ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к некоторым замещенным бициклическим соединениям, представляющим собой агонисты рексиноидных рецепторов (RXR) и, следовательно, подходящим для лечения некоторых расстройств, которые можно предотвратить или лечить активацией этого рецептора. Кроме того, настоящее изобретение относится к соединениям, способам их получения, фармацевтическим композициям, содержащим указанные соединения, и к применению указанных соединений при лечении некоторых расстройств. Ожидается, что соединения согласно настоящему изобретению найдут применение в лечении заболеваний, таких как инсулиннезависимый сахарный диабет 2 типа (NIDDM), резистентность к инсулину, ожирение, нарушенная гликемия натощак, нарушение толерантности к глюкозе, нарушения липидного обмена, таких как дислипидемия, гипертония, а также других заболеваний и состояний.
УРОВЕНЬ ТЕХНИКИ
Ретиноевая кислота (RA), которая представляет собой витамин А в виде карбоновой кислоты, представляет собой производное питательного вещества, обладающее замечательными биологическими эффектами при регуляции глюкозы, жирных кислот, холестерина и метаболизма аминокислот, и также в биологии жировой ткани и системе контроля энергетического баланса. Например, исследования показали, что у мышей лечение полностью транс-ретиноевыми кислотами (ATRA, англ. all trans RA) снижает массу тела и ожирение вне зависимости от каких-либо изменений в потреблении пищи, а также улучшает толерантность к глюкозе. Кроме того, также было показано, что вызванная ATRA потеря жира в теле коррелирует с активацией бурой жировой ткани наряду с увеличением окислительного метаболизма и термогенеза в белой жировой ткани. Также известно, что это сопровождается повышением уровня циркулирующих неэтерифицированных жирных кислот, что было интерпретировано следующим образом: жирные кислоты, мобилизованные из жировых запасов, подвергаются окислению внутри самих адипоцитов или в других тканях.
Считается, что ретиноевая кислота (RA) регулирует экспрессию гена преимущественно через активацию либо рецепторов ретиноевых кислот (RAR), которые, как выяснилось, активируются с помощью полностью транс-ретиноевых кислот (ATRA) и 9-цис-ретиноевых кислот, либо рексиноидными рецепторами (RXR). В отличие от рецепторов ретиноевых кислот (RAR) рексиноидные рецепторы (RXR) является частью суперсемейства ядерных рецепторов, которые функционируют как транскрипционный фактор, активируемый лигандами. RXR являются гетеродимерными партнерами многих ядерных рецепторов, таких как рецепторы ретиноевых кислот (RAR), рецепторы гормонов щитовидной железы (T3R), рецепторы печени (LXR), рецепторы, активируемые пролифератором пероксисом (PPAR), которые также образуют гомодимер с самими RXR. Активация этих рецепторов как таковая должна оказывать значительное влияние на экспрессию генов.
Модулируя множество факторов транскрипции, ретиноевые кислоты регулируют некоторые аспекты метаболических заболеваний, таких как гипергликемия, гипертриглицеридемия, гиперхолестеринемия, гипертония, усталость мышц, гемостаз и контроль массы тела, а также висцеральное ожирение, что делает их привлекательной мишенью для лечения ожирения и диабета, а также связанных с ними симптомов.
В дополнение к метаболическому гомеостазу ретиноевая кислота играет важную роль в регуляции здоровья тканей, а также регулирует воспалительные реакции. Действительно, было показано, что агонисты RXR обладают многими видами потенциальной биологической активности, включая активность в отношении лечения рака, кожных заболеваний, болезней обмена веществ, фиброза, нейродегенеративных заболеваний, сосудистых заболеваний, заболеваний глаз и воспалительных заболеваний.
Следовательно, имеющиеся данные говорят о том, что соединения, которые являются агонистами RXR, будут полезны при лечении ряда клинических состояний, и поэтому в настоящее время продолжается поиск подходящих агонистов RXR.
Желательно обеспечить соединения, являющиеся агонистами RXR. Ожидается, что эти соединения будут полезны для лечения состояний, которые можно лечить путем активации этих рецепторов.
Также желательно обеспечить фармацевтическую композицию, содержащую соединение, которое является агонистом RXR, фармацевтически приемлемые наполнитель, разбавитель или носитель.
Еще более желательно разработать способ профилактики или лечения состояния у млекопитающего, которое можно лечить путем активации RXR.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящем изобретении предложено соединение формулы (I):
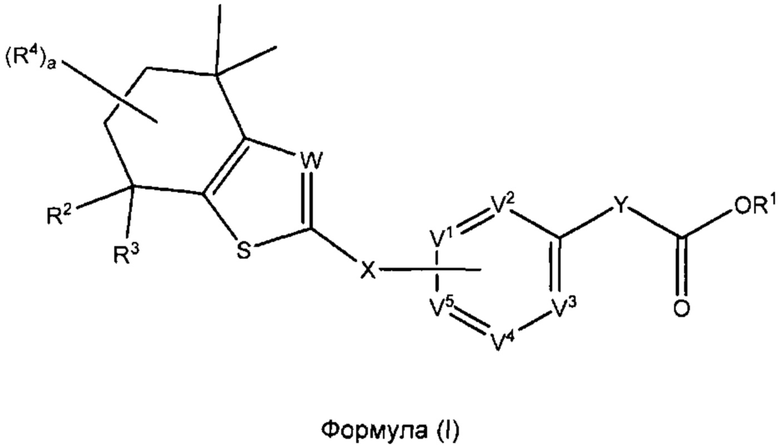
где
R1 выбран из группы, состоящей из Н, C1-C6 алкила и защитной группы карбоновой кислоты;
R2 и R3 независимо выбраны из группы, состоящей из Н и C1-C6 алкила;
каждый R4 независимо выбран из группы, состоящей из Н, галогена, ОН, NO2, CN, SH, NH2, CF3, OCF3, необязательно замещенного С1-С12алкила, необязательно замещенного С1-С12галогеналкила, необязательно замещенного С2-С12алкенила, необязательно замещенного С2-С12алкинила, необязательно замещенного С2-С12гетероалкила, необязательно замещенного С3-С12циклоалкила, необязательно замещенного С3-С12циклоалкенила, необязательно замещенного С2-С12гетероциклоалкила, необязательно замещенного С2-С12гетероциклоалкенила, необязательно замещенного С6-С18арила, необязательно замещенного С1-С18гетероарила, необязательно замещенного С1-С12алкилокси, необязательно замещенного С2-С12алкенилокси, необязательно замещенного С2-С12алкинилокси, необязательно замещенного С2-С10гетероалкилокси, необязательно замещенного С3-С12циклоалкилокси, необязательно замещенного С3-С12циклоалкенилокси, необязательно замещенного С2-С12гетероциклоалкилокси, необязательно замещенного С2-С12гетероциклоалкенилокси, необязательно замещенного С6-С18арилокси, необязательно замещенного С1-С18гетероарилокси, необязательно замещенного С1-С12алкиламино, SR5, SO3H, SO2NR5R5, SO2R5, SONR5R5, SOR5, COR5, COOH, COOR5, CONR5R5, NR5COR5, NR5COOR5, NR5SO2R5, NR5CONR5R5, NR5R5 и ацила, или два R4 на одном атоме углерода совместно образуют заместитель =O или группу формулы =NOH или два R4 на соседних атомах углерода совместно образуют двойную связь;
каждый R5 независимо выбран из группы, состоящей из Н, необязательно замещенного С1-С12алкила, необязательно замещенного С2-С10гетероалкила, необязательно замещенного С1-С12галогеналкила, необязательно замещенного С3-С12циклоалкила, необязательно замещенного С6-С18арила и необязательно замещенного С1-С18гетероарила;
а представляет собой целое число, выбранное из группы, состоящей из 0, 1, 2, 3 и 4;
X представляет собой связь или связующую группу;
V2, V3 и V4 каждый независимо выбраны из группы, состоящей из N и CR6;
каждый R6 независимо выбран из группы, состоящей из Н, галогена, ОН, NO2, CN, SH, NH2, CF3, OCF3, необязательно замещенного С1-С12алкила, необязательно замещенного С1-С12галогеналкила, необязательно замещенного С2-С12алкенила, необязательно замещенного С2-С12алкинила, необязательно замещенного С2-С12гетероалкила, необязательно замещенного С3-С12циклоалкила, необязательно замещенного С3-С12циклоалкенила, необязательно замещенного С2-С12гетероциклоалкила, необязательно замещенного С2-С12гетероциклоалкенила, необязательно замещенного С6-С18арила, необязательно замещенного С1-С18гетероарила, необязательно замещенного С1-С12алкилокси, необязательно замещенного С2-С12алкенилокси, необязательно замещенного С2-С12алкинилокси, необязательно замещенного С2-С10гетероалкилокси, необязательно замещенного С3-С12циклоалкилокси, необязательно замещенного С3-С12циклоалкенилокси, необязательно замещенного С2-С12гетероциклоалкилокси, необязательно замещенного С2-С12гетероциклоалкенилокси, необязательно замещенного С6-С18арилокси, необязательно замещенного С1-С18гетероарилокси, необязательно замещенного С1-С12алкиламино, SR5, SO3H, SO2NR5R5, SO2R5, SONR5R5, SOR5, COR5, COOH, COOR5, CONR5R5, NR5COR5, NR5COOR5, NR5SO2R5, NR5CONR5R5, NR5R5 и ацила;
V1 и V5 выбраны из группы, состоящей из N, CR7 и CR8 таким образом, что один из V1 и V5 представляет собой CR8 и другой представляет собой Н или CR7;
каждый R7 независимо выбран из группы, состоящей из Н, галогена, ОН, NO2, CN, SH, NH2, CF3, OCF3, необязательно замещенного С1-С12алкила, необязательно замещенного С1-С12галогеналкила, необязательно замещенного С2-С12алкенила, необязательно замещенного С2-С12алкинила, необязательно замещенного С2-С12гетероалкила, необязательно замещенного С3-С12циклоалкила, необязательно замещенного С3-С12циклоалкенила, необязательно замещенного С2-С12гетероциклоалкила, необязательно замещенного С2-С12гетероциклоалкенила, необязательно замещенного С6-С18арила, необязательно замещенного С1-С18гетероарила, необязательно замещенного С1-С12алкилокси, необязательно замещенного С2-С12алкенилокси, необязательно замещенного С2-С12алкинилокси, необязательно замещенного С2-С10гетероалкилокси, необязательно замещенного С3-С12циклоалкилокси, необязательно замещенного С3-С12циклоалкенилокси, необязательно замещенного С2-С12гетероциклоалкилокси, необязательно замещенного С2-С12гетероциклоалкенилокси, необязательно замещенного С6-С18арилокси, необязательно замещенного С1-С18гетероарилокси, необязательно замещенного С1-С12алкиламино, SR5, SO3H, SO2NR5R5, SO2R5, SONR5R5, SOR5, COR5, COOH, COOR5, CONR5R5, NR5COR5, NR5COOR5, NR5SO2R5, NR5CONR5R5, NR5R5 и ацила, или R7 совместно с атомом углерода, к которому он присоединен, образует циклический фрагмент с заместителем на X;
R8 представляет собой связь с X,
Y представляет собой связь или связующую группу;
W выбран из группы, состоящей из N и CR9;
R9 выбран из Н и С1-С6алкила;
или его фармацевтически приемлемая соль.
В еще одном аспекте настоящее изобретение относится к фармацевтической композиции, содержащей соединение согласно настоящему изобретению и фармацевтически приемлемые разбавитель, наполнитель или носитель.
В другом аспекте настоящее изобретение относится к способу профилактики или лечения состояния у млекопитающего, включающему введение эффективного количества соединения согласно настоящему изобретению. В одном варианте реализации настоящего изобретения состояние представляет собой состояние, которое можно предотвратить или лечить с помощью активации RXR.
В другом аспекте настоящее изобретение относится к применению соединения согласно настоящему изобретению для получения лекарственного средства для лечения состояния у млекопитающего. В одном варианте реализации настоящего изобретения состояние представляет собой состояние, которое можно предотвратить или лечить с помощью активации RXR.
В другом аспекте настоящее изобретение относится к применению соединения согласно настоящему изобретению для лечения состояния у млекопитающего. В одном варианте реализации настоящего изобретения состояние представляет собой состояние, которое можно предотвратить или лечить с помощью активации RXR.
Примеры состояний, которые можно лечить, включают рак, дерматологические заболевания, заболевания дыхательной и легочной системы, нарушения обмена веществ, воспалительные заболевания, заболевания почек, аутоиммунные заболевания и нейродегенеративные заболевания.
Примеры рака включают рак груди, рак поджелудочной железы, кожную Т-клеточную лимфому (рецидивирующую или резистентную кожную Т-клеточную лимфому), рак легкого, рак печени (гепатоцеллюлярную карциному), саркому Капоши (саркому Капоши, связанную со СПИДом), кожную Т-клеточную лимфому, промиелолейкоз, рак кожи (базально-клеточный рак), немелкоклеточный рак легкого, рак почки (распространенный почечно-клеточный рак), рак желудочно-кишечного тракта (желудка) (прогрессирующий рак пищеварительного тракта), мезотелиому, и немелкоклеточный рак легких.
Примеры дерматологических расстройств включают дерматит (тяжелая хроническая экзема рук у взрослых), псориаз (тяжелый бляшковидный псориаз), псориаз (от умеренного до тяжелого) и облысение.
Примеры расстройств дыхательной и легочной системы включают бронхиальную метаплазию и легочный фиброз (фиброз).
Примеры метаболических заболеваний включают преддиабет, диабет 2 типа, ожирение, гиперхолестеринемию, гипертриглицеридемию, гипертензию, дислипидемию, гиперинсулинемию, заболевания печени, неалкогольный стеатогепатит (НАСГ) и атеросклероз.
Примеры воспалительных заболеваний включают окислительный стресс, фиброз почек, заболевания печени, такие как стеатоз, стеатогепатит (алкогольный и неалкогольный), фиброз и цирроз печени, аутоиммунные заболевания и экспериментальный аутоиммунный энцефаломиелит.
Примером нейродегенеративного расстройства является болезнь Альцгеймера.
Примеры других состояний, которые можно лечить, включают ожирение и сердечно-сосудистые заболевания.
Соединения согласно настоящему изобретению также можно применять для обеспечения ряда полезных эффектов у млекопитающих. Примеры полезных эффектов, которые могут быть обеспечены, включают увеличение мышечной выносливости, улучшение сердечной функции и достижение эффекта, миметического физическим упражнениям.
Следовательно, в другом аспекте настоящего изобретения предложен способ увеличения мышечной выносливости у млекопитающего, причем указанный способ включает введение эффективного количества соединения согласно настоящему изобретению.
Следовательно, в другом аспекте настоящего изобретения предложен способ улучшения сердечной функции у млекопитающего, причем указанный способ включает введение эффективного количества соединения согласно настоящему изобретению. Примеры улучшения сердечной функции, включают систолическую работу, систолический объем крови, долю выброса и наполнение желудочка.
Следовательно, в другом аспекте настоящего изобретения предложен способ достижения эффекта, миметического физическим упражнениям, у млекопитающего, причем указанный способ включает введение эффективного количества соединения согласно настоящему изобретению.
Эти и другие принципы настоящего изобретения изложены в настоящем документе.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На фигуре 1А показано удержание холестерина в нейрональных клетках, обработанных соединением 40.
На фигуре 1В показаны уменьшенные уровни оксидативного стресса (ROS, англ. reduced oxidative stress) в нейрональных клетках, обработанных соединением 40.
На фигуре 1С показана активность каспазы в нейрональных клетках, обработанных соединением 40.
На фигуре 1D показана экспрессия АВСА1 в нейрональных клетках, обработанных соединением 40.
На фигуре 1Е показана экспрессия ABCG1 в нейрональных клетках, обработанных соединением 40.
На фигуре 2А показаны уменьшенные уровни оксидативного стресса (ROS) в астроцитах, обработанных соединением 40.
На фигуре 2В показаны уровни IL6 в астроцитах, обработанных соединением 40.
На фигуре 2С показана экспрессия IL6 в астроцитах, обработанных соединением 40.
На фигуре 2D показана экспрессия IL1B в астроцитах, обработанных соединением 40.
На фигуре 2Е показана экспрессия СОХ2 в астроцитах, обработанных соединением 40.
На фигуре 2F показана экспрессия МСР1 в астроцитах, обработанных соединением 40.
На фигуре 3А показан рост клеточной линии MiA РаСа2 на клетках, обработанных соединением 40.
На фигуре 3В показаны результаты анализа клеточной пролиферации (инкорпорирование BrdU) роста клеточной линии MiA РаСа2 на клетках, обработанных соединением 40.
На фигуре 4А показаны уменьшенные уровни оксидативного стресса (ROS) в MiA РаСа2, обработанных соединением 40.
На фигуре 4В показаны уровни каспазы в MiA РаСа2, обработанных соединением 40.
На фигуре 4С показана гибель клеток ELISA в Mia РаСа2, обработанных соединением 40.
На фигуре 5А показано влияние соединения 40 на клеточную пролиферацию клеток MCF7.
На фигуре 5В показано влияние соединения 40 на клеточную пролиферацию клеток MCF7 (инкорпорирование BrdU).
На фигуре 5С показаны уменьшенные уровни оксидативного стресса (ROS) в клетках MCF7, обработанных соединением 40.
На фигуре 5D показана гибель клеток ELISA в клетках MCF7, обработанных соединением 40.
На фигуре 6 показана дифференциация клеток HL60 PML, обработанных соединением 40.
На фигуре 7А показано влияние соединения 40 на глюкозу у мышей с алиментарным ожирением (DIO, англ. Diet Induced Obesity).
На фигуре 7В показано влияние соединения 40 на уровни инсулина у мышей с алиментарным ожирением.
На фигуре 8 показано влияние соединения 40 на индекс инсулинорезистентности (НОМА IR, англ. Homeostasis Model Assessment of Insulin Resistance) у мышей с алиментарным ожирением.
На фигуре 9 показано влияние соединения 40 на уровень триглицеридов в крови натощак у мышей с алиментарным ожирением.
На фигуре 10А показано влияние соединения 40 на общий уровень холестерина у мышей с алиментарным ожирением.
На фигуре 10В показано влияние соединения 40 на не-ЛПВП холестерин (холестерин липопротеинов высокой плотности) у мышей с алиментарным ожирением (DIO).
На фигуре 11А показано влияние соединения 40 на СЖК (свободные жирные кислоты) у мышей с алиментарным ожирением.
На фигуре 11В показано влияние соединения 40 на уровни глицерина у мышей с алиментарным ожирением.
На фигуре 12А показано влияние соединения 40 на массу тела у мышей с алиментарным ожирением.
На фигуре 12В показано влияние соединения 40 на уровни абсолютной массы жировой ткани у мышей с алиментарным ожирением.
На фигуре 13А показано влияние соединения 40 на термогенез у мышей с алиментарным ожирением.
На фигуре 13В показано влияние соединения 40 на размер мезентериальных адипоцитов у мышей с алиментарным ожирением.
На фигуре 13С показано влияние соединения 40 на экспрессию генов в WAT (белой жировой ткани).
На фигуре 14А показано влияние соединения 40 на упражнение на мышечную выносливость (дистанция) у мышей с алиментарным ожирением.
На фигуре 14В показано влияние соединения 40 на упражнение на мышечную выносливость (общее время бега) у мышей с алиментарным ожирением.
На фигуре 15 показано влияние соединения 40 на мышей ob/ob.
На фигуре 16А показано влияние соединения 40 на общий уровень холестерина у мышей ob/ob.
На фигуре 16В показано влияние соединения 40 на холестерин липопротеидов низкой плотности (ЛПНП) у мышей ob/ob.
На фигуре 17 показано влияние соединения 40 на мышей db/db.
На фигуре 18 показано влияние соединения 40 на профиль глюкозы у получающих питание мышей db/db.
На фигуре 19 показано влияние соединения 40 на уровни HbA1c с у мышей db/db.
На фигуре 20А показано влияние соединения 40 на уровни глюкозы у крыс линии Zucker с ожирением, которых кормили глюкозой.
На фигуре 20В показано влияние соединения 40 на уровни глюкозы у крыс Zucker с ожирением натощак.
На фигуре 21А показано влияние соединения 40 на уровни глюкозы у крыс Zucker с ожирением натощак.
На фигуре 21В показано влияние соединения 40 на уровни инсулина у крыс Zucker с ожирением.
На фигуре 22 показано влияние соединения 40 на уровни HbA1c у крыс Zucker с ожирением.
На фигуре 23 показано влияние соединения 40 на уровни триглицеридов в крови натощак у крыс Zucker с ожирением.
На фигуре 24 показано влияние соединения 40 на уровни С-реактивного белка (CRP) у крыс Zucker с ожирением.
На фигуре 25A-D показано влияние соединения 40 на сердечную функцию крыс Zucker с ожирением.
На фигурах 26A-D показано влияние соединения 40 на уровни биомаркеров сердечнососудистого риска у крыс Zucker с ожирением.
На фигуре 27 показано влияние соединения 40 на скорость распространения возбуждения у крыс Zucker с ожирением.
На фигуре 28А показано влияние соединения 40 на уровни азота мочевины (BUN) крови у крыс Zucker с ожирением.
На фигуре 28В показано влияние соединения 40 на уровни креатинина сыворотки крови у крыс Zucker с ожирением.
На фигуре 29А показано влияние соединения 40 на уровень глюкозы в крови натощак у крыс Wistar.
На фигуре 29В показано влияние соединения 40 на глюкозу у крыс линии Wistar.
На фигуре 30А показано влияние соединения 40 на еженедельную массу тела у крыс Wistar.
На фигуре 30В показано влияние соединения 40 на уровень триглицеридов в крови натощак у крыс Wistar.
На фигуре 31 показано влияние соединения 40 на абсолютную массу сердца у крыс Wistar.
На фигуре 32А показано влияние соединения 40 на толщину стенки левого желудочка у крыс Wistar.
На фигуре 32В показано влияние соединения 40 на толщину внутрижелудочковой перегородки у крыс Wistar.
На фигуре 33А показано влияние соединения 40 на систолический объем крови у крыс Wistar.
На фигуре 33В показано влияние соединения 40 на работу верхушечного толчка у крыс Wistar.
На фигурах 34А-С показано влияние соединения 40 на различные факторы, относящиеся к деятельности сердца, у крыс Wistar.
На фигурах 35А-В показано влияние соединения 40 на различные факторы, относящиеся к деятельности сердца, у крыс Wistar.
На фигуре 36 показано влияние соединения 40 на диастолический объем левого желудочка у крыс Wistar.
На фигурах 37A-D показано влияние соединения 40 на различные уровни триглицеридов у хомяков.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящем изобретении используются ряд терминов, которые хорошо известны специалисту в данной области техники. Тем не менее, для целей ясности ниже будет определен ряд терминов.
Термин «незамещенный», используемый в настоящем изобретении, обозначает, что заместители отсутствуют или представляют собой водород.
Термин «необязательно замещенный», используемый в настоящем изобретении, обозначает, что группа может быть или не быть дополнительно замещенной или конденсированной (таким образом, чтобы образовать конденсированную полициклическую систему) с одной или несколькими замещающими неводородными группами. В некоторых вариантах реализации настоящего изобретения замещающие группы представляют собой одну или несколько групп, независимо выбранных из группы, состоящей из галогена, =O, =S, -CN, -NO2, -CF3, -OCF3, алкила, алкенила, алкинила, галогеналкила, галогеналкенила, галогеналкинила, гетероалкила, циклоалкила, циклоалкенила, гетероциклоалкила, гетероциклоалкенила, арила, гетероарила, циклоалкилалкила, гетероциклоалкилалкила, гетероарилалкила, арилалкила, циклоалкилалкенила, гетероциклоалкилалкенила, арилалкенила, гетероарилалкенила, циклоалкилгетероалкила, гетероциклоалкилгетероалкила, арилгетероалкила, гетероарилгетероалкила, гидрокси, гидроксиалкила, алкилокси, алкилоксиалкила, алкилоксициклоалкила, алкилоксигетероциклоалкила, алкилоксиарила, алкилоксигетероарила, алкилоксикарбонила, алкиламинокарбонила, алкенилокси, алкинилокси, циклоалкилокси, циклоалкенилокси, гетероциклоалкилокси, гетероциклоалкенилокси, арилокси, фенокси, бензилокси, гетероарилокси, арилалкилокси, амино, алкиламино, ациламино, аминоалкила, ариламино, сульфониламино, сульфиниламино, сульфонила, алкилсульфонила, арилсульфонила, аминосульфонила, сульфинила, алкилсульфинила, арилсульфинила, аминосульфиниламиноалкила, С(=O)ОН, -C(=O)Re, C(=O)ORe, C(=O)NReRf, C(=NOH)Re, C(=NRe)NRfRg, NReRf, NReC (=O)Rf, NReC (=O)ORf, NReC (=O)NRfRg, NReC(=NRf)NRgRh, NReSO2RF, -SReSO2NReRf, -ORe, OC(=O)NReRf, OC(=O)Re и ацила,
где Re, Rf, Rg и Rh каждый независимо выбран из группы, состоящей из Н, С1-С12алкила, С1-С12галогеналкила, С2-С12алкенила, С2-С12алкинила, С1-С10гетероалкила, С3-С12циклоалкила, С3-С12циклоалкенила, С1-С12гетероциклоалкила, С1-С12гетероциклоалкенила, С6-С18арила, С1-С18гетероарила и ацила или любые два или более из Ra, Rb, Rc и Rd совместно с атомами, к которым они присоединены, образуют гетероциклическую кольцевую систему, содержащую от 3 до 12 атомов в кольце.
В некоторых вариантах реализации настоящего изобретения каждый необязательный заместитель независимо выбран из группы, состоящей из галогена, =O, =S, -CN, -NO2, -CF3, -OCF3, алкила, алкенила, алкинила, галогеналкила, галогеналкенила, галогеналкинила, гетероалкила, циклоалкила, циклоалкенила, гетероциклоалкила, гетероциклоалкенила, арила, гетероарила, гидрокси, гидроксиалкила, алкилокси, алкилоксиалкила, алкилоксиарила, алкилоксигетероарила, алкенилокси, алкинилокси, циклоалкилокси, циклоалкенилокси, гетероциклоалкилокси, гетероциклоалкенилокси, арилокси, гетероарилокси, арилалкила, гетероарилалкила, арилалкилокси, амино, алкиламино, ациламино, аминоалкила, ариламино, сульфонила, алкилсульфонила, арилсульфонила, аминосульфонила, аминоалкила, СООН, SH и ацила.
Примеры особенно подходящих необязательных заместителей включают F, Cl, Br, I, CH3, СН2СН3, ОН, ОСН3, CF3, OCF3, NO2, NH2 и CN.
В определениях ряда заместителей указанных ниже утверждается, что «группа может быть концевой группой или мостиковой группой». Это обозначает, что использование термина призвано охватывать ситуацию, когда указанная группа представляет собой линкер между двумя другими частями молекулы, а также ситуацию, когда группа представляет собой концевой фрагмент. Используя термин алкил в качестве примера, некоторые публикации будут использовать термин «алкилен» для мостиковой группы и, следовательно, в этих публикациях существует различие между терминами «алкил» (концевая группа) и «алкилен» (мостиковая группа). В настоящем изобретении таких различий не делается, и большинство групп могут представлять собой либо мостиковую группу, либо концевую группу.
Термин «ацил» обозначает группу RC(=O)-, в которой группа R может представлять собой алкильную, циклоалкильную, гетероциклоалкильную, арильную или гетероарильную группу, как определено в настоящем изобретении. Примеры ацила включают ацетил и бензоил. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через атом углерода карбонильной группы.
Термин «ациламино» обозначает группу RC(=O)-NH-, в которой группа R может представлять собой алкильную, циклоалкильную, гетероциклоалкильную, арильную или гетероарильную группу, как определено в настоящем изобретении. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через атом азота.
«Алкенил» как группа или часть группы обозначает алифатическую углеводородную группу, содержащую по меньшей мере одну двойную связь углерод-углерод, которая может быть неразветвленной или разветвленной, предпочтительно содержащей 2-12 атомов углерода, более предпочтительно 2-10 атомов углерода, наиболее предпочтительно 2-6 атомов углерода, в нормальной цепи. Группа может содержать множество двойных связей в нормальной цепи, и ориентация относительно каждой независимо представляет собой Е или Z. Алкенил предпочтительно представляет собой 1-алкенильную группу. Типичные алкенильные группы включают, но не ограничиваются ими, этенил, пропенил, бутенил, пентенил, гексенил, гептенил, октенил и ноненил. Указанная группа может представлять собой концевую группу или мостиковую группу.
Термин «алкенилокси» относится к группе алкенил-О-, в которой алкенил представляет собой такой, как определено в настоящем изобретении. Предпочтительные группы алкенилокси представляют собой группы С1-С6алкенилокси. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через атом кислорода.
«Алкил» как группа или часть группы, относится к неразветвленной или разветвленной алифатической углеводородной группе, предпочтительно С1-С12алкилу, более предпочтительно С1-С10алкилу, наиболее предпочтительно С1-С6, если не указано иное. Примеры подходящих неразветвленных или разветвленных С1-С6 алкильных заместителей включают метил, этил, н-пропил, 2-пропил, н-бутил, втор-бутил, трет-бутил, гексил и подобные. Указанная группа может представлять собой концевую группу или мостиковую группу.
Термин «алкиламино» включает как моно-алкиламино, так и диалкиламино, если не указано иное. Термин «моно-алкиламино» обозначает группу алкил-NH-, в которой алкил такой, как определено в настоящем изобретении. Термин «диалкиламино» обозначает группу (алкил)2N-, в которой каждый алкил может быть одинаковым или различным и каждый из них такой, как определено в настоящем изобретении для алкила. Алкильная группа предпочтительно представляет собой С1-С6алкильную группу. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через атом азота.
Термин «алкиламинокарбонил» относится к группе формулы (алкил)x(Н)yNС(=O)-, в которой алкил такой, как определено в настоящем изобретении, x равно 1 или 2 и сумма x+y=2. Указанная группа может представлять собой концевую группу или мостиковую группу. Если группа представляет собой концевую группу, она связана с остальной частью молекулы через атом углерода карбонильной группы.
Термин «алкилокси» относится к группе алкил-О-, в которой алкил такой, как определено в настоящем изобретении. Предпочтительный алкилокси представляет собой С1-С6алкилокси. Примеры включают, но не ограничиваются ими, метокси и этокси. Указанная группа может представлять собой концевую группу или мостиковую группу.
Термин «алкилоксиалкил» относится к группе алкилокси-алкил-, в которой алкилокси и алкильные фрагменты такие, как определено в настоящем изобретении. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через алкильную группу.
Термин «алкилоксиарил» относится к группе алкилокси-арил-, в которой алкилокси и арильные фрагменты такие, как определено в настоящем изобретении. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через арильную группу.
Термин «алкилоксикарбонил» относится к группе алкил-O-С(=O)-, в которой алкил представляет собой такой, как определено в настоящем изобретении. Алкильная группа предпочтительно представляет собой С1-С6алкильную группу. Примеры включают, но не ограничиваются ими, метоксикарбонил и этоксикарбонил. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через атом углерода карбонильной группы.
Термин «алкилоксициклоалкил» относится к группе алкилокси-циклоалкил-, в которой алкилокси и циклоалкильные фрагменты представляют собой такие, как определено в настоящем изобретении. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через циклоалкильную группу.
Термин «алкилоксигетероарил» относится к группе алкилокси-гетероарил-, в которой алкилокси и гетероарильные фрагменты представляют собой такие, как определено в настоящем изобретении. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через гетероарильную группу.
Термин «алкилоксигетероциклоалкил» относится к группе алкилокси-гетероциклоалкил-, в которой алкилокси и гетероциклоалкильные фрагменты представляют собой такие, как определено в настоящем изобретении. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через гетероциклоалкильную группу.
Термин «алкилсульфинил» обозначает группу алкил-S-(=O)-, в которой алкил такой, как определено в настоящем изобретении. Алкильная группа предпочтительно представляет собой С1-С6алкильную группу. Примерные алкилсульфинильные группы включают, но не ограничиваясь ими, метилсульфинил и этилсульфинил. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через атом серы.
Термин «алкилсульфонил» относится к группе алкил-S(=O)2-, в которой алкил представляет собой такой, как определено выше. Алкильная группа предпочтительно представляет собой С1-С6алкильную группу. Примеры включают, но не ограничиваются ими, метилсульфонил и этилсульфонил. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через атом серы.
«Алкинил» как группа или часть группы обозначает алифатическую углеводородную группу, содержащую тройную связь углерод-углерод, которая может быть неразветвленной или разветвленной, предпочтительно содержащей от 2-12 атомов углерода, более предпочтительно 2-10 атомов углерода, более предпочтительно 2-6 атомов углерода, в нормальной цепи. Примерные структуры включают, но не ограничиваются ими, этинил и пропинил. Указанная группа может представлять собой концевую группу или мостиковую группу.
Термин «алкинилокси» обозначает группу алкинил-О-, в которой алкинил такой, как определено в настоящем изобретении. Предпочтительные группы алкинилокси представляют собой группы С1-С6алкинилокси. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через атом кислорода.
Термин «аминоалкил» обозначает группу NH2-алкил-, в которой алкильная группа такая, как определено в настоящем изобретении. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через алкильную группу.
Термин «аминосульфонил» обозначает группу NH2-S(=O)2-. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через атом серы.
Термин «арил» как группа или часть группы обозначает (i) необязательно замещенный моноциклический или конденсированный полициклический, ароматический карбоцикл (кольцевую структуру, в которой все кольцевые атомы представляют собой углерод), предпочтительно содержащий от 5 до 12 атомов в кольце. Примеры арильных групп включают фенил, нафтил и подобные; (ii) необязательно замещенный частично насыщенный бициклический ароматический карбоциклический фрагмент, в котором фенильная и С5-7циклоалкильная или С5-7циклоалкенильная группа конденсированы с образованием циклической структуры, такой как тетрагидронафтил, инденил или инданил. Указанная группа может представлять собой концевую группу или мостиковую группу. Обычно арильная группа представляет собой С6-С18арильную группу.
Термин «арилалкенил» обозначает группу арил-алкенил-, в которой арил и алкенил представляют собой такие, как определено в настоящем изобретении. Примерные арилалкенильные группы включают фенилаллил. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через алкенильную группу.
Термин «арилалкил» обозначает группу арил-алкил-, в которой арильные и алкильные фрагменты представляют собой такие, как определено в настоящем изобретении. Предпочтительные арилалкильные группы содержат C1-5алкильный фрагмент. Типичные арилалкильные группы включают бензил, фенэтил, 1-нафталенметил и 2-нафталенметил. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через алкильную группу.
Термин «арилалкилокси» относится к группе арил-алкил-О-, в которой алкил и арил такие, как определено в настоящем изобретении. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через атом кислорода.
Термин «ариламино» включает как моно-ариламино, так и диариламино, если не указано иное. Термин «моноариламино» обозначает группу формулы арилNH-, в которой арил такой, как определено в настоящем изобретении. Термин «диариламино» обозначает группу (арил)2N-, в которой каждый арил может быть одинаковым или различным и каждый из них такой, как определено в настоящем изобретении для арила. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через атом азота.
Термин «арилгетероалкил» обозначает группу арил-гетероалкил-, в которой арильные и гетероалкильные фрагменты такие, как определено в настоящем изобретении. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через гетероалкильную группу.
Термин «арилокси» относится к группе алрил-О-, в которой арил такой, как определено в настоящем изобретении. Предпочтительно арилокси представляет собой С6-С18арилокси, более предпочтительно С6-С10арилокси. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через атом кислорода.
Термин «арилсульфонил» обозначает группу арил-S-(=O)2-, в которой арильная группа такая, как определено в настоящем изобретении. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через атом серы.
Термин «связь» представляет собой сцепление атомов в соединении или молекуле. Связь может представлять собой одинарную связь, двойную связь или тройную связь.
Термин «циклоалкенил» обозначает неароматическую моноциклическую или полициклическую кольцевую систему, содержащую по меньшей мере одну двойную углерод-углеродную связь и предпочтительно содержащую от 5-10 атомов углерода в кольце. Примерные моноциклические циклоалкенильные кольца включают циклопентенил, циклогексенил или циклогептенил. Циклоалкенильная группа может быть замещена одной или несколькими замещающими группами. Обычно циклоалкенильная группа представляет собой С3-С12алкенильную группу. Указанная группа может представлять собой концевую группу или мостиковую группу.
Термин «циклоалкил» относится к насыщенному моноциклическому или конденсированному или спиро-полициклическому карбоциклу, предпочтительно содержащему от 3 до 9 атомов углерода в кольце, такому как циклопропил, циклобутил, циклопентил, циклогексил и подобные, если не указано иное. Этот термин включает моноциклические системы, такие как циклопропил и циклогексил, бициклические системы, такие как декалин и полициклические системы, такие как адамантан. Обычно циклоалкильная группа представляет собой С3-С12алкильную группу. Указанная группа может представлять собой концевую группу или мостиковую группу.
Термин «циклоалкилалкил» обозначает группу циклоалкил-алкил-, в которой циклоалкильные и алкильные фрагменты такие, как определено в настоящем изобретении. Примерные моноциклоалкилалкильные группы включают циклопропилметил, циклопентилметил, циклогексилметил и циклогептилметил. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через алкильную группу.
Термин «циклоалкилалкенил» обозначает группу циклоалкил-алкенил-, в которой циклоалкильные и алкенильные фрагменты такие, как определено в настоящем изобретении. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через алкенильную группу.
Термин «циклоалкилгетероалкил» обозначает группу циклоалкил-гетероалкил-, в которой циклоалкильные и гетероалкильные фрагменты такие, как определено в настоящем изобретении. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через гетероалкильную группу.
Термин «циклоалкилокси» относится к группе циклоалкил-О-, в которой циклоалкил такой, как определено в настоящем изобретении. Предпочтительный циклоалкилокси представляет собой С1-С6циклоалкилокси. Примеры включают, но не ограничиваются ими, циклопропанокси и циклобутанокси. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через атом кислорода.
Термин «циклоалкенилокси» относится к группе циклоалкенил-О-, в которой циклоалкенил такой, как определено в настоящем изобретении. Предпочтительный циклоалкенилокси представляет собой С1-С6циклоалкенилокси. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через атом кислорода.
Термин «галогеналкил» относится к алкильной группе, определенной в настоящем изобретении, в которой один или несколько атомов водорода заменены на атом галогена, выбранный из группы, состоящей из фтора, хлора, брома и йода. Галогеналкильная группа обычно имеет формулу CnH(2n+1-m)Xm, где каждый X независимо выбран из группы, состоящей из F, Cl, Br и I. В группах этого типа n обычно имеет значение от 1 до 10, более предпочтительно от 1 до 6, наиболее предпочтительно от 1 до 3. m обычно имеет значение от 1 до 6, более предпочтительно от 1 до 3. Примеры галогеналкила включают фторметил, дифторметил и трифторметил.
Термин «галогеналкенил» относится к алкенильной группе, определенной в настоящем изобретении, в которой один или несколько атомов водорода заменены на атом галогена, независимо выбранный из группы, состоящей из F, Cl, Br и I.
Термин «галогеналкинил» относится к алкинильной группе, определенной в настоящем изобретении, в которой один или несколько атомов водорода заменены на атом галогена, независимо выбранный из группы, состоящей из F, Cl, Br и I.
Термин «галоген» обозначает хлор, фтор, бром или йод.
Термин «гетероалкил» относится к неразветвленной или разветвленной цепи алкильной группы, предпочтительно содержащей от 2 до 12 атомов углерода, более предпочтительно от 2 до 6 атомов углерода в цепи, в которой один или более атомов углерода (и любые связанные атомы водорода) каждый независимо друг от друга заменены на гетероатомную группу, выбранную из S, О, Р и NR', где R' выбран из группы, состоящей из Н, необязательно замещенного С1-С12алкила, необязательно замещенного С3-С12циклоалкила, необязательно замещенного С6-С18арила и необязательно замещенного С1-С18гетероарила. Примерные гетероалкилы включают алкильные эфиры, вторичные и третичные алкиламины, амиды, алкилсульфиды и подобные. Примеры гетероалкила также включают гидрокси С1-С6алкил, С1-С6алкилокси С1-С6алкил, аминоС1-С6алкил, С1-С6алкиламиноС1-С6алкил и ди(С1-С6алкил)аминоС1-С6алкил. Указанная группа может представлять собой концевую группу или мостиковую группу.
Термин «гетероалкилокси» относится к группе гетероалкил-О-, в которой гетероалкил такой, как определено в настоящем изобретении. Предпочтительный гетероалкилокси представляет собой С2-С6гетероалкилокси. Указанная группа может представлять собой концевую группу или мостиковую группу.
Термин «гетероарил» отдельно или как часть группы относится к группам, содержащим ароматическое кольцо (предпочтительно 5 или 6-членное ароматическое кольцо), содержащим один или более гетероатомов в качестве кольцевых атомов в ароматическом кольце, при этом остальные кольцевые атомы представляют собой атомы углерода. Подходящие гетероатомы включают азот, кислород и серу. Указанная группа может представлять собой моноциклическую или бициклическую гетероарильную группу. Примеры гетероарила включают тиофен, бензотиофен, бензофуран, бензимидазол, бензоксазол, бензотиазол, бензизотиазол, нафто[2,3-b]тиофен, фуран, изоиндолизин, ксантолен, феноксатин, пиррол, имидазол, пиразол, пиридин, пиразин, пиримидин, пиридазин, тетразол, индол, изоиндол, 1Н-индазол, пурин, хинолин, изохинолин, фталазин, нафтиридин, хиноксалин, циннолин, карбазол, фенантридин, акридин, феназин, тиазол, изотиазол, фенотиазин, оксазол, изооксазол, фуразан, феноксазин, 2-, 3- или 4-пиридил, 2-, 3-, 4-, 5- или 8-хинолил, 1-, 3-, 4-, 5-изохинолинил или 1-, 2- или 3-индолил и 2- или 3-тиенил. Гетероарильная группа обычно представляет собой С1-С18гетероарильную группу. Указанная группа может представлять собой концевую группу или мостиковую группу.
Термин «гетероарилалкил» обозначает группу гетероарил-алкил-, в которой гетероарильные и алкильные фрагменты представляют собой такие, как определено в настоящем изобретении. Предпочтительные гетероарилалкильные группы содержат низший алкильный фрагмент. Примерные гетероарилалкильные группы включают пиридилметил. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через алкильную группу.
Термин «гетероарилалкенил» обозначает группу гетероарил-алкенил-, в которой гетероарильные и алкенильные фрагменты такие, как определено в настоящем изобретении. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через алкенильную группу.
Термин «гетероарилгетероалкил» обозначает группу гетероарил-гетероалкил-, в которой гетероарильные и гетероалкильные фрагменты такие, как определено в настоящем изобретении. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через гетероалкильную группу.
Термин «гетероарилокси» относится к группе гетероарил-О-, в которой гетероарил такой, как определено в настоящем изобретении. Предпочтительный гетероарилокси представляет собой С1-С18гетероарилокси. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через атом кислорода.
Термин «гетероциклический» относится к насыщенной, частично ненасыщенной или полностью ненасыщенной моноциклической, бициклической или полициклической кольцевой системе, содержащей по меньшей мере один гетероатом, выбранный из группы, состоящей из азота, серы и кислорода в качестве кольцевого атома. Примеры гетероциклических групп включают гетероциклоалкил, гетероциклоалкенил и гетероарил.
Термин «гетероциклоалкенил» относится к гетероциклоалкильной группе, как определено в настоящем изобретении, но содержащей по меньшей мере одну двойную связь. Обычно гетероциклоалкенильная группа представляет собой С2-С12гетероциклоалкенильную группу. Указанная группа может представлять собой концевую группу или мостиковую группу.
Термин «гетероциклоалкил» относится к насыщенному моноциклическому, бициклическому или полициклическому кольцу, содержащему по меньшей мере один гетероатом, выбранный из азота, серы, кислорода, предпочтительно от 1 до 3 гетероатомов по крайней мере в одном кольце. Каждое кольцо является предпочтительно от 3 до 10-членным, более предпочтительно от 4 до 7-членным. Примеры подходящих гетероциклоалкильных заместителей включают пирролидил, тетрагидрофурил, тетрагидротиофуранил, пиперидил, пиперазил, тетрагидропиранил, морфилино, 1,3-диазапен, 1,4-диазапен, 1,4-оксазепан и 1,4-оксатиапан. Обычно гетероциклоалкильная группа представляет собой С2-С12гетероциклоалкильную группу. Указанная группа может представлять собой концевую группу или мостиковую группу.
Термин «гетероциклоалкилалкил» относится к группе гетероциклоалкил-алкил-, в которой гетероциклоалкильные и алкильные фрагменты такие, как определено в настоящем изобретении. Примерные гетероциклоалкилалкильные группы включают (2-тетрагидрофуранил)метил, (2-тетрагидротиофуранил)метил. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через алкильную группу.
Термин «гетероциклоалкилалкенил» относится к группе гетероциклоалкил-алкенил-, в которой гетероциклоалкильные и алкенильные фрагменты такие, как определено в настоящем изобретении. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через алкенильную группу.
Термин «гетероциклоалкилгетероалкил» обозначает группу гетероциклоалкил-гетероалкил-, в которой гетероциклоалкильные и гетероалкильные фрагменты такие, как определено в настоящем изобретении. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через гетероалкильную группу.
Термин «гетероциклоалкилокси» относится к группе гетероциклоалкил-О-, в которой гетероциклоалкил такой, как определено в настоящем изобретении. Предпочтительно, гетероциклоалкилокси представляет собой С1-С6гетероциклоалкилокси. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через атом кислорода.
Термин «гетероциклоалкенилокси» относится к группе гетероциклоалкенил-О-, в которой гетероциклоалкенил такой, как определено в настоящем изобретении. Предпочтительный гетероциклоалкенилокси представляет собой С1-С6гетероциклоалкенилокси. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через атом кислорода.
Термин «гидроксиалкил» относится к алкильной группе, как определено в настоящем изобретении, в которой один или несколько атомов водорода заменены ОН группой. Гидроксиалкильная группа обычно имеет формулу CnH(2n+1-x)(ОН)x. В группах этого типа n обычно имеет значение от 1 до 10, более предпочтительно от 1 до 6, наиболее предпочтительно от 1 до 3. x обычно имеет значение от 1 до 6, более предпочтительно от 1 до 3.
Термин «сульфинил» обозначает группу R-S(=O)-, в которой группа R может представлять собой ОН, алкил, циклоалкил, гетероциклоалкил; арильную или гетероарильную группу, как определено в настоящем изобретении. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через атом серы.
Термин «сульфиниламино» обозначает группу R-S(=O)-NH-, в которой группа R может представлять собой ОН, алкил, циклоалкил, гетероциклоалкил; арильную или гетероарильную группу, как определено в настоящем изобретении. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через атом азота.
Термин «сульфонил» обозначает группу R-S(=O)2-, в которой группа R может представлять собой ОН, алкил, циклоалкил, гетероциклоалкил; арильную или гетероарильную группу, как определено в настоящем изобретении. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через атом серы.
Термин «сульфониламино» обозначает группу RS(=O)2-NH-. Указанная группа может представлять собой концевую группу или мостиковую группу. Если указанная группа представляет собой концевую группу, она связана с остальной частью молекулы через атом азота.
Следует понимать, что в семейство соединения формулы (I) включены изомерные формы, включая диастереоизомеры, энантиомеры, таутомеры и геометрические изомеры в виде конфигурационных изомеров «Е» или «Z» или смесь изомеров Е и Z. Следует также понимать, что некоторые изомерные формы, такие как диастереомеры, энантиомеры и геометрические изомеры, могут быть разделены физическими и/или химическими методами и при помощи специалистов в данной области техники. Для тех соединений, где существует возможность геометрической изомерии, заявитель изображает тот изомер, которым, как полагают, представлено соединение, хотя следует понимать, что может присутствовать другой изомер.
Некоторые соединения, раскрытые в вариантах реализации настоящего изобретения, могут существовать в виде отдельных стереоизомеров, рацематов, и/или смесей энантиомеров и/или диастереомеров. Подразумевается, что все указанные отдельные стереоизомеры, рацематы и их смеси включены в рамки объекта изобретения, описанного и заявленного в настоящем изобретении.
Кроме того, формула (I), включает, где это применимо, сольватированные, а также несольватированные формы соединений. Таким образом, каждая формула включает соединения, имеющие указанную структуру, в том числе гидратированные и негидратированные формы.
Термин «фармацевтически приемлемые соли» относится к солям, которые сохраняют желательную биологическую активность идентифицированных выше соединений и включает фармацевтически приемлемые соли присоединения кислот и соли присоединения оснований. Подходящие фармацевтически приемлемые соли присоединения кислот соединений формулы (I) можно получить с помощью неорганической кислоты или органической кислоты. Примерами таких неорганических кислот являются соляная, серная и фосфорная кислота. Подходящие органические кислоты можно выбрать из алифатических, циклоалифатических, ароматических, гетероциклических карбоновых и сульфоновых органических кислот, примерами которых являются муравьиная, уксусная, пропионовая, янтарная, гликолевая, глюконовая, молочная, яблочная, винная, лимонная, фумаровая, малеиновая, алкилсульфоновая, арилсульфоновая. Аналогично, соль присоединения основания можно получить с помощью способов, хорошо известных в данной области техники, с применением органических или неорганических оснований. Пример подходящих органических оснований включает простые амины, такие как метиламин, этиламин, триэтиламин и подобные. Примеры подходящих неорганических оснований включают NaOH, КОН и подобные. Дополнительную информацию о фармацевтически приемлемых солях можно найти в Remington's Pharmaceutical Sciences Mack Publishing Co, Easton, PA 1995. В случае агентов, которые представляют собой твердые вещества, специалисты в данной области техники понимают, что соединения согласно настоящему изобретению, агенты и соли могут существовать в различных кристаллических или полиморфных формах, и все они включены в пределы объема настоящего изобретения и указанной формулы.
Термины «терапевтически эффективное количество» или «эффективное количество» означают количество, достаточное для получения полезных или желаемых клинических результатов. Эффективное количество можно вводить в один или несколько приемов. Эффективного количества обычно достаточно для временного облегчения, улучшения, стабилизирования, обращения вспять, замедления или задержания прогрессирования патологического состояния.
Как указано выше, соединения согласно настоящему изобретению имеют формулу (I):
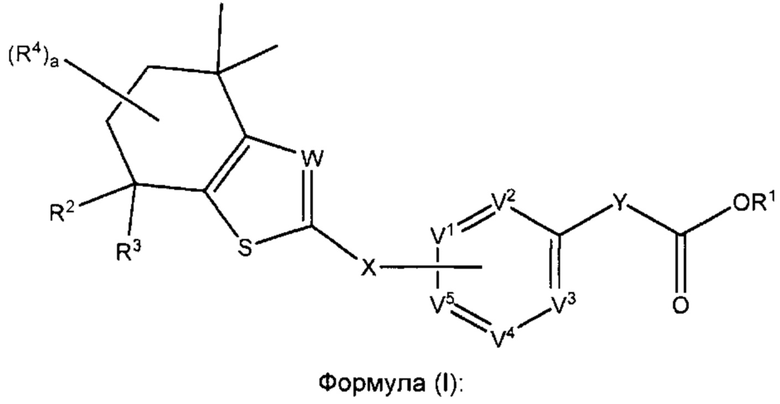
Как и с любой группой структурно родственных соединений, которые обладают особенной ценностью, некоторые варианты реализации переменных в соединении формулы (I) особенно полезны для конечного применения.
В некоторых вариантах реализации настоящего изобретения R1 представляет собой Н. В некоторых вариантах реализации настоящего изобретения R1 представляет собой С1-С6алкил. В некоторых вариантах реализации настоящего изобретения R1 представляет собой защитную группу карбоновой кислоты.
В некоторых вариантах реализации настоящего изобретения Y представляет собой связь. В некоторых вариантах реализации настоящего изобретения Y представляет собой связующий фрагмент. Связующий фрагмент может представлять собой любой подходящий фрагмент, который обеспечивает связь между ароматическим кольцом и карбоксильным фрагментом. Примеры подходящих связующих фрагментов включают необязательно замещенный С1-С12алкил, необязательно замещенный С2-С12алкенил, необязательно замещенный С2-С12алкинил, необязательно замещенный С1-С12галогеналкил, необязательно замещенный С2-С12гетероалкил, необязательно замещенный С3-С12циклоалкил, необязательно замещенный С3-С12циклоалкенил, необязательно замещенный С2-С12гетероциклоалкил и необязательно замещенный С2-С12гетероциклоалкенил. Конкретные примеры подходящих линкеров включают транс-этенил и циклопропиловую группу.
