионилинсулина в концентрации 15 мг/мл приготавливали в 250 мМ аммоний-ацетатном буфере, и рН доводили до 2,81 с помощью 1 М HCl. К данному раствору добавляли 0,040 мл жидкого фенола и 4,25 мл 95% EtOH. Затем к этой реакционной смеси добавляли 0,400 мл 4%-ного подкисленного раствора ZnCl2. рН раствора корректировали от 3,7 до 5,4, используя 50% NH4OH, и отбирали аликвоты объемом 1 мл при каждом из следующих желательных значений рН: 4,0; 4,2; 4,4; 4,6; 4,8; 5,0; 5,2; 5,4. Образцы оставляли стоять без перемешивания в течение 24 ч. На снимках, полученных с помощью микроскопа через 24 ч, были видны иглообразные кристаллы (смотри фиг.5) для рН в диапазоне от 4,0 до 5,2.
9.7.5. Получение кристаллического Zn-комплекса IN105 R-типа в 100 мМ аммоний-ацетатном буфере (30, 20 и 10% EtOH)
Свежий раствор соединения MPEG3-пропионилинсулина в концентрации 15 мг/мл приготавливали в 100 мМ аммоний-ацетатном буфере, и рН доводили до 2,8 с помощью 5 М HCl. К данному раствору добавляли 0,040 мл жидкого фенола и 4,25 мл 95% EtOH. Затем к этой реакционной смеси добавляли 0,400 мл 4%-ного подкисленного раствора ZnCl2. рН раствора корректировали от 2,9 до 5,6 с помощью 5 М NH4OH и отбирали аликвоты объемом 0,5 мл при каждом из следующих желательных значений рН: 4,2; 4,4; 4,6; 4,8; 5,0; 5,2; 5,4; 5,6. Эти образцы оставляли стоять без перемешивания в течение 24 ч. На снимках, полученных с помощью микроскопа через 24 ч, были видны иглообразные кристаллы для рН в диапазоне от 4,4 до 4,8.
Свежий раствор соединения MPEG3-пропионилинсулина в концентрации 15 мг/мл приготавливали в 100 мМ аммоний-ацетатном буфере, и рН доводили до 2,8 с помощью 5 М HCl. К данному раствору добавляли 0,040 мл жидкого фенола и 95% EtOH, 2,25 мл. Затем к этой реакционной смеси добавляли 0,400 мл 4%-ного подкисленного раствора ZnCl2. рН раствора корректировали от 2,9 до 5,6 с помощью 5 М NH4OH и отбирали аликвоты объемом 0,5 мл при каждом из следующих желательных значений рН: 4,2; 4,4; 4,6; 4,8; 5,0; 5,2; 5,4; 5,6. Эти образцы оставляли стоять без перемешивания в течение 24 ч. На снимках, полученных с помощью микроскопа через 24 ч, были видны сферообразные кристаллы для рН в диапазоне от 4,8 до 5,4.
Свежий раствор соединения MPEG3-пропионилинсулина в концентрации 15 мг/мл приготавливали в 100 мМ аммоний-ацетатном буфере, и рН доводили до 2,8 с помощью 5 М HCl. К данному раствору добавляли 0,040 мл жидкого фенола и 1,15 мл 95% EtOH. Затем к этой реакционной смеси добавляли 0,400 мл 4%-ного подкисленного раствора ZnCl2. рН раствора корректировали от 2,8 до 5,6 с помощью 5 М NH4OH и отбирали аликвоты объемом 0,5 мл при каждом из следующих желательных значений рН: 4,2; 4,4; 4,6; 4,8; 5,0; 5,2; 5,4; 5,6. Эти образцы оставляли стоять без перемешивания в течение 24 ч. На снимках, полученных с помощью микроскопа через 24 ч, были видны иглообразные кристаллы для рН в диапазоне от 5,0 до 5,6.
9.7.6. Получение кристаллического Zn-комплекса IN105 R-типа в 20% органического вещества с 0,1 и 0,2% фенола
Свежий раствор соединения MPEG3-пропионилинсулина в концентрации 15 мг/мл приготавливали в 100 мМ аммоний-ацетатном буфере, и рН доводили до 3,0 с помощью 5 М HCl. К данному раствору добавляли 0,010 мл жидкого фенола и 2,5 мл 95% EtOH. Затем к этой реакционной смеси добавляли 0,400 мл 4%-ного подкисленного раствора ZnCl2. рН раствора корректировали от 3,2 до 5,6 с помощью 5 М NH4OH и отбирали аликвоты объемом 0,5 мл при каждом из следующих желательных значений рН: 4,2; 4,4; 4,6; 4,8; 5,0; 5,2; 5,4; 5,6. Эти образцы оставляли стоять без перемешивания в течение 24 ч. На снимках, полученных с помощью микроскопа через 24 ч, были видны сферообразные кристаллы для рН в диапазоне от 4,4 до 5,4.
Свежий раствор соединения MPEG3-пропионилинсулина в концентрации 15 мг/мл приготавливали в 100 мМ аммоний-ацетатном буфере, и рН доводили до 3,0 с помощью 5 М HCl. К данному раствору добавляли 0,020 мл жидкого фенола и 2,5 мл 95% EtOH. Затем к этой реакционной смеси добавляли 0,400 мл 4%-ного подкисленного раствора ZnCl2. рН раствора корректировали от 3,3 до 5,6 с помощью 5 М NH4OH и отбирали аликвоты объемом 0,5 мл при каждом из следующих желательных значений рН: 4,2; 4,4; 4,6; 4,8; 5,0; 5,2; 5,4; 5,6. Эти образцы оставляли стоять без перемешивания в течение 24 ч. На снимках, полученных с помощью микроскопа через 24 ч, были видны сферообразные кристаллы для рН в диапазоне от 4,4 до 5,2.
9.7.7. Получение кристаллического Zn-комплекса IN105 R-типа в масштабе 8,0 граммов при рН 4,8 и комнатной температуре
Свежий раствор соединения MPEG3-пропионилинсулина в концентрации 15 мг/мл приготавливали в 250 мМ аммоний-ацетатном буфере, и рН доводили до 2,0 с помощью 5 М HCl. К данному раствору добавляли 2,13 мл жидкого фенола и 225 мл 95% EtOH. Затем к этой реакционной смеси добавляли 21,3 мл 4%-ного подкисленного раствора ZnCl2. рН раствора доводили до 4,8 с помощью 5 М NH4OH. Раствор оставляли стоять без перемешивания в течение 24 ч перед тем, как собирать кристаллы. На снимках, полученных с помощью микроскопа, были видны иглообразные кристаллы при Т=0.
Кристаллы собирали путем распределения реакционной смеси в 6×250 мл центрифужных пробирок. Пробирки центрифугировали при 10000 об/мин в течение 8 минут при 10°С перед тем, как декантировать супернатант. Затем, перед объединением содержимого 6 пробирок в 2 пробирках, в каждую пробирку добавляли 10 мл холодной H2O. Процесс центрифугирования повторяли еще раз с холодной водой и два раза с холодным EtOH. Кристаллы затем сушили в настольном лиофилизаторе в течение 2 суток. Данный способ давал выход 93% (масс./масс.) относительно исходного вещества.
9.7.8. Получение кристаллического Zn-комплекса IN105 R-типа в масштабе 1,5 грамма при рН 4,8 и комнатной температуре
Свежий раствор соединения MPEG3-пропионилинсулина (IN105) приготавливали путем растворения 1,52 г твердого IN105 в 100 мл 250 мМ ацетата аммония, рН 7,5. Раствор доводили до рН 2,8 с помощью 5 М HCl / 5 М NH4OH. Твердый фенол плавили в теплой водяной бане при 40-60°С. В реакционную колбу добавляли 400 мкл жидкого фенола и 42,5 мл 95% EtOH. Затем в реакционную колбу добавляли 4 мл 4%-ного подкисленного водного ZnCl2. Полученный раствор затем доводили до рН 4,8 с помощью 5 М NH4OH. Реакцию затем оставляли стоять без перемешивания в течение 48 ч перед тем, как собирать кристаллы. Образование иглообразных кристаллов наблюдали через 21 час через микроскоп.
Кристаллы собирали путем распределения реакционной смеси в 4×50 мл центрифужные пробирки. Сначала пробирки центрифугировали при 1000 об/мин в течение 8 минут. Затем декантировали супернатант. Кристаллы в каждой из побирок промывали 1×5 мл аликвотой ледяной Н2О, затем центрифугировали при 3000 об/мин в течение 8 минут. Затем декантировали супернатант. Процедуру промывки/центрифугирования повторяли с 1×5 мл аликвотой ледяной H2O, затем с 1×5 мл аликвотой ледяного EtOH. Кристаллы затем сушили в вакуумном эксикаторе в течение ночи. Способ давал выход 73% (масс./масс.) относительно исходного вещества.
9.7.9. Получение кристаллического Zn-комплекса IN105 R-типа в масштабе 1,5 грамма при рН 4,4 и комнатной температуре
Свежий раствор соединения MPEG3-пропионилинсулина (IN105) приготавливали путем растворения 1,50 г твердого IN105 в 100 мл 250 мМ ацетата аммония, рН 7,5. Раствор доводили до рН 2,6 с помощью 5 М HCl. Твердый фенол плавили в теплой водяной бане при 40-60°С. В реакционную колбу добавляли 400 мкл жидкого фенола и 42,5 мл 95% EtOH. Затем в реакционную колбу добавляли 4 мл 4%-ного подкисленного водного ZnCl2. Полученный раствор затем доводили до рН 4,4 с помощью 5 М NH4OH. Реакцию затем оставляли стоять без перемешивания в течение 22 ч перед тем, как собирать кристаллы. Смесь из образующихся иглообразных кристаллов и осадка наблюдали через 2 ч через микроскоп. Данная реакционная смесь, по-видимому, становится полностью кристаллической через 21 час (при наблюдении через микроскоп).
Кристаллы собирали путем переноса реакционной смеси в 1×250 мл центрифужную пробирку. Сначала пробирку центрифугировали при 10000 об/мин в течение 8 минут. Затем декантировали супернатант. Кристаллы промывали 1×20 мл аликвотой ледяной H2O, затем центрифугировали при 10000 об/мин в течение 8 минут. Затем декантировали супернатант. Процедуру промывки/центрифугирования повторяли с 1×20 мл аликвотой ледяной Н2О, затем с 2×20 мл аликвотами ледяного EtOH и в конце с 1×20 мл аликвотой ледяной H2O. Кристаллы затем сушили в вакуумном эксикаторе в течение ночи. Способ давал выход 67% (масс./масс.) относительно исходного вещества.
9.7.10. Получение кристаллического Zn-комплекса IN105 R-типа в масштабе 8 граммов при рН 4,8 и комнатной температуре
Свежий раствор соединения MPEG3-пропионилинсулина (IN105) приготавливали путем растворения 7,98 г твердого IN105 в 533 мл 250 мМ ацетата аммония, рН 7,5. Раствор доводили до рН 2,4 с помощью 5 М HCl. Твердый фенол плавили в теплой водяной бане при 40-60°С. В реакционную колбу добавляли 2,13 мл жидкого фенола и 225 мл 95% EtOH. Затем в реакционную колбу добавляли 21,3 мл 4%-ного подкисленного водного ZnCl2. Полученный раствор затем доводили до рН 4,8 с помощью 5 М NH4OH. Реакцию затем оставляли стоять без перемешивания в течение 21 ч перед тем, как собирать кристаллы. Данная реакционная смесь, по-видимому, становится полностью кристаллической через 2 ч (при наблюдении через микроскоп).
Кристаллы собирали путем распределения реакционной смеси в 6×250 мл центрифужных пробирок. Сначала пробирки центрифугировали при 10000 об/мин в течение 8 минут. Затем декантировали супернатант. Кристаллы в каждой пробирке промывали 1×10 мл аликвотой ледяной H2O, затем центрифугировали при 10000 об/мин в течение 8 минут. Затем декантировали супернатант. Процедуру промывки/центрифугирования повторяли с 1×10 мл аликвотой ледяной H2O, затем с 2×10 мл аликвотами ледяного EtOH и в конце с 1×10 мл аликвотой ледяной H2O. Кристаллы затем сушили в вакуумном эксикаторе в течение 2 суток. Способ давал выход 87% (масс./масс.) относительно исходного вещества.
9.7.11. Получение кристаллического Zn-комплекса IN105 R-типа в масштабе 10,0 граммов при рН 4,8 и комнатной температуре
Свежий раствор соединения MPEG3-пропионилинсулина (IN105) приготавливали путем растворения 10,06 г твердого IN105 в 670 мл 250 мМ ацетата аммония, рН 7,5. Раствор доводили до рН 2,6 с помощью 5 М HCl. Твердый фенол плавили в теплой водяной бане при 40-60°С. В реакционную колбу добавляли 2,7 мл жидкого фенола и 285 мл 95% EtOH. Затем в реакционную колбу добавляли 27 мл 4%-ного подкисленного водного ZnCl2. Полученный раствор затем доводили до рН 4,8 с помощью 5 М NH4OH. Реакцию затем оставляли стоять без перемешивания в течение 21 ч перед тем, как собирать кристаллы. Данная реакционная смесь, по-видимому, становится полностью кристаллической через 2,5 ч (при наблюдении через микроскоп).
Кристаллы собирали путем распределения реакционной смеси в 6×250 мл центрифужных пробирок. Сначала пробирки центрифугировали при 10°С, 10000 об/мин в течение 8 минут. Затем декантировали супернатант. Кристаллы в каждой пробирке промывали 1×10 мл аликвотой ледяной Н2О и объединяли в 2×250 мл центрифужных пробирках, затем центрифугировали при 10°С, 10000 об/мин в течение 8 минут. Затем декантировали супернатант. Процедуру промывки/центрифугирования повторяли с 1×30 мл аликвотой ледяной H2O, затем с 2×30 мл аликвотами ледяного EtOH и в конце с 1×30 мл аликвотой ледяной H2O. Кристаллы затем сушили с помощью настольного лиофилизатора в течение 3 суток. Способ давал выход 89% (масс./масс.) относительно исходного вещества.
9.8. Получение и анализ кристаллического Zn-комплекса HIM2 с использованием органического растворителя
9.8.1. Получение Zn-комплексов HIM2 R-типа
Свежий раствор HIM2 в концентрации 15 мг/мл приготавливали в 250 мМ аммоний-ацетатном буфере, и рН доводили до 2,95 с помощью 5 М HCl. К данному раствору добавляли 40 мкл жидкого фенола и 3,5 мл 95% EtOH. Затем к этой реакционной смеси добавляли 600 мкл 4%-ного подкисленного раствора ZnCl2. рН раствора корректировали от 3,14 до 6,0 с помощью 5 М NH4OH и отбирали аликвоты объемом 500 мкл при каждом из следующих желательных значений рН: 4,2; 4,4 (смотри фиг.6А), 4,6; 4,8; 5,0; 5,2; 5,4 (смотри Фиг.6Б); 5,6; 5,8; 6,0. Эти образцы оставляли стоять без перемешивания в течение 24 ч. На снимках, полученных с помощью микроскопа через 24 ч, были видны иглообразные кристаллы при рН 4,4. При рН в диапазоне от 4,6 до 6,0 видны большие кристаллообразные твердые частицы различных форм и размеров.
Свежий раствор HIM2 в концентрации 15 мг/мл приготавливали в 250 мМ аммоний-ацетатном буфере, и рН доводили до 2,95 с помощью 5 М HCl. К данному раствору добавляли 40 мкл жидкого фенола и 3,5 мл 95% EtOH. Затем к этой реакционной смеси добавляли 400 мкл 4%-ного подкисленного раствора ZnCl2. рН раствора корректировали от 3,22 до 6,0 с помощью 5 М NH4OH и отбирали аликвоты объемом 500 мкл при каждом из следующих желательных значений рН: 4,2; 4,4; 4,6; 4,8; 5,0; 5,2 (смотри Фиг.7А); 5,4; 5,6; 5,8; 6,0. Эти образцы оставляли стоять без перемешивания в течение 24 ч. На снимках, полученных с помощью микроскопа через 24 ч, видны кристаллообразные твердые частицы различных форм и размеров при рН 4,2-6,0.
Свежий раствор HIM2 в концентрации 15 мг/мл приготавливали в 250 мМ аммоний-ацетатном буфере, и рН доводили до 2,95 с помощью 5 М HCl. К данному раствору добавляли 40 мкл жидкого фенола и 3,5 мл 95% EtOH. Затем к этой реакционной смеси добавляли 200 мкл 4%-ного подкисленного раствора ZnCl2. рН раствора корректировали от 3,19 до 6,0 с помощью 5 М NH4OH и отбирали аликвоты объемом 500 мкл при каждом из следующих желательных значений рН: 4,2; 4,4; 4,6; 4,8; 5,0 (смотри фиг.7Б); 5,2; 5,4; 5,6; 5,8; 6,0. Эти образцы оставляли стоять без перемешивания в течение 24 ч. На снимках, полученных с помощью микроскопа через 24 ч, были видны кристаллообразные твердые частицы различных форм и размеров при рН 4,4-4,6. При рН в диапазоне от 4,8 до 5,2 видны более однородные иглообразные кристаллы.
Свежий раствор HIM2 в концентрации 15 мг/мл приготавливали в 250 мМ аммоний-ацетатном буфере, и рН доводили до 2,95 с помощью 5 М HCl. К данному раствору добавляли 40 мкл жидкого фенола и 2,6 мл 95% EtOH. Затем к этой реакционной смеси добавляли 600 мкл 4%-ного подкисленного раствора ZnCl2. рН раствора корректировали от 3,04 до 6,0 с помощью 5 М NH4OH и отбирали аликвоты объемом 500 мкл при каждом из следующих желательных значений рН: 4,2; 4,4; 4,6; 4,8; 5,0; 5,2; 5,4 (смотри фиг.8А); 5,6; 5,8; 6,0. Эти образцы оставляли стоять без перемешивания в течение 24 ч. На снимках, полученных с помощью микроскопа через 24 ч, были видны плоские, похожие на снежинки, кристаллы при рН 4,6-5,4.
Свежий раствор HIM2 в концентрации 15 мг/мл приготавливали в 250 мМ аммоний-ацетатном буфере, и рН доводили до 2,95 с помощью 5 М HCl. К данному раствору добавляли 40 мкл жидкого фенола и 2,6 мл 95% EtOH. Затем к этой реакционной смеси добавляли 400 мкл 4%-ного подкисленного раствора ZnCl2. рН раствора корректировали от 3,05 до 6,0 с помощью 5 М NH4OH и отбирали аликвоты объемом 500 мкл при каждом из следующих желательных значений рН: 4,2; 4,4; 4,6; 4,8; 5,0 (смотри Фиг.8Б); 5,2; 5,4; 5,6; 5,8; 6,0. Эти образцы оставляли стоять без перемешивания в течение 24 ч. На снимках, полученных с помощью микроскопа через 24 ч, были видны иглообразные кристаллы при рН 5,0, кристаллообразные твердые частицы при рН 5,2, и плоские, похожие на снежинки, кристаллы при рН 5,4.
Rxn 6 Свежий раствор HIM2 в концентрации 15 мг/мл приготавливали в 250 мМ аммоний-ацетатном буфере, и рН доводили до 2,95 с помощью 5 М HCl. К данному раствору добавляли 40 мкл жидкого фенола и 2,6 мл 95% EtOH. Затем к этой реакционной смеси добавляли 200 мкл 4%-ного подкисленного раствора ZnCl2. рН раствора корректировали от 3,09 до 6,0 с помощью 5 М NH4OH и отбирали аликвоты объемом 500 мкл при каждом из следующих желательных значений рН: 4,2; 4,4; 4,6; 4,8 (смотри фиг.9А); 5,0; 5,2; 5,4; 5,6; 5,8; 6,0. Эти образцы оставляли стоять без перемешивания в течение 24 ч. На снимках, полученных с помощью микроскопа через 24 ч, были видны иглообразные кристаллы и кристаллообразное твердое вещество при рН 4,8-5,6.
Свежий раствор HIM2 в концентрации 15 мг/мл приготавливали в 250 мМ аммоний-ацетатном буфере, и рН доводили до 2,76 с помощью 5 М HCl. К данному раствору добавляли 40 мкл жидкого фенола и 4,25 мл 95% EtOH. Затем к этой реакционной смеси добавляли 250 мкл 4%-ного подкисленного раствора ZnCl2. рН раствора корректировали от 2,97 до 5,8 с помощью 5 М NH4OH и отбирали аликвоты объемом 500 мкл при каждом из следующих желательных значений рН: 4,4; 4,6; 4,8; 5,0; 5,2; 5,4; 5,6; 5,8. Эти образцы оставляли стоять без перемешивания в течение 24 ч. На снимках, полученных с помощью микроскопа через 24 ч, было видно образование кристаллообразного осадка при рН 4,6-5,8.
Свежий раствор HIM2 в концентрации 15 мг/мл приготавливали в 250 мМ аммоний-ацетатном буфере, и рН доводили до 2,76 с помощью 5 М HCl. К данному раствору добавляли 40 мкл жидкого фенола и 4,25 мл 95% EtOH. Затем к этой реакционной смеси добавляли 200 мкл 4%-ного подкисленного раствора ZnCl2. рН раствора корректировали от 3,06 до 5,8 с помощью 5 М NH4OH и отбирали аликвоты объемом 500 мкл при каждом из следующих желательных значений рН: 4,4; 4,6; 4,8; 5,0; 5,2; 5,4 (смотри фиг.9Б); 5,6; 5,8. Эти образцы оставляли стоять без перемешивания в течение 24 ч. На снимках, полученных с помощью микроскопа через 24 ч, было видно образование кристаллообразного осадка при рН 4,6-5,6.
Свежий раствор HIM2 в концентрации 15 мг/мл приготавливали в 250 мМ аммоний-ацетатном буфере, и рН доводили до 2,76 с помощью 5 М HCl. К данному раствору добавляли 40 мкл жидкого фенола и 4,25 мл 95% EtOH. Затем к этой реакционной смеси добавляли 150 мкл 4%-ного подкисленного раствора ZnCl2. рН раствора корректировали от 3,09 до 5,8 с помощью 5 М NH4OH и отбирали аликвоты объемом 500 мкл при каждом из следующих желательных значений рН: 4,4; 4,6; 4,8; 5,0; 5,2; 5,4; 5,6; 5,8. Эти образцы оставляли стоять без перемешивания в течение 24 ч. На снимках, полученных с помощью микроскопа через 24 ч, были видны кристаллы различных размеров и форм при рН 5,0-5,2.
Свежий раствор HIM2 в концентрации 15 мг/мл приготавливали в 250 мМ аммоний-ацетатном буфере, и рН доводили до 2,76 с помощью 5 М HCl. К данному раствору добавляли 40 мкл жидкого фенола и 4,25 мл 95% EtOH. Затем к этой реакционной смеси добавляли 100 мкл 4%-ного подкисленного раствора ZnCl2. рН раствора корректировали от 3,09 до 5,8 с помощью 5 М NH4OH и отбирали аликвоты объемом 500 мкл при каждом из следующих желательных значений рН: 4,4; 4,6; 4,8; 5,0 (смотри фиг.10А); 5,2; 5,4 (смотри фиг.10Б); 5,6; 5,8. Эти образцы оставляли стоять без перемешивания в течение 24 ч. На снимках, полученных с помощью микроскопа через 24 ч, были видны иглообразные кристаллы при рН 5,0, и кристаллическое вещество различных форм и размеров при рН 5,2-5,6.
