Настоящее изобретение относится к фосфиновому комплексу ванадия.
Более конкретно, настоящее изобретение относится к фосфиновому комплексу ванадия и его применению в каталитической системе для (со)полимеризации сопряженных диенов.
Настоящее изобретение также относится к каталитической системе для (со)полимеризации сопряженных диенов, содержащей указанный фосфиновый комплекс ванадия.
Кроме того, настоящее изобретение относится к способу (со)полимеризации сопряженных диенов, в частности, к способу полимеризации 1,3-бутадиена или изопрена, отличающемуся тем, что в нем используют указанную каталитическую систему.
Известно, что стереоспецифическая (со)полимеризация сопряженных диенов является очень важным способом в химической промышленности для получения продуктов, которые являются наиболее широко используемыми типами резины.
Указанная стереоспецифическая (со)полимеризация может обеспечить полимеры с различной структурой, т.е. 1,4-транс структуру, 1,4-цис структуру, 1,2 структуру и, в случае асимметричных сопряженных диенов (например, изопрена), 3,4-структуру.
Каталитические системы на основе ванадия известны в течение уже некоторого времени в области (со)полимеризации сопряженных диенов своей способностью обеспечивать диеновые (со)полимеры с 1,4-транс структурой и являются наиболее важными системами для получения 1,4-транс полибутадиена, как описано, например, в: Porri L. et al., "Comprehensive Polymer Science" (1989), Eastmond G. C. et al. Eds., Pergamon Press, Oxford, UK, Vol. 4, Part II, pag. 53-108.
Гетерогенные каталитические системы, полученные путем каомбинации галогенидов ванадия (например, хлорида ванадия(III) (VCl3), хлорида ванадия(IV) (VCl4)) с соединениями алкилалюминия (например, триэтил-алюминием (AlEt3), хлоридом диэтилалюминия (AlEt2Cl)), обеспечивают кристаллический 1,4-транс полибутадиен (содержание 1,4-транс звеньев равно 97%-100%) с высокой молекулярной массой и температурой плавления (Тпл) около 145°С. Дополнительные подробности об указанных каталитических системах можно найти, например, в: Natta G. et al., "La Chimica e L'Industria" (1958), Vol. 40, pag. 362 и "Chemical Abstract" (1959), Vol. 53, pag. 195; Natta G. et al., "La Chimica e L'Industria" (1959), Vol. 41, pag. 116 и "Chemical Abstract" (1959), Vol. 53, pag. 15619.
Полибутадиен с высоким содержанием 1,4-транс звеньев, но с низкой молекулярной массой можно получить с гомогенными каталитическими системами, такими как, например, хлорид ванадия(III) - (три-тетрагидрофуран)/ хлорид диэтилалюминия (VCl3(TГФ)3/AlEt2Cl), ацетилацетонат ванадия(III) / хлорид диэтилалюминия [V(acac)3/AlEt2Cl] и ацетилацетонат ванадия(III) / метилалюмоксан [V(acac)3/MAO]. Дополнительные подробности об указанных каталитических системах можно найти, например, в: Natta G. et al., "Atti Accademia Nazionale dei Lincei - Classe di Scienze fisiche, matematiche e naturali" (1961), Vol. 31(5), pag. 189 и "Chemical Abstract" (1962), Vol. 57, pag. 4848; Porri L. et al., "Die Makromoleculare Chemie" (1963), Vol. 61(1), pag. 90-103; Ricci G. et al., "Polymer Communication" (1991), Vol. 32, pag. 514-517; Ricci G. et al., "Journal of Polymer Science Part A: Polymer Chemistry" (2007), Vol. 45(20), pag. 4635-4646.
Некоторые из указанных выше гомогенных каталитических систем, например, ацетилацетонат ванадия(III) /триэтилалюминий [V(acac)3/AlEt3], потенциально интересны для получения 1,2 полибутадиена, как описано, например, в Natta G. et al., "La Chimica e L'Industria " (1959), Vol. 41, pag. 526 и "Chemical Abstract" (1960), Vol. 54, pag. 1258.
Каталитические системы, полученные путем комбинирования производных циклопентадиенилванадия, как например, хлорид бис(циклопентадиенил)ванадия / метилалюмоксан (Cp2VCl/MAO) и трихлорид циклопентадиенилванадия три-триэтилфосфин/ метилалюмоксан [CpVCl3(PEt3)3/MAO], способны обеспечить полибутадиен с преобладанием 1,4-цис структуры (содержание 1,4-цис звеньев равно примерно 85%). Дополнительные подробности об указанных каталитических системах можно найти, например, в: Ricci G. et al., " Polymer " (1996), Vol. 37(2), pag. 363-365; Porri L. et al., " Metalorganic Catalyst for Synthesis and Polymerization " (1999), Kaminsky W. Ed., Springer-Verlag Berlin Heidelberg, pag. 519-530.
Кроме того, хорошо известно, что каталитические системы на основе ванадия также активны в полимеризации изопрена. В частности, каталитическая система триалкилалюминий / хлорид ванадия(III) (AlR3/VCl3, в которой R = метил, этил, пропил, бутил, предпочтительно этил) обеспечивает полиизопрен с высоким содержанием 1,4-транс звеньев, даже если степень активности является достаточно низкой. Предпочтительно, указанную полимеризацию осуществляют при молярном соотношении Al/V, предпочтительно находящемся в диапазоне от 3 до 6, в присутствии алифатического растворителя (например, н-гептана), при относительно низкой температуре, предпочтительно находящейся в диапазоне от 20°С до 50°С.
Комплексы ванадия с фосфином также известны в литературе.
Например, Bansemer R. L. et al., " Inorganic Chemistry " (1985), Vol. 24(19), pag. 3003-3006, приводят данные о синтезе и характеристиках комплекса VCl3(PMePh2)2, в котором Me = метил и Ph = фенил.
Bultitude G. et al., in "Journal of the Chemical Society, Dalton Transactions" (1986), Issue 10, pag. 2253-2258, приводят данные о синтезе и характеристиках комплекса VCl3(PMePh2)2, в котором Me = метил и Ph = фенил и его аддуктов из ацетонитрила.
Girolami S. G. et al., в "Journal of the Chemical Society, Dalton Transactions" (1985), Issue 7, pag. 1339-1348, приводят данные о синтезе и свойствах двухвалентных комплексов 1,2-бис(диметилфосфино)этана (dmpe), таких как, например, MCl2(dmpe)2 и MMe2(dmpe)2, в которых М = Ti, V, Сr, Мn или Fe.
Поскольку (со)полимеры сопряженных диенов, в частности полибутадиена и полиизопрена, с доминирующим содержанием 1,4-транс и 1,4-цис звеньев могут преимущественно использоваться для получения шин, в частности, покрышек для шин, а также в обувной промышленности (например, в производстве подошв для обуви), изучение новых каталитических систем, способных обеспечить указанные (со)полимеры, все еще представляет большой интерес.
Задача настоящего изобретения состоит в том, чтобы найти новый фосфиновый комплекс ванадия, который можно использовать в каталитической системе, способной обеспечить (со)полимеры сопряженных диенов, такие как, например, линейный или разветвленный полибутадиен или линейный или разветвленный полиизопрен, с доминирующим содержанием 1,4-транс и 1,4-цис звеньев, т.е. с содержанием 1,4-транс и 1,4-цис звеньев ≥ 60%, предпочтительно в диапазоне от 70% до 99%.
Заявитель обнаружил новый фосфиновый комплекс ванадия, имеющий общую формулу (I) или (II), представленную ниже, способный обеспечить (со)полимеры сопряженных диенов, такие как, например, линейный или разветвленный полибутадиен или полиизопрен, с доминирующим содержанием 1,4-транс и 1,4-цис звеньев, т.е. с содержанием 1,4-транс и 1,4-цис звеньев ≥ 60%, предпочтительно в диапазоне от 70% до 99%.
Следовательно, предмет настоящего изобретения представляет собой фосфиновый комплекс ванадия, имеющий общую формулу (I) или (II):
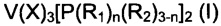
 ,
,
где
- X представляет собой анион, выбранный из галогенов, таких как, например, хлор, бром, иод, предпочтительно хлор; или выбран из следующих групп: тиоцианат, изоцианат, сульфат, кислый сульфат, фосфат, кислый фосфат, карбоксилат, дикарбоксилат;
- R1 одинаковые или отличающиеся друг от друга, представляют собой атом водорода или аллильную группу (СН2=СН-СН2-), или выбран из алкильных групп С1-С20, предпочтительно C1-C15, линейных или разветвленных, возможно галогенированных, возможно замещенных циклоалкильных групп;
- n - целое число в диапазоне от 0 до 3;
- R2, одинаковые или отличающиеся друг от друга, выбраны из возможно замещенных арильных групп;
- R3, одинаковые или отличающиеся друг от друга, представляют собой атом водорода или аллильную группу (СН2=СН-СН2-), или выбраны из алкильных групп C1-C20, предпочтительно С1-С15, линейных или разветвленных, возможно галогенированных, возможно замещенных циклоалкильных групп, возможно замещенных арильных групп;
- R4 представляет собой группу -NR5, в которой R5 представляет собой атом водорода или выбран из C1-C20 алкильных групп, предпочтительно С1-С15, линейных или разветвленных; или R4 представляет собой алкиленовую группу -(СН2) р-, в которой р представляет собой целое число в диапазоне от 1 до 5;
при условии, что, если в общей формуле (I) n равно 1 и R1 представляет собой метил, то R2 отличается от фенила.
Для целей настоящего описания и формулы изобретения, определения числовых диапазонов всегда включают крайние точки, если не указано иное.
Для целей настоящего описания и формулы изобретения, термин "содержащий" также включают термины "который по существу состоит из" или "который состоит из".
Термин "C1-C20 алкильные группы" означает алкильные группы, имеющие от 1 до 20 атомов углерода, линейные или разветвленные. Специфические примеры C1-C20 алкильных групп представляют собой: метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, пентил, гексил, гептил, октил, н-нонил, н-децил, 2-бутилоктил, 5-метилгексил, 4-этилгексил, 2-этилгептил, 2-этилгексил.
Термин "возможно галогенированные C1-C20 алкильные группы" означает алкильные группы, имеющие от 1 до 20 атомов углерода, линейные или разветвленные, насыщенные или ненасыщенные, в которых по меньшей мере один из атомов водорода замещен атомом галогена, таким как, например, фтор, хлор, бром, предпочтительно фтор, хлор. Специфическими примерами C1-C20 алкильных групп, возможно содержащих гетероатомы, являются фторметил, дифторметил, трифторметил, трихлорметил, 2,2,2-трифторэтил, 2,2,2-трихлорэтил, 2,2,3,3-тетрафторпропил, 2,2,3,3,3-пентафторпропил, перфторпентил, перфтороктил, перфтордецил.
Термин "циклоалкильные группы" означает циклоалкильные группы, имеющие от 3 до 30 атомов углерода. Указанные циклоалкильные группы могут быть возможно замещены одной или более группами, одинаковыми или отличающимися друг от друга, выбранными из атомов галогена, гидроксильных групп; C1-C12 алкильных групп; C1-C12 алкоксигрупп; цианогрупп; аминогрупп; нитрогрупп. Специфические примеры циклоалкильных групп представляют собой циклопропил, 2,2-дифторциклопропил, циклобутил, циклопентил, циклогексил, гексаметилциклогексил, пентаметилциклопентил, 2-циклооктилэтил, метил циклогексил, метоксициклогексил, фторциклогексил, фенилциклогексил.
Термин "арильные группы" означает карбоциклические ароматические группы. Указанные карбоциклические ароматические группы могут быть возможно замещены одной или более группами, одинаковыми или отличающимися друг от друга, выбранными из атомов галогена, таких как, например, фтор, хлор, бром; гидроксильных групп; С1-С12 алкильных групп; С1-С12 алкоксигрупп; цианогрупп; аминогрупп; нитрогрупп. Специфические примеры арильных групп представляют собой: фенил, метилфенил, триметилфенил, метоксифенил, гидроксифенил, фенилоксифенил, фторфенил, пентафторфенил, хлорфенил, бромфенил, нитрофенил, диметиламинофенил, нафтил, фенилнафтил, фенантрен, антрацен.
Согласно предпочтительному воплощению настоящего изобретения, в указанном фосфиновом комплексе ванадия, имеющем общую формулу (I) или (II):
- X представляет собой анион, выбранный из галогена, такого как, например, хлор, бром, иод, предпочтительно хлор;
- R1, одинаковые или отличающиеся друг от друга, представляют собой атом водорода или выбраны из С1-С20 алкильных групп, предпочтительно С1-С15, линейных или разветвленных, предпочтительно представляют собой метил, этил, изопропил, трет-бутил; или выбраны из возможно замещенных циклоалкильных групп, предпочтительно представляют собой циклопентил, циклогексил;
- n - целое число от 0 до 3;
- R2, являясь одинаковыми, выбраны из возможно замещенных арильных групп, предпочтительно представляют собой фенил;
- R3, являясь одинаковыми, выбраны из С1-С20 алкильных групп, предпочтительно С1-С15, линейных или разветвленных, предпочтительно представляют собой метил, этил; или выбраны из возможно замещенных арильных групп, предпочтительно представляют собой фенил;
- R4 представляет собой группу -NR5, в которой R5 представляет собой атом водорода; или R4 представляет собой группу - (СН2) р-, в которой р равно 2.
Фосфиновый комплекс ванадия, имеющий общую формулу (I) или (II), можно рассматривать, в соответствии с настоящим изобретением, в любом физическом состоянии, таком как, например, выделенная и очищенная твердая форма, сольватированная форма в подходящем растворителе или нанесенная на подходящие органические или неорганические твердые вещества, предпочтительно имеющие зернистую или порошкообразную физическую форму.
Фосфиновый комплекс ванадия, имеющий общую формулу (I) или (II), можно получить в соответствии со способами, известными в области техники. Например, указанный фосфиновый комплекс ванадия можно получить путем реакции между соединениями ванадия, имеющими общую формулу V(Х)3, в которой X представляет собой атом галогена, такой как, например, хлор, бром, иод, предпочтительно хлор, как таковой или в составе комплексов с простыми эфирами (например, диэтиловым эфиром, тетрагидрофураном (ТГФ), диметоксиэтаном), предпочтительно в составе комплексов с тетрагидрофураном (ТГФ), с фосфинами, выбранными, например, из: трифенилфосфина, трис(пентафторфенил)фосфина, трис(п-трифторметилфенил)фосфина, трис(2,4,6-триметоксифенил)фосфина, трис(2,4,6-триметилфенил)фосфина, дифенилфосфина, трис(о-толил)фосфина, трис(м-толил)фосфина, трис(п-толил)фосфина, трис(о-метоксифенил)фосфина, трис(м-метоксифенил)фосфина, трис(п-метоксифенил)фосфина, трис(2,4-диметилфенил)фосфина, три-1-нафтилфосфина, (о-толил)дифенилфосфина, (метил)дифенилфосфина, (этил)дифенилфосфина, (н-пропил)дифенилфосфина, (изопропил)дифенилфосфина, (аллил)дифенилфосфина, (трет-бутил)дифенилфосфина, (циклогексил)дифенилфосфина, (триметилсилил)дифенилфосфина, ди(метил)фенилфосфина, ди(этил)фенилфосфина, ди(н-пропил)фенилфосфина, ди(трет-бутил)фенилфосфина, ди(циклогексил)фенилфосфина, триэтилфосфина, три(н-пропил)фосфина, три(изопропил)фосфина, три(н-бутил)фосфина, три(аллил)фосфина, три(изобутил)фосфина, три(трет-бутил)фосфина, три(циклопентил)фосфина, три(циклогексил)фосфина, трис(триметилсилил)фосфина, ди(трет-бутил)фосфина, метилди(трет-бутил)фосфина, ди(трет-бутил)изопропилфосфина, ди(трет-бутил)неопентилфосфина, ди(циклопентил)фосфина, ди(циклогексил)фосфина, ди(2-норборнил)фосфина, ди(изобутил)фосфина, трет-бутилди(циклогексил)фосфина, ди(трет-бутил)циклогексилфосфина, бис(диметилфосфино)метана, 1,2-бис(диметилфосфино)этана, 1,2-бис(диэтилфосфино)этана, 1,3-бис(диэтилфосфино)пропана, 1,3-бис(диизопропилфосфино)пропана, бис(дициклогексилфосфино)метана, 1,2-бис(дициклогексилфосфино)этана, 1,3-бис(дициклогексилфосфино)пропана, бис(дифенил-фосфино)метана, 1,2-бис(дифенилфосфино)этана, 1,3-бис(дифенилфосфино)пропана, N,N-бис(дифенилфосфино)амина, 1,2-бис(фенилфосфино)этана, 1,3-бис(фенилфосфино)пропана, причем указанные фосфины используют в стехиометрических количествах, работая, предпочтительно, в присутствии по меньшей мере одного растворителя, который можно выбрать, например, из углеводородных растворителей (например, толуола), хлорированных растворителей (например, дихлорметана), растворителей на основе простых эфиров (например, тетрагидрофурана (ТГФ)), или их смесей, при температуре в диапазоне от комнатной температуры до 110°С, предпочтительно при температуре кипячения растворителя с обратным холодильником. Фосфиновый комплекс ванадия, полученный таким образом, можно впоследствии выделить с помощью способов, известных в данной области техники, таких как, например, осаждение с помощью растворителя с обратной полярностью (например, пентана) с последующим отделением путем фильтрования или декантации и возможным последующим растворением в подходящем растворителе с последующей кристаллизацией при низкой температуре.
Для целей описания и формулы настоящего изобретения выражение "комнатная температура" означает температуру в диапазоне от 20°С до 25°С.
Как указано выше, настоящее изобретение также относится к каталитической системе для (со)полимеризации сопряженных диенов, содержащей указанный фосфиновый комплекс ванадия, имеющий общую формулу (I) или (II).
Следовательно, настоящее изобретение также относится к каталитической системе для (со)полимеризации сопряженных диенов, содержащей:
(a) по меньшей мере один фосфиновый комплекс ванадия, имеющий общую формулу (I) или (II);
(b) по меньшей мере один со-катализатор, выбранный из органических производных алюминия, предпочтительно из:
(b1) соединений алюминия, имеющих общую формулу (III):
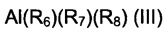 ,
,
где R6 представляет собой атом водорода, или атом фтора, или выбран из С1-С20 алкильных групп, линейных или разветвленных, циклоалкильных групп, арильных групп, алкиларильных групп, арилалкильных групп, алкоксигрупп; R7 и R8, одинаковые или отличающиеся друг от друга, выбраны из С1-С20 алкильных групп, линейных или разветвленных, циклоалкильных групп, арильных групп, алкиларильных групп, арилалкильных групп;
(b2) алюмоксанов, имеющих общую формулу (IV):
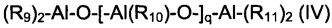 ,
,
где R9, R10 и R11, одинаковые или отличающиеся друг от друга, представляют собой атом водорода, или атом галогена, такой как, например, хлор, бром, иод, фтор; или выбраны из С1-С20 алкильных групп, линейных или разветвленных, циклоалкильных групп, арильных групп, причем указанные группы возможно замещены одним или более атомов кремния или германия; и q - целое число в диапазоне от 0 до 1000;
(b3) органических производных алюминия, частично гидролизованных;
(b4) галогенидов алкилалюминия, имеющих общую формулу (V)
или (VI):
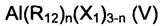
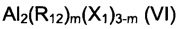 ,
,
где n представляет собой 1 или 2; m - целое число в диапазоне от 1 до 5; R12, одинаковые или отличающиеся друг от друга, выбраны из С1-С20 алкильных групп, линейных или разветвленных; X1 представляет собой атом хлора или брома, предпочтительно хлора; или их смесей.