В соединениях согласно настоящему изобретению а представляет собой целое число, выбранное из группы, состоящей из 0, 1, 2, 3, и 4. В некоторых вариантах реализации настоящего изобретения а равно 0. В некоторых вариантах реализации настоящего изобретения а равно 1. В некоторых вариантах реализации настоящего изобретения а равно 2. В некоторых вариантах реализации настоящего изобретения а равно 3. В некоторых вариантах реализации настоящего изобретения а равно 4.
В некоторых вариантах реализации соединения согласно настоящему изобретению R1 представляет собой Н, Y представляет собой связь и а равно 0. Это позволяет получить соединение формулы (II):
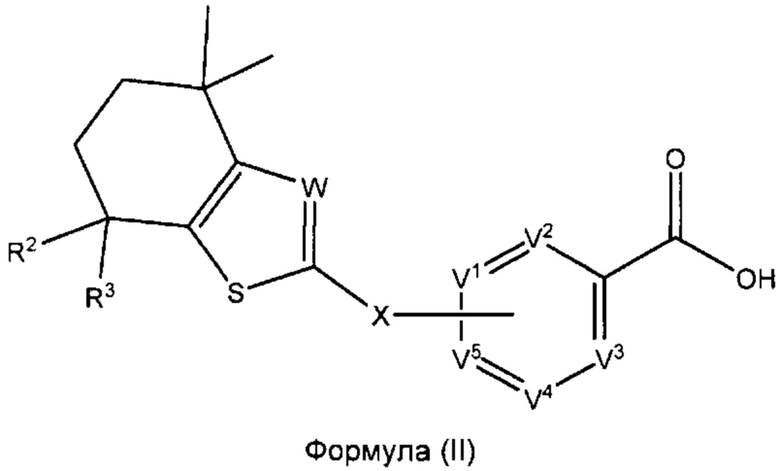
или его фармацевтически приемлемую соль:
где R2, R3, W, X, V1, V2, V3, V4 и V5 такие, как описано выше.
В соединениях согласно настоящему изобретению R2 и R3 выбраны из группы, состоящей из Н и С1-С6 алкила. Примеры подходящих значений для R2 и R3 представляют собой Н, метил, этил, изопропил, пропил, 2-этил-пропил, 3,3-диметил-пропил, бутил, изобутил, 3,3-диметил-бутил, 2-этил-бутил, пентил, 2-метил, пентил и гексил. В некоторых вариантах реализации настоящего изобретения R2 и R3 представляют собой Н. В некоторых вариантах реализации настоящего изобретения R2 и R3 представляют собой метил.
В соединениях согласно настоящему изобретению W выбран из N или CR9. В некоторых вариантах реализации настоящего изобретения W представляет собой N. В некоторых вариантах реализации настоящего изобретения W представляет собой CR9.
В некоторых вариантах реализации соединений согласно настоящему изобретению R1 представляет собой Н, Y представляет собой связь, а равно 0 и W представляет собой N. Это позволяет получить соединение формулы (IIа):
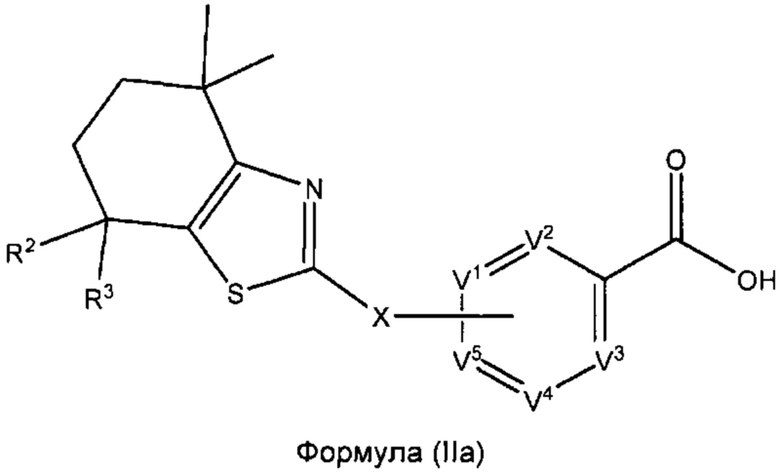
или его фармацевтически приемлемую соль:
где R2, R3, X, V1, V2, V3, V4 и V5такие, как описано выше.
В некоторых вариантах реализации соединений согласно настоящему изобретению R1 представляет собой Н, Y представляет собой связь, а равно 0 и W представляет собой CR9. Это позволяет получить соединение формулы (IIb):
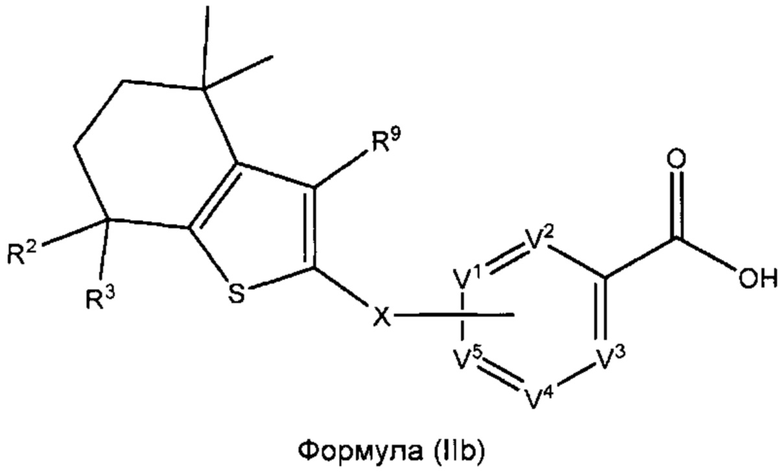
или его фармацевтически приемлемую соль:
где R2, R3, R9, X, V1, V2, V3, V4 и V5 такие, как описано выше.
В соединениях согласно настоящему изобретению X представляет собой связь или связующий фрагмент. В некоторых вариантах реализации настоящего изобретения X представляет собой связь. В некоторых вариантах реализации настоящего изобретения X представляет собой связующий фрагмент. В некоторых вариантах реализации соединений согласно настоящему изобретению X представляет собой связующую группу, выбранную из группы, состоящей из связи, -О-, -S-, -NR10-, NR10C(=O)-, -С(=R11)- и -(CR12R13)b-;
где R10 выбран из группы, состоящей из Н, необязательно замещенного С1-С12алкила, необязательно замещенного С2-С12алкенила, необязательно замещенного С2-С12алкинила, необязательно замещенного С1-С12галогеналкила, необязательно замещенного С2-С12гетероалкила, необязательно замещенного С3-С12циклоалкила, необязательно замещенного С3-С12циклоалкенила, необязательно замещенного С2-С12гетероциклоалкила, необязательно замещенного С2-С12гетероциклоалкенила, необязательно замещенного С6-С18арила, необязательно замещенного С1-С18гетероарила, необязательно замещенного С3-С12циклоалкилС1-С12алкила, необязательно замещенного С2-С12гетероциклоалкилС1-С12алкила, необязательно замещенного С6-С18арилС1-С12 алкила, необязательно замещенного С1-С18гетероарилС1-С12алкила, SO3H, SO2NR5R5, SO2R5, SONR5R5, SOR5, COR5, COOH, COOR5, CONR5R5 и ацила;
где R11 выбран из группы, состоящей из О, NR14, NOR14 и CR14R15, где каждый из R14 и R15 независимо выбран из группы, состоящей из Н, необязательно замещенного С1-С12алкила, необязательно замещенного С2-С12алкенила, необязательно замещенного С2-С12алкинила, необязательно замещенного С1-С12галогеналкила, необязательно замещенного С2-С12гетероалкила, необязательно замещенного С3-С12циклоалкила, необязательно замещенного С3-С12циклоалкенила, необязательно замещенного С2-С12гетероциклоалкила, необязательно замещенного С2-С12гетероциклоалкенила, необязательно замещенного С6-С18арила, необязательно замещенного С1-С18гетероарила, необязательно замещенного С3-С12циклоалкилС1-С12алкила, необязательно замещенного С2-С12гетероциклоалкилС1-С12алкила, необязательно замещенного С6-С18арилС1-С12алкила, необязательно замещенного С1-С18гетероарилС1-С12алкила и ацила;
каждый R12 и R13 независимо выбран из группы, состоящей из Н, необязательно замещенного С1-С12алкила, необязательно замещенного С2-С12алкенила, необязательно замещенного С2-С12алкинила, необязательно замещенного С1-С12галогеналкила, необязательно замещенного С2-С12гетероалкила, необязательно замещенного С3-С12циклоалкила, необязательно замещенного С3-С12циклоалкенила, необязательно замещенного С2-С12гетероциклоалкила, необязательно замещенного С2-С12гетероциклоалкенила, необязательно замещенного С6-С18арила, необязательно замещенного С1-С18гетероарила, необязательно замещенного С3-С12циклоалкилС1-С12алкила, необязательно замещенного С2-С12гетероциклоалкилС1-С12алкила, необязательно замещенного С6-С18арилС1-С12алкила, необязательно замещенного С1-С18гетероарилС1-С12алкила и ацила или R12 и R13 на одном атоме углерода совместно образуют циклическую группу или R12 и R13 на соседних атомах углерода совместно образуют двойную связь;
b представляет собой целое число, выбранное из группы, состоящей из 0, 1, 2, 3 и 4;
В некоторых вариантах реализации настоящего изобретения X представляет собой связь. В некоторых вариантах реализации настоящего изобретения X представляет собой -О-, в некоторых вариантах реализации настоящего изобретения X представляет собой -S-. В некоторых вариантах реализации настоящего изобретения X представляет собой -NR10-. В некоторых вариантах реализации настоящего изобретения X представляет собой NR10C(=O)-. В некоторых вариантах реализации настоящего изобретения X представляет собой С(=R11). В некоторых вариантах реализации настоящего изобретения X представляет собой -(CR12R13)b-.
Соединения формулы (I) содержат фрагмент формулы (В).
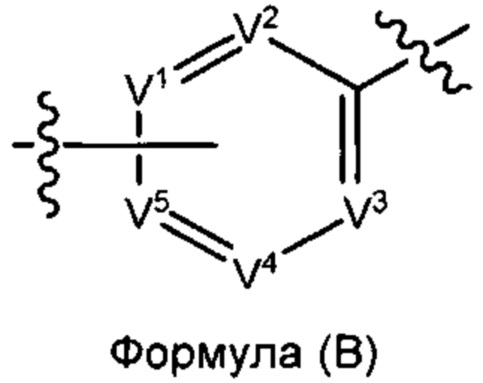
В некоторых вариантах реализации настоящего изобретения V2 представляет собой CR6. В некоторых вариантах реализации настоящего изобретения V2 представляет собой N. В некоторых вариантах реализации настоящего изобретения V3 представляет собой CR6. В некоторых вариантах реализации настоящего изобретения V3 представляет собой N. В некоторых вариантах реализации настоящего изобретения V4 представляет собой CR6. В некоторых вариантах реализации настоящего изобретения V4 представляет собой N.
V1 и V5 выбраны из группы, состоящей из N, CR7 и CR8 таким образом, что один из V1 и V5 представляет собой CR8, а другой представляет собой Н или CR7. То есть точка присоединения группы X к ароматическому кольцу имеет место либо при V1, либо при V5. В некоторых вариантах реализации настоящего изобретения V1 представляет собой CR7. В некоторых вариантах реализации настоящего изобретения V1 представляет собой N. В некоторых вариантах реализации настоящего изобретения V1 представляет собой CR8. В некоторых вариантах реализации настоящего изобретения V5 представляет собой CR7. В некоторых вариантах реализации настоящего изобретения V5 представляет собой CR8. В некоторых вариантах реализации настоящего изобретения V5 представляет собой N.
В некоторых вариантах реализации соединений согласно настоящему изобретению R1 представляет собой Н, Y представляет собой связь, а равно 0, W представляет собой N и V1 представляет собой CR8. Это позволяет получить соединение формулы (IIIа):
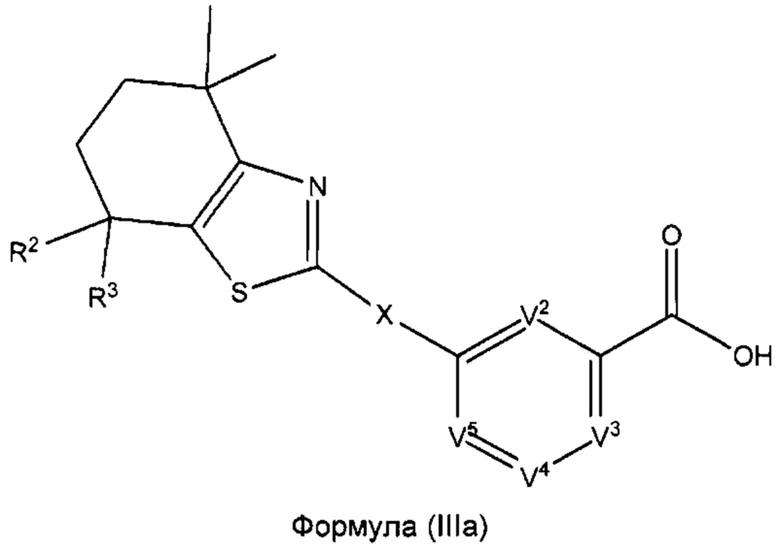
или его фармацевтически приемлемую соль:
где R2, R3, X, V2, V3, V4 и V5 такие, как описано выше.
В некоторых вариантах реализации соединений согласно настоящему изобретению R1 представляет собой Н, Y представляет собой связь, а равно 0, W представляет собой N и V5 представляет собой CR8. Это позволяет получить соединение формулы (IIIb):
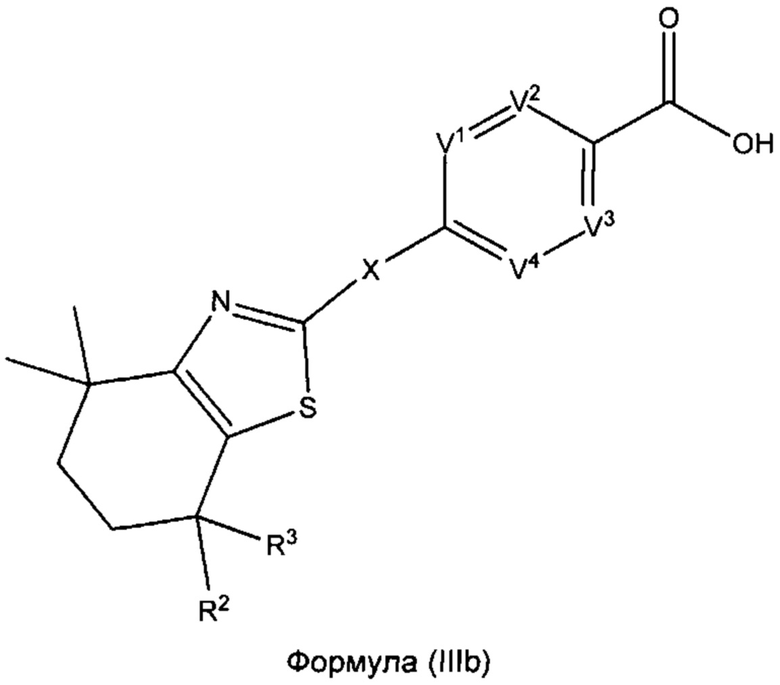
или его фармацевтически приемлемую соль:
где R2, R3, X, V1, V2, V3, V4 и V5 такие, как описано выше.
В некоторых вариантах реализации соединений согласно настоящему изобретению R1 представляет собой Н, Y представляет собой связь, а равно 0, W представляет собой CR9 и V1 представляет собой CR8. Это позволяет получить соединение формулы (IIIс):
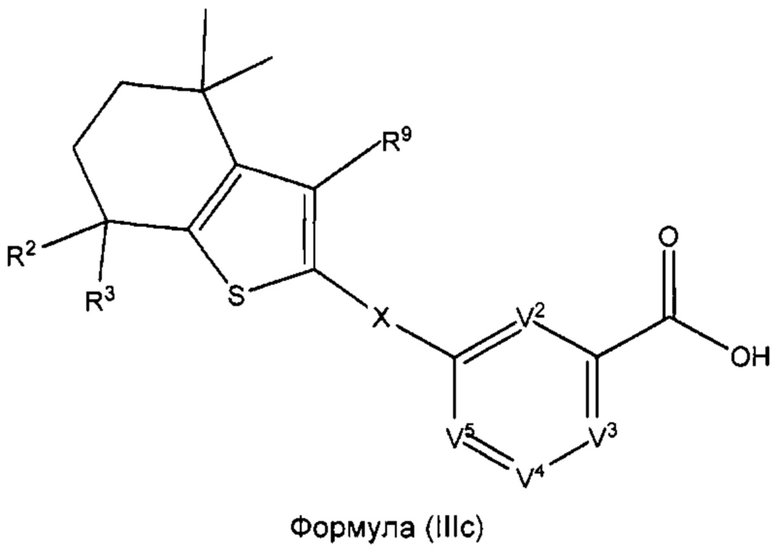
или его фармацевтически приемлемую соль:
где R2, R3, R9, X, V2, V3, V4 и V5 такие, как описано выше.
В некоторых вариантах реализации соединений согласно настоящему изобретению R1 представляет собой Н, Y представляет собой связь, а равно 0, W представляет собой CR9 и V5 представляет собой CR8. Это позволяет получить соединение формулы (IIId):
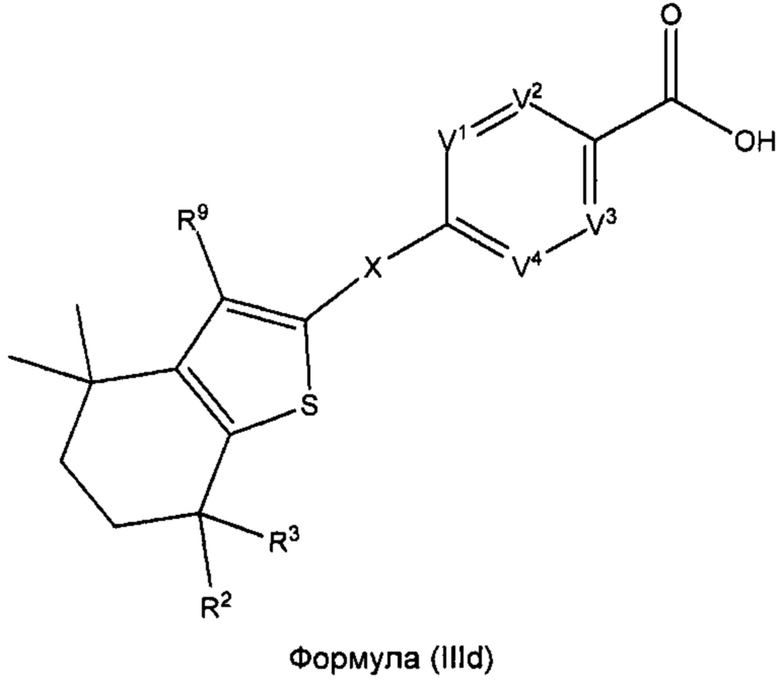
или его фармацевтически приемлемую соль:
где R2, R3, R9, X, V1, V2, V3 и V4 такие, как описано выше.
В результате комбинации V1, V2, V3, V4 и V5 в некоторых вариантах реализации настоящего изобретения фрагмент формулы (В) выбран из группы, состоящей из:
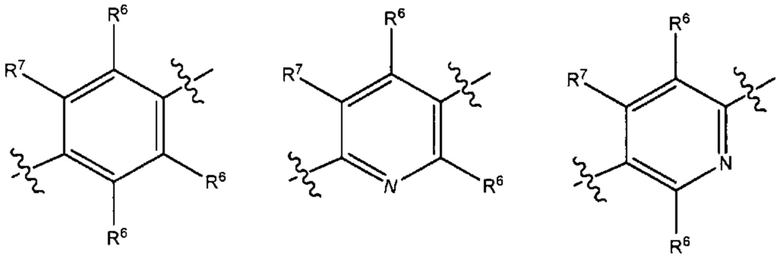
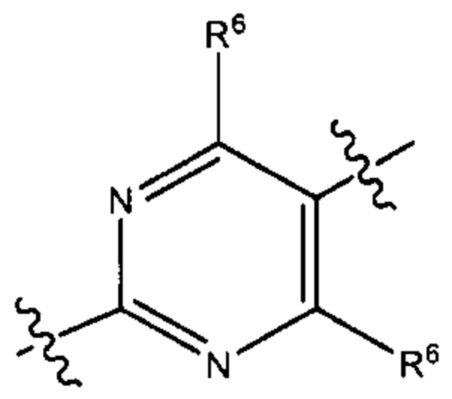 и
и 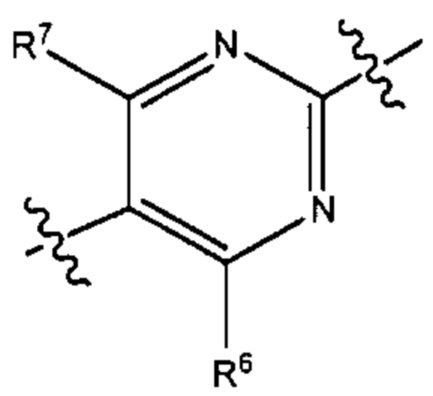
где R6 и R7 такие, как описано выше.
В некоторых вариантах реализации настоящего изобретения фрагмент формулы (В) представляет собой группу формулы
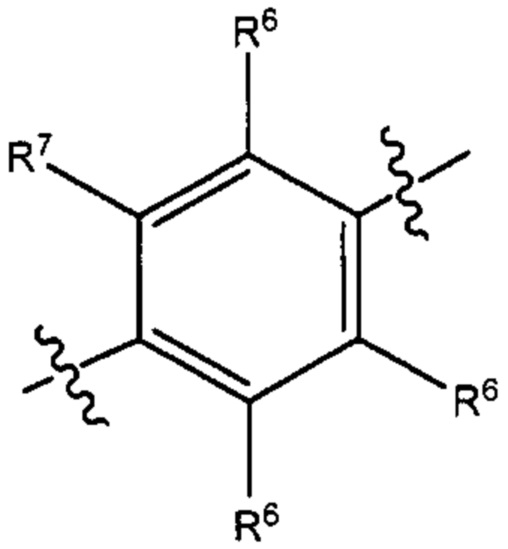
где R6 и R7 такие, как описано выше.
В некоторых вариантах реализации настоящего изобретения фрагмент формулы (В) представляет собой группу формулы
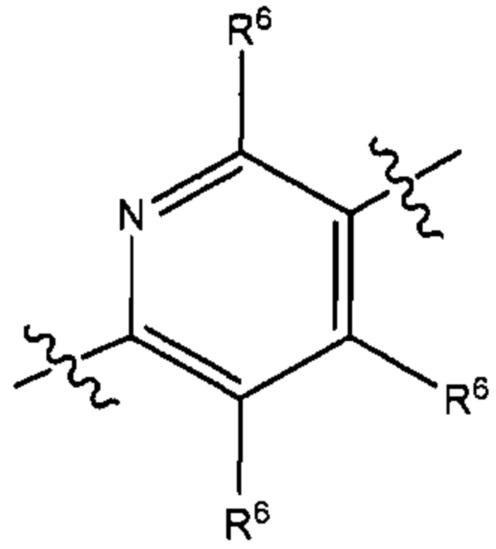
где R6 такой, как описано выше.
В некоторых вариантах реализации настоящего изобретения фрагмент формулы (В) представляет собой группу формулы
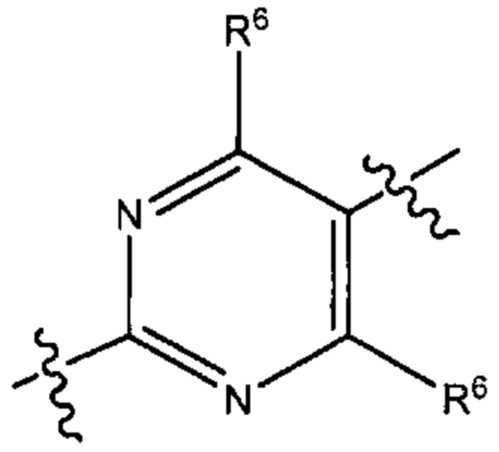
где R6 такой, как описано выше.
В некоторых вариантах реализации настоящего изобретения, содержащих фрагмент R4, каждый из R4 независимо выбран из группы, состоящей из Н, галогена, ОН, NO2, CN, SH, NH2, CF3, OCF3, С1-С12алкила и С1-С12алкокси, или два R4 на одном атоме углерода совместно образуют группу =O. В некоторых вариантах реализации настоящего изобретения каждый R4 независимо выбран из группы, состоящей из Н, ОН, ОСН3, ОСН2 СН3 и NHBn. В особенно предпочтительном варианте реализации настоящего изобретения каждый R4 представляет собой Н.
В некоторых вариантах реализации настоящего изобретения, содержащих фрагмент R5, каждый R5 независимо выбран из группы, состоящей из Н и С1-С12алкила.
В некоторых вариантах реализации настоящего изобретения каждый R6 независимо выбран из группы, состоящей из Н, галогена, ОН, NO2, CN, SH, NH2, CF3, OCF3, С1-С12алкила и С1-С12алкилокси.
В некоторых вариантах реализации настоящего изобретения каждый R6 независимо выбран из группы, состоящей из Н, СН3, СН2СН3, СН2СН2СН3, СН(СН3)2, (СН2)3СН3, Cl, Br, F, I, ОН, NO2, NH2, NHSO2CH2CH2CH3, CN, OCH3, OCH2CH2CH3, ОС6Н5, ОСН2ССН, ОСН2циклопропила, CF3 и OCF3.
В некоторых вариантах реализации настоящего изобретения каждый R7 независимо выбран из группы, состоящей из Н, СН3, СН2СН3, СН2СН2СН3, СН(СН3)2, (СН2)3СН3, Cl, Br, F, I, ОН, NO2, NH2, CN, ОСН3, ОСН2СН2СН3, CF3 и OCF3.
В некоторых вариантах реализации соединений согласно настоящему изобретению, содержащих группу R9, группа R9 выбрана из группы, состоящей из Н, метила, этила, изопропила, пропила, 3,3-диметил-пропила, бутила, изобутила, 3,3-диметил-бутила, 2-этил-бутила, пентила и гексила. В некоторых вариантах реализации настоящего изобретения R9 выбран из группы, состоящей из Н, метила и этила. В некоторых вариантах реализации настоящего изобретения R9 представляет собой Н. В некоторых вариантах реализации настоящего изобретения R9 представляет собой метил. В некоторых вариантах реализации настоящего изобретения R9 представляет собой этил.
В некоторых вариантах реализации соединений согласно настоящему изобретению, содержащих группу R10, группа R10 выбрана из группы, состоящей из Н, метила, циклопропилметила, этила, изопропила, пропила, циклопропила, 3,3-диметил-пропила, бутила, изобутила, циклобутила, 3,3-диметил-бутила, 2-этил-бутила, пентила гексила, фенила и пиридин-2-ила. В некоторых вариантах реализации настоящего изобретения R10 выбран из группы, состоящей из Н, метила и этила. В некоторых вариантах осуществления R10 представляет собой Н. В некоторых вариантах реализации настоящего изобретения R10 представляет собой метил. В некоторых вариантах реализации настоящего изобретения R10 представляет собой этил.
В некоторых вариантах осуществления R11 представляет собой О. В некоторых вариантах реализации настоящего изобретения R11 представляет собой NR14. В некоторых вариантах реализации настоящего изобретения R11 представляет собой NOR14. В некоторых вариантах реализации настоящего изобретения R11 представляет собой CR14R15.
В некоторых вариантах реализации соединений согласно настоящему изобретению, содержащих группу R12 или группу R13, группы R12 или R13 независимо выбраны из группы, состоящей из Н, метила, циклопропилметила, этила, изопропила, пропила, циклопропила, 3,3-диметил-пропила, бутила, изобутила, 3,3-диметил-бутила, 2-этил-бутила, пентила и гексила.
В некоторых вариантах реализации соединений согласно настоящему изобретению, содержащих группу R14 или группу R15, группы R14 или R15 независимо выбраны из группы, состоящей из Н, метила, циклопропилметила, этила, изопропила, пропила, циклопропила, 3,3-диметил-пропила, бутила, изобутила, 3,3-диметил-бутила, 2-этил-бутила, пентила и гексила.
Многие, если не все переменные, обсужденные выше, могут быть необязательно замещенными. Если переменная представляет собой необязательно замещенную, тогда в некоторых вариантах реализации настоящего изобретения каждый необязательный заместитель независимо выбран из группы, состоящей из галогена, =O, =S, -CN, -NO2, -CF3, -OCF3, алкила, алкенила, алкинила, галогеналкила, галогеналкенила, галогеналкинила, гетероалкила, циклоалкила, циклоалкенила, гетероциклоалкила, гетероциклоалкенила, арила, гетероарила, циклоалкилалкила, гетероциклоалкилалкила, гетероарилалкила, арилалкила, циклоалкилалкенила, гетероциклоалкилалкенила, арилалкенила, гетероарилалкенила, циклоалкилгетероалкила, гетероциклоалкилгетероалкила, арилгетероалкила, гетероарилгетероалкила, гидрокси, гидроксиалкила, алкилокси, алкилоксиалкила, алкилоксициклоалкила, алкилоксигетероциклоалкила, алкилоксиарила, алкилоксигетероарила, алкилоксикарбонила, алкиламинокарбонила, алкенилокси, алкинилокси, циклоалкилокси, циклоалкенилокси, гетероциклоалкилокси, гетероциклоалкенилокси, арилокси, фенокси, бензилокси, гетероарилокси, арилалкилокси, амино, алкиламино, ациламино, аминоалкила, ариламино, сульфониламино, сульфиниламино, сульфонила, алкилсульфонила, арилсульфонила, аминосульфонила, сульфинила, алкилсульфинила, арилсульфинила, аминосульфиниламиноалкила, С(=O)ОН, -C(=O)Re, C(=O)ORe, C(=O)NReRf, C(=NOH)Re, C(=NRe)NRfRg, NReRf, NReC(=O)Rf, NReC(=O)ORf, NReC(=O)NRfRg, NReC(=NRf)NRgRh, NReSO2Rf, -SRe, SO2NReRf, -ORe, OC(=O)NReRf, OC(=O)Re и ацила,
где Re, Rf, Rg и Rh, каждый независимо выбран из группы, состоящей из Н, С1-С12алкила, С1-С12 галогеналкила, С2-С12алкенила, С2-С12алкинила, С1-С10гетероалкила, С3-С12циклоалкила, С3-С12циклоалкенила, С1-С12гетероциклоалкила, С1-С12гетероциклоалкенила, С6-С18арила, С1-С18гетероарила и ацила, или любые два или более из Ra, Rb, Rc и Rd совместно с атомами, к которым они присоединены, образуют гетероциклическую кольцевую систему, содержащую от 3 до 12 атомов в кольце.
В некоторых вариантах реализации настоящего изобретения каждый необязательный заместитель независимо выбран из группы, состоящей из: F, Cl, Br, =O, =S, -CN, -NO2, алкила, алкенила, гетероалкила, галогеналкила, алкинила, арила, циклоалкила, гетероциклоалкила, гетероарила, гидрокси, гидроксиалкила, алкокси, алкиламино, аминоалкила, ациламино, фенокси, алкоксиалкила, бензилокси, алкилсульфонила, арилсульфонила, аминосульфонила, C(O)ORa или СООН, SH и ацила.
В некоторых вариантах реализации настоящего изобретения каждый необязательный заместитель независимо выбран из группы, состоящей из: F, Br, Cl, =O, =S, -CN, метила, трифторметила, этила, 2,2,2-трифторэтила, изопропила, пропила, 2-этил-пропила, 3,3-диметил-пропила, бутила, изобутила, 3,3-диметил-бутила, 2-этил-бутила, пентила, 2-метил-пентила, пент-4-енила, гексила, гептила, октила, фенила, NH2, -NO2, фенокси, гидрокси, метокси, трифтор-метокси, этокси и метилендиокси.
В некоторых вариантах реализации настоящего изобретения каждый необязательный заместитель независимо выбран из группы, состоящей из Н, СН3, СН2СН3, СН2СН2СН3, СН(СН3)2, (СН2)3СН3, Cl, Br, F, I, ОН, NO2, NH2, CN, ОСН3, ОСН2СН2СН3, CF3 и OCF3.
В качестве альтернативы, два необязательных заместителя на одном фрагменте совместно можно образовать конденсированный циклический заместитель, присоединенный к фрагменту, который является необязательно замещенным. Соответственно термин «необязательно замещенный» включает конденсированное кольцо, такое как циклоалкильное кольцо, гетероциклоалкильное кольцо, арильное кольцо или гетероарильное кольцо.
В дополнение к соединениям формулы I, раскрытые варианты реализации настоящего изобретения также направлены на фармацевтически приемлемые соли, фармацевтически приемлемые N-оксиды, фармацевтически приемлемые пролекарства и фармацевтически активные метаболиты таких соединений и фармацевтически приемлемые соли таких метаболитов.
Настоящее изобретение также относится к фармацевтическим композициям, содержащим соединение согласно настоящему изобретению и фармацевтически приемлемый носитель, разбавитель или наполнитель.
Конкретные соединения согласно настоящему изобретению включают следующее:
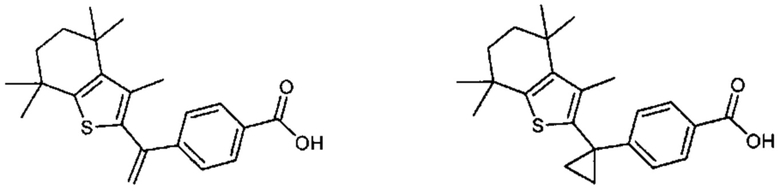
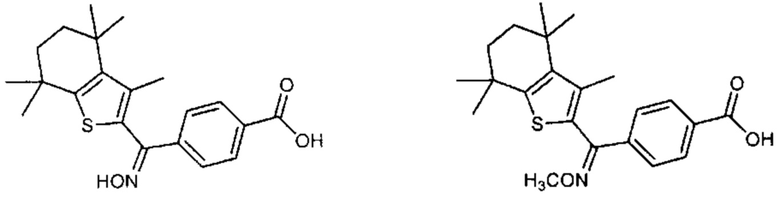
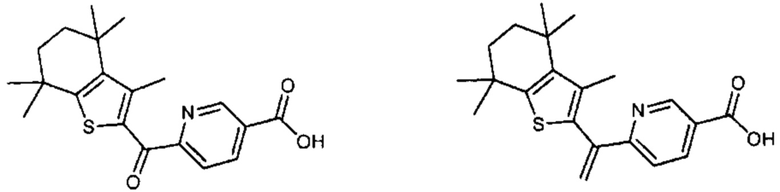
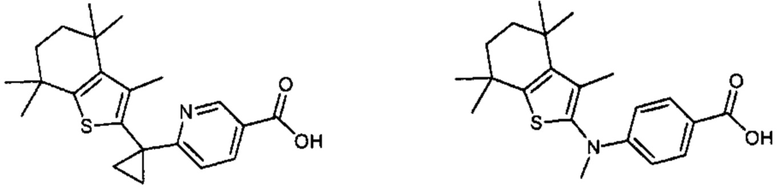
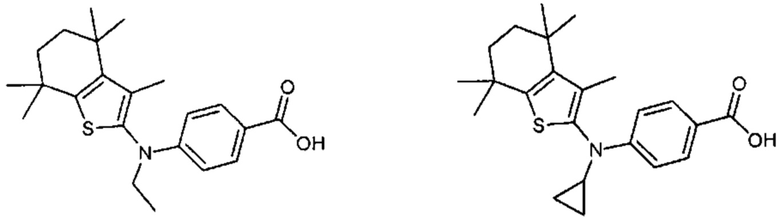
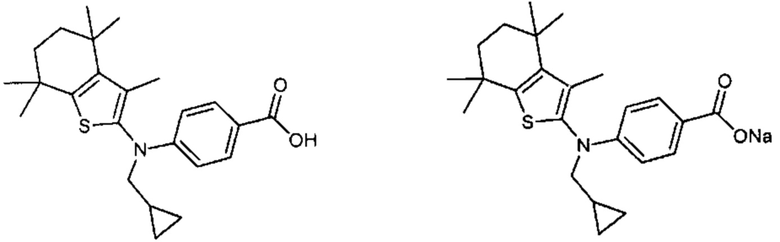
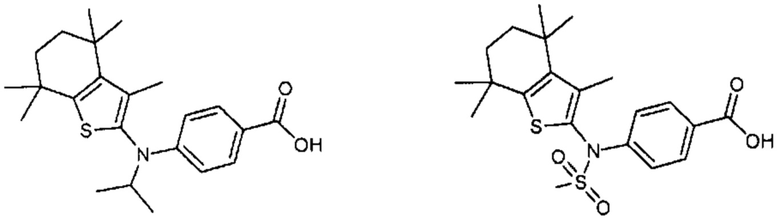
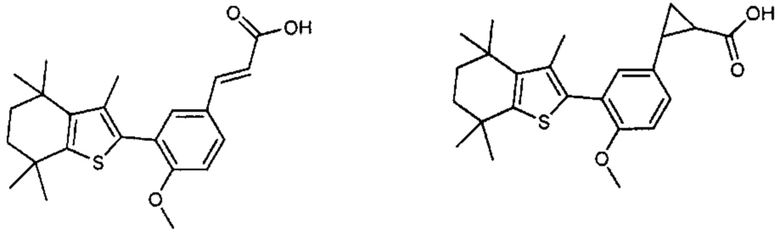
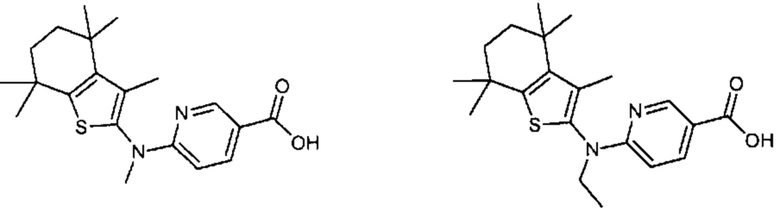
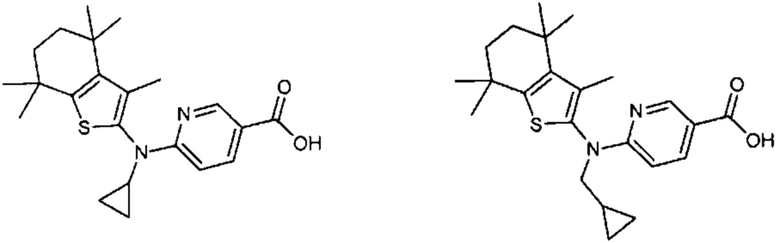
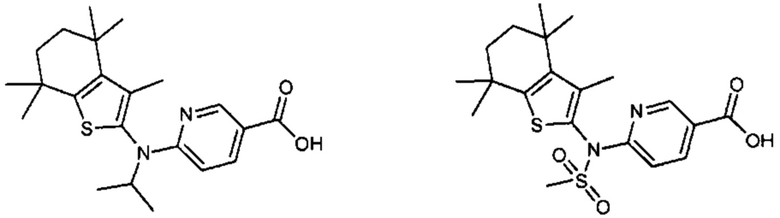
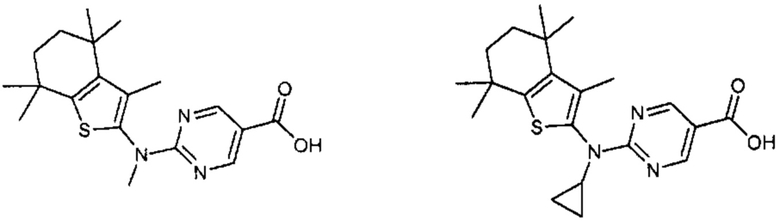
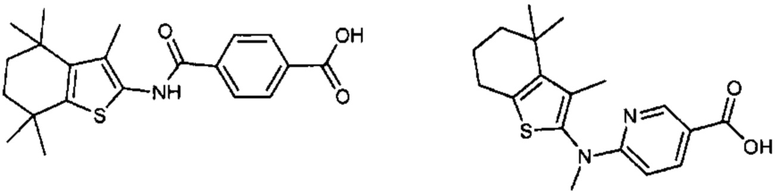
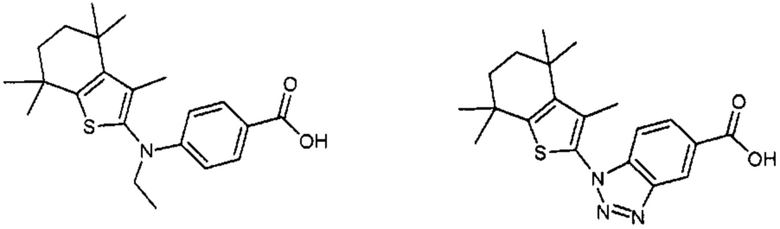
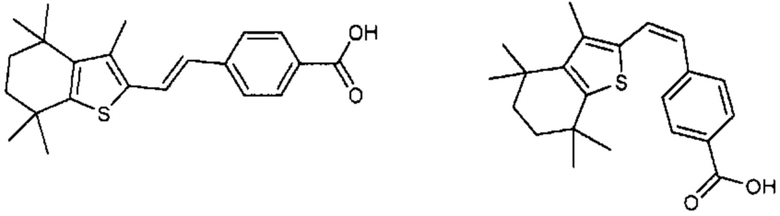
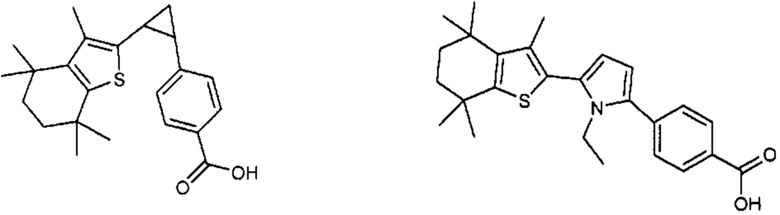
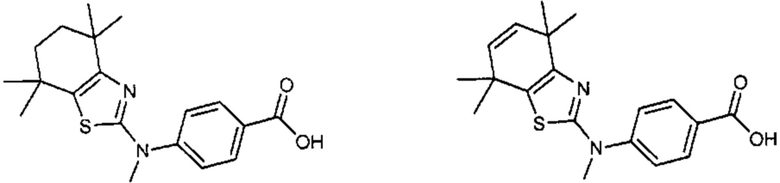
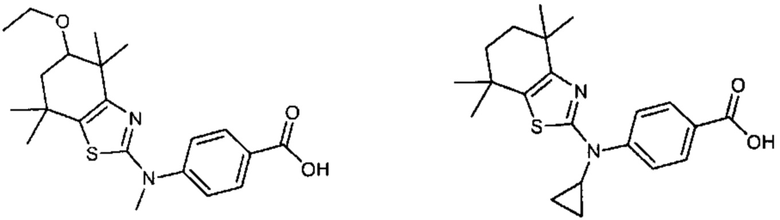
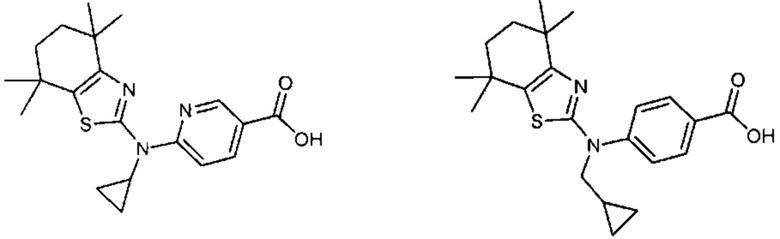
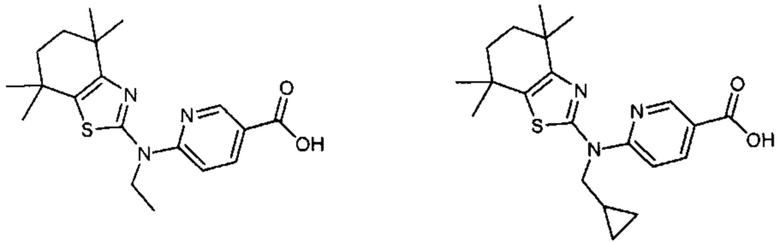
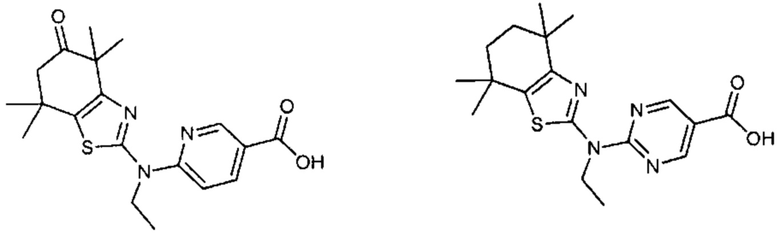
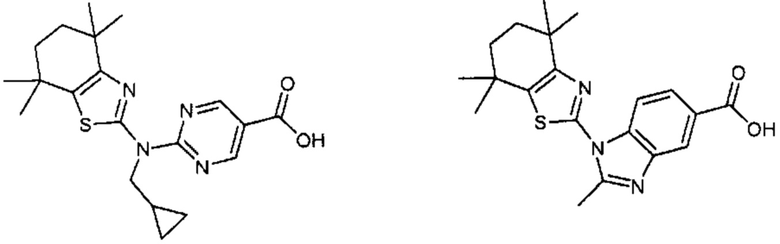
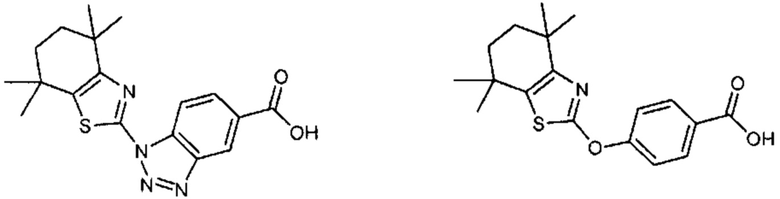
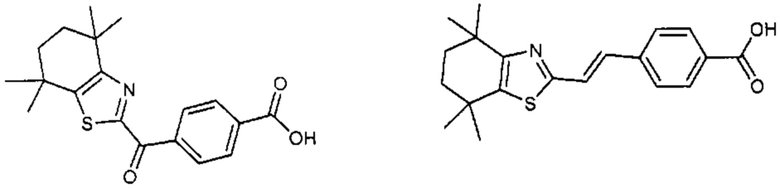
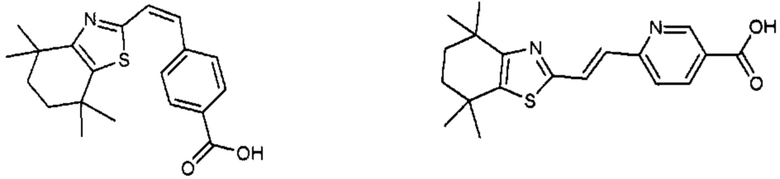
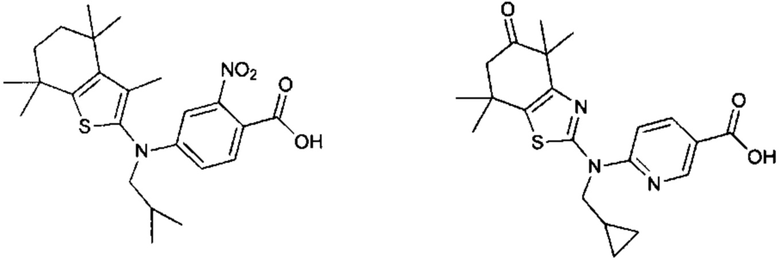
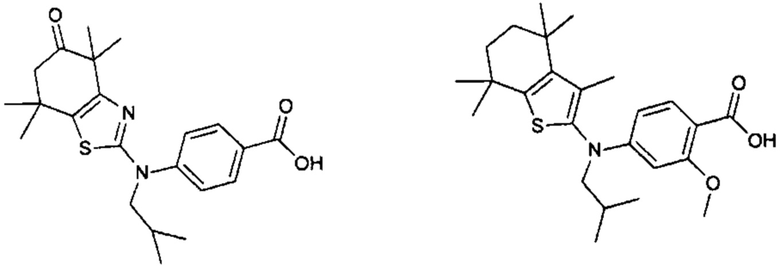
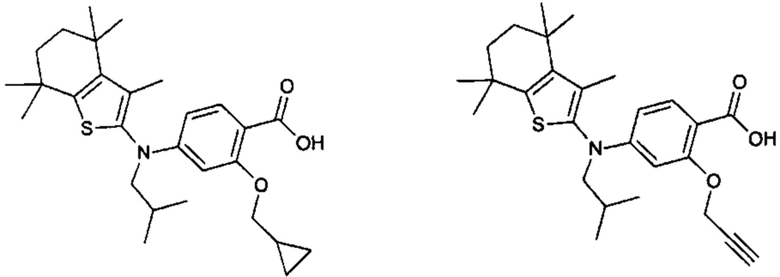
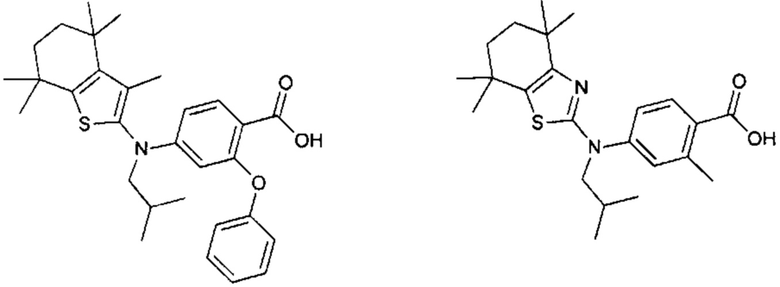
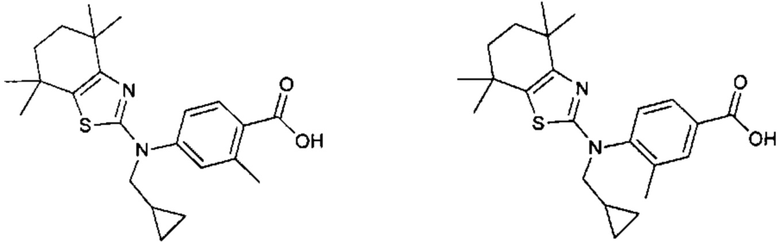
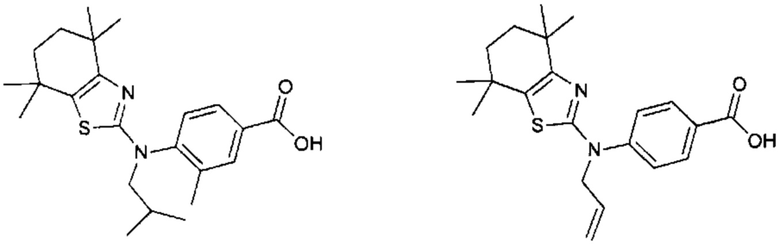
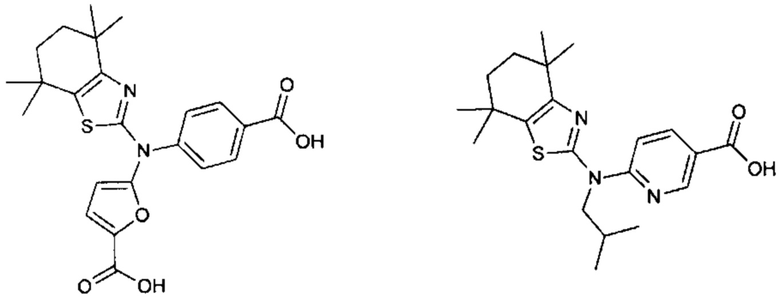
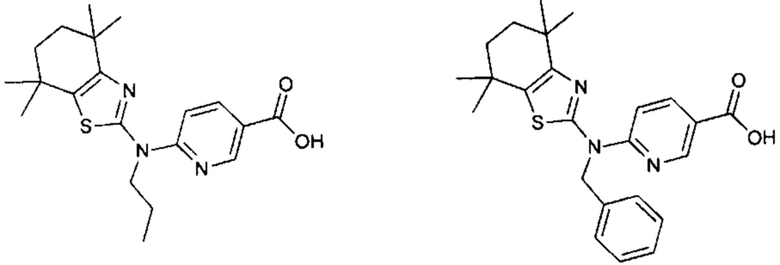
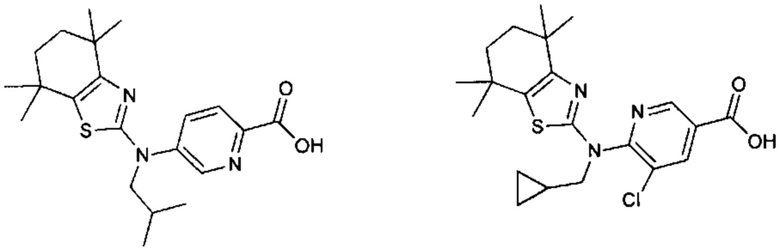
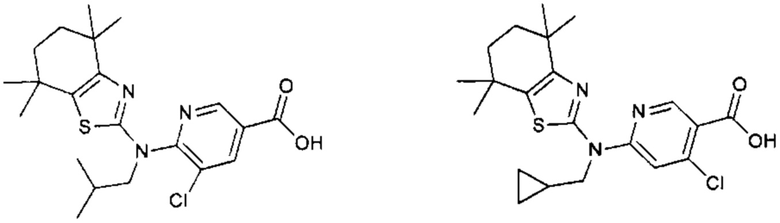
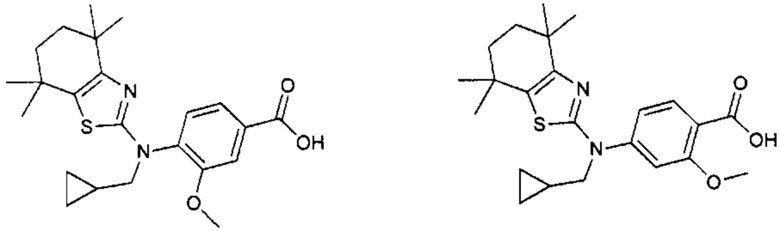
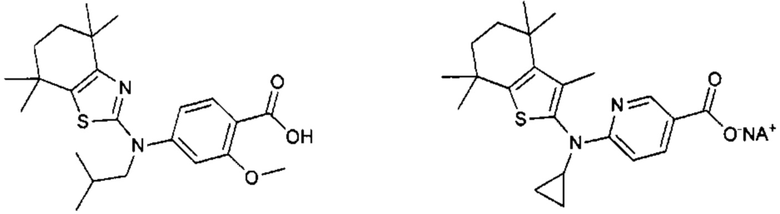
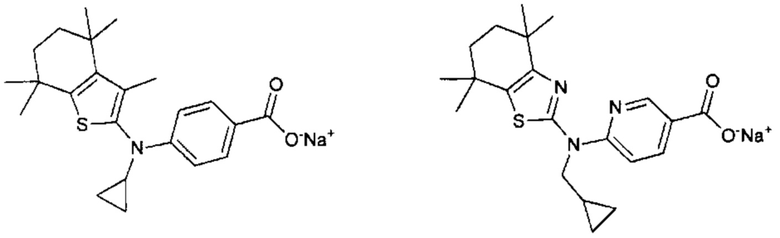
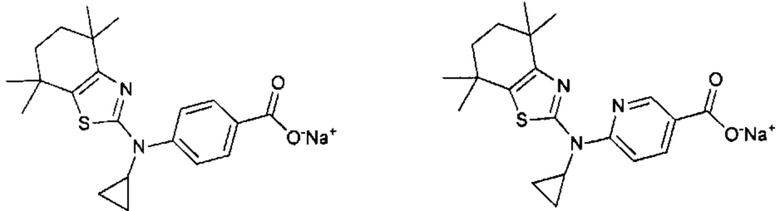
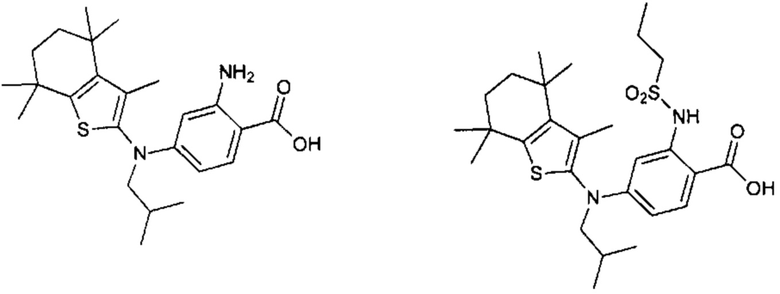
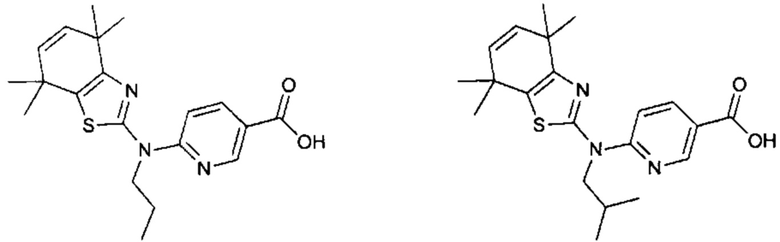
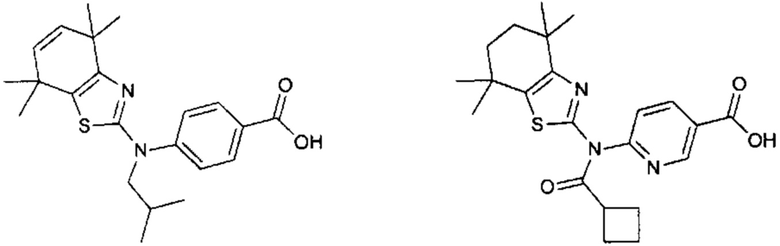
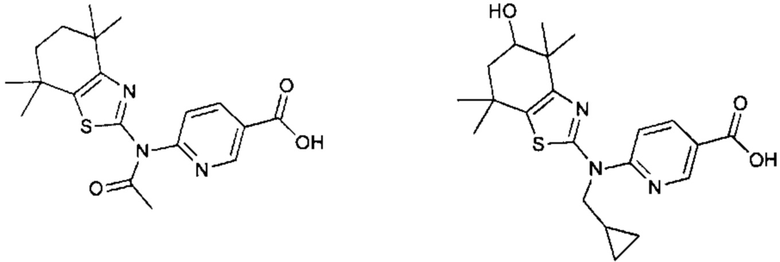
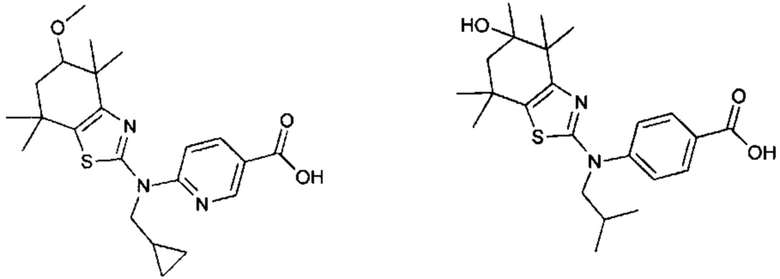
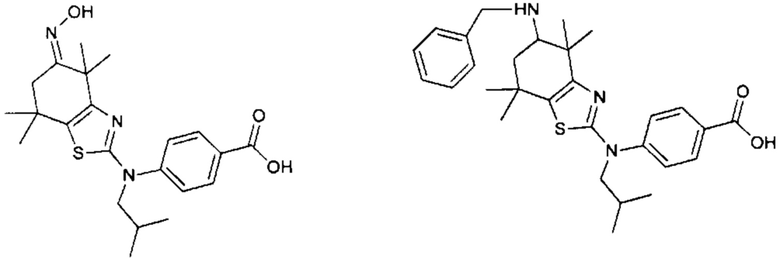
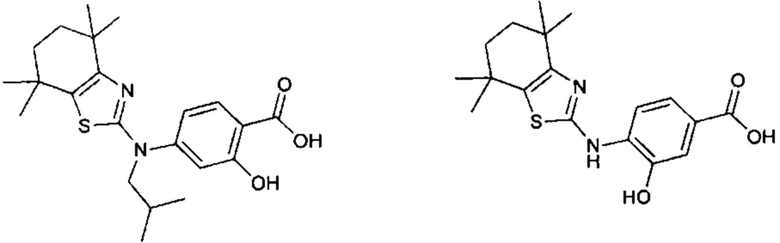
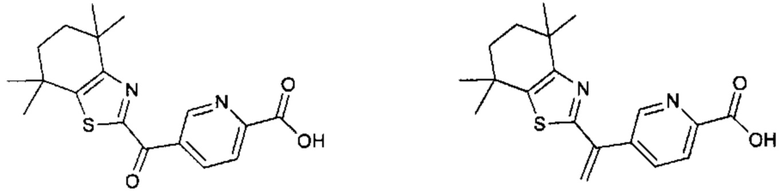
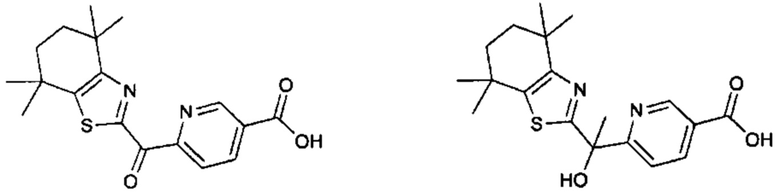
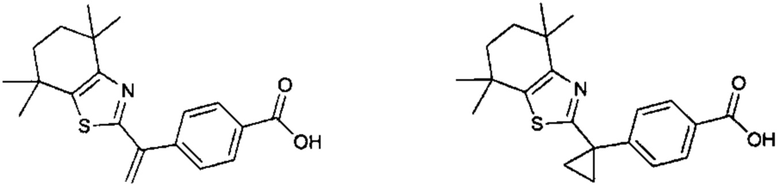
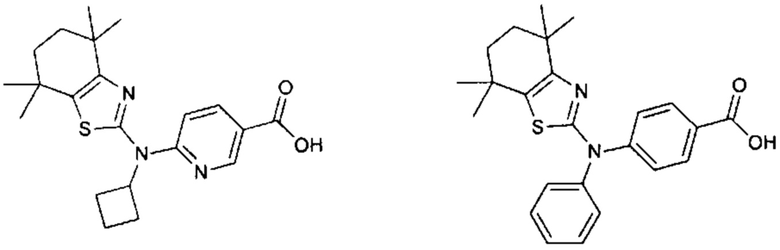
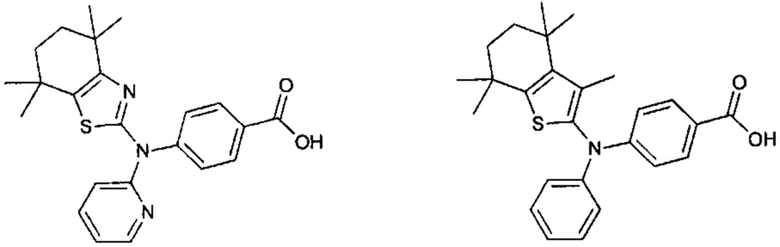
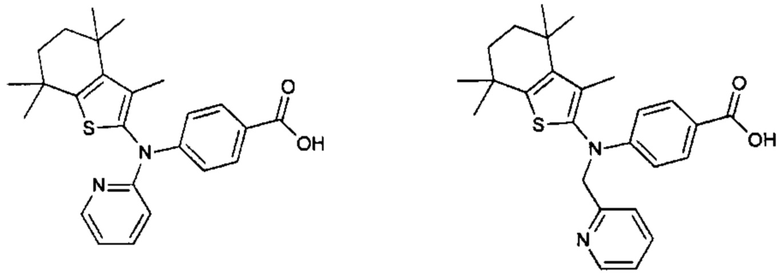
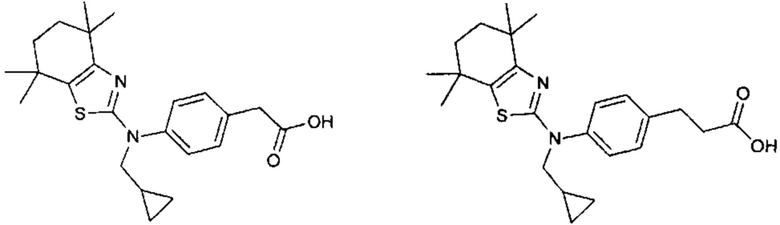
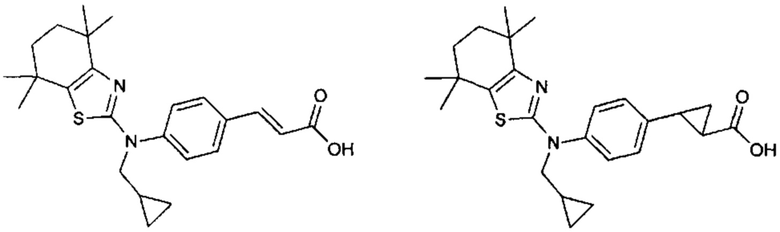
или фармацевтически приемлемые соли или пролекарства указанных соединений.