Свежий раствор HIM2 в концентрации 15 мг/мл приготавливали в 250 мМ аммоний-ацетатном буфере, и рН доводили до 2,76 с помощью 5 М HCl. К данному раствору добавляли 20 мкл жидкого фенола и 4,25 мл 95% EtOH. Затем к этой реакционной смеси добавляли 250 мкл 4%-ного подкисленного раствора ZnCl2. рН раствора корректировали от 3,08 до 5,8 с помощью 5 М NH4OH и отбирали аликвоты объемом 500 мкл при каждом из следующих желательных значений рН: 4,4; 4,6; 4,8; 5,0; 5,2; 5,4; 5,6; 5,8. Эти образцы оставляли стоять без перемешивания в течение 24 ч. На снимках, полученных с помощью микроскопа через 24 ч, было видно образование кристаллообразного осадка при рН 4,8-5,8.
Свежий раствор HIM2 в концентрации 15 мг/мл приготавливали в 250 мМ аммоний-ацетатном буфере, и рН доводили до 2,76 с помощью 5 М HCl. К данному раствору добавляли 20 мкл жидкого фенола и 4,25 мл 95% EtOH. Затем к этой реакционной смеси добавляли 200 мкл 4%-ного подкисленного раствора ZnCl2. рН раствора корректировали от 3,05 до 5,8 с помощью 5 М NH4OH и отбирали аликвоты объемом 500 мкл при каждом из следующих желательных значений рН: 4,4; 4,6; 4,8; 5,0; 5,2; 5,4; 5,6; 5,8. Эти образцы оставляли стоять без перемешивания в течение 24 ч. На снимках, полученных с помощью микроскопа через 24 ч, были видны очень маленькие кристаллообразные твердые частицы.
Свежий раствор HIM2 в концентрации 15 мг/мл приготавливали в 250 мМ аммоний-ацетатном буфере, и рН доводили до 2,76 с помощью 5 М HCl. К данному раствору добавляли 20 мкл жидкого фенола и 4,25 мл 95% EtOH. Затем к этой реакционной смеси добавляли 200 мкл 4%-ного подкисленного раствора ZnCl2. рН раствора корректировали от 3,05 до 5,8 с помощью 5 М NH4OH и отбирали аликвоты объемом 500 мкл при каждом из следующих желательных значений рН: 4,4; 4,6; 4,8; 5,0; 5,2; 5,4; 5,6; 5,8. Эти образцы оставляли стоять без перемешивания в течение 24 ч. На снимках, полученных с помощью микроскопа через 24 ч, были видны очень маленькие кристаллообразные твердые частицы.
Свежий раствор HIM2 в концентрации 15 мг/мл приготавливали в 250 мМ аммоний-ацетатном буфере, и рН доводили до 2,76 с помощью 5 М HCl. К данному раствору добавляли 20 мкл жидкого фенола и 4,25 мл 95% EtOH. Затем к этой реакционной смеси добавляли 100 мкл 4%-ного подкисленного раствора ZnCl2. рН раствора корректировали от 3,06 до 5,8 с помощью 5 М NH4OH и отбирали аликвоты объемом 500 мкл при каждом из следующих желательных значений рН: 4,4; 4,6; 4,8; 5,0; 5,2; 5,4; 5,6; 5,8. Эти образцы оставляли стоять без перемешивания в течение 24 ч. На снимках, полученных с помощью микроскопа через 24 ч, были видны очень маленькие кристаллообразные твердые частицы.
9.9. Сокристаллизация HIM2 и IN105c цинком
9.9.1. Получение сокристаллизованных Zn-комплексов HIM2 и IN105 R-типа
9.9.2. 50:50 (HIM2.IN105)
Свежий раствор HIM2 и IN105 приготавливали путем растворения 37,3 мг HIM2 и 36,4 мг IN105 в 4 мл 250 мМ ацетата аммония, рН 7,5. Раствор доводили до рН 2,84 с помощью 5 М HCl. Твердый фенол плавили в теплой водяной бане при 40-60°С. В реакционную колбу добавляли 16 мкл жидкого фенола и 1,75 мл 95% EtOH. Затем в реакционную колбу добавляли 80 мкл 4%-ного подкисленного водного ZnCb. Затем рН раствора корректировали от 3,19 до 5,60 с помощью 5 М NH4OH и отбирали аликвоты объемом 0,500 мл при каждом из следующих желательных значений рН: 4,4; 4,6; 4,8; 5,0; 5,2; 5,4 и 5,6. Эти образцы оставляли стоять без перемешивания в течение 4 ч. На снимках, полученных с помощью микроскопа через 4 ч, видны кристаллы различных размеров и форм в диапазоне рН от 4,4 до 5,6.
Свежий раствор HIM2 и IN105 приготавливали путем растворения 37,1 мг HIM2 и 35,9 мг IN105 в 4 мл 250 мМ ацетата аммония, рН 7,5. Раствор доводили до рН 3,03 с помощью 5 М HCl. Твердый фенол плавили в теплой водяной бане при 40-60°С. В реакционную колбу добавляли 16 мкл жидкого фенола и 1,75 мл 95% EtOH. Затем в реакционную колбу добавляли 40 мкл 4%-ного подкисленного водного ZnCl2. Затем рН раствора корректировали от 3,38 до 5,60 с помощью 5 М NH4OH и отбирали аликвоты объемом 0,500 мл при каждом из следующих желательных значений рН: 4,4; 4,6; 4,8; 5,0 (смотри фиг.11А); 5,2 (смотри фиг.11Б); 5,4 и 5,6 (смотри фиг.12А). Эти образцы оставляли стоять без перемешивания в течение 4 ч. На снимках, полученных с помощью микроскопа через 4 ч, видны в основном короткие иглообразные кристаллы в диапазоне рН от 4,6 до 5,6.
9.9.3. 70:30 (HIM2:IN105)
Свежий раствор HIM2 и IN105 приготавливали путем растворения 53,4 мг HIM2 и 23,2 мг IN105 в 4 мл 250 мМ ацетата аммония, рН 7,5. Раствор доводили до рН 2,62 с помощью 5 М HCl. Твердый фенол плавили в теплой водяной бане при 40-60°С. В реакционную колбу добавляли 16 мкл жидкого фенола и 1,75 мл 95% EtOH. Затем в реакционную колбу добавляли 80 мкл 4%-ного подкисленного водного ZnCl2. Затем рН раствора корректировали от 3,02 до 5,60 с помощью 5 М NH4OH и отбирали аликвоты объемом 0,500 мл при каждом из следующих желательных значений рН: 4,4; 4,6; 4,8; 5,0; 5,2 (смотри фиг.12Б); 5,4 и 5,6. Эти образцы оставляли стоять без перемешивания в течение 1 ч. На снимках, полученных с помощью микроскопа через 1 час, видно образование кристаллообразного осадка различных размеров и форм в диапазоне рН от 4,6 до 5,6.
Свежий раствор HIM2 и IN105 приготавливали путем растворения 53,6 мг HIM2 и 23,5 мг IN105 в 4 мл 250 мМ ацетата аммония, рН 7,5. Раствор доводили до рН 2,89 с помощью 5 М HCl. Твердый фенол плавили в теплой водяной бане при 40-60°С. В реакционную колбу добавляли 16 мкл жидкого фенола и 1,75 мл 95% EtOH. Затем в реакционную колбу добавляли 40 мкл 4%-ного подкисленного водного ZnCl2. Затем рН раствора корректировали от 3,28 до 5,60 с помощью 5 М NH4OH и отбирали аликвоты объемом 0,500 мл при каждом из следующих желательных значений рН: 4,4; 4,6; 4,8; 5,0; 5,2 (смотри фиг.13А); 5,4 и 5,6. Эти образцы оставляли стоять без перемешивания в течение 1 ч. На снимках, полученных с помощью микроскопа через 1 час, видно образование в основном кристаллообразного осадка различных размеров и форм в диапазоне рН от 4,6 до 4,8 и большое количество коротких иглообразных кристаллов в диапазоне рН от 5,0 до 5,4.
9.9.4. 30:70 (HIM2:IN105)
Свежий раствор HIM2 и IN105 приготавливали путем растворения 23,3 мг HIM2 и 54,7 мг IN105 в 4 мл 250 мМ ацетата аммония, рН 7,5. Раствор доводили до рН 2,84 с помощью 5 М HCl. Твердый фенол плавили в теплой водяной бане при 40-60°С. В реакционную колбу добавляли 16 мкл жидкого фенола и 1,75 мл 95% EtOH. Затем в реакционную колбу добавляли 80 мкл 4%-ного подкисленного водного ZnCl2. Затем рН раствора корректировали от 3,27 до 5,60 с помощью 5 М NH4OH и отбирали аликвоты объемом 0,500 мл при каждом из следующих желательных значений рН: 4,4; 4,6; 4,8; 5,0; 5,2; 5,4 и 5,6. Эти образцы оставляли стоять без перемешивания в течение 1 ч. На снимках, полученных с помощью микроскопа через 1 час, видно образование в основном кристаллообразного осадка различных размеров и форм в диапазоне рН от 4,4 до 5,0 и несколько иглообразных кристаллов в диапазоне рН от 5,2 до 5,6.
Свежий раствор HIM2 и IN105 приготавливали путем растворения 24,8 мг HIM2 и 54,9 мг IN105 в 4 мл 250 мМ ацетата аммония, рН 7,5. Раствор доводили до рН 3,09 с помощью 5 М HCl. Твердый фенол плавили в теплой водяной бане при 40-60°С. В реакционную колбу добавляли 16 мкл жидкого фенола и 1,75 мл 95% EtOH. Затем в реакционную колбу добавляли 40 мкл 4%-ного подкисленного водного ZnCl2. Затем рН раствора корректировали от 3,47 до 5,60 с помощью 5 М NH4OH и отбирали аликвоты объемом 0,500 мл при каждом из следующих желательных значений рН: 4,4; 4,6; 4,8; 5,0; 5,2; 5,4 и 5,6 (смотри фиг.13Б). Эти образцы оставляли стоять без перемешивания в течение 1 ч. На снимках, полученных с помощью микроскопа через 1 час, видно образование в основном кристаллообразного осадка различных размеров и круглой формы в диапазоне рН от 4,4 до 5,0 и кристаллы различных форм и размеров в диапазоне рН от 5,2 до 5,6.
9.9.5. Получение сокристаллизованных Zn-комплексов HIM2 и IN105 R-типа
9.9.6. 50:50 (HIM2:IN105)
Свежий раствор HIM2 и IN105 приготавливали путем растворения 37,4 мг HIM2 и 35,9 мг IN105 в 4 мл 250 мМ ацетата аммония, рН 7,5. Раствор доводили до рН 2,60 с помощью 5 М HCl. Твердый фенол плавили в теплой водяной бане при 40-60°С. В реакционную колбу добавляли 16 мкл жидкого фенола и 1,75 мл 95% EtOH. Затем в реакционную колбу добавляли 40 мкл 4%-ного подкисленного водного ZnCl2. Затем рН раствора корректировали от 2,15 до 5,60 с помощью 5 М NH4OH и отбирали аликвоты объемом 0,500 мл при каждом из следующих желательных значений рН: 4,4; 4,6; 4,8; 5,0; 5,2; 5,4 и 5,6. Эти образцы оставляли стоять без перемешивания в течение 24 ч. На снимках, полученных с помощью микроскопа через 24 ч, были видны кристаллические твердые частицы различных форм и размеров при рН=4,6-5,6.
9.9.7. 70:30 (HIM2.IN105)
Свежий раствор HIM2 и IN105 приготавливали путем растворения 57,0 мг HIM2 и 24,5 мг IN105 в 4 мл 250 мМ ацетата аммония, рН 7,5. Раствор доводили до рН 2,43 с помощью 5 М HCl. Твердый фенол плавили в теплой водяной бане при 40-60°С. В реакционную колбу добавляли 16 мкл жидкого фенола и 1,75 мл 95% EtOH. Затем в реакционную колбу добавляли 40 мкл 4%-ного подкисленного водного ZnCl2. Затем рН раствора корректировали от 2,92 до 5,60 с помощью 5 М NH4OH и отбирали аликвоты объемом 0,500 мл при каждом из следующих желательных значений рН: 4,4; 4,6 (смотри фиг.14А); 4,8; 5,0; 5,2; 5,4 и 5,6. Эти образцы оставляли стоять без перемешивания в течение 24 ч. На снимках, полученных с помощью микроскопа через 24 ч, были видны иглообразные кристаллы при рН 5,0-5,2.
9.9.8. 30:70 (HIM2:IN105)
Свежий раствор HIM2 и IN105 приготавливали путем растворения 24,1 мг HIM2 и 53,8 мг IN105 в 4 мл 250 мМ ацетата аммония, рН 7,5. Раствор доводили до рН 2,35 с помощью 5 М HCl. Твердый фенол плавили в теплой водяной бане при 40-60°С. В реакционную колбу добавляли 16 мкл жидкого фенола и 1,75 мл 95% EtOH. Затем в реакционную колбу добавляли 40 мкл 4%-ного подкисленного водного ZnCl2. Затем рН раствора корректировали от 2,60 до 5,60 с помощью 5 М NH4OH и отбирали аликвоты объемом 0,500 мл при каждом из следующих желательных значений рН: 4,4; 4,6; 4,8; 5,0 (смотри фиг.14Б); 5,2; 5,4 и 5,6. Эти образцы оставляли стоять без перемешивания в течение 24 ч. На снимках, полученных с помощью микроскопа через 24 ч, были видны иглообразные кристаллы при рН 5,0-5,2.
9.9.9. Получение сокристаллизованных Zn-комплексов HIM2 и инсулина человека R-типа
9.9.10. 50:50 (HIM2:инсулин)
Свежий раствор HIM2 и инсулина приготавливали путем растворения 39,2 мг HIM2 и 38,7 мг инсулина в 4 мл 250 мМ ацетата аммония, рН 7,5. Раствор доводили до рН 2,53 с помощью 5 М HCl. Твердый фенол плавили в теплой водяной бане при 40-60°С. В реакционную колбу добавляли 16 мкл жидкого фенола и 1,75 мл 95% EtOH. Затем в реакционную колбу добавляли 40 мкл 4%-ного подкисленного водного ZnCl2. Затем рН раствора корректировали от 2,82 до 5,60 с помощью 5 М NH4OH и отбирали аликвоты объемом 0,500 мл при каждом из следующих желательных значений рН: 4,4; 4,6; 4,8; 5,0; 5,2 (смотри фиг.15А); 5,4 и 5,6. Эти образцы оставляли стоять без перемешивания в течение 24 ч. На снимках, полученных с помощью микроскопа через 24 ч, было видно кристаллообразное твердое вещество различных форм и размеров при рН 5,2 и 5,4. Большое количество очень маленьких иглообразных кристаллов наблюдали при рН 5,6.
9.9.11. 70:30 (HIM2:инсулин)
Свежий раствор HIM2 и инсулина приготавливали путем растворения 56,5 мг HIM2 и 20,2 мг инсулина в 4 мл 250 мМ ацетата аммония, рН 7,5. Раствор доводили до рН 3,23 с помощью 5 М HCl. Твердый фенол плавили в теплой водяной бане при 40-60°С. В реакционную колбу добавляли 16 мкл жидкого фенола и 1,75 мл 95% EtOH. Затем в реакционную колбу добавляли 40 мкл 4%-ного подкисленного водного ZnCl2. Затем рН раствора корректировали от 2,82 до 5,60 с помощью 5 М NH4OH и отбирали аликвоты объемом 0,500 мл при каждом из следующих желательных значений рН: 4,4; 4,6; 4,8; 5,0; 5,2; 5,4 и 5,6. Эти образцы оставляли стоять без перемешивания в течение 24 ч. На снимках, полученных с помощью микроскопа через 24 ч, было видно кристаллообразное твердое вещество различных форм и размеров при рН 5,2 и 5,6.
9.9.12. 30:70 (HIM2:инсулин)
Свежий раствор HIM2 и инсулина приготавливали путем растворения 21,8 мг HIM2 и 49,2 мг инсулина в 4 мл 250 мМ ацетата аммония, рН 7,5. Раствор доводили до рН 3,23 с помощью 5 М HCl. Твердый фенол плавили в теплой водяной бане при 40-60°С. В реакционную колбу добавляли 16 мкл жидкого фенола и 1,75 мл 95% EtOH. Затем в реакционную колбу добавляли 40 мкл 4%-ного подкисленного водного ZnCl2. Затем рН раствора корректировали от 2,93 до 5,60 с помощью 5 М NH4OH и отбирали аликвоты объемом 0,500 мл при каждом из следующих желательных значений рН: 4,4; 4,6; 4,8; 5,0; 5,2; 5,4 (смотри фиг.15Б) и 5,6. Эти образцы оставляли стоять без перемешивания в течение 24 ч. На снимках, полученных с помощью микроскопа через 24 ч, были видны плоские, похожие на снежинки, кристаллы при рН 4,8. При рН 5,0 получали смесь иглообразных и похожих на снежинки кристаллов. При рН 5,2-5,6 наблюдали большое количество очень маленьких иглообразных кристаллов.
10. Растворимость в воде Zn-комплексов
Двести мкл 0,1 М фосфатно-солевого буфера (PBS, фильтрованный, рН=7,4) добавляли в коническую реакционную побирку объемом 1 мл. В данную пробирку медленно добавляли небольшое количество образца до тех пор, пока не наблюдали насыщения. Этот раствор периодически встряхивали. После достижения насыщения данную пробирку помещали в маленькую центрифужную пробирку, и образец центрифугировали при 2000 об/мин в течение 3 минут при комнатной температуре. После центрифугирования из супернатанта отбирали 10 мкл образца и разбавляли в 490 мкл буфера (0,1 М PBS). Разведенный образец анализировали посредством ВЭЖХ для определения его концентрации.
11. Примеры ферментативной устойчивости in vitro Zn-комплексов IN105
Конъюгаты соединений инсулинов (IN105) присутствовали в 10 мМ натрий-фосфатном буфере (рН приблизительно 7,4), и их концентрацию определяли посредством ВЭЖХ (растворы разводили буфером, чтобы можно было сделать эквимолярные сравнения исходного соединения и конъюгатов, примерно 0,6 мг/мл). Лиофилизированный фермент химотрипсин ресуспендировали в 1 мМ HCl до концентрации примерно 7,53 ед./мл. Аликвоту каждого образца объемом 1,53 мл добавляли в пробирки для образцов и в контрольные пробирки объемом 0,850 мл. Образцы тестировали с двойной повторностью вместе с четырьмя контрольными пробирками на образец. Аликвоты инкубировали при 37°С в термомиксере в течение 15 минут. Затем в каждую пробирку с образцом добавляли 17 мкл фермента химотрипсина. В каждую контрольную пробирку добавляли пять мкл HCl. Сразу после этих добавлений из пробирок с образцами и контрольных пробирок брали 200 мкл и помещали в 50 мкл 1% TFA (трифторуксусной кислоты), аликвоты которой предварительно вносили в центрифужные пробирки. Данный образец служит в качестве Т=0.
Отбор образцов соединения инсулина (без цинка), IN105 (без цинка) и соединения инсулина (стандартного соединения инсулина) повторяли в следующие моменты времени: 0, 2, 5, 8, 12, 15 и 30 минут. Процедуру контроля повторяли в следующие моменты времени: 0, 8, 15, 30 минут. Для образцов Т-типа и R-типа процедуру повторяли в следующие моменты времени: 0, 5, 8, 12, 30, 40 и 60 минут. Процедуру контроля для Zn-комплексов повторяли в следующие моменты времени: 0, 12, 40 и 60 минут. Образцы хранили при -20°С до тех пор, пока не осуществляли их анализ посредством ВЭЖХ. ВЭЖХ осуществляли для определения процента деградации относительно соответствующего Т=0 минут для каждого перевара. Строили график зависимости натурального логарифма количества оставшегося образца, выраженного в процентах, от времени, и для каждого перевара осуществляли линейную регрессию. Период полужизни вычисляли с помощью уравнения t½=-0,693/угловой коэффициент.
Результаты для концентрации белка 0,6 мг/мл
12. Примеры препаратов
12.1.1. Примеры жидких препаратов
12.1.1. Буферный раствор для Zn-HIM2 R6-типа. Исследование буфера
** рН может быть подведено 10% HCl и/или 10% гидроксида натрия
12.1.2. Приготовление перорального жидкого разбавителя для препарата с каприновой кислотой/лауриновой кислотой
Приблизительно 60% требуемого объема стерильной воды переносили в подходящий контейнер. В данный контейнер добавляли соответствующее количество (как указанно в таблице ниже) трометамина, троламина, безводной лимонной кислоты и гранул гидроксида натрия и хорошо перемешивали до тех пор, пока не растворятся. Температуру доводили до 21-25°С (или до комнатной температуры) и измеряли рН этой жидкости. В случае необходимости, рН доводили до 7,7-7,9 с помощью 1 н. гидроксида натрия или 1 н. соляной кислоты. Затем температуру доводили до 45-50°С путем нагревания на плитке и поддерживали эту температуру. Затем к теплому раствору добавляли каприновую кислоту и перемешивали до тех пор, пока она не растворится. Температуру доводили до 21-25°С (или до комнатной температуры) и измеряли рН этой жидкости. При необходимости, рН доводили до 7,7-7,9 с помощью 1 н. гидроксида натрия или 1 н. соляной кислоты. Затем раствор перемешивали в течение 5 минут. Добавляли соответствующее количество стерильной воды до объема, равного 100% требуемого объема, и хорошо перемешивали.
IN105, HIM2 или ZnHIM2 взвешивали в количествах, необходимых для достижения соответствующей концентрации в исследованиях доз, например взвешивали 1 мг IN105 (белка) и смешивали с 1 мл препарата с получением 1 мг/мл IN105 в препарате.
12.1.3. Приготовление перорального жидкого разбавителя для препарата с олеиновой кислотой/лауриновой кислотой/холатом
Приготавливали пероральный жидкий препарат Zn HIM2 R-типа, содержащий компоненты, приведенные в таблице ниже:
Приготавливали пероральные жидкие образцы, содержащие 1 мг/мл белкового эквивалента Zn HIM2 R-типа (ZnHIM2-R). ZnHIM2-R извлекали из морозильной камеры (-20°С), помещали в эксикатор и оставляли нагреваться до комнатной температуры. 1 мг/мл белкового эквивалента ZnHIM2-R приготавливали в растворе перорального жидкого разбавителя следующим образом. Взвешивали 6,4 мг ZnHIM2-R. Затем 5,0 мл перорального жидкого разбавителя переносили в контейнер и мягко вращали для перемешивания. Растворение происходило в течение приблизительно 45 минут. Полученный раствор представлял собой суспензию (мутную по виду). Перед дозированием раствор мягко вращали в течение 60 секунд, чтобы убедиться, что раствор является гомогенным раствором. Для ZnHIM2-R содержание белка составляло 78,6%, 1 мг/мл белкового эквивалента, количество = 5 мл. Количество ZnHIM2-R=(1 мг/мл)/(0,786)×(5,0 мл)=6,4 мг. Концентрация ZnHIM2-R=(6,4 мг)/(5,0 мл)=1,28 мг/мл (эквивалентно 1 мг/мл в пересчете на содержание белка).