Специфические примеры соединений алюминия, имеющих общую формулу (III), использующихся в частности для целей настоящего изобретения, представляют собой: гидрид диэтилалюминия, гидрид ди-н-пропил-алюминия, гидрид ди-н-бутил-алюминия, гидрид ди-изо-бутил-алюминия (DIBAH), гидрид дифенилалюминия, гидрид ди-р-толил-алюминия, гидрид дибензилалюминия, гидрид диэтилалюминия, гидрид фенил-н-пропил-алюминия, гидрид р-толил-этил-алюминия, гидрид р-толил-н-пропил-алюминия, гидрид р-толил-изо-пропил-алюминия, гидрид бензилэтилалюминия, гидрид бензил-н-пропил-алюминия, гидрид бензил-изопропилалюминия, этилат диэтилалюминия, этилат диизобутилалюминия, этилат дипропилалюминия, триметилалюминий, триэтилалюминий (TEA), три-н-пропил-алюминий, три-изо-бутил-алюминий (TIBA), три-н-бутил-алюминий, трипентилалюминий, тригексилалюминий, трициклогексилалюминий, триоктилалюминий, трифенилалюминий, три-р-толилалюминий, трибензилалюминий, этилдифенилалюминий, этил-ди-р-толилалюминий, этилдибензилалюминий, диэтилфенилалюминий, ди-этил-р-толилалюминий, диэтилбензилалюминий. Триэтилалюминий (TEA), три-н-пропилалюминий, триизобутилалюминий (TIBA), тригексилалюминий, гидрид диизобутилалюминия (DIBAH), фторид диэтилалюминия являются особенно предпочтительными.
Как известно, алюмоксаны представляют собой соединения, содержащие Al-O-Al связи, с различным соотношением O/Аl, получаемые в соответствии с методиками, известными в уровне техники, такими как, например, путем реакции алкилалюминия или галогенида алкилалюминия в контролируемых условиях с водой или с другими соединениями, содержащими заранее заданные количества доступной воды, как, например, в случае реакции триметилалюминия с гексагидратом сульфата алюминия, пентагидратом сульфата меди или пентагидратом сульфата железа.
Указанные алюмоксаны и, в частности, метилалюмоксан (МАО), представляют собой соединения, которые можно получить путем известных превращений металлоорганических химических веществ, как, например, путем добавления триметилалюминия к суспензии гексагидрата сульфата алюминия в гексане.
Специфические примеры алюмоксанов, имеющих общую формулу (IV), использующиеся в частности для целей настоящего изобретения, представляют собой: метилалюмоксан (МАО), этилалюмоксан, н-бутил-алюмоксан, тетраизобутилалюмоксан (TIBAO), трет-бутилалюмоксан, тетра-(2,4,4-триметилпентил)алюмоксан (TIOАО), тетра-(2,3-диметилбутил)алюмоксан (TDMBAO), тетра-(2,3,3-триметилбутил)алюмоксан (ТТМВАО). Метилалюмоксан (МАО), как таковой, или в "сухой" форме (МАО-сухой), является особенно предпочтительным.
Дополнительные подробности об алюмоксанах, имеющих общую формулу (IV), можно найти в международной патентной заявке WO 2011/061151.
Предпочтительно, частично гидролизованные органические производные алюминия (b3) выбраны из соединений алюминия, имеющих общую формулу (III), насыщенный по меньшей мере одним протон-донорным соединением, причем соединение алюминия, имеющее общую формулу (III), и протон-донорное соединение используют в молярном соотношении от 0,001:1 до 0,2:1. Предпочтительно, указанное протон-донорное соединение можно выбрать, например, из воды, спиртов, таких как, например, метанол, этанол, изопропиловый спирт, н-пропиловый спирт, трет-бутанол, изобутиловый спирт, н-бутиловый спирт; спирты с более высокой молекулярной массой, такие как, например, 1-деканол, 2-ундеканол; карбоновой кислоты, такой как, например, стеариновая кислота; или из их смесей. Вода является наиболее предпочтительной.
Специфические примеры галогенидов алкилалюминия, имеющих общую формулу (V) или (VI), представляют собой: хлорид диэтилалюминия (AlEt2Cl), хлорид диметилалюминия (AlMe2Cl), дихлорид этилалюминия (AlEtCl2), хлорид диизобутилалюминия [Al(i-Bu)2Cl); сесквихлорид этилалюминия (Al2Et3Cl3), сесквихлорид метилалюминия (Аl2Ме3Сl3).
В целом, образование каталитической системы, содержащей фосфиновый комплекс ванадия, имеющий общую формулу (I) или (II), и со-катализатор (b), предпочтительно проводят в инертной жидкой среде, более предпочтительно в углеводородном растворителе. Выбор фосфинового комплекса ванадия, имеющего общую формулу (I) или (II), и со-катализатора (b), а также конкретной используемой методики может меняться в зависимости от молекулярных структур и целевого результата, согласно известному из релевантной литературы, доступной специалисту в данной области техники, для комплексов других переходных металлов с лигандами различной природы, как например, в: Ricci G. et al., "Advances in Organometallic Chemistry Research" (2007), Yamamoto K. Ed., Nova Science Publisher, Inc., USA, pg. 1-36; Ricci G. et al., "Coordination Chemistry Reviews"(2010), Vol. 254, pg. 661-676; Ricci G. et al., "Ferrocenes: Compounds, Properties and Applications" (2011), Elisabeth S. Phillips Ed., Nova Science Publisher, Inc., USA, pg. 273-313; Ricci G. et al., "Chromium: Environmental, Medical and Material Studies" (2011), Margaret P. Salden Ed., Nova Science Publisher, Inc., USA, pg. 121-1406; Ricci G. et al., "Cobalt: Characteristics, Compounds, and Applications" (2011), Lucas J. Vidmar Ed., Nova Science Publisher, Inc., USA, pg. 39-81; Ricci G. et al., “Phosphorus: Properties, Health effects and Environment” (2012), Ming Yue Chen и Da-Xia Yang Eds., Nova Science Publisher, Inc., USA, pg. 53-94.
Предпочтительно, если, в соответствии с настоящим изобретением, использовать для образования каталитической системы для (со)полимеризации (со)катализаторы (b), их можно привести в контакт с фосфиновым комплексом ванадия, имеющим общую формулу (I) или (II), в такой пропорции, что молярное соотношение между ванадием, присутствующим в фосфиновом комплексе ванадия, имеющем общую формулу (I) или (II), и алюминием, присутствующим в (со)катализаторах (b), может составлять от 1 до 10000, предпочтительно от 50 до 1000. Последовательность, в которой фосфиновый комплекс ванадия, имеющий общую формулу (I) или (II), и (со)катализатор приводят в контакт один с другим, не является особенно критичной.
Для целей описания и формулы изобретения настоящего изобретения термины "моль" и "молярное соотношение" используют и в отношении соединений, состоящих из молекул, и в отношении атомов и ионов, не учитывая, что для последних термины грамм-атом или атомное соотношение более уместны и несмотря на то что они более точны с точки зрения научной терминологии.
Для целей настоящего изобретения, к указанной выше каталитической системе можно добавлять другие добавки или компоненты так, чтобы она удовлетворяла специфическим требованиям на практике. Следовательно, каталитические системы, полученные таким образом, можно считать включенными в объем настоящего изобретения. Добавки и/или компоненты, которые можно добавлять в процессе получения или смешивания каталитической системы по настоящему изобретению, представляют собой, например, инертные растворители, такие как, например, алифатические и/или ароматические углеводороды; алифатические и/или ароматические простые эфиры; слабо координирующие добавки (например, основания Льюиса), выбранные, например, из неполимеризуемых олефинов; стерически затрудненные или обедненные электронной плотностью простые эфиры; галогенирующие агенты, такие как, например, галогениды кремния, галогенированные углеводороды, предпочтительно хлорированные; или их смеси.
Указанную каталитическую систему можно получить, как уже указано выше, в соответствии со способами, известными в уровне техники.
Например, указанную каталитическую систему можно получить отдельно (предварительно) и впоследствии ввести в среду (со)полимеризации. В этом случае, указанную каталитическую систему можно получить путем взаимодействия по меньшей мере одного фосфинового комплекса ванадия (а), имеющего общую формулу (I) или (II), с по меньшей мере одним со-катализатором (b), возможно в присутствии других добавок или компонентов, выбранных из указанных выше, в присутствии растворителя, такого как, например, толуол, гептан, при температуре от 20°С до 60°С, в течение времени от 10 секунд до 10 часов, предпочтительно от 30 секунд до 5 часов.
Альтернативно, указанную каталитическую систему можно получить in situ, т.е. непосредственно в среде (со)полимеризации. В этом случае, указанную каталитическую систему можно получить путем раздельного введения фосфинового комплекса ванадия (а), имеющего общую формулу (I) или (II), со-катализатора (b) и заранее выбранного сопряженного диена(ов) для участия в (со)полимеризации, работая в условиях, в которых проводят (со)полимеризацию.
Дополнительные подробности по получению указанной каталитической системы можно найти в приведенных ниже примерах.
Для целей настоящего изобретения, указанные выше каталитические системы можно также наносить на подложку из инертных твердых веществ, предпочтительно включающих оксиды кремния и/или алюминия, такие как, например, диоксид кремния, оксид алюминия или силикоалюминаты. Для нанесения на подложку указанных каталитических систем можно использовать известные методики нанесения на подложку, обычно включающие контакт в подходящей инертной жидкой среде между подложкой, возможно активированной путем нагревания до температур выше 200°С, и одним или обоими компонентами (а) и (b) каталитической системы согласно настоящему изобретению. Для целей настоящего изобретения нет необходимости, чтобы оба компонента были нанесены на подложку, так как только фосфиновый комплекс ванадия (а), имеющий общую формулу (I) или (II), или со-катализатор (b) может присутствовать на поверхности подложки. В последнем случае, компонент, отсутствующий на поверхности, впоследствии приводится в контакт с компонентом, нанесенным на подложку, когда нужно получить активный катализатор путем полимеризации.
Объем настоящего изобретения также включает фосфиновый комплекс ванадия, имеющий общую формулу (I) или (II), и каталитические системы на его основе, которые нанесены на подложку из твердого вещества путем функциональной модификации последнего и образования ковалетной связи между твердым веществом и фосфиновым комплексом ванадия, имеющим общую формулу (I) или (II).
Кроме того, настоящее изобретение относится к способу (со)полимеризации сопряженных диенов, отличающемуся тем, что он использует указанную каталитическую систему.
Количество фосфинового комплекса ванадия (а), имеющего общую формулу (I) или (II), и со-катализатора (b), которые можно использовать в (со)полимеризации сопряженных диенов изменяется в соответствии с проводимым способом (со)полимеризации. Указанное количество, однако, должно быть таким, чтобы молярное соотношение ванадия (V), присутствующего в фосфиновом комплексе ванадия, имеющем общую формулу (I) или (II), к металлу, присутствующему в со-катализаторе (b), т.е. алюминию, находилось в пределах значений, указанных выше.
Специфические примеры сопряженных диенов, которые можно (со)полимеризовать с использованием каталитической системы по настоящему изобретению, представляют собой: 1,3-бутадиен, 2-метил-1,3-бутадиен (изопрен), 2,3-диметил-1,3-бутадиен, 1,3-пентадиен, 1,3-гексадиен, цикло-1,3-гексадиен. 1,3-Бутадиен и изопрен являются предпочтительными. Указанные выше (со)полимеризуемые сопряженные диены можно использовать по отдельности или в смеси с двумя или более диенами. В последнем случае, т.е. при использовании смеси двух или более диенов, получают сополимер.
Согласно особенно предпочтительному воплощению, настоящее изобретение относится к способу полимеризации 1,3-бутадиена или изопрена, отличающемуся тем, что он использует указанную каталитическую систему.
В целом, указанную (со)полимеризацию можно проводить в присутствии растворителя полимеризации, обычно выбранного из инертных органических растворителей, таких как, например, насыщенные алифатические углеводороды, такие как, например, бутан, пентан, гексан, гептан или их смеси; насыщенные циклоалифатические углеводороды, такие как, например, циклопентан, циклогексан или их смеси; моноолефины, такие как, например, бутен-1, бутен-2 или их смеси; ароматические углеводороды, такие как, например, бензол, толуол, ксилол или их смеси; галогенированные углеводороды, такие как, например, метиленхлорид, хлороформ, четыреххлористый углерод, трихлорэтилен, перхлорэтилен, 1,2-дихлорэтан, хлорбензол, бромбензол, хлортолуол или их смеси. Предпочтительно (со)растворитель полимеризации выбран из ароматических или галогенированных углеводородов.
Альтернативно, указанную (со)полимеризацию можно проводить с использованием в качестве (со)растворителя полимеризации того же сопряженного диена(ов), который нужно (со)полимеризовать, в соответствии со способом, известным как "объемный способ".
Обычно, концентрация (со)полимеризуемого диена в указанном (со)растворителе полимеризации составляет от 5% масс, до 50% масс, предпочтительно от 10% масс, до 20% масс, относительно общей массы смеси сопряженного диена и инертного органического растворителя.
Обычно указанную (со)полимеризацию можно проводить при температуре от -70°С до +100°С, предпочтительно от -20°С до +80°С.
Что касается давления, предпочттельно работать при давлении (со)полимеризуемых компонентов смеси.
Указанную (со)полимеризацию можно проводить непрерывно и партиями.
Как указано выше, указанный способ позволяет получить (со)полимеры сопряженных диенов, такие как, например, линейный или разветвленный полибутадиен или линейный или разветвленный полиизопрен с преимущественным содержанием 1,4-транс и 1,4-цис звеньев, т.е. имеющие содержание 1,4-транс и 1,4-цис звеньев ≥ 60%, предпочтительно от 70% до 99%.
Для целей более ясного понимания и практического осуществления настоящего изобретения ниже приведены некоторые его иллюстративные и неограничивающие примеры.
ПРИМЕРЫ
Реагенты и материалы
Ниже приведены реагенты и материалы, используемые в следующих примерах изобретения, виды их возможной предварительной обработки и их производитель:
- трихлортрис(тетрагидрофуранил)ванадий [VСl3(ТГФ)3]: получен, как описано Manzer L. Е. et al.," Inorganic S ynthesis " (1982), Vol. 21, pag. 135-140;
- (метил)дифенилфосфин (Strem): степень чистоты 99%, использовали как есть;
- (этил)дифенилфосфин (Strem): степень чистоты 99%, использовали как есть;
- (изо-пропил)дифенилфосфин (Aldrich): степень чистоты 97%, использовали как есть;
- (циклогексил)дифенилфосфин (Strem): степень чистоты 98%, использовали как есть;
- трифенилфосфин (Strem): степень чистоты 99%, использовали как есть;
- три(циклогексил)фосфин (Strem): степень чистоты 97%, использовали как есть;
- три(циклопентил)фосфин (Strem): степень чистоты ≥ 95%, использовали как есть;
- ди(циклогексил)фенилфосфин (Aldrich): степень чистоты 95%, использовали как есть;
- три(трет)-бутил)фосфин (Strem): степень чистоты 99%, использовали как есть;
- 1,2-бис(диметилфосфино)этан (Strem): степень чистоты 98%, использовали как есть;
- 1,2-бис(диэтилфосфино)этан (Strem): степень чистоты 98%, использовали как есть;
- N,Н-бис(дифенилфосфино)амин (Strem): степень чистоты мин. 98%, использовали как есть;
- толуол (Fluka): степень чистоты > 99,5%, кипятили над натрием (Na) с обратным холодильником в течение примерно 8 часов, затем перегоняли и хранили над молекулярными ситами под азотом;
- пентан (Fluka): степень чистоты 99%, кипятили над натрием/калием (Na/K) в течение примерно 8 часов, затем перегоняли и хранили над молекулярными ситами под азотом;
- гептан (Aldrich): использовали как есть;
- 1,3-бутадиен (Air Liquide): чистый, ≥ 99,5%, отбирали из баллона испарением перед каждым получением, сушили путем пропускания через колонку, набитую молекулярными ситами, и конденсировали внутри реактора, который предварительно охлаждали до -20°С;
- изопрен (Aldrich): чистый, ≥ 99%, кипятили с обратным холодильником над гидридом кальция в течение 2 часов, затем перегоняли из ловушки в ловушку (англ. - "trap-to-trap") и хранили в атмосфере азота при 4°С в холодильнике;
- метилалюмоксан (МАО) (раствор в толуоле 10% масс.) (Aldrich): использовали как есть, или в "сухой" форме (МАО-сухой), полученной путем удаления свободного триметилалюминия одновременно с растворителем из указанного раствора в толуоле под вакуумом и сушки остатка, также полученного под вакуумом;
- метанол (Carlo Erba, RPE): использовали как есть или возможно обезвоженным путем перегонки над магнием (Мг);
- хлороводородная кислота в водном растворе концентрацией 37% (Aldrich): использовали как есть;
- 1,2-дихлорбензол (Aldrich): степень чистоты 99%, кипятили с обратным холодильником над гидридом кальция (СаН2) в течение примерно 8 часов, затем перегоняли и хранили над молекулярными ситами под азотом;
- дейтерированный тетрахлорэтилен (C2D2Cl4) (Acros): использовали как есть;
- дейтерированный хлороформ (CDCl3) (Acros): использовали как есть.
Использовали методики анализа и характеристики, приведенные ниже.
Элементный анализ
а) Определение ванадия (V)
Чтобы определить массовое содержание ванадия (V) в фосфиновых комплексах ванадия по настоящему изобретению, точно взвешенную навеску примерно 30 мг - 50 мг образца, работая в сухом боксе под током азота, помещали в платиновый тигль объемом приблизительно 30 мл вместе со смесью из 1 мл 40%-ной плавиковой кислоты (HF) (Aldrich), 0,25 мл 96%-ной серной кислоты (H2SO4) и 1 мл 70%-ной азотной кислоты (HNO3) (Aldrich). Затем тигль нагревали на горячем нагревательном столике, повышая температуру до появления белых сернистых паров (около 200°С). Полученную таким образом смесь охлаждали до комнатной температуры (20°С-25°С), добавляли 1 мл 70%-ной азотной кислоты (HNO3) (Aldrich) и снова оставляли до появления паров. После повторения указанной последоватлеьности еще дважды получили прозрачный, практически бесцветный раствор. Затем на холоду добавляли 1 мл 70%-ной азотной кислоты (HNO3) (Aldrich) и потом около 15 мл воды, затем нагревали до 80°С в течение около 30 минут. Полученный таким образом образец разбавляли водой чистоты MilliQ до массы 50 г, определяли точную массу и получали раствор, который подвергали аналитическому определению с помощью инструментального метода с использованием ИСП-ОЭС спектрометра (ионизационно связанная плазма с оптической эмиссией) Thermo Optek IRIS Advantage Duo путем сравнения с растворами известной концентрации. Для этой цели, для каждого аналита получали калибовочную кривую в диапазоне концентраций 0 ррm (млн долей) - 10 ррm (млн долей) путем измерения растворов с известным титром, полученных путем разбавления сертифицированных растворов по массе.
Потом перед проведением спектрофотометрических измерений раствор образца, полученный, как описано выше, снова разбавляли по массе для получения концентраций, близких к концентрациям стандартных образцов. Все образцы получали в двойном количестве. Результаты считали допустимыми, если относительное результаты отдельных испытаний отличались не более, чем на 2% от их среднего значения.
b) Определение хлора
Для осуществления указанной цели, образцы фосфиновых комплексов ванадия по настоящему изобретению, около 30 мг - 50 мг, были точно взвешены в стеклянные лабораторные стаканы объемом 100 мл в сухом боксе под током азота. Добавляли 2 г карбоната натрия (Nа2СО3) (Aldrich) и, вне сухого бокса, 50 мл воды MilliQ. Доводили до кипения на горячем нагревательном столике при перемешивании магнитной мешалкой в течение примерно 30 минут. Оставляли охлаждаться, затем добавляли разбавленную 1/5 серную кислоту (H2SO4) (Aldrich) до кислой среды и затем титровали 0,1 н-ным раствором нитрата серебра (AgNO3) (Aldrich) с помощью потенциометрического титратора.