Соединения согласно настоящему изобретению представляют собой агонисты RXR и, следовательно, обладают способностью активировать эти рецепторы. Способность активировать рецепторы может являться результатом активности соединений, действующих непосредственно и исключительно на рецептор с модулированием/стимуляцией биологической активности. Тем не менее, следует понимать, что соединения могут также действовать, по меньшей мере частично, на другие факторы, связанные с активностью рецептора.
Активацию RXR можно осуществлять любым из ряда способов, известных в данной области техники. Например, если требуется активация in vitro, соответствующее количество соединения можно добавлять к раствору, содержащему RXR. В случаях, когда необходимо активировать RXR у млекопитающего, активация RXR обычно включает в себя введение млекопитающему соединения, содержащего RXR.
В другом аспекте настоящее изобретение обеспечивает способ профилактики или лечения состояния у млекопитающего, причем указанный способ включает введение эффективного количества соединения согласно настоящему изобретению. В одном варианте реализации настоящего изобретения состояние представляет собой состояние, которое можно лечить с помощью активации RXR.
В другом аспекте настоящего изобретения предложено применение соединения согласно настоящему изобретению для приготовления лекарственного средства для лечения состояния у млекопитающего. В одном варианте реализации настоящего описание состояние представляет собой состояние, которое можно лечить с помощью активации RXR.
В другом аспекте настоящего изобретения предложено применение соединения согласно настоящему изобретению для лечения состояния у млекопитающего. В одном варианте реализации настоящего изобретения указанное состояние представляет собой состояние, которое можно лечить с помощью активации RXR.
В некоторых вариантах реализации настоящего изобретения указанное состояние выбрано из группы, состоящей из раковых заболеваний, таких как предзлокачественные и злокачественные гиперпролиферативные заболевания, такие как рак груди, поджелудочной железы, кожи, простаты, шейки матки, матки, толстой кишки, мочевого пузыря, пищевода, желудка, легких, гортани, печени, полости рта, поджелудочной железы, крови и лимфатической системы, метаплазии, дисплазии, неоплазии, лейкоплакии и папилломы слизистых оболочек и для лечения саркомы Капоши, кожной Т-клеточной лимфомы, промиелоцитарного лейкоза, немелкоклеточного рака легкого, рака почки (распространенного почечно-клеточного рака), рака желудочно-кишечного тракта, мезотелиомы, бронхиальной метаплазии; кожных расстройств, таких как дерматит (тяжелая хроническая экзема рук у взрослых), псориаз (тяжелый бляшковидный псориаз), алопеция (выпадение волос), старческих кератозов, мышьяковых кератозов, воспалительной и невоспалительной угревой сыпи, псориаза, ихтиозов и других расстройств кератинизации и гиперпролиферативных расстройств кожи и расстройств кератинизации кожи, экземы, атопического дерматита, болезни Дарье, красного плоского лишая, для профилактики и обращения вспять глюкокортикоидных повреждений (стероидная атрофия), в качестве местного противомикробного агента, в качестве агентов пигментации кожи и для лечения и обращения вспять влияния возраста и светового повреждения кожи; метаболических заболеваний, таких как диабет, диабет II типа, ожирение, гипергликемия, гиперхолестеринемия, гипертриглицеридемия; гиперинсулинемия, сердечно-сосудистых заболеваний, включая заболевания связанные с метаболизмом липидов, таких как дислипидемии, для профилактики рестеноза и в качестве агента для повышения уровня циркулирующего тканевого активатора плазминогена (ТРА), НАСГ (неалкогольного стеатогепатита), стеатоза, стеатогепатита, цирроза; заболевания печени, фиброза почек и фиброза печени, заболеваний глаз, таких как пролиферативная витреоретинопатия (PVR), отслойка сетчатки, сухость глаз и другие поражения роговой оболочки глаза; и воспалительных заболеваний, воспаления, окислительного стресса, легочного фиброза, илеита, колита и болезни Крона; нейродегенеративных заболеваний, таких как болезнь Альцгеймера, болезнь Паркинсона и боковой амиотрофический склероз (ALS), множественный склероз, неправильной функции гипофиза, включая недостаточное производство гормона роста, модуляции апоптоза, включая как индукцию апоптоза так и ингибирование активированного Т-клеточного апоптоза, восстановления роста волос и рассеянного склероза.
В некоторых вариантах реализации настоящего изобретения указанное состояние представляет собой диабет. В некоторых вариантах реализации настоящего изобретения указанное состояние представляет собой диабет II типа. В некоторых вариантах реализации настоящего изобретения указанное состояние представляет собой ожирение. В некоторых вариантах реализации настоящего изобретения указанное состояние представляет собой сердечно-сосудистое заболевание.
Соединения согласно настоящему изобретению также можно применять для обеспечения ряда полезных эффектов у млекопитающих. Примеры полезных эффектов, которые могут быть предоставлены, включают увеличение мышечной выносливости, улучшение сердечной функции и достижение эффекта, миметического физическим упражнениям.
В одном варианте реализации настоящего изобретения положительный эффект представляет собой увеличение мышечной выносливости у млекопитающего. В одном варианте реализации настоящего изобретения положительный эффект представляет собой улучшение сердечной функции у млекопитающего. Примеры сердечной функции, которую можно улучшить, включают работу верхушечного толчка, систолический объем крови, долю выброса и наполнение желудочка. В одном варианте реализации настоящего изобретения положительный эффект представляет собой достижение эффекта, миметического физическим упражнениям, у млекопитающих.
Введение человеку соединений, попадающих под формулу (I), может осуществляться любым из приемлемых способов для энтерального введения, таким как пероральный или ректальный, или для парентерального введения, таким как подкожно, внутримышечно, внутривенно и внутрикожно. Инъекции могут быть болюсными или через постоянную или интермиттирующую инфузию. Активное соединение обычно содержится в фармацевтически приемлемом носителе или разбавителе и в таком количестве, которого достаточно для доставки пациенту терапевтически эффективной дозы. В различных вариантах реализации настоящего изобретения активирующее соединение может быть селективно токсичными или более токсичным по отношению к быстро пролиферирующим клеткам, например, клеткам раковых опухолей, чем к нормальным клеткам.
При использовании соединений согласно настоящему изобретению они могут быть введены в любой форме или любым способом, которые делают соединение биодоступным. Специалист в области приготовления композиций может легко выбрать подходящую форму и способ введения в зависимости от конкретных характеристик выбранного соединения, состояния, подлежащего лечению, стадии состояния, подлежащего лечению, и других значимых обстоятельств. С целью получения дополнительной информации, авторы настоящего изобретения предлагают читателю ознакомиться с Remingtons Pharmaceutical Sciences, 19th edition, Mack Publishing Co. (1995).
Соединения согласно настоящему изобретению можно вводить отдельно или в виде фармацевтической композиции в сочетании с фармацевтически приемлемыми носителем, разбавителем или наполнителем. Соединения согласно настоящему изобретению, которые сами по себе являются эффективными, обычно приготовляют и вводят в форме их фармацевтически приемлемых солей, поскольку эти формы обычно более стабильны, легко кристаллизуется и обладают повышенной растворимостью.
Однако эти соединения обычно используют в форме фармацевтических композиций, которые приготовлены в зависимости от желаемого способа введения. Таким образом, в некоторых вариантах реализации настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы (I) и фармацевтически приемлемые носитель, разбавитель или наполнитель. Указанные композиции получают способами, хорошо известными в данной области техники.
В других вариантах реализации настоящего изобретения предложена фармацевтическая упаковка или набор, содержащий один или более контейнеров, заполненных одним или несколькими ингредиентами фармацевтических композиций согласно настоящему изобретению. В такой упаковке или наборе можно найти контейнер, содержащий стандартную дозировку агента(ов). Наборы могут содержать композицию, содержащую эффективный агент либо в виде концентратов (в том числе лиофилизированные композиции), которые можно в дальнейшем разбавлять перед применением, либо они могут быть предоставлены в той концентрации, которая используется для применения, при этом флаконы могут содержать одну или более дозировок. В наборах единичные дозировки могут быть предоставлены в удобной форме стерильных флаконов, таким образом, что врач может применять флаконы напрямую, при этом указанные флаконы содержат необходимое количество и концентрацию агента(ов). К такому (таким) контейнеру(ам) могут быть приложены различные письменные материалы, такие как инструкции по применению или уведомление в форме, предписанной правительственным органом, регулирующим производство, применение или продажу фармацевтических или биологических продуктов, при этом такое уведомление отражает одобрение государственным органом производство, применение или продажу указанных продуктов для введения человеку.
Соединения согласно настоящему изобретению можно применять или вводить в комбинации с одним или несколькими дополнительным(и) препаратом(ами) для лечения упомянутых расстройств/заболеваний. Компоненты можно вводить в той же композиции или в отдельных составах. При введении в виде отдельных составов соединения согласно настоящему изобретению можно вводить последовательно или одновременно с другим лекарственным средством(ами).
В дополнение к возможности введения в комбинации с одним или несколькими дополнительными препаратами, соединения согласно настоящему изобретению можно применять в комбинированной терапии. При комбинированной терапии соединения обычно вводят в комбинации друг с другом. Таким образом, одно или более из соединений согласно настоящему изобретению можно вводить либо одновременно (в виде комбинированного препарата), либо последовательно, с тем, чтобы достичь желаемого эффекта. Это особенно желательно в тех случаях, когда терапевтический профиль каждого соединения отличается, чтобы комбинированное действие двух препаратов обеспечивало улучшенный терапевтический результат.
Фармацевтические композиции согласно настоящему изобретению для парентерального введения содержат фармацевтически приемлемые стерильные водные или неводные растворы, дисперсии, суспензии или эмульсии, а также стерильные порошки для восстановления до стерильных инъекционных растворов или дисперсий непосредственно перед использованием. Примеры подходящих водных и неводных носителей, разбавителей, растворителей или наполнителей включают воду, этанол, полиолы (такие как глицерин, пропиленгликоль, полиэтиленгликоль и т.п.) и их подходящие смеси, растительные масла (такие как оливковое масло) и инъецируемые органические сложные эфиры, такие как этилолеат. Требуемая текучесть может поддерживаться, например, путем применения материалов для покрытий, таких как лецитин, путем поддержания требуемого размера частиц в случае дисперсий, и при применении поверхностно-активных веществ.
Такие композиции могут также содержать вспомогательные вещества, такие как консерванты, увлажняющие агенты, эмульгаторы и диспергирующие агенты. Предотвращение действия микроорганизмов можно обеспечить с помощью включения в состав различных антибактериальных и противогрибковых агентов, например, парабена, хлорбутанола, фенола, сорбиновой кислоты и подобных. Кроме того, может быть желательным включение в состав изотонических агентов, таких как сахара, хлорид натрия и подобных. Пролонгированную абсорбцию инъецируемой фармацевтической формы можно осуществить путем включения агентов, которые замедляют абсорбцию, таких как моностеарат алюминия и желатин.
Если необходимо, а также для более эффективного распределения, соединения согласно настоящему изобретению можно включать в системы доставки медленноговысвобождения или высвобождения нацеленного действия, такие как полимерные матрицы, липосомы и микросферы.
Инъекционные составы можно стерилизовать, например, с помощью фильтрации через бактериальный фильтр или включением стерилизующих агентов в форме твердых стерильных композиций, которые можно растворять или диспергировать в стерильной воде или другой стерильной среде для инъекций непосредственно перед использованием.
Твердые лекарственные формы для перорального введения включают капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых лекарственных формах активное соединение смешано с по меньшей мере одним инертным, фармацевтически приемлемым наполнителем или носителем, таким как цитрат натрия или дикальцийфосфат и/или а) наполнителями или сухими разбавителями, такими как крахмалы, лактоза, сахароза, глюкоза, маннит и кремниевая кислота, b) связующими агентами, такими как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и гуммиарабик, с) увлажнителями, такими как глицерин, d) дезинтегрирующими агентами, такими как агар-агар, карбонат кальция, картофельный крахмал или крахмал из тапиоки, альгиновая кислота, некоторые силикаты и карбонат натрия, е) замедляющими растворение агентами, такими как парафин, f) ускорителями абсорбции, такими как четвертичные аммониевые соединения, g) смачивающими агентами, такими как, например, цетиловый спирт и моностеарат глицерина, h) абсорбентами, такими как каолин и бентонитовая глина, и i) смазывающими агентами, такими как тальк, стеарат кальция, стеарат магния, твердыми полиэтиленгликолями, лаурилсульфатом натрия и их смесями. В случае капсул, таблеток и пилюль лекарственная форма может также содержать буферные агенты.
Твердые композиции подобного типа можно также использовать в качестве наполнителей в мягких и твердых заполненных желатиновых капсулах с применением таких наполнителей, как лактоза или молочный сахар, а также полиэтиленгликолей с высокой молекулярной массой и подобными.
Твердые лекарственные формы таблеток, драже, капсул, пилюль и гранул можно получать с покрытиями и оболочками, такими как энтеросолюбильные покрытия и другие покрытия, хорошо известные в данной области приготовления фармацевтических препаратов. Они могут необязательно содержать замутняющие агенты и могут также иметь такой состав, что они высвобождают активный ингредиент(ы) только, или предпочтительно, в определенной части кишечного тракта, необязательно, замедленным способом. Примеры капсулирующих композиций, которые можно применять, включают полимерные вещества и воски.
Активные соединения могут быть также в микроинкапсулированной форме, если это необходимо, с одним или несколькими из указанных выше наполнителей.
Жидкие лекарственные формы для перорального введения содержат фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к активным соединениям жидкие лекарственные формы могут содержать инертные разбавители, обычно применяемые в данной области техники, такие как, например, вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, хлопковое, арахисовое, кукурузное, зародышевое, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры сорбитана и жирных кислот и их смеси.
Кроме инертных разбавителей, указанные композиции для перорального приема могут также содержать адъюванты, такие как смачивающие агенты, эмульгаторы и суспендирующие агенты, подсластители, вкусовые и ароматизирующие агенты.
Суспензии, помимо активных соединений, могут содержать суспендирующие агенты, как, например, этоксилированные изостеариловые спирты, сложные эфиры полиоксиэтилена сорбитола и сорбитана, микрокристаллическую целлюлозу, метагидроксид алюминия, бентонит, агар-агар и трагакант и также их смеси.
Композиции для ректального или вагинального введения предпочтительно представляют собой суппозитории, которые можно получить путем смешивания соединения согласно настоящему изобретению с подходящими не раздражающими эксципиентами или носителями, такими как масло какао, полиэтиленгликоль или воск для суппозиториев, которые являются твердыми при комнатной температуре, но становятся жидкими при температуре тела и, следовательно, расплавляются в прямой кишке или вагинальной полости и высвобождают активное соединение.
Лекарственные формы для местного введения соединения согласно настоящему изобретению включают порошки, пластыри, спреи, мази и ингаляторы. Активное соединение смешивают в стерильных условиях с фармацевтически приемлемым носителем и любыми необходимыми консервантами, буферами или пропеллентами, которые могут потребоваться.
Количество вводимого соединения будет предпочтительно лечить и ослаблять или облегчить состояние. Терапевтически эффективное количество можно легко определить с помощью специалиста диагноста путем применения обычных способов и наблюдением результатов, полученных в аналогичных условиях. При определении терапевтически эффективного количества необходимо рассматривать ряд факторов, в том числе, но не ограничиваясь ими, вид животного, его размер, возраст и общее состояние здоровья, наличие особых условий, тяжесть состояния, реакции пациента на лечение, конкретное вводимое соединение, способ введения, биодоступность вводимого состава, режим приема выбранного лекарственного средства, применение других лекарственных препаратов и другие соответствующие обстоятельства.
Предпочтительная дозировка будет находиться в диапазоне от примерно 0,01 до 300 мг в день на килограмм массы тела. Более предпочтительная дозировка будет находиться в диапазоне от 0,1 до 100 мг в день на килограмм массы тела, более предпочтительная от 0,2 до 80 мг в день на килограмм массы тела, еще более предпочтительная от 0,2 до 50 мг в день на килограмм массы тела. Подходящую дозу можно вводить в виде нескольких поддоз в день.
СИНТЕЗ СОЕДИНЕНИЙ
Агенты согласно различным вариантам реализации настоящего изобретения можно получить с применением способов реакции и схем синтеза, как описано ниже, с применением способов доступных в данной области техники, с применением исходных материалов, которые легко доступны. Получение конкретных соединений согласно вариантам реализации настоящего изобретения подробно описано в следующих примерах, но специалистам в данной области техники будет понятно, что описанные химические реакции можно легко адаптировать для получения ряда других агентов согласно различным вариантам реализации настоящего изобретения. Например, синтез не являющихся примерами соединений, можно успешно выполнить с помощью модификаций, очевидных для специалистов в данной области техники, например, с помощью подходящей защиты соответствующих групп, путем изменения на другие подходящие реагенты, известные в данной области техники, или путем обычных модификаций реакционных условий. Список подходящих защитных групп в органическом синтезе можно найти в TW Greene's Protective Groups in Organic Synthesis, 3-е издание, John Wiley & Sons, 1991. В качестве альтернативы, другие реакции, описанные в настоящем изобретении или известные в данной области техники, признаны как имеющие применимость для получения других соединений согласно различным вариантам реализации настоящего изобретения.
Реагенты, применяемые для синтеза соединений, можно получить или приготовить в соответствии со способами, известными в данной области техники.
Символы, сокращения и соглашения в способах, схемах и примерах соответствуют тем, которые используются в современной научной литературе. В частности, но не в качестве ограничения, следующие сокращения использованы в примерах и по всему тексту настоящего изобретения.
- г (граммы)
- л (литры)
- Гц (герцы)
- моль (моли)
- RT, комнатная температура
- мин (минуты)
- МеОН (метанол)
- СНСl3 (хлороформ)
- DCM (дихлорметан)
- ДМСО (диметилсульфоксид)
- EtOAc (этилацетат)
- мг (миллиграммы)
- мл (миллилитры)
- фунты на квадратный дюйм
- мМ (миллимоль)
- МГц (мегагерц)
- ч (часы)
- ТСХ (тонкослойная хроматография)
- EtOH (этанол)
- CDCl3 (дейтерированный хлороформ)
- HCl (соляная кислота)
- ДМФА (N,N-диметилформамид)
- ТГФ (тетрагидрофуран)
- K2CO3 (карбонат калия)
- Na2SO4 (сульфат натрия)
- RM реакционная смесь.
Если не указано иное, все температуры выражены в °С (градусах по Цельсию). Все реакции проводили при комнатной температуре, если не оговорено иное.
Все применяемые растворители и реагенты являются коммерчески доступными и могут быть приобретены у фирм Sigma Aldrich, Fluka, Acros, Spectrochem, Alfa Aesar, Avra, Qualigens, Merck, Rankem и Leonid Chemicals.
Спектры 1Н-ЯМР регистрировали на спектрометре Bruker AV 300. Химические сдвиги выражали в частях на миллион (ppm, δ единицы). Постоянные взаимодействия приведены в герцах (Гц). Структуры расщепления описывают мультиплетности и обозначены как s (синглет-), d (дублет), t (триплет), q (квартет), m (мультиплет), или br (широкий).
Масс-спектры получали на одноквадрупольном ЖХ-МС 6120 производства Agilent technologies, с применением либо атмосферной химической ионизации (APCI), либо ионизации электрораспылением (ESI), или комбинации этих двух источников.
Все образцы исследовали на системе SHIMADZU с насосом LC-20 AD, диодным матричным детектором SPD-M20A, автоматическим пробоотборником SIL-20A.
СХЕМЫ СИНТЕЗА
Схема для получения некоторых соединений согласно настоящему изобретению показана на схеме 1, приведенной ниже. Таким образом, применяя стандартные способы хлорирования 2,5-диметил-2,5-гександиола в присутствии HCl с получением 2,5-дихлор-2,5-диметилгексана (промежуточное соединение 1) и последующим алкилированием Фриделя-Крафтса 3-метилтиофена с получением 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофена (промежуточное соединение 2). Алкилирование Фриделя-Крафтса 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофена с метил 4-хлоркарбонил)бензоатом (промежуточное соединение 3) с получением метил 4-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)карбонил]бензоата (промежуточное соединение 4). Образование алкена (промежуточное соединение 4) в условиях Виттига с получением соответствующего сложного эфира метил 4-[1-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)этенил]бензоата (промежуточное соединение 5) и с последующим гидролизом с получением соответствующей кислоты.
Схема 1:
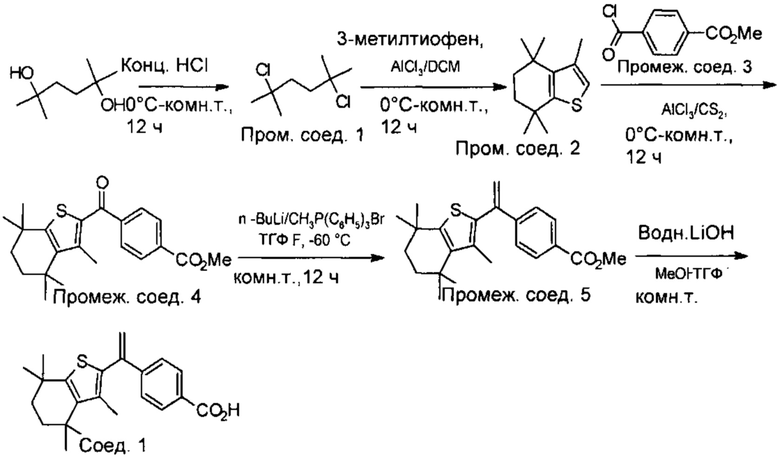
Пример 1
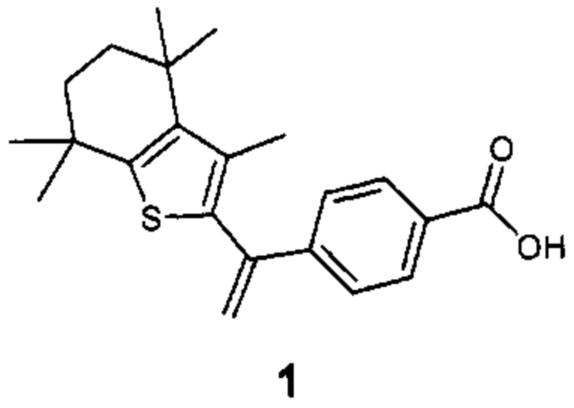
Соединение 1 синтезировали из 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофена и метил 4-(хлоркарбонил)бензоата, следуя способу, описанному в схеме 1; чистота: 98,87%
Промежуточное соединение 1: 2,5-дихлор-2,5-диметилгексан
В круглодонную колбу 250 мл с двумя горлышками, загруженную 2,5-диметил-2,5-гександиолом (15 г, 102,6 ммоль), медленно добавляли концентрированную HCl (60 мл) при 0°С и перемешивали при комнатной температуре в течение ночи. Реакционную смесь гасили льдом, полученную гетерогенную смесь фильтровали и промывали водой и сушили в вакууме с получением целевого соединения в виде твердого вещества белого цвета (15,0 г, выход: 79,91%): 1Н ЯМР (300 МГц, CDCl3): 1,88 (s, 4Н), 1,53 (s, 12Н).
Промежуточное соединение 2: 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен
В круглодонную колбу 250 мл с тремя горлышками, загруженную 2,5-дихлор-2,5-диметилгексаном (6,7 г, 36,6 ммоль) и 3-метилтиофеном (3,6 г, 36,6 ммоль) в сухом DCM (50 мл), снабженную конденсатором с водяным холодильником, небольшими порциями добавляли AlCl3 (4,9 г, 36,6 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение ночи и затем нагревали с обратным холодильником в течение 15 мин. Реакционную смесь охлаждали до комнатной температуры, гасили льдом и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали с помощью колоночной хроматографии с получением целевого соединения в виде бледно-желтого маслянистого продукта (3,5 г, выход: 46,0%).
Промежуточное соединение 3: Метил 4-(хлоркарбонил)бензоат
В круглодонную колбу 100 мл с одним горлышком, снабженную конденсатором с водяным холодильником, загруженную 4- (метоксикарбонил)бензойной кислотой (7,0 г, 38,8 ммоль) и сухим DCM (70 мл), добавляли по каплям SOCl2 (9,2 г, 77,7 ммоль) при 0°С с последующим добавлением ДМФА (0,5 мл). Затем реакционную смесь нагревали с обратным холодильником в течение 1 ч (или до тех пор, пока реакционная смесь не станет прозрачным раствором). Реакционную смесь концентрировали в атмосфере N2 с получением неочищенного продукта в виде желтого масла (6,9 г, выход: 89,5%), которое использовали для следующего этапа без очистки.
Промежуточное соединение 4: Метил 4-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)карбонил]бензоат
В круглодонную колбу 250 мл с двумя горлышками загружали метил 4-(хлор карбонил)бензоат (6,9 г, 34,6 ммоль), растворенный в CS2 (30,0 мл), добавляли AlCl3 (11,5 г, 86,5 ммоль) при 0°С. Указанный 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен (6,0 г, 28,8 ммоль), растворенный в CS2 (30 мл), медленно добавляли по каплям к реакционной смеси. Реакционную смесь перемешивали при комнатной температуре в течение ночи и затем нагревали с обратным холодильником в течение 15 мин. Реакционную смесь гасили льдом и экстрагировали этилацетатом, промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме с получением неочищенного масла, которое очищали с помощью колоночной хроматографии с получением целевого соединения в виде желтого маслянистого продукта (1,5 г, выход: 14,0%).
Промежуточное соединение 5: Метил 4-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)этенил]бензоат
В круглодонную колбу 100 мл с двумя горлышками, загруженную метилтрифенилфосфонийбромидом (0,965 г, 2,7 ммоль) и сухим ТГФ (10 мл), добавляли по каплям н-бутиллитий [0,8 мл (1,6 М раствор), 2,7 ммоль] при температуре -60°С в атмосфере N2. Реакционную смесь перемешивали при комнатной температуре (RT) в течение 1 часа и наблюдали образование раствора желтого цвета, который затем медленно добавляли в круглодонную колбу 50 мл, содержащую (0,5 г, 1,35 ммоль) соединения метил 4-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)карбонил]бензоата (0,5 г, 1,35 ммоль) в сухом ТГФ (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь гасили холодной водой и экстрагировали этилацетатом. Органический слой промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме; полученный неочищенный продукт очищали с помощью колоночной хроматографии с получением целевого соединения в виде бледно-желтого твердого вещества. (0,11 г, выход: 22,0%): 1Н ЯМР (300 МГц, CDCl3): 7,90-7,93 (d, 2Н), 7,33-7,36 (d, 2Н), 5,65 (s, 1Н), 5,38 (s, 1Н), 3,85 (s, 3Н), 1,88 (s, 3Н), 1,50 (s, 4Н), 1,25 (s, 6Н), 1,21 (s, 6Н).
Соединение 1: 4-[1-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)этенил]бензойная кислота
В круглодонную колбу объемом 25 мл, загруженную метил 4-[1-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)этенил]бензоатом (0,06 г, 0,16 ммоль) и ТГФ (3 мл), добавляли водный раствор NaOH (0,013 г, 0,32 ммоль) и метанол (3 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали; полученный остаток растворяли в минимальном количестве воды, подкисляли до рН 3 при охлаждении, экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над безводным Na2SO4, фильтровали и концентрировали. Полученный неочищенный продукт очищали растиранием с гексаном с получением целевого соединения в виде твердого вещества бледно-желтого цвета (0,02 г, выход: 34,7%): МС (ESI, 120 эВ): m/z=353,1 (М-Н)+.
Схема 2:
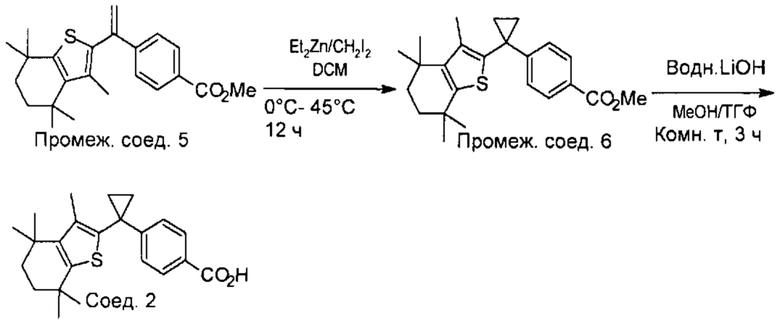
Пример 2
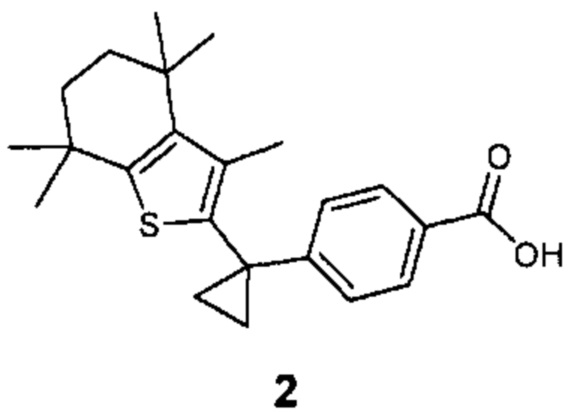
Соединение 2 синтезировали из метил 4-[1-(3,4,4,7,7-пентаметил-4, 5,6,7-тетрагидро-1-бензотиофен-2-ил)этенил]бензота и гидролиза метил 4-[1-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)циклопропил]бензоата, следуя способу, описанному в схеме 2. Чистота: 95,69%
Промежуточное соединение 6: Метил 4-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)циклопропил]бензоат
В круглодонную колбу 50 мл с двумя горлышками загружали метил 4-[1-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)этенил]бензоат (0,1 г, 0,27 ммоль) в DCM (10 мл). К перемешиваемому раствору добавляли по каплям диэтилцинк (1,6 мл, 1,6 ммоль) при 0°С и через 10 мин. добавляли CH2I2 (0,87 г, 3,2 ммоль), и затем реакционную смесь нагревали при комнатной температуре в течение ночи. Реакционную смесь гасили льдом, экстрагировали с DCM и объединяли органические экстракты, промывали водой и рассолом, сушили над безводным Na2SO4 и концентрировали. Полученный неочищенный продукт очищали с помощью препаративной ТСХ с получением целевого соединения в виде бесцветного маслянистого продукта (0,015 г, выход: 15,0%).
Соединение 2: 4-[1-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)циклопропил]бензойная кислота
В круглодонную колбу 50 мл, загруженную метил 4-[1-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)циклопропил]бензоатом (0,15 г, 0,04 ммоль) и ТГФ (2 мл), добавляли водный раствор LiOH (0,0037 г, 0,16 ммоль) и метанол (2 мл). Реакционный раствор перемешивали при комнатной температуре в течение примерно 3 часов. Растворитель удаляли при пониженном давлении. Полученный остаток растворяли в минимальном количестве воды, подкисляли до рН 3 при охлаждении, экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Полученное твердое вещество дополнительно очищали растиранием с гексаном с получением соединения в виде твердого вещества бледно-желтого цвета (0,01 г, выход: 69,2%): МС (ESI, 120 эВ): m/z=367,1 (М-Н)+.
Схема 3
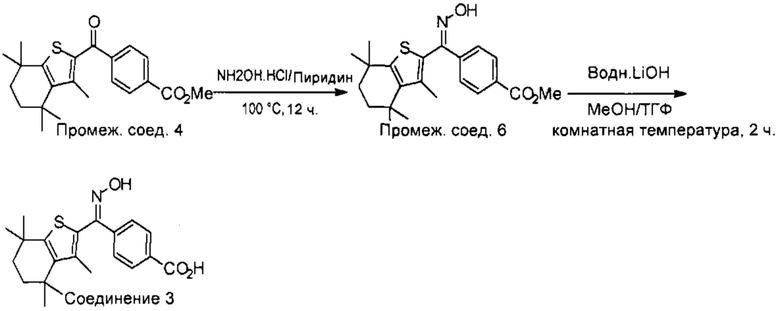
Пример 3
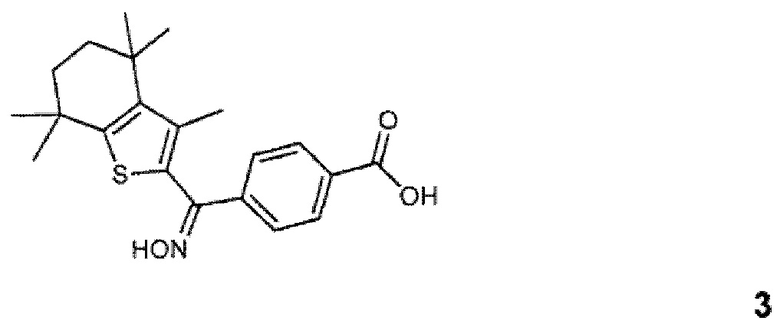
Соединение 3 синтезировали из метил 4-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)карбонил]бензота и гидролиза метил 4-[(Е)-(гидроксиимино)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)метил]бензоата, следуя способу, описанному в схеме 3; чистота: 97,84%
Промежуточное соединение 7: Метил 4-[(E,Z)-(гидроксиимино)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)метил]бензоат
К перемешиваемому раствору метил 4-[(3,4,4,7,-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)карбонил]бензоата (0,3 г, 0,8 ммоль) в пиридине (4,9 г, 62,0 ммоль) добавляли NH4OH.HCl (0,11 г, 1,6 ммоль) в атмосфере N2. Затем реакционную смесь нагревали при 100°С в течение ночи. Пиридин удаляли полностью под вакуумом; остаток растворяли в этилацетате, промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Сырой продукт, полученный таким образом, очищали с помощью колоночной хроматографии на силикагеле с использованием петролейного эфира (60-80) и этилацетата в качестве элюента. Продукт (изомер-1) отделяли и получали в виде желтой жидкости (0,2 г, выход: 64,85%): МС (ESI, 120 эВ): m/z=386,0 (М+Н)+.
Соединение 3: 4-[(Е)-(гидроксиимино)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)метил]бензойная кислота
Круглодонную колбу 50 мл загружали метил 4-[(Е,Z)-(гидроксиимино)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)метил]бензоат (0,2 г, 0,5 ммоль) и ТГФ (1 мл), перемешивали и к перемешиваемому раствору добавляли водный раствор 5М NaOH (0,2 г, 10,0 ммоль) и метанол (1 мл). Далее реакционную смесь перемешивали при комнатной температуре в течение примерно 2 часов. Реакционную смесь концентрировали; остаток растворяли в минимальном количестве воды, подкисляли до рН 3 при охлаждении, экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Один изомер был отделен при помощи кристаллизации; растворяли соединение в минимальном количестве этилацетата, добавили несколько капель эфира и затем гексана, с получением целевого соединения в виде желтого твердого вещества (0,17 г, выход: 91,52%): МС (ESI, 120 эВ): m/z=372,2 (М+Н+).
Пример 4: 4-[(E,Z)-(метоксиимино)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)метил]бензойная кислота
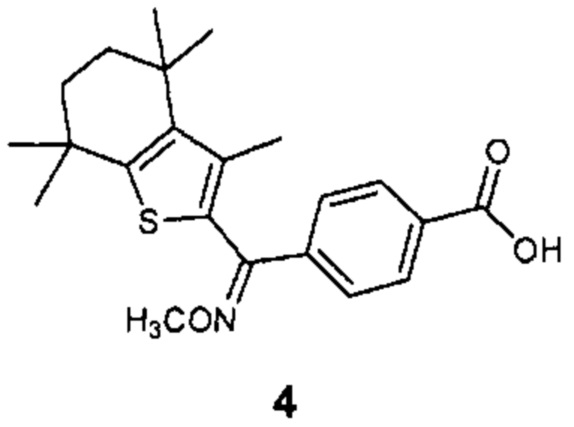
Соединение 4 синтезировали из метил 4-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)карбонил]бензота и гидролиза метил 4-[(E,Z)-(метоксиимино) (3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)метил]бензоата, следуя способу, аналогичному описанному в схеме 3 (0,025 г. выход: 26,0%); чистота: 98,96%
Пример 5: 6-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)карбонил]пиридин-3-карбоновая кислота
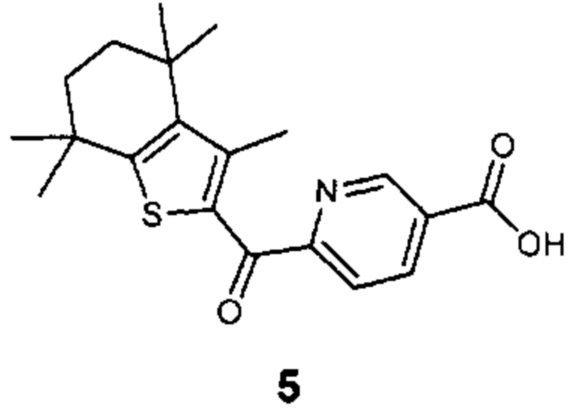
Соединение 5 синтезировали из 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофена и метил 6-(хлоркарбонил)пиридин-3-карбоксилата, следуя способу, аналогичному описанному в схеме 1 (0,008 г, выход: 55,47%); чистота: 99,38%
Пример 6: 6-[1-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)этенил]пиридин-3-карбоновая кислота
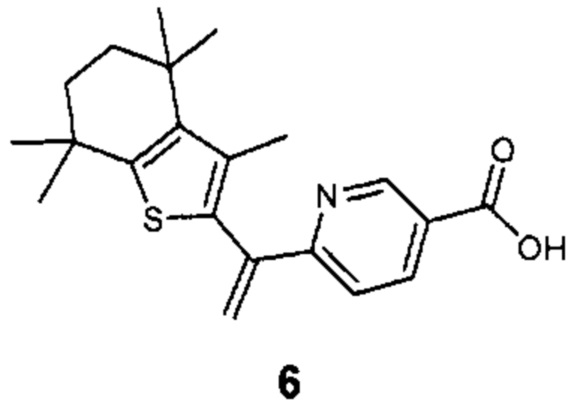
Соединение 6 синтезировали из 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофена и метил 6-(хлоркарбонил)пиридин-3-карбоксилата, следуя способу, аналогичному описанному в схеме 1 (0, 0012 г, выход: 62,44%); чистота: 98,37%
Пример 7: 6-[1-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)циклопропил]пиридин-3-карбоновая кислота
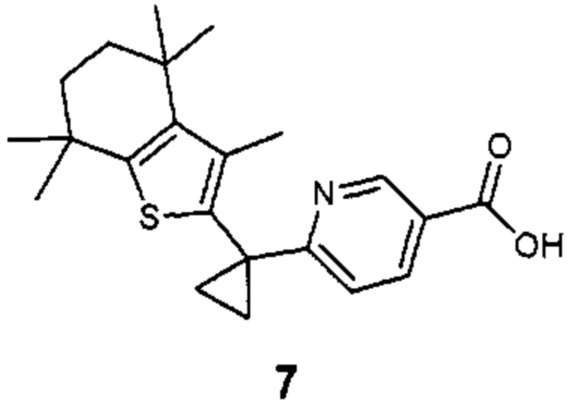
Соединение 7 синтезировали из метил 6-[1-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)этенил]пирдин-3-карбоксилата и гидролиза метил 6-[1-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)циклопропил]пиридин-3-карбоксилата, следуя способу, описанному в схеме 2. (0,015 г, выход: 40,6%); чистота: 99,26%
Схема 4
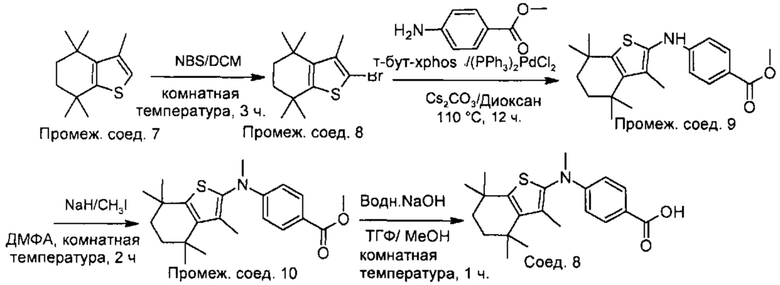
Пример 8:
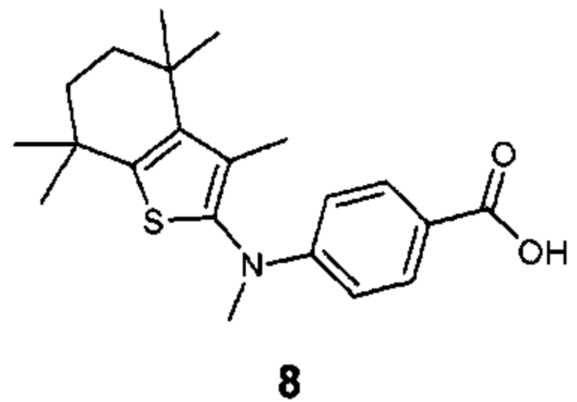
Соединение 8 синтезировали из 2-бром-3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофена и метил 4-аминобензоата, следуя способу, описанному в схеме 4; чистота: 97,45%
Промежуточное соединение 8: 2-бром-3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен
К перемешиваемому раствору 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофена (1,2 г, 5,8 ммоль) в DCM (5 мл) добавляли NBS (1 г, 5,6 ммоль) и перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь гасили льдом, экстрагировали с DCM и объединяли органические экстракты, промывали водой и рассолом, сушили над безводным Na2SO4 и концентрировали в вакууме. Полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением целевого соединения в виде бесцветной жидкости (1,0 г, выход: 62,5%): 1Н-ЯМР (300 МГц, CDCl3): δ 2,16 (s, 3Н), 1,58 (s, 4Н), 1,19-1,20 (d, 12Н).