12.1.4. Приготовление жидких препаратов с каприновой кислотой
Приблизительно 60% требуемого объема стерильной воды переносили в подходящий контейнер. В данный контейнер добавляли соответствующее количество (как указанно в таблице ниже) трометамина, троламина, безводной лимонной кислоты и гранул гидроксида натрия и хорошо перемешивали до тех пор, пока не растворятся. Температуру доводили до 21-25°С (или до комнатной температуры) и измеряли рН этой жидкости. В случае необходимости, рН доводили до 7,7-7,9 с помощью 1 н. гидроксида натрия или 1 н. соляной кислоты. Затем температуру доводили до 45-50°С путем нагревания на плитке и поддерживали эту температуру. Затем к теплому раствору добавляли каприновую кислоту и перемешивали до тех пор, пока каприновая кислота не растворится. Температуру доводили до 21-25°С (или до комнатной температуры) и измеряли рН этой жидкости. При необходимости, рН доводили до 7,7-7,9 с помощью 1 н. гидроксида натрия или 1 н. соляной кислоты. Затем раствор перемешивали в течение 5 минут. Добавляли соответствующее количество стерильной воды до объема, равного 100% требуемого объема, и хорошо перемешивали.
IN105 взвешивали в количествах, необходимых для достижения соответствующей концентрации в исследованиях дозирования, например взвешивали 1 мг IN105 (белка) и смешивали с 1 мл препарата с получением 1 мг/мл IN105 в препарате.
12.1.5. Капрат и/или лаурат в жидких препаратах с фосфатным буфером
Приготовление 100 мМ натрий-фосфатного буфера, рН 7,8 или 8,2. 1,17 грамма моногидрата мононатрия фосфата переносили в колбу объемом 1 л. Добавляли приблизительно 500 мл стерильной воды и хорошо перемешивали до тех пор, пока не растворится. Затем добавляли 24,58 грамма гептагидрата фосфата натрия двухосновного и хорошо перемешивали до тех пор, пока не растворится. Разбавляли до соответствующего объема стерильной водой и хорошо перемешивали. Фильтровали через 0,22 мкм фильтр. рН доводили до 7,8 или 8,2 с помощью 1 н. HCl или 1 н. NaOH.
Для IN105 60% соответствующего объема фосфатного буфера рН 7,8 или 8,2 переносили в подходящий контейнер. Затем добавляли капрат в количестве, рассчитанном таким образом, чтобы получить концентрацию 3% (масс./об. конечного раствора), и хорошо перемешивали до тех пор, пока не растворится. Затем доводили рН до 7,8 или 8,2 с помощью 1 н. HCl или 1 н. NaOH. Разбавляли до соответствующего объема (например до 100 мл) фосфатным буфером рН 7,8 или 8,2.
Для BN-054 взвешивали 400 граммов 100 мМ натрий-фосфатного буфера, рН 7,8, в подходящем контейнере. Добавляли 9,7 грамма капрата натрия и 11,1 грамма лаурата натрия и хорошо перемешивали до тех пор, пока не растворятся. Добавляли соответствующее количество 100 мМ натрий-фосфатного буфера, рН 7,8, так чтобы чистый вес был равен 500 граммам.
12.1.6. Жидкий препарат с аргинином или троламином
Приготовление 100 мМ натрий-фосфатного буфера, рН 7,8. 1,17 грамма моногидрата мононатрия фосфата переносили в колбу объемом 1 л. Добавляли приблизительно 500 мл стерильной воды и хорошо перемешивали до тех пор, пока не растворится. Затем добавляли 24,58 грамма гептагидрата фосфата натрия двухосновного и хорошо перемешивали до тех пор, пока не растворится. Разбавляли до соответствующего объема стерильной водой и хорошо перемешивали. Фильтровали через 0,22 мкм фильтр. рН доводили до 7,8 с помощью 1 н. HCl или 1 н. NaOH.
60% соответствующего объема фосфатного буфера рН 7,8 переносили в подходящий контейнер. В данный контейнер добавляли соответствующее количество (как указанно в таблице ниже) аргинина или троламина и хорошо перемешивали до тех пор, пока не растворятся. Затем добавляли капрат в количестве, рассчитанном таким образом, чтобы получить концентрацию 3% (масс./об. конечного раствора), и хорошо перемешивали до тех пор, пока не растворится. Затем доводили рН до 7,8 с помощью 1 н. HCl или 1 н. NaOH. Разбавляли до соответствующего объема (например до 100 мл) фосфатным буфером рН 7,8.
IN105 взвешивали в количестве, необходимом для достижения соответствующей концентрации в исследованиях дозирования, например взвешивали 1 мг IN105 (белка) и смешивали с 1 мл препарата с получением 1 мг/мл IN105 в препарате.
12.1.7. Жидкий препарат с каприловой кислотой
Приблизительно 60% требуемого объема стерильной воды переносили в подходящий контейнер. В данный контейнер добавляли соответствующее количество (как указанно в таблице ниже) трометамина, троламина, безводной лимонной кислоты и гранул гидроксида натрия и хорошо перемешивали до тех пор, пока не растворятся. Температуру доводили до 21-25°С (или до комнатной температуры) и измеряли рН этой жидкости. В случае необходимости, рН доводили до 7,7-7,9 с помощью 1 н. гидроксида натрия или 1 н. соляной кислоты. Затем температуру доводили до 45-50°С путем нагревания на плитке и поддерживали эту температуру. Затем к теплому раствору добавляли каприловую кислоту и перемешивали до тех пор, пока каприловая кислота не растворится. Температуру доводили до 21-25°С (или до комнатной температуры) и измеряли рН этой жидкости. При необходимости, рН доводили до 7,7-7,9 с помощью 1 н. гидроксида натрия или 1 н. соляной кислоты. Затем раствор перемешивали в течение 5 минут. Добавляли соответствующее количество стерильной воды до объема, равного 100% требуемого объема, и хорошо перемешивали.
IN105 взвешивали в количестве, необходимом для достижения соответствующей концентрации в исследованиях дозирования, например взвешивали 1 мг IN105 (белка) и смешивали с 1 мл препарата с получением 1 мг/мл IN105 в препарате.
12.1.8. Жидкий препарат с линолевой кислотой
Приготовление 100 мМ натрий-фосфатного буфера, рН 7,8. 1,17 грамма моногидрата мононатрия фосфата переносили в колбу объемом 1 л. Добавляли приблизительно 500 мл стерильной воды и хорошо перемешивали до тех пор, пока не растворится. Затем добавляли 24,58 грамма гептагидрата фосфата натрия двухосновного и хорошо перемешивали до тех пор, пока не растворится. Разбавляли до соответствующего объема стерильной водой и хорошо перемешивали. Фильтровали через 0,22 мкм фильтр. рН доводили до 7,8 с помощью 1 н. HCl или 1 н. NaOH.
60% соответствующего объема фосфатного буфера рН 7,8 переносили в подходящий контейнер. Затем добавляли натриевую соль линолевой кислоты в количестве, рассчитанном таким образом, чтобы получить концентрацию 3% (масс./об. конечного раствора), и хорошо перемешивали до тех пор, пока не растворится. Затем доводили рН до 7,8 с помощью 1 н. HCl или 1 н. NaOH. Разбавляли до соответствующего объема (например до 100 мл) фосфатным буфером рН 7,8.
12.1.9. Приготовление жидких препаратов с каприновой кислотой и лауриновой кислотой
Приблизительно 60% требуемого объема стерильной воды переносили в подходящий контейнер. В данный контейнер добавляли соответствующее количество (как указанно в таблице ниже) трометамина, троламина, безводной лимонной кислоты и гранул гидроксида натрия и хорошо перемешивали до тех пор, пока не растворятся. Температуру доводили до 21-25°С (или до комнатной температуры) и измеряли рН этой жидкости. В случае необходимости, рН доводили до 7,7-7,9 с помощью 1 н. гидроксида натрия или 1 н. соляной кислоты. Затем температуру доводили до 45-50°С путем нагревания на плитке и поддерживали эту температуру. Затем к теплому раствору добавляли каприновую кислоту и/или лауриновую кислоту и перемешивали до тех пор, пока каприновая кислота и/или лауриновая кислота не растворялись. Температуру доводили до 21-25°С (или до комнатной температуры) и измеряли рН этой жидкости. При необходимости, рН доводили до 7,7-7,9 с помощью 1 н. гидроксида натрия или 1 н. соляной кислоты. Затем раствор перемешивали в течение 5 минут. Добавляли соответствующее количество стерильной воды до объема, равного 100% требуемого объема, и хорошо перемешивали.
IN105 взвешивали в количестве, необходимом для достижения соответствующей концентрации в исследованиях дозирования, например взвешивали 1 мг IN105 (белка) и смешивали с 1 мл препарата с получением 1 мг/мл IN105 в препарате.
12.2. Примеры твердых лекарственных препаратов
12.2.1. Приготовление и профиль растворения твердого лекарственного препарата с капратом/лауратом с использованием Nobex-IN105-[854]
Приблизительно 58 мг капрата натрия, 57 мг лаурата натрия, 286 мг маннита, 30 мг натрия крахмал гликолята и 6 мг (белка) Nobex-IN105 переносили на кусок бумаги для взвешивания и тщательно перемешивали. Смесь переносили в пресс и прессовали под давлением 350 psi (2413,25 кПа) с получением таблетки.
Препарат Nobex-IN105-[854] в твердой лекарственной форме (таблетки), 58 мг капрата и 57 мг лаурата на таблетку
Тестирование растворения осуществляли с использованием USP аппарата с 2 секциями для растворения. В качестве среды использовали воду, скорость вращения составляла 50 об/мин, и объем среды составлял 500 мл. Образцы для растворения анализировали посредством ВЭЖХ с использованием градиентной системы. Подвижные фазы представляли собой воду с 0,1% TFA (подвижная фаза А) и ацетонитрил с 0,1% TFA (подвижная фаза Б). Использовали следующий градиент: 0 минут - 100% подвижная фаза А, 11 минут - 65% подвижная фаза А, 15 минут - 20% подвижная фаза А, 16 минут - 20% подвижная фаза А, 17 минут - 100% подвижная фаза А. Длина волны составляла 214 нм, колонка представляла собой С18 (150×2 мм). Нижеследующие таблицы и графики суммируют данные по растворению, полученные при тестировании растворения таблетированного препарата Nobex-Zn-IN105 [854], содержащего 6 мг Zn-IN105 (белка), 286 мг маннита, 58 мг капрата натрия, 57 мг лаурата натрия и 30 мг натрия крахмал гликолята (Explotab):
Суммарные данные для профиля растворения таблеток Nobex-Zn-IN105 [854], % растворенного IN105
Суммарные данные для профиля растворения таблеток Nobex-Zn-IN105 [854], % растворенного капрата
Суммарные данные для профиля растворения таблеток Nobex-Zn-IN105 [854], % растворенного лаурата
12.2.2. Приготовление препарата в твердой лекарственной форме (таблетка), 143 мг капрата и 140 мг лаурата на таблетку
Приготовление препарата Nobex-IN105 [856]
Приблизительно 143 мг капрата натрия, 140 мг лаурата натрия, 150 мг маннита, 30 мг натрия крахмал гликолята и 6 мг (белка) Nobex-IN105 переносили на кусок бумаги для взвешивания и тщательно перемешивали. Смесь переносили в пресс и прессовали под давлением 350 psi (2413,25 кПа) с получением таблетки.
Препарат Nobex-IN105-r856l в твердой лекарственной форме (таблетки), 143 мг капрата и 140 мг лаурата на таблетку
Тестирование растворения осуществляли с использованием USP аппарата с 2 секциями для растворения. В качестве среды использовали воду, скорость вращения составляла 50 об/мин, и объем среды составлял 500 мл. Образцы для растворения анализировали посредством ВЭЖХ с использованием градиентной системы. Подвижные фазы представляли собой воду с 0,1% TFA (подвижная фаза А) и ацетонитрил с 0,1% TFA (подвижная фаза Б). Использовали следующий градиент: 0 минут - 100% подвижная фаза А, 11 минут - 65% подвижная фаза А, 15 минут - 20% подвижная фаза А, 16 минут - 20% подвижная фаза А, 17 минут - 100% подвижная фаза А. Длина волны составляла 214 нм, колонка представляла собой С18 (150×2 мм). Нижеследующие таблицы и графики суммируют данные по растворению, полученные при тестировании растворения таблеток Nobex-Zn-IN105, содержащих 6 мг Zn-IN105 (белка), 150 мг маннита, 143 мг капрата натрия, 140 мг лаурата натрия и 30 мг натрия крахмал гликолята (Explotab):
Суммарные данные для профиля растворения таблеток Nobex-Zn-IN105 [856], % растворенного IN105
Суммарные данные для профиля растворения таблеток Nobex-Zn-IN105 [856], % растворенного капрата
Суммарные данные для профиля растворения таблеток Nobex-Zn-IN105 [856], % растворенного лаурата
12.2.3. Приготовление препарата в твердой лекарственной форме (таблетка), 143 мг капрата на таблетку
Приготовление препарата Nobex-IN105 [859]
Приблизительно 143 мг капрата натрия, 150 мг маннита, 30 мг натрия крахмал гликолята и 6 мг (белка) Nobex-IN105 переносили на кусок бумаги для взвешивания и тщательно перемешивали. Данную смесь переносили в пресс и прессовали под давлением 350 psi (2413,25 кПа) с получением таблетки.
Препарат Nobex-IN105-r8591 в твердой лекарственной форме (таблетки), 143 мг капрата на таблетку
Тестирование растворения осуществляли с использованием USP аппарата с 2 секциями для растворения. В качестве среды использовали воду, скорость вращения составляла 50 об/мин, и объем среды составлял 500 мл. Образцы для растворения анализировали посредством ВЭЖХ с использованием градиентной системы. Подвижные фазы представляли собой воду с 0,1% TFA (подвижная фаза А) и ацетонитрил с 0,1% TFA (подвижная фаза Б). Использовали следующий градиент: 0 минут - 100% подвижная фаза А, 11 минут - 65% подвижная фаза А, 15 минут - 20% подвижная фаза А, 16 минут - 20% подвижная фаза А, 17 минут - 100% подвижная фаза А. Длина волны составляла 214 нм, колонка представляла собой С18 (150×2 мм). Нижеследующие таблицы и графики суммируют данные по растворению, полученные при тестировании растворения таблеток Nobex-Zn-IN105, содержащих 6 мг Zn-IN105 (белка), 150 мг маннита, 143 мг капрата натрия и 30 мг натрия крахмал гликолята (Explotab):
Суммарные данные для профиля растворения таблеток Nobex-Zn-IN105 [859], % растворенного IN105
Суммарные данные для профиля растворения таблеток Nobex-Zn-IN105 [859], % растворенного капрата
12.2.4. Приготовление препарата в твердой лекарственной форме (таблетка), 286 мг капрата на таблетку
Приготовление препарата Nobex-IN105 [860]
Приблизительно 286 мг капрата натрия, 150 мг маннита, 30 мг натрия крахмал гликолята и 6 мг (белка) Nobex-IN105 переносили на кусок бумаги для взвешивания и тщательно перемешивали. Данную смесь переносили в пресс и прессовали под давлением 350 psi (2413,25 кПа) с получением таблетки.
Препарат Nobex-IN105-[860] в твердой лекарственной форме (таблетки), 286 мг капрата на таблетку
Тестирование растворения осуществляли с использованием USP аппарата с 2 секциями для растворения. В качестве среды использовали воду, скорость вращения составляла 50 об/мин, и объем среды составлял 500 мл. Образцы для растворения анализировали посредством ВЭЖХ с использованием градиентной системы. Подвижные фазы представляли собой воду с 0,1% TFA (подвижная фаза А) и ацетонитрил с 0,1% TFA (подвижная фаза Б). Использовали следующий градиент: 0 минут - 100% подвижная фаза А, 11 минут - 65% подвижная фаза А, 15 минут - 20% подвижная фаза А, 16 минут - 20% подвижная фаза А, 17 минут - 100% подвижная фаза А. Длина волны составляла 214 нм, колонка представляла собой С18 (150×2 мм). Нижеследующие таблицы и графики суммируют данные по растворению, полученные при тестировании растворения таблеток Nobex-Zn-IN105, содержащих 6 мг Zn-IN105 (белка), 150 мг маннита, 286 мг капрата натрия и 30 мг натрия крахмал гликолята (Explotab):
Суммарные данные для профиля растворения таблеток Nobex-Zn-IN105 [860], % растворенного IN105
Суммарные данные для профиля растворения таблеток Nobex-Zn-IN105 [860], % растворенного капрата
12.2.5. Приготовление препарата в твердой лекарственной форме (таблетка), 100 мг капрата на таблетку
Приготовление препарата Nobex-IN105 [861]
Приблизительно 100 мг капрата натрия, 150 мг маннита, 25 мг натрия крахмал гликолята и 6 мг (белка) Nobex-IN105 переносили на кусок бумаги для взвешивания и тщательно перемешивали. Данную смесь переносили в пресс и прессовали под давлением 350 psi (2413,25 кПа) с получением таблетки.
Препарат Nobex-IN105-[861] в твердой лекарственной форме (таблетки), 100 мг капрата на таблетку
Тестирование растворения осуществляли с использованием USP аппарата с 2 секциями для растворения. В качестве среды использовали воду, скорость вращения составляла 50 об/мин, и объем среды составлял 500 мл. Образцы для растворения анализировали посредством ВЭЖХ с использованием градиентной системы. Подвижные фазы представляли собой воду с 0,1% TFA (подвижная фаза А) и ацетонитрил с 0,1% TFA (подвижная фаза Б). Использовали следующий градиент: 0 минут - 100% подвижная фаза А, 11 минут - 65% подвижная фаза А, 15 минут - 20% подвижная фаза А, 16 минут - 20% подвижная фаза А, 17 минут - 100% подвижная фаза А. Длина волны составляла 214 нм, колонка представляла собой С18 (150×2 мм). Нижеследующие таблицы и графики суммируют данные по растворению, полученные при тестировании растворения таблеток Nobex-Zn-IN105, содержащих 6 мг Zn-IN105 (белка), 150 мг маннита, 100 мг капрата натрия и 25 мг натрия крахмал гликолята (Explotab):
Суммарные данные для профиля растворения таблеток Nobex-Zn-IN105 [861], % растворенного IN105
Суммарные данные для профиля растворения таблеток Nobex-Zn-IN105 [861], % растворенного капрата
12.2.6. Приготовление препарата в твердой лекарственной форме (таблетка), 150 мг капрата на таблетку
Приготовление препарата Nobex-IN105 [862]
Приблизительно 150 мг капрата натрия, 150 мг маннита, 25 мг кроскармеллозы натрия и 6 мг (белка) Nobex-IN105 переносили на кусок бумаги для взвешивания и тщательно перемешивали. Данную смесь переносили в пресс и прессовали под давлением 350 psi (2413,25 кПа) с получением таблетки.
Препарат Nobex-IN105-[862] в твердой лекарственной форме (таблетки), 150 мг капрата на таблетку
Тестирование растворения осуществляли с использованием USP аппарата с 2 секциями для растворения. В качестве среды использовали воду, скорость вращения составляла 50 об/мин, и объем среды составлял 500 мл. Образцы для растворения анализировали посредством ВЭЖХ с использованием градиентной системы. Подвижные фазы представляли собой воду с 0,1% TFA (подвижная фаза А) и ацетонитрил с 0,1% TFA (подвижная фаза Б). Использовали следующий градиент: 0 минут - 100% подвижная фаза А, 11 минут - 65% подвижная фаза А, 15 минут - 20% подвижная фаза А, 16 минут - 20% подвижная фаза А, 17 минут - 100% подвижная фаза А. Длина волны составляла 214 нм, колонка представляла собой С18 (150×2 мм). Нижеследующие таблицы и графики суммируют данные по растворению, полученные при тестировании растворения таблеток Nobex-Zn-IN105, содержащих 6 мг Zn-IN105 (белка), 150 мг маннита, 150 мг капрата натрия и 25 мг кроскармеллозы натрия (Explotab):
Суммарные данные для профиля растворения таблеток Nobex-Zn-IN105 [862], % растворенного IN105
Суммарные данные для профиля растворения таблеток Nobex-Zn-IN105 [862], % растворенного капрата
13. Примеры in vitro устойчивости к ферменту
Конъюгаты соединений инсулинов (HIM2) присутствовали в 10 мМ натрий-фосфатном буфере (рН приблизительно 7,4), и их концентрацию определяли посредством ВЭЖХ (данные растворы разводили буфером для того, чтобы можно было осуществить эквимолярные сравнения исходного соединения и конъюгатов, примерно 0,6 мг/мл). Лиофилизированный фермент химотрипсин ресуспендировали в 1 мМ HCl до концентрации 7,53 ед./мл. Аликвоту каждого образца объемом 1,53 мл добавляли в пробирки для образцов и в контрольные пробирки объемом 0,850 мл. Образцы тестировали с двойной повторностью вместе с четырьмя контрольными пробирками на образец. Аликвоты инкубировали при 37°С в термомиксере в течение 15 минут. Затем в каждую пробирку с образцом добавляли 17 мкл фермента химотрипсина. В каждую контрольную пробирку добавляли пять мкл HCl. Сразу после добавления из пробирок с образцами и из контрольных пробирок брали 200 мкл и помещали в 50 мкл 1% TFA, аликвоты которой предварительно вносили в центрифужные пробирки. Этот образец служит в качестве Т=0.
Процедуру отбора образцов инсулина (без цинка), HIM2 (без цинка) и инсулина (стандартного соединения инсулина) повторяли в следующие моменты времени: 0, 2, 5, 8, 12, 15 и 30 минут. Процедуру контроля повторяли в следующие моменты времени: 0, 8, 15, 30 минут. Для образцов Т-типа и R-типа эту процедуру повторяли в следующие моменты времени: 0, 5, 8, 12, 30, 40 и 60 минут. Процедуру контроля для Zn-комплексов повторяли в следующие моменты времени: 0, 12, 40 и 60 минут. Образцы хранили при -20°С до тех пор, пока не осуществляли их анализ посредством ВЭЖХ. ВЭЖХ осуществляли для определения процента деградации относительно соответствующего Т=0 минут для каждого перевара. Строили график зависимости натурального логарифма количества оставшегося образца, выраженного в процентах, от времени, и для каждого перевара осуществляли линейную регрессию. Период полужизни вычисляли с помощью уравнения t½=-0,693/угловой коэффициент.
Результаты для концентрации белка 0,6 мг/мл
Результаты для концентрации белка 0,3 мг/мл
14. Примеры in vivo
14.1. Пролонгированный анализ глюкозы в крови у мышей (MBGA)
Шесть групп для парного дозирования из 5 самцов мышей CF-1 (Charles River Laboratories; 25-30 г) получали подкожные инъекции либо конъюгата соединения инсулина (тестируемый продукт), либо рекомбинантного инсулина человека. Тестируемый продукт растворяли в фосфатном буфере (0,01 М, рН приблизительно 7,4), содержащем 0,1% (масс./масс.) бычьего сывороточного альбумина, и вводили в количестве 100; 66,6; 43,3; 30; 20 и 13,3 мкг/кг. Инсулин растворяли в фосфатном буфере (0,01 М, рН приблизительно 7,4), содержащем 0,1% (масс./масс.) бычьего сывороточного альбумина, и вводили в количестве 50; 33,3; 21,7; 15; 10 и 6,7 мкг/кг. После того, как животные получали подкожную дозу в углубление, образованное бедром и пахом, их возвращали в клетки на 30 минут при комнатной температуре и затем быстро анестезировали и брали кровь. Образцы крови собирали в пробирки с гепарином для анализа глюкозы. Если анализ глюкозы откладывался, то пробирки хранили в ледяной воде и перед анализом снова нагревали до комнатной температуры.