с) Определение углерода, водорода и азота
Определение содержания углерода, водорода и азота в фосфиновых комплексах ванадия по настоящему изобретению проводили с помощью автоматического анализатора Carlo Erba Mod. 1106.
Рентгеноструктурный анализ (XRD)
Для этой цели образцы фосфиновых комплексов ванадия по настоящему изобретению, около 1 г, загружали на пористую перегородку горячего экстрактора для твердых веществ и непрерывно экстрагировали кипящим пентаном в течение примерно 2 дней в получением кристаллических веществ (отдельных кристаллов), которые анализировали методом рентгеноструктурного анализа (XRD) с помощью дифрактометра Bruker AXS Smart Apex II, снабженного ПЗС (CCD) детектором (детектор, основой которого является прибор с зарядовой связью) и секцией Oxford Cryostram для потока азота, присоединенной к основанию гониометра, для обеспечения сбора данных при различных температурах, т.е. в диапазоне температур от 100 K (-173.15°С) до 300 K (26.85°С): рабочие условия приведены в таблице 1 и в таблице 2.
Таблица 1 и таблица 2 также содержат кристаллографические характеристики анализируемых образцов.
Спектры ЯМР 13С и 1Н
Спектры ЯМР 13С и 1Н записывали на спектрометре ядерного магнитного резонанса Bruker Avance 400, используя дейтерированные тетрахлорэтилен (C2D2Cl4) при 103°С и гексаметилдисилоксан (ГМДС) (Aldrich) в качестве внутреннего стандарта или используя дейтерированный хлороформ (CDCl3) при 25°С и тетраметилсилан (ТМС) (Aldrich) в качестве внутреннего стандарта. Для этой цели полимерные растворы использовали с концентрацией, равной 10% масс, относительно общей массы полимерного раствора.
Микроструктуру полимеров определяли путем анализа вышеуказанных спектров на основании литературных данных Mochel, V. D., в "Journal of Polymer Science Part A-1: Polymer Chemistry" (1972), Vol. 10, Issue 4, pag. 1009-1018, для полибутадиена и Sato H. et al., в "Journal of Polymer Science: Polymer Chemistry Edition" (1979), Vol. 17, Issue 11, pag. 3551-3558, для полиизопрена.
ИК спектры с Фурье преобразованием
ИК спектры с Фурье преобразованием записывали на спектрометрах Thermo Nicolet Nexus 670 и Bruker IFS 48.
ИК спектры с Фурье преобразованием полимеров получали для полимерных пленок, нанесенных на таблетки бромида калия (KВr), причем указанные пленки получали путем нанесения раствора анализируемого полимера в горячем 1,2-дихлорбензоле. Концентрация анализруемых полимерных растворов была равна 10% масс. относительно общей массы полимерного раствора.
Определение молекулярной массы
Определение молекулярной массы (MW) полученных полимеров осуществляли методом гель-проникающей хроматографии ГПХ в следующих рабочих условиях:
- насос Agilent 1100;
- детектор Agilent 1100 I.R.;
- колонки PL Mixed-A;
- растворитель/элюент: тетрагидрофуран (ТГФ) (Aldrich);
- расход: 1 мл/мин;
- температура: 25°С;
- способ вычисления молекулярной массы: унверсальный способ калибровки.
В результате получали среднемассовую молекулярную массу (Mw) и коэффициент полидисперсности (PDI), соответствующий соотношению Mw/Mn (Мn = среднечисловая молекулярная масса).
ПРИМЕР 1
Синтез VCl3(PMePh2)2 [образец ММ261]
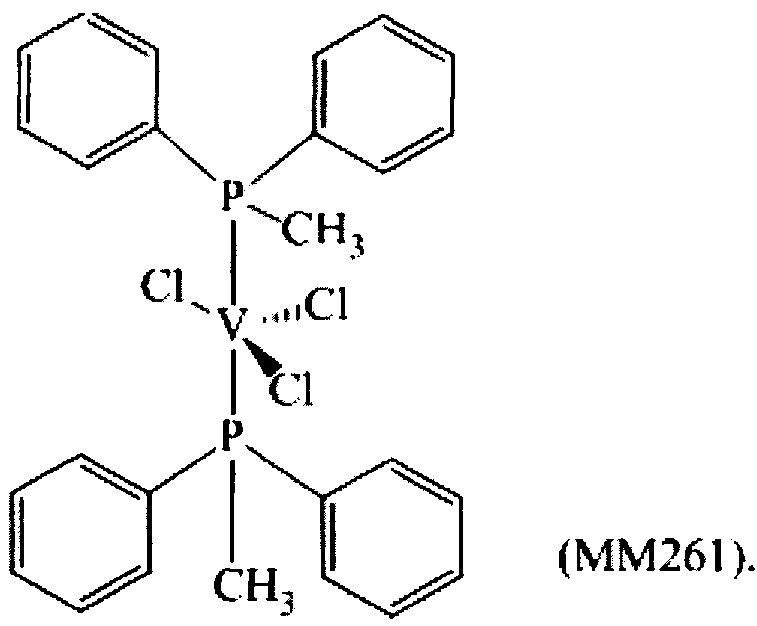
1,02 г (2,75×100-3 моль) трихлортрис(тетрагидрофуран)ванадия [VСl3(ТГФ)3], 15 мл толуола и затем 2,19 г (1,10×10-2 моль) (метил)дифенилфосфина (P/V молярное соотношение = 4) помещали в колбу с отводом объемом 100 мл. Полученную смесь выдерживали при интенсивном перемешивании при комнатной температуре в течение 15 минут и затем нагревали при кипячении с обратным холодильником в течение 3 часов. Полученную суспензию отфильтровывали в горячем виде (60°С) и собранную фракцию концентрировали под вакуумом при комнатной температуре. Затем по каплям при перемешивании добавляли около 50 мл пентана, получая осаждение порошка лилового цвета. Примерно через 3 часа все отфильтровывали и полученный твердый остаток светло-лилового цвета промывали пентаном (50 мл) и сушили под вакуумом при комнатной температуре с получением 1,476 г (конверсия относительно исходного [VСl3(ТГФ)3]=96,3%) комплекса VCl3(PMePh2)2 (молекулярная масса = 557,53 гхмоль-1).
Элементный анализ [получено (вычислено)] С: 56,20% (55,99%); Н: 4,60% (4,70%); Cl: 19,20% (19,07%); Р: 11,10% (11,11%); V: 9,20% (9,13%).
На Фиг. 1 показана структурная формула, полученная методом рентгено-структурного анализа полученного комплекса VСl3(РМеРh2)2.
В Таблице 1 и Таблице 2 представлены кристаллографические данные полученного комплекса VСl3(РМеРh2)2.
ПРИМЕР 2
Синтез VCl3(PEtPh2)2 [образец G1298]
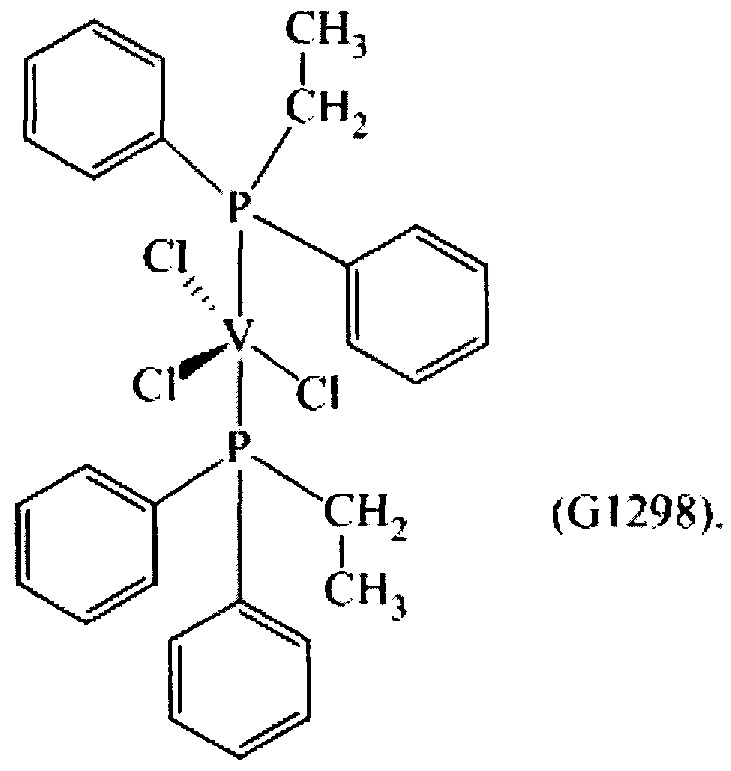
1,28 г (3,42×10-3 моль) трихлортрис(тетрагидрофуран)ванадия [VСl3(ТГФ)3], 15 мл толуола и затем 2,90 г (1,37×10-2 моль) (этил)дифенилфосфина (Р/V молярное соотношение = 4) помещали в колбу с отводом объемом 100 мл. Полученную смесь выдерживали при интенсивном перемешивании при комнатной температуре в течение 15 минут и затем нагревали при кипячении с обратным холодильником в течение 1 часа. Полученную суспензию отфильтровывали в горячем виде (60°С) и собранную фракцию концентрировали под вакуумом при комнатной температуре. Затем по каплям при перемешивании добавляли около 50 мл пентана, получая осаждение порошка сиреневого цвета. Примерно через 3 часа все отфильтровывали и полученный твердый остаток серо-розового цвета промывали пентаном (50 мл) и сушили под вакуумом при комнатной температуре с получением 1,8226 г (конверсия относительно исходного [VСl3(ТГФ)3]=91,0%) комплекса VCl3(PEtPh2)2 (молекулярная масса = 585,79 гхмоль-1).
Элементный анализ [получено (вычислено)] С: 57,40% (57,41%); Н: 5,10% (5,16%); Cl: 18,20% (18,16%); Р: 10,07% (10,58%); V: 8,60% (8,70%).
На Фиг. 2 показана структурная формула, полученная методом рентгено-структурного анализа полученного комплекса VCl3(PEtPh2)2.
В Таблице 1 и Таблице 2 представлены кристаллографические данные полученного комплекса VCl3(PEtPh2)2.
ПРИМЕР 3
Синтез VCl3(PiPrPh2)2 [образец G1325]
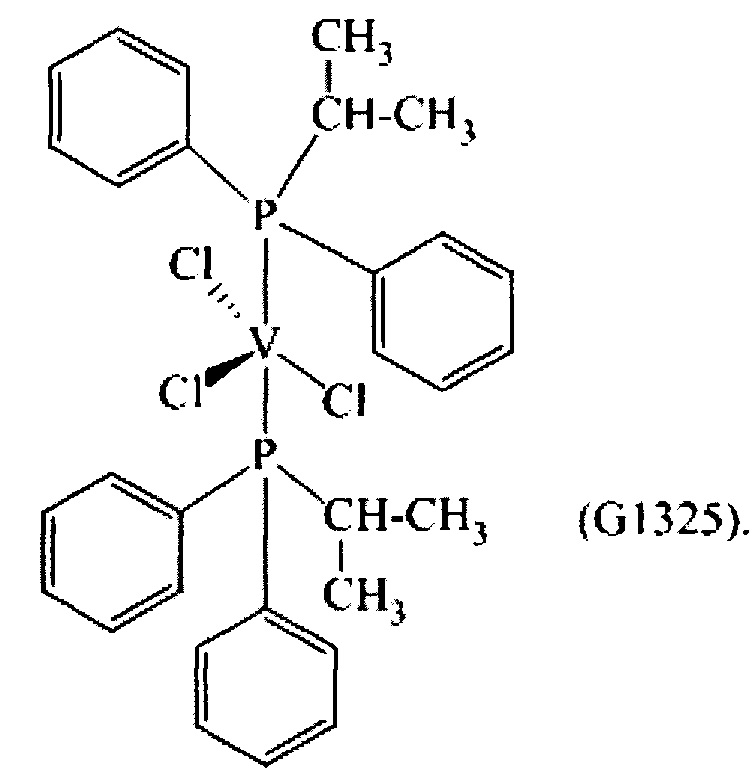
1,28 г (3,42×10-3 моль) трихлортрис(тетрагидрофуран)ванадия [VСl3(ТГФ)3], 15 мл толуола и затем 2,90 г (1,37×10-2 моль) (изо-пропил)дифенилфосфина (P/V молярное соотношение = 4) помещали в колбу с отводом объемом 100 мл. Полученную смесь выдерживали при интенсивном перемешивании при комнатной температуре в течение 15 минут и затем нагревали при кипячении с обратным холодильником в течение 1 часа. Полученную суспензию отфильтровывали в горячем виде (60°С) и собранную фракцию концентрировали под вакуумом при комнатной температуре. Затем по каплям при перемешивании добавляли около 50 мл пентана, получая осаждение порошка сиреневого цвета. Примерно через 3 часа все отфильтровывали и полученный твердый остаток серо-розового цвета промывали пентаном (50 мл) и сушили под вакуумом при комнатной температуре с получением 1,8226 г (конверсия относительно исходного [VСl3(ТГФ)3]=91,0%) комплекса VCl3(PiPh2)2 (молекулярная масса = 585,79 гхмоль-1).
Элементный анализ [получено (вычислено)] С: 57,40% (57,41%); Н: 5,10% (5,16%); Cl: 18,20% (18,16%); Р: 10,07% (10,58%); V: 8,60% (8,70%).
ПРИМЕР 4
Синтез VCl3(РСуРh2)2 [образец ММ300]
0,86 г (2,30×10-3 моль) трихлортрис(тетрагидрофуран)ванадия [VСl3(ТГФ)3], 20 мл толуола и затем 2,40 г (9,0×10-3 моль) дифенил(циклогексил)фосфина (P/V молярное соотношение = 4) помещали в колбу с отводом объемом 100 мл. Полученную смесь выдерживали при интенсивном перемешивании при комнатной температуре в течение 60 минут и затем нагревали при кипячении с обратным холодильником в течение 1 часа. Полученную суспензию отфильтровывали в горячем виде (60°С) и собранную фракцию концентрировали под вакуумом при комнатной температуре. Затем по каплям при перемешивании добавляли около 50 мл пентана, получая осаждение темного порошка. Примерно через 3 часа все отфильтровывали и полученный твердый остаток светлого серо-голубого цвета промывали пентаном (50 мл) и сушили под вакуумом при комнатной температуре с полуением 1,30 г (конверсия относительно исходного [VСl3(ТГФ)3]=81,4%) комплекса VCl3(РСуРh2)2 (молекулярная масса = 693,97 гхмоль-1).
Элементный анализ [получено (вычислено)] С: 62,40% (62,31%); Н: 6,30% (6,10%); Cl: 15,50% (15,33%); Р: 9,0% (8,93%); V: 7,20% (7,34%).
ПРИМЕР 5
Синтез VCl3(PPh3)2 [образец ММ295]
1,0 г (2,66×10-3 моль) трихлортрис(тетрагидрофуран)ванадия [VСl3(ТГФ)3], 10 мл толуола и затем 2,80 г (1,06×10-2 моль) трифенилфосфина (Р/V молярное соотношение = 4) помещали в колбу с отводом объемом 100 мл. Полученную смесь выдерживали при интенсивном перемешивании при комнатной температуре в течение 60 минут и затем нагревали при кипячении с обратным холодильником в течение 3 часов. Полученную суспензию отфильтровывали в горячем виде (60°С) и собранную фракцию концентрировали под вакуумом при комнатной температуре. Затем по каплям при перемешивании добавляли около 50 мл пентана, получая осаждение темного порошка. Примерно через 3 часа все отфильтровывали и полученный твердый остаток насыщенного темно-лилового цвета промывали пентаном (50 мл) и сушили под вакуумом при комнатной температуре с получением 1,50 г (конверсия относительно исходного [VСl3(ТГФ)3]=82,7%) комплекса VCl3(PPh3)2 (молекулярная масса = 681,87 гхмоль-1).
Элементный анализ [получено (вычислено)] С: 63,30% (63,41%); Н: 4,50% (4,43%); Cl: 15,50% (15,60%); Р: 9,0% (9,08%); V: 7,60% (7,47%).
ПРИМЕР 6
Синтез VСl3(РСу3)2 [образец ММ370]
0,827 г (2,20×10-3 моль) трихлортрис(тетрагидрофуран)ванадия VCl3(ТГФ)3], 18 мл толуола и затем 2,47 г (8,82×10-2 моль) три(циклогексил)фосфина (P/V молярное соотношение = 4) помещали в колбу с отводом объемом 100 мл. Полученную смесь выдерживали при интенсивном перемешивании при комнатной температуре в течение 24 часов. Полученную суспензию отфильтровывали в горячем виде (60°С) и собранную фракцию концентрировали под вакуумом при комнатной температуре. Затем по каплям при перемешивании добавляли около 50 мл пентана, получая осаждение темного порошка. Примерно через 3 часа все отфильтровывали и полученный твердый остаток лилового цвета промывали пентаном (50 мл) и сушили под вакуумом при комнатной температуре с получением 0,387 г (конверсия относительно исходного [VСl3(ТГФ)3]=25,6%) комплекса VCl3(PCy3)2 (молекулярная масса = 718,16 гхмоль-1).
Элементный анализ [получено (вычислено)] С: 60,30% (60,21%); Н: 9,20% (9,26%); Cl: 14.70% (14.81%); Р: 8,70% (8,63%); V: 7,30% (7,09%).
ПРИМЕР 7
Синтез VCl3(PCyp3)2 [образец G1286]
0,88 г (2,34×10-3 моль) трихлортрис(тетрагидрофуран)ванадия [VСl3(ТГФ)3], 10 мл толуола и затем 2,23 г (9,36×10-3 моль) три(циклопентил)фосфина (Р/V молярное соотношение = 4) помещали в колбу с отводом объемом 100 мл. Полученную смесь выдерживали при интенсивном перемешивании при комнатной температуре в течение 15 минут и затем нагревали при кипячении с обратным холодильником в течение 3 часов. Полученную суспензию отфильтровывали в горячем виде (60°С) и собранную фракцию концентрировали под вакуумом при комнатной температуре. Затем по каплям при перемешивании добавляли около 50 мл пентана, получая осаждение лилового порошка. Примерно через 3 часа все отфильтровывали и полученный твердый остаток лилового цвета промывали пентаном (50 мл) и сушили под вакуумом при комнатной температуре с получением 0,802 г (конверсия относительно исходного [VСl3(ТГФ)3]=54,1%) комплекса VСl3(РСур3)2 (молекулярная масса = 634,0 гхмоль-1).
Элементный анализ [получено (вычислено)] С: 56,90% (56,83%); Н: 8,70% (8,59%); Cl: 16,70% (16,78%); Р: 9,80% (9,77%); V: 8,0% (8,03%).
На Фиг. 3 показана структурная формула, полученная методом рентгено-структурного анализа полученного комплекса VСl3(РСур3)2.
В Таблице 1 и Таблице 2 представлены кристаллографические данные полученного комплекса VCl3(РСур3)2.