Промежуточное соединение 9: Метил 4-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоат
К перемешиваемому раствору 2-бром-3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофена (0,26 г, 0,9 ммоль) в диоксане (5 мл) добавляли метил 4-аминобензоат (0,16 г, 1,1 ммоль), CS2CO3 (0,88 г, 6,5 ммоль) в атмосфере аргона. После продувки реакционной смеси аргоном в течение приблизительно 10 минут, добавляли трет-бутиловый xphos (0,007 г, 0,02 ммоль) и Pd (PPh3)2Cl2 (0,031 г, 0,044 ммоль), и реакционную смесь нагревали при 110°С в течении ночи. Реакционную смесь гасили водой и экстрагировали этилацетатом, органические экстракты промывали водой и рассолом, сушили над безводным Na2SO4 и концентрировали в вакууме. Полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с использованием петролейного эфира (60-80) и этилацетата в качестве элюента с получением целевого продукта в виде желтого твердого вещества (0,09 г, выход: 27,97%).
Промежуточное соединение 10: Метил 4-[метил(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоат
К перемешиваемому раствору NaH (0,042 г, 0,31 ммоль) в ДМФА (2 мл) добавляли 6-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]пиридин-3-карбоксилат (0,07 г, 0,2 ммоль) в ДМФА (2 мл) по каплям при 0°С. Через 30 мин добавляли CH3I (0,045 г, 0,31 ммоль) к указанному выше раствору и полученный раствор перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь гасили насыщенным раствором NH4Cl и экстрагировали этилацетатом. Органический экстракт промывали водой и рассолом, сушили над безводным Na2SO4 и концентрировали в вакууме. Полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с использованием петролейного эфира (60-80) и этилацетата в качестве элюента с получением целевого продукта в виде твердого вещества белого цвета (0,025 г, выход: 33,65%): 1Н ЯМР (300 МГц, CDCl3): δ 7,80-7,83 (d, 2Н), 6,52-6,55 (d, 2Н), 3,78 (s, 3Н), 3,20 (s, 3Н), 1,90 (s, 3Н), 1,63 (s, 4Н), 1,22-1,23 (d, 12Н).
Соединение 8: 4-[1-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензойная кислота
В круглодонную колбу 25 мл загруженную метил 4-[метил (3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоатом (0,025 г, 0,0672 ммоль) и ТГФ (0,5 мл) добавляли водный раствор 5М NaOH (0,04 г, 1,7 ммоль) и метанол (0,5 мл). Реакционный раствор перемешивали при комнатной температуре в течение примерно 1 часа. Реакционную смесь концентрировали; остаток растворяли в минимальном количестве воды, подкисляли до рН 3 при охлаждении и экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное твердое вещество дополнительно очищали растиранием с гексаном с получением целевого соединения в виде твердого вещества желтого цвета (0,011 г, выход: 45,83%): МС (ESI, 120 эВ): m/z=358,1 (М+Н+).
Пример 9: 4-[этил (3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензойная кислота
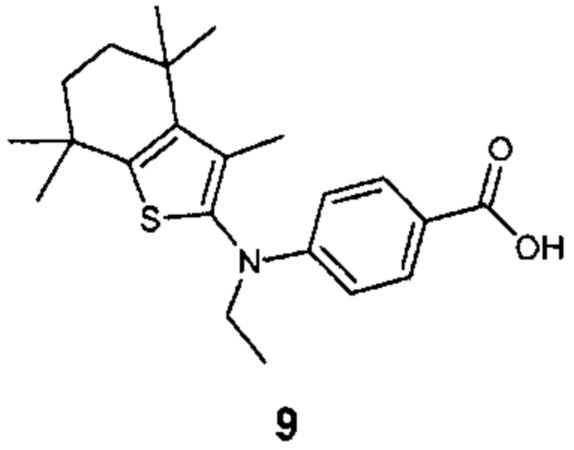
Соединение 9 синтезировали из 2-бром-3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофена и метил 4-аминобензоата, следуя способу, аналогичному описанному в схеме 4 (0,03 г, выход: 50,25%); чистота: 99,17%
Схема 5
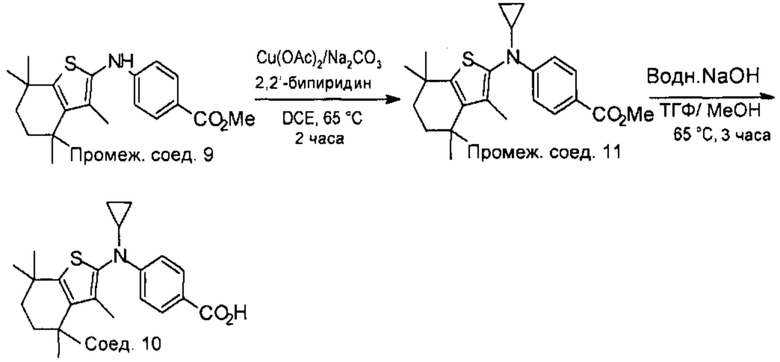
Пример 10: 4-[циклопропил (3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензойная кислота
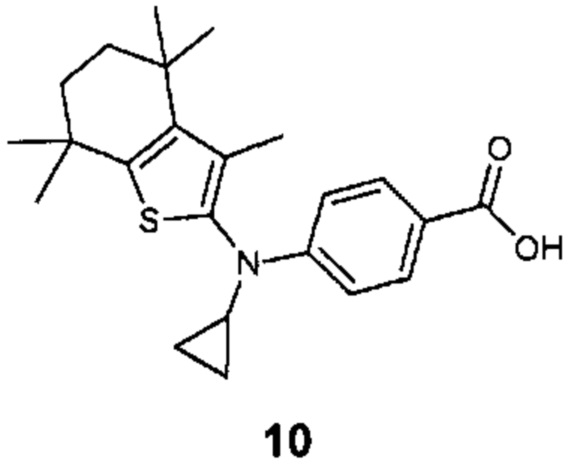
Соединение 10 синтезировали из 2-бром-3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен и метил-4-аминобензоата и N-алкилированием эфира (промежуточный соединение 9) с циклопропилбороновой кислотой в присутствии ацетата меди и с последующей процедурой гидролиза, описанной в схеме 5; чистота: 98,42%
Промежуточное соединение 11: Метил 4-[циклопропил(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоат
В круглодонную колбу 25 мл с двум горлышками, загруженную ацетатом меди (II) (0,11 г, 0,56 ммоль), добавляли 2,2'-бипиридин (0,087 г, 0,56 ммоль) в DCE (5 мл). Реакционную смесь нагревали при 70°С в течение 30 мин, после чего добавляли циклопропилбороновую кислоту (0,096 г, 1,12 ммоль), метил 4-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоат (0,2 г, 0,6 ммоль), Na2CO3 (0,12 г, 1,12 ммоль) и молекулярных сит (0,2 г) при 70°С и продолжали нагревать в течение ночи. Реакционную смесь гасили водой, экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали препаративной ТСХ с получением целевого соединения в виде полутвердого вещества желтого цвета (0,04 г, выход: 18,06%): 1Н ЯМР (300 МГц, CDCl3): δ 7,80-7,83 (d, 2Н), 6,73-6,76 (d, 2Н), 3,78 (s, 3Н), 2,69-2,74 (m, 1Н), 1,83 (s, 3Н), 1,63 (s, 4Н), 1,21-1,23 (d, 12Н), 0,80-0,82 (m, 2Н), 0,60-0,61 (m, 2Н).
Соединение 10: 4-[циклопропил (3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензойная кислота
В круглодонную колбу объемом 25 мл, загруженную метил 4-[циклопропил (3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоатом (0,04 г, 0,1 ммоль) и ТГФ (0,5 мл), добавляли водный раствор гидроксида натрия (0,024 г, 0,6 ммоль) и метанол (0,5 мл). Указанный реакционный раствор нагревали при 50°С в течение ночи. Реакционную смесь концентрировали; остаток растворяли в минимальном количестве воды, подкисляли до рН 3 при охлаждении и экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное твердое вещество дополнительно очищали растиранием с гексаном с получением целевого соединения в виде твердого вещества белого цвета (0,012 г, выход: 31,3%): МС (ESI, 120 эВ): m/z=384,2 (М+Н)+.
Пример 11: 4-[(циклопропилметил)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензойная кислота
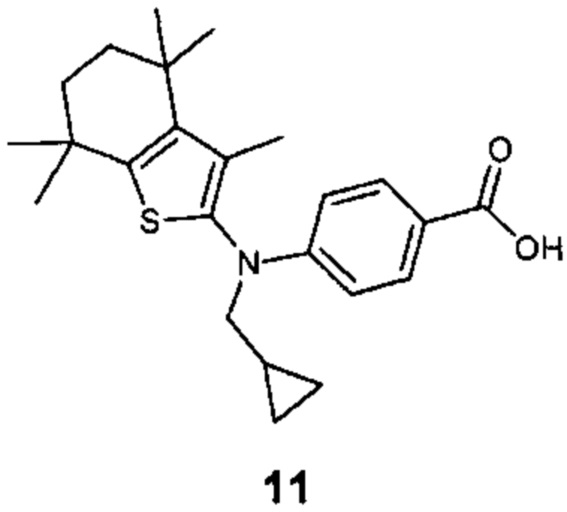
Соединение 11 синтезировали из 2-бром-3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофена и метил 4-аминобензоата, следуя способу, аналогичному описанному в схеме 4 (0,025 г, выход: 52,08%). чистота: 98,83%
Схема 6
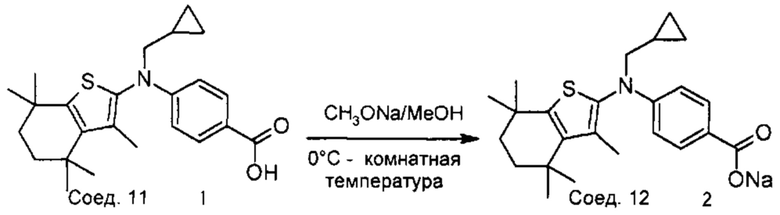
Соединение 12 синтезировали из 4-[(циклопропилметил)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензойной кислоты и метоксида натрия в метаноле, следуя способу, описанному в схеме 6; чистота: 96,75%
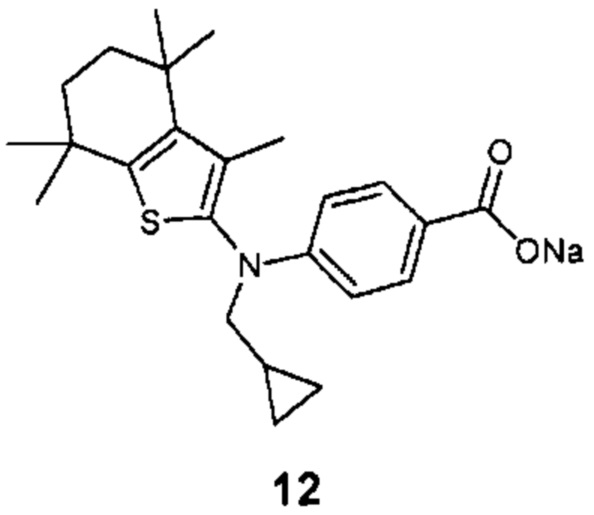
Соединение 12: Натрий 4-[(циклопропилметил)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоат
К перемешиваемому раствору 4-[(циклопропилметил)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензойной кислоты (0,1 г, 0,25 ммоль) в МеОН медленно добавляли раствор 1М CH3ONa (0,014 г, 0,25 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Растворитель удаляли при пониженном давлении. Метанол добавляли к полученному вязкому веществу и удаляли полностью под высоким вакуумом. Этот процесс повторяли до тех пор, пока совсем не осталось воды. Полученное вязкое вещество кристаллизовали этилацетатом и н-гексаном с получением твердого вещества почти белого цвета (0,04 г, выход: 32,00%): МС (ESI, 120 эВ): m/z=398,1 (М-22)+.
Пример 13: 4-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)(пропан-2-ил)амино]бензойная кислота
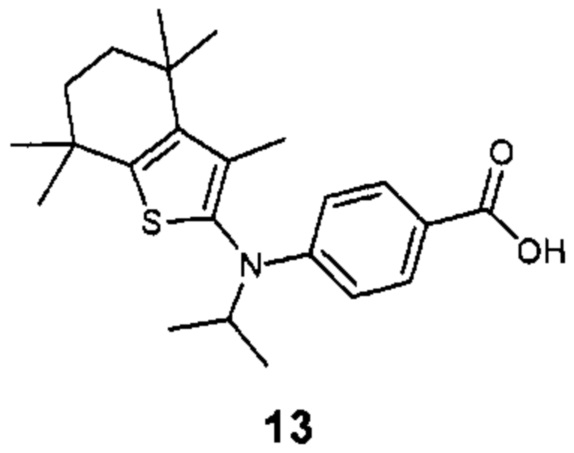
Соединение 13 синтезировали из 2-бром-3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофена и метил 4-аминобензоата, следуя способу, аналогичному описанному в схеме 4 (0,007 г, выход: 72,72%); чистота: 85,93%
Пример 14: 4-[(метилсульфонил)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензойная кислота
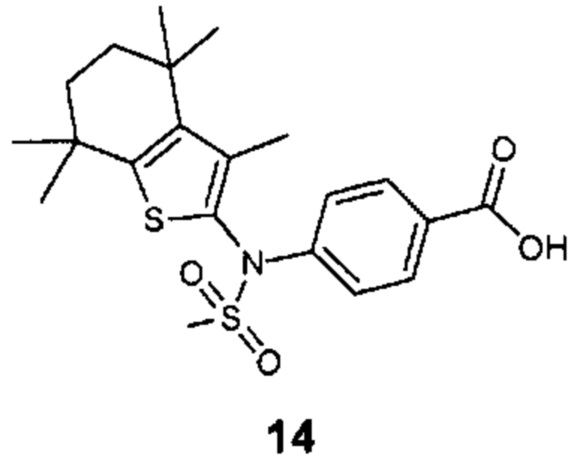
Соединение 14 синтезировали из 2-бром-3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофена и метил 4-аминобензоата, следуя способу, аналогичному описанному в схеме 4 (0,020 г, выход: 58,99%); чистота: 91,67%.
Схема 7
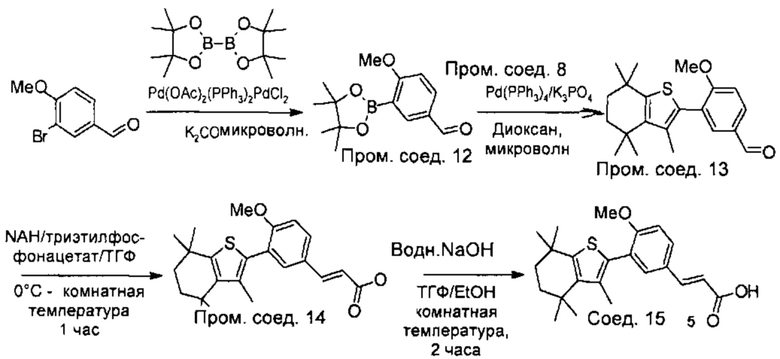
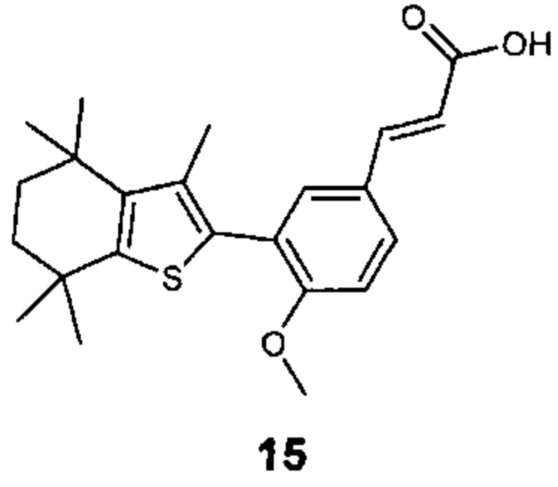
Соединение 15 синтезировали из 2-бром-3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофена и 4-метокси-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензальдегида, следуя способу, описанному в схеме 7; чистота: 94,87%
Промежуточное соединение 12: 4-метокси-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензальдегид
К перемешиваемому раствору 3-бром-4-метоксибензальдегида (2,0 г, 9,3 ммоль) в диоксане (5 мл) добавляли бис (пинаколато)дибор (2,82 г, 11,1 ммоль) в смеси 1,4 диоксана (10,0 мл) и ацетата калия (2,73 г, 27,9 ммоль) при 0°С в атмосфере аргона. Затем добавили (PPh3)2PdCl2 (0,38 г, 0,465 ммоль) и реакционную смесь оставляли в условиях микроволновой печи. Полученную таким образом реакционную смесь разбавляли этилацетатом, промывали водой и рассолом сушили над безводным Na2SO4 и концентрировали в вакууме. Полученный неочищенный продукт очищали с помощью CombiFlash с получением целевого соединения в виде твердого вещества бледно-розового цвета (1,3 г, выход: 53,76%): 1Н-ЯМР (300 МГц, CDCl3): δ 9,83 (s, 1Н), 8,13 (s, 1Н), 7,88-7,91 (d, 1Н), 6,89-6,92 (d, 1Н), 3,86 (s, 3Н), 1,3 (s, 12Н).
Промежуточное соединение 13: 4-метокси-3-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)бензальдегид
К перемешиваемому раствору 4-метокси-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензальдегида (1,0 г, 3,8 ммоль) в диоксане (10 мл) добавляли 2 бромо-3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен (1,2 г, 4,1 ммоль) в 1, 4 диоксане (10 мл), воду (3 мл) и трехосновный фосфат калия (2,42 г, 11,4 ммоль). Реакционную смесь продували аргоном в течение примерно 10 мин с последующим добавлением Pd (PPh3)4 (0,2 г, 0,1 ммоль). Реакцию завершали в микроволновых условиях. Реакционную смесь разбавляли этилацетатом, промывали водой и рассолом, сушили над безводным Na2SO4 и концентрировали в вакууме. Полученный неочищенный продукт очищали с помощью системы очистки CombiFlash с получением целевого соединения в виде полутвердого вещества желтого цвета (0,7 г, выход: 52,63%).
Промежуточное соединение 14: Этил (2Е)-3-[4-метокси-3-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)фенил]проп-2-эноат
В круглодонную колбу 50 мл, загруженную NaH (0,098 г, 4,0 ммоль) и ТГФ (20,0 мл), добавляли 4-метокси-3- (3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензо-тиофен-2-ил)бензальдегид (0,7 г, 2,0 ммоль) при 0°C с последующим добавлением триэтилфосфинацетата (0,92 г, 4,0 ммоль) и колбу перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь гасили водой и экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над безводным Na2SO4 и концентрировали в вакууме. Полученный неочищенный продукт очищали с помощью системы очистки CombiFlash с получением целевого соединения в виде бесцветного масла (0,74 г, выход: 89,68%).
Соединение 15: (2Е)-3-[4-метокси-3-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)фенил]проп-2-вая кислота
В круглодонную колбу 50 мл, загруженную этилом (2Е)-3-[4-метокси-3-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)фенил] проп-2-еновой кислоты 0,065 г, 0,15 ммоль) и ТГФ (2 мл), добавляли NaOH (0,012 г, 0,31 ммоль) и этанол (2 мл) и перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении и промывали эфиром. Полученный остаток подкисляли 1N HCl и экстрагировали этилацетатом. Объединенный экстракт затем промывали водой, рассолом, и сушили над безводным Na2SO4, фильтровали и концентрировали. Полученный продукт растирали в порошок с н-гексаном с получением целевого продукта в виде твердого вещества белого цвета (0,035 г, выход: 45,5%): МС (ESI, 120 эВ): m/z=385,2 (М+Н+).
Схема 8
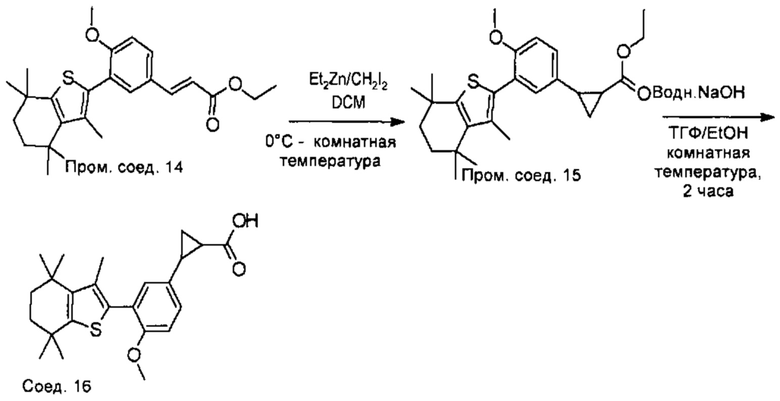
Соединение 16 синтезировали из этил (2E)-3-[4-метокси-3-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)фенил]проп-2-онат и гидролизом этил 2-[4-метокси-3-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)фенил]циклопропанкарбоксилата, следуя способу, описанному в схеме 8; чистота: 96,17%
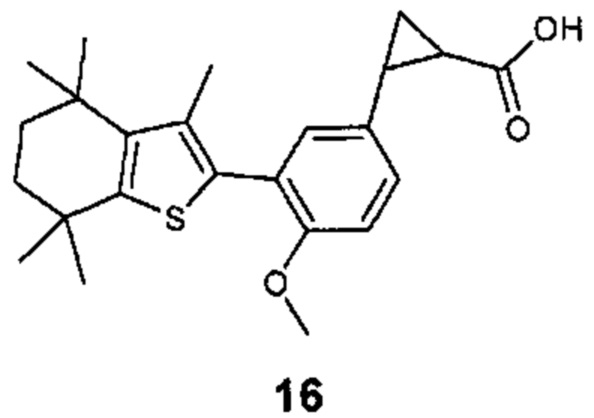
Промежуточное соединение 15: Этил 2-[4-метокси-3-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)фенил]циклопропанкарбоксилат
К перемешиваемому раствору этил (2Е)-3-[3-метокси-4-(3,4,4,7,7-пентаметил-4,5,6,-тетрагидро-1-бензотиофен-2-ил)фенил]проп-2-еноата (0,1 г, 0,24 ммоль) в DCM (2 мл), добавляли CH2I2 (0,78 г, 2,9 ммоль) с последующим добавлением диэтилового цинка [1,45 мл (1М раствор в гептане), 1,4 ммоль] при 0°С. Реакционную смесь перемешивали при комнатной температуре до завершения реакции. Реакционную смесь затем гасили льдом и экстрагировали этилацетатом и промывали водой и рассолом, сушили над безводным Na2SO4 и концентрировали в вакууме. Полученный продукт очищали с помощью препаративной ТСХ с получением целевого соединения в виде бесцветного масла (0,015 г, выход: 19,9%).
Соединение 16: 2-[4-метокси-3-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)фенил]циклопропанкарбоновая кислота
В круглодонную колбу 25 мл, загруженную этил 2-[4-метокси-3-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)фенил]циклопропанкарбоксилатом (0,017 г, 0,04 ммоль) в ТГФ (1 мл), добавляли NaOH (0,008 г, 0,2 ммоль) в воде и этаноле (1 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении и промывали эфиром. Полученный остаток подкисляли 1N HCl и экстрагировали этилацетатом. Объединенный экстракт промывали водой, рассолом, и сушили над безводным Na2SO4 и концентрировали. Полученный продукт очищали с помощью препаративной ТСХ с получением целевого продукта в виде полутвердого вещества бледно-желтого цвета (0,0011 г, выход: 6,27%): МС (ESI, 120 эВ): m/z=399,2 (М+Н+).
Схема 9
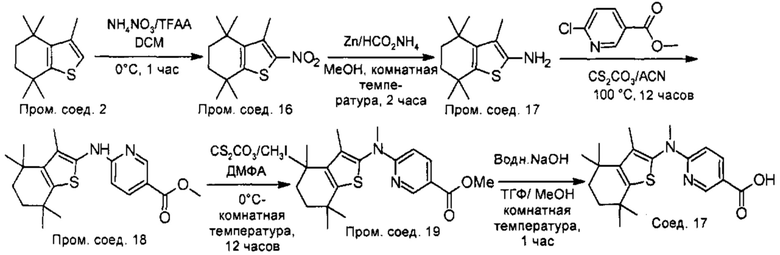
Соединение 17 синтезировали из 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-амина и метил 6-хлорпиридин-3-карбоксилата, следуя способу, описанному в схеме 9; чистота: 98,40%
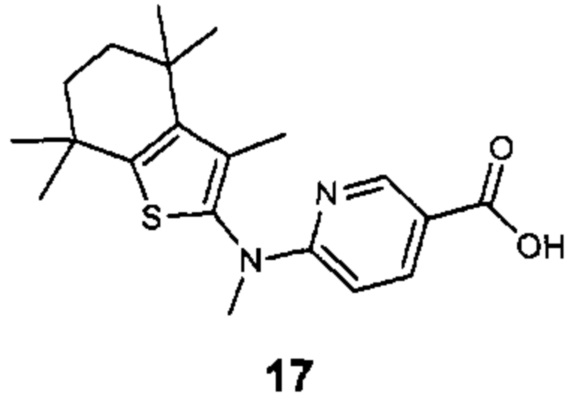
Промежуточное соединение 16: 3,4,4,7,7-пентаметил-2-нитро-4,5,6,7-тетрагидро-1-бензотиофен
К перемешиваемому раствору 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофена (4,8 г, 23,1 ммоль) в DCM (5 мл) добавляли NH4NO3 (2,22 г, 27,7 ммоль) и TFFA (14,5 г, 69,2 ммоль) по каплям при 0°С. Реакционную смесь выдерживали при температуре 0°С в течение 2 часов. Смесь затем гасили льдом и экстрагировали этилацетатом, промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме с получением неочищенного масла, которое очищали с помощью колоночной хроматографии на силикагеле с получением целевого соединения в виде желтой жидкости (1,5 г, выход: 20,44%).
Промежуточное соединение 17: 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-амин
К перемешиваемому раствору 3,4,4,7,7-пентаметил-2-нитро-4,5,6,7-тетрагидро-1-бензотиофена (1,6 г, 6,3 ммоль) в метаноле (15 мл) добавляли формиат аммония (1,99 г, 31,6 ммоль) с последующим добавлением цинковой пыли (2,0 г, 30,6 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении. Полученный остаток промывали водой и рассолом, сушили над безводным Na2SO4 и концентрировали в вакууме. Неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением целевого соединения в виде желтой жидкости (0,8 г, выход: 57,14%).
Промежуточное соединение 18: Метил 6-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]пиридин-3-карбоксилат
К перемешиваемому раствору 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-амина (0,25 г, 1,17 ммоль) в ацитонитриле (3 мл) добавляли метил 6-хлорпиридин-3-карбоксилат (0,1 г, 0,58 ммоль) и CS2CO3 (0,6 г, 1,74 ммоль). Реакционную смесь нагревали с обратным холодильником при 100°С в течение ночи. Реакционную смесь гасили водой и экстрагировали этилацетатом, промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме с получением неочищенного масла, которое очищали с помощью колоночной хроматографии на силикагеле с получением целевого соединения в виде твердого вещества желтого цвета (0,15 г, выход: 72,58%).
Промежуточное соединение 19: Метил 6-[метил(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]пиридин-3-карбоксилат
К перемешиваемому раствору NaH (0,011 г, 0,45 ммоль) в ДМФА (4 мл) добавляли метил 6-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]пиридин-3-карбоксилат (0,1 г, 0,28 ммоль) в ДМФА (4 мл) при 0°С. Через 30 мин добавляли CH3I (0,063 г, 0,45 ммоль) при 0°С. Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 1 часа. Реакционную смесь гасили льдом и экстрагировали этилацетатом, промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме с получением целевого соединения в виде желтой жидкости, соединение использовали в таком виде на следующем этапе (0,1 г, выход: 89,47%).
Соединение 17: 6-[метил (3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]пиридин-3-карбоновая кислота
К перемешиваемому раствору метил 6-[метил (3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино] пиридин-3-карбоксилат (0,1 г, 0,3 ммоль) в ТГФ (2 мл) добавляли метанол (2 мл) и воду (2 мл), NaOH (0,043 г, 1,0 ммоль) при 0°С. Затем смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали. Полученную таким образом соль промывали эфиром. Остаток растворяли в минимальном количестве воды, подкисляли 1,5 N HCl и экстрагировали этилацетатом, сушили и концентрировали с получением неочищенного продукта. Полученный продукт очищали с помощью препаративной ТСХ с получением целевого соединения в виде твердого вещества желтого цвета (0,02 г, выход: 18,6%). МС (ESI, 120 эВ): m/z=359,1 (М+Н+).
Пример 18: 6-[этил (3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]пиридин-3-карбоновая кислота
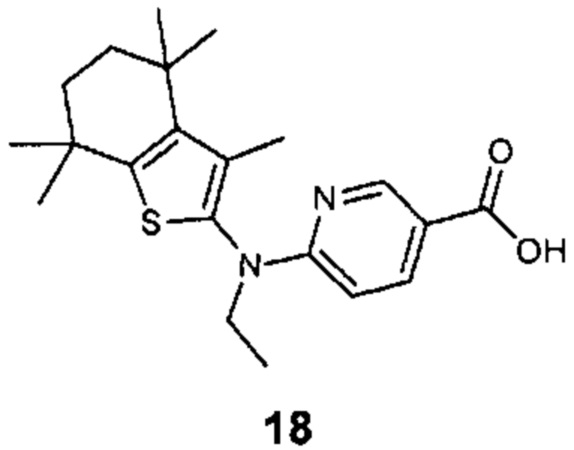
Соединение 18 синтезировали из 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-амина и метил 6-хлорпиридин-3-карбоксилата, следуя способу, аналогичному описанному в схеме 9 (0,025 г, выход: 21,57%); чистота: 94,38%
Пример 19: 6-[циклопропил (3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]пиридин-3-карбоновая кислота
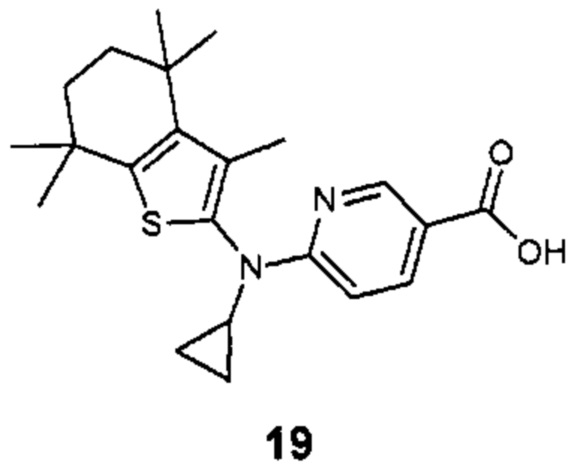
Соединение 19 синтезировали из 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-амина и метил 6-хлорпиридин-3-карбоксилата, следуя способу, аналогичному описанному в схеме 9. Далее, проводили N-алкилирование эфира (промежуточное соединение 18) циклопропилбороновой кислотой с помощью способа, аналогичного описанному для получения промежуточного соединения 11 в схеме 5 (0,109 г, выход: 52,02%); чистота: 98,35%
Пример 20: 6-[(циклопропилметил)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]пиридин-3-карбоновая кислота
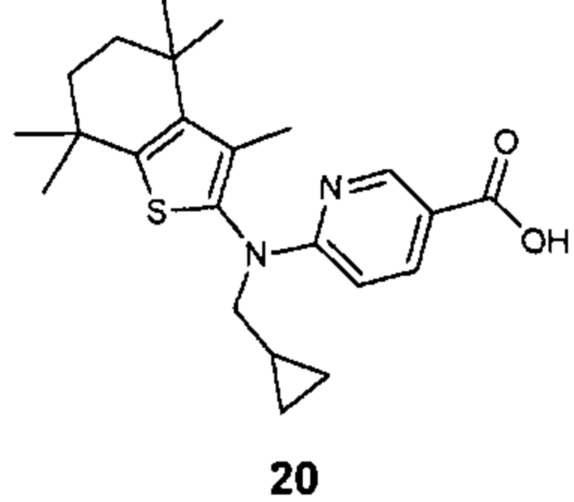
Соединение 20 синтезировали из 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-амина и метил 6-хлорпиридин-3-карбоксилата, следуя способу, аналогичному описанному в схеме 9 (0,025 г, выход: 17,85%); чистота: 98,96%
Пример 21: 6-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)(пропан-2-ил)амино]пиридин-3-карбоновая кислота
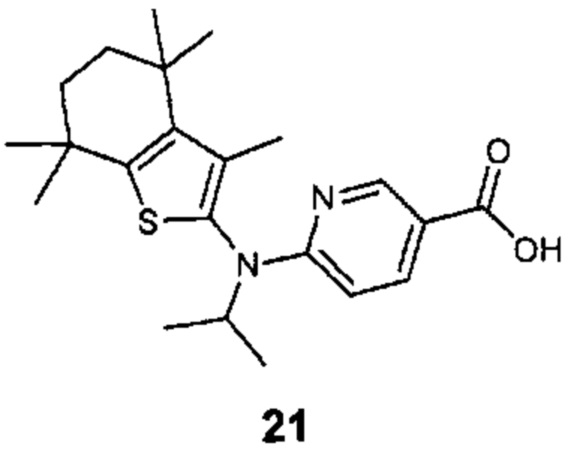
Соединение 21 синтезировали из 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-амина и метил 6-хлорпиридин-3-карбоксилата, следуя способу, аналогичному описанному в схеме 9 (0,0012 г, выход: 25,94%); чистота: 95,69%
Пример 22: 6-[(метилсульфонил)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]пиридин-3-карбоновая кислота
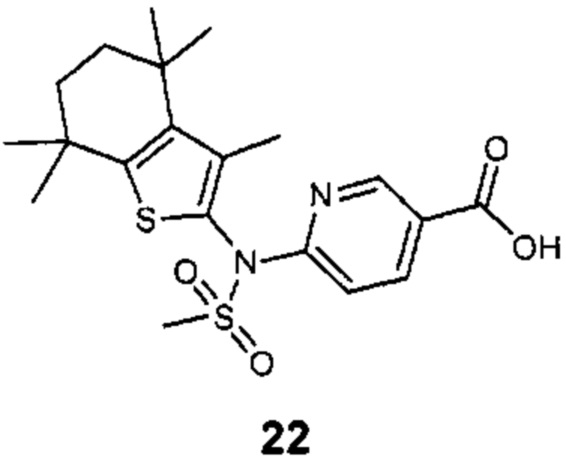
Соединение 22 синтезировали из 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-амина и метил 6-хлорпиридин-3-карбоксилата, следуя способу, аналогичному описанному в схеме 9 (0,02 г, выход: 64,52%); чистота: 98,13%: чистота: 98,13%
Схема 10

Пример 23: 2-[метил(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]пиримидин-5-карбоновая кислота
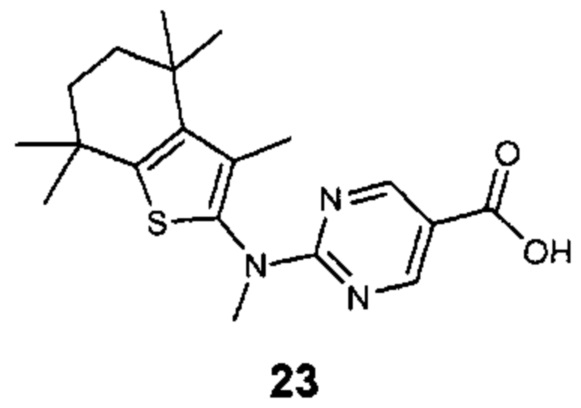
Соединение 23 синтезировали из 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-амина и этил 2-(метилсульфонил)пиримидин-5-карбоксилата, следуя способу, описанному в схеме 10; чистота: 90,31%
Промежуточное соединение 20: Этил 2-(метилсульфанил)пиримидин-5-карбоксилат
К перемешиваемому раствору этил 4-хлор-2-(метилсульфанил)пиримидин-5-карбоксилата (4,0 г, 17,2 ммоль) в этаноле в сосуд для гидрогенизации добавляли Na2СО3 (1,4 г, 16,7 ммоль) и 10% Pd-C (2,0 г) и дегазировали в течение примерно 10 мин. Реакционную смесь выдерживали в шейкере Парра для гидрогенизации (60 фунтов на квадратный дюйм, примерно 0,413685 МПа) в течение 48 часов. Реакционную смесь фильтровали через целит, промывали этанолом, концентрировали при пониженном давлении с получением целевого продукта в виде бледно-желтой жидкости (3,3 г. выход: 98,2 г, %). m/z=199,1 (М+Н)+.
Промежуточное соединение 21: Этил 2-(метилсульфонил) пиримидин-5-карбоксилат
К перемешиваемому раствору этил 2-(метилсульфанил) пиримидин-5-карбоксилата (3,3 г, 16,6 ммоль) в DCM (30 мл) добавляли по каплям m-СРВА (11,49 г, 66,5 ммоль) в DCM (30 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь гасили водой и экстрагировали этилацетатом, промывали насыщенным раствором NaНСО3, водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме с получением продукта в виде почти белого твердого вещества. Продукт использовали в таком виде для следующего этапа (2,0 г, выход: 52,0%); 1Н ЯМР (300 МГц, ДМСО-d6): δ 9,48 (s, 2Н), 4,39-4,46 (q, 2Н), 3,48 (s, 3Н), 1,33-1,39 (t, 3Н).
Промежуточное соединение 22: Этил 2-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]пиримидин-5-карбоксилат
К перемешиваемому раствору NaH (0,31 г, 12,5 ммоль) в ТГФ (10,0 мл) добавляли 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-амин (2,0 г, 8,6 ммоль) и этил-2-(метилсульфонил)пиримидин-5-карбоксилат (1,93 г, 8,6 ммоль) в ТГФ (10 мл) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь гасили водой и экстрагировали этилацетатом, промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме с получением неочищенного продукта в виде жидкости оранжевого цвета. Продукт использовали в таком виде для следующего этапа (1,4 г, выход: 44,5%); m/z=374,2 (М+Н)+.
Промежуточное соединение 23: Этил 2-[метил (3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]пиримидин-5-карбоксилат
К перемешиваемому раствору NaH (0,11 г, 4,6 ммоль) в ТГФ (3 мл), медленно добавляли этил 2-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино] пиримидин-5-карбоксилат (0,7 г, 1,87 ммоль) в ТГФ при 0°С с последующим добавлением CH3I (0,43 г, 2,9 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь гасили льдом и экстрагировали этилацетатом, промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме с получением неочищенного продукта в виде жидкости оранжевого цвета.
Продукт использовали в таком виде для следующего этапа (0,04 г, выход: 5,4%); m/z=388,3 (М+Н)+.
Соединение 23: 2-[метил(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]пиримидин-5-карбоновая кислота
К перемешиваемому раствору этил 2-[метил (3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]пиримидин-5-карбоксилата (0,007 г, 0,018 ммоль) в ТГФ (1 мл) добавляли водный раствор LiOH (0,0017 г, 0,074 ммоль) и метанол (1 мл) и раствор перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали; полученную соль промывали эфиром. Остаток растворяли в минимальном количестве воды, подкисляли 1,5 N HCl и экстрагировали этилацетатом, сушили и концентрировали с получением целевого продукта в виде твердого вещества белого цвета (0,002 г, выход: 30,93%). МС (ESI, 120 эВ): m/z=360,1 (М+Н+).
Пример 24: 2-[циклопропил (3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]пиримидин-5-карбоновая кислота
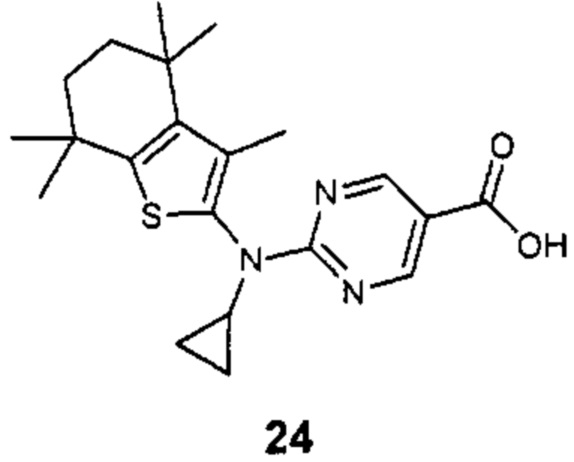
Соединение 24 синтезировали из 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-амина и этил 2-(метилсульфонил)пиримидин-5-карбоксилата, следуя способу, аналогичному описанному в схеме 10. Далее проводили N-алкилирование эфира (промежуточное соединение 22) циклопропилбороновой кислотой с помощью способа, аналогичного описанному для получения промежуточного соединения 11 в схеме 5 (0,0035 г, выход: 15,05%); чистота: 95,10%.
Схема 11
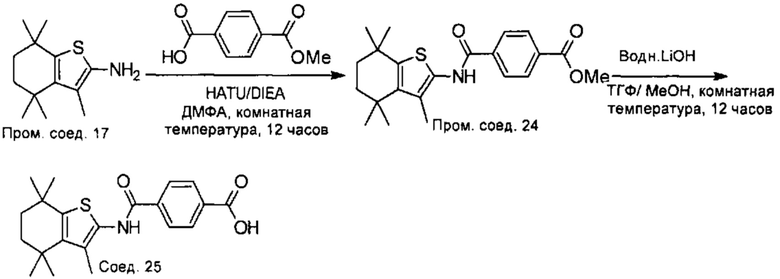
Пример 25: 4-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)карбамоил]бензойная кислота
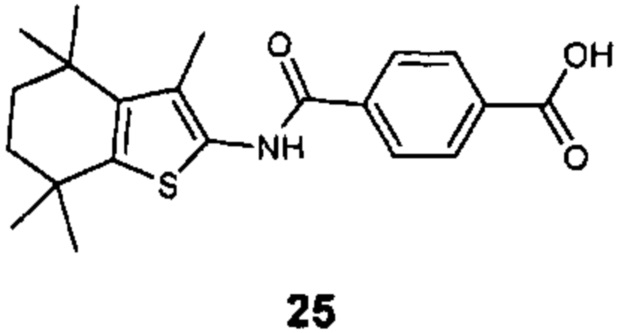
Соединение 25 синтезировали из 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-амина и 4-(метилсульфонил)бензойной кислоты, следуя способу, описанному в схеме 11; чистота: 98,24%
Промежуточное соединение 24: Метил 4-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)карбамоил]бензоат
К перемешиваемому раствору 4-(метоксикарбонил)бензойной кислоты (0,16 г, 0,9 ммоль) в ДМФА (3 мл) добавляли 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-амин (0,2 г, 0,9 ммоль) и HATU (0,41 г, 1,1 ммоль) в атмосфере N2. Затем добавляли DIEA (0,69 г, 5,4 ммоль) при 0°С. Реакционную смесь оставляли перемешиваться при комнатной температуре в течение ночи. Реакционную смесь гасили льдом и экстрагировали этилацетатом, промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии с получением целевого соединения в виде твердого вещества желтого цвета (0,4 г, выход: 11,53%). 1Н ЯМР (300 МГц, ДМСО-d6): δ 10,21 (s, 1Н), 8,02-8,10 (m, 4Н), 3,90 (s, 3Н), 2,14 (s, 3Н), 1,66 (s, 4Н), 1,23-1,26 (d, 12Н).
Соединение 25: 4-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)карбамоил]бензойная кислота
К перемешиваемому раствору метил 4-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)карбамоил]бензоата (0,04 г, 0,1 ммоль) в ТГФ (1 мл) добавляли метанол (1 мл) и воду (1 мл), LiOH (0,025 г, 1,0 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь гасили разбавленной HCl. Указанный водный раствор экстрагировали при помощи этилацетата и органические экстракты объединяли, промывали водой и рассолом, сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное твердое вещество дополнительно очищали с помощью препаративной ТСХ с получением целевого соединения в виде твердого вещества желтого цвета (0,009 г, выход: 2,42%): МС (ESI, 120 эВ): m/z=372,2 (М+Н+).
Схема 12
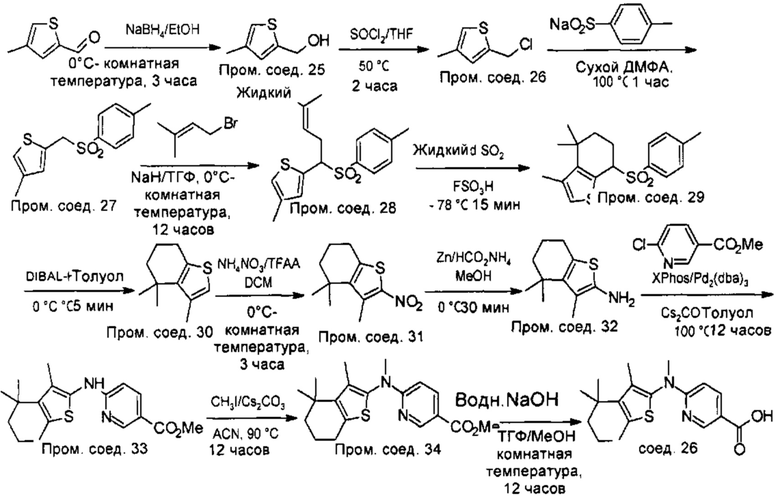
Пример 26: 6-[метил (3,4,4-триметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]пиридин-3-карбоновая кислота
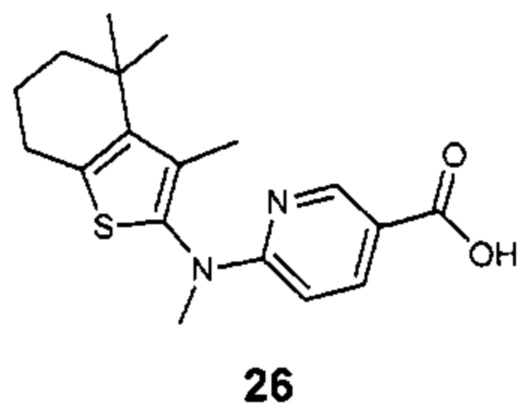
Соединение 26 синтезировали из 3,4,4-триметил-4,5,6,7-тетрагидро-1-бензотиофен-2-амина и метил 6-хлорпиридин-3-карбоксилата, следуя способу в схеме 12; чистота: 92,31%
Промежуточное соединение 25: (4-метилтиофен-2-ил) метанол
К перемешиваемому раствору 4-метилтиофен-2-карбальдегида (6,0 г, 47,6 ммоль) в этаноле (15 мл) добавляли NaBH4 (2,1 г, 57,0 ммоль) при 0°С в атмосфере N2 и продолжали перемешивать при комнатной температуре в течение 12 ч. Реакционную смесь гасили льдом и экстрагировали этилацетатом, промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме с получением целевого продукта в виде жидкости оранжевого цвета (5,2 г, выход: 85,2%). 1Н ЯМР (300 МГц, ДМСО-d6): δ 6,96 (s, 1Н), 6,76 (s, 1Н), 5,34-5,38 (t, 1Н), 4,55-4,56 (d, 2Н), 2,16 (t, 3Н).
Промежуточное соединение 26: 2-(хлорметил)-4-метилтиофен
К перемешиваемому раствору (4-метилтиофен-2-ил) метанола (5,2 г, 40,5 ммоль) в ТГФ (50 мл) добавляли SOCl2 (5,78 г, 48,6 ммоль) и нагревали при 50°С в течение 3 часов. Реакционную смесь гасили льдом и экстрагировали этилацетатом, промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме с получением целевого продукта в виде жидкости светло-коричневого цвета (5,0 г, выход: 83,9%). 1Н ЯМР (300 МГц, ДМСО-d6): δ 7,14 (s, 1Н), 7,00 (s, 1Н), 4,97 (s 2Н), 2,16 (t, 3Н).
Промежуточное соединение 27: 4-метил-2-{[(4-метилфенил)сульфонил]метил}тиофен
К перемешиваемому раствору 2-(хлорметил)-4-метилтиофен (5,0 г, 33,3 ммоль) в ДМФАА (50 мл) добавляли п-толуол сульфинат (5,9 г, 33,3 ммоль). Реакционную смесь нагревали при температуре 100°С в течение 2 часов. Реакционную смесь гасили водой и экстрагировали диэтиловым эфиром, затем промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме с получением неочищенного продукта. Полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле, с использованием петролейного эфира (60-80) и этилацетата в качестве элюента с получением целевого продукта в виде твердого вещества почти белого цвета (5,2 г, выход: 58,9%): 1Н ЯМР (300 МГц, CDCl3): δ 7,52-7,55 (d, 2Н), 7,19-7,22 (d, 2Н), 6,76 (s, 1Н), 6,65 (s, 1Н), 4,36 (s, 2Н), 2,36 (s, 3Н), 2,11 (s, 3Н).
Промежуточное соединение 28: 4-Метил-1-(4-метилтиофен-2-ил)пент-3-ен-1-ил 4-метилфенилсульфон
К суспензии NaH (0,16 г, 6,8 ммоль) в ТГФ (3 мл) добавляли 4-метил-2-{[(4-метилфенил)сульфонил]метил}тиофен (0,9 г, 3,4 ммоль) в ТГФ (3 мл) и полученную массу перемешивали при температуре 0°С в течение 30 мин. К полученному выше раствору медленно добавляли 1-бром-3-метилбут-2-ен (0,51 г, 3,4 ммоль) в ТГФ (3 мл) при 0°С и полученную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь гасили водой и экстрагировали этилацетатом, промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали. Полученный неочищенный продукт очищали с помощью CombiFlash с получением целевого соединения в виде твердого вещества белого цвета (1,0 г, выход: 88,0%).