Глюкозу в плазме крови измеряли с помощью глюкометра (например One Touch® Basic; Lifescan), который калибровали в начале каждого дня, когда его использовали, в соответствии с инструкциями производителя. Затем активность конъюгата соединения инсулина рассчитывали относительно стандартной кривой, которую строили для ответа на рекомбинантный инсулин человека. Вычисления основаны на предположении, что рекомбинантный инсулин человека имеет активность 27,4 МЕ/мг.
Результаты приведены на фиг.16-20. На фиг.16 показаны MBGA-профили биологической активности HIM2. На фиг.17 показаны MBGA-профили биологической активности продукта соединения инсулина Zn-HIM2 R-типа. На фиг.18 показаны MBGA-профили биологической активности продукта соединения инсулина Zn-HIM2 Т-типа. На фиг.19 показаны MBGA-профили биологической активности продукта соединения инсулина Zn-HIM2 с протамином. На фиг.20 показано снижение глюкозы при действии протаминового комплекса R-типа через 30 и 90 минут после введения дозы. Эти результаты показывают, что биологическая активность HIM2 уменьшается незначительно в результате образования комплекса с Zn++. Протаминовый комплекс R-типа (смотри фиг.17 [7]) показывает более значительное снижение глюкозы через 30 и 90 минут.
Дополнительно, на фиг.21-24 показаны MBGA-профили биологической активности IN-186, IN-192, IN-190, IN-191, IN-189, IN-178, IN-193, IN-194, IN-185, IN-196 и IN-197, имеющих следующие структуры:
В1 моноконъюгат, IN-186:
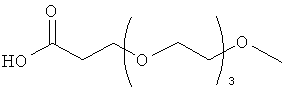
В29 моноконъюгат, IN-194:
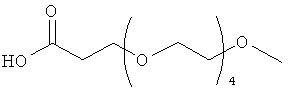
В29 моноконъюгат, IN-197:
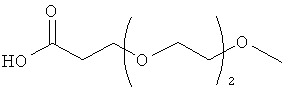
В29 моноконъюгат, IN-178:
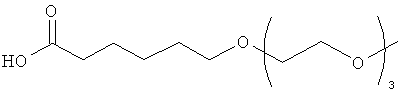
В29 моноконъюгат, IN-190:
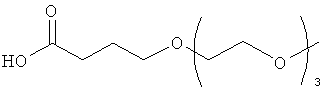
В29 моноконъюгат, IN-196:
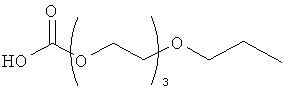
В29 моноконъюгат.IN-191:
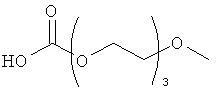
В29 моноконъюгат, IN-189:
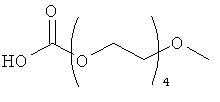
14.2. Исследования с фиксированным уровнем глюкозы у собак
14.2.1. Первоначальные исследования HIM2
Хирургическая подготовка собак (n=3 или 6) заключалась в установке катетера в бедренной артерии (анастезия изофлураном). Животным давали возможность восстановиться в течение 16-17 дней, после чего им не давали корм в течение ночи и проводили исследования на животных, находящихся в сознании. После 60-минутного периода установления равновесия следовал контрольный период 20 минут, после которого через рот вводили лекарство. Соединение инсулин Zn++-HIM2 R-типа тестировали в буферном растворе, приготовленном, как показано в Примере Ошибка! Ссылка не найдена. Кроме того, комплексы R-типа и NPH-типа тестировали в пероральном жидком препарате, который содержит каприновую кислоту и лауриновую кислоту, приготовленном, как показано в Примере Ошибка! Ссылка не найдена.
Все три анализируемых образца тестировали с использованием только одного дозового уровня (дозовый уровень определяли на основе предварительных экспериментальных результатов). Уровень глюкозы в плазме крови затем фиксировали на эугликемическом уровне путем инфузии D-20 через ножную вену в течение 4 ч. Для измерения глюкозы, соединения инсулина и С-пептида образцы крови (4 мл) отбирали в моменты времени -20, 0, 5, 10, 20, 30, 45, 90, 120, 180 и 240 минут. Образцы артериальной крови получали по мере необходимости, чтобы поддерживать фиксированный уровень глюкозы в плазме. В итоге в каждом эксперименте отбирали 72 мл крови.
Осуществляли следующие измерения: скорости инфузии глюкозы, концентрации соединения инсулина, концентрации С-пептида и уровней С-пептида в плазме (что позволяло оценить эндогенное высвобождение соединения инсулина). Скорость инфузии глюкозы, необходимая для поддержания эугликемии, является показателем действия соединения инсулина.
После эксперимента свободный конец катетера скрывали в подкожной ткани и собакам давали возможность восстановиться в течение двух недель перед другим исследованием, в котором использовали другой анализируемый продукт. Животных рандомизировали для введения дозы и использовали в целом 3 раза. Общее число собак равнялось 6.
Данные результаты приведены на фиг.25 и 26.
14.2.2. Первоначальные исследования IN105
Данное исследование проводили на шести (6) нечистокровных собаках, не получавших корм в течение ночи, находящихся в сознании, которые до этого получали корм, содержащий 34% белка, 46% углеводов, 14,5% жира и 5,5% волокон по массе на сухой вес. Каждое животное имело силиконовый катетер, вставленный в бедренную артерию, как описано ранее (1), приблизительно за три недели до эксперимента. В день эксперимента катетер извлекали из подкожного углубления под местной анестезией. Тестируемый продукт: Nobex-IN105 (Lot.# KJ-173-095 & KJ-173-116) присутствовал в пероральном препарате жирных кислот (Nobex-IN-[753]-040422) в концентрации 1,0 мг/мл. Каждая собака получала 0,25 мг/кг пероральной дозы Nobex-IN105 (объем вводимых доз составлял 0,25 мл/кг; 1,0 мг/мл). Nobex-IN105 давали в момент времени t=0, и глюкозу (D-20) вводили через головную вену для поддержания эугликемии. Образцы артериальной крови отбирали для измерения инсулина и глюкозы, как описано ранее (1). После завершения эксперимента артериальный катетер скрывали в подкожной ткани, где он был размещен во время первичной хирургической манипуляции.
Во время эксперимента у одной из собак была рвота сразу после введения дозы, и была введена только часть дозы. Поэтому результаты данного эксперимента представлены с учетом данных, полученных на этой собаке, и без них.
После перорального введения Nobex-IN105 уровни инсулина в артериальной плазме повышались у всех шести собак, включая выброс (Oral-2i). Средний уровень артериального инсулина повышался от 6,0±1,4 мкед./мл (6,3±1,7 мкед./мл, n=5) до максимального значения 109,4±31,4 мкед./мл (127,8±30,1 мкед./мл, n=5) через 10 минут после введения и затем падал, так что к 150 минуте у всех собак уровни инсулина вернулись к базальному уровню (Фиг.27 и 28).
Эугликемию поддерживали путем инфузии глюкозы. Скорость инфузии глюкозы, необходимая для поддержания эугликемии, была наиболее высокой у животных, у которых наблюдался больший подъем артериального инсулина (Фиг.27 и 28). Средняя площадь под кривой скорости инфузии глюкозы (AUC0-240) составляла 578,5±144,5 мг/кг/мин (669,4±137,7 мг/кг/мин, n=5)
14.2.3. Твердые препараты
Скрининговые исследования препаратов с использованием модели с фиксированным уровнем глюкозы проводили на нечистокровных собаках, не получавших корм в течение ночи, находящихся в сознании, которые до этого получали корм, содержащий 34% белка, 46% углеводов, 14,5% жира и 5,5% волокон по массе на сухой вес. Каждое животное имело силиконовый катетер, вставленный в бедренную артерию, как описано ранее (1), приблизительно за три недели до эксперимента. В день эксперимента катетер извлекали из подкожного углубления под местной анестезией. Анализируемый продукт содержал 1,0 мг/мл Zn IN105 в различных жидких препаратах или 5-6 мг IN105 на капсулу или таблетку. Каждая собака в каждом эксперименте получала пероральную жидкую дозу приблизительно 0,25 мг/кг или капсулу или таблетку, содержащую 5 или 6 мг IN105 два раза подряд: первый раз в момент времени t=0 и второй раз в момент времени t=120 минут. Для поддержания эугликемии через головную вену вводили глюкозу (D-20). В некоторых случаях, когда после введения дозы эффект длился более 120 минут, время исследования удлиняли. Образцы артериальной крови отбирали для измерения инсулина, глюкозы и С-пептида, как описано ранее (1). После завершения эксперимента артериальный катетер возвращали обратно в подкожную ткань.
Препараты, как в виде раствора, так и в виде твердых лекарственных форм, приготавливали с использованием различных уровней жирных кислот, буферов, разбавителей и разрыхлителей. Чтобы свести к минимуму количество переменных, жидкие и твердые препараты содержали постоянные уровни IN105 и каждого эксципиента (капринового, лауринового, каприлового, миристинового и линолевого), соответственно, варьировали только относительные количества жирных кислот, буфера, разбавителей (маннита или микрокристаллической целлюлозы) и/или разрыхлителя (Explotab). Для каждого дозированного препарата проводили оценку и сравнение скорости инфузии глюкозы и данных по абсорбции IN105 (иммунореактивности инсулина плазмы).
Сначала осуществляли эксперименты для упрощения и улучшения оптимизированного жидкого препарата (ссылка 2) до жидкого препарата, который можно было бы легко превращать в твердую лекарственную форму. Это осуществляли путем замены свободной жирной кислоты на соответствующую натриевую соль (например, каприновую кислоту заменяли на капрат натрия), а также путем удаления буферных компонентов (лимонной кислоты, троламина, трометамина, гидроксида натрия), которые, как считали, больше не требуются. Кроме того, исследовали влияние других жирных кислот, таких как линолевая, каприловая и миристиновая кислоты, и аминокислоты аргинина на абсорбцию IN105.
После скрининга первоначальной группы протипов с использованием одной или двух собак на эксперимент, выбрали несколько препаратов-прототипов и тестировали их с использованием дополнительных собак для того, чтобы лучше определить вариабельность и согласованность действия препаратов на индивидуальных животных.
Разработали способ растворения, и исследования по растворению осуществляли на нескольких препаратах-кандидатах для оценки профилей растворения IN105 и содержания жирных кислот.
РЕЗУЛЬТАТЫ
В первоначальных экспериментах собаки, которым вводили IN105 в 3% (масс./об.) натриевой соли каприновой кислоты в фосфатном буфере без дополнительных эксципиентов, продемонстрировали (Фиг.29-30) похожий ответ при сравнении с оптимизированным жидким препаратом, содержащим 3% (масс./об.) каприновой кислоты в буфере троламин/лимонная кислота/трометамин/гидроксид натрия. Это показывает, что жирная кислота в форме натриевой соли ведет себя сравнительно так же, как в кислотной форме, и что дополнительные компоненты буфера не вносят вклад в данный препарат.
В отдельных исследованиях, где оценивали альтернативные жирные кислоты, заменяя 3% капрата либо 3% каприловой кислотой, либо 3% линолиновой кислотой, ни одна из данных альтернатив не показала значительных эффектов. Применение каприловой кислоты привело к необходимости в уровнях глюкозы от низких до средних, тогда как применение линолиновой кислоты не вызывало необходимости инфузии глюкозы, что свидетельствовало об отсутствии эффекта. Оба препарата продемонстрировали относительно низкие уровни артериального инсулина. Жидкий препарат, содержащий аргинин, не показал никаких преимуществ в отношении GIR (скорости инфузии глюкозы) или уровней IN105.
В предварительных исследованиях твердые препараты оценивали как в виде порошковой смеси, которой заполнены твердые желатиновые капсулы, так и в виде таблеток, прессованных вручную с использованием Carver Press. Капсулы с порошковой смесью, содержащей 6 мг IN105 (инсулиновый эквивалент, 0,25 мг/кг) и 57 мг капрата/57 мг лаурата, не продемонстрировали никаких значительных эффектов при первом введении вплоть до 120 минут, а при повторном введении наблюдался значительный эффект с GIR, согласованной с абсорбцией IN105, где уровни были намного выше базального уровня в период времени от 0 до 120 минут. Эти данные свидетельствуют о непостоянной, но потенциальной замедленной реакции на IN105 в капсульной лекарственной форме. Растворение только слегка замедляется капсулой, если сравнивать с таблетками, хотя корреляция in-vitrolin-vivo может быть небольшой или отсутствует.
В первоначальных скрининговых исследованиях таблеток-прототипов, таблетки, содержащие 6 мг IN105 и 150 мг маннита, 30 мг натрия крахмал гликолята с 143 мг капрата и 143 мг лаурата или без него, демонстрировали значительно более высокую GIR и абсорбцию IN105, чем такие же таблетки с 54 мг капрата и/или 54 мг лаурата (Фиг.31, 32 и 33). Это свидетельствует о надлежащем дозовом ответе на более высокие GIR и уровни IN105 относительно увеличивающихся уровней капрата и лаурата. Данные таблетки демонстрировали быстрый и согласованный GIR ответ во времени относительно капсул, наполненных IN105. Кроме того, таблетки с IN105 демонстрировали увеличение инсулина в артериальной плазме у всех собак, которым вводили дозу.
В серии заключительных исследований были выбраны три препарата таблеток-прототипов [препарат [856] содержал 143 мг капрата и 140 мг лаурата, [860] содержал 286 мг капрата, и [862] содержал 150 мг капрата] для оценки у 5 собак (три разных собаки для каждого препарата), чтобы определить согласованность действия (Фиг.34-37). Уровни инсулина в артериальной плазме повышались у всех трех собак и для всех 18 доз (3 собаки × 3 таблетки × 2 дозы на каждую) с соответствующим GIR-ответом после перорального введения 6 мг (инсулиновый эквивалент) IN105, приготовленного в виде таблеток, содержащих либо 150 мг или 280 мг капрата, либо 143 мг/140 мг капрата/лаурата (Фиг.31-32 и 38-40). Уровни С-пептида (нг/мл), для таблеток с 150 мг и 280 мг капрата, в среднем (n=2), показали снижение от первоначального уровня 0,30±0,05 до 0,22±0,02 во время первого введения дозы и от 0,1±0,05 до 0,02±0,0 при повторном введении дозы, и, для таблеток с 140 мг/140 мг капрата/лаурата, показали снижение от 0,21±0,05 до 0,05±0,02 во время первого введения дозы и от 0,18±0,05 до 0,18±0,1. Это указывает на подавление секреции С-пептида поджелудочной железой в результате введения экзогенного IN105.
Все три препарата таблеток-прототипов IN105 показали согласованные уровни абсорбции IN105 и полученной скорости инфузии глюкозы между дозами и для одной собаки и между собаками, в том числе в разные дни.
Во время заключительных исследований, в которых использовали группу из 6 собак, одна собака (собака #3) давала меньший ответ на все жидкие и твердые дозировки. Для более точного представления результатов данные представлены как с учетом результатов, полученных на собаке #3, так и без них. Данные, полученные на собаках, которые не получили полную дозу (плохое кормление через желудочный зонд, рвота и так далее) или имели эндогенный инсулин, исключены.
Изучение растворимости: типичные образцы таблеток и капсул тестировали на растворимость (описано выше).
Обсуждение
В данных исследованиях показано, что таблетка-прототип IN105, содержащая капрат или капрат и лаурат (натриевые соли) с маннитом и разрыхлитель натрия крахмал гликолят и содержащая 6 мг IN105 (приблизительно 0,25 мг/кг), доставляемая пероральным путем, вызывала значительное и согласованное повышение уровня инсулина в артериальной плазме, которое для сохранения эугликемии требует инфузии глюкозы.
Данные таблетки-прототипы приводили к таким уровням IN105 и скоростям GIR, которые являются по меньшей мере такими же хорошими или, возможно, лучшими, чем жидкие препараты, содержащие сравнимые уровни капрата или лаурата. Данные таблетки-прототипы сохраняют профиль абсорбции перорального жидкого препарата. Относительная пероральная биоэффективность выбранных препаратов таблеток-прототипов (например таблеток, содержащих 280 мг и 150 мг капрата, n=6, AUC для GIR=496±117 и 500±275), по-видимому, лучше, чем у жидких препаратов (например у жидкого препарата с 3% каприновой кислоты, n=5, AUC для GIR=182±92 и 198±119).
Эти данные свидетельствуют о том, что таблетки, содержащие капрат натрия в качестве единственной жирной кислоты вместе с маннитом и разрыхлителем, натрия крахмал гликолятом, были бы полезны для дальнейшей разработки твердых лекарственных форм для использования в клинических исследованиях. Эти данные также свидетельствуют о том, что уровни инсулина после перорального введения IN105 в выбранных таблетированных формах-прототипах с капратом (капрат натрия в количестве либо 150 мг, либо 286 мг) монотонно повышаются, с типичным Tmax около 20 минут после введения дозы и Cmax приблизительно 59,0±20,1 и 62,9±25,4 мкед./мл, для обеих доз. Уровни инсулина в плазме остаются повышенными и близкими к уровню Cmax в течение 10-15 минут и выше базальных уровней в течение 120 минут после каждой дозы. GIR, необходимая для поддержания эугликемии, при использовании данных таблеток достигала Tmax в момент времени или около 30-40 минут для обеих доз, и GIR Cmax достигала среднего значения 8,4±1,99 и 7,41±2,18 мг/кг/мин. Таблетированные лекарственные формы требовали более высокого GIR Cmax (7,4-8,4 против 4,5-5,4) и требовали инфузии глюкозы в течение более продолжительного периода (100-120 минут против 60-90 минут) для поддержания эугликемии, чем оптимизированный жидкий препарат.
При сравнении с уровнями инсулина в артериальной плазме исторического инсулина, вводимого SQ (подкожным) или ингаляционным путем, оказывается, что данные таблетки-прототипы обеспечивают максимальные уровни инсулина, схожие с теми, которые имеют место при доставке SQ или ингаляционным путем, и имеют профиль инсулина, сравнимый с профилем инсулина для инсулина, вводимого ингаляционным путем (Фиг.34-37).
Эксперименты с таблетками-прототипами свидетельствуют о том, что выбранные таблетки-прототипы подходят в качестве твердых лекарственных препаратов для дальнейшей оценки IN105, при этом дальнейшая разработка должна быть сфокусирована на создании клинического препарата, который может быть получен с использованием таблет-пресса.