ПРИМЕР 8
Синтез VCl3(PCy2H)2 [образец G1303]
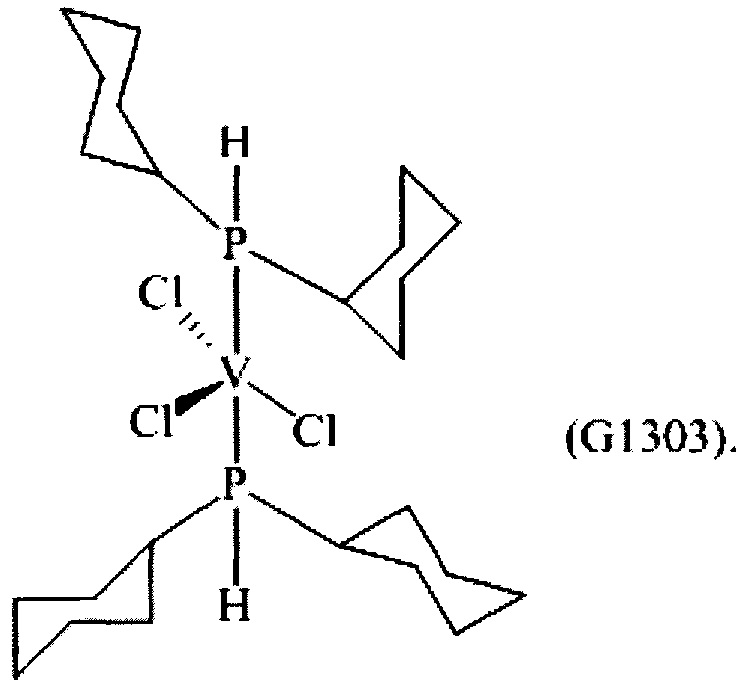
0,955 г (2,0×10-3 моль) трихлортрис(тетрагидрофуран)ванадия [VСl3(ТГФ)3], 10 мл толуола и затем 1,5863 г (8,0×10-3 моль) ди(циклогексил)фосфина (P/V молярное соотношение = 4) помещали в колбу с отводом объемом 100 мл. Полученную смесь выдерживали при интенсивном перемешивании при комнатной температуре в течение 60 минут и затем нагревали при кипячении с обратным холодильником в течение 3 часов. Полученную суспензию отфильтровывали в горячем виде (60°С) и собранную фракцию концентрировали под вакуумом при комнатной температуре. Затем по каплям при перемешивании добавляли около 50 мл пентана, получая осаждение темного порошка. Примерно через 3 часа все отфильтровывали и полученный твердый остаток светло-коричневого цвета промывали пентаном (50 мл) и сушили под вакуумом при комнатной температуре с получением 0,3768 г (конверсия относительно исходного [VСl3(ТГФ)3]=42,0%) комплекса VСl3(РСу2Н)2 (молекулярная масса = 553,87 гхмоль-1).
Элементный анализ [получено (вычислено)] С: 52,20% (52,04%); Н: 8,50% (8,37%); Cl: 19,30% (19,20%); Р: 11.10% (11,18%); V: 9,40% (9,20%).
ПРИМЕР 9
Синтез VCl3(PtBu3)2 [образец G1299]
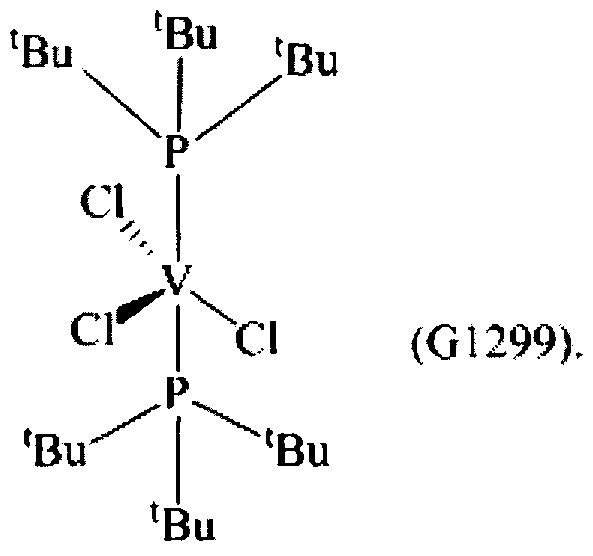
0,466 г (2,16×10-3 моль) трихлортрис(тетрагидрофуран)ванадия [VСl3(ТГФ)3], 4 мл толуола и затем 1,74 г (8,64×10-2 моль) три(трет-бутил)фосфина (Р/V молярное соотношение = 4) помещали в колбу с отводом объемом 100 мл. Полученную смесь выдерживали при интенсивном перемешивании при комнатной температуре в течение 15 минут и затем нагревали при кипячении с обратным холодильником в течение 3 часов. Полученную суспензию отфильтровывали в горячем виде (60°С) и собранную фракцию концентрировали под вакуумом при комнатной температуре. Затем по каплям при перемешивании добавляли около 50 мл пентана, получая осаждение порошка сиреневого цвета. Примерно через 3 часа все отфильтровывали и полученный твердый остаток серо-фиолетового цвета промывали пентаном (50 мл) и сушили под вакуумом при комнатной температуре с получением 0,3768 г (конверсия относительно исходного [VСl3(ТГФ)3]=31,0%) комплекса VСl3(РtВu3)2 (молекулярная масса = 561,93 гхмоль-1).
Элементный анализ [получено (вычислено)] С: 51,50% (51,30%); Н: 9,50% (9,69%); Cl: 19,10% (18,93%); Р: 11,20% (11,02%); V: 9,30% (9,07%).
ПРИМЕР 10
Синтез VCl3(dmpe) [образец G1275]
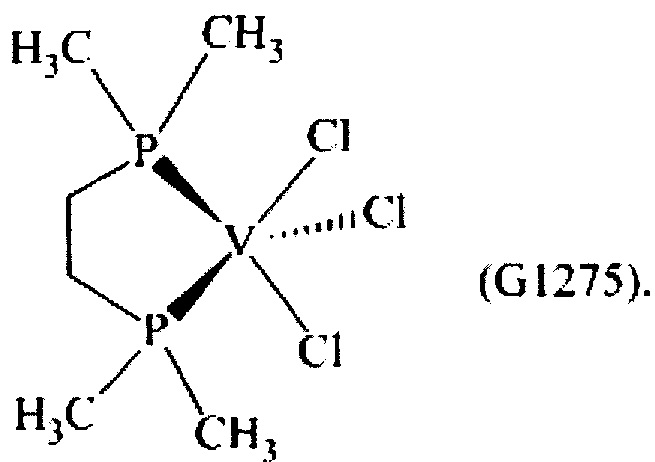
1,25 г (3,33×10-3 моль) трихлортрис(тетрагидрофуран)ванадия [VСl3(ТГФ)3], 14 мл толуола и затем 1,0 г (0,68×10-2 моль) 1,2-бис(диметилфосфино)этана (Р/V молярное соотношение = 2) помещали в колбу с отводом объемом 100 мл. Полученную смесь выдерживали при интенсивном перемешивании при комнатной температуре в течение 15 минут и затем нагревали при кипячении с обратным холодильником в течение 3 часов. Полученную суспензию отфильтровывали в горячем виде (60°С) и собранную фракцию концентрировали под вакуумом при комнатной температуре. Затем по каплям при перемешивании добавляли около 50 мл пентана, получая осаждение высокотонкодисперсного порошка. Примерно через 3 часа все отфильтровывали и полученный твердый остаток довольно темного цвета промывали пентаном (50 мл) и сушили под вакуумом при комнатной температуре с получением 0,895 г (конверсия относительно исходного [VСl3(ТГФ)3]=87,6%) комплекса VCl3(dmpe) (молекулярная масса = 307,44 гхмоль-1).
Элементный анализ [получено (вычислено)] С: 23,20% (23,44%); Н: 5,30% (5,25%); Cl: 34,40% (34,60%); Р: 20,40% (20,15%); V: 16,80% (16,57%).
ПРИМЕР 11
Синтез VCl3(depe) [образец G1274]
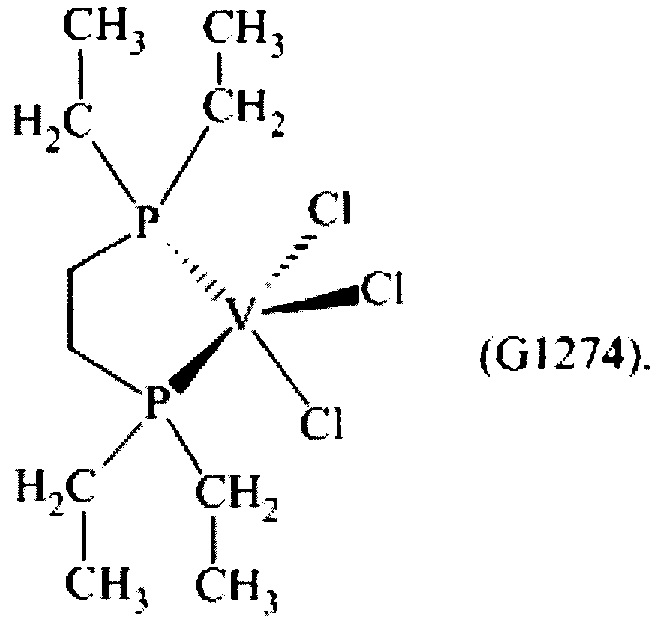
0,443 г (1,22×10-3 моль) трихлортрис(тетрагидрофуран)ванадия [VСl3(ТГФ)3], 5 мл толуола и затем 1,0 г (4,90×10-3 моль) 1,2-бис(диэтилфосфино)этана (P/V молярное соотношение = 4) помещали в колбу с отводом объемом 100 мл. Полученную смесь выдерживали при интенсивном перемешивании при комнатной температуре в течение 15 минут и затем нагревали при кипячении с обратным холодильником в течение 3 часов. Полученную суспензию отфильтровывали в горячем виде (60°С) и собранную фракцию концентрировали под вакуумом при комнатной температуре. Затем о каплям при перемешивании добавляли около 25 мл пентана, получая осаждение высокотонкодисперсного порошка. Примерно через 3 часа все отфильтровывали и полученный твердый остаток зеленого цвета промывали пентаном (50 мл) и сушили под вакуумом при комнатной температуре с получением 0,411 г (конверсия относительно исходного [VСl3(ТГФ)3]=92,6%) комплекса VCl3(depe) (молекулярная масса = 363,55 гхмоль-1).
Элементный анализ [получено (вычислено)] С: 32,90% (33,04%); Н: 6,40% (6,55%); Cl: 29,56% (29,26%); Р: 17,24% (17,04%); V: 14,03% (14,01%).
ПРИМЕР 12
Синтез VCl3(dppa) [образец G1281]
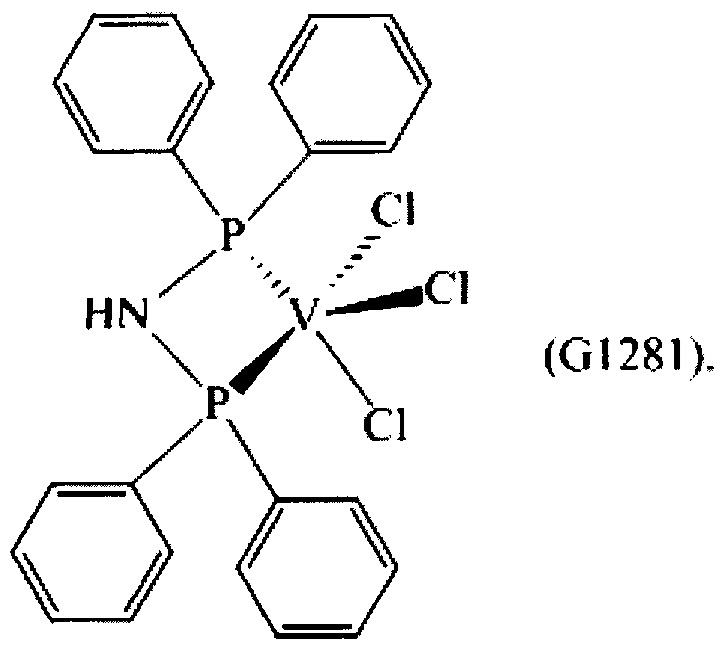
0,748 г (2,09×10-3 моль) трихлортрис(тетрагидрофуран)ванадия [VСl3(ТГФ)3], 10 мл толуола и затем 1,444 г (3,75×10-3 моль) N,N-бис(дифенилфосфино)амина (P/V молярное соотношение = 1,8) помещали в колбу с отводом объемом 100 мл. Полученную смесь выдерживали при интенсивном перемешивании при комнатной температуре в течение 15 минут и затем нагревали при кипячении с обратным холодильником в течение 2 часов. Полученную суспензию отфильтровывали в горячем виде (60°С) и собранную фракцию концентрировали под вакуумом при комнатной температуре. Затем по каплям при перемешивании добавляли около 50 мл пентана, получая осаждение высокотонкодисперсного порошка. Примерно через 3 часа все отфильтровывали и полученный твердый остаток горчичного цвета промывали пентаном (50 мл) и сушили под вакуумом при комнатной температуре с получением 0,356 г (конверсия относительно исходного [VСl3(ТГФ)3]=31,4%) комплекса VCl3(dppa) (молекулярная масса = 542,68 гхмоль-1).
Элементный анализ [получено (вычислено)] С: 53,23% (53,12%); Н: 3,90% (3,90%); Cl: 19,88% (19,60%); N: 2,75% (2,58%); Р: 11,50% (11,42%); V: 9,50% (9,39%).
ПРИМЕР 13 (ММ267)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем добавляли 9,14 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли метилалюмоксан (МАО) в растворе в толуоле (1,26 мл; 2,0×10-3 моль, что эквивалентно примерно 1,45 г) и затем комплекс VCl3(PMePh2)2 [образец MM261] (5,6 мл суспензии в толуоле с концентрацией 2 мг/мл; 2×10-5 моль, что эквивалентно примерно 11,2 мг), полученный, как описано в примере 1. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 72 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,241 г полибутадиена со смешанной цис/транс/1,2-структурой с содержанием 1,4-транс и 1,4-цис звеньев 77,2%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
На Фиг. 4 приведен ИК спектр с Фурье преобразованием полученного полибутадиена.
ПРИМЕР 14 (ММ268)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем добавляли 4,1 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли метилалюмоксан (МАО) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VCl3(PMePh2)2 [образец ММ261] (5,6 мл суспензии в толуоле с концентрацией 2 мг/мл; 2×10-5 моль, что эквивалентно примерно 11,2 мг), полученный, как описано в примере 1. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 4,5 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,203 г полибутадиена со смешанной цис/транс/1,2-структурой с содержанием 1,4-транс и 1,4-цис звеньев 85,8%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
На Фиг. 5 приведен ИК спектр с Фурье преобразованием полученного полибутадиена.
ПРИМЕР 15 (ММ281)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем добавляли 11,6 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе в толуоле (1,6 мл; 2,5×10-3 моль, что эквивалентно примерно 0,145 г) и затем комплекс VCl3(PMePh2)2 [образец ММ261] (2,8 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 5,6 мг), полученный, как описано в примере 1. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 5 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,498 г полибутадиена со смешанной цис/транс/1,2-структурой с содержанием 1,4-транс и 1,4-цис звеньев 60%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
На Фиг. 6 приведен ИК спектр с Фурье преобразованием полученного полибутадиена.
ПРИМЕР 16 (ММ275)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем 7 мл толуола добавляли и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VСl3(РМеРh2)2 [образец ММ261] (2,8 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 5,6 мг), полученный, как описано в примере 1. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 2 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0.845 г полибутадиена со смешанной цис/транс/1,2-структурой с содержанием 1,4-транс и 1,4-цис звеньев 74,8%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
ПРИМЕР 17 (G1282)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем 7 мл толуола добавляли и и температуру полученного таким образом раствора доводили до -30°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VСl3(РМеРh2)2 [образец ММ261] (2,8 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 5,6 мг), полученный, как описано в примере 1. Все вместе выдерживали при перемешивании магнитной мешалкой при -30°С в течение 24 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,364 г полибутадиена с преимущественно 1,4-транс структурой с содержанием 1,4-транс звеньев 95,1%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
На Фиг. 7 приведен ИК спектр с Фурье преобразованием полученного полибутадиена.
ПРИМЕР 18 (ММ319)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем добавляли 6,75 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли метилалюмоксан (МАО) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VCl3(PEtPh2)2 [образец G1298] (2,95 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 5,9 мг), полученный, как описано в примере 2. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С, в течение 20 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,364 г полибутадиена со смешанной цис/транс/1,2-структурой с содержанием 1,4-транс и 1,4-цис звеньев 85,4%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
На Фиг. 8 приведен ИК спектр с Фурье преобразованием полученного полибутадиена.
ПРИМЕР 19 (ММ320)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем добавляли 6,75 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VCl3(PEtPh2)2 [образец G1298] (2,95 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 5,9 мг), полученный, как описано в примере 2. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 3 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0.815 г полибутадиена со смешанной цис/транс/1,2-структурой с содержанием 1,4-транс и 1,4-цис звеньев of 71,3%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
На Фиг. 9 приведен ИК спектр с Фурье преобразованием полученного полибутадиена.
ПРИМЕР 20 (ММ393)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем добавляли 9,9 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе в толуоле (3,15 мл; 5×10-3 моль, что эквивалентно примерно 0,29 г) и затем комплекс VCl3(PEtPh2)2 [образец G1298] (2,95 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 5,9 мг), полученный, как описано в примере 2. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 2,5 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 1,17 г полибутадиена со смешанной цис/транс/1,2-структурой с содержанием 1,4-транс и 1,4-цис звеньев 62,7%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
На Фиг. 10 приведен ИК спектр с Фурье преобразованием полученного полибутадиена.
ПРИМЕР 21 (ММ394)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем добавляли 12,4 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе в толуоле (0,63 мл; 1×10-3 моль, что эквивалентно примерно 0,058 г) и затем комплекс VCl3(PEtPh2)2 [образец G1298] (2,95 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 5,9 мг), полученный, как описано в примере 2. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 5 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0.483 г полибутадиена со смешанной цис/транс/1,2-структурой с содержанием 1,4-транс и 1,4-цис звеньев 61,7%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
На Фиг. 11 приведен ИК спектр с Фурье преобразованием полученного полибутадиена.
ПРИМЕР 22 (ММ395)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем добавляли 9,9 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли метилалюмоксан (МАО) в растворе в толуоле (3,15 мл; 5×10-3 моль, что эквивалентно примерно 0,29 г) и затем комплекс VCl3(PEtPh2)2 [образец G1298] (2,95 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 5,9 мг), полученный, как описано в примере 2.
Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 24 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,281 г полибутадиена со смешанной цис/транс/1,2-структурой с содержанием 1,4-транс и 1,4-цис звеньев 81,8%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
На Фиг. 12 приведен ИК спектр с Фурье преобразованием полученного полибутадиена.
ПРИМЕР 23 (ММ396)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем добавляли 12,4 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли метилалюмоксан (МАО) в растворе в толуоле (0,63 мл; 1×10-2 моль, что эквивалентно примерно 0,058 г) и затем комплекс VCl3(PEtPh2)2 [образец G1298] (2,95 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 5,9 мг), полученный, как описано в примере 2. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 24 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,203 г полибутадиена со смешанной цис/транс/1,2-структурой с содержанием 1,4-транс и 1,4-цис звеньев 80,2%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
На Фиг. 13 приведен ИК спектр с Фурье преобразованием полученного полибутадиена.
ПРИМЕР 24 (ММ398)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем добавляли 9,9 мл 1,2-дихлорбензола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе в 1,2-дихлорбензоле (3,15 мл; 5×10-3 моль, что эквивалентно примерно 0,29 г) и затем комплекс VCl3(PEtPh2)2 [образец G1298] (2,95 мл раствора 1,2-дихлорбензола с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 5,9 мг), полученный, как описано в примере 2. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 2,16 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,778 г полибутадиена со смешанной цис/транс/1,2-структурой с содержанием 1,4-транс и 1,4-цис звеньев 75,5%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
На Фиг. 14 приведен ИК спектр с Фурье преобразованием полученного полибутадиена.
ПРИМЕР 25 (ММ374)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем добавляли 6,65 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли метилалюмоксан (МАО) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VCl3(PiPrPh2)2 [образец G1325] (3,05 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 6,1 мг), полученный, как описано в примере 3. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 2 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,235 г полибутадиена со смешанной цис/транс/1,2-структурой с содержанием 1,4-транс и 1,4-цис звеньев 84%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
На Фиг. 15 приведен ИК спектр с Фурье преобразованием полученного полибутадиена.
ПРИМЕР 26 (ММ341)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем добавляли 6,65 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VCl3(PiPrPh2)2 [образец G1325] (3,05 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 6,1 мг), полученный, как описано в примере 3. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 2 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,684 г полибутадиена со смешанной цис/транс/1,2-структурой с содержанием 1,4-транс и 1,4-цис звеньев 73,2%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
На Фиг. 16 приведен ИК спектр с Фурье преобразованием полученного полибутадиена.