Промежуточное соединение 29: 3,4,4-триметил-7-[(4-метилфенил)сульфонил]-4,5,б,7-тетрагидро-1-бензотиофен
К раствору 4-метил-1- (4-метилтиофен-2-ил)пент-3-ен-1-ил-4-метилфенил сульфона (2,5 г, 7,23 моль) в жидком SO2 (40 мл) добавляли FSO3H (0,4 мл) при -78°С и перемешивали при той же температуре в течение 15 мин. Реакционную смесь гасили CH3ONA, затем нейтрализовали раствором NaOH и экстрагировали диэтиловым эфиром. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали. Полученный неочищенный продукт очищали с помощью системы очистки CombiFlash с получением целевого соединения в виде бесцветного вязкого материала (1,5 г, выход: 60,0%).
Промежуточное соединение 30: 3,4,4-триметил-4,5,6,7-тетрагидро-1-бензотиофен
К раствору 3,4,4-триметил-7-[(4-метилфенил)сульфонил]-4,5,6,7-тетрагидро-1-бензотиофена (1,5 г, 4,4 ммоль) в толуоле (10 мл) одной порцией добавляли DIBAL-H (6,5 мл, 1М р-р. 1,5 экв) при комнатной температуре. Затем смесь перемешивали при 0°С в течение 5 минут и осторожно гасили этанолом (0,5 мл), водой (1,0 мл) и концентрированным HCl (0,5 мл). Реакционную смесь экстрагировали диэтиловым эфиром, промывали водным раствором NaOH, водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали. Полученный неочищенный продукт очищали с помощью CombiFlash с получением целевого продукта в виде маслянистого продукта (0,5 г, выход: 61,2%): 1Н ЯМР (300 МГц, CDCl3): δ 6,64 (s, 1Н), 2,70-2,74 (m, 2Н), 2,31 (s, 3Н), 1,78-1,86 (m, 2Н), 1,59-1,63 (m, 2Н), 1,28 (s, 6Н).
Промежуточное соединение 31: 3,4,4-триметил-2-нитро-4,5,6,7-тетрагидро-1-бензотиофен
К раствору 3,4,4-триметил-4,5,6,7-тетрагидро-1-бензотиофена (0,25 г, 1,4 ммоль) в DCM (2 мл) добавляли NH4NO3 (0,13 г, 1,62 ммоль) при 0°С с последующим медленным добавлением TFAA (0,58 г, 2,8 ммоль). Полученный раствор перемешивали при 0°С в течение 30 мин с последующим перемешиванием при комнатной температуре в течение 3 часов. Реакционную смесь затем гасили ледяной водой и экстрагировали с DCM. Органический слой, полученный таким образом, промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали с получением черной массы. Полученную массу очищали с помощью препаративной ТСХ с получением целевого соединения в виде бесцветного маслянистого продукта (0,07 г, выход: 22,4%): 1Н ЯМР (300 МГц, CDCl3): δ 2,71-2,75 (m, 2Н), 2,69 (s, 3Н), 1,79-1,87 (m, 2Н), 1,63-1,67 (m, 2Н), 1,33 (s, 6Н).
Промежуточное соединение 32: 3,4,4-триметил-4,5,6,7-тетрагидро-1-бензотиофен-2-амин
К раствору 3,4,4-триметил-2-нитро-4,5,6,7-тетрагидро-1-бензотиофена (0,14 г, 0,62 ммоль) в метаноле (3 мл) добавляли формиат аммония (0,19 г, 3 ммоль) и цинковую пыль (0,2 г, 3,0 ммоль) и перемешивали при 0°С в течение 30 мин. Реакционную смесь гасили водой и затем фильтровали через целит. Метанол удаляли в вакууме и неочищенный продукт разбавляли этилацетатом, органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения в виде коричневого маслянистого продукта (0,1 г, выход: 82,3%); МС (ESI, 120 эВ): m/z=196,1 (М+Н+).
Промежуточное соединение 33: Метил 6-[(3,4,4-триметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]пиридин-3-карбоксилат
Пробирку для проведения реакций под давлением объемом 20 мл с завинчивающейся крышкой загружали 3,4,4-триметил-4,5,6,7-тетрагидро-1-бензотиофен-2-амином (0,1 г, 0,5 ммоль), метил-6-хлорпиридин-3-карбоксилатом (0,043 г, 0,25 ммоль), CS2CO3 (0,325 г, 1,0 ммоль), толуолом и ксантфосом (0,014 г, 0,025 ммоль). Через реакционную массу в течение 15 мин продували аргон с последующим добавлением Рd2 (dba)3 (0,022 г, 0,025 ммоль) и реакционную смесь затем нагревали при 100°С в течение ночи. Реакционную массу разбавляли водой и экстрагировали этилацетатом, органический слой и промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали. Полученный неочищенный продукт очищали с помощью системы очистки CombiFlash с получением целевого соединения в виде коричневого маслянистого продукта (0,5 г, выход: 24,2%); МС (ESI, 120 эВ): m/z=331,1 (М+Н+).
Промежуточное соединение 34: Метил 6-[метил(3,4,4-триметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]пиридин-3-карбоксилат
В пробирку для проведения реакций под давлением объемом 20 мл с завинчивающейся крышкой загружали метил 6-[(3,4,4-триметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]пиридин-3-карбоксилат (0,04 г, 0,12 ммоль), CS2CO3 (0,078 г, 0,24 ммоль), CH3l (0,03 г, 0,24 ммоль) и ДМФА (2 мл) и полученную смесь нагревали при 80°С в течение ночи. Реакционную массу разбавляли водой и экстрагировали диэтиловым эфиром. Органический слой, полученный таким образом, промывали водой и рассолом, сушили над Na2SO4 и концентрировали. Неочищенную массу использовали в таком виде на следующем этапе (0,05 г); МС (ESI, 120 эВ): m/z=345,0 (М+Н)+.
Соединение 26: 6-[метил(3,4,4-триметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]пиридин-3-карбоновая кислота
Круглодонную колбу объемом 25 мл, снабженную магнитной мешалкой, загружали метанолом (1 мл) и ТГФ (2 мл). К перемешиваемому растворителю добавляли метил 6-метил (3,4,4-триметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]пиридин-3-карбоксилат (0,05 г, 0,14 ммоль) и вод. NaOH (0,011 г, 0,28 ммоль) и колбу перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь полностью концентрировали и неочищенный продукт промывали эфиром и разбавляли водой, нейтрализовали лимонной кислотой и экстрагировали этилацетатом. Органический слой, полученный таким образом, промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали. Полученный продукт очищали с помощью препаративной ТСХ с получением целевого продукта в виде бесцветного твердого вещества (0,01 г, выход: 30,3%): МС (ESI, 120 эВ): m/z=331,1 (М+Н+).
Схема 13
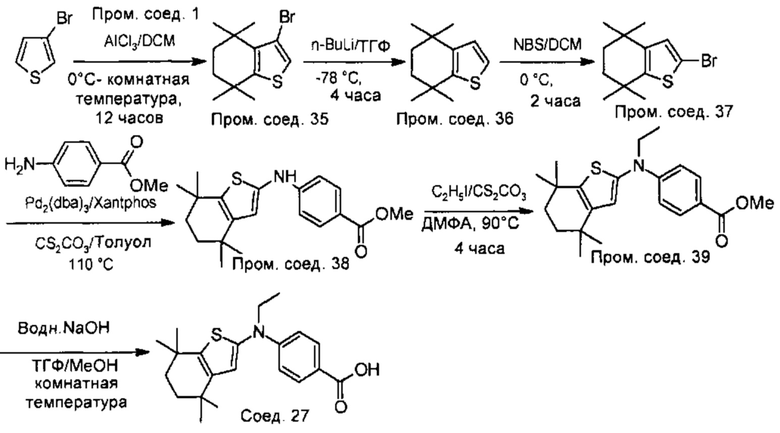
Пример 27: 4-[этил (4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензойная кислота
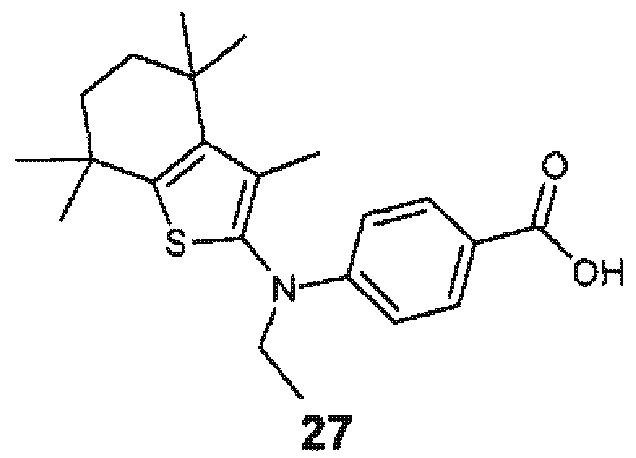
Соединение 27 синтезировали из 2-бром-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1-бензотиофена и метил-4-аминобензоата, следуя способу, описанному в схеме 13; чистота: 96,71%.
Промежуточное соединение 35: 3-бром-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1-бензотиофен
К суспензии AlCl3 (12,2 г, 92,0 ммоль) в DCM (100 мл) при 0°С медленно добавляли раствор 2,5-дихлор-2,5-диметилгексана (16,8 г, 92,0 ммоль) и 3-бромтиофен (15,0 г, 92,0 ммоль) в DCM (50 мл) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь выливали в химический стакан, содержащий лед и экстрагировали с DCM. Комбинированный органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали с получением черной массы. Полученный продукт очищали с помощью колоночной хроматографии на силикагеле (230-400 меш) с получением соединения в виде бледно-желтого маслянистого продукта (8,0 г, выход: 31,84%). 1Н ЯМР (300 МГц, CDCl3): δ 6,99 (s, 1Н), 1,62 (s, 4Н), 1,34 (s, 6Н), 1,23 (s, 6Н).
Промежуточное соединение 36: 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1-бензотиофен
К перемешиваемому раствору 3-бром-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1-бензотиофена (4,0 г, 14,0 моль. 0,029 ммоль) добавляли н-бутиллитий (18,0 мл. 1,6М в гексане) при -78°С в атмосфере N2 и перемешивали при той же температуре в течение 4 часов. Реакционную смесь гасили насыщенным раствором NH4Cl и экстрагировали диэтиловым эфиром. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали. Полученный продукт очищали с помощью колоночной хроматографии на силикагеле (230-400 меш) с получением целевого соединения в виде бледно-желтого маслянистого продукта (8,0 г, выход: 88,0%). 1Н ЯМР (300 МГц, CDCl3): δ 6,98-6,99 (d, 1Н), 6,76-6,78 (d, 1Н), 1,59-1,67 (m, 4Н), 1,24 (s, 6Н), 1,15 (s, 6Н).
Промежуточное соединение 37: 2-бром-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1-бензотиофен
К раствору 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1-бензотиофена (0,5 г, 2,5 ммоль) в DCM (5 мл) порциями добавляли NBS (0,5 г, 2,8 ммоль) при 0°С и перемешивали при той же температуре в течение 3 часов. Реакционную смесь гасили водой и экстрагировали с DCM. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали. Неочищенный материал очищали с помощью системы очистки CombiFlash с получением бесцветного маслянистого продукта (0,55 г, выход: 76,9%). 1Н ЯМР (300 МГц, CDCl3): δ 6,69 (s, 1Н), 1,55-1,64 (m, 4Н), 1,20 (s, 6Н), 1,11 (s, 6Н).
Промежуточное соединение 38: метил 4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоат
Пробирку для проведения реакций под давлением объемом 25 мл с завинчивающейся крышкой загружали 2-бром-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1-бензотиофеном (0,55 г, 2,0 ммоль), метил-4-аминобензатом (0,61 г, 4,0 ммоль), CS2CO3 (1,3 г, 4,0 ммоль), толуолом (10 мл) и XPhos (0,06 г, 0,1 ммоль). Через реакционную массу в течение 15 мин продували аргон с последующим добавлением Pd2 (dba)3 (0,036 г, 0,04 ммоль) и указанную массу, полученную таким образом нагревали при 100°С в течение ночи. Реакционную массу разбавляли водой и экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали, затем очищали с помощью системы очистки CombiFlash с получением твердого вещества бледно-желтого цвета (0,17 г, выход: 24,5%); МС (ESI, 120 эВ): m/z=344,1 (М+Н).
Промежуточное соединение 39: метил 4-[этил(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоат
Пробирку для проведения реакций под давлением объемом 20 мл загружали метил 4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоатом (0,17 г, 0,5 ммоль), CS2CO3 (0,48 г, 1,5 ммоль) и ДМФА (3 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. с последующим добавлением C2H5l (0,11 г, 0,74 ммоль) и нагревали при 90°С в течение 4 часов. Реакционную смесь разбавляли водой и экстрагировали диэтиловым эфиром. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали до коричневого масла, затем очищали с помощью системы очистки CombiFlash с получением твердого вещества бледно-желтого цвета (0,17 г, выход: 59,8%). 1Н ЯМР (300 МГц, CDCl3): 6 7,77-7,80 (d, 2Н), 6,65-6,68 (d, 2Н), 6,44 (s, 1Н), 3,78 (s, 3Н), 3,61-3,68 (q, 2Н), 1,60-1,68 (m, 4Н), 1,24 (s, 6Н), 1,18-1,20 (t, 3Н), 1,13 (s, 6Н).
Соединение 27: 4-[этил(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензойная кислота
Круглодонную колбу объемом 25 мл, снабженную магнитной мешалкой, загружали метанолом (1 мл) и ТГФ (2 мл). К перемешиваемому растворителю добавляли метил 4-[этил (4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоат (0,11 г, 0,3 ммоль) и водный NaOH (0,023 г, 0,6 ммоль) и перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь концентрировали; полученный неочищенный продукт промывали эфиром и разбавляют водой и затем нейтрализовали с помощью 1N HCl. Экстрагировали этилацетатом, промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали. Полученное твердое вещество дополнительно очищали с помощью препаративной ТСХ с получением целевого соединения в виде твердого вещества желтого цвета (0,05 г, выход: 50,0%); МС (ESI, 120 эВ): m/z=358,2 (М+Н+).
Схема 14
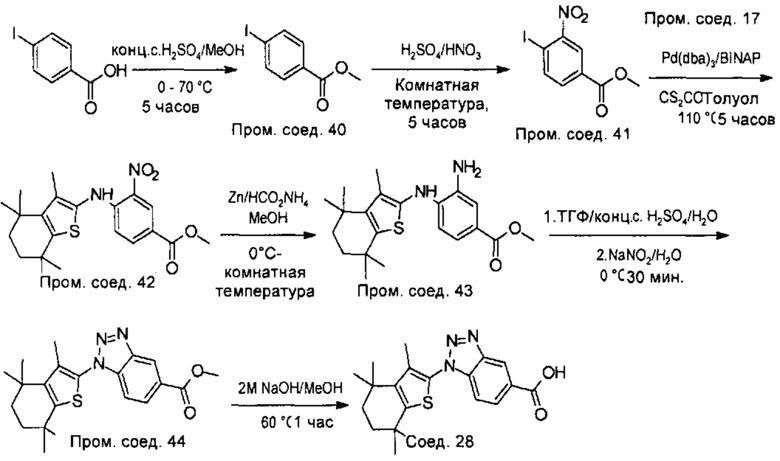
Пример 28: 1-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)-1H-бензотриазол-5-карбоновая кислота
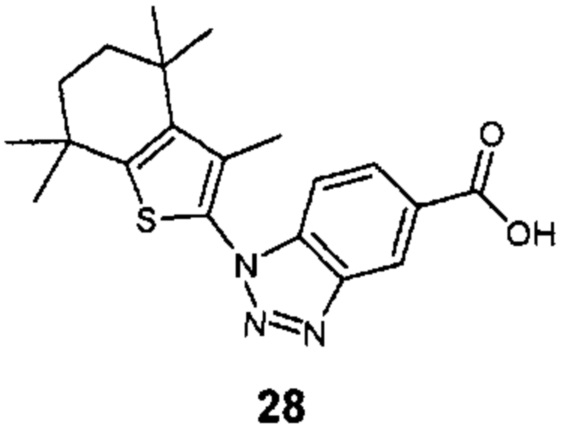
Соединение 28 синтезировали из 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-6ензотиофен-2-амина и метил 4-иод-3-нитробензоата, следуя способу, описанному в схеме 14; чистота: 86,18%.
Промежуточное соединение 40: метил 4-иодбензоата
К раствору 4-иодбензойной кислоты (8,0 г, 32,2 ммоль) в МеОН (80 мл) добавляли H2SO4 (2,0 мл) при 0°С. Реакционную смесь нагревали с обратным холодильником при 70°С в течение 5h. Растворитель удаляли в вакууме, полученный остаток растворяли в этилацетате, промывали насыщенным раствором NaHCO3 и рассолом, сушили над безводным Na2SO4 и концентрировали. Очищали с помощью кристаллизации со смесью DCM/н-гексана с выходом целевого соединения в виде твердого вещества белого цвета (8,0 г, выход: 94,0%): 1Н ЯМР (300 МГц, CDCl3): δ 7,73-7,82 (m, 2Н), 3,91 (s, 3Н).
Промежуточное соединение 41: Метил-4-иод-3-нитробензоат
К охлажденному льдом раствору метил 4-иодбензоата (8,0 г, 30,5 ммоль) в концентрированного H2SO4 (57 мл) добавляли по каплям раствор концентрированного HNO3 (45 мл) и концентрированного H2SO4 (38,0 мл). Реакционную смесь перемешивали при комнатной температуры в течение 5 ч, затем гасили льдом и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором NaHCO3 и рассолом (50 мл), сушили над безводным Na2SO4 и выпаривали при пониженном давлении. Остаток перекристаллизовывали из DCM/н-гексана с получением целевого соединения в виде твердого вещества желтого цвета (7,0 г, выход: 74,71%).
Промежуточное соединение 42: Метил-4-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоат
К раствору метил 4-иод-3-нитробензоата (1,0 г, 3,26 ммоль) и промежуточного соединения 17 (1,5 г, 6,5 ммоль) в сухом толуоле (20 мл), в инертной атмосфере добавляли Pd2 (dba)3 (0,15 г, 0,16 ммоль), ксантфос (0,16 г, 0,33 ммоль) и Cs2CO3 (3,2 г, 9,8 ммоль). Смесь нагревали с обратным холодильником при 110°С в атмосфере аргона в течение 5 ч и затем фильтровали через целит. Фильтрат промывали водой и рассолом, сушили над безводным Na2SO4 и выпаривали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии и кристаллизовали из н-гексана с получением целевого соединения в виде жидкий продукта красного цвета (0,9 г, выход: 68,7%).
Промежуточное соединение 43: Метил 3-амино-4-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоат
К раствору метил 3-нитро-4-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоата (0,9 г, 2,2 ммоль) в метаноле (10 мл) добавляли цинковую пыль (0,71 г, 11,2 ммоль) при 0°С. Смесь перемешивали при комнатной температуре до завершения реакции и затем фильтровали через целит. Фильтрат выпаривали при пониженном давлении. Остаток экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением целевого соединения в виде рыхлого твердого вещества желтого цвета (0,8 г, выход: 96,39%).
Промежуточное соединение 44: Метил 1-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)-1Н-бензотриазол-5-карбоксилат
К охлажденному льдом раствору метил 3-амино-4-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоата (0,4 г, 1,08 ммоль) в ТГФ (5,0 мл) добавляли смесь концентрированного H2SO4 (2,0 мл) в Н2O (20 мл) с последующим добавлением по каплям раствора NaNO2 (0,12 г, 1,73 ммоль) в воде (2,0 мл). Смесь перемешивали при 0°С в течение 30 мин, затем выливали в воду и экстрагировали этилацетатом. Органический слой промывали водой и рассолом (30 мл), сушили над безводным Na2SO4 и выпаривали при пониженном давлении. Остаток очищали с помощью колоночной флэш-хроматографии с получением целевого соединения в виде твердого вещества бледно-желтого цвета (0,1 г, выход: 24,33%): МС (ESI, 120 эВ): m/z=(М+Н)+; 1Н ЯМР (300 МГц, CDCl3): 8,85 (s, 1Н), 8,21-8,24 (d, 1Н), 7,50-7,53 (d, 1Н), 4,00 (s, 3Н), 2,04 (s, 3Н), 1,77 (s, 4Н), 1,36-1,37 (d, 12Н).
Соединение 28: 1-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)-1H-бензотриазол-5-карбоновая кислота
К раствору метил 1-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)-1H-бензотриазол-5-карбоксилат (0,1 мг, 0,26 ммоль) в МеОН (2 мл) и ТГФ (2 мл) добавляли водн. NaOH (0,052 г, 1,3 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часов. Растворители удаляли при пониженном давлении полностью; полученный остаток растворяли в минимальном количестве воды, подкисляли до рН 3 при охлаждении, экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное твердое вещество дополнительно очищали растиранием с гексаном с получением целевого соединения в виде твердого вещества белого цвета (0,025 г, выход: 26,04%): МС (ESI, 120 эВ): m/z=370,1 (М+Н+).
Схема 15
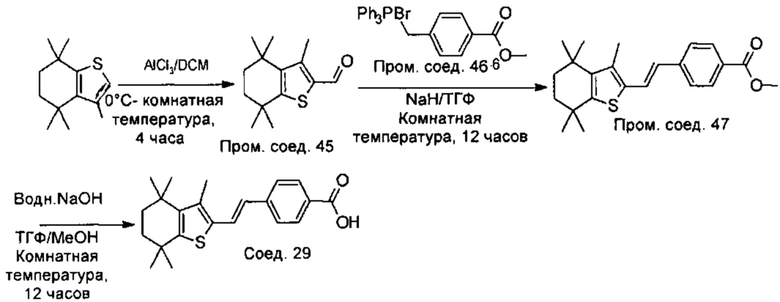
Пример 29: 4-(E)[-2-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)этенил]бензойная кислота
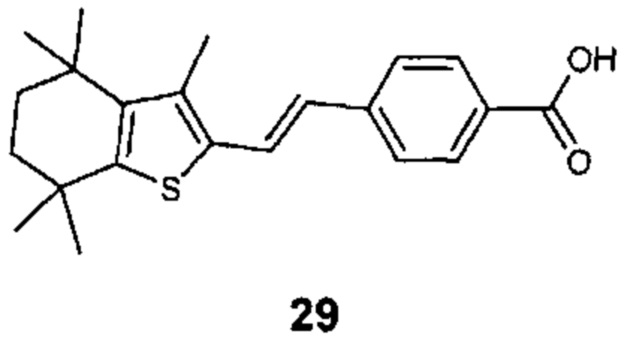
Соединение 29 синтезировали из 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-карбальдегида и метил 4-{[бром (трифенил)-l5-фосфанил]метил}бензоата, следуя способу, описанному в схеме 15; чистота: 97,20%.
Промежуточное соединение 45: 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-карбальдегид
В круглодонную колбу с двумя горлышками объемом 250 мл, загруженную AlCl3 (1,18 г, 5,6 ммоль) и DCM (60 мл), при перемешивании добавляли 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен (0,96 г, 5,6 ммоль) и дихлорметиловый эфир (0,5 мл) при 0°С и содержимое колбы продолжали перемешивать при комнатной температуре в течение 4 часов. Реакционную смесь доводили до комнатной температуры, гасили льдом и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением целевого соединения в виде твердого вещества белого цвета (0,6 г, выход: 47,0%): МС (ESI, 120 эВ): m/z=237,1 (М+Н+). 1Н ЯМР (300 МГц, CDCl3): δ 9,94 (s, 1Н), 2,55 (s, 3Н), 1,62 (s, 4Н), 1,26-1,27 (s, 12Н).
Промежуточное соединение 46: Метил 4-{[бром(трифенил)-λ5-phosphanyl]метил}бензоат
К перемешиваемому раствору метил 4-(бромметил)бензоата (1,0 г, 4,3 ммоль) в ацетонитриле (10 мл) добавляли PPh3 (1,71 г, 6,5 ммоль) и нагревали при 60°С в течение 2 часов. Реакционную смесь концентрировали. Полученный остаток промывали водой и рассолом и сушили над безводным Na2SO4 и концентрировали. Полученный продукт очищали с помощью кристаллизации с толуолом получая целевое соединение в виде твердого вещества белого цвета (1,5 г, выход: 70,4%).
Промежуточное соединение 47: Метил 4-[(E)-2-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)этенил]бензоат
К перемешиваемому раствору NaH (0,067 г, 2,6 ммоль) в ТГФ (3 мл) медленно добавляли метил 4-{[бром(трифенил)-λ5-фосфанил]метил}бензоат (0,81 г, 1,65 ммоль) при 0°С в атмосфере N2 и продолжали перемешивание при той же температуре в течение 45 мин. К полученному выше раствору добавляли 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-карбальдегида (0,3 г, 1,2 ммоль) в ТГФ (3 мл) и раствор продолжали перемешивать при комнатной температуре в течение 12 ч. Реакционную смесь затем гасили водой и экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над безводным Na2SO4 и концентрировали в вакууме. Полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением целевого соединения в виде твердого вещества желтого цвета (1,0 г, выход: 32,0%): МС (ESI, 120 эВ): m/z=369,1 (М+Н+).
Соединение 29: 4-(E)[-2-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)этенил]бензойная кислота
К перемешиваемому раствору метил 4-[(Е)-2-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)этенил]бензоата (0,05 г, 0,13 ммоль) и ТГФ (2 мл) добавляли водный раствор NaOH (0,0065 г, 0,27 ммоль). Смесь перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь концентрировали и остаток растворяли в минимальном количестве воды, подкисляли до рН 3 при охлаждении, экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над безводным Na2SO4 и концентрировали в вакууме. Полученное твердое вещество дополнительно очищали с помощью препаративной ТСХ с получением целевого соединения в виде твердого вещества желтого цвета (0,004 г, выход: 8,31%): МС (ESI, 120 эВ): m/z=353,0 (М-Н)+.
Пример 30: 4-[(Z)-2-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)этенил]бензойная кислота
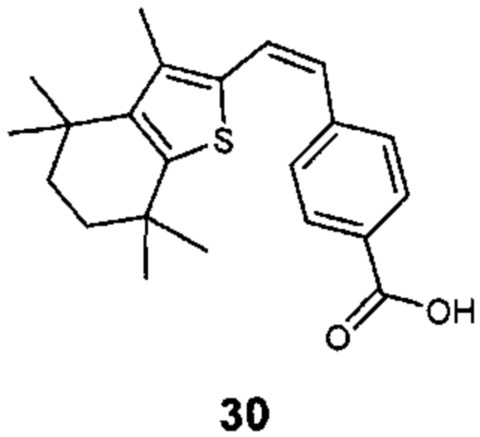
Соединение 30 синтезировали из 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-карбальдегида и метил 4-{[бром (трифенил)-15-фосфанил]метил}бензоата, следуя способу, описанному в схеме 15 (0,0028 г, выход: 5,82%); чистота: 98,45%.
Пример 31: 4-[2-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)циклопропил]бензойная кислота
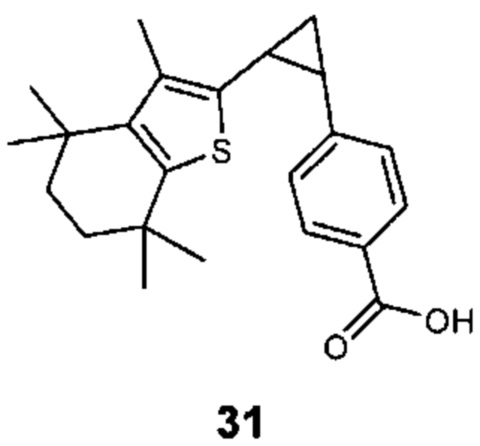
Соединение 31 синтезировали из 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-карбальдегида и метил 4-{[бром (трифенил)-15-фосфанил]метил}бензоата, следуя способу, описанному в схеме 15. Затем циклопропанирование проводили в соответствии с таким же способом, как описано для промежуточного соединения 6 в схеме 2 с последующим гидролизом с получением целевого продукта (0,004 г, выход: 4,17%); чистота: 96,84%.
Схема 16
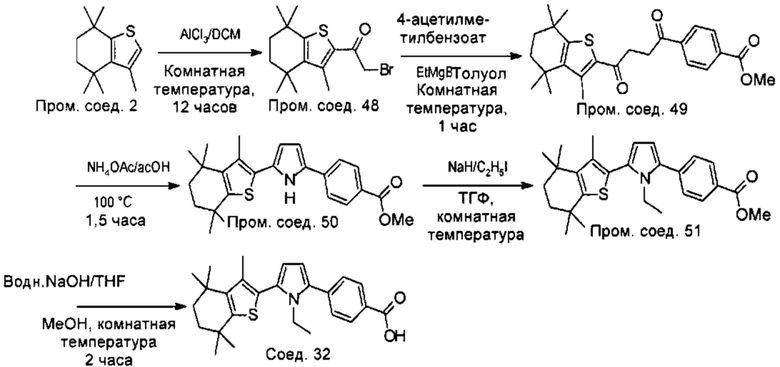
Пример 32: 4-[1-Этил-5-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)-1H-пиррол-2-ил]бензойная кислота
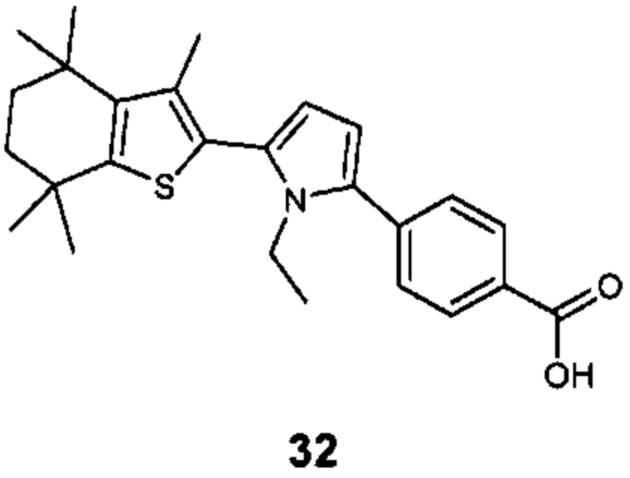
Соединение 32 синтезировали из 2-бром-1-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофена-2-ил)-этанона и 4-ацетилметилбензоата, следуя способу, описанному в схеме 16; чистота: 96,40%.
Промежуточное соединение 48: 2-бром-1-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)этанон
К перемешиваемому раствору AlCl3 (1,54 г, 11,5 ммоль) в DCM (10 мл) добавляли бромацетилхлорид (0,76 г, 1,1 ммоль) при 0°С. Через 25 минут к указанному выше раствору добавляли по каплям 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен (2,4 г, 1,15 ммоль) в ДХМ. Реакционную смесь перемешивали при комнатной температуре в течение ночи и затем нагревали с обратным холодильником в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры, гасили льдом и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением целевого соединения в виде жидкости коричневого цвета (1,8 г, выход: 48,65%).
Промежуточное соединение 49: Метил 4-[4-оксо-4-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)бутаноил]бензоат
К перемешиваемому раствору этилмагнийбромида [(1,0 мл, 3М раствор), 3,2 ммоль)] в толуоле (10 мл) добавляли диэтиламин (0,24 г, 3,24 ммоль) и реакционную смесь перемешивали при комнатной температуре 15 минут. Затем к вышеуказанному раствору добавляли 2-бром-1-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)этанона (0,9 г, 2,7 ммоль) и метил-4-ацетилбензоат (0,39 г, 2,2 ммоль) в толуоле (10 мл) при 0°С. Реакционную смесь охлаждали до комнатной температуры, гасили водой и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением целевого соединения в виде твердого вещества желтого цвета (0,35 г, выход: 37,29%): 1Н ЯМР (300 МГц, DMSO-d6): δ 8,11 (s, 4Н), 3,90 (s, 3Н), 3,37-3,41 (t, 2Н), 3,18-3,22 (t, 2Н), 2,54 (s, 3Н), 1,68 (s, 4Н), 1,29-1,33 (d, 12Н).
Промежуточное соединение 50: Метил 4-[5-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)-1Н-пиррол-2-ил]бензоат
К перемешиваемому раствору метил 4-[4-оксо-4-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)бутаноил]бензоата (0,29 г, 0,68 ммоль) в уксусной кислоте (5 мл) добавляли ацетат аммония (2,63 г, 34,0 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 1,5 ч. Реакционную смесь затем гасили водой и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением целевого соединения в виде рыхлого твердого вещества желтого цвета (0,1 г, выход: 35,06%): 1Н ЯМР (300 МГц, CDCl3): δ 8,39 (s, 1Н), 7,93-7,95 (d, 2Н), 7,43-7,46 (d, 2Н), 6,61-6,62 (d, 1Н), 6,27-6,28 (d, 1Н), 3,83 (s, 3Н), 2,32 (s, 3Н), 1,63 (s, 4Н), 1,25-1,26 (d, 12Н).
Промежуточное соединение 51: Метил 4-[1-этил-5-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)-1H-пиррол-2-ил]бензоат
К перемешиваемому раствору NaH (0,007 г, 0,31 ммоль) в ДМФА (2 мл) добавляли по каплям метил 4-[5-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)-1Н-пиррол-2-ил]бензоат в ДМФА (1 мл) (0,09 г, 0,22 ммоль) при 0°С. Через 20 мин добавляли этилиодид (0,048 г, 0,31 ммоль). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 1 часа. Реакционную смесь гасили водой, экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением целевого соединения в виде рыхлого твердого вещества желтого цвета (0,06 г, выход: 68,85%): 1Н ЯМР (300 МГц, CDCl3): δ 7,99-8,02 (d, 2Н), 7,43-7,46 (d, 2Н), 6,27-6,28 (d, 1Н), 6,16-6,17 (d, 1Н), 3,89-3,93 (q, 2Н), 3,87 (s, 3Н), 2,12 (s, 3Н), 1,65 (s, 4Н), 1,26-1,27 (d, 12Н), 0,79-0,86 (t, 3Н).
Соединение 32: 4-[1-Этил-5-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)-1H-пиррол-2-ил] бензойная кислота
К перемешиваемому раствору 4-[5-(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)-1Н-пиррол-2-ил]бензоата (0,06 г, 0,14 ммоль) в ТГФ (1 мл) и метанола (1 мл) добавляли водный раствор NaOH (0,033 г, 0,83 ммоль) при 0°С. Смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали и остаток растворяли в минимальном количестве воды, подкисляли до рН 3 при охлаждении, экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над безводным Na2SO4 и концентрировали. Полученный продукт очищали с помощью растирания с н-гексаном с получением целевого соединения в виде твердого вещества желтого цвета (0,034 г, выход: 78,0%): МС (ESl, 120 эВ): m/z=420,2 (М-Н)+.
Схема 17
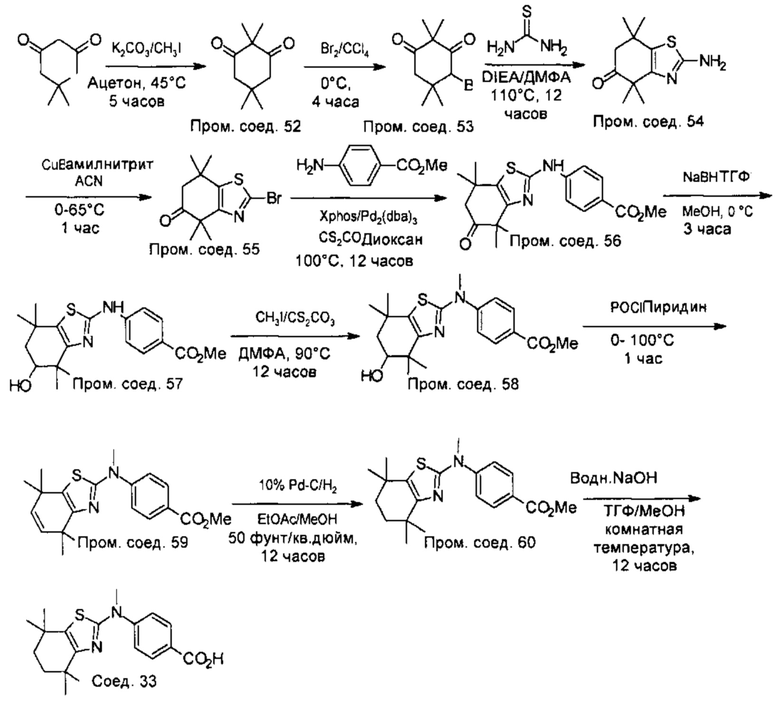
Пример 33: 4-[метил(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензойная кислота
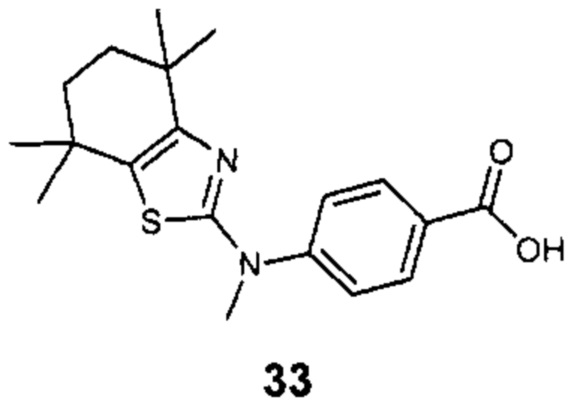
Соединение 33 синтезировали из 2-бром-4,4,7,7-тетраметил-,6,7-дигидро-1,3-бензотиазол-5(4H)-она и метил-4-аминобензоата, следуя способу, описанному в схеме 17; чистота: 92,66%.
Промежуточное соединение 52: 2,2,5,5-тетраметилциклогексан-1,3-диона
К раствору 5,5-диметилциклогексан-1,3-диона (100,0 г, 713,4 ммоль) в ацетоне (1 л) добавляли карбонат калия (294,3 г, 2140,2 ммоль) и смесь перемешивали при комнатной температуре в течение 30 мин. Реакционную массу охлаждали до 0°С и добавляли метилиодид (404,9 г, 2852,6 ммоль) и реакционную смесь нагревали при 45°С в течение 5 ч. Реакционную смесь затем концентрировали, разбавляли водой и экстрагировали этилацетатом. Комбинированный органический слой промывали водой и рассолом, сушили над Na2SO4 и концентрировали. Полученный продукт очищали с помощью кристаллизации с помощью 2% этилацетата в н-гексане с получением целевого соединения в виде бесцветных игольчатых кристаллов (55,0 г, выход: 45,83%): 1Н ЯМР (300 МГц, CDCl3): δ 2,54 (s, 4Н), 1,23 (s, 6Н), 0,92 (s, 6Н).
Промежуточное соединение 53: 4-бром-2,2,5,5-тетраметилциклогексан-1,3-дион
К раствору 2,2,5,5-тетраметилциклогексан-1,3-диона (20,0 г, 118,9 ммоль) в CCl4 добавляли бром (17,1 г, 107,0 ммоль) в CCl4 (50 мл) при 0°С и перемешивали при той же температуре в течение 4 часов. Реакционную смесь гасили водой, экстрагировали с DCM. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле (230-400 меш) с получением целевого соединения в виде твердого вещества белого цвета (0,15 г, выход: 51,04%).
Промежуточное соединение 54: 2-амино-4,4,7,7-тетраметил-6,7-дигидро-1,3-нафталин-5 (4Н)-он
Пробирку для проведения реакций под давлением объемом 200 мл загружали раствором 4-бром-2,2,5,5-тетраметилциклогексан-1,3-диона (15,0 г, 60,0 ммоль) в ДМФА (150 мл), к которому добавляли тиомочевину (4,6 г, 60,0 ммоль) и N,N-диизопропилэтиламин (23,2 г 180,0 ммоль) и нагревали при 100°С в течение ночи. Реакционную смесь затем гасили водой, экстрагировали с DCM. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали с помощью системы очистки CombiFlash с получением целевого соединения в виде твердого вещества черного цвета (2,0 г, выход: 14,6%).
Промежуточное соединение 55: 2-бром-4,4,7,7-тетраметил-6,7-дигидро-1,3-нафталин-5(4Н)-он
К раствору бромида меди (II) (1,29 г, 5,8 ммоль) в ацетонитриле добавляли изоамилнитрит (1,01 г, 8,7 ммоль) при 0°С и затем перемешивали в течение 15 мин. К перемешиваемой массе ацетонитрила медленно добавляли раствор 2-амино-4,4,7,7-тетраметил-6,7-дигидро-1,3-бензотиазол-5 (4Н)-она (1,3 г, 5,8 ммоль) при 0°С и реакционную смесь дополнительно перемешивали в течение 15 мин и затем нагревали при 65°С в течение 1 ч. Реакционную смесь фильтровали через целит и фильтрат промывали водой и рассолом, сушили над Na2SO4 и концентрировали с получением целевого продукта в виде твердого вещества коричневого цвета (1,3 г, выход: 77,7%): 1Н ЯМР (300 МГц, CDCl3): δ 2,59 (s, 2Н), 1,37 (s, 6Н), 1,26 (s, 6Н).
Промежуточное соединение 56: метил 4-[(4,4,7,7-тетраметил-5-оксо-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензоат
Пробирку для проведения реакций под давлением объемом 20 мл с завинчивающейся крышкой загружали 2-бром-4,4,7,7-тетраметил-6,7-дигидро-1,3-бензотиазол-5 (4Н)-оном (1,2 г, 4,2 ммоль), метил 4-аминобензоатом (1,25 г, 8,3 ммоль), CS2CO3 (2,69 г, 8,3 ммоль), XPhos (0,12 г, 0,25 ммоль) и 1,4-диоксаном (10 мл). Затем смесь продували аргоном в течение 15 мин. К реакционной смеси добавляли Pd2 (dba)3 (0,078 г, 0,084 ммоль) и смесь нагревали при 100°С в течение ночи. Реакционную смесь разбавляли водой и экстрагировали этилацетатом; органический слой промывали водой и рассолом, сушили над Na2SO4 и концентрировали. Неочищенный продукт перекристаллизовывали из метанола с получением целевого продукта в виде бесцветного твердого вещества (0,5 г, выход: 33,3%): 1Н ЯМР (300 МГц, DMSO-d6): δ 10,61 (s, 1Н), 7,89-7,92 (d, 2Н), 7,69-7,72 (d, 2Н), 3,81 (s, 3Н), 2,70 (s, 2Н), 1,35 (s, 6Н), 1,26 (s, 6Н).
Промежуточное соединение 57: метил 4-[(5-гидрокси-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензоат
К раствору метил 4-[(4,4,7,7-тетраметил-5-оксо-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензоата (0,05 г, 0,13 ммоль) в метаноле (2 мл) и ТГФ (1 мл) добавляли NaBH4 (0,005 г, 0,13 ммоль) при 0°С и перемешивали при той же температуре в течение 3 часов. Реакционную массу нейтрализовали насыщенным раствором NH4Cl и концентрировали. Полученную массу разбавляли этилацетатом и органический слой промывали водой и рассолом, сушили над Na2SO4 и концентрировали с получением целевого продукта в виде бесцветного твердого вещества (0,05 г, выход: 99,4%): 1Н ЯМР (300 МГц, DMSO-d6): δ 10,44 (s, 1Н), 7,87-7,90 (d, 2Н), 7,67-7,70 (d, 2Н), 4,73-4,74 (d, 1Н), 3,80 (s, 3Н), 3,69-3,73 (m, 1Н), 1,69-1,75 (m, 2Н), 1,32 (s, 3Н), 1,25-1,27 (d, 6Н), 1,05 (s, 3Н).
Промежуточное соединение 58: метил 4-[(5-гидрокси-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил) (метил)амино]бензоат
В пробирку для проведения реакций под давлением объемом 20 мл с завинчивающейся крышкой загружали метил-4-[(5-гидрокси-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензоат (0,05 г, 0,14 ммоль), CS2CO3 (0,13 г, 0,4 ммоль), CH3l (0,039 г, 0,28 ммоль) и ДМФА (2 мл). Реакционную смесь нагревали при 90°С в течение ночи. Реакционную смесь разбавляли водой и экстрагировали диэтиловым эфиром, органический слой промывали водой и рассолом, сушили над Na2SO4 и концентрировали с получением целевого продукта в виде твердого вещества бледно-коричневого цвета (0,04 г, выход: 77,01%).
Промежуточное соединение 59: метил 4-[метил(4,4,7,7-тетраметил-4,7-дигидро-1,3-бензотиазол-2-ил)амино]бензоат
К раствору метил 4-[(5-гидрокси-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)(метил)амино]бензоата (0,1 г, 0,26 ммоль) в пиридине (5 мл) добавляли POCl3 (0,08 г, 0,53 ммоль) при 0°С и нагревали при 100°С в аппарате Дина-Старка в течение 1 ч. Реакционную смесь гасили водой, экстрагировали с DCM. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением целевого соединения в виде бесцветного рыхлого твердого вещества (0,08 г, выход: 74,8%): 1Н ЯМР (300 МГц, CDCl3): δ 7,94-7,97 (d, 2Н), 7,41-7,44 (d, 2Н), 5,38-5,48 (dd, 2Н), 3,84 (s, 3Н), 3,50 (s, 3Н), 1,24-1,25 (d, 6Н).
Промежуточное соединение 60: метил 4-[метил(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензоат
Сосуд шейкер Парра загружали метил 4-[метил(4,4,7,7-тетраметил-4,7-дигидро-1,3-бензотиазол-2-ил)амино]бензоатом (0,06 г, 0,16 ммоль), этилацетатом (5 мл) и метанолом (2 мл) и продували N2 в течение приблизительно 2 мин. Затем в сосуд добавляли 10% Pd-С (0,02 г) и выдерживали для гидрогенизации (50 фунтов на квадратный дюйм) в течение ночи. Образование продукта контролировали с помощью ЖХ-МС. Затем добавляли дополнительные 10% Pd-C (50,0 мг) и реакционную смесь вновь выдерживали для гидрогенизации при 50 фунтов на квадратный дюйм в течение 2-х дней. Растворитель удаляли при пониженном давлении. Полученный продукт очищали с помощью препаративной ВЭЖХ с получением целевого соединения в виде твердого вещества белого цвета (0,012 г, выход: 19,9%).
Соединение 33: 4-[метил(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензойная кислота
Круглодонную колбу объемом 10 мл, снабженную магнитной мешалкой, загружали метанолом (1 мл) и ТГФ (2 мл). К перемешиваемому растворителю добавляли метил 4-[метил(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензоат (0,15 г, 0,41 ммоль) и водный NaOH (0,016 г, 0,41 ммоль) и перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь полностью выпаривали, неочищенный продукт промывали эфиром и разбавляли водой, затем нейтрализовывали 1 NHCl и затем экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4 и концентрировали. Полученный продукт очищали с помощью растирания с н-гексаном с получением целевого соединения в виде твердого вещества белого цвета (0,01 г, выход: 69,4%): МС (ESI, 120 эВ): m/z=345,2 (М+Н)+.
Пример 34: 4-[метил(4,4,7,7-тетраметил-4,7-дигидро-1,3-бензотиазол-2-ил)амино]бензойная кислота
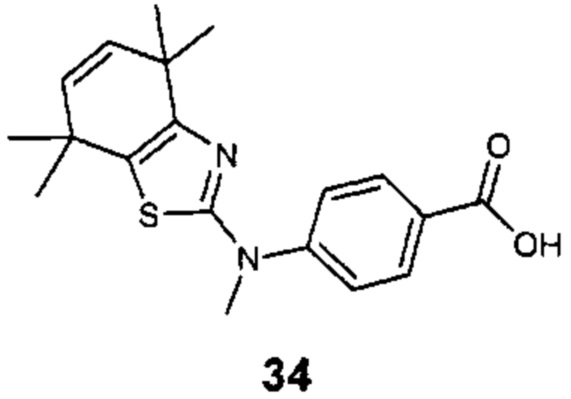
Соединение 34 синтезировали из 2-бром-4,4,7,7-тетраметил-,6,7-тетрагидро-1,3-бензотиазол-5 (4Н)-она и метил-4-аминобензоата, следуя способу, описанному в схеме 15 (0,01 г, выход: 52,05%); чистота: 93,68%
Пример 35: 4-[(5-Этокси-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)(метил)амино]бензойная кислота
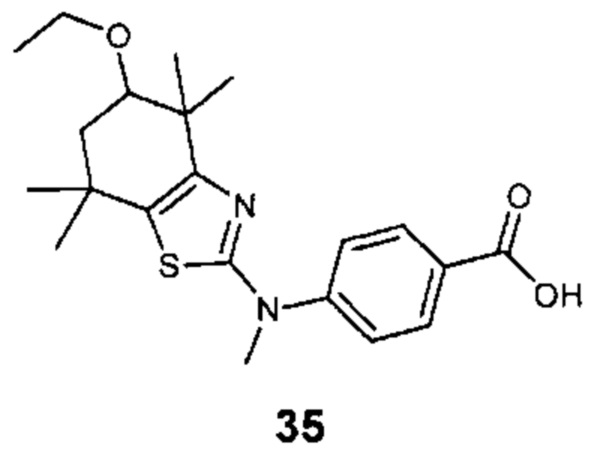
Соединение 35 синтезировали из 2-бром-4,4,7,7-тетраметил-,6,7-дигидро-1,3-бензотиазол-5 (4Н)-она и метил-4-аминобензоата, следуя способу, описанному в схеме 16 (0,005 г, выход: 43,28%); чистота: 84,49%.
Схема 18
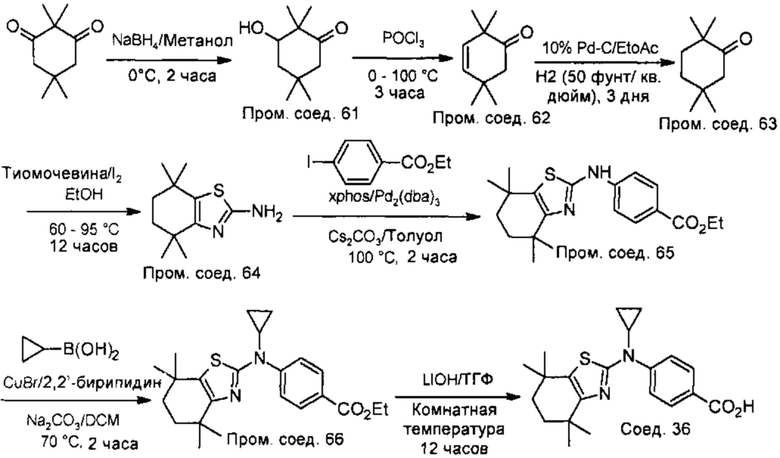
Пример 36: 4-[циклопропил(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензойная кислота
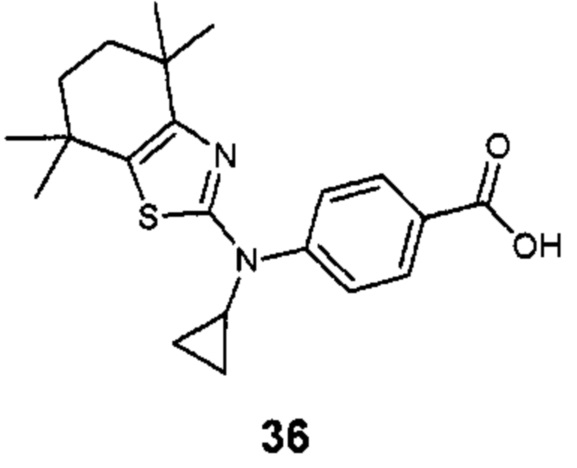
Соединение 36 синтезировали из 3,4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина и этил 4-иодбензоата, следуя способу, описанному в схеме 18; чистота: 98,84%.
Промежуточное соединение 61: 3-гидрокси-2,2,5,5-тетраметилциклогексанон
К перемешиваемому раствору 2,2,5,5-тетраметилциклогексан-1,3-диона (10 г, 59,5 ммоль) в метаноле (100 мл) порциями добавляли NaBH4 (0,67 г, 17,86 ммоль) при 0°С. Реакционную смесь оставляли перемешиваться при температуре 0°С в течение 2 часов. Реакционную смесь гасили льдом. Метанол удаляли и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии с получением целевого соединения в виде вязкого твердого вещества белого цвета (7,0 г, выход: 100,0%).