Данное описание разделено на разделы с предметным указателем только для удобства при ссылке. Предполагается, что разделы и предметный указатель не ограничивают объем изобретения. Воплощения, описанные в данном изобретении, имеют цель проиллюстрировать многие аспекты и признаки изобретения, и предполагается, что они не ограничивают объем изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНСУЛИН-ОЛИГОМЕРНЫЕ КОНЪЮГАТЫ, ИХ ПРЕПАРАТЫ И ПРИМЕНЕНИЯ | 2005 |
|
RU2393168C2 |
| УЛУЧШЕННЫЕ ХЕЛАТНЫЕ КОНЪЮГАТЫ | 2002 |
|
RU2298012C2 |
| ТВЕРДАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ (ВАРИАНТЫ) И СПОСОБ КОНТРОЛЯ КОНЦЕНТРАЦИИ ГЛЮКОЗЫ С ЕЕ ПОМОЩЬЮ, СПОСОБ ПОЛУЧЕНИЯ ТВЕРДОЙ ФАРМАЦЕВТИЧЕСКОЙ КОМПОЗИЦИИ (ВАРИАНТЫ), ТАБЛЕТКА (ВАРИАНТЫ) И СПОСОБ ПОЛУЧЕНИЯ АМФОРНЫХ ЧАСТИЦ | 2008 |
|
RU2453332C2 |
| УСОВЕРШЕНСТВОВАННЫЕ КОНЪЮГАТЫ N4 ХЕЛАТООБРАЗУЮЩИХ АГЕНТОВ | 2005 |
|
RU2360701C2 |
| ПРОЛЕКАРСТВА, СОДЕРЖАЩИЕ КОНЪЮГАТ ИНСУЛИНА И ЛИНКЕРА | 2010 |
|
RU2574667C2 |
| КОМПОЗИЦИЯ ИНСУЛИНА ДЛИТЕЛЬНОГО ДЕЙСТВИЯ | 2010 |
|
RU2556340C2 |
| ПРОИЗВОДНЫЕ ОКСАДИАЗОЛОНА В КАЧЕСТВЕ АГОНИСТОВ PPAR-ДЕЛЬТА | 2005 |
|
RU2374243C2 |
| РЕЖИМЫ ДОЗИРОВАНИЯ ДЛЯ СОЕДИНЕНИЙ КЛАССА ЭХИНОКАНДИНОВ | 2013 |
|
RU2719579C2 |
| ПРОЛЕКАРСТВА, СОДЕРЖАЩИЕ КОНЪЮГАТ ЭКСЕНДИН-ЛИНКЕР | 2011 |
|
RU2593774C2 |
| ПРОЛЕКАРСТВА, СОДЕРЖАЩИЕ КОНЬЮГАТ ГИАЛУРОНОВОЙ КИСЛОТЫ, ЛИНКЕРА И ДВОЙНОГО АГОНИСТА GLP-1/ГЛЮКАГОНА | 2016 |
|
RU2719482C2 |



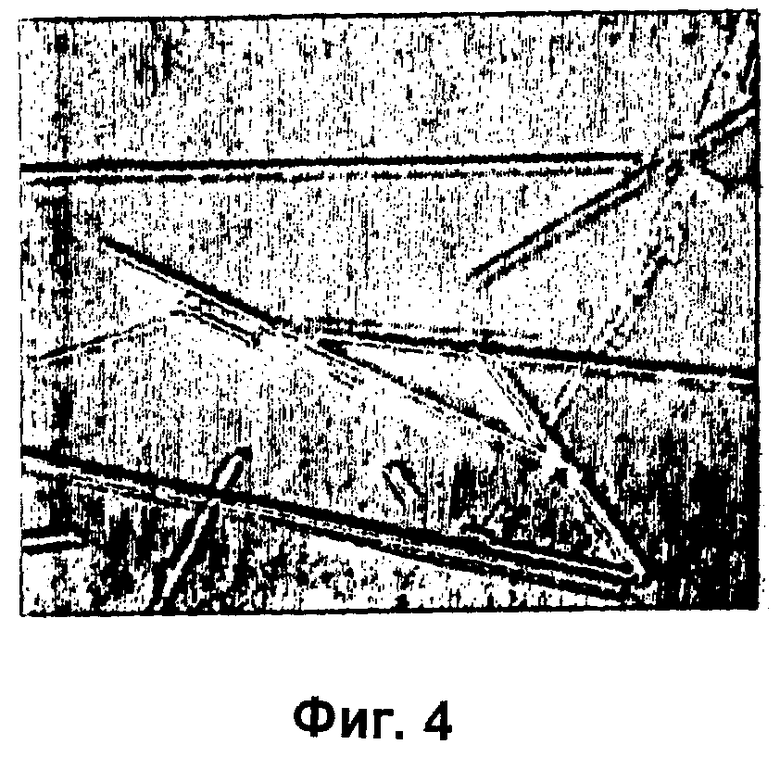

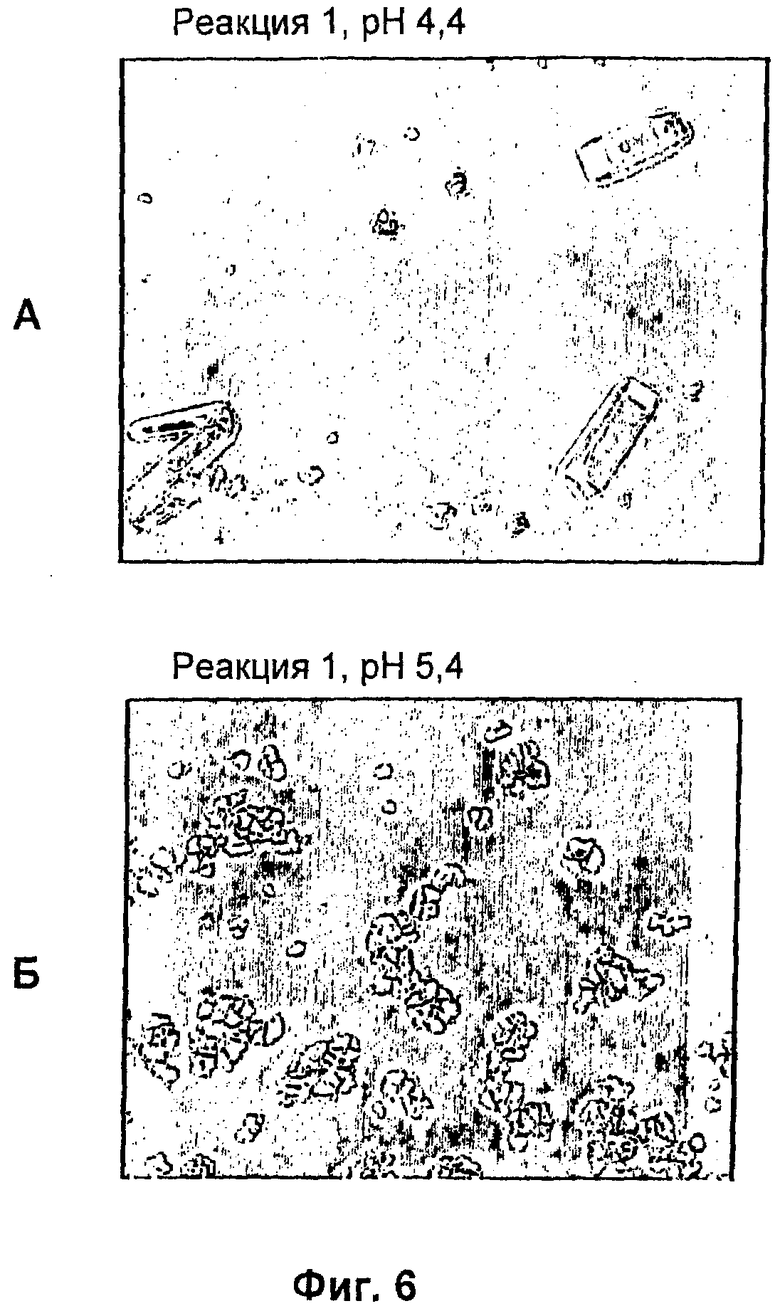
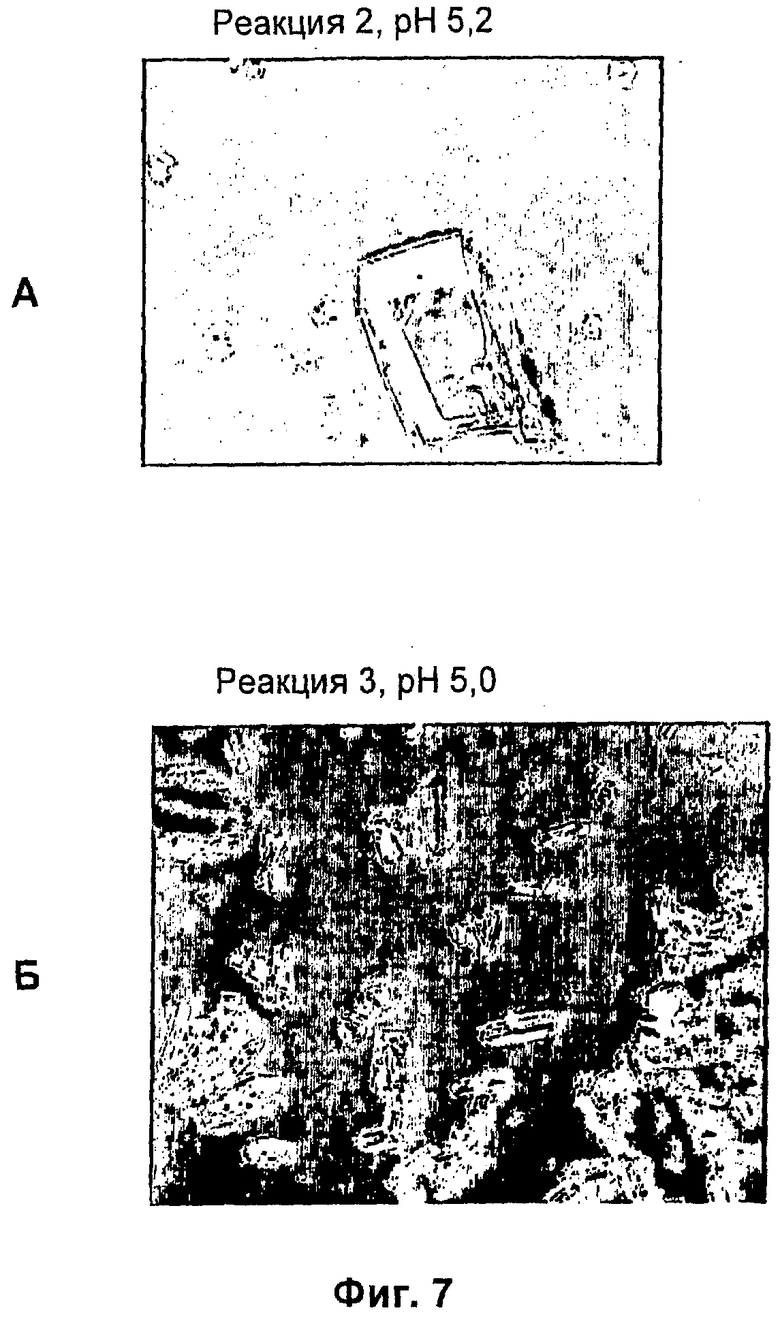
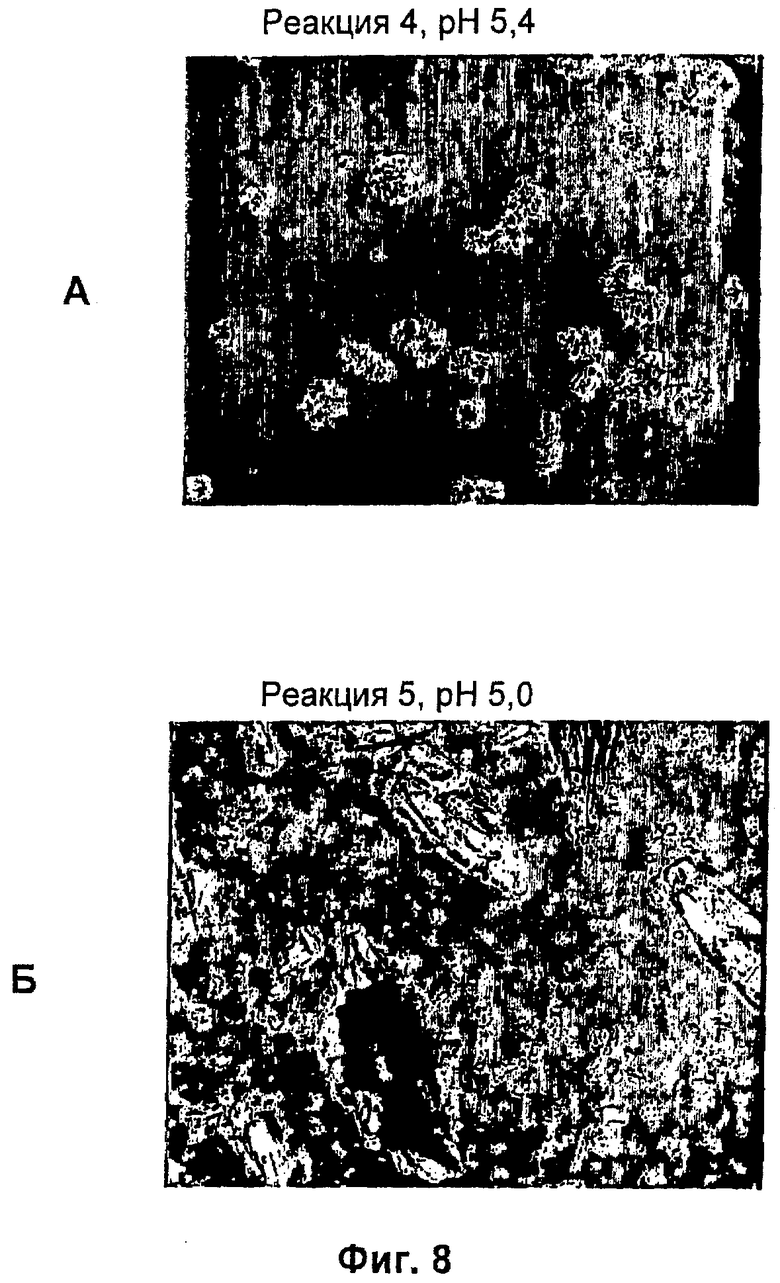
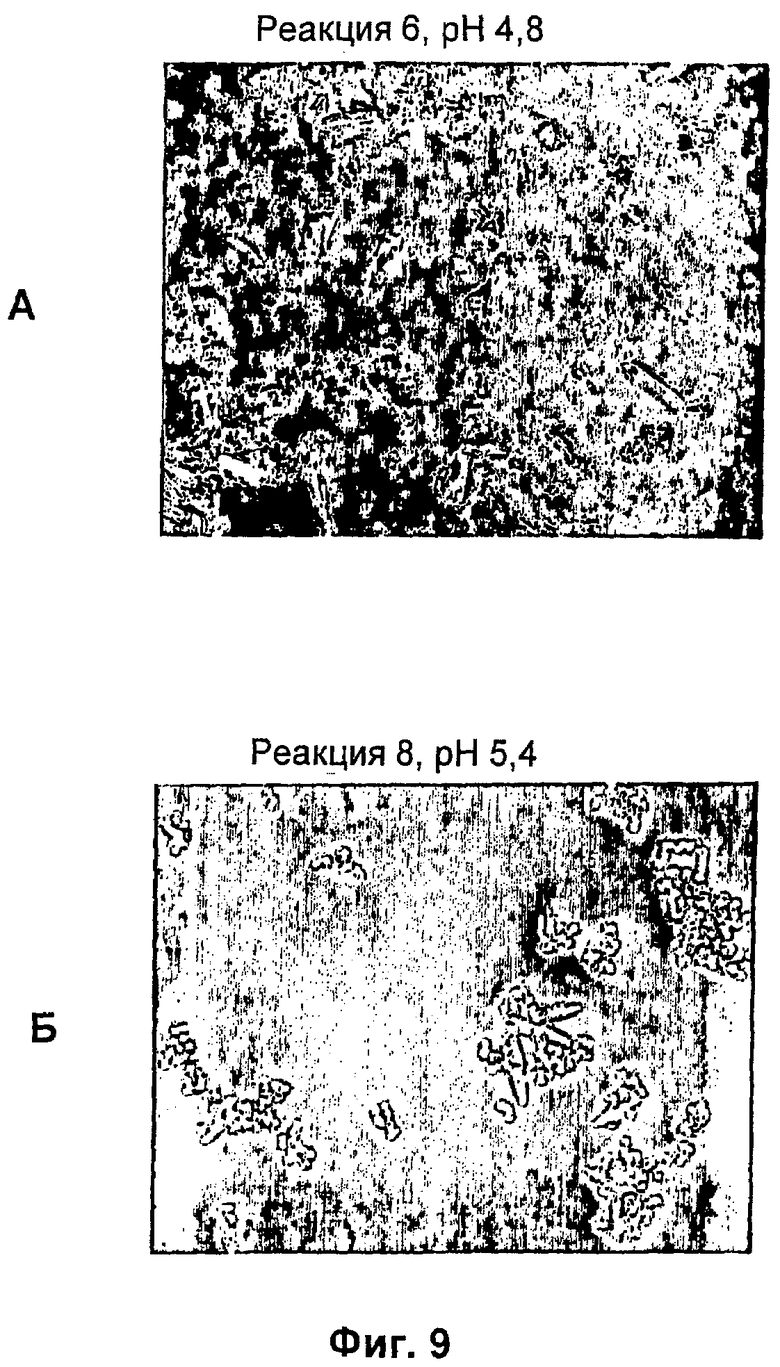