ПРИМЕР 27 (ММ335)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем добавляли 6,25 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VСl3(РСуРh2)2 [образец ММ300] (3,45 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 6, 9 мг), полученный, как описано в примере 4. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 2 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 1,1 г полибутадиена со смешанной цис/транс/1,2-структурой с содержанием 1,4-транс и 1,4-цис звеньев 68,8%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
На Фиг. 17 приведен ИК спектр с Фурье преобразованием полученного полибутадиена.
ПРИМЕР 28 (ММ336)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем добавляли 6,25 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли метилалюмоксан (МАО) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VCl3(PCyPh2)2 [образец ММ300] (3,45 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 6,9 мг), полученный, как описано в примере 4. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 72 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,607 г полибутадиена со смешанной цис/транс/1,2-структурой с содержанием 1,4-транс и 1,4-цис звеньев 82%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
На Фиг. 18 приведен ИК спектр с Фурье преобразованием полученного полибутадиена.
ПРИМЕР 29 (ММ338)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем добавляли 6,25 мл толуола и температуру полученного таким образом раствора доводили до -30°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VСl3(РСуРh2)2 [образец ММ300] (3,45 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 6,9 мг), полученный, как описано в примере 4. Все вместе выдерживали при перемешивании магнитной мешалкой при -30°С в течение 24 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,449 г полибутадиена с преимущественно 1,4-транс структурой с содержанием 1,4-транс звеньев 95,8%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
ПРИМЕР 30 (G1306)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем добавляли 6,25 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли метилалюмоксан (МАО) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VCl3(PPh3)2 [образец ММ295] (3,4 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 6,8 мг), полученный, как описано в примере 5. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 21 часа. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,742 г полибутадиена со смешанной цис/транс/1,2-структурой с содержанием 1,4-транс и 1,4-цис звеньев 81%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
ПРИМЕР 31 (G1307)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем добавляли 6,3 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VCl3(PPh3)2 [образец ММ295] (3,4 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 6,8 мг), полученный, как описано в примере 5. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 21 часа. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 1,301 г полибутадиена со смешанной цис/транс/1,2-структурой с содержанием 1,4-транс и 1,4-цис звеньев 68,8%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
На Фиг. 19 приведен ИК спектр с Фурье преобразованием полученного полибутадиена.
ПРИМЕР 32 (ММ317)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем добавляли 6,9 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли метилалюмоксан (МАО) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VСl3(РtBu3)2 [образец G1299] (2,8 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 5,6 мг), полученный, как описано в примере 9. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 20 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0.819 г полибутадиена со смешанной цис/транс/1,2-структурой с содержанием 1,4-транс и 1,4-цис звеньев 86,5%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
На Фиг. 20 приведен ИК спектр с Фурье преобразованием полученного полибутадиена.
ПРИМЕР 33 (ММ318)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем добавляли 6,9 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VСl3(РtBu3)2 [образец G1299] (2.8 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 5,6 мг), полученный, как описано в примере 9. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 20 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,692 г полибутадиена со смешанной цис/транс/1,2-структурой с содержанием 1,4-транс и 1,4-цис звеньев 64,8%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
На Фиг. 21 приведен ИК спектр с Фурье преобразованием полученного полибутадиена.
ПРИМЕР 34 (ММ365)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем добавляли 6?55 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VСl3(РСур3)2 [образец G1286] (3?15 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 6,3 мг), полученный, как описано в примере 7. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 2 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,67 г полибутадиена со смешанной цис/транс/1,2-структурой с содержанием 1,4-транс и 1,4-цис звеньев 76,3%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
На Фиг. 22 приведены спектры ЯМР 1Н и 13С полученного полибутадиена.
ПРИМЕР 35 (G1376)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем добавляли 6,25 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VCl3(PCy3)2 [образец ММ370] (3,45 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 6,9 мг), полученный, как описано в примере 6. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 15 минут. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,461 г полибутадиена со смешанной цис/транс/1,2-структурой с содержанием 1,4-транс и 1,4-цис звеньев 81%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
ПРИМЕР 36 (ММ378)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем добавляли 6,9 мл гептана и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе гептана (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VCl3(PCy2H)2 [образец G1303] (2,77 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 5,5 мг), полученный, как описано в примере 8. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 20 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,338 г полибутадиена со смешанной цис/транс/1,2-структурой с содержанием 1,4-транс и 1,4-цис звеньев 83,5%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
ПРИМЕР 37 (ММ379)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем добавляли 6,9 мл гептана и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе гептана (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс Сl3(РСу2Н)2 [образец G1303] (2,77 мл суспензии в гептане с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 5,5 мг), полученный, как описано в примере 8. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 2 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,268 г полибутадиена со смешанной цис/транс/1,2-структурой с содержанием 1,4-транс и 1,4-цис звеньев 62,3%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
На Фиг. 23 приведен ИК спектр с Фурье преобразованием полученного полибутадиена.
ПРИМЕР 38 (ММ279)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем добавляли 8,25 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VCl3(dmpe) [образец G1275] (1,53 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 3,06 мг), полученный, как описано в примере 10. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 72 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% of антиоксиданта Irganox® 1076 (производства Ciba) с получением 0.113 г полибутадиена со смешанной цис/транс/1,2-структурой с содержанием 1,4-транс и 1,4-цис звеньев 64,6%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
На Фиг. 24 приведен ИК спектр с Фурье преобразованием полученного полибутадиена.
ПРИМЕР 39 (G1284)
2 мл 1,3-бутадиена, что эквивалентно примерно 1,4 г, конденсировали на холоду (-20°С) в тестовой пробирке объемом 25 мл. Затем добавляли 7 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VСl3(dрра) [образец G1281] (2,72 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 5,5 мг), полученный, как описано в примере 12. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 3,5 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,445 г полибутадиена со смешанной цис/транс/1,2-структурой с содержанием 1,4-транс и 1,4-цис звеньев 73,1%: другие характеристики способа полученного полибутадиена приведены в таблице 3.
На Фиг. 25 приведен ИК спектр с Фурье преобразованием полученного полибутадиена.
ПРИМЕР 40 (G1314)
2 мл изопрена, что эквивалентно примерно 1,36 г, помещали в тестовую пробирку объемом 25 мл. Затем добавляли 6,75 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VCl3(PEtPh2)2 [образец G1298] (2,95 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 5,9 мг), полученный, как описано в примере 2. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 18 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,860 г полиизопрена со смешанной цис/транс/3,4-структурой с содержанием 1,4-транс и 1,4-цис звеньев 81,4%: другие характеристики способа и полученного полиизопрена приведены в таблице 4.
На Фиг. 26 приведен ИК спектр с Фурье преобразованием полученного полиизопрена.
ПРИМЕР 41 (ММ401)
2 мл изопрена, что эквивалентно примерно 1,36 г, помещали в тестовую пробирку объемом 25 мл. Затем добавляли 6,75 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VСl3(РЕtРh2)2 [образец G1298] (2,95 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 5,9 мг), полученный, как описано в примере 2. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 1,15 часа. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,104 г полиизопрена со смешанной цис/транс/3,4-структурой с содержанием 1,4-транс и 1,4-цис звеньев 70,4%: другие характеристики способа и полученного полиизопрена приведены в таблице 4.
На Фиг. 27 приведен ИК спектр с Фурье преобразованием полученного полиизопрена.
ПРИМЕР 42 (ММ402)
2 мл изопрена, что эквивалентно примерно 1,36 г, помещали в тестовую пробирку объемом 25 мл. Затем добавляли 6,75 мл 1,2-дихлорбензола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе 1,2-дихлорбензола (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VCl3(PEtPh2)2 [образец G1298] (2,95 мл суспензии в 1,2-дихлорбензоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 5,9 мг), полученный, как описано в примере 2. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 1,15 часа. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,207 г полиизопрена со смешанной цис/транс/3,4-структурой с содержанием 1,4-транс и 1,4-цис звеньев 63,5%: другие характеристики способа и полученного полиизопрена приведены в таблице 4.
На Фиг. 28 приведен ИК спектр с Фурье преобразованием полученного полиизопрена.
ПРИМЕР 43 (ММ343)
2 мл изопрена, что эквивалентно примерно 1,36 г, помещали в тестовую пробирку объемом 25 мл. Затем добавляли 6,65 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VСl3(РiРrРh2)2 [образец G1325] (3,05 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 6.1 мг), полученный, как описано в примере 3. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 24 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 1,02 г полиизопрена со смешанной цис/транс/3,4-структурой с содержанием 1,4-транс и 1,4-цис звеньев 77,3%: другие характеристики способа и полученного полиизопрена приведены в таблице 4.
На Фиг. 29 приведен ИК спектр с Фурье преобразованием полученного полиизопрена.
ПРИМЕР 44 (ММ346)
2 мл изопрена, что эквивалентно примерно 1,36 г, помещали в тестовую пробирку объемом 25 мл. Затем добавляли 6,65 мл 1,2-дихлорбензола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе в 1,2-дихлорбензоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VCl3(PiPrPh2)2 [образец G1325] (3,05 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 6,1 мг), полученный, как описано в примере 3. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 30 минут. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 1 г полиизопрена со смешанной цис/транс/3,4-структурой с содержанием 1,4-транс и 1,4-цис звеньев 68,9%: другие характеристики способа и полученного полиизопрена приведены в таблице 4.
ПРИМЕР 45 (ММ371)
2 мл изопрена, что эквивалентно примерно 1,36 г, помещали в тестовую пробирку объемом 25 мл. Затем добавляли 6,65 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VСl3(РiРrРh2)2 [образец G1325] (3,05 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 6,1 мг), полученный, как описано в примере 3. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 5 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,249 г полиизопрена со смешанной цис/транс/3,4-структурой с содержанием 1,4-транс и 1,4-цис звеньев 74,2%: другие характеристики способа и полученного полиизопрена приведены в таблице 4.
На Фиг. 30 приведен ИК спектр с Фурье преобразованием полученного полиизопрена.
ПРИМЕР 46 (ММ372)
2 мл изопрена, что эквивалентно примерно 1,36 г, помещали в тестовую пробирку объемом 25 мл. Затем добавляли 6,65 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли метилалюмоксан (МАО) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VCl3(PiPrPh2)2 [образец G1325] (3,05 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 6,1 мг), полученный, как описано в примере 3. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 96 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,764 г полиизопрена с доминирующей 1,4-цис-структурой с содержанием 1,4-цис звеньев 87%: другие характеристики способа и полученного полиизопрена приведены в таблице 4.
На Фиг. 31 приведен ИК спектр с Фурье преобразованием полученного полиизопрена.
ПРИМЕР 47 (ММ337)
2 мл изопрена, что эквивалентно примерно 1,36 г, помещали в тестовую пробирку объемом 25 мл. Затем добавляли 6,25 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VСl3(РСyРh2)2 [образец ММ300] (3,45 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 6,9 мг), полученный, как описано в примере 4. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 2 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,387 г полиизопрена со смешанной цис/транс/3,4-структурой с содержанием 1,4-транс и 1,4-цис звеньев 76,2%: другие характеристики способа и полученного полиизопрена приведены в таблице 4.
На Фиг. 32 приведен ИК спектр с Фурье преобразованием полученного полиизопрена.
На Фиг. 33 приведены спектры ЯМР 1Н и 13С полученного полиизопрена.
ПРИМЕР 48 (G1310)
2 мл изопрена, что эквивалентно примерно 1,36 г, помещали в тестовую пробирку объемом 25 мл. Затем добавляли 6,3 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VСl3(РРh3)2 [образец ММ295] (3,4 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 6,8 мг), полученный, как описано в примере 5. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 2 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,12 г полиизопрена со смешанной цис/транс/3,4-структурой с содержанием 1,4-транс и 1,4-цис звеньев 75%: другие характеристики способа и полученного полиизопрена приведены в таблице 4.
На Фиг. 34 приведен ИК спектр с Фурье преобразованием полученного полиизопрена.
ПРИМЕР 49 (ММ332)
2 мл изопрена, что эквивалентно примерно 1,36 г, помещали в тестовую пробирку объемом 25 мл. Затем добавляли 6,9 мл толуола и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VСl3(РtBu3)2 [образец G1299] (2,8 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 5,6 мг), полученный, как описано в примере 9. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 24 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,415 г полиизопрена со смешанной цис/транс/3,4-структурой с содержанием 1,4-транс и 1,4-цис звеньев 86,2%: другие характеристики способа и полученного полиизопрена приведены в таблице 4.
На Фиг. 35 приведен ИК спектр с Фурье преобразованием полученного полиизопрена.
ПРИМЕР 50 (ММ375)
2 мл изопрена, что эквивалентно примерно 1,36 г, помещали в тестовую пробирку объемом 25 мл. Затем 6.25 мл толуола добавляли и и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе в толуоле (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VСl3(РСy3)2 [образец ММ370] (3,45 мл суспензии в толуоле с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 6,9 мг), полученный, как описано в примере 6. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 19 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,358 г полиизопрена со смешанной цис/транс/3,4-структурой с содержанием 1,4-транс и 1,4-цис звеньев 76,8%: другие характеристики способа и полученного полиизопрена приведены в таблице 4.
На Фиг. 36 приведен ИК спектр с Фурье преобразованием полученного полиизопрена.
ПРИМЕР 51 (ММ377)
2 мл изопрена, что эквивалентно примерно 1,36 г, помещали в тестовую пробирку объемом 25 мл. Затем добавляли 6,25 мл гептана и температуру полученного таким образом раствора доводили до 20°С. Затем добавляли сухой метилалюмоксан (МАО-сухой) в растворе гептана (6,3 мл; 1×10-2 моль, что эквивалентно примерно 0,58 г) и затем комплекс VСl3(РСy2Н)2 [образец G1303] (2,77 мл суспензии в гептане с концентрацией 2 мг/мл; 1×10-5 моль, что эквивалентно примерно 5,5 мг), полученный, как описано в примере 8. Все вместе выдерживали при перемешивании магнитной мешалкой при 20°С в течение 20 часов. Полимеризацию затем останавливали путем добавления 2 мл метанола, содержащего несколько капель соляной кислоты. Полученный полимер затем подвергали коагуляции путем добавления 40 мл раствора метанола, содержащего 4% антиоксиданта Irganox® 1076 (производства Ciba) с получением 0,674 г полиизопрена со смешанной цис/транс/3,4-структурой с содержанием 1,4-транс и 1,4-цис звеньев 82,7%: другие характеристики способа и полученного полиизопрена приведены в таблице 4.
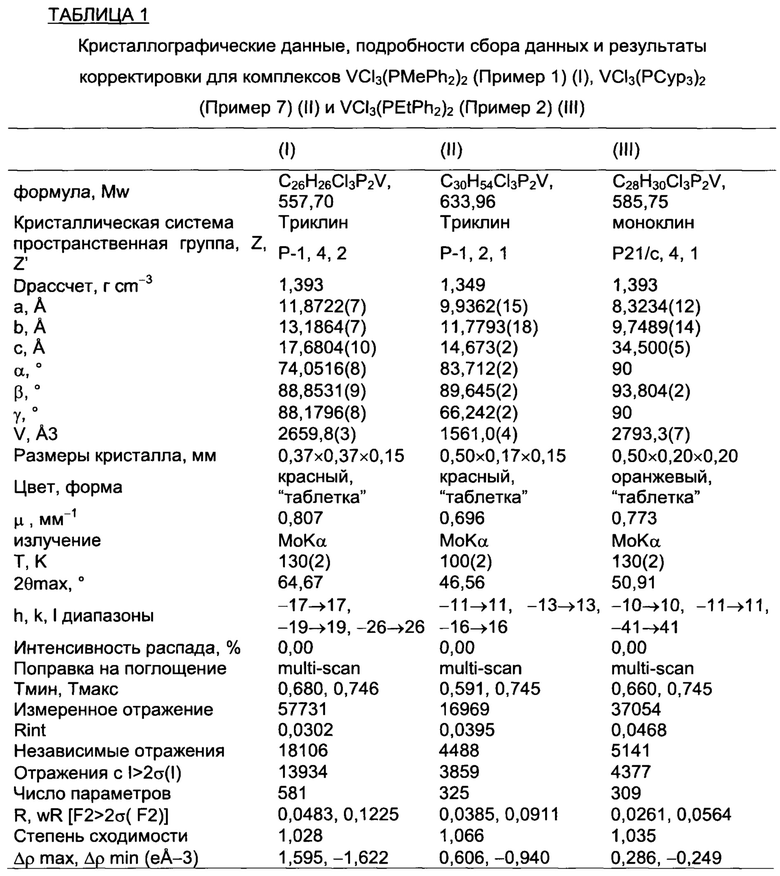
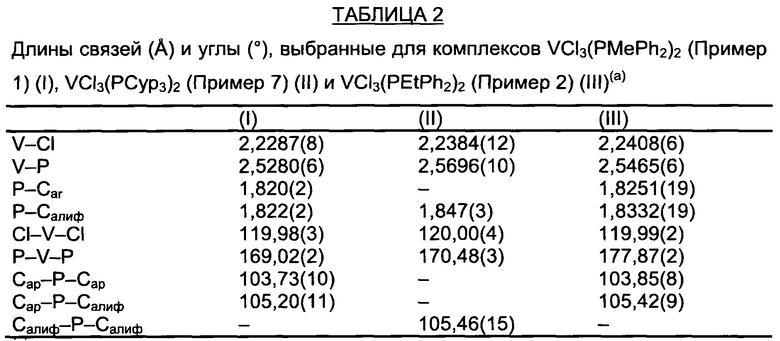
(a): каждое приведенное значение получали в виде средней величины всех соответствующих параметров, имеющихся в стрктуре.