Промежуточное соединение 62: 2,2,5,5-тетраметилциклогекс-3-ен-1-он
К перемешиваемому раствору 3-гидрокси-2,2,5,5-тетраметилциклогексанона (8 г, 47,1 ммоль) в пиридине (30 мл) добавляли фосфороксихлорид (14,43 г, 94,1 ммоль) при 0°С. После добавления реакционную смесь нагревали до 100°С в аппарате Дина-Старка в течение 2 часов. Реакционную смесь гасили 1,5N HCl и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали с получением целевого соединения в виде жидкости желтого цвета (8,0 г, выход: 100,0%).
Промежуточное соединение 63: 2,2,5,5-тетраметилциклогексанон
Перемешиваемый раствор 2,2,5,5-тетраметилциклогекс-3-ен-1-он (8 г, 52,6 ммоль) в этилацетате (80 мл) продували азотом в течение 10 мин. К полученному раствору добавляли 10% Pd-C (3,0 г). Реакционную смесь выдерживали в шейкере Парра для гидрогенизации (50 фунтов на квадратный дюйм) в течение 3-х дней. Реакционную смесь фильтровали через целит, промывали этилацетатом и концентрировали с получением целевого соединения в виде жидкости зеленого цвета (3,8 г, выход: 46,76%): 13C-DEPT ЯМР (300 МГц, DMSO-d6): δ 50,6, 36,2, 33,8, 28,1, 24,8.
Промежуточное соединение 64: 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амин
В пробирку для проведения реакций под давлением объемом 250 мл с завинчивающейся крышкой загружали 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амин (6,0 г, 38,9 ммоль) в этаноле (30 мл), добавляли тиомочевину (8,9 г, 116 ммоль) и нагревали при 60°С в течение 15 мин. К полученному раствору добавляли йод (9,8 г, 38,6 ммоль) и нагревали при 95°С в течение ночи. Реакционную смесь гасили водой и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением целевого соединения в виде твердого вещества желтого цвета (0,03 г, выход: 7,51%): МС (ESl, 120 эВ): m/z=211,1 (М+Н+).
Промежуточное соединение 65: Этил 4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензоат
В пробирку для проведения реакций под давлением объемом 100 мл с завинчивающейся крышкой загружали 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-амин (0,3 г, 1,4 ммоль) в толуоле (5 мл), добавляли этил 4-иодбензоат (0,2 г, 0,72 ммоль), CS2CO3 (0,7 г, 2,16 ммоль) и XPhos (6,0 г, 38,9 ммоль). Реакционную смесь продували аргоном в течение 20 мин, после чего добавляли Pd2 (dba)3 (0,009 г, 0,04 ммоль). Реакционную смесь нагревали при 100°С в течение ночи. Реакционную смесь гасили водой и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4, концентрировали и восстанавливали. Полученный неочищенный продукт очищали с помощью системы очистки CombiFlash с получением целевого соединения в виде твердого вещества желтого цвета (0,2 г, выход: 39,85%).
Промежуточное соединение 66: Этил 4-[(циклопропилметил) (4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензоат
В круглодонную колбу 25 мл с двум горлышками, загруженную ацетатом меди (II) (0,122 г, 0,61 ммоль), добавляли 2,2'-бипиридин (0,095 г, 0,61 ммоль) в DCE (5 мл). Реакционную смесь нагревали при 70°С в течение 30 мин, затем добавляли циклопропилбороновую кислоту (0,1 г, 1,22 ммоль), этил 4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензоат (0,22 г, 0,61 ммоль), Na2CO3 (0,128 г, 1,22 ммоль), молекулярные сита (0,25 г) добавляли при 70°С и продолжали нагрев в течение ночи. Реакционную смесь гасили водой, экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением целевого соединения в виде полутвердого вещества желтого цвета (0,17 г, выход: 71,0%).
Соединение 36: 4-[циклопропилметил(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензойная кислота
К перемешиваемому раствору этил 4-[циклопропил-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензоата (0,17 г, 0,43 ммоль) в ТГФ (2 мл) добавляли водный раствор LiOH (0,1 г, 4,3 ммоль) и этанол (92 мл). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение ночи. Реакционную смесь концентрировали, подкисляли 1,5N HCl и экстрагировали этилацетатом. Органический слой промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении с получением целевого соединения в виде твердого вещества белого цвета (0,12 г, выход: 81,0%): МС (ESI, 120 эВ): m/z=371,2 (М+Н+).
Пример 37: 6-[Циклопропил(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновая кислота
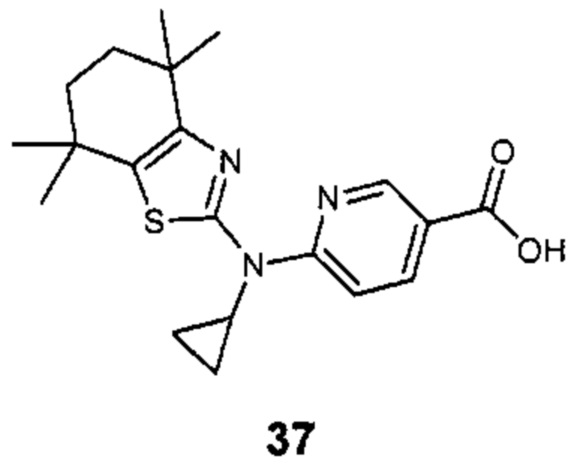
Соединение 37 синтезировали из 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина и метил 6-хлорпиридин-3-карбоксилата, следуя способу, аналогичному описанному в схеме 18 (0,030 г, выход: 40,4%); чистота: 98,66%.
Пример 38: 4-[циклопропил метил(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензойная кислота
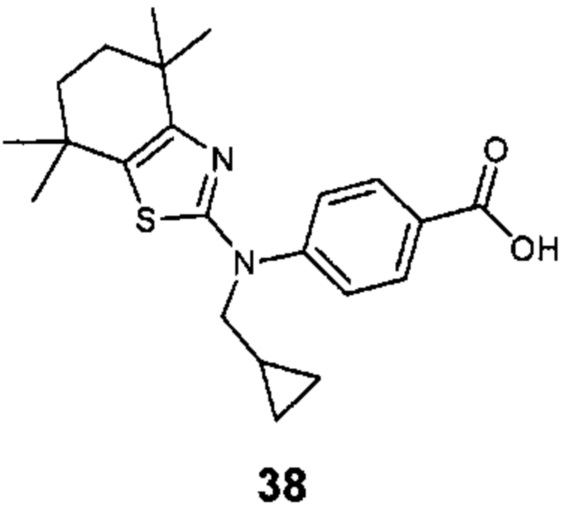
Соединение 38 синтезировали из 3,4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина и этил 4-иодбензоата, следуя способу, аналогичному описанному в схеме 18 (0,006 г, выход: 36,4%); чистота: 94,93%.
Пример 39: 6-[этил(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновая кислота
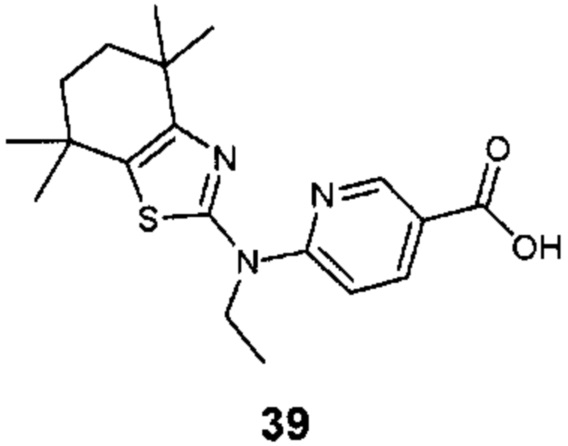
Соединение 39 синтезировали из 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина и метил 6-хлорпиридин-3-карбоксилата, следуя способу, аналогичному описанному в схеме 18 (0,015 г, выход: 55,63%); чистота: 94,07%.
Пример 40: 6-[(циклопропилметил)(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновая кислота
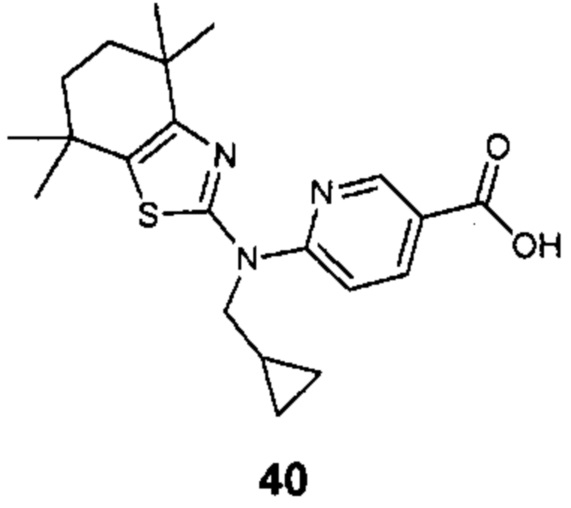
Соединение 40 синтезировали из 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина и метил 6-хлорпиридин-3-карбоксилата, следуя способу, аналогичному описанному в схеме 18 (0,04 г, выход: 51,9%); чистота: 97,94%.
Схема 19
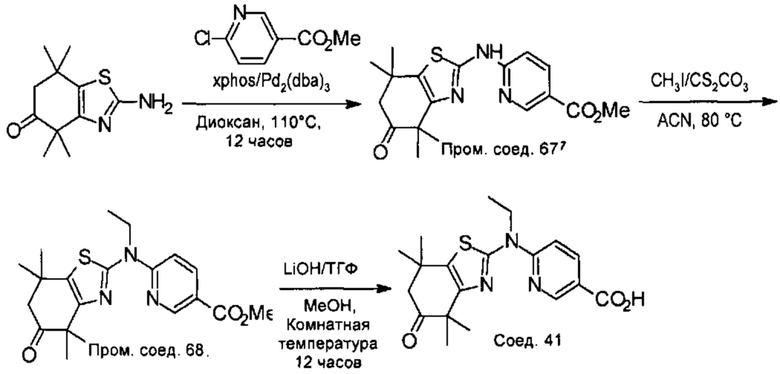
Пример 41: 6-[этил(4,4,7,7-тетраметил-5-оксо-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновая кислота
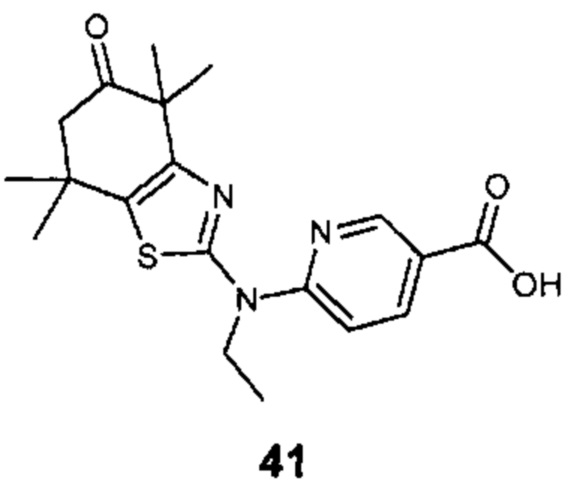
Соединение 41 синтезировали из 2-амино-4,4,7,7-тетраметил-,6,7-дигидро-1,3-бензотиазол-5 (4Н)-она и метил-6-хлорпиридин-3-карбоксилата, следуя способу, описанному в схеме 19; чистота: 91,07%.
Промежуточное соединение 67: метил 6-[(4,4,7,7-тетраметил-5-оксо-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоксилат
В пробирку объемом 100 мл загружали 2-амиино-4,4,7,7-тетраметил-6,7-дигидро-1,3-бензотиазол-5 (4Н)-он (0,38 г, 1,75 ммоль) и толуол (5 мл), добавляли метил 6-хлорпиридин-3-карбоксилат (0,15 г, 0,87 ммоль), CS2CO3 (0,6 г, 1,75 ммоль), и XPhos (0,05 г, 0,08 ммоль) в атмосфере аргона. Реакционную смесь продували аргоном в течение 20 минут. Затем к полученному раствору добавляли Pd2 (dba)3 (0,032 г, 0,035 ммоль). Реакционную смесь нагревали до 100°С в течение ночи. Реакционную смесь гасили водой и экстрагировали этилацетатом. Органический слой промывали водой, рассолом и сушили над безводным Na2SO4, фильтровали и концентрировали. Полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением целевого продукта в виде твердого желтого цвета вещества (0,025 г, выход: 2,5%).
Промежуточное соединение 68: метил 6-[этил(4,4,7,7-тетраметил-5-оксо-4)5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоксилат
К перемешиваемому раствору метил 6-[(4,4,7,7-тетраметил-5-оксо-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоксилата (0,15 г, 0,42 ммоль) в ацетонитриле (3 мл) добавляли Cs2CO3 (0,41 г, 1,25 ммоль) и C2H5l (0,1 г, 0,84 ммоль). Реакционную смесь нагревали с обратным холодильником при 90°С в течение ночи. Реакционную смесь гасили водой и экстрагировали этилацетатом, промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме с получением неочищенного масла, которое очищали с помощью колоночной хроматографии на силикагеле с получением целевого соединения в виде твердого вещества бледно-желтого цвета (0,04 г, выход: 25,00%). 1Н ЯМР (300 МГц, CDCl3): δ 8,98-8,99 (d, 1Н), 8,16-8,20 (dd, 1Н), 6,98-7,01 (d, 1Н), 4,35-4,42 (q, 2Н), 3,86 (s, 3Н), 2,59 (s, 2Н), 1,37 (s, 6Н), 1,32-1,37 (t, 3Н), 1,31 (s, 6Н).
Соединение 41: 6-[этил(4,4,7,7-тетраметил-5-оксо-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновая кислота
К перемешиваемому раствору метил 6-[этил (4,4,7,7-тетраметил-5-оксо-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоксилата (0,04 г, 0,1 ммоль) в ТГФ (1 мл) и метанолу (1 мл) добавляли LiOH (0,024 г, 1,03 ммоль) в воде (1 мл). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение ночи. Реакционную смесь полностью концентрировали и неочищенный продукт промывали эфиром и разбавляли водой и затем нейтрализовали лимонной кислотой и экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали. Полученный продукт очищали с помощью растирания с н-гексаном с получением продукта в виде твердого вещества белого цвета (0,02 г, выход: 53,6%): МС (ESI, 120 эВ): m/z=374,2 (М+Н)+.
Схема 20
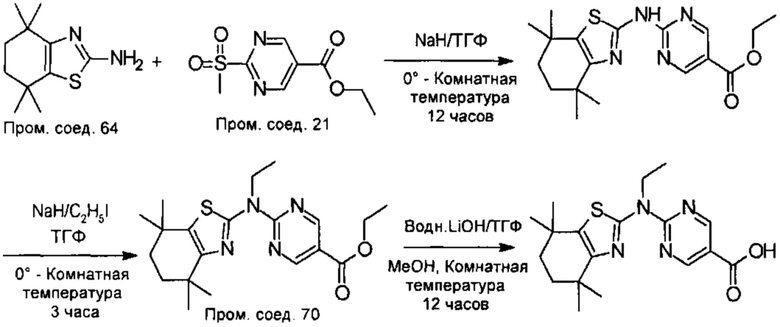
Пример 42: 2-[этил(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиримидин-5-карбоновая кислота
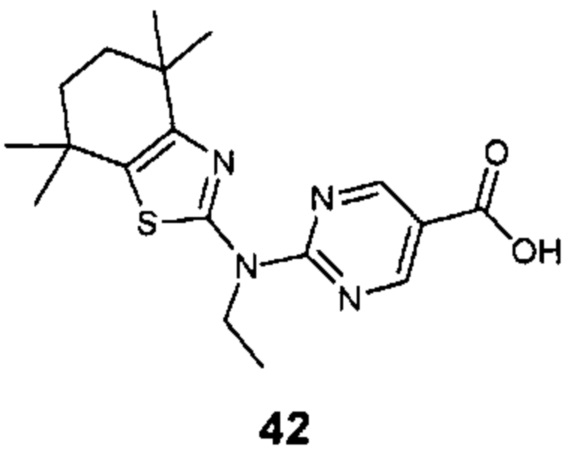
Соединение 42 синтезировали из 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина и этил 2-(метилсульфонил)пиримидин-5-карбоксилата, следуя способу, описанному в схеме 20; чистота: 93,88%.
Промежуточное соединение 69: этил 2-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиримидин-5-карбоксилат
К перемешиваемому раствору NaH (0,2 г, 8,57 ммоль) в ТГФ (20 мл) добавляли 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амин (1,2 г, 5,71 ммоль) и этил 2-(метилсульфонил)пиримидин-5-карбоксилат (2,62 г, 11,43 ммоль) в ТГФ (10 мл) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь гасили водой и экстрагировали этилацетатом, промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме с получением неочищенного продукта в виде вязкого твердого вещества желтого цвета (5,0 г, выход: 97,00%).
Промежуточное соединение 70: этил 2-[этил(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиримидин-5-карбоксилат
К перемешиваемому раствору этил 2-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]пиримидин-5-карбоксилата (0,13 г, 0,36 ммоль) и Cs2CO3 (0,29 г, 0,9 ммоль) в ТГФ (3 мл) по каплям добавляли C2H5l (0,084 г, 0,54 ммоль) при 0°С. Реакционную смесь нагревали при температуре 80°С в течение 12 часов. Реакционную смесь гасили льдом и экстрагировали этилацетатом, промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме. Полученный неочищенный продукт очищали с помощью системы очистки CombiFlash с получением целевого соединения в виде жидкости бледно-желтого цвета (0,06 г, выход: 38,6%). m/z=389,1 (М+Н)+. 1Н ЯМР (300 МГц, CDCl3): δ 9,04 (s, 2Н), 4,58-4,65 (q, 2Н), 4,28-4,36 (q, 2Н), 1,64 (s, 4Н), 1,30-1,35 (t, 3Н), 1,20 (s, 6Н), 1,19 (s, 6Н).
Соединение 42: 2-[этил(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиримидин-5-карбоновая кислота
К перемешиваемому раствору этил 2-[этил(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиримидин-5-карбоксилата (0,06 г, 0,155 ммоль) в ТГФ (1 мл) добавляли водный NaOH (0,062 г, 1,55 ммоль) и метанол (1 мл). Затем смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали; полученную соль промывали эфиром. Остаток растворяли в минимальном количестве воды, подкисляли 1,5 N HCl и экстрагировали этилацетатом, сушили и концентрировали с получением целевого продукта в виде твердого вещества почти белого цвета (0,025 г, выход: 34,67%). МС (ESI, 120 эВ): m/z=361,1 (М+Н)+.
Пример 43: 2-[(циклопропилметил)(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиримидин-5-карбоновая кислота
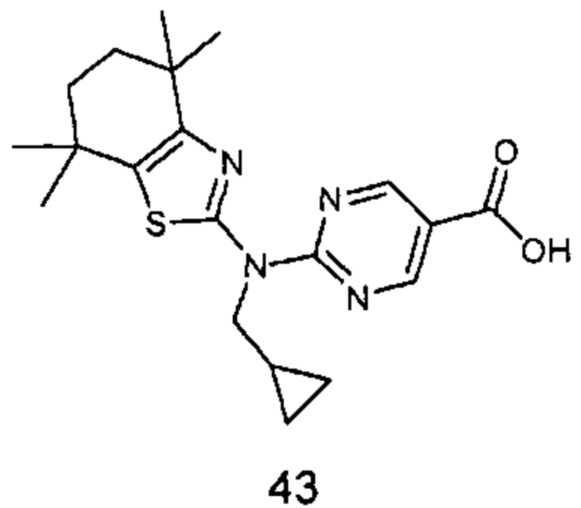
Соединение 43 синтезировали из 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина и этил 2-(метилсульфонил)пиримидин-6-карбоксилата, следуя способу, описанному в схеме 20; чистота: 97,57%.
Схема 21
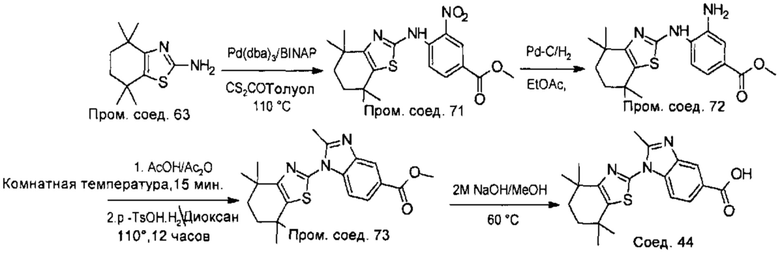
Пример 44: 2-метил-1-(3,4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)-1Н-бензимидазол-5-карбоновая кислота
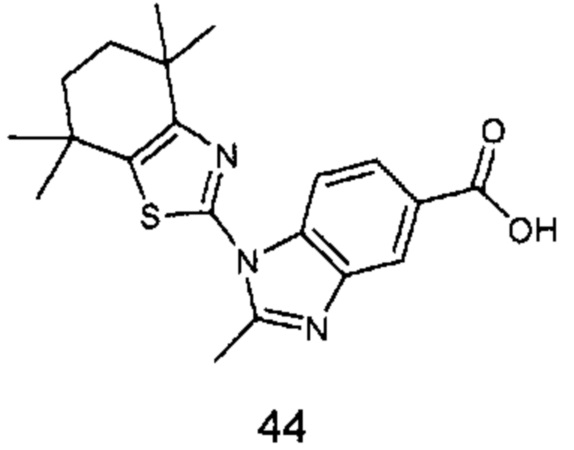
Соединение 44 синтезировали из 3,4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина и метил 4-иод-3-нитробензоата следуя способу описанному в схеме 21; чистота; 94,22%.
Промежуточное соединение 71: Этил 3-нитро-4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензоат
В пробирку для проведения реакций под давлением объемом 100 мл с завинчивающейся крышкой загружали 4-иод-3-нитробензоат (1,0 г, 3,26 ммоль) в толуоле (10 мл); добавляли 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амин (1,3 г, 6,5 ммоль), CS2CO3 (3,2 г, 9,8 ммоль) и ксантфос (0,2 г, 0,3 ммоль) и продували через реакционную массу аргон в течение 15 мин. Затем добавляли Pd2 (dba)3 (0,04 г, 0,16 ммоль) и нагревали при 110°С в течение ночи. Реакционную массу разбавляли водой и экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии с получением целевого соединения в виде твердого вещества красного цвета (0,2 г, выход: 15,87%). 1Н ЯМР (300 МГц, CDCl3): δ 10,73 (s, 1Н), 8,92-8,93 (d, 1Н), 8,74-8,77 (d, 1Н), 8,20-8,24 (dd, 1Н), 3,94 (s, 3Н), 1,74 (s, 4Н), 1,30-1,31 (d, 12Н).
Промежуточное соединение 72: Этил 3-амино-4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензоат
К перемешиваемому раствору метил 3-нитро-4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензоата (0,5 г, 1,3 ммоль) в метаноле (5 мл) добавляли формиат аммония (0,41 г, 6,5 ммоль) с последующим добавлением цинковой пыли (0,41 г, 6,4 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 1 часов. Реакционную смесь концентрировали при пониженном давлении. Полученный остаток промывали водой, рассолом и сушили над безводным Na2SO4 и концентрировали в вакууме с получением целевого соединения в виде вязкого твердого вещества желтого цвета (0,47 г, выход: 100,0%). МС (ESI, 120 эВ): m/z=360,1 (М+Н+);
Промежуточное соединение 73: метил 2-метил-1-(3,4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)-1Н-бензимидазол-5-карбоксилат
К раствору метил 3-амино-4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензоата (0,16 г, 0,45 ммоль) в АсОН (4 мл) добавляли Ас2O (0,5 г) при комнатной температуре и перемешивали при той же температуре в течение 15 минут, выливали в воду (10 мл) и экстрагировали EtoAc. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и выпаривали при пониженном давлении. Полученный остаток растворяли в диоксане (3 мл) добавляли p-TsOH⋅H2O (0,25 г, 1,35 ммоль) и сухой пиридин (0,1 мл, 1,35 ммоль). Смесь нагревали с обратным холодильником при 110°С в течение ночи с использованием аппарата Дина-Старка. Реакционную смесь гасили водой и экстрагировали с EtOAc. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и выпаривали при пониженном давлении. Полученный неочищенный продукт очищали с помощью колоночной хроматографии с получением целевого соединения в виде жидкости желтого цвета (0,5 г, выход: 29,00%). МС (ESI, 120 эВ): m/z=384,1 (М+Н)+; 1Н ЯМР (300 МГц, CDCl3): δ 8,34 (s, 1Н), 7,94-7,97 (dd, 1Н), 7,63-7,66 (d, 1Н), 3,88 (s, 3Н), 2,71 (s, 3Н), 1,73 (s, 4Н), 1,27-1,32 (d, 12Н).
Соединение 44: 2-метил-1-(3,4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)-1H-бензимидазол-5-карбоновая кислота
К перемешиваемому раствору метил 2-метил-1-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)1Н-бензимидазол-5-карбоксилата (0,05 г, 0,13 ммоль) в ТГФ (2 мл) добавляли водный NaOH (0,03 г, 0,65 ммоль) и метанол (1 мл). Затем смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали; полученную соль промывали эфиром. Остаток растворяли в минимальном количестве воды, подкисляли 1,5 N HCl и экстрагировали этилацетатом, сушили и концентрировали с получением целевого продукта в виде твердого вещества желтого цвета (0,02 г, выход: 41,66%). МС (ESI, 120 эВ): m/z=370,1 (М+Н)+.
Схема 22:
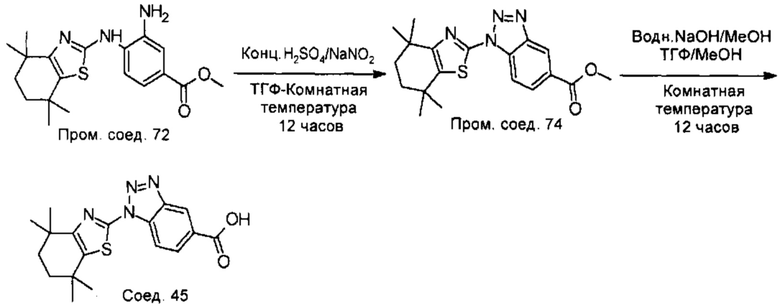
Пример 45: 1-(3,4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)-1Н-бензотриазол-5-карбоновая кислота
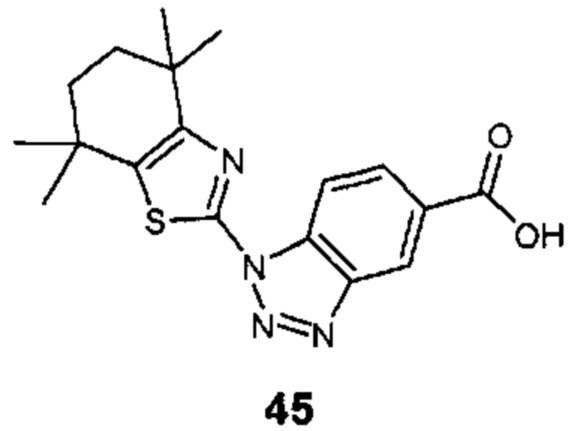
Соединение 45 синтезировали из метил 3-амино-4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензоата и гидролиза эфира (промежуточное соединение 74), следуя способу, описанному в схеме 22; чистота: 99,81%.
Промежуточное соединение 74: метил 1-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)-1Н-бензтриазол-5-карбоксилат
К перемешиваемому раствору метил 3-амино-4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензоата (0,24 г, 0,67 ммоль) в ТГФ (5 мл) добавляли концентрированную H2SO4 (1,0 мл в 10 мл воды) и NaNO2 (0,075 г, 1,07 ммоль) в воде при 0°С. Реакционную смесь перемешивали при той же температуре в течение 1 часа и при комнатной температуре в течение ночи. Реакционную смесь гасили раствором NaOH (раствор 2М) и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением целевого соединения в виде твердого вещества желтого цвета (0,09 г, выход: 36,0%). 1Н ЯМР (300 МГц, CDCl3): δ 8,73 (s, 1Н), 8,39-8,72 (d, 1Н), 8,21-8,25 (dd, 1Н), 3,92 (s, 3Н), 1,71 (s, 4Н), 1,29 (s, 12Н).
Соединение 45: 1-(3,4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)-1Н-бензотриазол-5-карбоновая кислота
К перемешиваемому раствору метил 1-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)1Н-бензотриазол-5-карбоксилата (0,09 г, 0,24 ммоль) в ТГФ (2 мл) добавляли водный NaOH (0,048 г, 1,2 ммоль) и метанол (2 мл). Затем смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали; полученную соль промывали эфиром. Остаток растворяли в минимальном количестве воды, подкисляли 1,5 N HCl и экстрагировали этилацетатом, сушили и концентрировали с получением целевого продукта в виде твердого вещества белого цвета (0,05 г, выход: 58,13%). МС (ESI, 120 эВ): m/z=357,0 (М+Н)+.
Схема 23
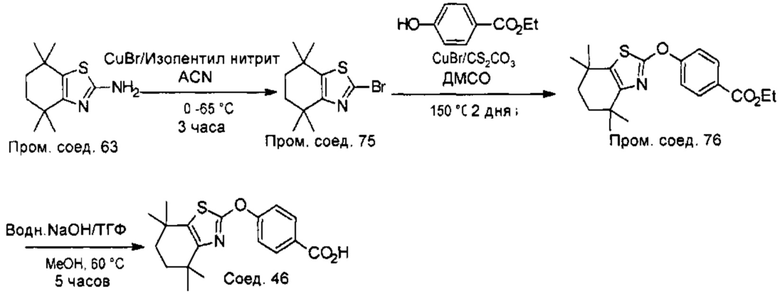
Пример 46: 4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)окси]бензойная кислота
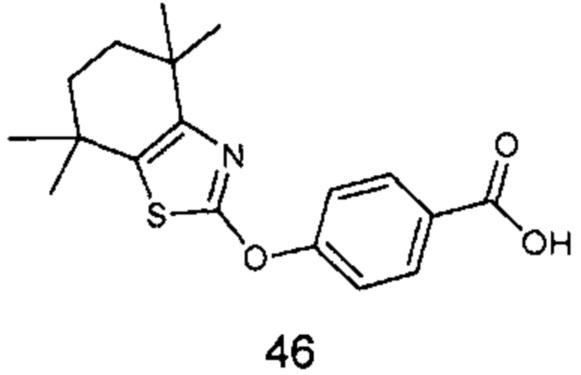
Соединение 46 синтезировали из 2-бром-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазола и метил-4-гидроксибензоата, следуя способу, описанному в схеме 23; чистота: 95,55%.
Промежуточное соединение 75: 2-бром-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол
В круглодонную колбу 250 мл с двумя горлышками загруженную бромидом меди (II) (1,6 г, 7,14 ммоль) и ацетонитрилом (10 мл) добавляли изопентилнитрит (1,25 г, 10,7 ммоль) при 0°С. Через 15 минут по каплям добавляли 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1 и 3-нафталин-2-амин (1,5 г, 7,14 ммоль) в ацетонитриле. Смесь перемешивали при 0°С 15 мин и нагревали при 65°С в течение 3 часов. Реакционную смесь разбавляли водой и экстрагировали этилацетатом, органический слой промывали водой и рассолом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали с получением целевого соединения в виде жидкости желтого цвета (1,5 г, выход: 77,46%). МС (ESI, 120 эВ): m/z=276,0 (М+Н+);
Промежуточное соединение 76: метил 4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)окси]бензоат
К перемешиваемому раствору этил 4-гидроксибензоата (0,44 г, 2,6 ммоль) в ДМСО (10 мл) добавляли CS2CO3 (2,1 г, 6,6 ммоль). Через 10 минут добавляли 2-бром-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол (0,6 г, 2,2 ммоль). Затем к полученному раствору добавляли бромид меди (I) (0,063 г, 4,4 ммоль). Реакционную смесь нагревали при температуре 150°С в течение 2-х дней. Реакционную смесь гасили водой и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4, концентрировали и восстанавливали. Полученный неочищенный продукт очищали с помощью колоночной хроматографии на силикагеле с получением целевого соединения в виде жидкости желтого цвета (0,4 г, выход: 25,34%).
Соединение 46: 4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)окси]бензойная кислота
К перемешиваемому раствору этил 4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)окси] бензоата (0,04 г, 1,0 ммоль) в ТГФ (1 мл) и метаноле (1 мл) добавляли NaOH (0,018 г, 0,45 ммоль) в воде (1 мл). Реакционную смесь выдерживали при комнатной температуре в течение ночи и затем нагревали до 60°С в течение 5 часов. Реакционную смесь концентрировали; полученную соль промывали эфиром. Остаток растворяли в минимальном количестве воды, подкисляли 1,5 N HCl и экстрагировали этилацетатом, сушили и концентрировали. Полученный продукт очищали с помощью препаративной ТСХ с получением целевого соединения в виде твердого вещества желтого цвета (0,01 г, выход: 30,3%). МС (ESI, 120 эВ): m/z=332,1 (М+Н)+.
Схема 24
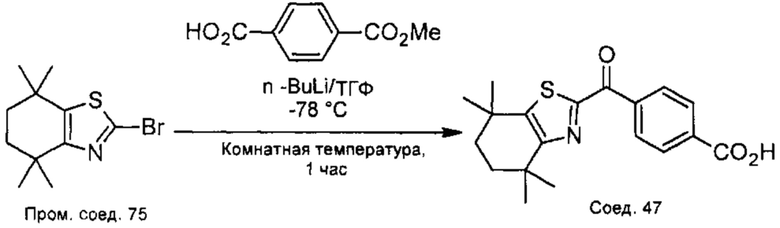
Пример 47: 4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)карбонил]бензойная кислота
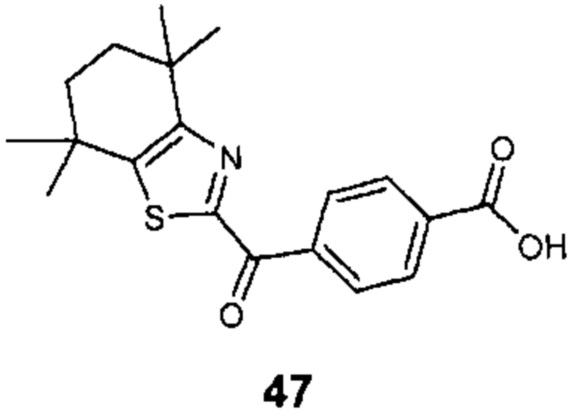
Соединение 47 синтезировали из 2-бром-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазола и 4-(метоксикарбонил)бензойной кислоты следуя способу, описанному в схеме 24; чистота: 99,25%.
Соединение 47: 4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)карбонил]бензойная кислота
К перемешиваемому раствору 4-(метоксикарбонил)бензойной кислоты (0,55 г, 2,0 ммоль) в ТГФ (10 мл) добавляли н-бутиллитий [1,25 мл (1,6М раствор)] при -78°С в атмосфере N2. Затем к полученному раствору добавляли 2-бром-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол (0,3 г, 0,0011 моль) в ТГФ (10 мл) и перемешивали при той же температуре примерно полтора часа и при комнатной температуре в течение 1 ч. Реакционную смесь гасили насыщенным раствором NH4Cl и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали с помощью препаративной ТСХ с получением целевого соединения в виде твердого вещества белого цвета (0,005 г, выход: 0,88%).
Схема 25
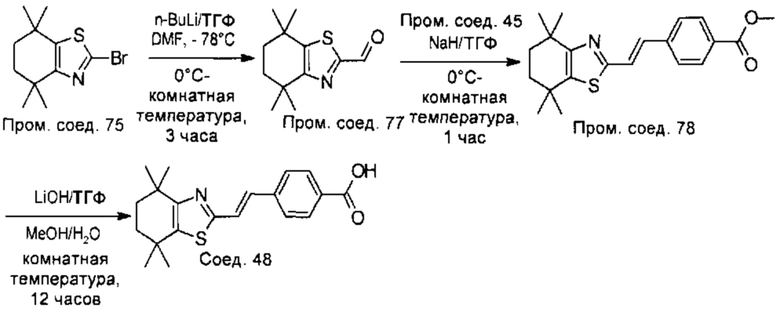
Пример 48: 4-(E)[-2-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)этенил]бензойная кислота
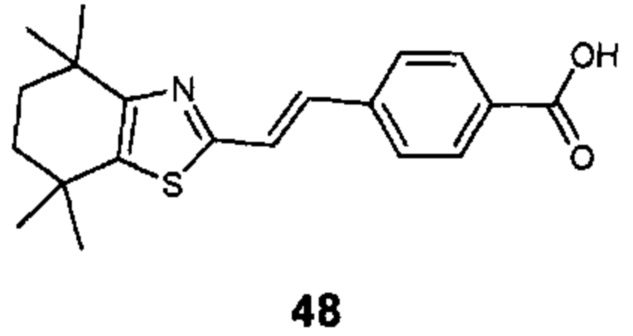
Соединение 48 синтезировали из 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазолн-2-карбальдегида и метил 4-{[бром (трифенил)-15-фосфанил]метил}бензоата следуя способу описанному в схеме 23; чистота: 98,05%.
Промежуточное соединение 77: 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-карбальдегид
В круглодонную колбу 250 мл с двумя горлышками, загруженную 2-бром-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазолом (1,0 г, 3,6 ммоль) в сухом ТГФ (10 мл), медленно добавляли н-бутиллитий [(2,5 мл (1,6 М), 4,0 ммоль)] при -78°С, затем перемешивали при -50°С в течение 1 ч. Затем к полученному раствору добавляли ДМФА (0,5 мл) при той же температуре и перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь гасили насыщенным раствором NH4Cl и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали с помощью системы очистки CombiFlash с получением целевого соединения в виде маслянистого продукта (0,65 г, выход: 80,55%). 1Н ЯМР (300 МГц, CDCl3): δ 9,83 (s, 1Н), 1,70 (s, 4Н), 1,27-1,29 (d, 2Н).
Промежуточное соединение 78: метил 4-[(E)-2-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)этенил]бензоат
К перемешиваемому раствору NaH (0,42 г, 1,7 ммоль) в сухом ТГФ (2 мл) добавляли метил 4-{[бром(трифенил)-15-фосфанил]метил}бензоат (0,57 г, 1,1 ммоль) в ТГФ (2 мл) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 45 мин, затем добавляли 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-карбальдегида (0,2 г, 0,8 ммоль) в ТГФ (1 мл) при 0°С и перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь гасили ледяной водой и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали с помощью системы очистки CombiFlash с получением целевого соединения в виде твердого вещества бледно-желтого цвета (0,226 г, выход: 83,14%).
Соединение 48: 4-(Е)[-2-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)этенил]бензойная кислота
Круглодонную колбу объемом 10 мл, снабженную магнитной мешалкой, загружали метанолом (3 мл) и ТГФ (3 мл). К перемешиваемому растворителю добавляли метил 4-[(Е)-2-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)этенил]бензоат (0,26 г, 0,7 ммоль) и водный LiOH (0,067 г, 2,8 ммоль) и перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь полностью выпаривали и неочищенный продукт промывали эфиром и разбавляли водой и затем нейтрализовали 1N HCl, затем экстрагировали этилацетатом, органический слой промывали водой и рассолом, сушили над Na2SO4 и концентрировали. Полученный продукт очищали с помощью препаративной ВЭЖХ с получением целевого соединения в виде твердого вещества белого цвета (0,0022 г, выход: 0,92%): МС (ESI, 120 эВ): m/z=342,1 (М+Н)+.
Пример 49: 4-(2)[-2-(4,4,717-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)этенил]бензойная кислота
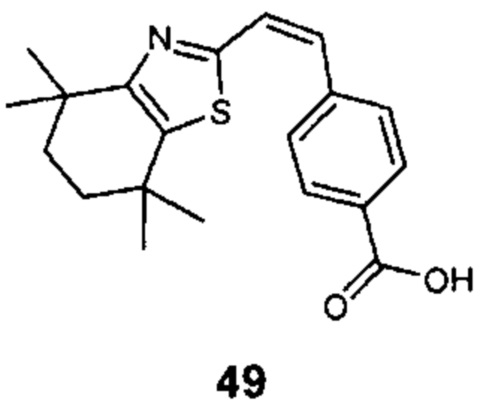
Соединение 49 синтезировали из 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазолн-2-карбальдегида и метил 4-{[бром (трифенил)-15-фосфанил]метил}бензоата, следуя способу, аналогичному описанному в схеме 25 (0,012 г, выход: 5,02%); чистота: 94,64%.
Схема 26
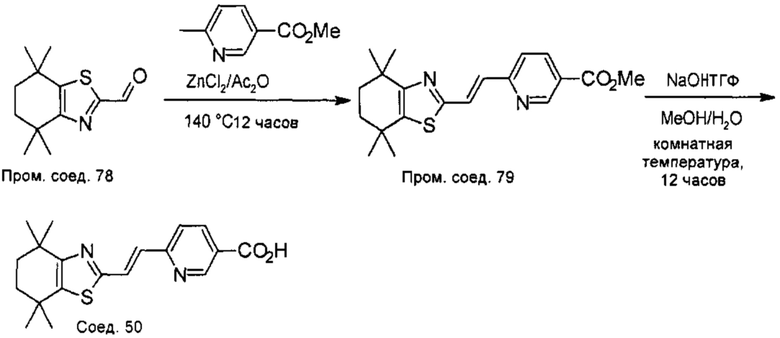
Пример 50: 6-(Е)[-2-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)этенил]пиридин-3-карбоновая кислота
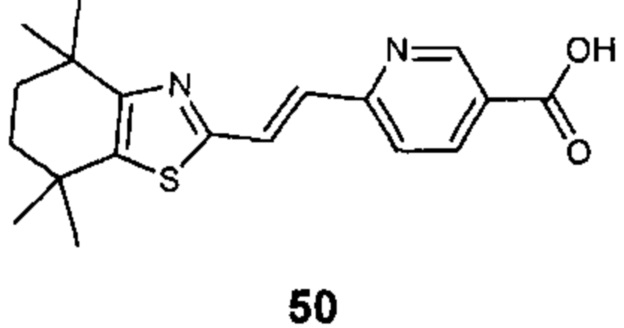
Соединение 50 синтезировали из 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-карбальдегида и метил 6-метилпиридин-3-карбоксилата, следуя способу, описанному в схеме 26; чистота: 91,66%.
Промежуточное соединение 79: метил 6-[(E)-2-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)этенил]пиридин-3-карбоксилат
К перемешиваемому раствору 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1-бензотиофен-2-карбальденида (0,155 г, 0,6 ммоль) в уксусном ангидриде (0,142 г, 1,3 ммоль) добавляли метил 6-метилпиридин-3-карбоксилат (0,105 г, 0,6 ммоль) и каталитическое количество ZnCl2 (0,004 г, 0,03 ммоль). Затем реакционную смесь нагревали при 140°С в течение ночи. Реакционную смесь гасили ледяной водой и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали с помощью системы очистки CombiFlash с получением целевого соединения в виде маслянистого продукта бледно-желтого цвета (0,16 г, выход: 40,07%). 1Н ЯМР (300 МГц, CDCl3): δ 9,11 (s, 1Н), 8,17-8,20 (d, 1Н), 7,73-7,78 (d, 1Н), 7,40-7,43 (d, 1Н) 7,28-7,33 (d, 1Н), 3,89 (s, 3Н), 1,67 (s, 4Н), 1,26 (s, 12Н).
Соединение 50: 6-(Е)[-2-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)этенил]пиридин-3-карбоновая кислота
К перемешиваемому раствору 4-[(Е)-2-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)этенил] бензоата (0,1 г, 0,28 ммоль) в ТГФ (3 мл) добавляли водный NaOH (0,022 г, 0,56 ммоль) и метанол (3 мл). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь полностью концентрировали и неочищенный продукт промывали эфиром и разбавляли водой и затем нейтрализовали 1N HCl, затем экстрагировали этилацетатом, органический слой промывали водой и рассолом, сушили над Na2SO4 и концентрировали. Полученный продукт очищали с помощью препаративной ТСХ с получением целевого соединения в виде твердого вещества желтого цвета (0,01 г, выход: 9,73%): МС (ESl, 120 эВ): m/z=343,1 (М+Н)+.
Схема 27
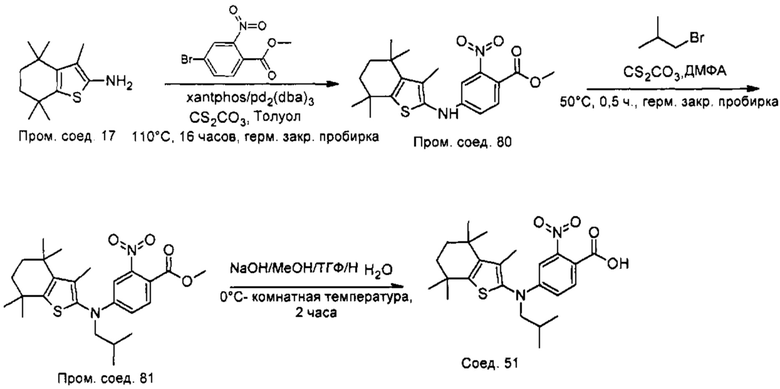
Пример 51: 4-[(2-метилпропил)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]нитробензойная кислота
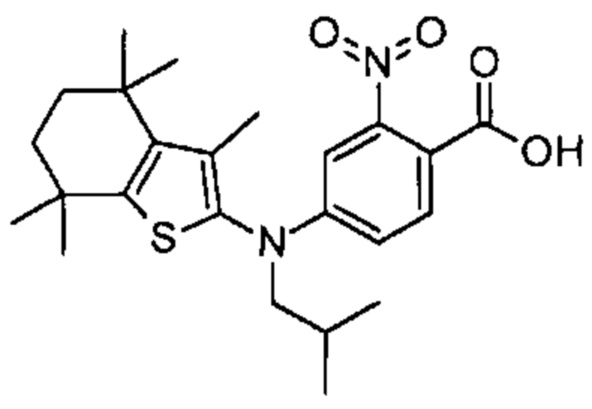
Соединение 51 синтезировали из метил 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-амина и метил 4-бром-2-нитробензоата, следуя способу, описанному в схеме 27; чистота: 94,91%.
Промежуточное соединение 80: метил 2-нитро-4-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоат
К перемешиваемому раствору 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-амина (0,31 г, 1,4 ммоль) в толуоле (3 мл) добавляли метил 4-бром-2-нитробензоат (0,18 г, 0,69 ммоль) и CS2CO3 (0,67 г, 2,1 ммоль). Полученный раствор продували аргоном в течение 15 мин. К перемешиваемому раствору добавляли ксантфос (0,019 г, 0,03 ммоль) с последующим добавлением Pd2 (dba)3 (0,012 г, 0,014 ммоль). Реакционную смесь нагревали с обратным холодильником при 110°С в течение 16 часов. Затем реакционную смесь охлаждали до комнатной температуры, выливали в воду и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме. Полученный неочищенный продукт очищали с помощью колоночной хроматографии с получением целевого соединения в виде вязкого продукта желтого цвета (3,5 г, выход: 25,93%).
Промежуточное соединение 81: метил 4-[(2-метилпропил) (3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]-2-нитробензоат
К перемешиваемому раствору метил 2-нитро-4-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоата (0,07 г, 0,17 ммоль) в ДМФА (3 мл) добавляли карбонат цезия (0,17 г, 0,51 ммоль) и изобутилбромид (0,05 г, 0,34 ммоль) при 0°С в герметично закрытую пробирку. Реакционную смесь нагревали при 50°С в течение 30 минут. Затем реакционную смесь охлаждали до комнатной температуры, выливали в воду и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме. Полученный неочищенный продукт очищали с помощью колоночной хроматографии с получением целевого соединения в виде вязкого продукта желтого цвета (0,015 г, выход: 19,48%).
Соединение 51: 4-[(2-метилпропил) (3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]нитробензойная кислота
Круглодонную колбу объемом 25 мл, снабженную магнитной мешалкой, загружали метанолом (0,5 мл) и ТГФ (0,5 мл). К перемешиваемому раствору добавляли метил 4-[(2-метилпропил) (3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]-2-нитробензоат (0,015 г, 0,03 ммоль) и водный NaOH (0,006 г, 0,16 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь полностью концентрировали и неочищенный продукт промывали эфиром и разбавляли водой и затем нейтрализовали 1.5N соляной кислотой, затем экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали. Полученное твердое вещество растирали с гексаном с получением целевого соединения в виде твердого вещества желтого цвета (0,01 г, выход: 68,49%): МС (ESI, 120 эВ): m/z=445,2 (М+Н+).
Пример 52: 6-[(циклопропил метил(4,4,7,7-тетраметил-5-оксо-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновая кислота
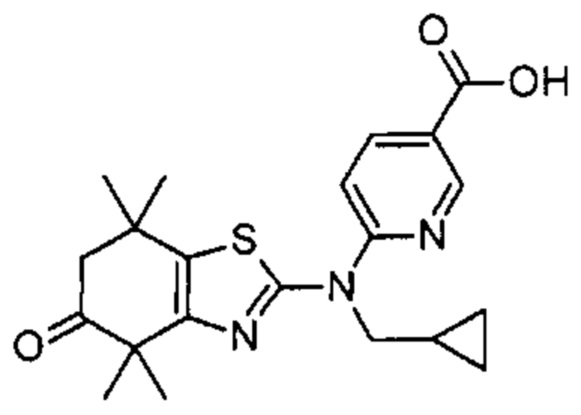
Соединение 52 синтезировали из метил 6-[(4,4,7,7-тетраметил-5-оксо-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоксилата (промежуточное соединение-67) и (бромметил)циклопропана, следуя способу, описанному в схеме 27; чистота: 96,73%.
Пример 53: 4-[(2-метилпропил)(4,4,7,7-тетраметил-5-оксо-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензойная кислота
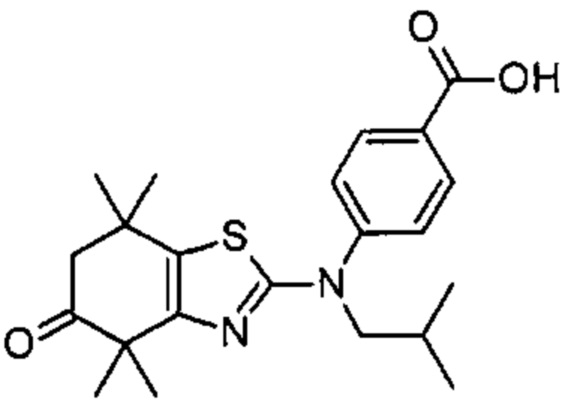
Соединение 53 синтезировали из метил 4-[(4,4,7,7-тетраметил-5-оксо-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензоата (промежуточное соединение-56) и 1-бром-2-метилпропана, следуя способу, описанному в схеме 27; чистота: 98,52%.
Пример 54: 2-метокси-4-[(2-метилпропил)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензойная кислота
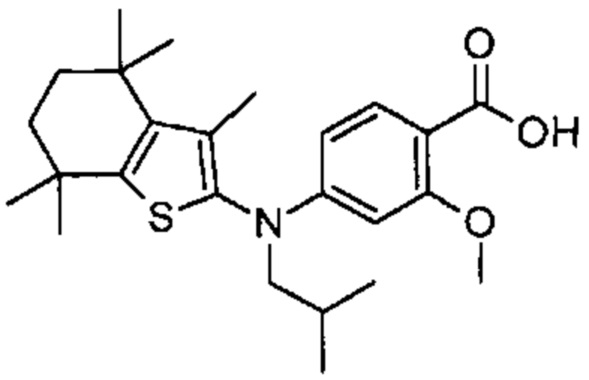
Соединение 54 синтезировали посредством реакции сочетания 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-амина (промежуточное соединение-17) и метил 4-иод-2-метоксибензоата с последующей реакцией с 1-бром-2-метилпропаном, следуя способу, описанному в схеме 27; чистота: 78,65%.