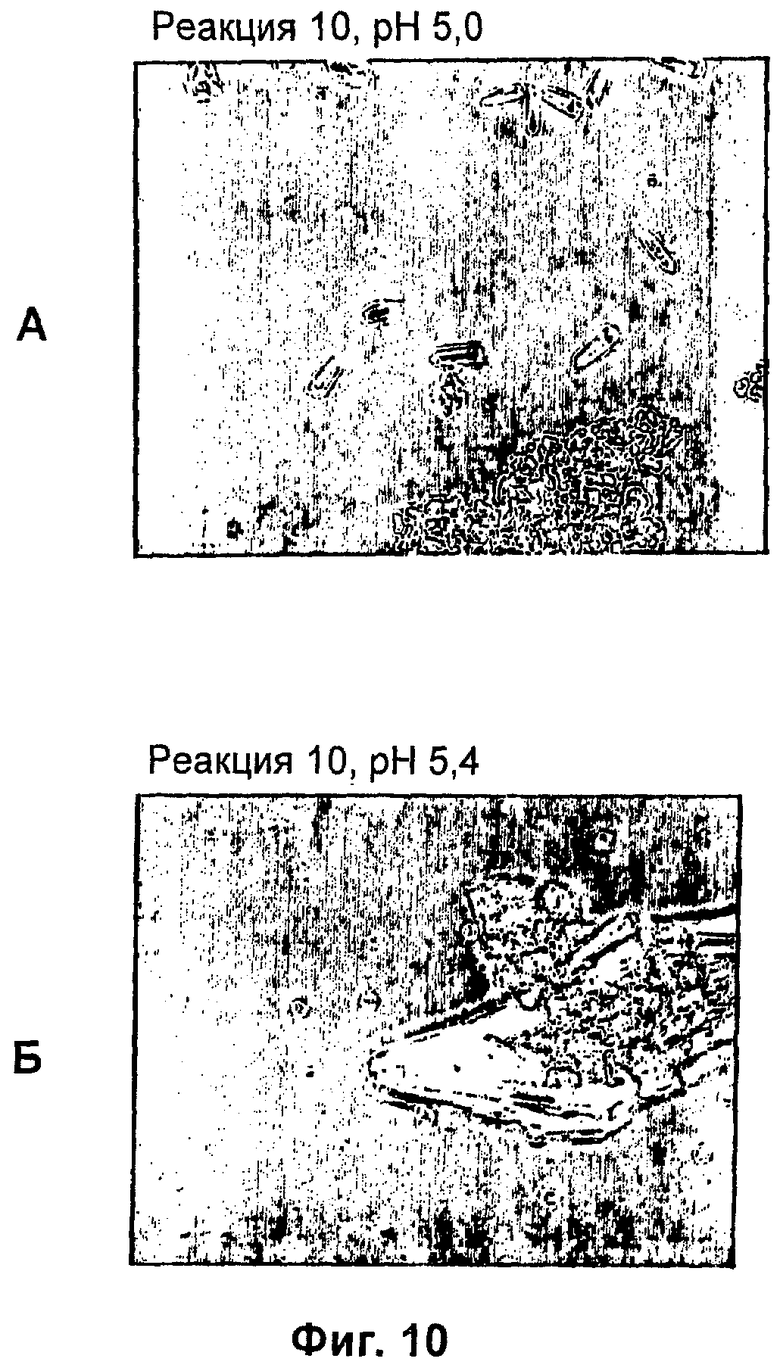
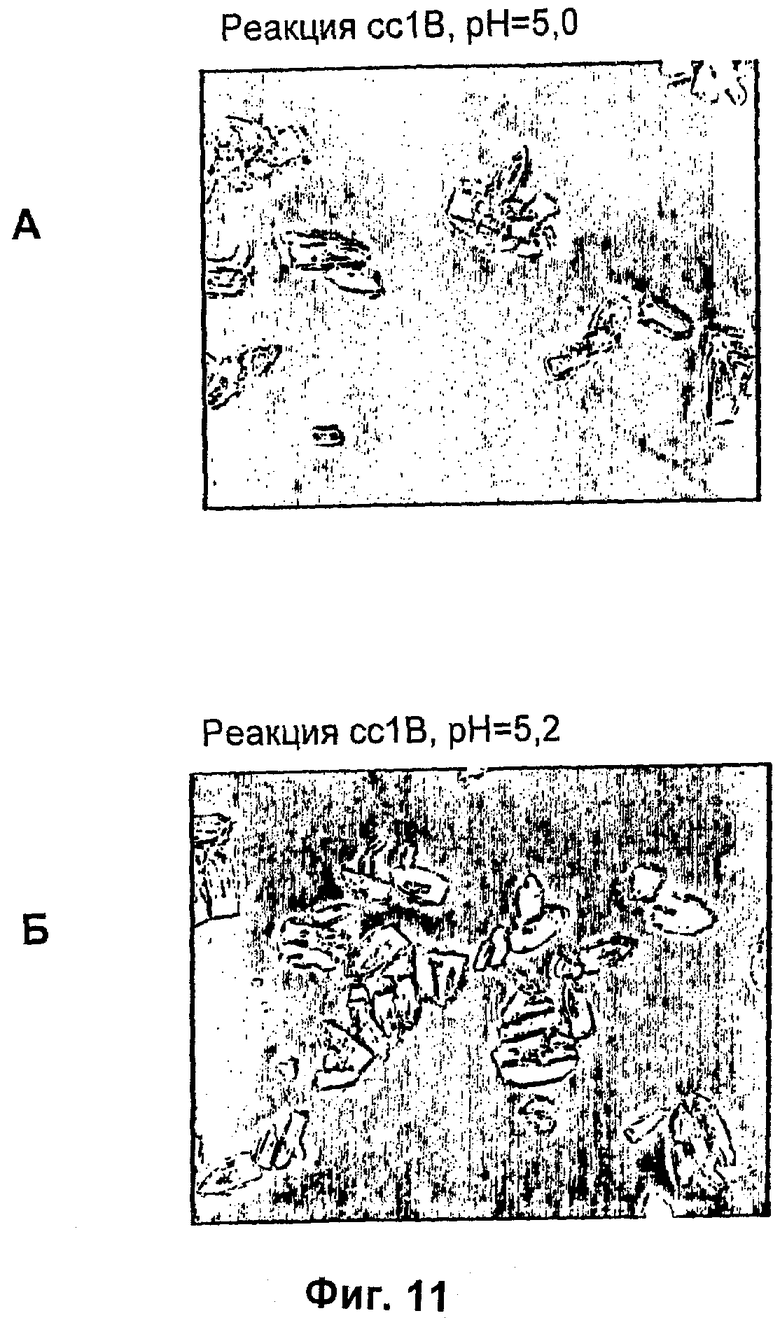


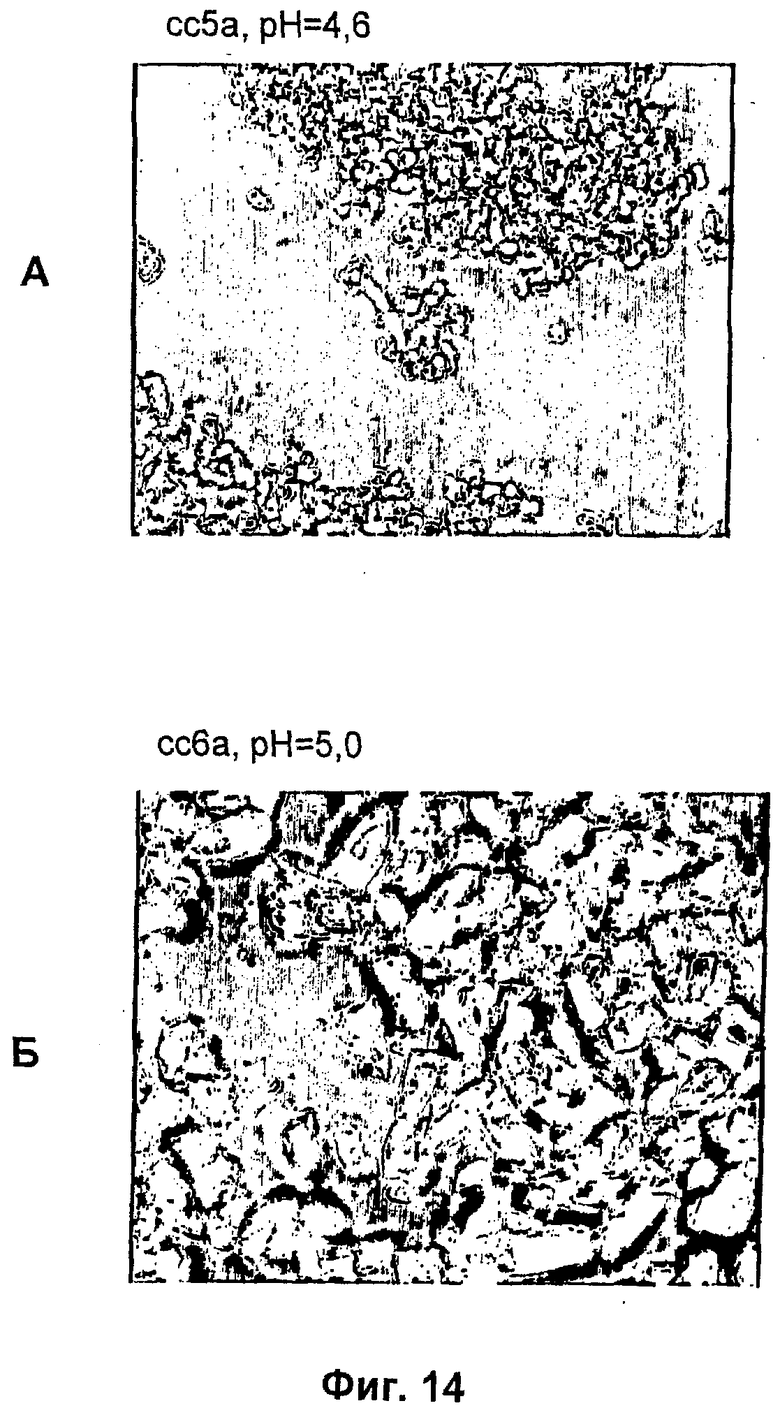

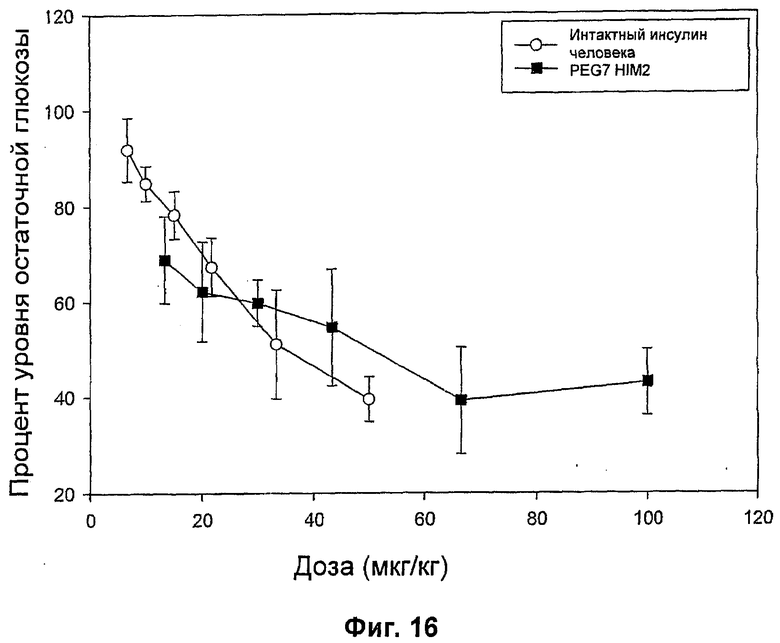
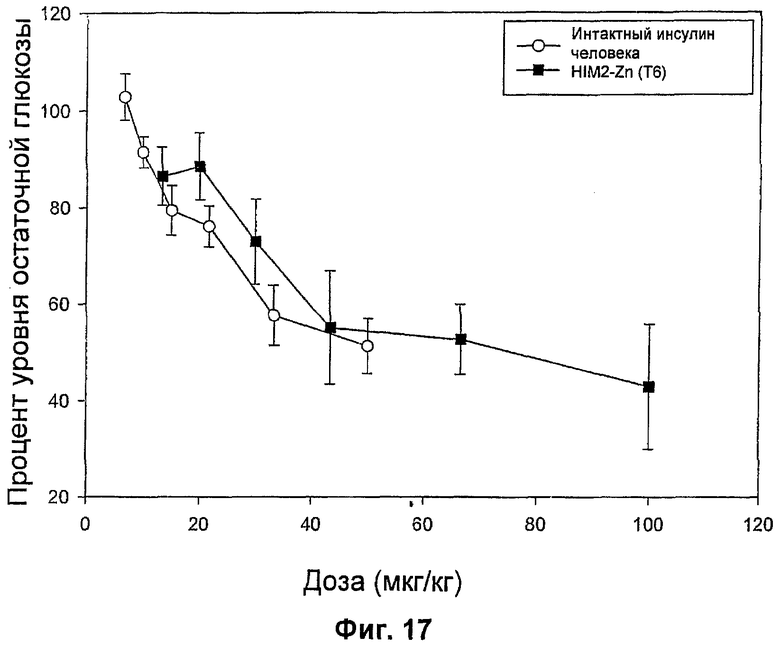
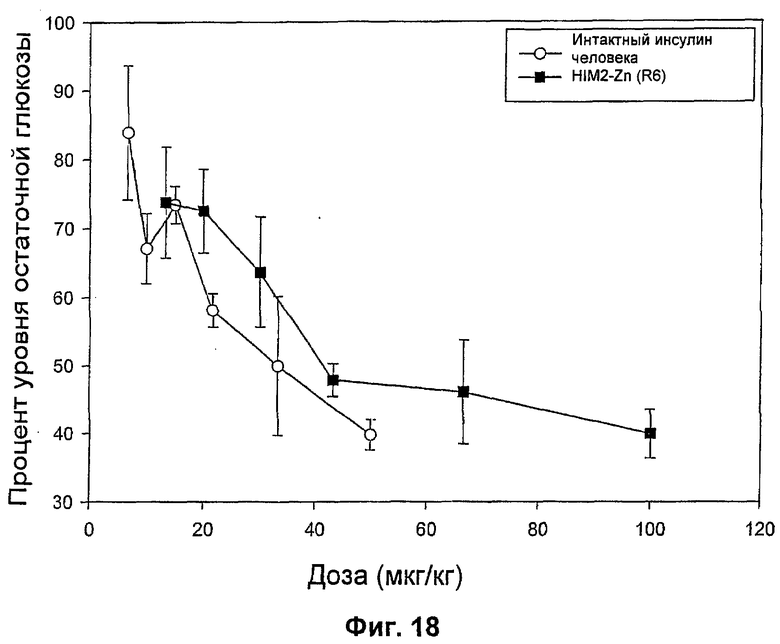
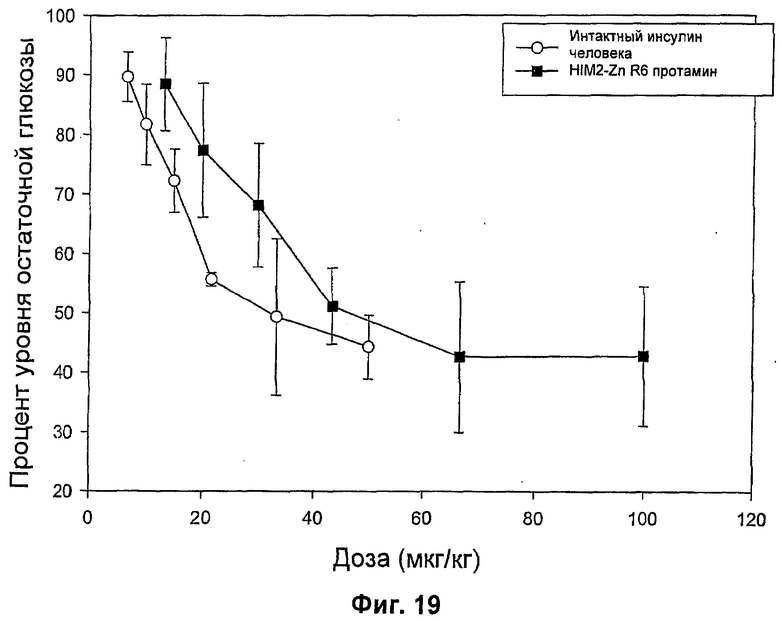
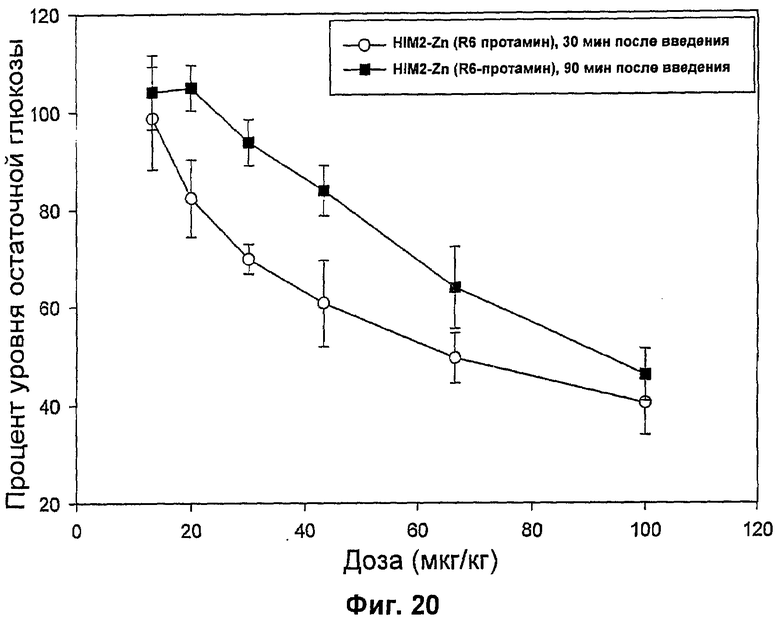
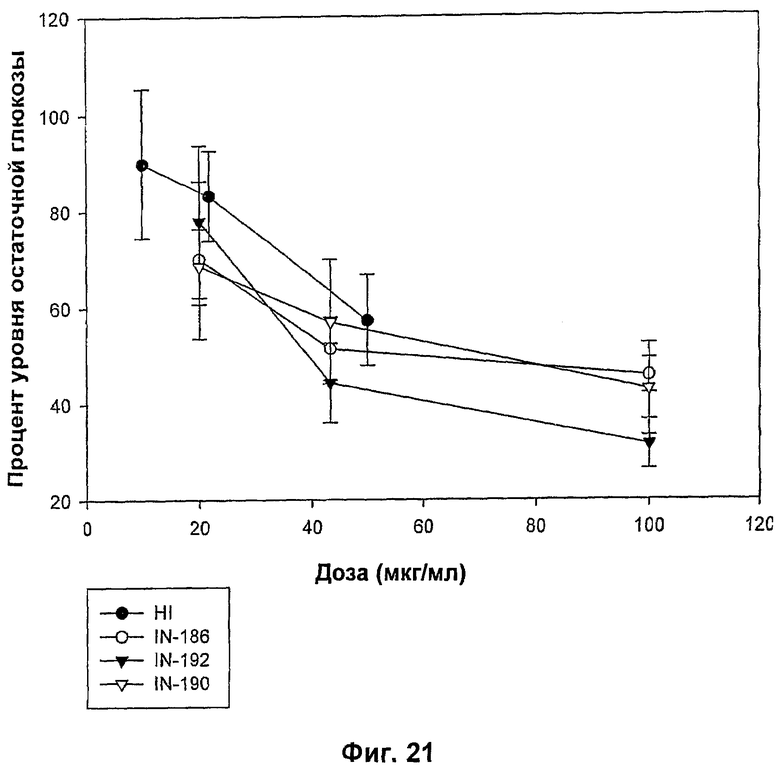
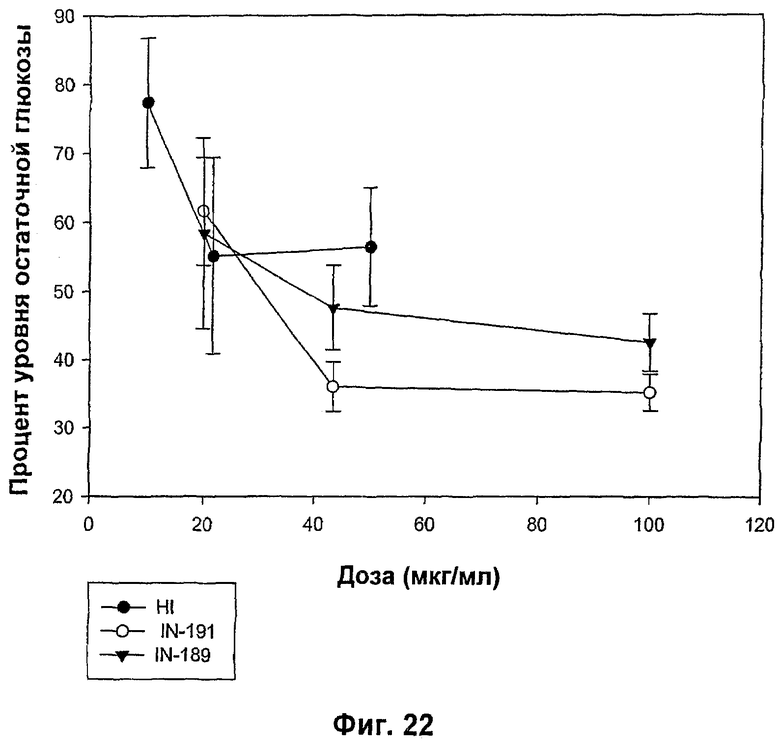
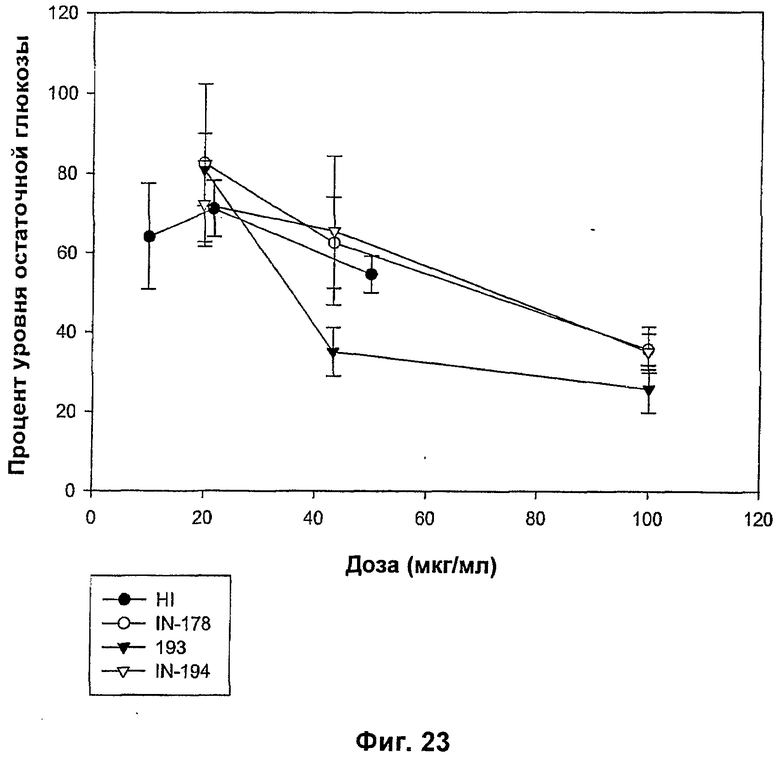
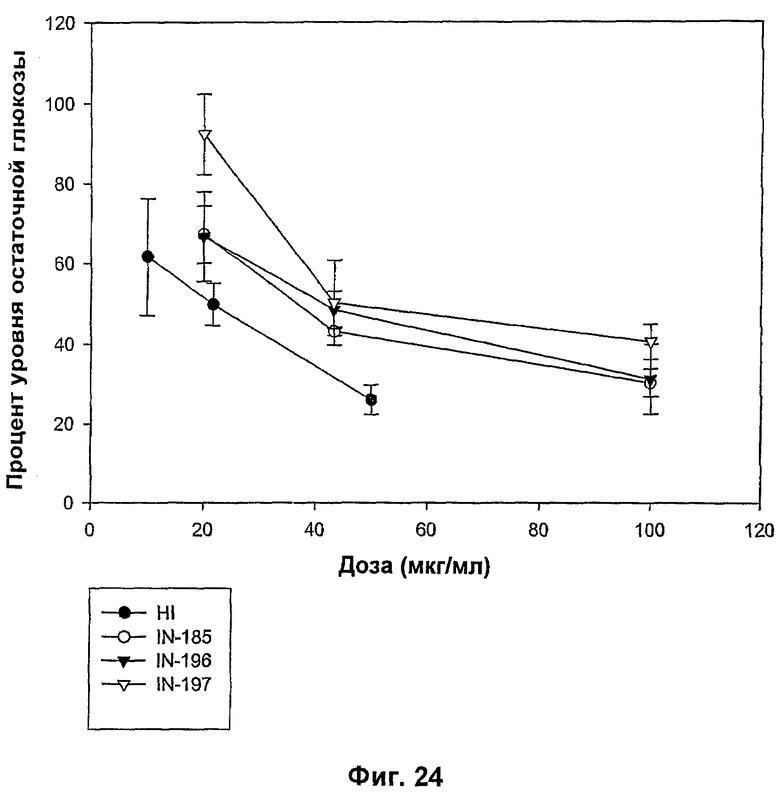
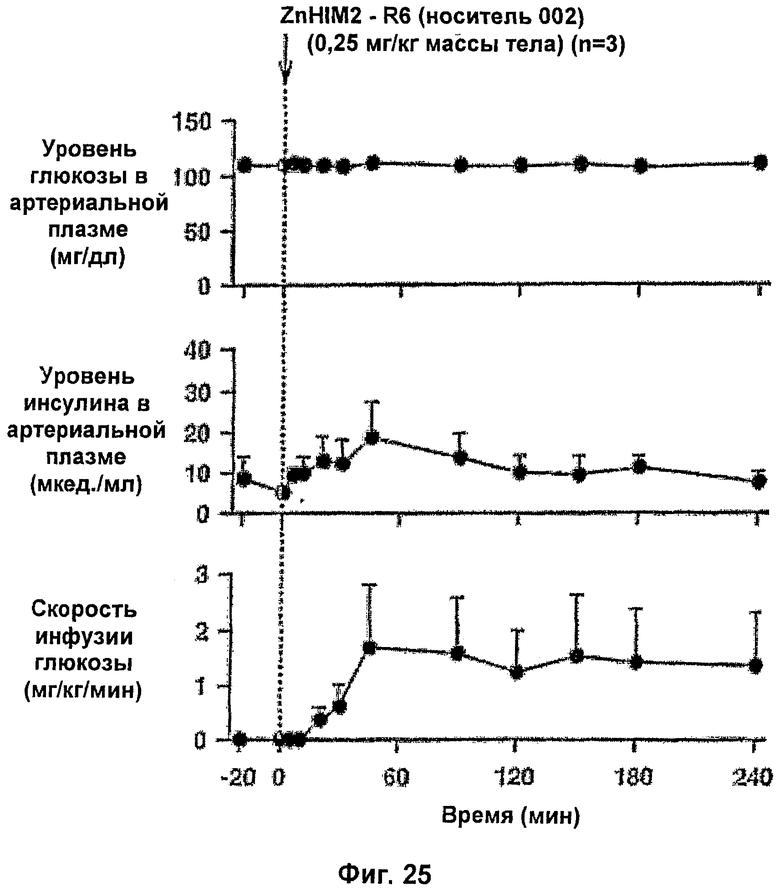
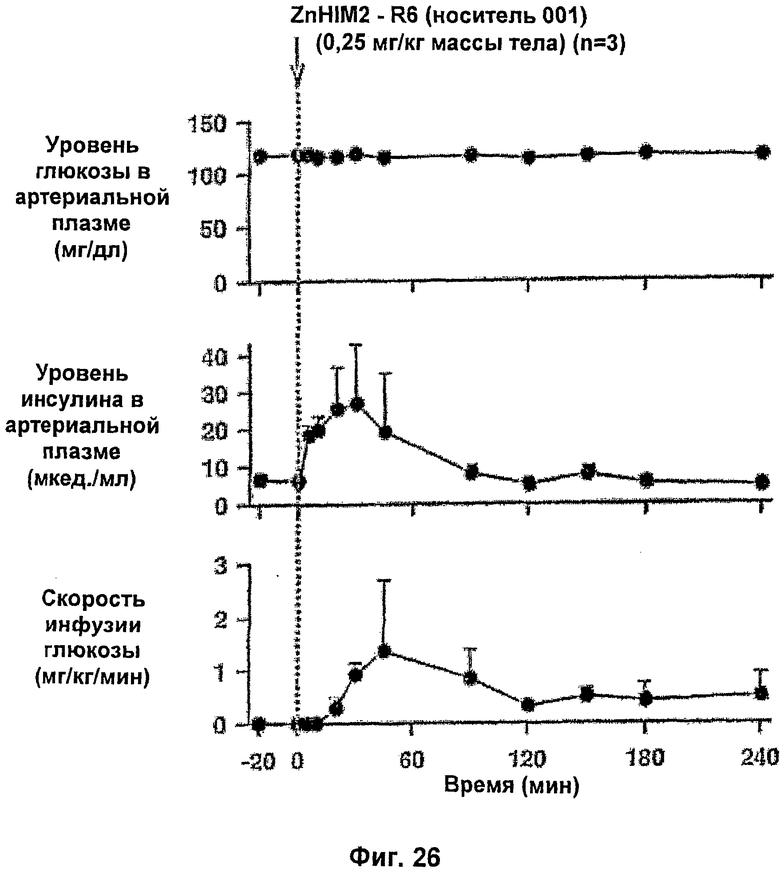
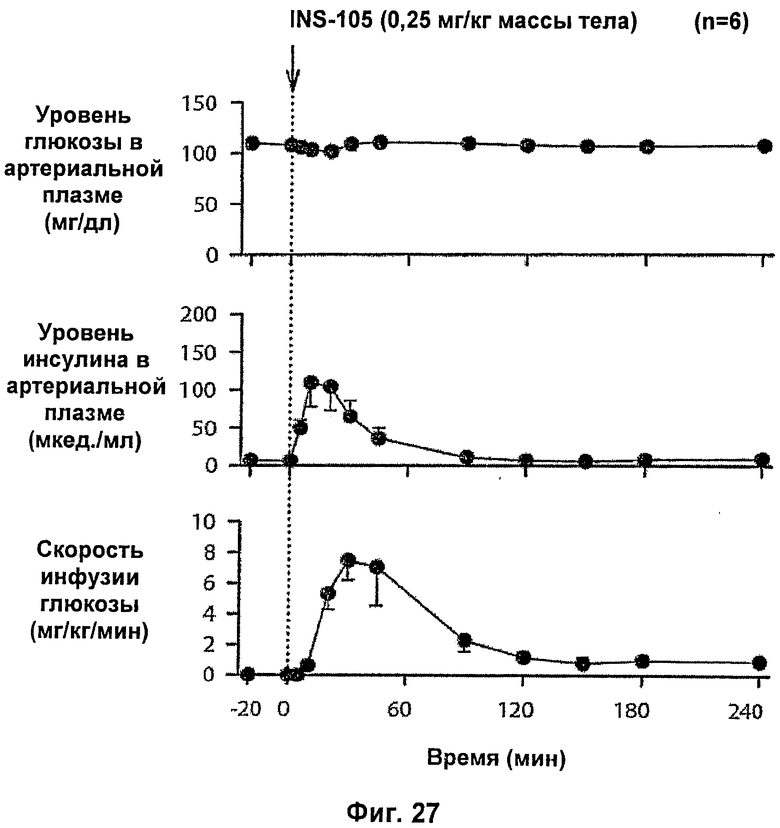
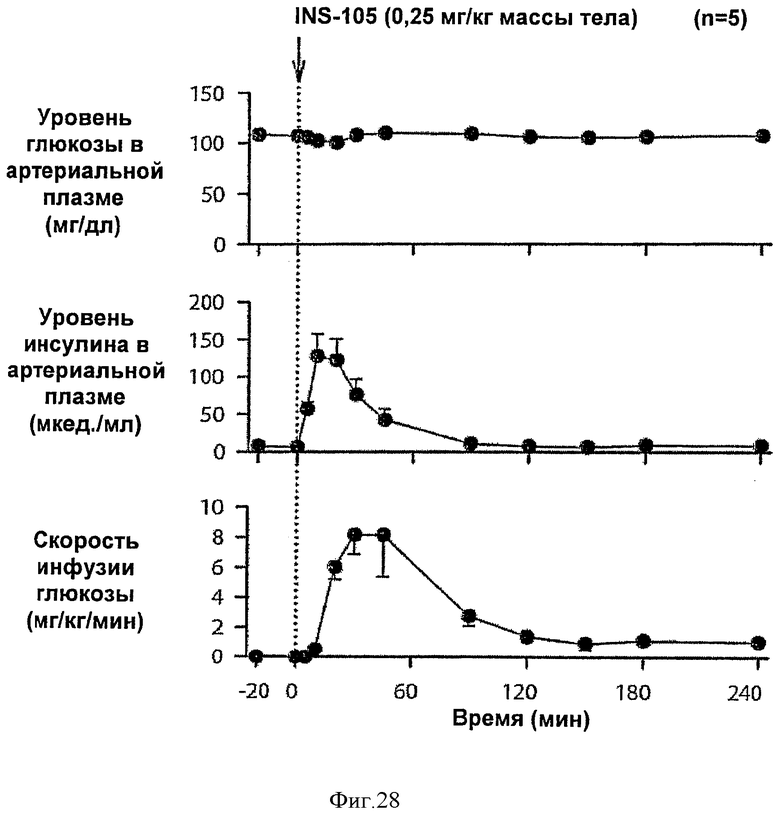
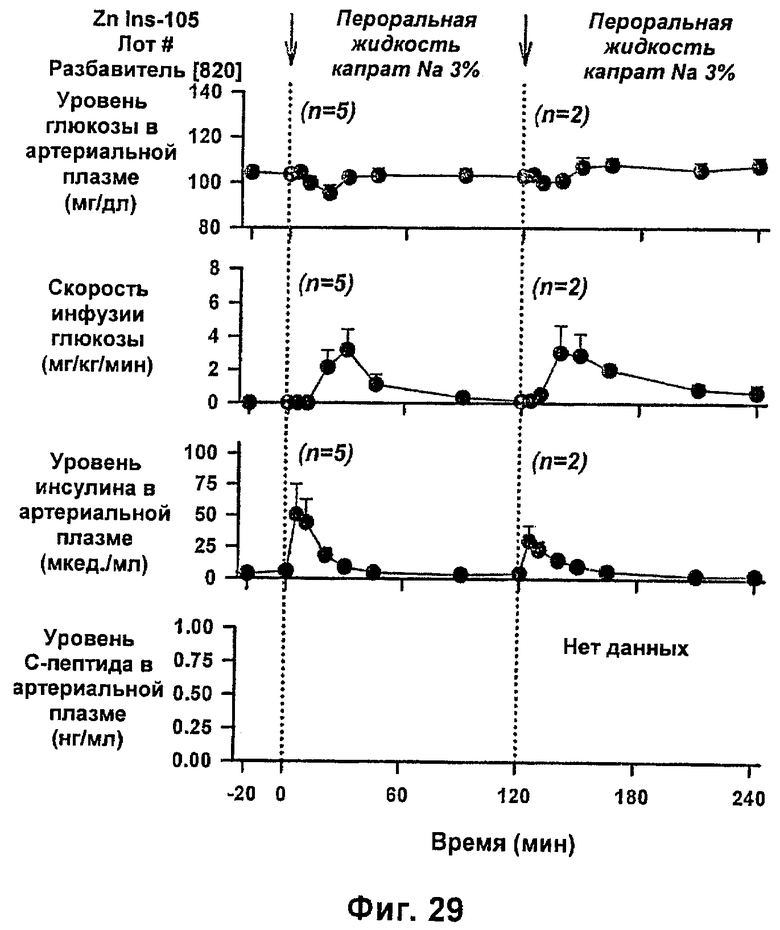
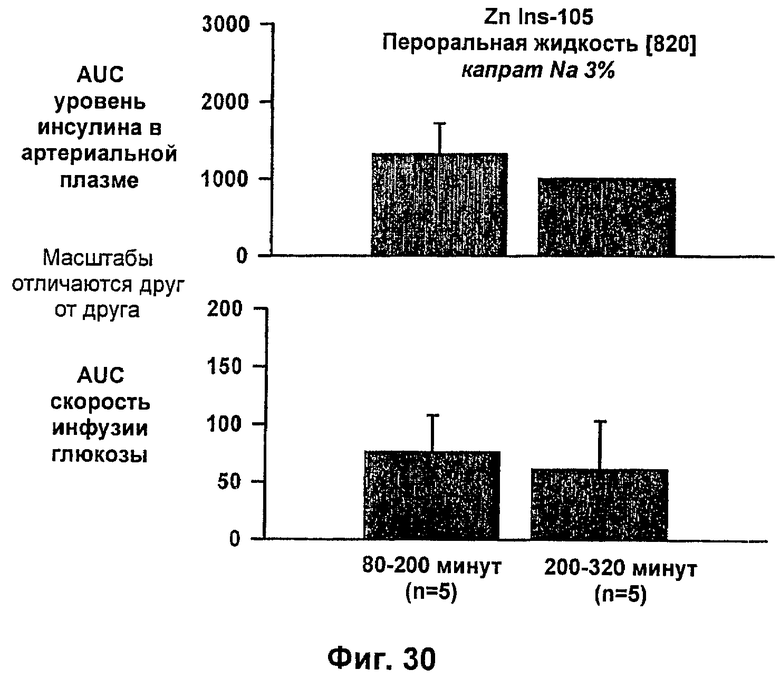
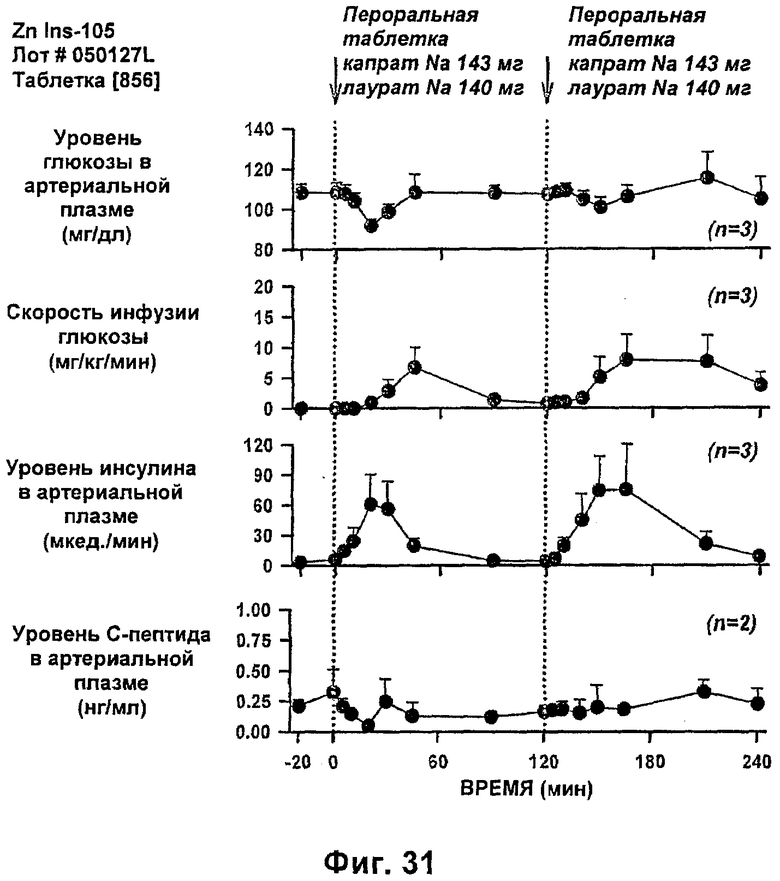
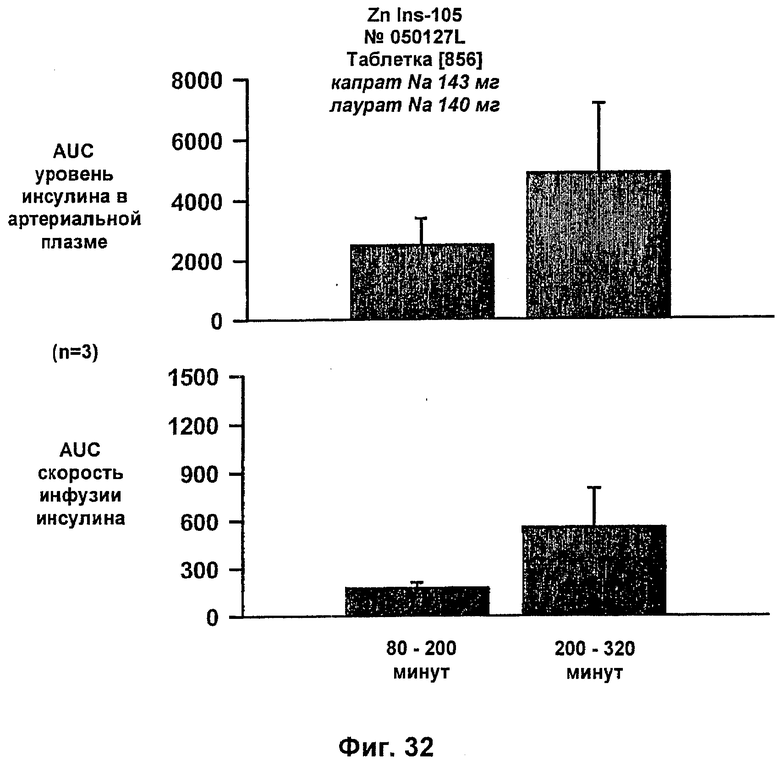
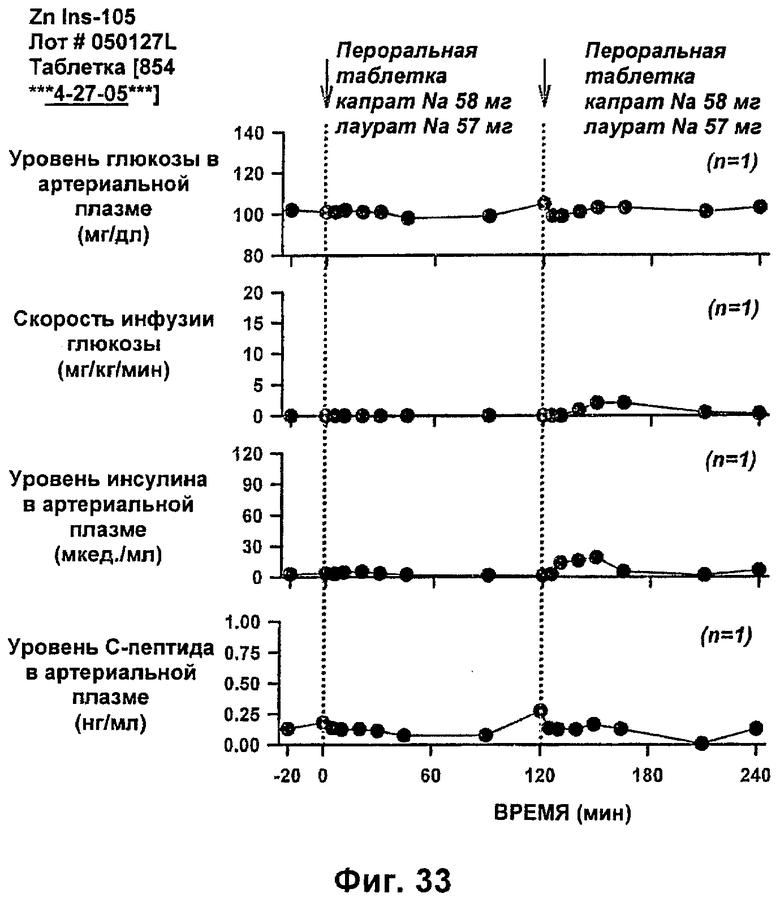
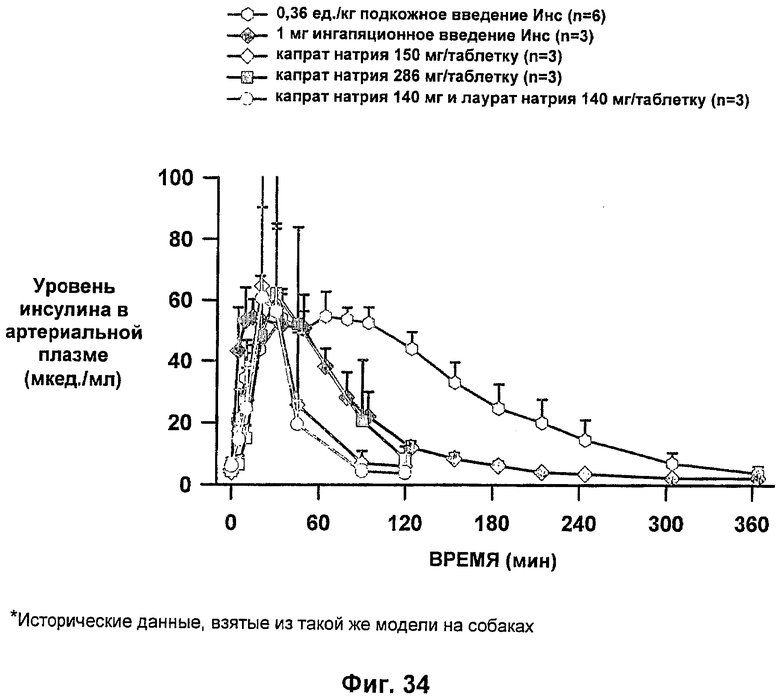
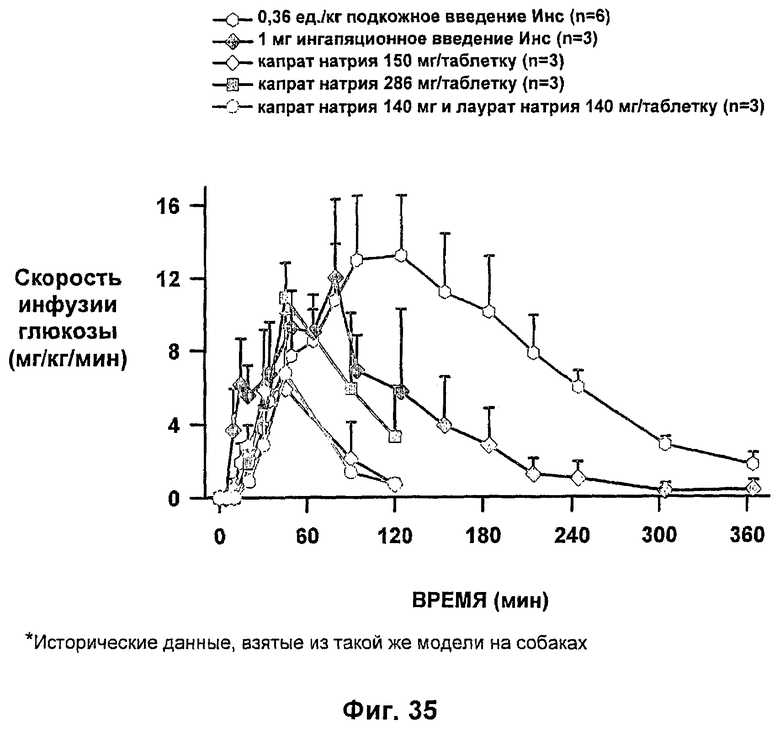
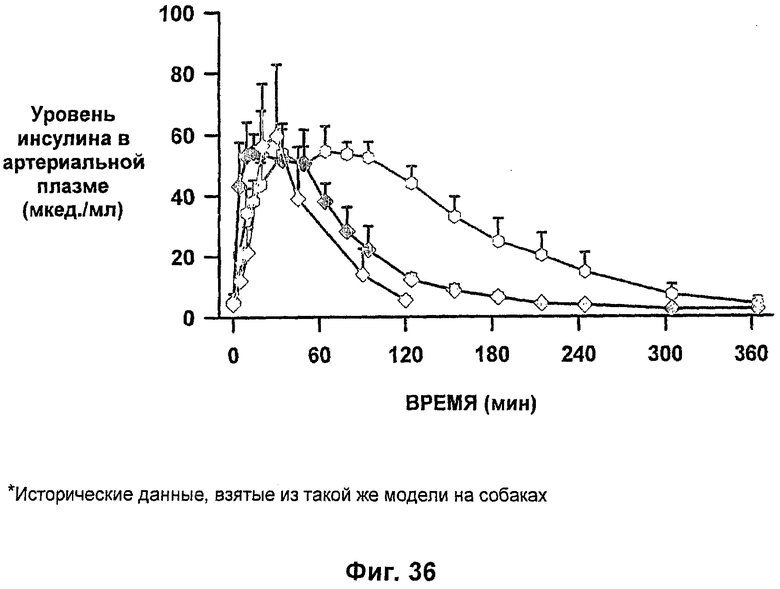
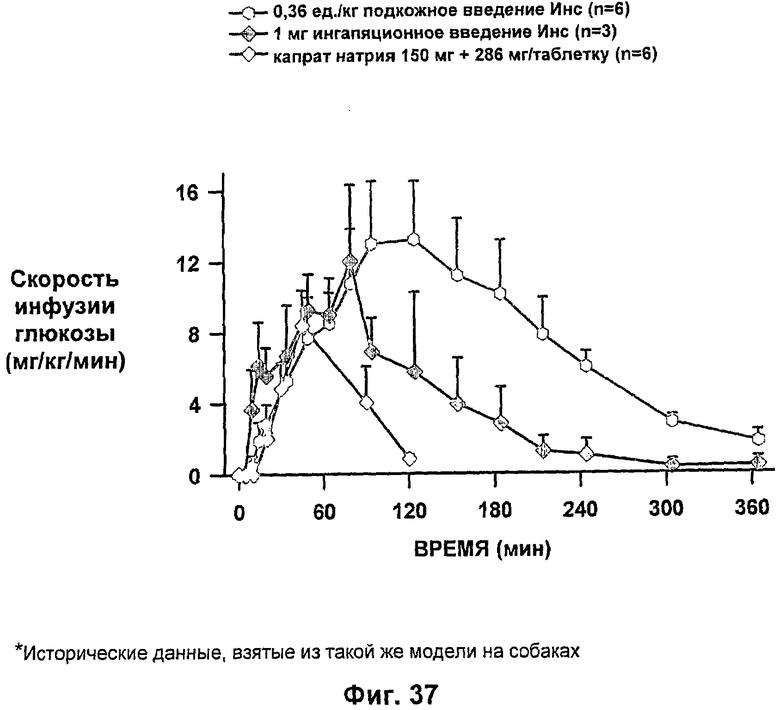
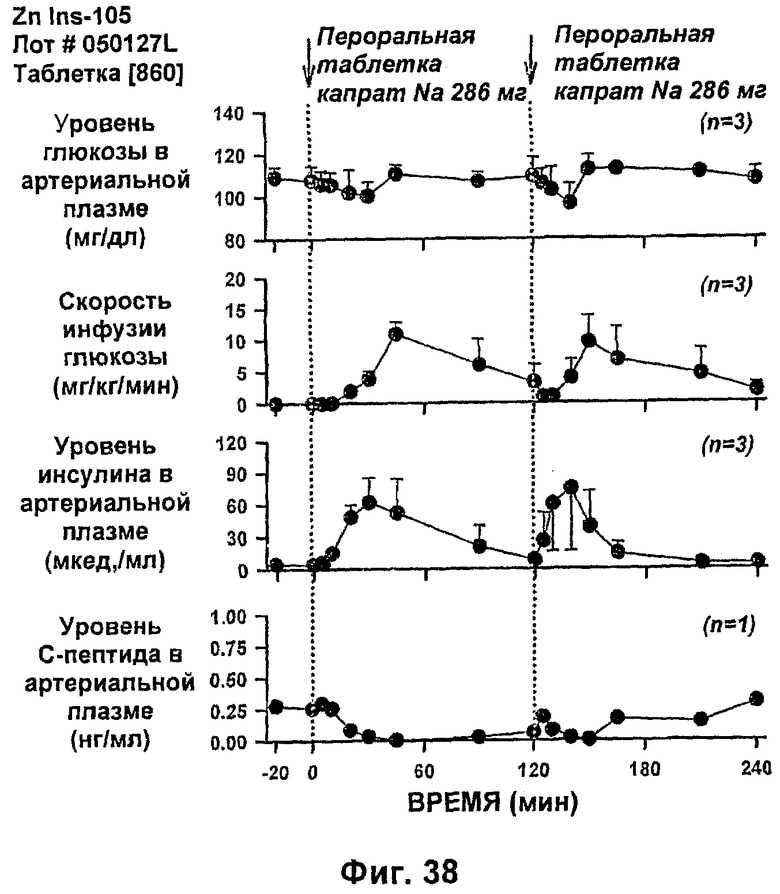
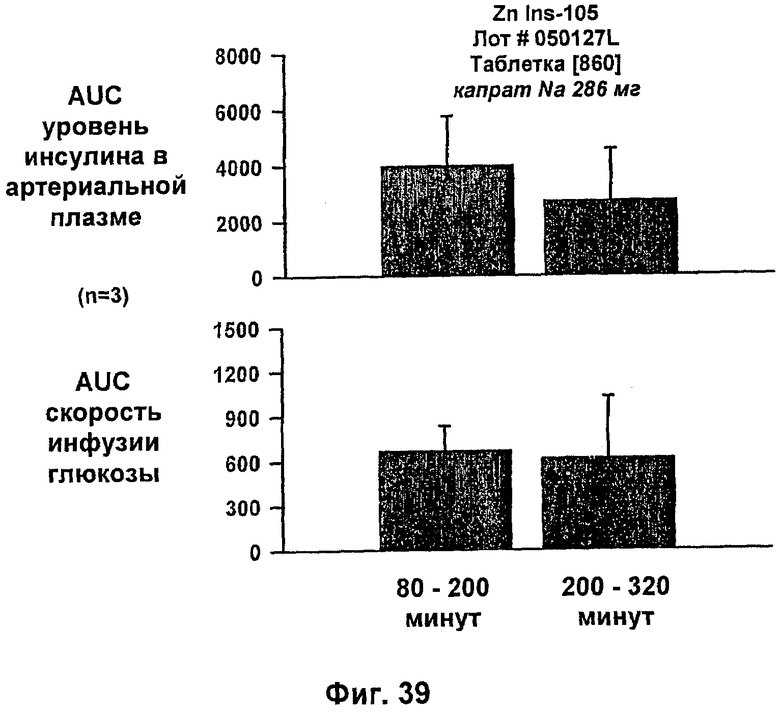
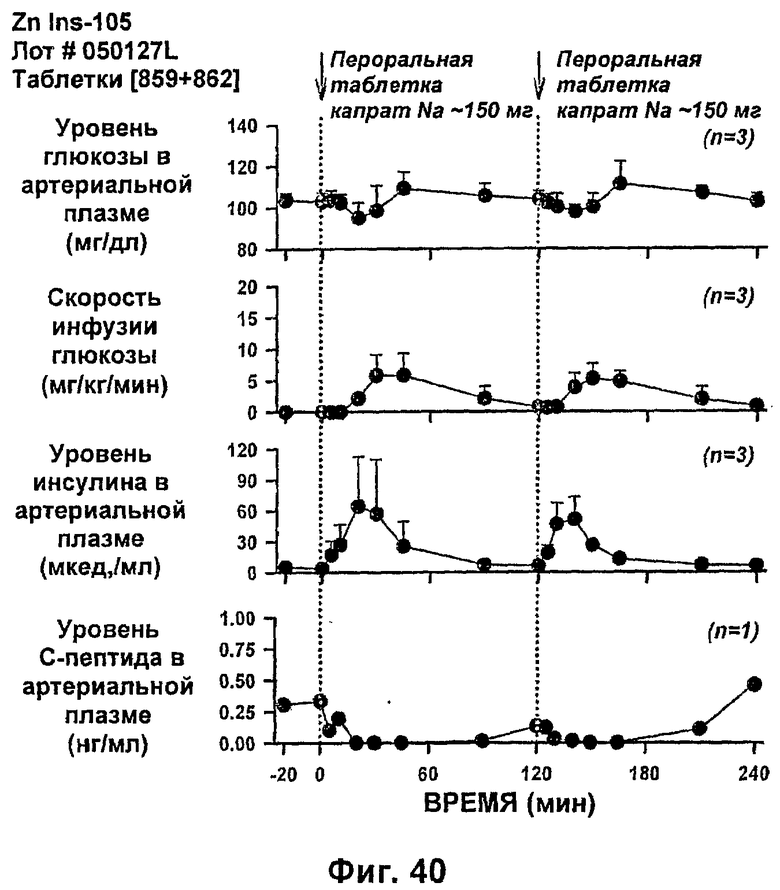
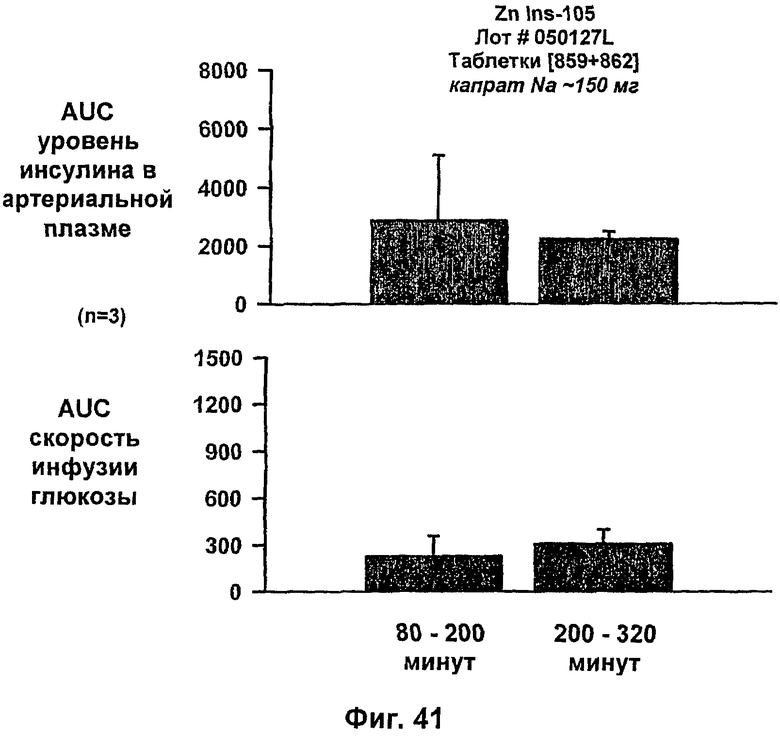
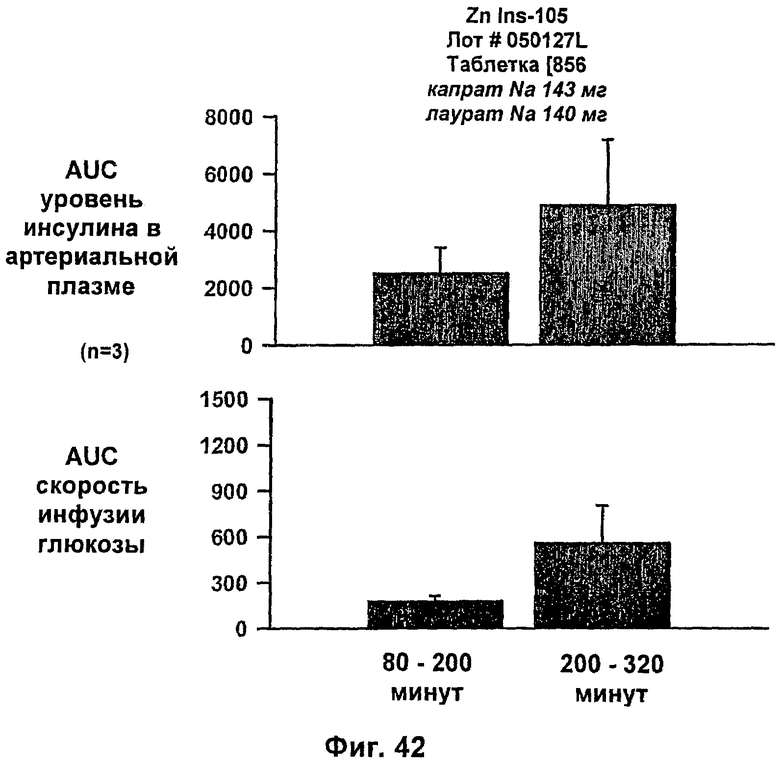
Группа изобретений относится к области биотехнологии и может быть использована при производстве препаратов инсулинов для терапевтических целей. Предложен комплекс конъюгата инсулина с двухвалентным катионом металла группы II или переходного металла, где инсулин конъюгирован с модифицирующей группировкой: -X-R1-Y-PAG-Z-R2 (IV), в которой: X, Y и Z представляют собой независимо