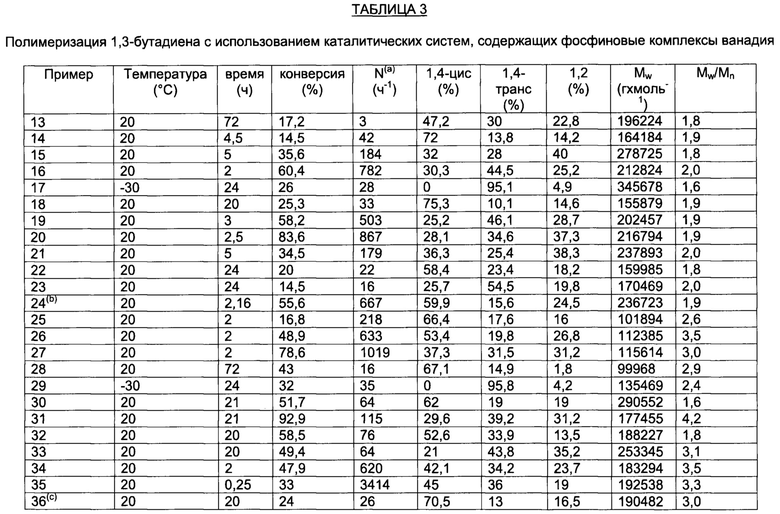

(a): количество моль полимеризованного 1,3-бутадиена в час на моль ванадия;
(b): растворитель полимеризации 1,2-дихлорбензол;
(c): растворитель полимеризации гептан
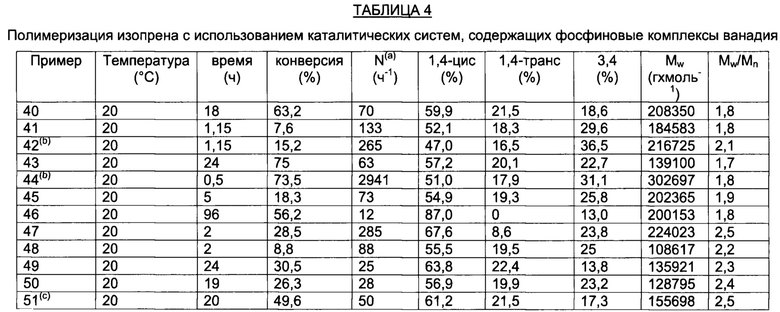
(a): количество моль полимеризованного изопрена в час на моль ванадия;
(b): растворитель полимеризации 1,2-дихлорбензол;
(с): растворитель полимеризации гептан
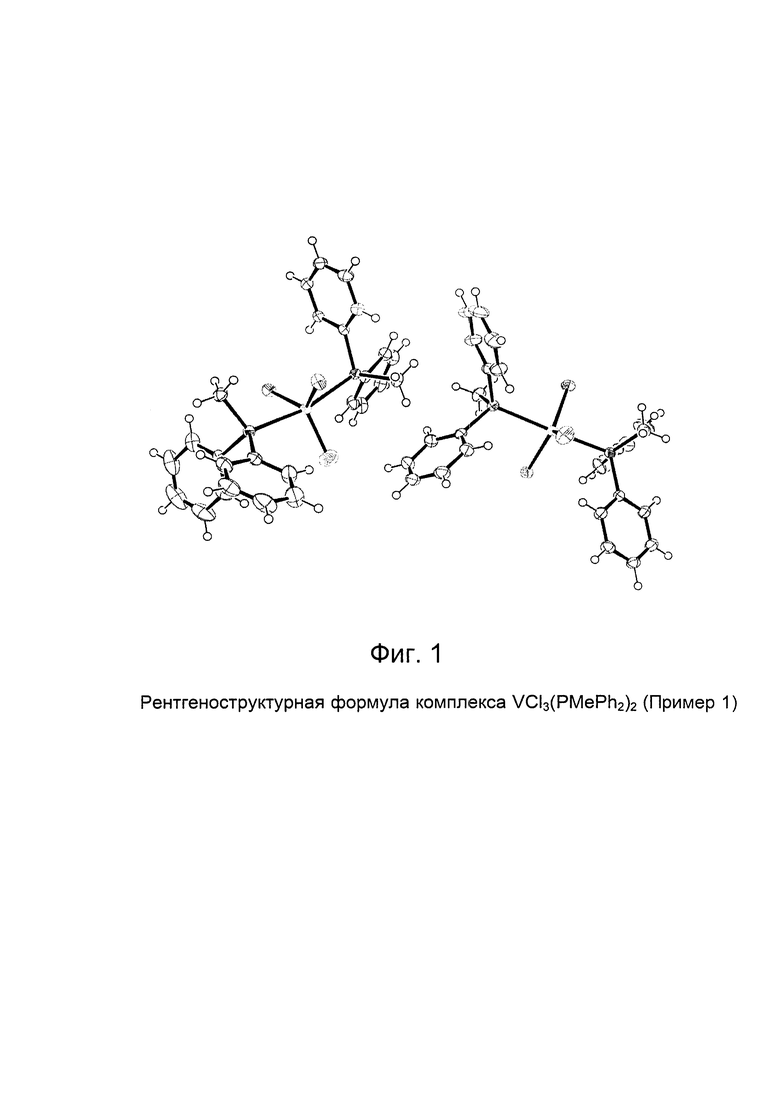
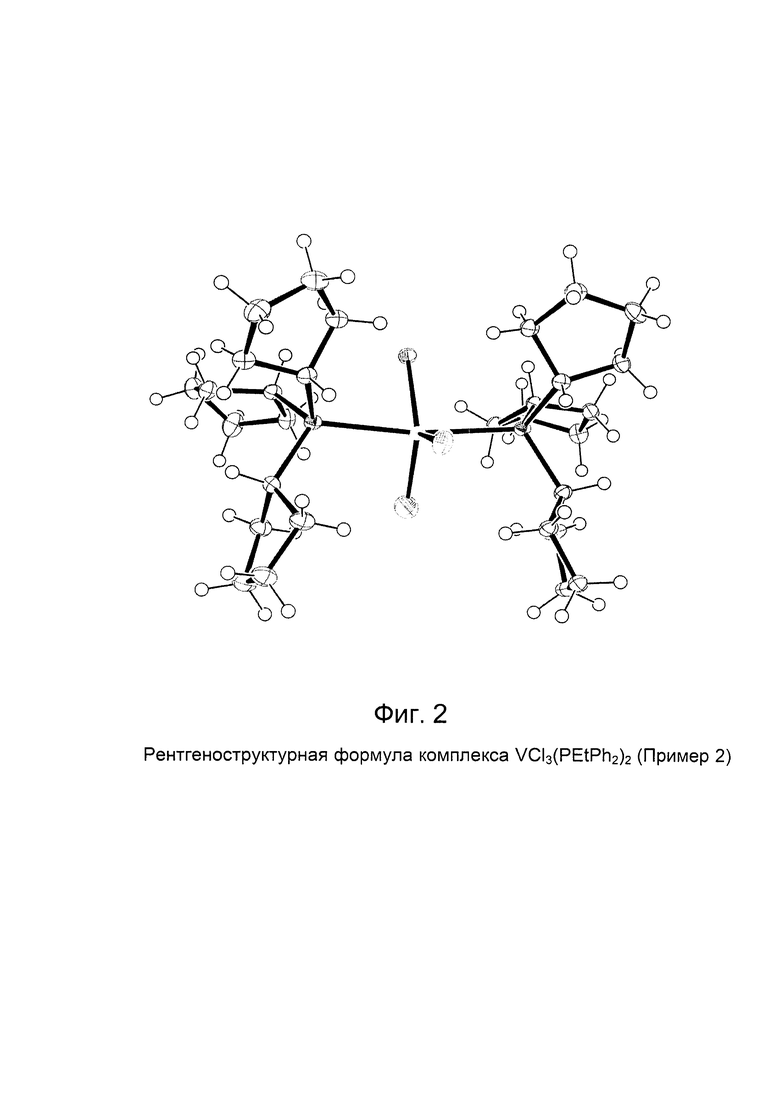
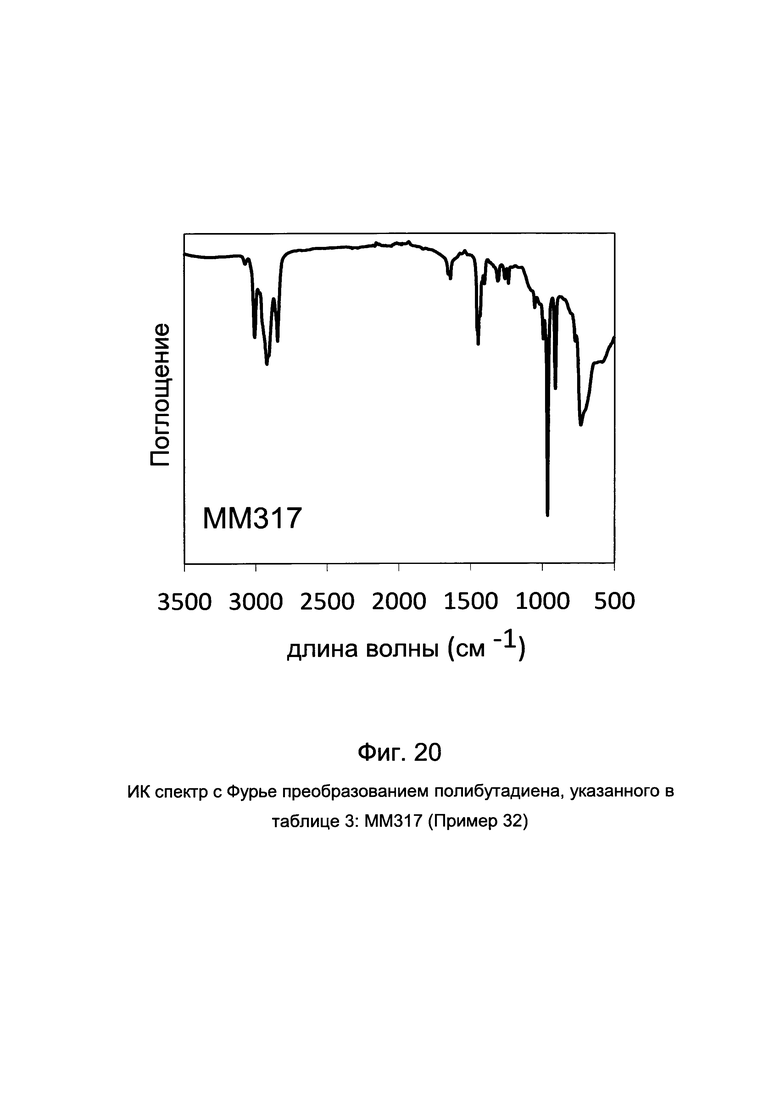
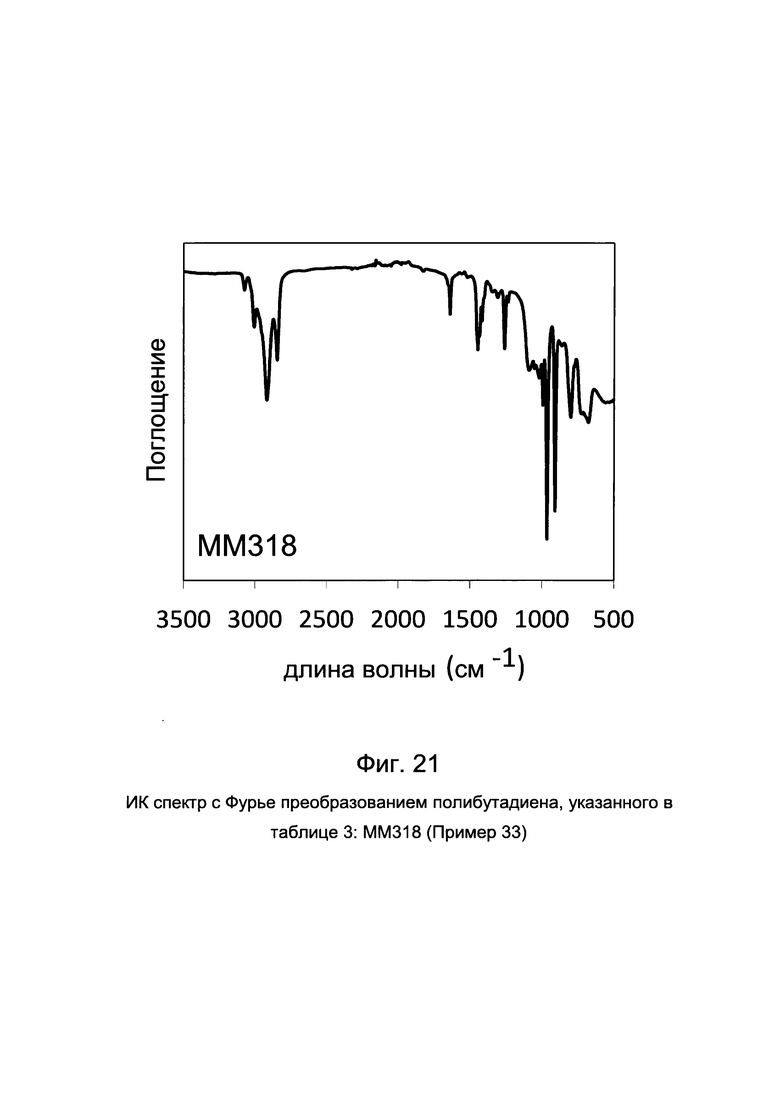
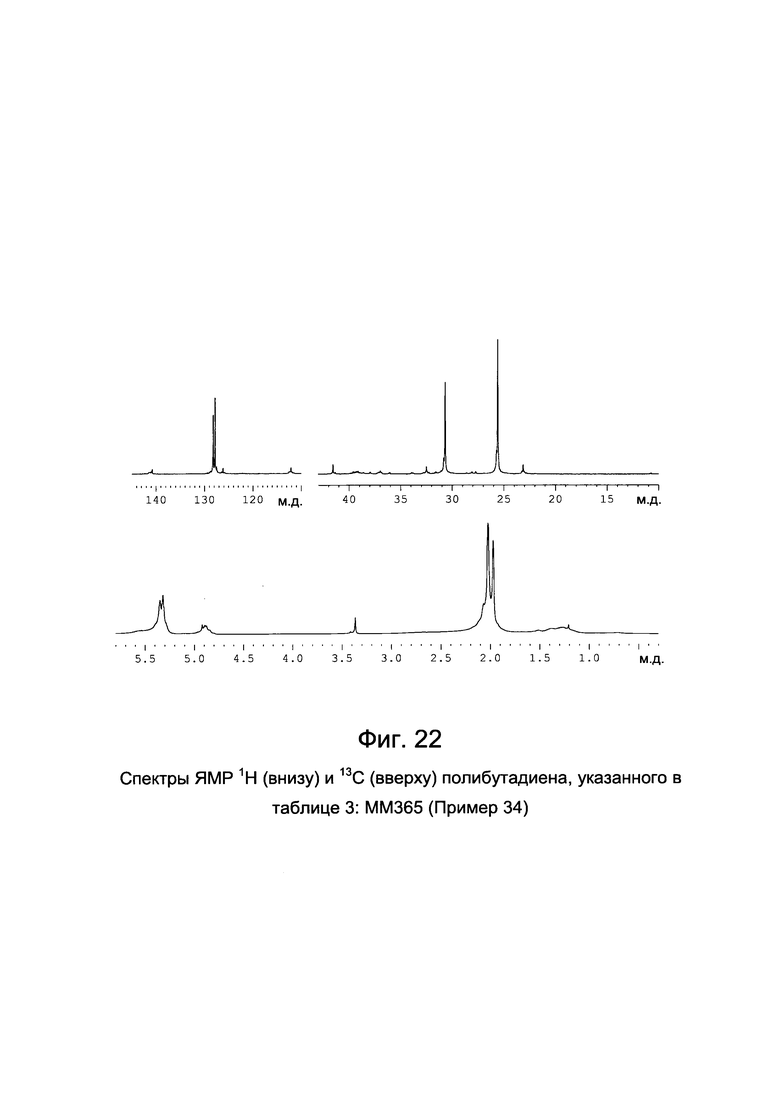
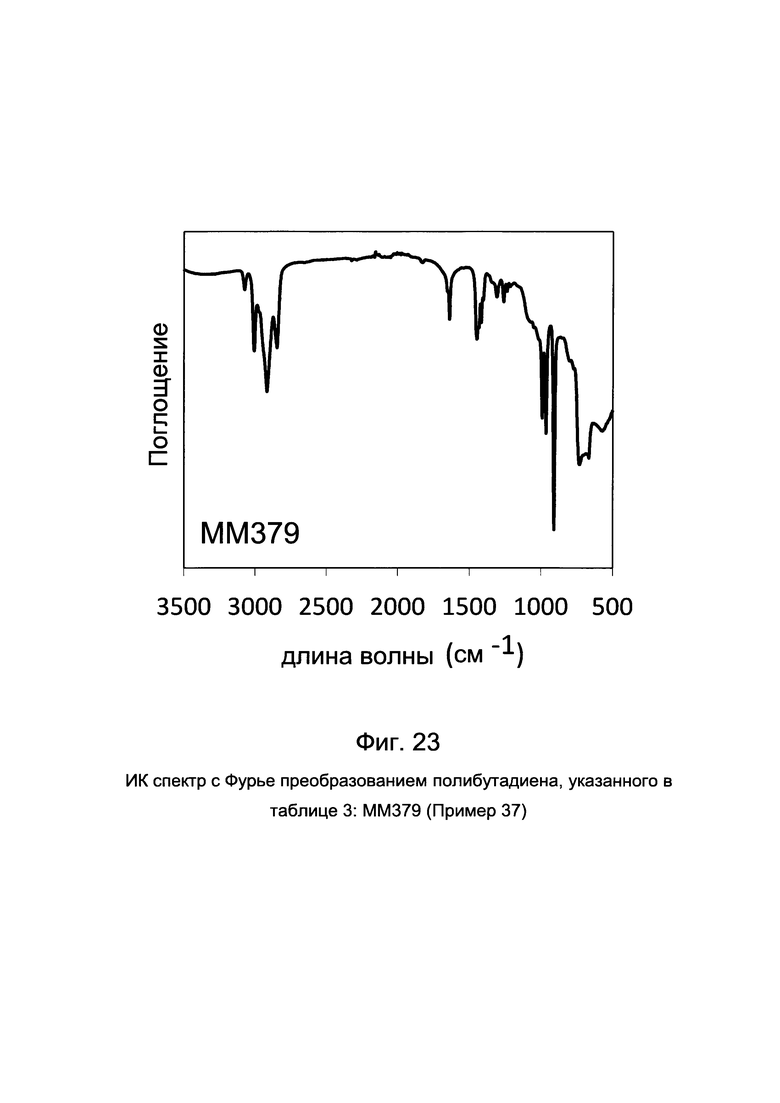
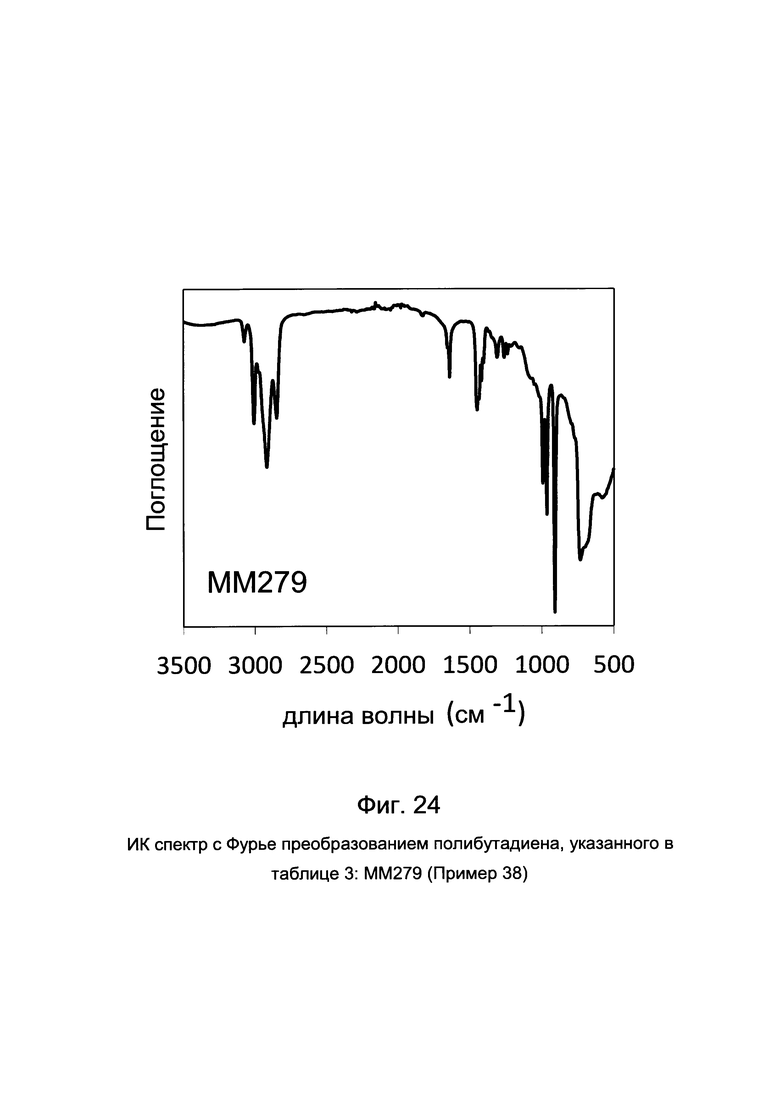
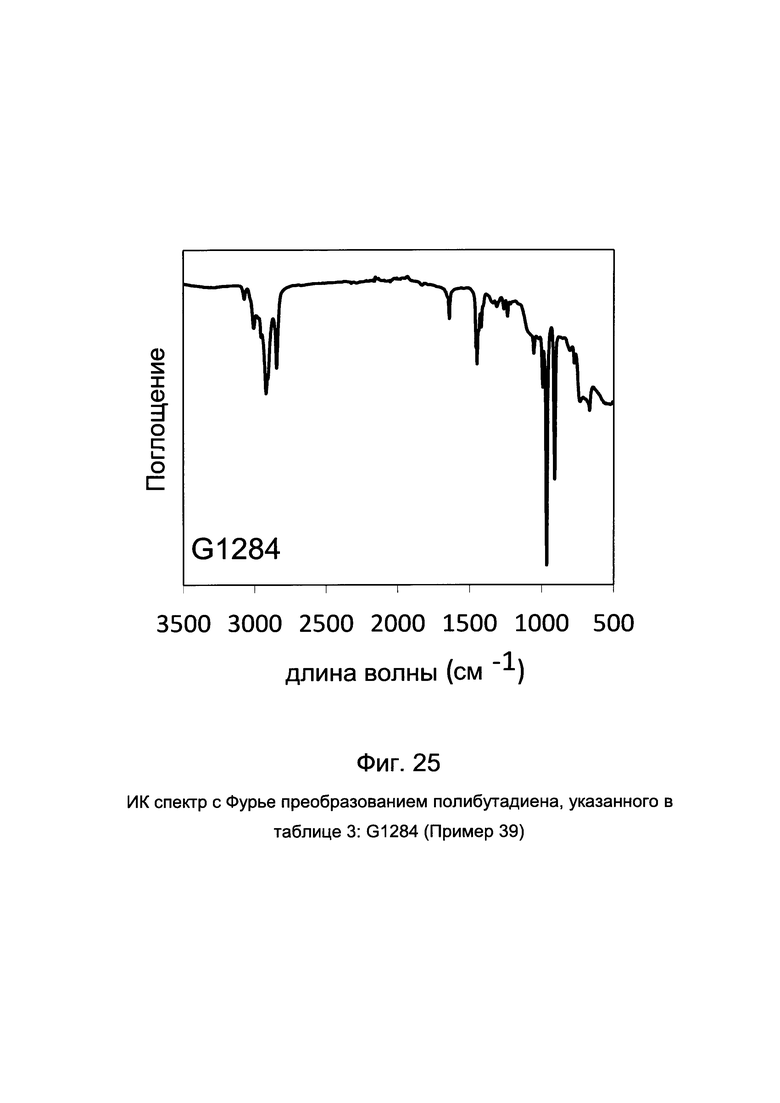
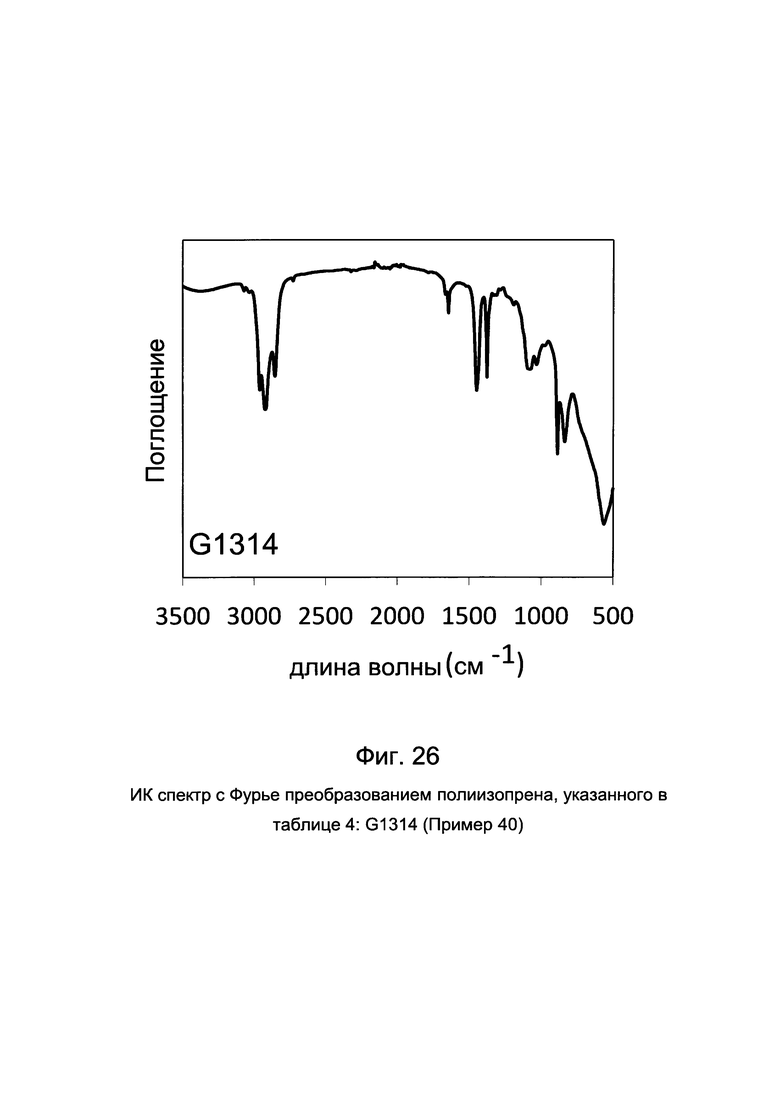
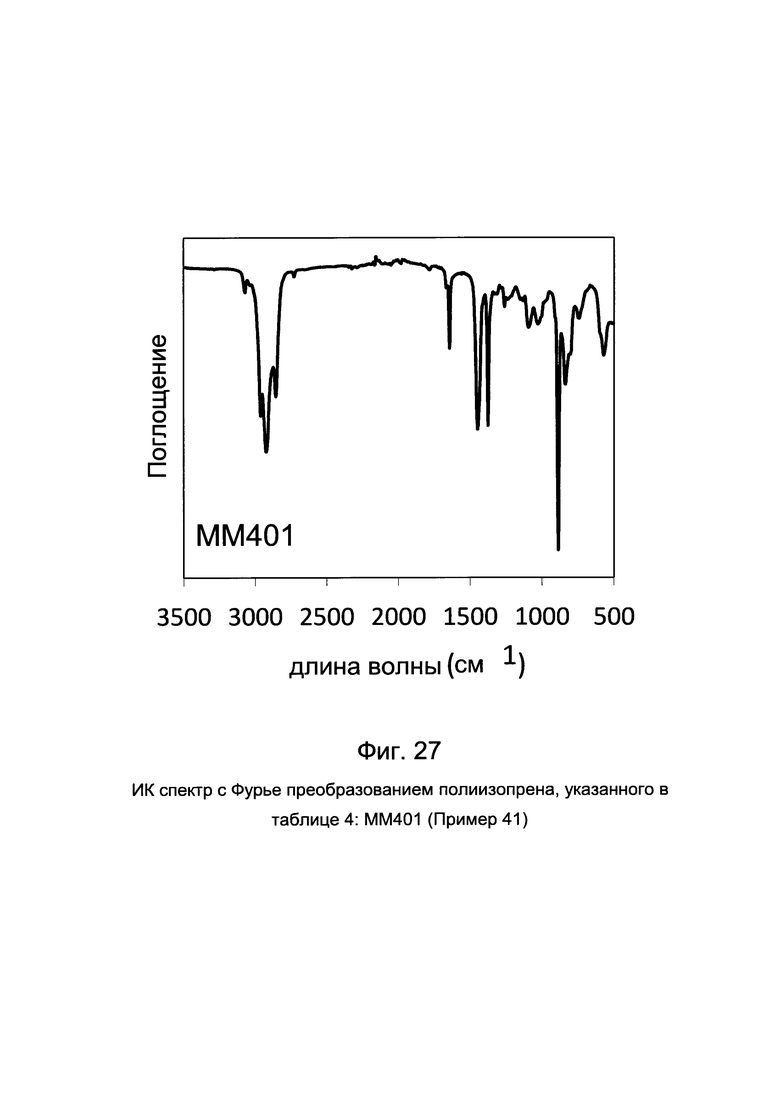
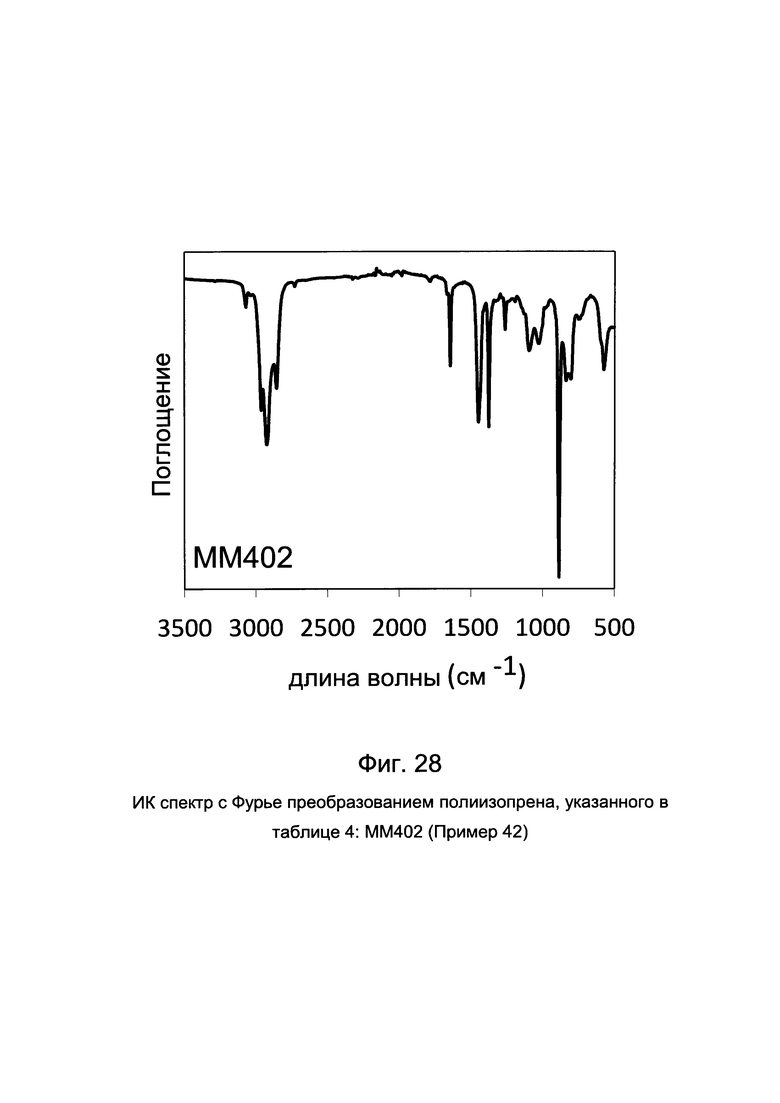
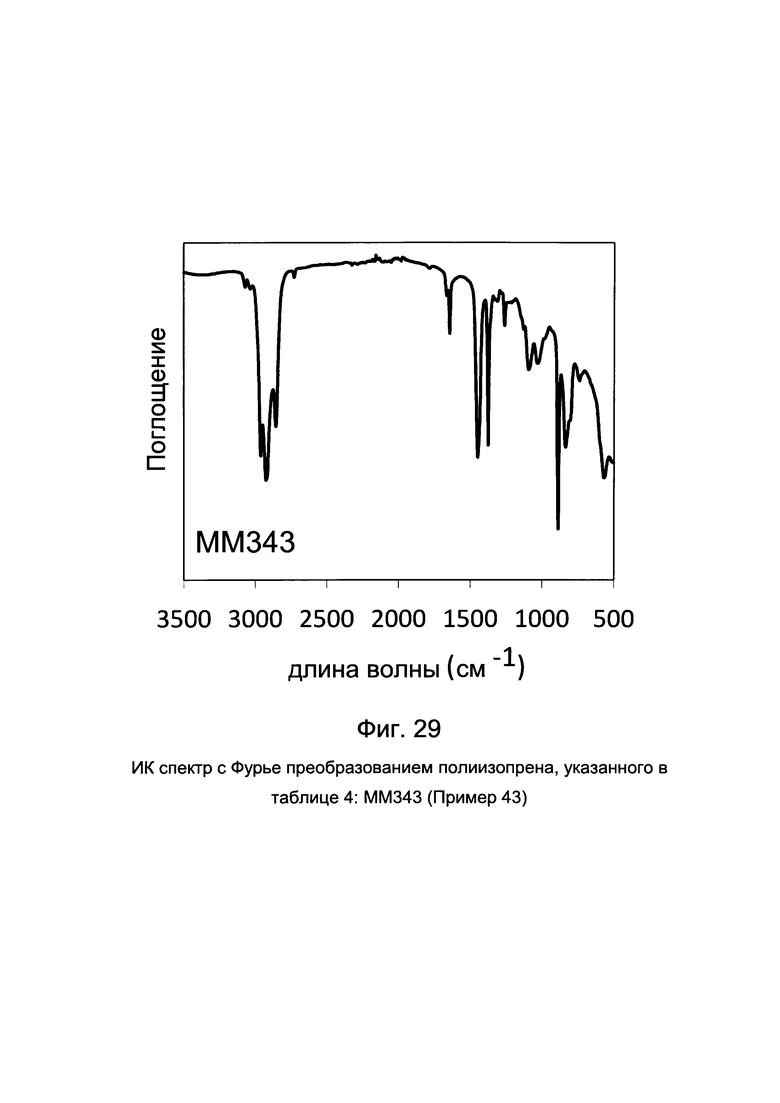
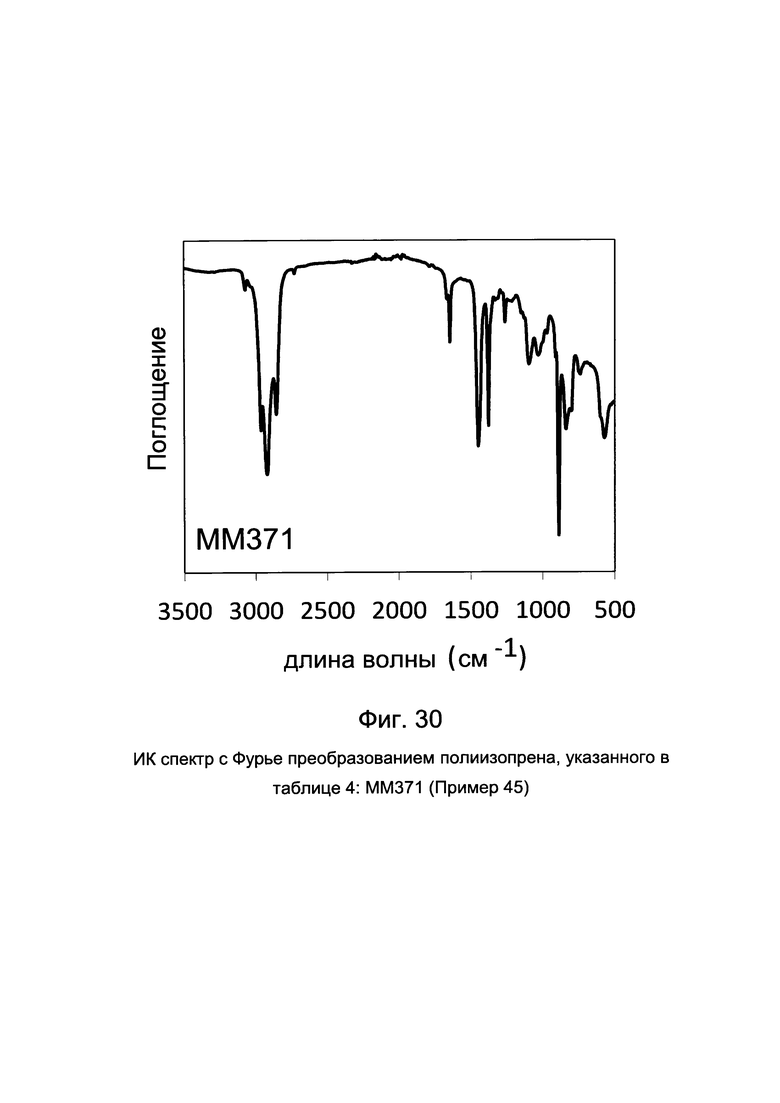
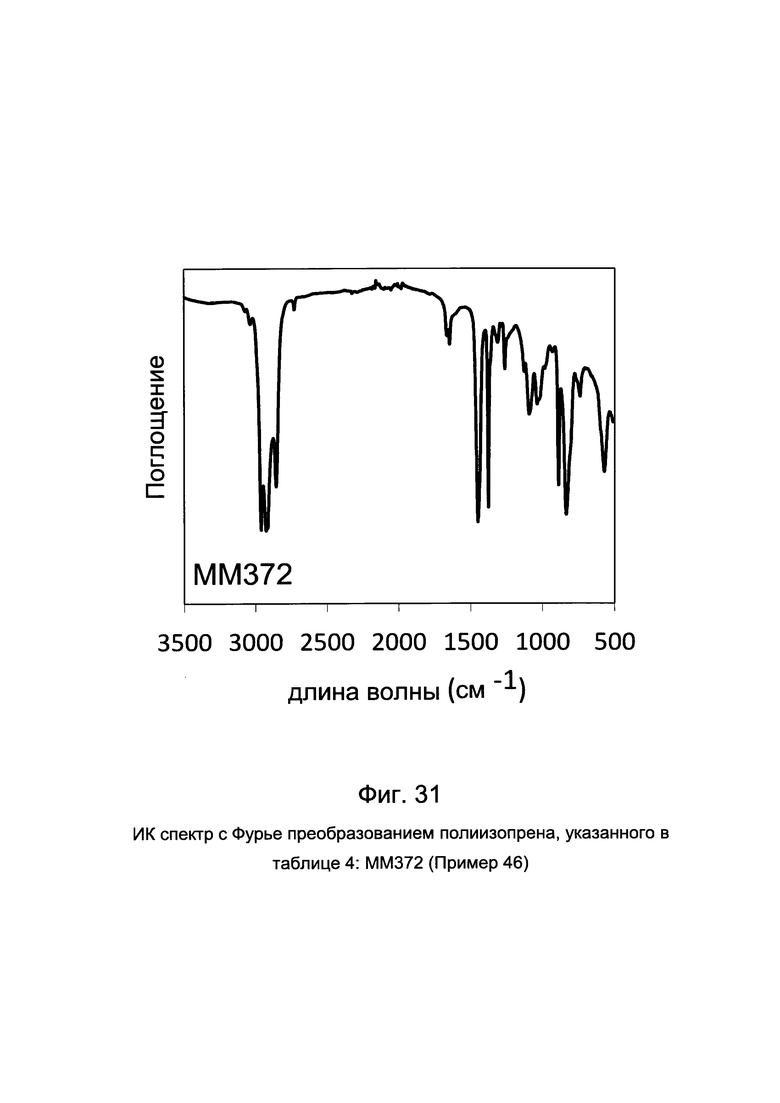
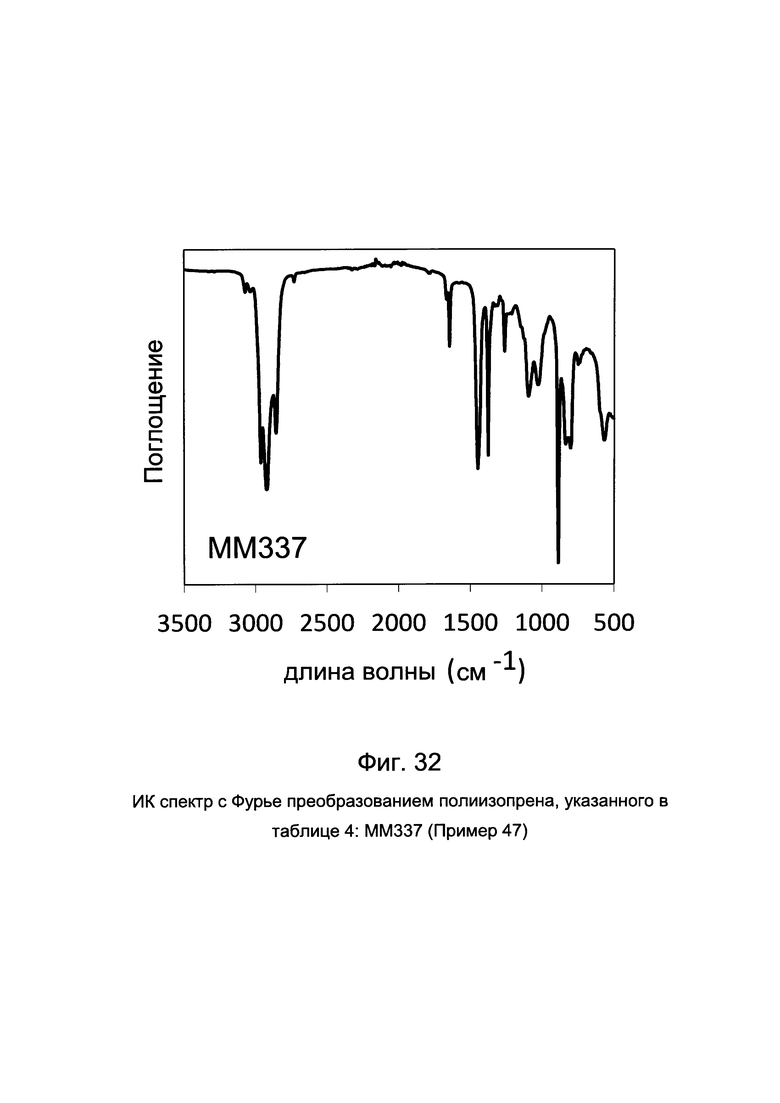
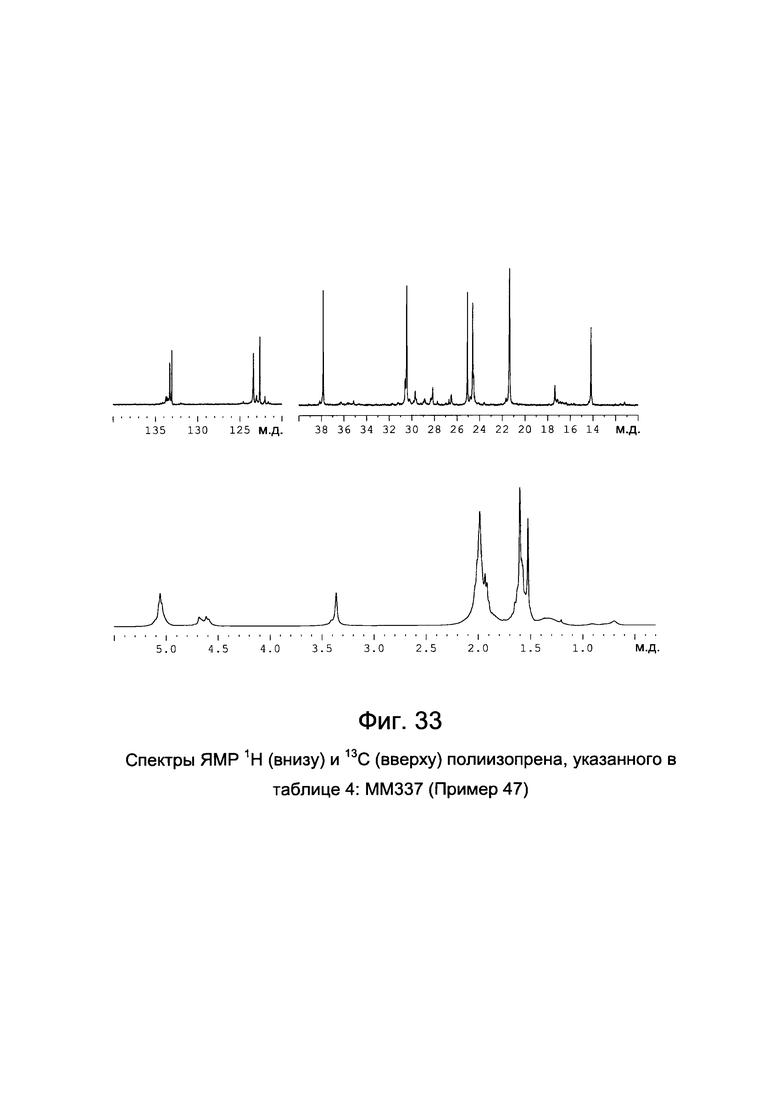
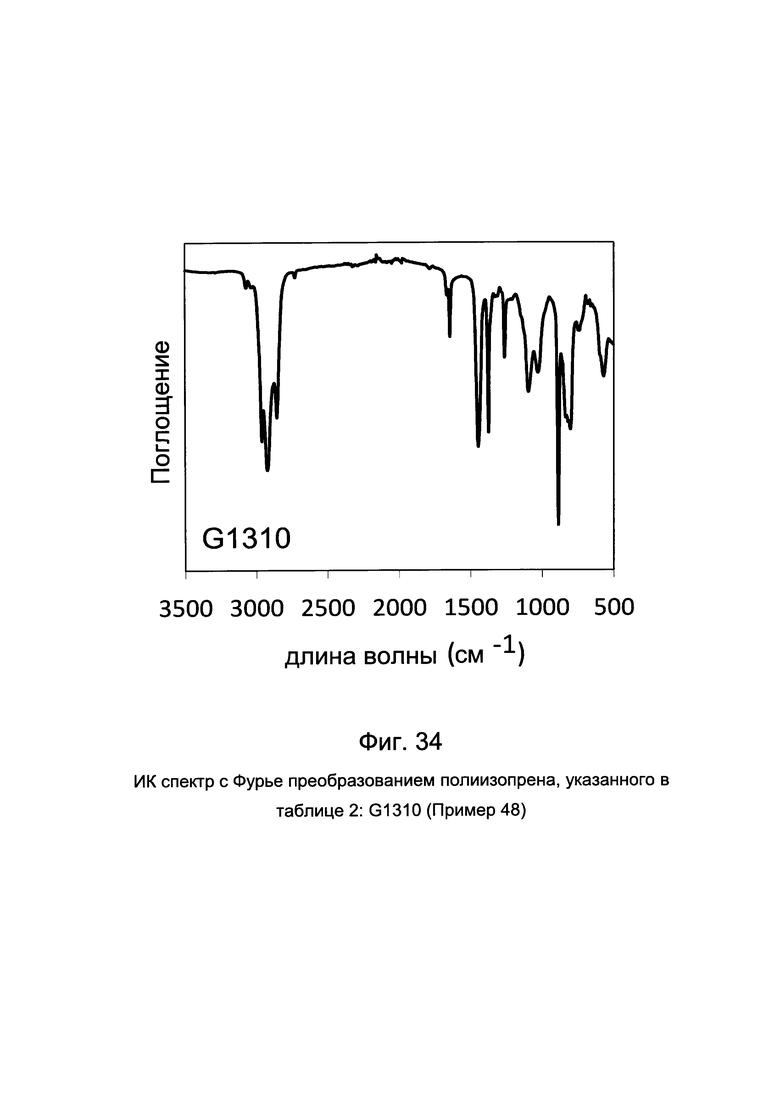
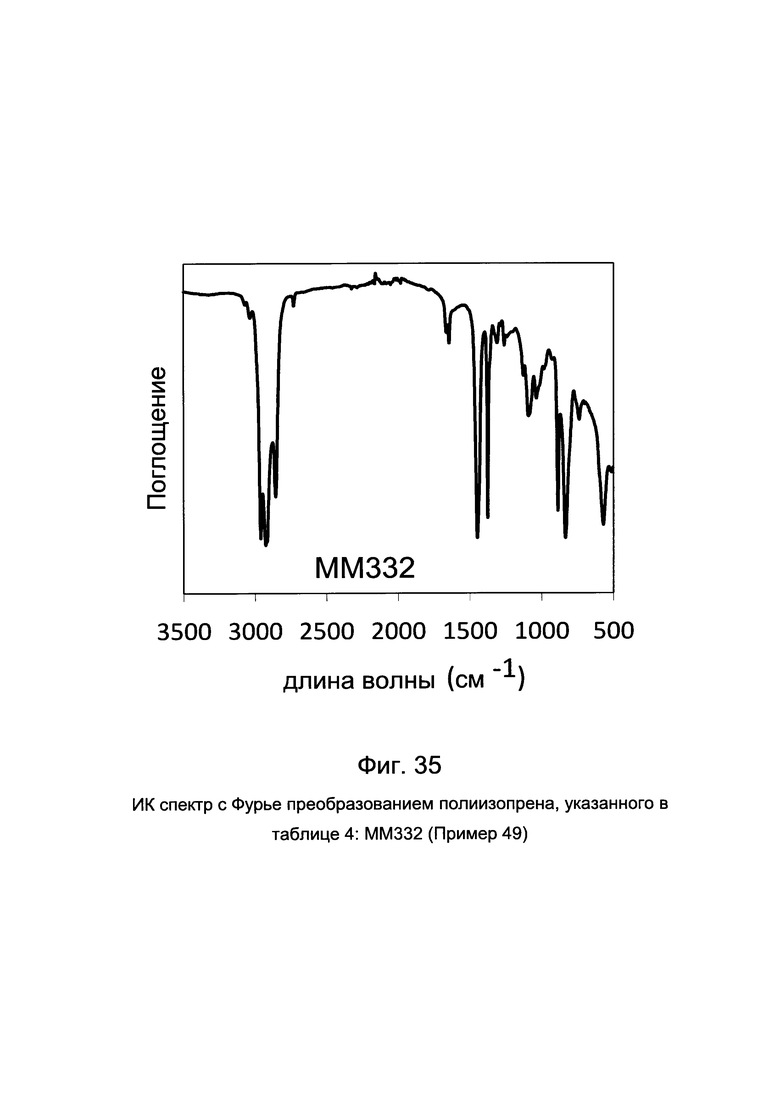
Изобретение относится к каталитической системе для (co)полимеризации сопряженных диенов. Каталитическая система включает: (a) по меньшей мере один фосфиновый комплекс ванадия, имеющий общую формулу (I) или (II): V(X)3[P(R1)n(R2)3-n]2 (I), V(X)3[(R3)2P(R4)P(R3)2] (II), где X представляет собой анион, выбранный из галогена, такого как хлор, бром, йод; R1, одинаковые или отличающиеся друг от друга, представляют собой атом водорода или выбраны из алкильных групп C1-C20, линейных или разветвленных, и С3-С6 циклоалкильных групп, n целое число в диапазоне от 0 до 3; R2, одинаковые или отличающиеся друг от друга, выбраны из арильных групп, представляющих собой карбоциклические ароматические группы, выбранные из фенила, нафтила, фенантрена и антрацена; R3, одинаковые или отличающиеся друг от друга, представляют собой атом водорода или выбраны из алкильных групп C1-C20, линейных или разветвленных, С3-С6 циклоалкильных групп и арильных групп, представляющих собой карбоциклические ароматические группы, выбранные из фенила, нафтила, фенантрена и антрацена; R4 представляет собой группу -NR5, в которой R5 представляет собой атом водорода или выбран из C1-C20 алкильных групп, линейных или разветвленных, или R4 представляет собой алкиленовую группу - (CH2)p-, в которой p представляет собой целое число в диапазоне от 1 до 5; при условии, что если в общей формуле (I) n равно 1 и R1 является метилом, то R2 отличается от фенила, и (b) по меньшей мере один co-катализатор, выбранный из органических производных алюминия, представляющих собой алюмоксаны, имеющие общую формулу (IV): (R9)2-Al-O-[-Al(R10)-O-]q-Al-(R11)2 (IV). В формуле (IV) R8, R9 и R10, одинаковые или отличающиеся друг от друга, представляют собой атом водорода или атом галогена, такого как хлор, бром, йод, фтор, или выбраны из C1-C20 алкильных групп, линейных или разветвленных, циклоалкильных групп, арильных групп, причем указанные группы возможно замещены одним или более атомов кремния или германия, и q - целое число в диапазоне от 0 до 1000. Также предложены способ (со)полимеризации сопряженных диенов и способ полимеризации 1,3-бутадиена или изопрена. Предложенная каталитическая система обеспечивает получение (со)полимеров сопряженных диенов с доминирующим содержанием 1,4-транс и 1,4-цис звеньев. 3 н. и 2 з.п. ф-лы, 36 ил., 4 табл., 51 пр.