Пример 55: 2-(циклопропил метокси)-4-[(2-метилпропил)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензойная кислота
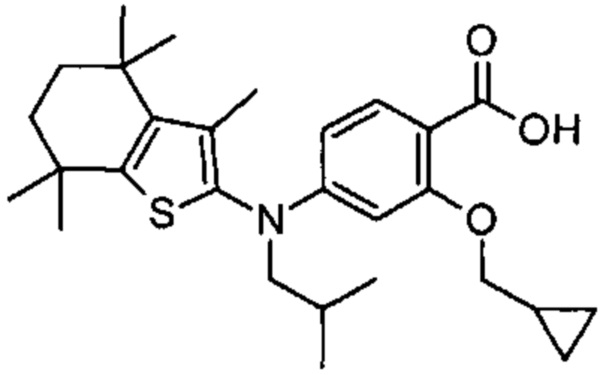
Соединение 55 синтезировали посредством реакции сочетания 3,4,4,7,4,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-амина (промежуточное соединение-17) и метил 2-(циклопропил метокси)-4-иодбензоата с последующей реакцией с 1-бром-2-метилпропаном, следуя способу, описанному в схеме 27; чистота: 93,67%.
Пример 56: 4-[(2-метилпропил)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]-2-(проп-2-ин-1-илокси) бензойная кислота
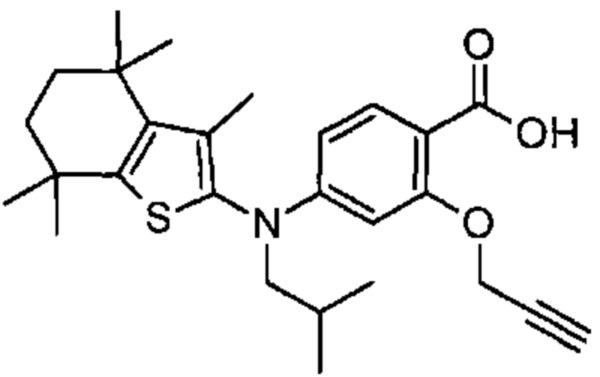
Соединение 56 синтезировали посредством реакции сочетания 3,4,4,7,4,7-пентаметил-4,5,5,7-тетрагидро-1-бензотиофен-2-амина (промежуточное соединение-17) и метил 4-иод-2-(проп-2-ин-1-илокси)бензоата с последующей реакцией с 1-бром-2-метилпропаном, следуя способу, описанному в схеме 27; чистота: 77,64%.
Пример 57: 4-[(2-метилпропил)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]-2-феноксибензойная кислота
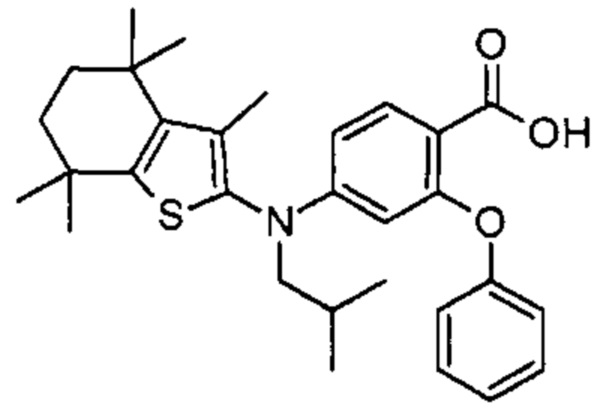
Соединение 57 синтезировали из реакции сочетания 3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-амина (промежуточное соединение-17) и метил 4-иод-2-феноксибензоата с последующей реакцией с 1-бром-2-метилпропаном, следуя способу, описанному в схеме 27; чистота: 93,79%.
Пример 58: 2-метил-4-[(2-метилпропил)(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензойная кислота
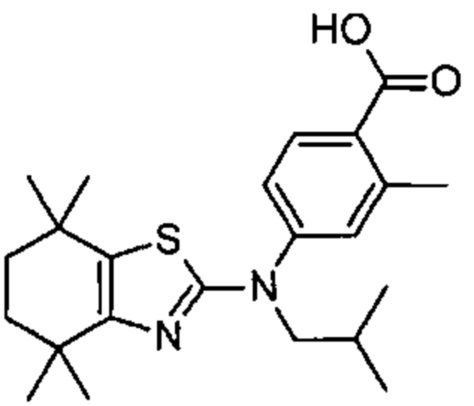
Соединение 58 синтезировали посредством реакции сочетания 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина (промежуточное соединение-64) и метил 4-бром-2-метилбензоата с последующей реакцией с 1-бром-2-метилпропаном, следуя способу, описанному в схеме 27; чистота: 97,99%.
Пример 59: 4-[(циклопропил метил)(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]-2-метилбензойная кислота
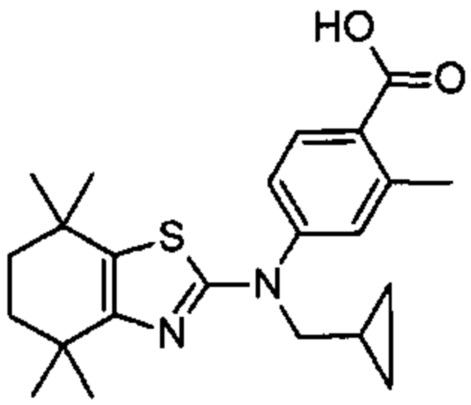
Соединение 59 синтезировали посредством реакции сочетания 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина (промежуточное соединение 64) и метил 4-бром-2-метилбензоата, с последующей реакцией с 1-бром-2-метилпропаном, следуя способу, описанному в схеме 27; чистота: 98,80%.
Пример 60: 4-[(циклопропил метил)(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]-3-метилбензойная кислота
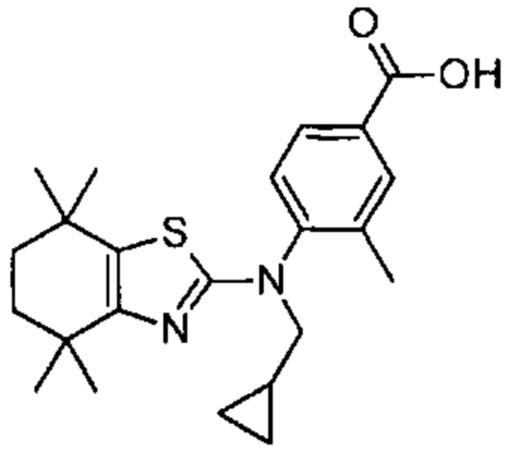
Соединение 60 синтезировали посредством реакции сочетания 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина (промежуточное соединение 64) и метил 4-бром-3-метилбензоата с последующей реакцией с (бромметил)циклопропаном, следуя способу, описанному в схеме 27; чистота: 93,08%.
Пример 61: 3-метил-4-[(2-метилпропил)(4,4,7(7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензойная кислота
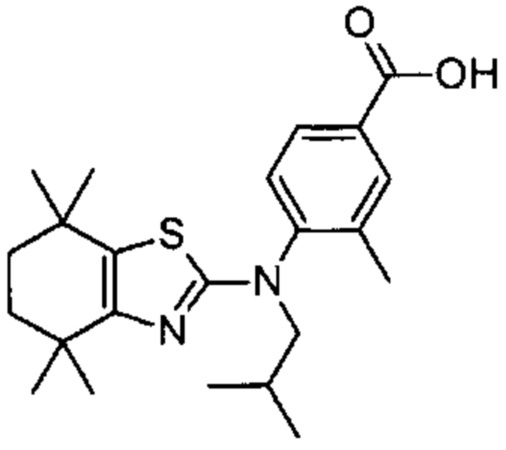
Соединение 61 синтезировали посредством реакции сочетания 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина (промежуточное соединение 64) и метил 4-бром-3-метилбензоата с последующей реакцией с 1-бром-2-метилпропаном, следуя способу, описанному в схеме 27; чистота: 98,15%.
Пример 62: 4-[проп-2-ен-1-ил(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензойная кислота
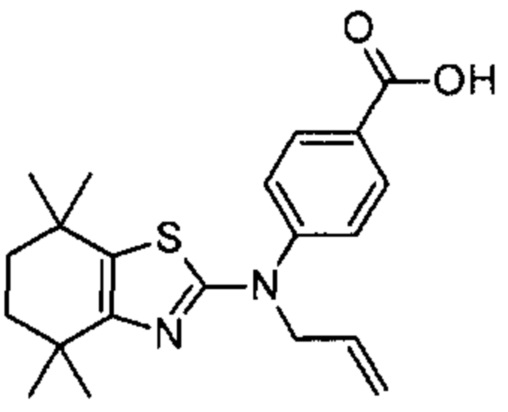
Соединение 62 синтезировали из этил 4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензоата (промежуточное соединение-65) и 3-бромпро-1-ена, следуя способу, описанному в схеме 27; чистота: 99,06%.
Пример 63: 5-[(4-карбоксифенил)(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]фуран-2-карбоновая кислота
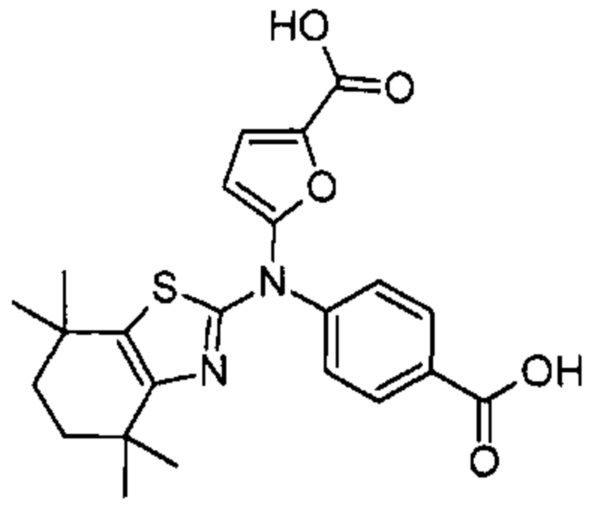
Соединение 63 синтезировали из этил 4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензоата (промежуточное соединение-65) и метил 5-бромфуран-2-карбоксилата, следуя способу, описанному в схеме 27; чистота: 89,73%.
Пример 64: 6-[(2-метилпропил)(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновая кислота
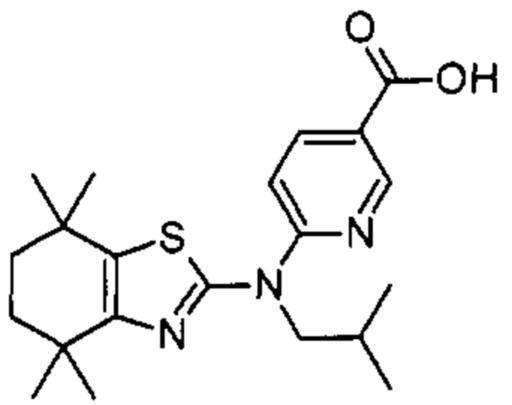
Соединение 64 синтезировали посредством реакции сочетания 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина (промежуточное соединение 64) и этил 6-хлорпиридин-3-карбоксилата с последующей реакцией с 1-бром-2-метилпропаном, как в способе, описанном в схеме 27; чистота: 95,24%.
Пример 65: 6-[пропил(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновая кислота
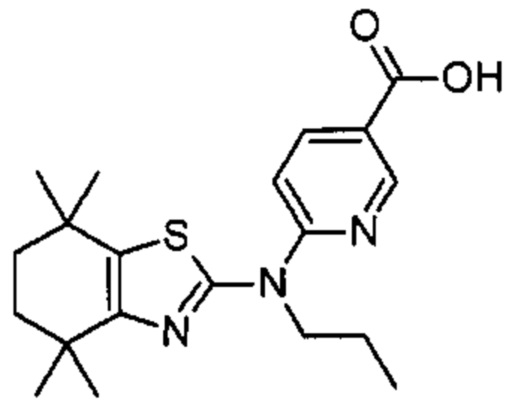
Соединение 65 синтезировали посредством реакции сочетания 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина (промежуточное соединение 64) и этил 6-хлорпиридин-3-карбоксилата с последующей реакцией с 1-бромпропаном, как в способе, описанном в схеме 27; чистота: 92,04%.
Пример 66: 6-[бензил(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновая кислота
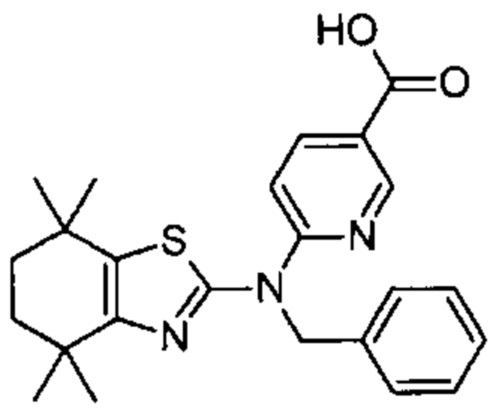
Соединение 66 синтезировали посредством реакции сочетания 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина (промежуточное соединение 64) и этил 6-хлорпиридин-3-карбоксилата с последующей реакцией с (бромметил)бензеном, следуя способу, описанному в схеме 27; чистота: 94,27%.
Пример 67: 5-[(2-метилпропил)(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-2-карбоновая кислота
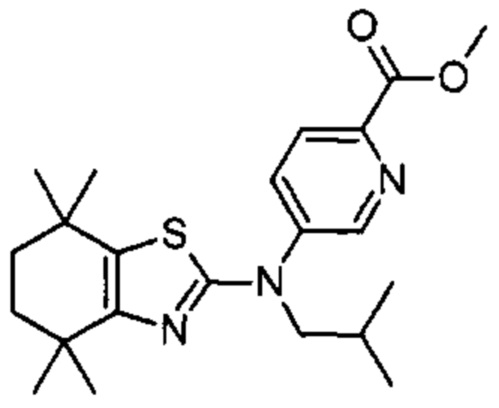
Соединение 67 синтезировали посредством реакции сочетания 4,4,4,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина (промежуточное соединение 64) и метил 5-хлорпиридин-2-карбоксилата с последующей реакцией с 1-бром-2-метилпропаном, следуя способу, описанному в схеме 27; чистота: 82,46%.
Пример 68: 6-[бензил (4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]-5-хлорпиридин-3-карбоновая кислота
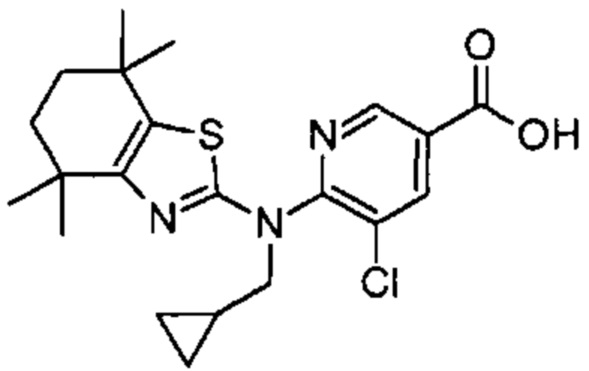
Соединение 68 синтезировали посредством реакции сочетания 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина (промежуточное соединение 64) и метил 5,6-дихлорпиридин-3-карбоксилата с последующей реакцией с (бромметил)циклопропаном, следуя способу, описанному в схеме 27; чистота: 92,59%.
Пример 69: 5-хлор-6-[(2-метилпропил)(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновая кислота
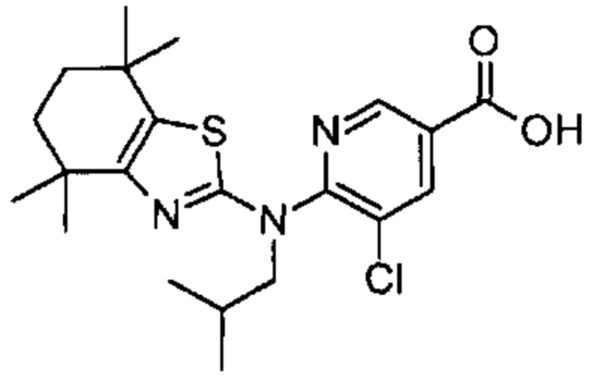
Соединение 69 синтезировали посредством реакции сочетания 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина (промежуточное соединение 64) и метил 5,6-дихлорпиридин-3-карбоксилата с последующей реакцией с 1-бром-2-метилпропаном, следуя способу, описанному в схеме 27; чистота: 91,69%.
Пример 70: 4-хлор-6-[(циклопропилметил)(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновая кислота
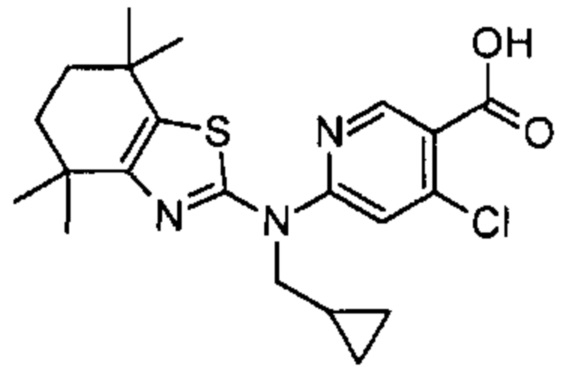
Соединение 70 синтезировали посредством реакции сочетания 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина (промежуточное соединение 64) и метил 4,6-дихлорпиридин-3-карбоксилата с последующей реакцией с (бромметил)циклопропаном, следуя способу, описанному в схеме 27; чистота: 93,32%.
Пример 71: 4-[(циклопропил метил)(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]-3-метоксибензойная кислота
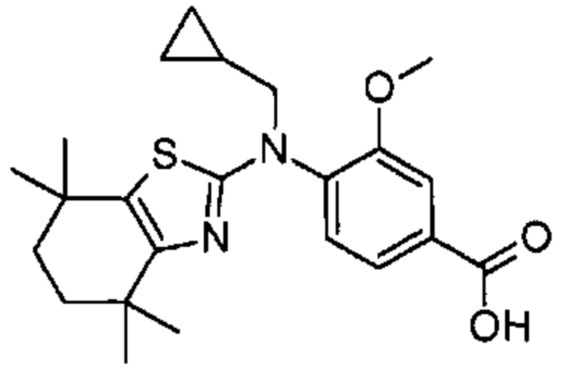
Соединение 71 синтезировали посредством реакции сочетания 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина (промежуточное соединение 64) и метил 4-иод-3-метоксибензоата с последующей реакцией с (бромметил)циклопропаном, следуя способу, описанному в схеме 27; чистота: 92,71%.
Пример 72: метил 4-[(циклопропил метил)(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]-2-метилбензоат
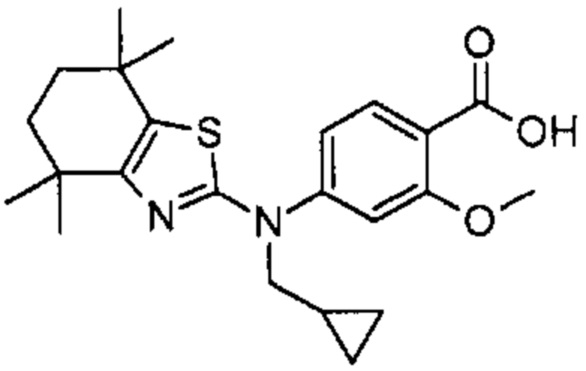
Соединение 72 синтезировали посредством реакции сочетания 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина (промежуточное соединение 64) и метил 4-иод-2-метоксибензоата с последующей реакцией с (бромметил)циклопропаном, следуя способу, описанному в схеме 27; чистота: 97,96%.
Пример 73: 2-метокси-4-[(2-метилпропил)(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензойная кислота
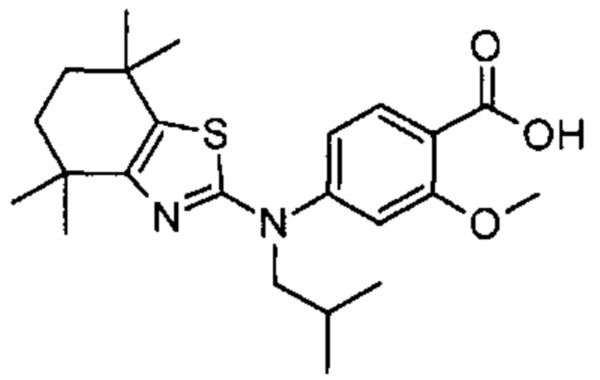
Соединение 73 синтезировали посредством реакции сочетания 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина (промежуточное соединение 64) и метил 4-иод-2-метоксибензоата с последующей реакцией с 1-бром-2-метилпропаном, следуя способу, описанному в схеме 27; чистота: 99,39%.
Схема 28
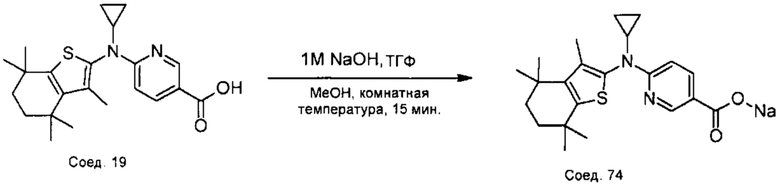
Пример 74: натрий 6-[циклопропил(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]пиридин-3-карбоксилат
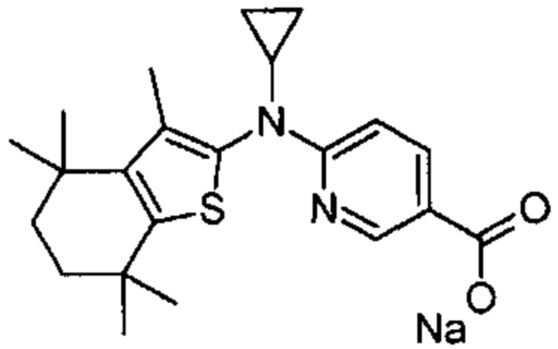
Соединение 74 синтезировали из 6-[циклопропил(3,4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]пиридин-3-карбоновой кислоты и NaOH, следуя способу, описанному в схеме 28; чистота: 98,63%.
Соединение 74: натрий 6-[циклопропил(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]пиридин-3-карбоксилат
К перемешиваемому раствору 6-[циклопропил(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]пиридин-3-карбоновой кислоты (0,02 г, 0,05 ммоль) в МеОН (0,5 мл) и ТГФ (0,5 мл) медленно добавляли раствор 1М NaOH (0,05ы г, 0,05 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 15 минут. Растворитель удаляли при пониженном давлении. В полученное вязкое вещество добавляли метанол и снова полностью удаляли его под высоким вакуумом. Этот процесс повторяли до тех пор, пока не оставалось воды. Полученное вязкое вещество кристаллизовали этилацетатом и н-гексаном с получением твердого вещества белого цвета (0,01 г, выход: 50%): МС (ESI, 120 эВ): m/z=348,18 (М-22)+.
Пример 75: натрий 4-[циклопропил (3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоат
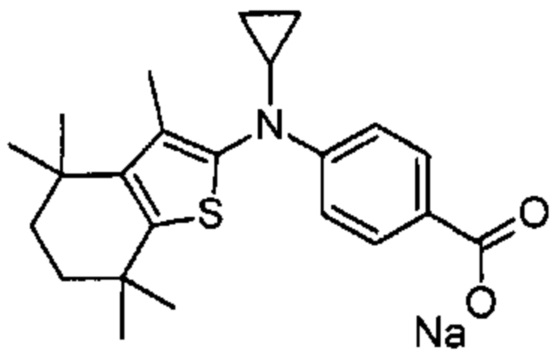
Соединение 75 синтезировали из 4-[циклопропил(3,4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензойной кислоты (соединение 10) и NaOH, следуя способу, описанному в схеме 28; чистота: 91,34%.
Пример 76: натрий 6-[(циклопропил метил)(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоксилат
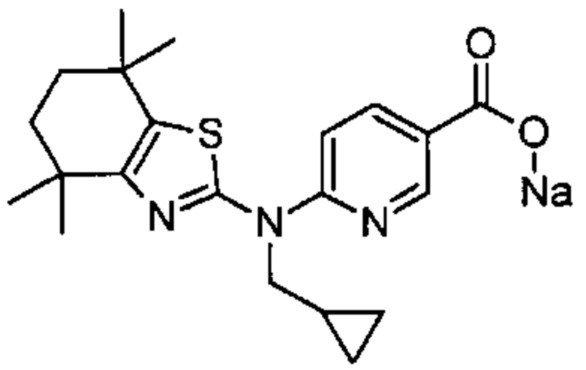
Соединение 76 синтезировали из 6-[(циклопропил метил) (4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновой кислоты (соединение 40) и NaOH, следуя способу, описанному в схеме 28; чистота: 99,83%.
Пример 77: натрий 4-[циклопропил (4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензоат
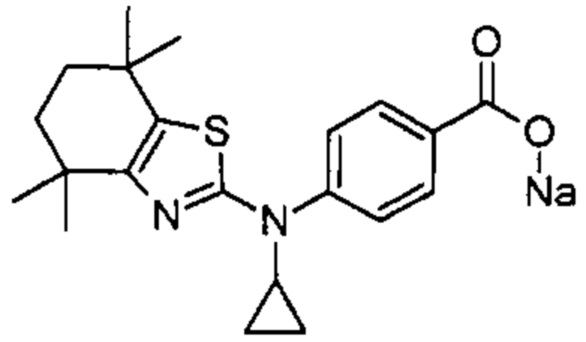
Соединение 77 синтезировали из 4-[циклопропил(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензойной кислоты (соединение 40) и NaOH, следуя способу, аналогичному описанному в схеме 28; чистота: 98,63%.
Пример 78: натрий 6-[циклопропил(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоксилат
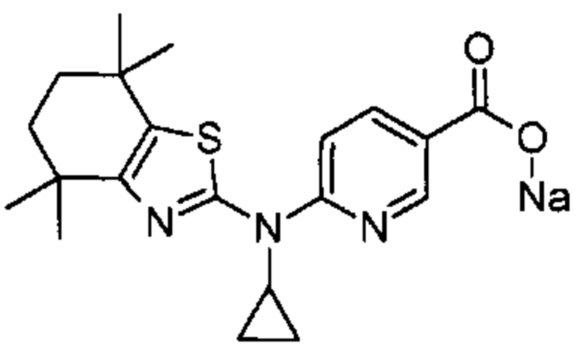
Соединение 78 синтезировали из 6-[циклопропил (4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновой кислоты и NaOH, следуя способу, аналогичному описанному в схеме 28; чистота: 95,47%.
Схема 29
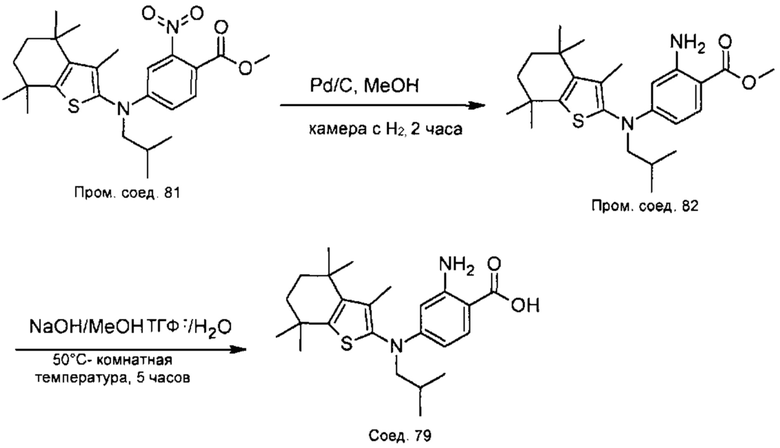
Пример 79: 2-амино-4-[(2-метилпропил)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензойная кислота
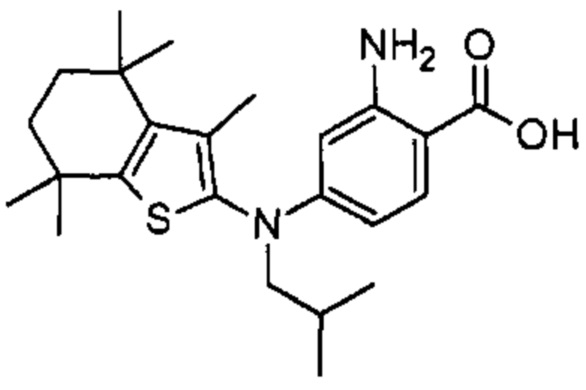
Соединение 79 синтезировали из метил 4-[(2-метилпропил)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]-2-бетробензоата (промежуточное соединение 81), следуя способу, описанному в схеме 29; чистота: 95,94%.
Промежуточное соединение 82: Метил 2-амино-4-[(2метилпропил)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоат
В круглодонную колбу объемом 25 мл с тремя горлышками, снабженную резиновой камерой, заполненной газообразным водородом, загружали метанол (5 мл). К перемешиваемому раствору метил 4-[(2-метилпропил) (3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]-2-нитробензоата (0,05 г, 0,11 ммоль) добавляли палладий на угле (0,005 г, 10%). Реакционную смесь перемешивали в течение 2 ч. Затем реакционную смесь фильтровали через слой целита, сушили над безводным Na2SO4 и концентрировали в вакууме. Полученный неочищенный продукт очищали с помощью колоночной хроматографии с получением целевого соединения в виде вязкого продукта желтого цвета (0,003 г, выход: 63,83%).
Соединение 79: 2-амино-4-[(2-метилпропил)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензойная кислота
Круглодонную колбу объемом 25 мл, снабженную магнитной мешалкой, загружали метанолом (2 мл) и ТГФ (2 мл). К перемешиваемому растворителю добавляли метил 2-амино-4-[(2-метилпропил)(4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоат (0,03 г, 0,07 ммоль) и водный NaOH (0,02 г, 0,35 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь полностью концентрировали и неочищенный продукт промывали эфиром и разбавляли водой и затем нейтрализовали 1,5 N НСl, затем экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали. Полученное твердое вещество растирали с гексаном с получением целевого соединения в виде вязкого вещества желтого цвета (0,015 г, выход: 51,72%): МС (ESI, 120 эВ): m/z=415,2 (М+Н+).
Схема 30
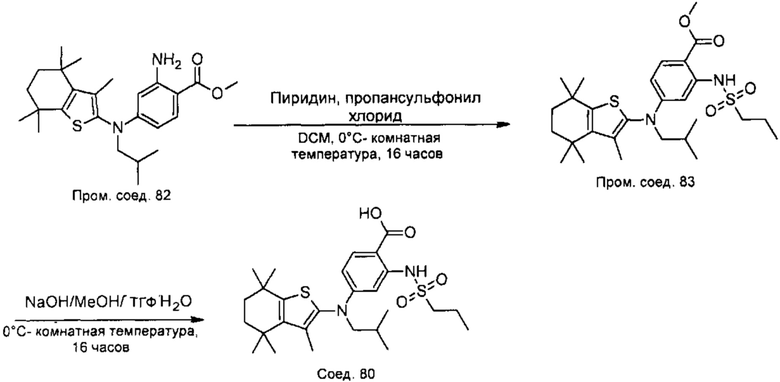
Пример 80: 4-[(2-метилпропил)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]-2-[(пропилсульфонил)амино]бензойная кислота
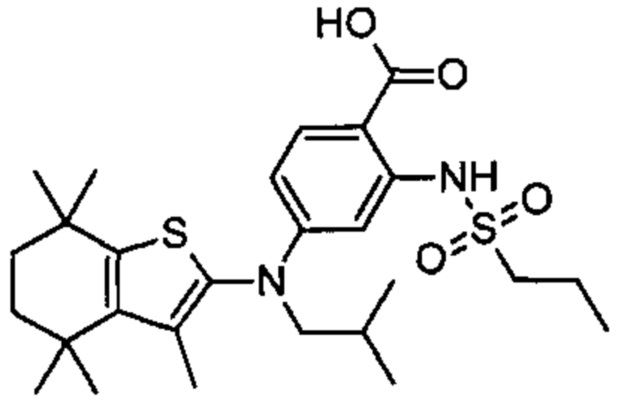
Соединение 80 синтезировали из метил 2-амино-4-[(2-метилпропил) (3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоата (промежуточное соединение-82), следуя способу, описанному в схеме 30; чистота: 88,10%.
Промежуточное соединение 83: метил 4-[(2-метилпропил) (3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]-2-[(пропилсульфонил)амино]бензоат
В круглодонную колбу 25 мл с двум горлышками, закруженную метил 2-амино-4-[(2-метилпропил)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоатом (0,05 г, 0,11 ммоль) в DCM (3 мл), по каплям добавляли пиридин (0,03 г, 0,33 ммоль) при 0°С. Затем к полученному раствору добавляли пропансульфонилхлорид (0,017 г, 0,12 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь выливали в воду и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме. Полученный неочищенный продукт очищали с помощью колоночной хроматографии с получением целевого соединения в виде жидкости желтого цвета (0,019 г, выход: 33,33%).
Соединение 80: 4-[(2-метилпропил)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]-2-[(пропилсульфонил)амино]бензойная кислота
Круглодонную колбу объемом 25 мл, снабженную магнитной мешалкой, загружали метанолом (2 мл) и ТГФ (2 мл). К этому перемешиваемому растворителю добавляли метил 2-амино-4-[(2-метилпропил) (4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоат (0,03 г, 0,07 ммоль) и водный NaOH (0,02 г, 0,35 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь полностью концентрировали и неочищенный продукт промывали эфиром и разбавляли водой и затем нейтрализовали 1,5 N HCl, затем экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали. Полученное твердое вещество растирали с гексаном с получением целевого соединения в виде вязкого вещества желтого цвета (0,015 г, выход: 51,72%): МС (ESI, 120 эВ): m/z=415,2 (М+Н+).
Схема 31
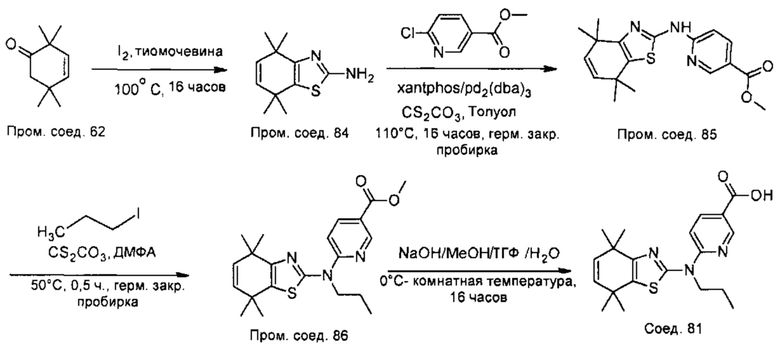
Пример 81: 6-[пропил (4,4,7,7-тетраметил-4,7-дигидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновая кислота
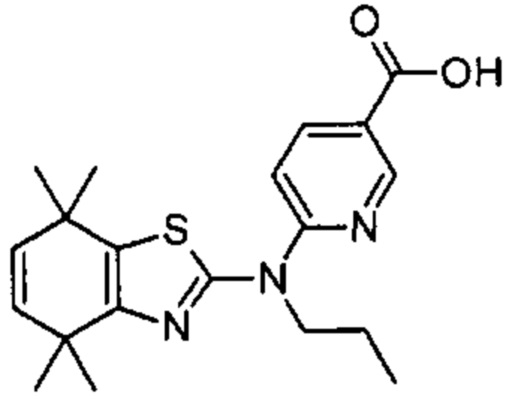
Соединение 81 синтезировали из 4,4,7,7-тетраметил-4,7-дигидро-1,3-бензотиазол-2-амина и метил 6-хлорпиридин-3-карбоксилата, следуя способу, описанному в схеме 31; чистота: 99,83%.
Промежуточное соединение 84: 4,4,7,7-тетраметил-4,7-дигидро-1,3-бензотиазол-2-амин
В круглодонную колбу 100 мл с одним горлышком, снабженную магнитной мешалкой и конденсатором, загруженную 2,2,5,5-тетраметилциклогекс-3-ен-1-оном (8,0 г, 53 ммоль), добавляли йод (13,36 г, 53 ммоль) и тиомочевину (10,0 г, 131 ммоль). Реакционную смесь нагревали при 100°С в течение 20 часов. Реакционную смесь охлаждали до комнатной температуры, выливали в водный раствор NaOH и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии с получением целевого соединения в виде твердого вещества желтого цвета (2 г, выход: 18,28%)
Промежуточное соединение 85: метил 6-[(4,4,7,7-тетраметил-4,7-дигидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоксилат
В пробирку для проведения реакций под давлением объемом 100 мл с завинчивающейся крышкой, загруженную 4,4,7,7-тетраметил-4,7-дигидро-1,3-бензотиазол-2-амином (1,7 г, 8,2 ммоль) в толуоле (6 мл), добавляли метил 6-хлорникотинат (0,7 г, 4,1 ммоль). К этому перемешиваемому растворителю добавляли CS2CO3 (3,9 г, 12,32 ммоль) и ксантфос (0,11 г, 0,21 ммоль). Полученный раствор дегазировали азотом в течение 10 минут. Затем в реакционную смесь добавляли Pd2 (dba)3 (0,075 г, 0,08 ммоль). Реакционную смесь нагревали при 100°С в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры и выливали в воду и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме. Полученный неочищенный продукт очищали на колонне с получением целевого соединения в виде твердого вещества бледно-желтого цвета (0,6 г, выход: 42,85%).
Промежуточное соединение 86: метил 6-[пропил(4,4,7,7-тетраметил-4,7-дигидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоксилат
К перемешиваемому раствору метил 6-[(4,4,7,7-тетраметил-4,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоксилат (0,14 г, 0,41 ммоль) в ДМФА (3 мл) добавляли карбонат цезия (0,4 г, 1,2 ммоль) и 1-иодпропан (0,11 г, 0,8 ммоль) при 0°С в герметично закрытую пробирку. Реакционную смесь нагревали при 50°С в течение 30 минут. Затем реакционную смесь охлаждали до комнатной температуры, выливали в воду и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме. Полученный неочищенный продукт очищали с помощью колоночной хроматографии с получением целевого соединения в виде вязкого продукта желтого цвета (0,1 г, выход: 62,5%).
Соединение 81: 6-[пропил(4,4,7,7-тетраметил-4,7-дигидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновая кислота
Круглодонную колбу объемом 25 мл, снабженную магнитной мешалкой, загружали метанолом (1 мл) и ТГФ (1 мл). К перемешиваемому растворителю добавляли метил 6-[пропил(4,4,7,7-тетраметил-4,7-дигидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоксилат (0,1 г, 0,25 ммоль) и водный NaOH (0,05 г, 1,2 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь полностью концентрировали и неочищенный продукт промывали эфиром и разбавляли водой и затем нейтрализовали 1,5 N HCl, затем экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали. Полученное твердое вещество растирали с гексаном с получением целевого соединения в виде вязкого вещества желтого цвета (0,05 г, выход: 52,63%): МС (ESI, 120 эВ): m/z=372,1 (М+Н+).
Пример 82: 6-[(2-метилпропил)(4,4,7,7-тетраметил-4,7-дигидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновая кислота
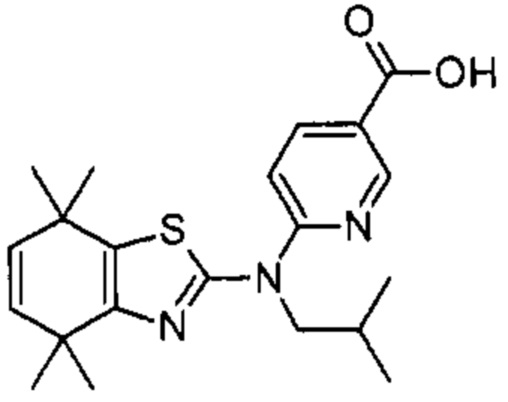
Соединение 82 синтезировали из метил 6-[(4,4,7,7-тетраметил-4,7-дигидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоксилата (промежуточное соединение 85) и 1-бром-2-метилпропана, следуя способу, описанному в схеме 31 (0,06 г, выход: 62,5%); чистота: 99,65%.
Пример 83: 4-[(2-метилпропил)(4,4,7,7-тетраметил-4,7-дигидро-1,3-бензотиазол-2-ил)амино]бензойная кислота
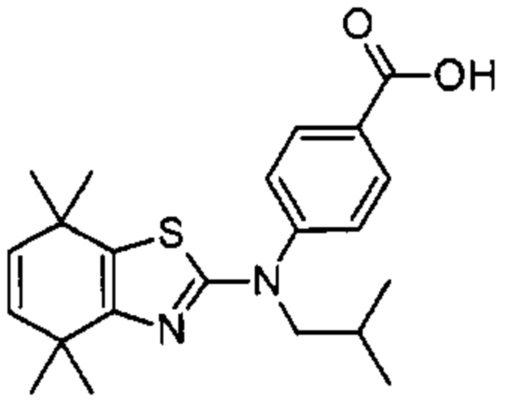
Соединение 83 синтезировали посредством реакции сочетания 4,4,4,7-тетраметил-4,7-дигидро-1,3-бензотиазол-2-амина (промежуточное соединение 84) и метил 4-иодбензоата с последующей реакцией с 1-бром-2-метилпропаном, следуя способу, описанному в схеме 31 (0,14 г, 29,78%); чистота: 98,91%.
Схема 32
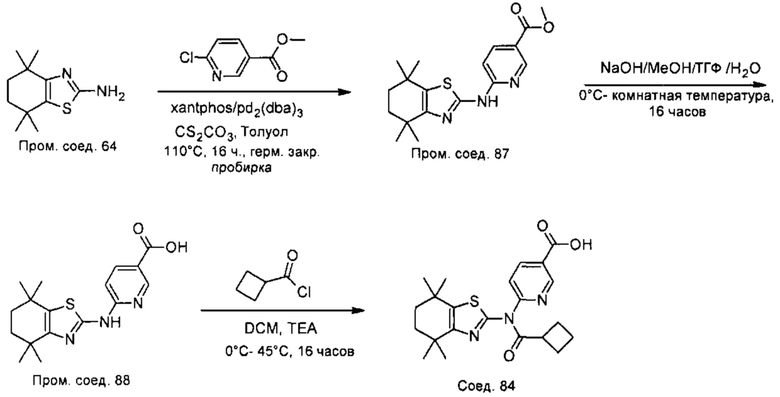
Пример 84: 6-[(циклобутилкарбонил)(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновая кислота
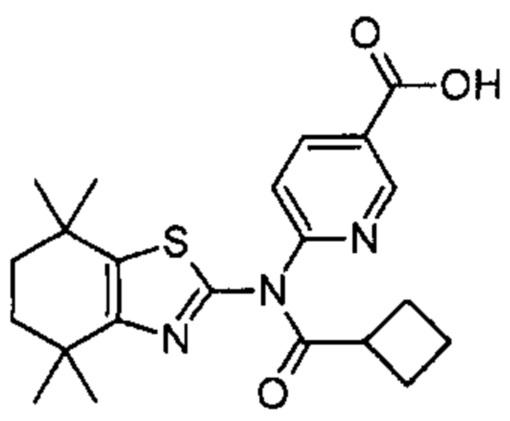
Соединение 84 синтезировали из 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина и метил 6-хлорпиридин-3-карбоксилата, следуя способу, описанному в схеме 32; чистота: 98,33%.
Промежуточное соединение 87: метил 6-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоксилат
В пробирку для проведения реакций под давлением объемом 100 мл с завинчивающейся крышкой, загруженную 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амином (1,8 г, 8,2 ммоль) в толуоле (6 мл), добавляли метил 6-хлорникотинат (0,7 г, 4,1 ммоль). К перемешиваемому растворителю добавляли CS2CO3 (3,9 г, 12,32 ммоль) и ксантфос (0,11 г, 0,21 ммоль). Полученный раствор дегазировали азотом в течение 10 минут. Затем в реакционную смесь добавляли Pd2 (dba)3 (0,075 г, 0,08 ммоль). Реакционную смесь нагревали при 100°С в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры и выливали в воду и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме. Полученный неочищенный продукт очищали на колонне с получением целевого соединения в виде твердого вещества желтого цвета (0,6 г, выход: 42,86%).
Промежуточное соединение 88: 6-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновая кислота
Круглодонную колбу объемом 25 мл, снабженную магнитной мешалкой, загружали метанолом (15 мл) и ТГФ (5 мл). К перемешиваемому растворителю добавляли метил 6-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоксилат (0,35 г, 1,0 ммоль) и водный NaOH (0,02 г, 5,0 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь полностью концентрировали и неочищенный продукт промывали эфиром и разбавляли водой и затем нейтрализовали 1,5 N HCl, затем экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали. Полученное твердое вещество растирали с гексаном с получением целевого соединения в виде твердого вещества бледно-желтого цвета (0,25 г, выход: 75,75%).
Соединение 84: 6-[(циклобутилкарбонил)(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновая кислота
В круглодонную колбу 50 мл с двумя горлышками, загруженную 6-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновой кислотой (0,12 г, 0,36 ммоль) в сухом DCM (4 мл), по каплям добавляли триэтиламин (0,25 г, 2,5 ммоль) при 0°С. Затем к полученному раствору добавляли циклобутилкарбонилхлорид (0,17 г, 1,44 ммоль) при 0°С. Реакционную смесь нагревали при 45°С в течение 16 часов. Реакционную смесь выливали в воду и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме. Полученный неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением целевого соединения в виде твердого вещества желтого цвета (0,002 г, выход: 1,43%). МС (ESI, 120 эВ): m/z=414,2 (М+Н+).
Пример 85: 6-[ацетил(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновая кислота
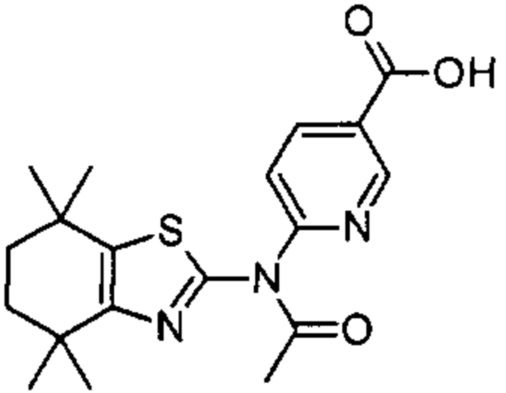
Соединение 85 синтезировали из реакции сочетания 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина и метил 6-хлорпиридин-3-карбоксилата с последующей реакцией с ацетилхлоридом, следуя способу, описанному в схеме 32; чистота: 99,17%.
Схема 33
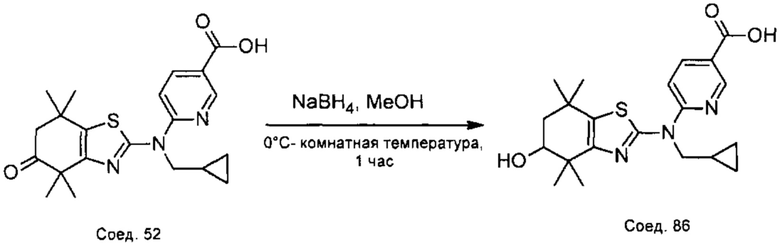
Пример 86: 6-[(циклопропилметил)(5-гидрокси-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновая кислота
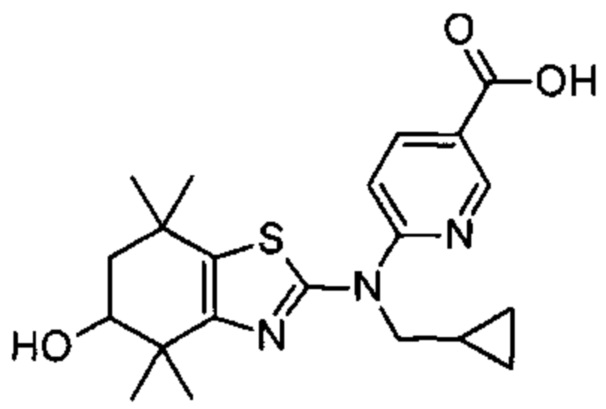
Соединение 86 синтезировали из 6-[(циклопропилметил)(4,4,7,7-тетраметил-5-оксо-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновой кислоты, следуя способу, описанному в схеме 33; чистота: 97,76%.
Соединение 86: 6-[(циклопропилметил)(5-гидрокси-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновая кислота
К перемешиваемому раствору 6-[(циклопропилметил)(5-гидрокси-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновой кислоты (0,75 г, 0,18 ммоль) в метаноле (3 мл) добавляли NaBH4 (0,0068 г, 0,18 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь гасили льдом и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме с получением чистого продукта в виде твердого вещества желтого цвета (0,058 г, выход: 85,71%). МС (ESI, 120 эВ): m/z=402,1 (М+Н+).
Схема 34
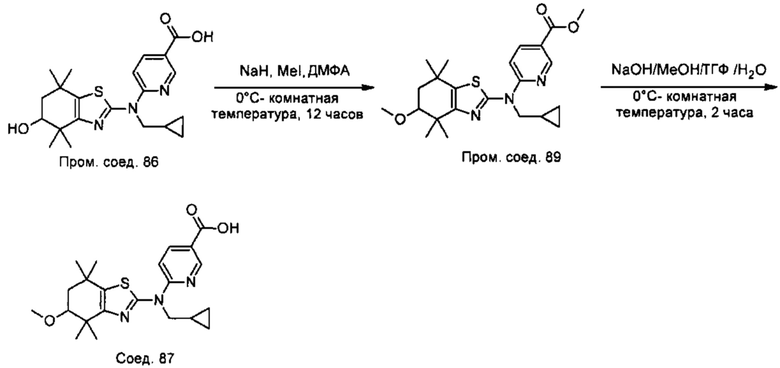
Пример 87: 6-[(циклопропилметил)(5-метокси-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновая кислота
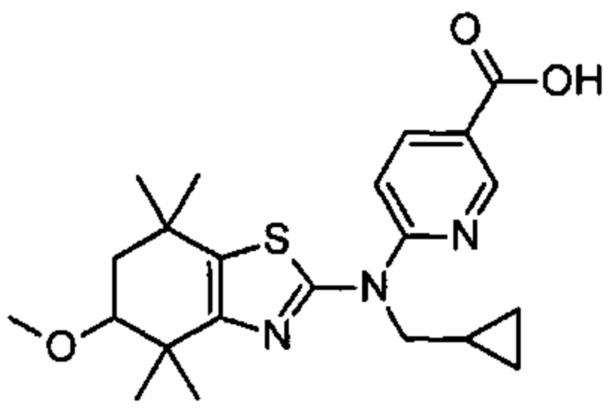
Соединение 87 синтезировали из 6-[(циклопропилметил)(5-гидрокси4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновой кислоты, следуя способу, описанному в схеме 34; чистота: 98,09%.
Промежуточное соединение 89: метил 6-[(циклопропилметил)(5-метокси-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоксилат
К перемешиваемому раствору NaH (0,012 г, 0,49 ммоль) в сухом ДМФА (3 мл) добавляли 6-[(циклопропилметил)(5-гидрокси-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновую кислоту (0,05 г, 0,12 ммоль) в ДМФА (2 мл) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 30 минут. К перемешиваемому раствору добавляли метилиодид (0,09 г, 0,6 ммоль) в ДМФА (1 мл) при 0°С и перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь гасили 1,5N HCl и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали с помощью CombiFlash с получением целевого соединения в виде бесцветной жидкости (0,02 г, выход: 40%).
Соединение 87: 6-[(циклопропилметил)(5-метокси-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновая кислота
Круглодонную колбу объемом 25 мл, снабженную магнитной мешалкой, загружали метанолом (1 мл) и ТГФ (1 мл). К перемешиваемому растворителю добавляли метил 6-[(циклопропилметил)(5-метокси-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоксилат (0,2 г, 0,46 ммоль) и водный NaOH (0,01 г, 2,3 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь полностью концентрировали и неочищенный продукт промывали эфиром и разбавляли водой и затем нейтрализовали 1,5 N HCl и экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали. Полученное твердое вещество растирали с гексаном с получением целевого соединения в виде твердого вещества желтого цвета (0,017 г, выход: 89,47%): МС (ESI, 120 эВ): m/z=416,2 (М+Н+).