выбранные связывающие группы и каждая из них возможно присутствует, и X, когда присутствует, связан с соединением инсулином ковалентной связью, где X, Y, Z независимо могут быть выбраны из -S-, -O-, -NH- и -С(O)-; по меньшей мере один из R1 или R2 присутствует и представляет собой низший алкил, возможно включающий карбонильную группу, и когда R1 представляет собой низший алкил, тогда R2 представляет собой блокирующую группу, выбранную из -CH3, -H, тозилата или -С(O)ОН; PAG представляет собой линейную или разветвленную углеродную цепь, включающую одну или более группировок PEG, содержащих от 2 до 10 (СН2СН2О)-субъединиц, и возможно включающую одну или более дополнительных группировок, выбранных из -S-, -O-, -NH- и -С(O)-; и где модифицирующая группировка имеет максимальное число от 3 до 25 тяжелых атомов, выбранных из C, S, N и O, и связана с лизином в положении, выбранном из группы, состоящей из положений В26, B27, В28, В29 и В30 инсулина в пределах 5 аминокислот C-конца B-цепи, тем самым давая моноконъюгат. Конъюгат инсулина является таким же или более растворимым, чем соответствующий неконъюгированный инсулин, и растворимость в воде конъюгата инсулина уменьшена путем добавления цинка. Полученные комплексы проявляют улучшенную химическую стабильность по сравнению с не находящимися в комплексе конъюгатами инсулина. Изобретения также включают способ получения таких комплексов, фармацевтические композиции для лечения недостаточности инсулина у человека, содержащие такие комплексы, и их применение в изготовлении лекарства для лечения диабета. 5 н. и 46 з.п. ф-лы, 42 ил., 40 табл., 86 пр.