1. Каталитическая система для (co)полимеризации сопряженных диенов, включающая:
(a) по меньшей мере один фосфиновый комплекс ванадия, имеющий общую формулу (I) или (II):
V(X)3[P(R1)n(R2)3-n]2 (I),
V(X)3[(R3)2P(R4)P(R3)2] (II),
где
- X представляет собой анион, выбранный из галогена, такого как хлор, бром, йод;
- R1, одинаковые или отличающиеся друг от друга, представляют собой атом водорода или выбраны из алкильных групп C1-C20, линейных или разветвленных, и С3-С6 циклоалкильных групп;
- n - целое число в диапазоне от 0 до 3;
- R2, одинаковые или отличающиеся друг от друга, выбраны из арильных групп, представляющих собой карбоциклические ароматические группы, выбранные из фенила, нафтила, фенантрена и антрацена;
- R3, одинаковые или отличающиеся друг от друга, представляют собой атом водорода или выбраны из алкильных групп C1-C20, линейных или разветвленных, С3-С6 циклоалкильных групп и арильных групп, представляющих собой карбоциклические ароматические группы, выбранные из фенила, нафтила, фенантрена и антрацена;
- R4 представляет собой группу -NR5, в которой R5 представляет собой атом водорода или выбран из C1-C20 алкильных групп, линейных или разветвленных; или R4 представляет собой алкиленовую группу - (CH2)p-, в которой p представляет собой целое число в диапазоне от 1 до 5;
при условии, что если в общей формуле (I) n равно 1 и R1 является метилом, то R2 отличается от фенила;
(b) по меньшей мере один co-катализатор, выбранный из органических производных алюминия, представляющих собой алюмоксаны, имеющие общую формулу (IV):
(R9)2-Al-O-[-Al(R10)-O-]q-Al-(R11)2 (IV),
в которой R8, R9 и R10, одинаковые или отличающиеся друг от друга, представляют собой атом водорода или атом галогена, такого как хлор, бром, йод, фтор, или выбраны из C1-C20 алкильных групп, линейных или разветвленных, циклоалкильных групп, арильных групп, причем указанные группы возможно замещены одним или более атомов кремния или германия; и q - целое число в диапазоне от 0 до 1000.
2. Каталитическая система для (co)полимеризации сопряженных диенов по п. 1, в которой в указанном фосфиновом ванадиевом комплексе, имеющем общую формулу (I) или (II):
- X представляет собой анион, выбранный из галогена, такого как хлор, бром, йод, предпочтительно хлор;
- R1, одинаковые или отличающиеся друг от друга, представляют собой атом водорода или выбраны из C1-C20 алкильных групп, предпочтительно C1-C15, линейных или разветвленных, предпочтительно представляют собой метил, этил, изопропил, трет-бутил, или выбраны из С3-С6 циклоалкильных групп, предпочтительно представляют собой циклопентил, циклогексил;
- n - целое число в диапазоне от 0 до 3;
- R2, одинаковые или отличающиеся друг от друга, выбраны из арильных групп, представляющих собой карбоциклические ароматические группы, выбранные из фенила, нафтила, фенантрена и антрацена, предпочтительно представляют собой фенил;
- R3, одинаковые или отличающиеся друг от друга, выбраны из C1-C20 алкильных групп, предпочтительно C1-C15, линейных или разветвленных, предпочтительно представляют собой метил, этил, или выбраны из арильных групп, представляющих собой карбоциклические ароматические группы, выбранные из фенила, нафтила, фенантрена и антрацена, предпочтительно представляют собой фенил;