1. Комплекс конъюгата инсулина и катиона, содержащий:
конъюгат соединения инсулина, содержащий соединение нативный инсулин или его полипептидный аналог, проявляющий частичную, полную или повышенную активность относительно соответствующего нативного инсулина, конъюгированные с модифицирующей группировкой, имеющей формулу:
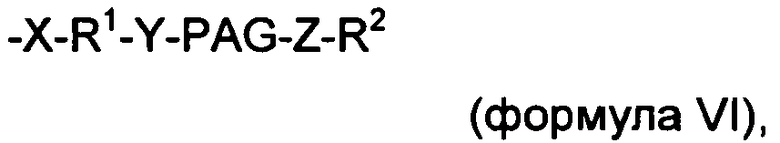
где:
X, Y и Z представляют собой независимо выбранные связывающие группы, и каждая из них возможно присутствует, и X, когда присутствует, связан с соединением инсулином ковалентной связью, где X, Y, Z независимо могут быть выбраны из -S-, -O-, -NH- и -С(O)-,
по меньшей мере один из R1 или R2 присутствует и представляет собой низший алкил, возможно включающий карбонильную группу, и когда R1 представляет собой низший алкил, тогда R2 представляет собой блокирующую группу, выбранную из -CH3, -H, тозилата или -С(O)ОН; и
PAG представляет собой линейную или разветвленную углеродную цепь, включающую одну или более алкиленгликолевых группировок, где алкиленгликолевая группировка представляет собой молекулу PEG (полиэтиленгликоль), содержащую от 2 до 10 PEG-субъединиц, имеющих формулу (CH2CH2O), и возможно включающую одну или более дополнительных группировок, выбранных из группы, состоящей из -S-, -O-, -NH- и -С(O)-, и
где модифицирующая группировка имеет максимальное число от 3 до 25 тяжелых атомов, выбранных из C, S, N и O, и где модифицирующая группировка связана с лизином в пределах 5 аминокислот C-конца B-цепи нативного инсулина или полипептидного аналога инсулина, и где лизин находится в положении, выбранном из группы, состоящей из положений В26, B27, В28, В29 и В30 инсулина, тем самым давая моноконъюгат;
и катион, где конъюгат соединения инсулина образует комплекс с двухвалентным катионом металла группы II или переходного металла.
2. Комплекс по п.1, где аналог соединения инсулина выбран из группы, состоящей из соединения нативного проинсулина, соединения искусственного проинсулина, соединения препроинсулина, содержащего соединение проинсулин и лидерный пептид или белок-носитель, соединенный с С-концом или N-концом соединения проинсулина; и соединения минипроинсулина.
3. Комплекс по п.2, где аналог соединения инсулина имеет лидерный пептид, который является отщепляемым или неотщепляемым.
4. Комплекс по п.1, где аналог соединения инсулина представляет собой одноцепочечный предшественник соединения инсулина, содержащий А-цепь-В-цепь соединения инсулина, где модифицирующая группировка связана с лизином на С-конце В-цепи, тем самым давая моноконъюгат.
5. Комплекс по п.1, где модифицирующая группировка выбрана так, чтобы сделать конъюгат соединения инсулина более гидрофильным или более амфифильным, чем соответствующее неконъюгированное соединение инсулин.
6. Комплекс по п.1, где гидрофильность конъюгата соединения инсулина уменьшена путем добавления цинка.
7. Комплекс по п.6, где:
модифицирующая группировка выбрана так, чтобы сделать конъюгат соединения инсулина таким же или более растворимым, чем соответствующее неконъюгированное соединение инсулин; и
растворимость в воде конъюгата соединения инсулина уменьшена путем добавления цинка.
8. Комплекс по п.1, где относительная липофильность конъюгата соединения инсулина по отношению к соответствующему исходному соединению инсулину равна 1 или меньше 1.
9. Комплекс по п.1, который представляет собой твердое вещество или жидкость.
10. Комплекс по п.1, где:
(а) растворимость комплекса конъюгата соединения инсулина с катионом при рН 7,4 меньше растворимости конъюгата соединения инсулина в растворе при рН 7,4, и
(б) конъюгат соединения инсулина с катионом остается растворимым при концентрации более чем приблизительно 1 г/л в водном растворе в диапазоне pH от 5,8 до 8,5.
11. Комплекс по п.1, где:
(а) конъюгат соединения инсулина представляет собой моноконъюгат; и
(б) комплекс образует кристаллы в водном растворе при рН от 4 до менее 6,5.
12. Комплекс по п.1, где конъюгат соединения инсулина образует кристаллы в водном растворе при рН, равном pI плюс или минус 2,5.
13. Комплекс по п.1, где катионный компонент содержит катион металла, выбранный из группы, состоящей из Zn++, Mn++, Ca++, Fe++, Ni++, Cu++, Co++ и Mg++.
14. Комплекс по п.1, где катионный компонент содержит Zn++.
15. Комплекс по п.1, где молярное отношение конъюгата соединения инсулина к катионному компоненту составляет от 1:2 до 1:12.
16. Комплекс по п.1, где устойчивость к расщеплению химотрипсином находящегося в комплексе с катионом конъюгата соединения инсулина выше, чем устойчивость к расщеплению химотрипсином соответствующего не находящегося в комплексе конъюгата соединения инсулина.
17. Комплекс по п.1, где устойчивость к расщеплению химотрипсином находящегося в комплексе с катионом конъюгата соединения инсулина выше, чем устойчивость к расщеплению химотрипсином соответствующего находящегося в комплексе, но неконъюгированного соединения инсулина.
18. Комплекс по п.9, где в сухом состоянии твердое вещество содержит:
более чем 71% (масс./масс.) конъюгата соединения инсулина, и от 0,5 до 4% (масс./масс.) Zn++ и возможно от 0,1 до 5% (масс./масс.) фенола.
19. Комплекс по п.1, где модифицирующая группировка выбрана так, чтобы сделать конъюгат соединения инсулина таким же или более растворимым, чем соответствующее неконъюгированное соединение инсулин.
20. Комплекс по п.1, где растворимость в воде конъюгата соединения инсулина уменьшена путем добавления цинка.
21. Комплекс по п.1, где модифицирующая группировка имеет структуру, выбранную из группы, состоящей из:
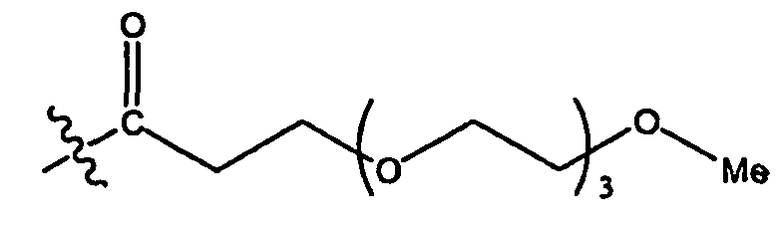 ;
;
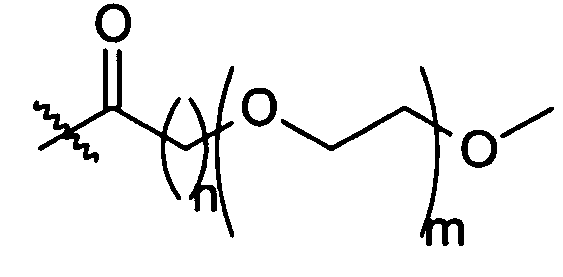 ,
,
где n равно от 1 до 4 и m равно от 2 до 4;
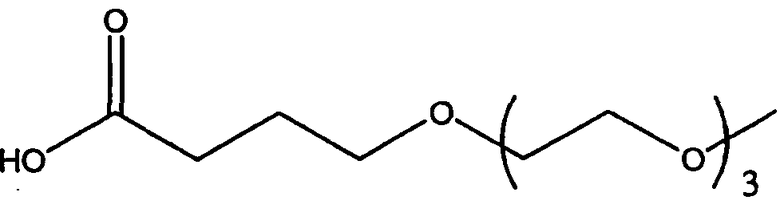 ;
;
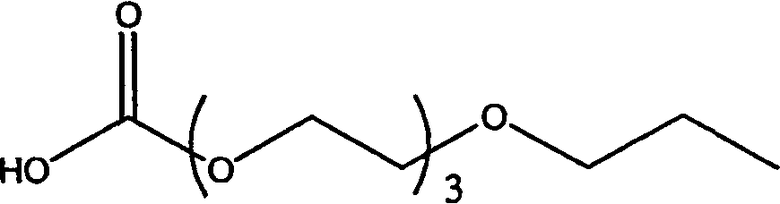 ;
;
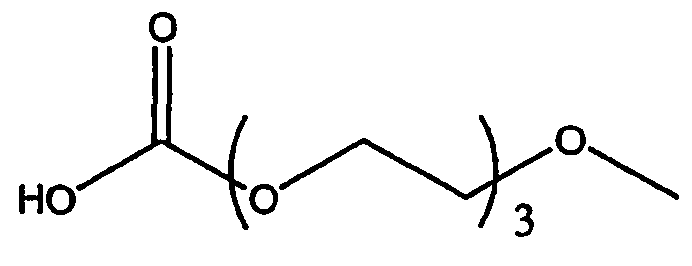 ;
;
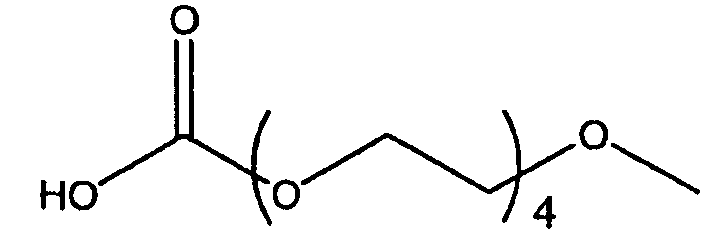 ;
;
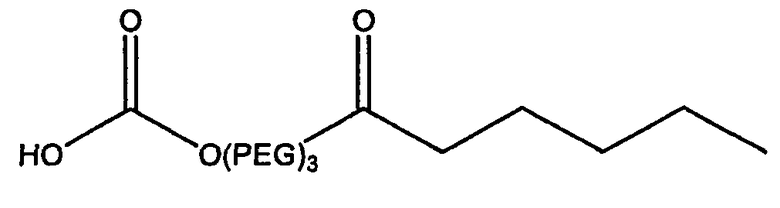 и
и
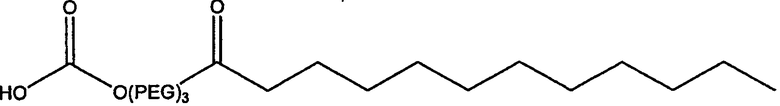 .
.
22. Комплекс по п.1, где модифицирующая группировка имеет формулу, выбранную из группы, состоящей из:
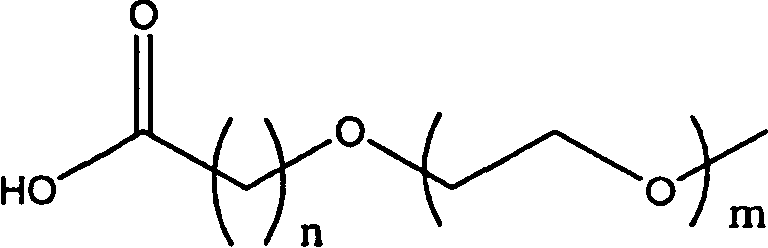
где n равно от 1 до 4 и m равно от 2 до 5; и
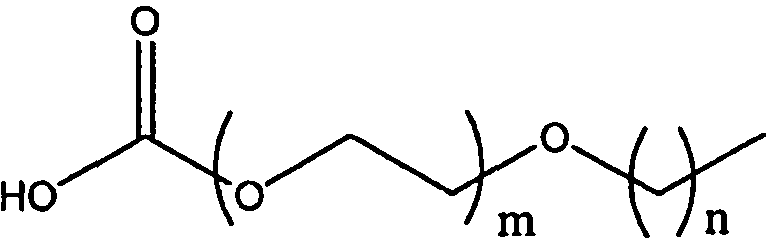 ,
,
где n равно 1, 2, 3 или 4 и m равно 2, 3, 4 или 5.
23. Комплекс по п.1, присутствующий в виде компонента сокристалла, содержащего гидрофильный конъюгат соединения инсулина и липофильный конъюгат соединения инсулина.
24. Комплекс по п.1, присутствующий в виде компонента сокристалла, содержащего гидрофильный конъюгат соединения инсулина и неконъюгированное соединение инсулин.
25. Комплекс по п.1, присутствующий в виде компонента сокристалла, содержащего IN105, кристаллизованный вместе с неконъюгированным соединением инсулин.
26. Комплекс по п.1, присутствующий в виде компонента сокристалла, содержащего IN105, кристаллизованный вместе с гидрофильным конъюгатом соединения инсулина.
27. Способ получения комплекса конъюгата соединения инсулина с катионом по п.1, включающий объединение в водном растворе конъюгата соединения инсулина и катиона.
28. Применение комплекса по любому из пп.1-26 в изготовлении лекарства для лечения диабета.
29. Композиция для лечения недостаточности инсулина у человека, содержащая терапевтически эффективное количество по меньшей мере одного из комплексов по любому из пп.1-22 и фармацевтически приемлемый носитель.
30. Композиция по п.29, дополнительно содержащая второй компонент, содержащий конъюгат инсулина или неконъюгированный инсулин.
31. Композиция по п.30, где комплекс представляет собой форму, выбранную из группы, состоящей из кристаллического твердого вещества, аморфного твердого вещества и сокристалла, содержащего по меньшей мере два конъюгата инсулина, причем данные конъюгаты инсулина являются одинаковыми или разными.
32. Композиция по п.29, представленная в виде лиофилизованного порошка, твердой смеси или гибридного комплекса.
33. Композиция по п.29, содержащая более одного комплекса, где конъюгаты инсулина данных комплексов содержат разные инсулины или разные конъюгаты инсулина.
34. Композиция по п.33, где разные конъюгаты инсулина имеют разные характеристики, выбранные из группы, состоящей из разных растворимостей, разных периодов полувыведения из кровотока и разных модифицирующих группировок.
35. Композиция по п.34, где один из комплексов имеет профиль быстрого действия; а другой из комплексов имеет профиль от среднего до длительного действия.
36. Композиция по п.34, где один из комплексов имеет профиль, подходящий для базального контроля инсулина, а другой из комплексов имеет профиль, подходящий для постпрандиального контроля глюкозы.
37. Композиция по п.31, представляющая собой сокристалл, содержащий IN105.
38. Композиция по п.31, представляющая собой сокристалл, имеющий PK/PD (фармакокинетический/фармакодинамический) профиль, подходящий для постпрандиального контроля глюкозы или для ночного базального контроля инсулина.
39. Композиция по п.30, содержащая неконъюгированный инсулин, выбранный из группы, состоящей из инсулина человека или лизпроинсулина.
40. Композиция по п.29, дополнительно содержащая комплексообразующий агент.
41. Композиция по п.40, где комплексообразующий агент выбран из группы, состоящей из протаминов, сурфена, глобиновых белков, спермина, спермидина, альбумина, аминокислот, карбоновых кислот, поликатионных полимерных соединений, катионных полипептидов, полилизина, анионных полипептидов, нуклеотидов и антисмысловых последовательностей.
42. Композиция по п.29, по существу не содержащая протамина.
43. Композиция по п.31, где комплекс находится в форме кристаллического твердого вещества, палочковидного кристалла, кристалла, имеющего нерегулярную морфологию, смеси аморфных и кристаллических твердых веществ или порошка.
44. Композиция по п.29 в форме порошка, содержащего твердый конъюгат инсулина с катионом и один или более дополнительных фармацевтически приемлемых компонентов.
45. Композиция по п.29, где в сухом состоянии твердое вещество содержит:
более чем приблизительно 71% (масс./масс.) конъюгата инсулина, и
от приблизительно 0,5 до приблизительно 4% (масс./масс.) Zn++, и возможно от приблизительно 0,1 до приблизительно 5% (масс./масс.) фенола.
46. Композиция по п.44, содержащая стабилизирующий агент, выбранный из группы, состоящей из фенола, мета-крезола и парабена.
47. Композиция по п.29, дополнительно содержащая от приблизительно 40 до приблизительно 60% (масс./масс.) жирнокислотного компонента, который содержит насыщенные или ненасыщенные C4-12 жирные кислоты и/или соли таких жирных кислот.
48. Композиция по п.29 в форме твердой или жидкой фармацевтической композиции, приготовленной в виде препарата для перорального введения путем проглатывания внутрь.
49. Композиция по п.47, где жирнокислотный компонент представляет собой каприновую кислоту и/или лауриновую кислоту, и/или соли каприновой кислоты и/или лауриновой кислоты.
50. Применение композиции по любому из пп.29-49 в изготовлении лекарства для лечения диабета.
51. Применение по п.50, где композицию приготавливают для доставки путем, выбранным из группы, состоящей из перорального, перорального проглатывания внутрь, парентерального, трансбуккального, сублингвального, назального, ингаляционного и трансдермального.
| WO 03022996 A2, 20.03.2003 | |||
| Устройство для автоматической остановки поезда с пути | 1934 |
|
SU43034A1 |
| Синхронизованное реле времени | 1958 |
|
SU121197A1 |
| US 5438040 A, 01.08.1995 | |||
| US 2001041786 A1, 15.11.2001 | |||
| US 2003027748 A1, 06.02.2003 | |||
| СПОСОБ ПОЛУЧЕНИЯ КОМПОЗИЦИОННОГО МАТЕРИАЛА ИЗ ВОЛЬФРАМСОДЕРЖАЩЕГО МИНЕРАЛЬНОГО СЫРЬЯ | 1995 |
|
RU2098232C1 |
| US 2003083232 A1, 05.01.2003 | |||
| АГРЕГАТЫ ПРОИЗВОДНЫХ ИНСУЛИНА ЧЕЛОВЕКА | 1998 |
|
RU2218349C2 |
| ТВЕРДОЕ ИНСУЛИНСОДЕРЖАЩЕЕ ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1997 |
|
RU2117488C1 |
| UCHIO T | |||
| ET AL | |||
| "Site-specific insulin conjugates with enhanced | |||