- R4 представляет собой группу -NR5, в которой R5 представляет собой атом водорода; или R4 представляет собой группу - (CH2)p-, в которой p равно 2.
3. Каталитическая система для (со)полимеризации сопряженных диенов по п. 1 или 2, в которой указанный co-катализатор выбран из алюмоксанов, имеющих общую формулу (IV), предпочтительно метилалюмоксана (MAO) как такового или в "сухой" форме (MAO-сухой).
4. Способ (со)полимеризации сопряженных диенов, в котором (со)полимеризацию сопряженных диенов проводят в присутствии каталитической системы по любому из пп. 1-3.
5. Способ полимеризации 1,3-бутадиена или изопрена, в котором полимеризацию 1,3-бутадиена или изопрена проводят в присутствии каталитической системы по любому из пп. 1-3.
| МЕХАНИЧЕСКАЯ ЧАСТЬ ТОРМОЗА ЖЕЛЕЗНОДОРОЖНОГО ГРУЗОВОГО ВАГОНА БУНКЕРНОГО ТИПА | 1992 |
|
RU2039677C1 |
| HOLT D.G.L | |||
| et al | |||
| Synthesis and structural studies of complexes of vanadium(II) and vanadium(III) halides with tertiary phosphines, Inorganica Chimica Acta, 1993, v | |||
| Станок для изготовления из дерева круглых палочек | 1915 |
|
SU207A1 |
| Походная разборная печь для варки пищи и печения хлеба | 1920 |
|
SU11A1 |
| NIEMAN J | |||
| et al | |||
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Organomet | |||
| Chem., 1983, v | |||
| Гудок | 1921 |
|
SU255A1 |
| Приспособление для градации давления в воздухопроводе воздушных тормозов | 1921 |
|
SU193A1 |
| BRADLEY S | |||
| et al | |||
| Synthesis and Structure of Amino-functionalized Cyclopentadienyl Vanadium Complexes and Evaluation of Their Butadiene Polymerization Behavior, Organometallics, 2002, v | |||
| Выбрасывающий ячеистый аппарат для рядовых сеялок | 1922 |
|
SU21A1 |
| ЭЛЕКТРИЧЕСКАЯ ЛАМПА НАКАЛИВАНИЯ С ДВУМЯ ПРЕДНАЗНАЧЕННЫМИ ДЛЯ ПАРАЛЛЕЛЬНОГО И ПОСЛЕДОВАТЕЛЬНОГО СОЕДИНЕНИЯ НИТЯМИ | 1925 |
|
SU3443A1 |
| HILLS A | |||
| et al | |||
| Compounds with Vanadium-Nitrogen and Vanadium-Oxygen Multiple Bonds, J | |||
| Chem | |||
| Soc | |||
| Dalton Trans., 1993, p | |||
| ПРЕЗЕРВАТИВ (КОНДОМ) | 1924 |
|
SU3609A1 |
| ISSLEIB K | |||
| et al | |||
| Beitrage zur Komplexchemie tertiarer Phosphine und Phosphinoxyde | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Прибор для исправления снимков рельефа местности | 1921 |
|
SU301A1 |
| Поршень для воздушных тормозов с сжатым воздухом | 1921 |
|
SU188A1 |
| HERMES A.R | |||
| et al | |||
| Phosphine Complexes of Trivalent Early Transition Metals | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Солесос | 1922 |
|
SU29A1 |
| Способ получения древесного угля | 1921 |
|
SU313A1 |
| TSAGKALIDIS W | |||
| et al | |||
| Hydrazine complexes of vanadium(II and III), Inorganica Chimica Acta, 1993, v | |||
| Автоматическая акустическая блокировка | 1921 |
|
SU205A1 |
| Коловратный насос с кольцевым поршнем, перемещаемым эксцентриком | 1921 |
|
SU239A1 |
| RICCI G | |||
| et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Пишущая машина | 1922 |
|
SU37A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Способ получения бензонафтола | 1920 |
|
SU363A1 |
| КАТАЛИТИЧЕСКИЕ СИСТЕМЫ ДЛЯ ОЛИГОМЕРИЗАЦИИ ЭТИЛЕНА, ИМЕЮЩИЕ ПОВЫШЕННУЮ СЕЛЕКТИВНОСТЬ | 2007 |
|
RU2456077C2 |
| Способ получения сополимеров изобутилена | 1972 |
|
SU505370A3 |