Схема 35
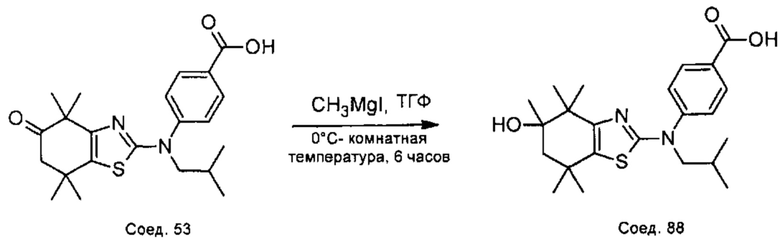
Пример 88: 4-[(5-гидрокси-4,4,5,7,7-пентаметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)(2-метилпропил)амино]бензойная кислота
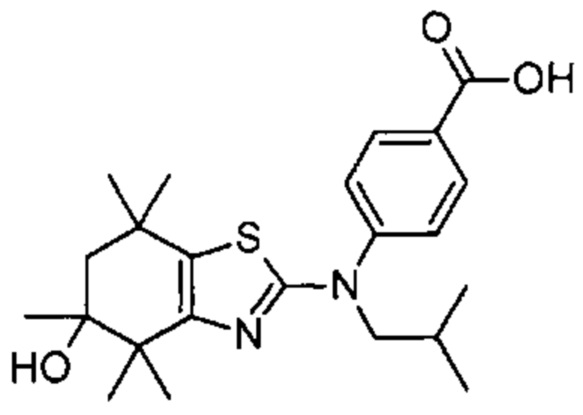
Соединение 88 синтезировали с помощью обработки 4-[(2-метилпропил)(4,4,7,7-тетраметил-5-оксо-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензойной кислоты реактивом Гриньяра, следуя способу, описанному в схеме 35; чистота: 94,80%.
Соединение 88: 4-[(5-гидрокси-4,4,5,7,7-пентаметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)(2-метилпропил)амино]бензойная кислота
В круглодонную колбу объемом 25 мл с двумя горлышками, загруженную 4-[(2-метилпропил)(4,4,7,7-тетраметил-5-оксо-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензойной кислотой (0,14 г, 0,35 ммоль), добавляли ТГФ (4 мл). К перемешиваемому раствору по каплям добавляли метилмагнийиодид (0,5 мл, 1,39 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 6 часов. Реакционную смесь гасили 1,5N HCl и экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии с получением целевого соединения в виде твердого вещества желтого цвета (0,007 г, выход: 4,66%): МС (ESI, 120 эВ): m/z=417,2 (М+Н+).
Схема 36
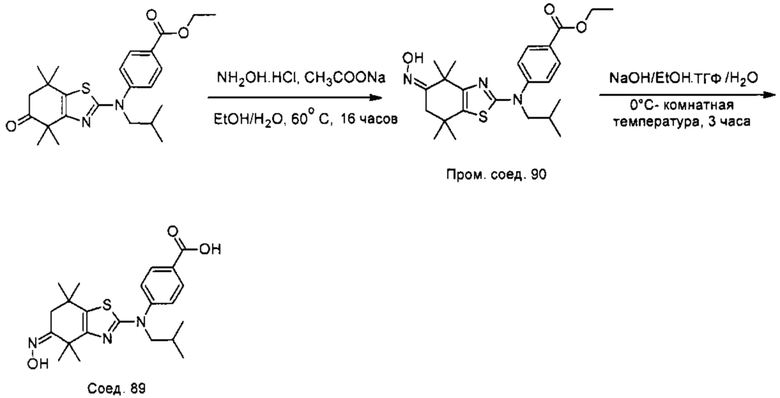
Пример 89: 4-{[(5Z)-5-(гидроксиимино)-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил](2-метилпропил)амино}бензойная кислота
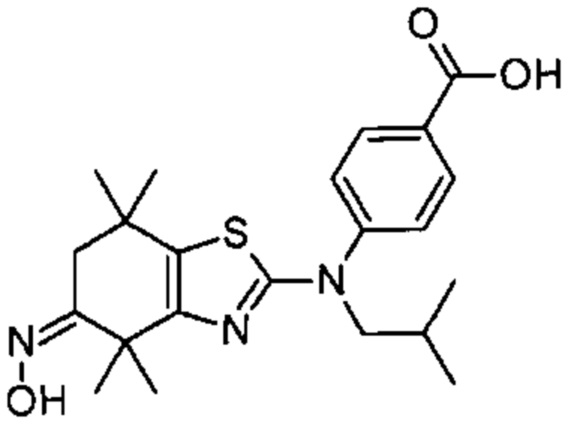
Соединение 89 синтезировали из этил 4-[(2-метилпропил) (4,4,7,7,-5-тетраметил-5-оксо-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензоата, следуя способу, описанному в схеме 36; чистота: 93,04%.
Промежуточное соединение 90: этил 4-{[(5Z)-5-(гидроксиимино)-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил](2-метилпропил)амино}бензоат
К перемешиваемому раствору этил 4-[(4,4,7,7-тетраметил-5-оксо-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензоата (0,1 г, 0,23 ммоль) в этаноле (1 мл) и воде (1 мл) добавляли ацетат натрия (0,037 г, 0,46 ммоль) с последующим добавлением хлоргидрата гидроксиламина (0,03 г, 0,46 ммоль) при 0°С. Реакционную смесь нагревали при 60°С в течение 16 часов. После завершения реакции к реакционной смеси добавляли воду и экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии с получением целевого соединения в виде твердого вещества белого цвета (0,05 г, выход: 50%): МС (ESI, 120 эВ): m/z=417,2 (М+Н+).
Соединение 89: 4-{[(5Z)-5-(гидроксиимино)-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил](2-метилпропил)амино}бензойная кислота
Круглодонную колбу объемом 25 мл, снабженную магнитной мешалкой, загружали этанолом (1 мл) и ТГФ (1 мл). К перемешиваемому растворителю добавляли этил 4-{[(5Z)-5-(гидроксиимино)-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил](2-метилпропил)амино}бензоат (0,05 г, 0,12 ммоль) и водный NaOH (0,02 г, 0,56 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь полностью концентрировали, неочищенный продукт промывали эфиром и разбавляли водой, а затем нейтрализовали 1,5 N HCl, после чего экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали. Полученное твердое вещество растирали с гексаном с получением целевого соединения в виде твердого вещества белого цвета (0,03 г, выход: 60%): МС (ESI, 120 эВ): m/z=416,2 (М+Н+).
Схема 37
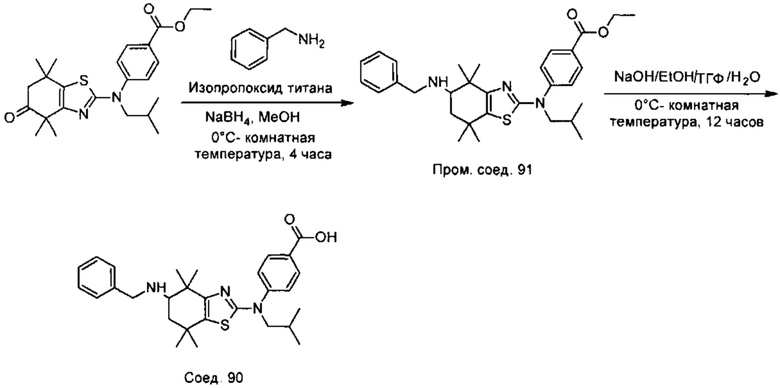
Пример 90: 4-{[5-(бензиламино)-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил](2-метилпропил)амино}бензойная кислота
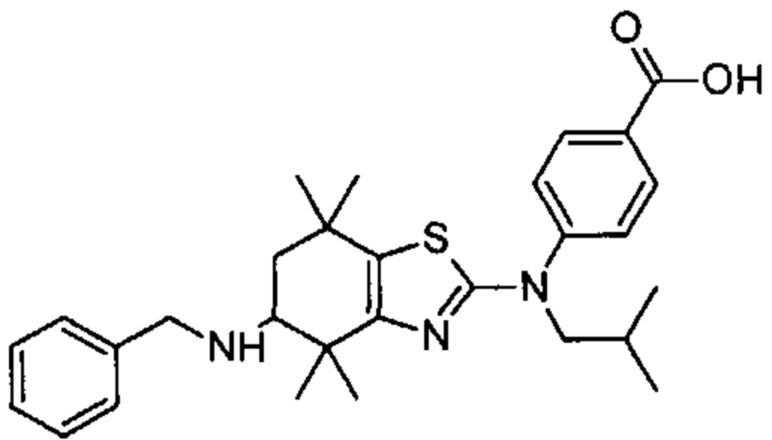
Соединение 90 синтезировали из этил 4-[(2-метилпропил) (4,4,7,7,-5-тетраметил-5-оксо-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензоата, следуя способу, описанному в схеме 37; чистота: 98,76%.
Промежуточное соединение 91: этил 4-{[5-(бензиламино)-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил](2-метилпропил)амино}бензоат
В круглодонную колбу объемом 25 мл с двумя горлышками, снабженную магнитной мешалкой, загруженную этил 4-[(2-метилпропил) (4,4,7,7-тетраметил-5-оксо-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензоатом (1 г, 0,23 ммоль), добавляли бензиламин (0,032 г, 0,34 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. К перемешиваемому раствору добавляли метанол (10 мл) с последующим добавлением NaBH4 (0,017 г, 0,46 ммоль) при 0°С. Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 4 часов. Реакционную смесь выливали на лед и экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали. Неочищенный продукт очищали с помощью колоночной хроматографии с получением целевого соединения в виде твердого вещества почти белого цвета (0,05 г, выход: 41,66%).
Соединение 90: 4-{[5-(бензиламино)-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил](2-метилпропил)амино}бензойная кислота
Круглодонную колбу объемом 25 мл, снабженную магнитной мешалкой, загружали этанолом (6 мл) и ТГФ (12 мл). К перемешиваемому растворителю добавляли этил 4-{[5-(бензилмино)-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил](2-метилпропил)амино}бензоат (0,025 г, 0,05 ммоль) и водный NaOH (0,01 г, 0,22 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь полностью концентрировали и неочищенный продукт промывали эфиром и разбавляли водой и затем нейтрализовали 1,5 N HCl, затем экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта. Полученный неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением целевого соединения в виде твердого вещества белого цвета (0,003 г, выход: 60%): МС (ESI, 120 эВ): m/z=492,2 (М+Н+).
Схема 38
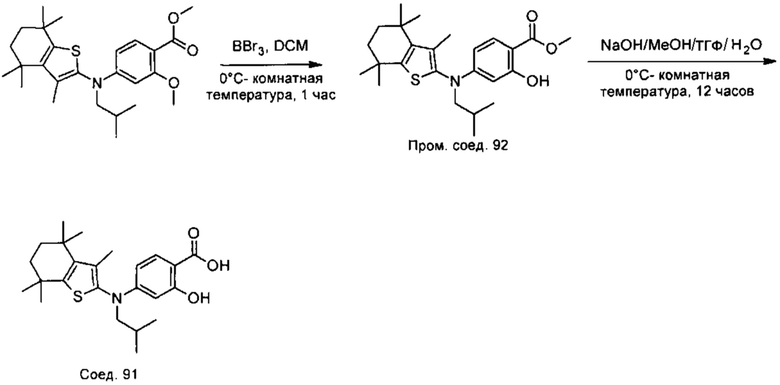
Пример 91: 2-гидрокси-4-[(2-метилпропил)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензойная кислота
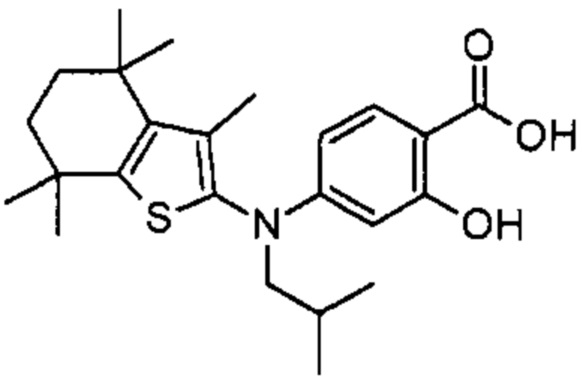
Соединение 91 синтезировали из метил 2-метокси-4-[(2-метилпропил)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоата, следуя способу, описанному в схеме 38; чистота: 90,27%.
Промежуточное соединение 92: метил 2-метокси-4-[(2метилпропил)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоат
К перемешиваемому раствору метил 2-метокси-4-[(2-метилпропил)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоата (0,02 г, 0,45 ммоль) в DCM (2 мл) по каплям добавляли трибромид бора (0,02 г, 0,9 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь гасили раствором бикарбоната и экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта в виде жидкости коричневого цвета (0,012 г, выход: 63%).
Соединение 91: 2-гидрокси-4-[(2-метилпропил)(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензойная кислота
Круглодонную колбу объемом 25 мл, снабженную магнитной мешалкой, загружали метанолом (1 мл) и ТГФ (1 мл). К перемешиваемому растворителю добавляли метил 2-метокси-4-[(2-метилпропил)(4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоат (0,012 г, 0,03 ммоль) и водный NaOH (0,006 г, 0,14 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь полностью концентрировали, неочищенный продукт промывали эфиром и разбавляли водой, а затем нейтрализовали 1,5 N HCl, после чего экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали. Полученный продукт растирали с гексаном с получением целевого соединения в виде твердого вещества бледно-желтого цвета (0,006 г, выход: 54,44%): МС (ESI, 120 эВ): m/z=414,2 (М-Н+);
Схема 39
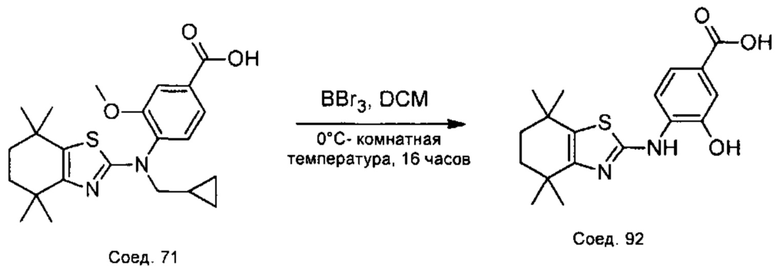
Пример 92: 3-гидрокси-4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензойная кислота
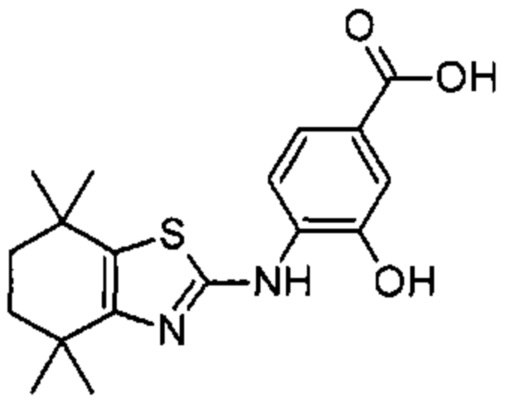
Соединение 92 синтезировали из 4-[(циклопропилметил)(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]-3-метоксибензойной кислоты, следуя способу, описанному в схеме 39; чистота: 94,26%.
Соединение 92: 3-гидрокси-4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензойная кислота
К суспензии 4-[(циклопропилметил)(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]-3-метоксибензойной кислоты (0,14 г, 0,34 ммоль) в DCM (5 мл) добавляли ВВr3 (0,17 г, 0,68 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Затем реакционную смесь гасили льдом и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта в виде жидкости коричневого цвета (0,55 г, 40%). МС (ESI, 120 эВ): m/z=(М+Н)+. 347,1.
Схема 40
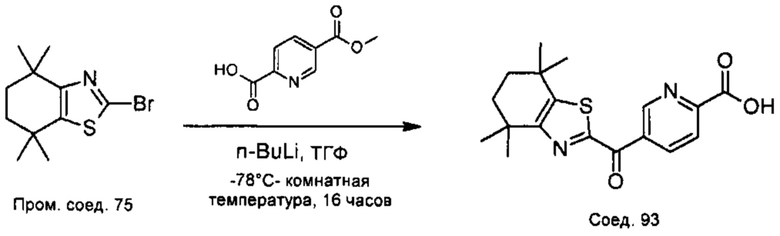
Пример 93: 5-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)карбонил]пиридин-2-карбоновая кислота
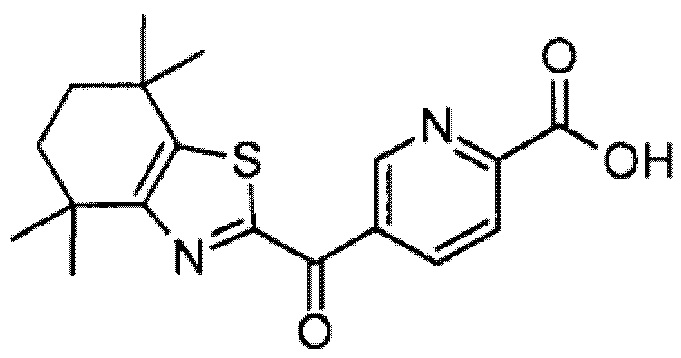
Соединение 93 синтезировали из 2-бром-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазола и 4-(метоксикарбонил)пиридин-2-карбоновой кислоты, следуя способу, описанному в схеме 40; чистота: 90,19%.
Соединение 93: 5-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)карбонил]пиридин-2-карбоновая кислота
В круглодонную колбу объемом 100 мл с двумя горлышками снабженную магнитной мешалкой, загруженную 2-бром-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол (2,5 г, 9,12 ммоль) в ТГФ (30 мл), по каплям добавляли н-бутиллитий (7,3 мл, 18,24 ммоль, 2,5 М раствор) при -78°С. Полученный раствор перемешивали при -78°С в течение 1 часа. Затем к полученному раствору по каплям добавляли 5-(метоксикарбонил)пиридин-2-карбоновую кислоту (1,4 г, 7,7 ммоль) в ТГФ (20 мл). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 16 часов. Реакционную смесь гасили раствором насыщенным хлоридом аммония и экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта. Полученный продукт очищали с помощью колоночной хроматографии с получением целевого соединения в виде твердого вещества белого цвета (2,5 г, выход: 96,15%). МС (ESI, 120 эВ): m/z=345,12 (М+Н+).
Схема 41
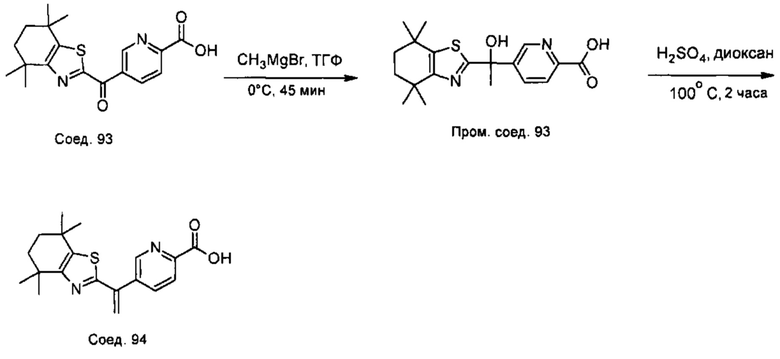
Пример 94: 5-[1-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)этенил]пиридин-2-карбоновая кислота
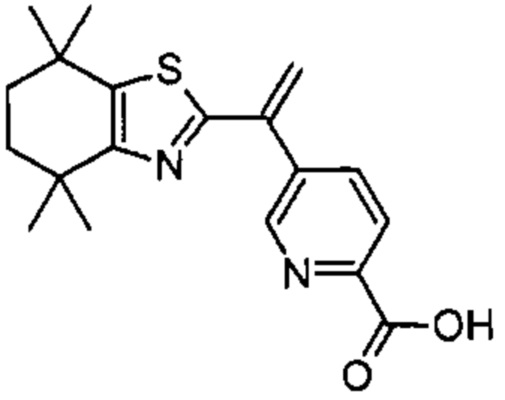
Соединение 94 синтезировали из 5-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)карбонил]пиридин-2-карбоновой кислоты, следуя способу, описанному в схеме 41; чистота: 87,82%.
Промежуточное соединение 93: 5-[1-гидрокси-1 -(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)этил]пиридин-2-карбоновая кислота
В круглодонную колбу объемом 100 мл с двумя горлышками, снабженную магнитной мешалкой, загруженную 5-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)карбонил]пиридин-2-карбоновой кислотой (0,8 г, 2,3 ммоль) в ТГФ (20 мл), по каплям добавляли метилмагнийиодид (1,74 мл, 6,9 ммоль, 4М раствор) при 0°С. Реакционную смесь перемешивали при 0°С в течение 40 минут. После завершения реакции реакционную смесь гасили HCL и экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта. Полученный продукт очищали с помощью колоночной хроматографии с получением целевого соединения в виде вязкого вещества желтого цвета (0,65 г, выход: 78,31%).
Соединение 94: 5-[1-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)этенил]пиридин-2-карбоновая кислота
К перемешиваемому раствору 5-[1-гидрокси-1-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)этил]пиридин-2-карбоновой кислоты (0,5 г, 1,39 ммоль) в диоксане (20 мл) добавляли концентрированную H2SO4. Реакционную смесь нагревали при 100°С в течение 4 часов. Развитие реакции контролировали с помощью ЖХ-МС. К реакции добавляли воду и экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта. Полученный продукт очищали с помощью препаративной ВЭЖХ с получением целевого соединения в виде твердого вещества желтого цвета (0,045 г, выход: 9,37%). МС (ESI, 120 эВ): m/z=343,15 (М+Н+).
Схема 42
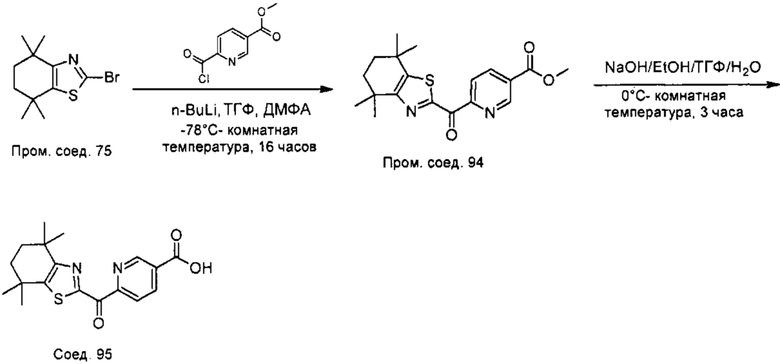
Пример 95: 6-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)карбонил]пиридин-3-карбоновая кислота
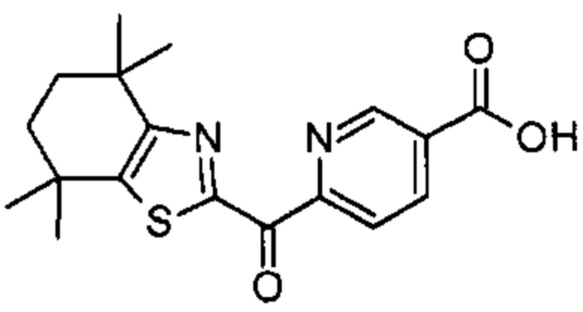
Соединение 95 синтезировали из 2-бром-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол и метил 6-(хлоркарбонил)пиридин-3-карбоксилата, следуя способу, описанному в схеме 42; чистота: 90,94%.
Промежуточное соединение 94: метил 6-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)карбонил]пиридин-3-карбоксилат
К перемешиваемому раствору 2-бром-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол (1 г, 3,64 ммоль) в ТГФ (10 мл) по каплям добавляли н-бутил лития при -78°С. Полученный раствор перемешивали при -78°С в течение 45 минут. К перемешиваемому раствору добавляли метил 6-(хлоркарбонил)пиридин-3-карбоксилат (1,32 г, 7,3 ммоль) [который получали взаимодействием хлористого тионила (15 мл) с 5-(метоксикарбонил)пиридин-2-карбоновой кислоты]. Реакционную смесь нагревали при 75°С в течение 3 часов, затем при комнатной температуре 16 часов. Реакционную смесь гасили насыщенным раствором хлорида аммония и экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта. Полученный продукт очищали с помощью колоночной хроматографии с получением целевого соединения в виде вязкого вещества коричневого цвета (0,06 г, выход: 4,6%). МС (ESI, 120 эВ): m/z=345,1 (M+H+).
Соединение 95: 6-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)карбонил]пиридин-3-карбоновая кислота
Круглодонную колбу объемом 25 мл, снабженную магнитной мешалкой, загружали метанолом (10 мл) и ТГФ (5 мл). К перемешиваемому растворителю добавляли метил 6-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)карбонил]пиридин-3-карбоксилат (0,06 г, 0,16 ммоль) и водный NaOH (0,067 г, 1,6 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь полностью концентрировали и неочищенный продукт промывали эфиром и разбавляли водой и затем нейтрализовали 1,5 N HCl, затем экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали с получением продукта. Полученный продукт растирали с гексаном с получением целевого соединения в виде вязкого вещества коричневого цвета (0,015 г, выход: 26,3%): МС (ESI, 120 эВ): m/z=345,1 (М+Н+).
Схема 43
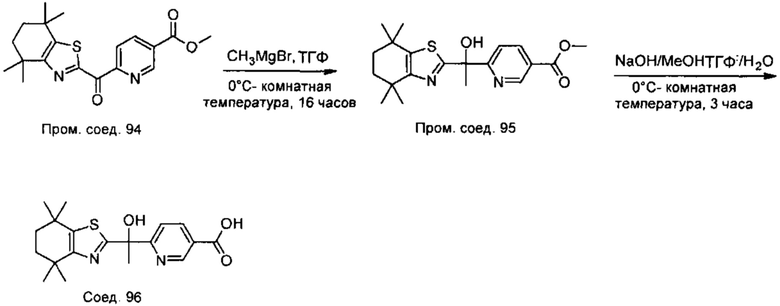
Пример 96: 6-[1-гидрокси-1-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)этил]пиридин-3-карбоновая кислота
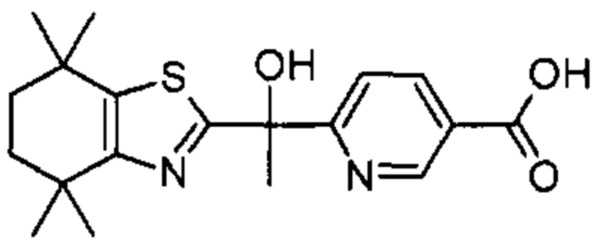
Соединение 96 синтезировали из метил 6-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)карбонил]пиридин-3-карбоксилата, следуя способу, описанному в схеме 43; чистота: 89,16%.
Промежуточное соединение 95: метил 6-[1-гидрокси-1-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)этил]пиридин-3-карбоксилат
В круглодонную колбу 100 мл с одним горлышком, снабженную магнитной мешалкой, загруженную 6-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол)карбонил]пиридин-3-карбоксилатом (0,6 г, 0,17 ммоль) в ТГФ (5 мл), по каплям добавляли бромметилмагний (0,17 мл, 0,5 ммоль, 4М раствор) при 0°С. Реакционную смесь перемешивали в течение 12 часов. После завершения реакции реакционную смесь гасили льдом и экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта. Полученный продукт очищали с помощью препаративной ТСХ с получением целевого соединения в виде твердого вещества белого цвета (0,025 г, выход: 43,8%).
Соединение 96: 6-[1-гидрокси-1-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)этил]пиридин-3-карбоновая кислота
Круглодонную колбу объемом 25 мл, снабженную магнитной мешалкой, загружали метанолом (10 мл) и ТГФ (5 мл). К перемешиваемому растворителю добавляли метил 6-[1-гидрокси-1-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)этил]пиридин-3-карбоксилат (0,012 г, 0,03 ммоль) и водный NaOH (0,13 г, 0,32 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь полностью концентрировали и неочищенный продукт промывали эфиром и разбавляли водой и затем нейтрализовали 1,5 N HCl, затем экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали с получением продукта. Полученный продукт растирали с гексаном с получением целевого соединения в виде твердого вещества коричневого цвета (0,01 г, выход: 90,90%): МС (ESI, 120 эВ): m/z=361,1 (М+Н+).
Схема 44
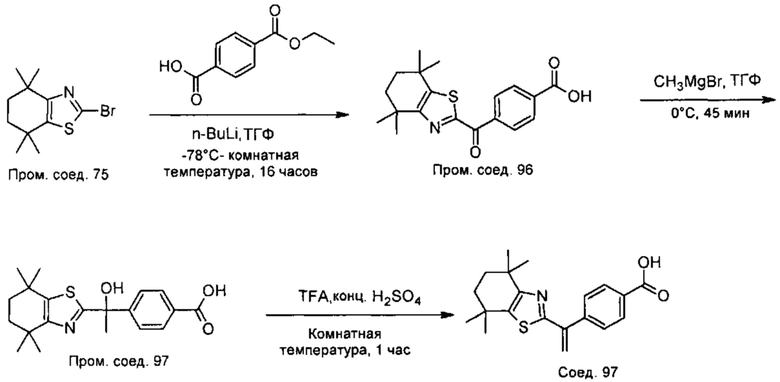
Пример 97: 4-[1-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)этенил]бензойная кислота
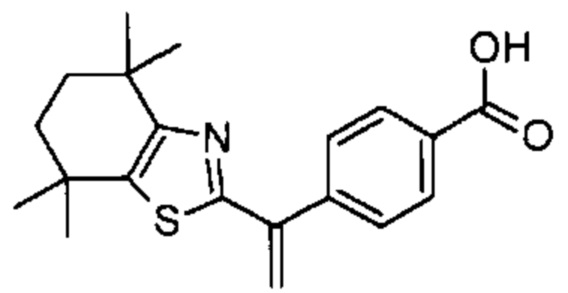
Соединение 97 синтезировали из 2-бром-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол- и 4-(этоксикарбонил)бензойной кислоты, следуя способу, указанному в схеме 44; чистота: 99,77%.
Промежуточное соединение 96: 4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)карбонил]бензойная кислота
В круглодонную колбу 100 мл с двумя горлышками, снабженную магнитной мешалкой, загруженную 2-бром-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол (0,91 г, 3,3 ммоль) в ТГФ (5 мл), по каплям добавляли н-бутиллитий (2,2 мл, 5,5 ммоль, 2,5М раствор) при -78°С. Полученный раствор перемешивали при -78°С в течение 1 часа. Затем полученному раствору по каплям добавляли 4-(этоксикарбонил)бензойную кислоту (0,5 г, 2,7 ммоль) в ТГФ (5 мл). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 16 часов. Реакционную смесь гасили насыщенным раствором хлорида аммония и экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта. Полученный продукт очищали с помощью колоночной хроматографии с получением целевого соединения в виде твердого вещества белого цвета (0,1 г, выход: 10,5%). МС (ESI, 120 эВ): m/z=344,1 (М+Н+).
Промежуточное соединение 97: 4-[1-гидрокси-1-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)этил]бензойная кислота
В круглодонную колбу объемом 100 мл с одним горлышком, снабженную магнитной мешалкой, загруженную 4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)карбонил]бензойной кислотой (0,1 г, 0,29 ммоль) в ТГФ (5 мл), по каплям добавляли метилмагнийиодид (0,15 мл, 6,0 ммоль, 4М раствор) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. После завершения реакции реакционную смесь гасили 1N HCL и экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта. Полученный продукт очищали с помощью препаративной ТСХ с получением целевого соединения в виде твердого вещества коричневого цвета (0,05 г, выход: 50%).
Соединение 97: 4-[1-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)этенил]бензойная кислота
К перемешиваемому раствору 4-[1-гидрокси-1-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)этил]бензойной кислоты (0,04 г, 0,1 ммоль) в TFA (0,2 мл) добавляли концентрированный H2SO4 (6,8 мл). Реакционную смесь выдерживали при комнатной температуре в течение 1 часа. Затем реакционную смесь гасили насыщенным раствором NaHCO3 и экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта. Полученный продукт очищали с помощью препаративной ВЭЖХ с получением целевого соединения в виде твердого вещества белого цвета (0,015 г, выход: 39,5%). МС (ESI, 120 эВ): m/z=342,15 (М+Н+).
Схема 45
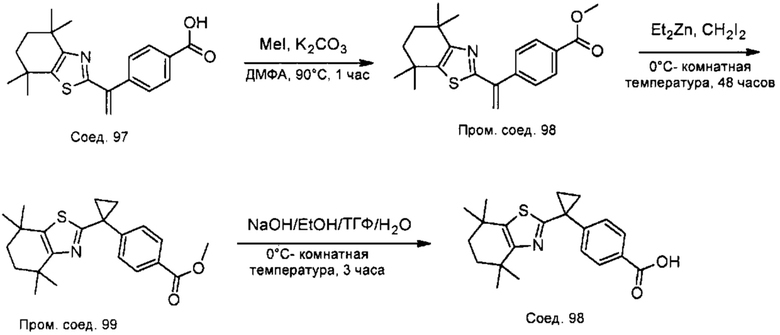
Пример 98: 4-[1-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)циклопропил]бензойная кислота
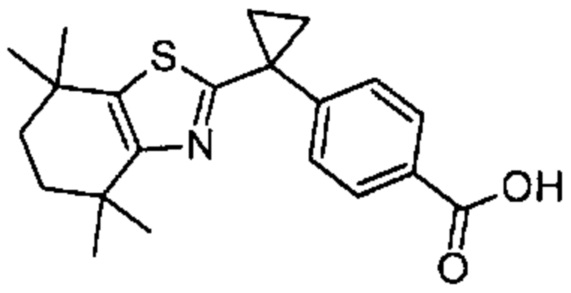
Соединение 98 синтезировали из 4-[1-4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)этенил]бензойной кислоты, следуя способу, указанному в схеме 45; чистота: 98,93%.
Промежуточное соединение 98: метил 4-[1-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)этенил]бензоат
К перемешиваемому раствору 4-[1-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)этенил]бензойной кислоты (0,5 г, 1,46 ммоль) в ДМФА (3 мл) добавляли K2СО3 (0,8 г, 5,85 ммоль). Затем к вышеуказанному перемешиваемому раствору добавляли метилиодид (0,3 мл, 4,4 ммоль). Реакционную смесь нагревали при 90°С в течение 1 часа. Реакционную смесь выливали в воду и экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта. Полученный продукт очищали с помощью колоночной хроматографии с получением целевого соединения в виде масла бледно-желтого цвета (0,13 г, выход: 25%).
Промежуточное соединение 99: метил 4-[1-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)циклопропил]бензоат
В круглодонную колбу 25 мл с двумя горлышками, снабженную магнитной мешалкой, загруженную 4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)этенил]бензойной кислотой (0,13 г, 0,4 ммоль) в DCM (5 мл), добавляли дииодометан (0,35 мл, 4,3 ммоль) при 0°С. К перемешиваемому раствору по каплям добавляли диэтилцинк (2,2 мл, 2,1 ммоль) при той же температуре. Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 48 часов. Реакционную смесь гасили 1N HCl и экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта. Полученный неочищенный продукт (бледно-желтое масло) использовали в таком виде на следующем этапе без дальнейшей очистки (0,15 г, выход: 25%).
Соединение 98: 4-[1-(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)циклопропил]бензойная кислота
Круглодонную колбу объемом 25 мл, снабженную магнитной мешалкой, загружали метанолом (10 мл) и ТГФ (2 мл). К перемешиваемому растворителю добавляли метил 4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)циклопропил]бензоат (0,15 г, 0,41 ммоль) и водный NaOH (0,08 г, 2,2 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь полностью концентрировали и неочищенный продукт промывали эфиром и разбавляли водой и затем нейтрализовали 1,5 N HCl, затем экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали с получением продукта. Полученный продукт снова очищали с помощью препаративной ВЭЖХ с получением целевого соединения в виде твердого вещества белого цвета (0,002 г, выход: 1,5%): МС (ESl, 120 эВ): m/z=356,1 (М+Н+).
Пример 99: 6-[циклобутил(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]пиридин-3-карбоновая кислота
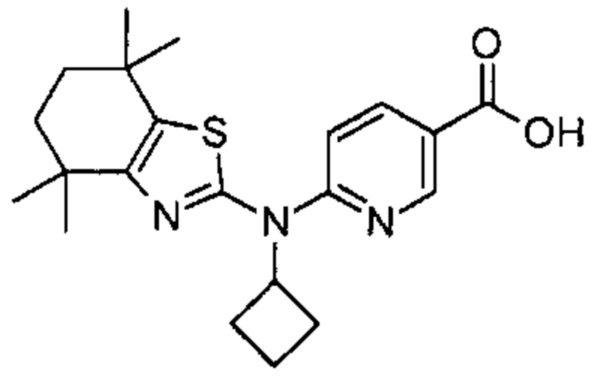
Соединение 99 синтезировали из 3,4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина и метил 6-хлорпиридин-3-карбоксилата, следуя способу, аналогичному описанному в схеме 18; чистота: 97,43%.
Пример 100: 4-[фенил(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензойная кислота
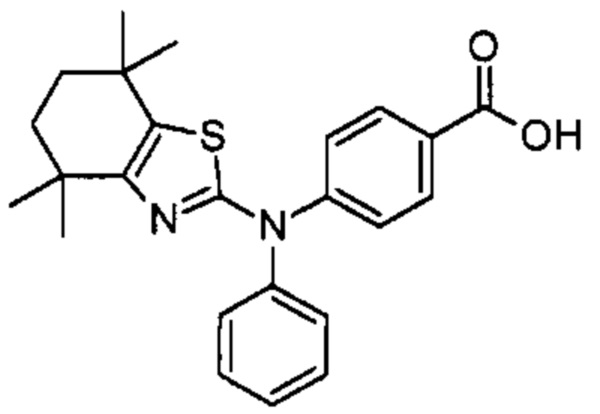
Соединение 100 синтезировали из 4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензоата (соединение 65) и иодбензена, следуя способу, аналогичному описанному в схеме 27; чистота: 96,86%.
Пример 101: 4-[пиридин-2-ил(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-6ензотиазол-2-ил)амино]бензойная кислота
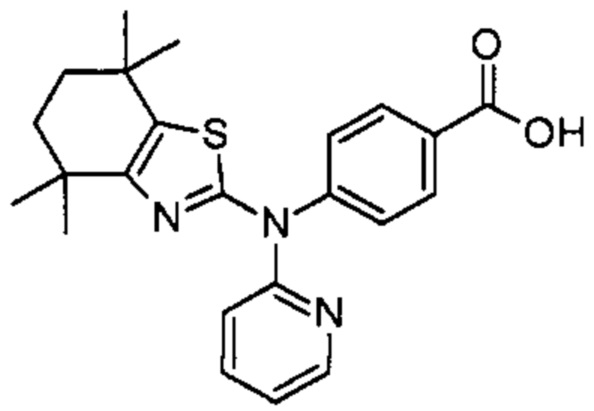
Соединение 101 синтезировали из 4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]бензоата (соединение 65) и 2-бромпиридина, следуя способу, аналогичному описанному в схеме 27; чистота: 97,95%.
Пример 102: 4-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)(фенил)амино]бензойная кислота
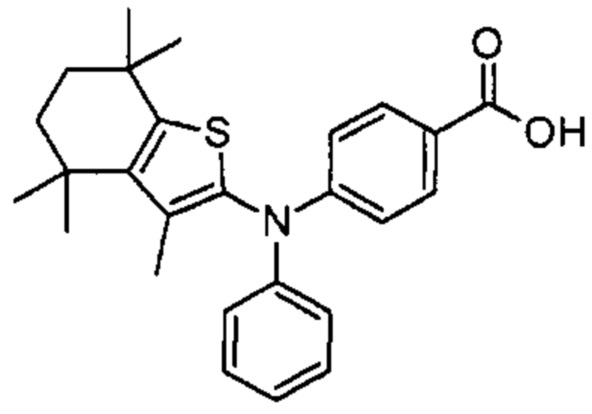
Соединение 102 синтезировали из метил 4-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоата (промежуточное соединение-9) и иодбензена, следуя способу, аналогичному описанному в схеме 27; чистота: 99,04%.
Пример 103: 4-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)(пиридин-2-ил)амино]бензойная кислота
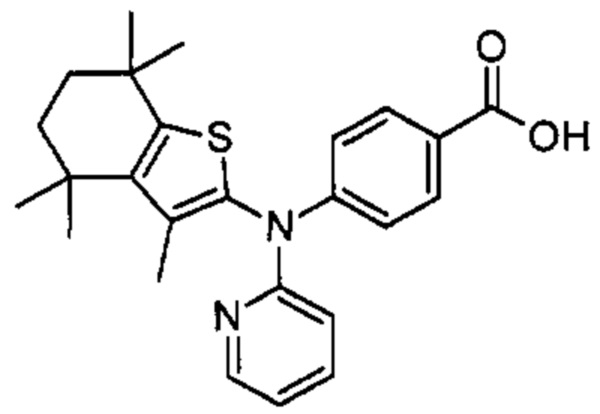
Соединение 103 синтезировали из метил 4-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоата (промежуточное соединение-9) и 2-бромпиридина, следуя способу, описанному в схеме 27; чистота: 91,19%.
Пример 104: 4-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)(пиридин-2-илметил)амино]бензойная кислота
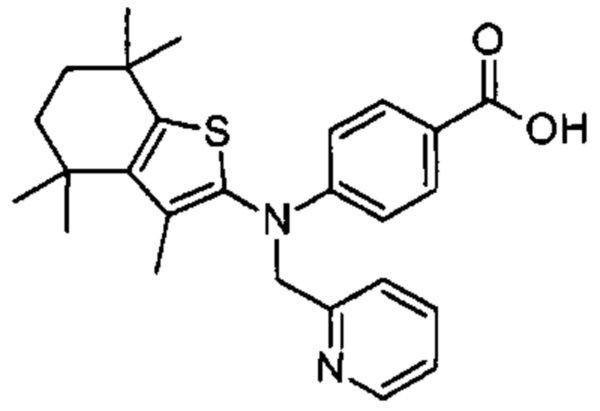
Соединение 104 синтезировали из метил 4-[(3,4,4,7,7-пентаметил-4,5,6,7-тетрагидро-1-бензотиофен-2-ил)амино]бензоата (промежуточное соединение-9) и 2-(бромметил)пиридина, следуя способу, аналогичному описанному в схеме 27; чистота: 99,23%.
Схема 46
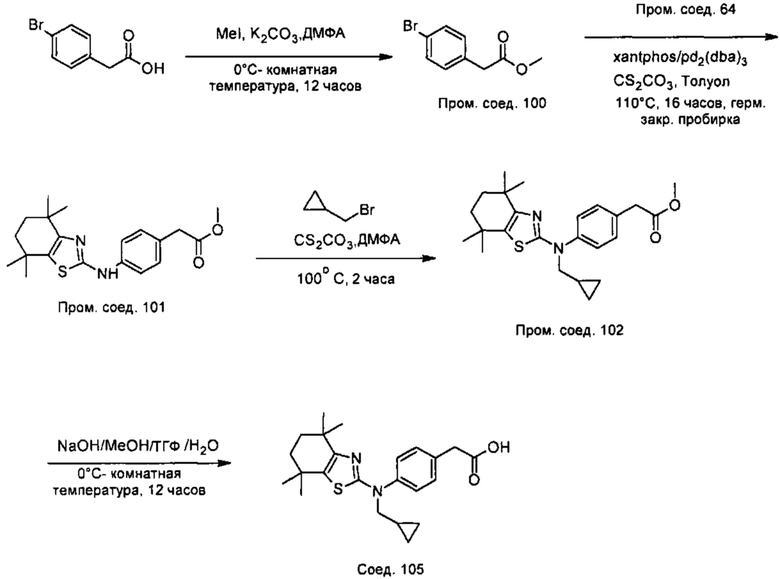
Пример 105: {4-[(Циклопропилметил)(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]фенил}уксусная кислота
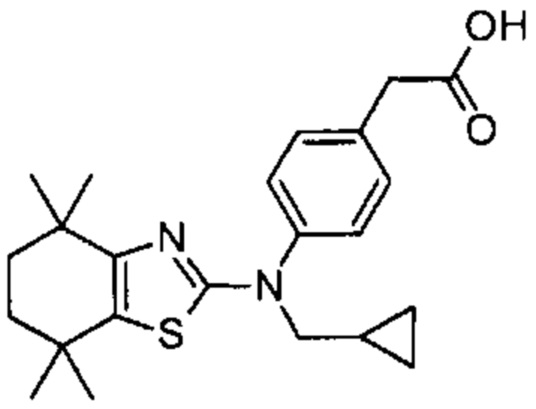
Соединение 105 синтезировали из 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина и метил (4-бромфенил)ацетата, следуя способу, описанному в схеме 46; чистота: 98,65%.
Промежуточное соединение 100: метил (4-бромфенил)ацетат
К перемешиваемому в круглодонной колбе раствору (4-бромфенил)уксусной кислоты (1,5 г, 6,9 ммоль) в ДМФА (10 мл) добавляли карбонат калия (1,92 г, 14 ммоль) и метилиодид (1,46 г, 10 ммоль) при 0°С. Реакционную смесь выдерживали при комнатной температуре в течение 2 часов. Затем реакционную смесь охлаждали до комнатной температуры, выливали в воду и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме. Полученный неочищенный продукт очищали с помощью колоночной хроматографии с получением целевого соединения в виде жидкости желтого цвета (1 г, выход: 63,69%).
Промежуточное соединение 101: Метил {4-[(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]фенил}ацетат
В пробирку для проведения реакций под давлением объемом 100 мл с завинчивающейся крышкой, загруженную 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амином (0,23 г, 1 ммоль) в толуоле (5 мл), добавляли метил (4-бромфенил)ацетат (0,42 г, 2 ммоль). К перемешиваемому растворителю добавляли CS2CO3 (0,97 г, 3 ммоль) и ксантфос (0,3 г, 0,05 ммоль). Полученный раствор дегазировали азотом в течение 10 минут. Затем в реакционную смесь добавляли Pd2 (dba)3 (0,02 г, 0,02 ммоль). Реакционную смесь нагревали при 100°С в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры и выливали в воду и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме. Полученный неочищенный продукт очищали на колонне с получением целевого соединения в виде твердого вещества желтого цвета (0,2 г, выход: 55,86%).
Промежуточное соединение 102: метил {4-[(циклопропилметил) (4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]фенил}ацетат
К перемешиваемому в круглодонной колбе раствору метил {4-[(4,4,7,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]фенил}ацетата (0,2 г, 0,55 ммоль) в ДМФА (5 мл) добавляли карбонат цезия (0,53 г, 1,65 ммоль) и циклопропилметил (0,11 г, 0,82 ммоль) при 0°С. Реакционную смесь нагревали при 100°С в течение 2 часов. Затем реакционную смесь охлаждали до комнатной температуры, выливали в воду и экстрагировали этилацетатом. Объединенные экстракты промывали водой и рассолом. Органический слой сушили над безводным Na2SO4 и концентрировали в вакууме. Полученный неочищенный продукт очищали с помощью колоночной хроматографии с получением целевого соединения в виде жидкости желтого цвета (0,018 г, выход: 78,26%).
Соединение 105: {4-[(циклопропилметил)(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]фенил}уксусная кислота
Круглодонную колбу объемом 25 мл, снабженную магнитной мешалкой, загружали метанолом (0,5 мл) и ТГФ (0,5 мл). К перемешиваемому растворителю добавляли метил {4-[(циклопропилметил)(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]фенил}ацетат (0,18 г, 0,04 ммоль) и водный NaOH (0,005 г, 0,13 ммоль) при 0°С. Реакционную смесь перемешивали при комнатной температуре в течение 20 минут. Реакционную смесь полностью концентрировали, неочищенный продукт промывали эфиром и разбавляли водой, а затем нейтрализовали 1,5 N HCl, после чего экстрагировали этилацетатом. Органический слой промывали водой и рассолом, сушили над Na2SO4, фильтровали и концентрировали. Полученное твердое вещество растирали с гексаном с получением целевого соединения в виде твердого вещества белого цвета (0,01 г, выход: 58,82%): МС (ESI, 120 эВ): m/z=398,2 (М+Н+).
Пример 106: 3-{4-[(Циклопропилметил)(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]фенил}пропановая кислота
Соединение 106 синтезировали из 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина и метил 3-(4-бромфенил)пропаноата, следуя способу, описанному в схеме 46; чистота: 90,67%.
Пример 107: ((2E)-3-{4-[(циклопропилметил)(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]фенил}проп-2-еновая кислота
Соединение 3 синтезировали из 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина и метил (2Е)-3-(4-иодфенил)проп-2-аноата, следуя способу, описанному в схеме 46; чистота: 95,98%.
Пример 108: 2-{4-[(Циклопропилметил)(4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-ил)амино]фенил}циклопропанкарбоновая кислота
Соединение 108 синтезировали из 4,4,7,7-тетраметил-4,5,6,7-тетрагидро-1,3-бензотиазол-2-амина и метил 2-(4-иодфенил)циклопропанкарбоксилата, следуя способу, описанному в схеме 46; чистота: 97,59%.
Соединения, описанные в Таблице 1, были синтезированы в соответствии со способами, изложенными выше, или их вариантами.
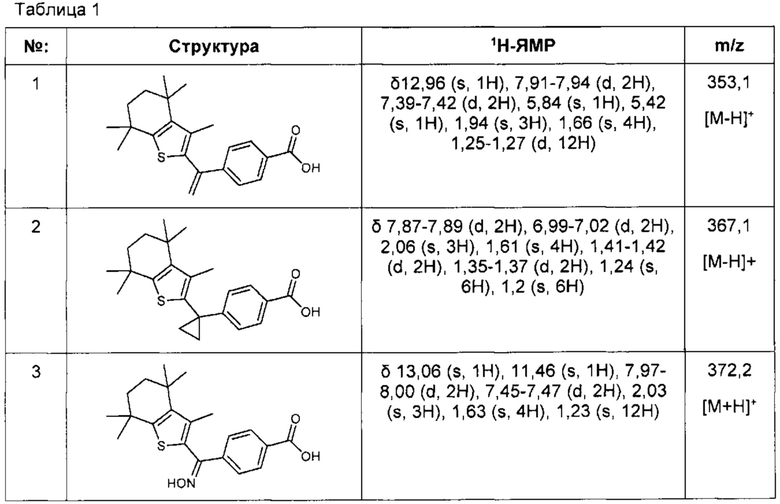
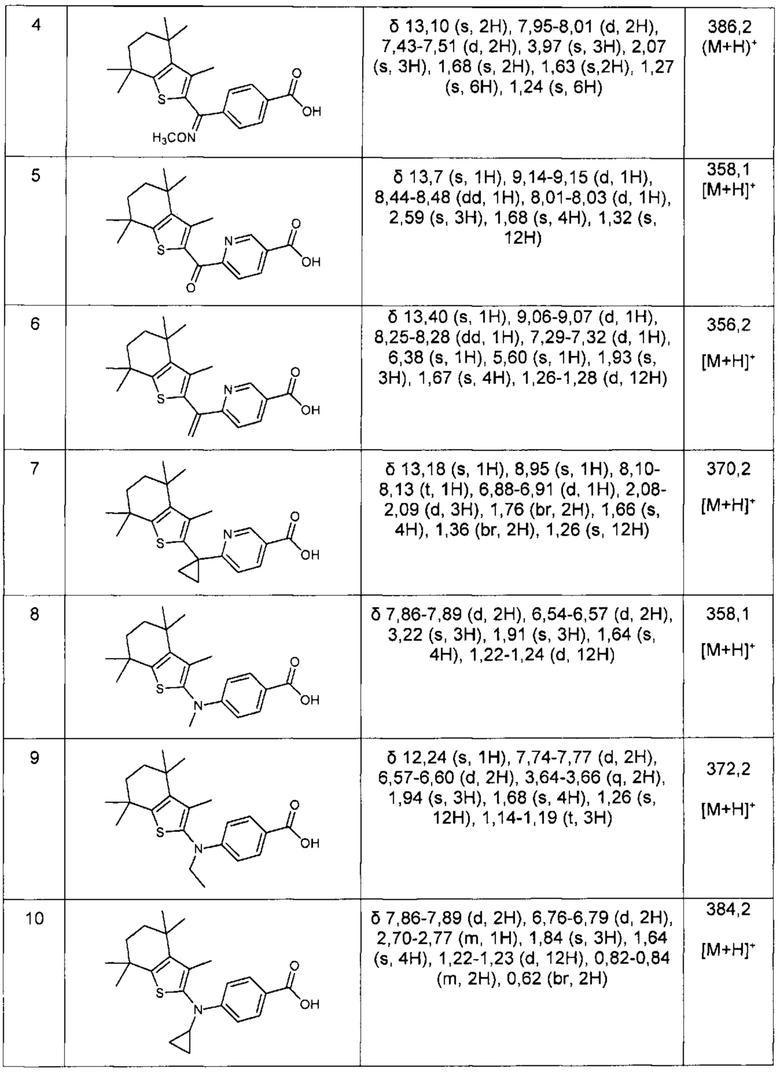
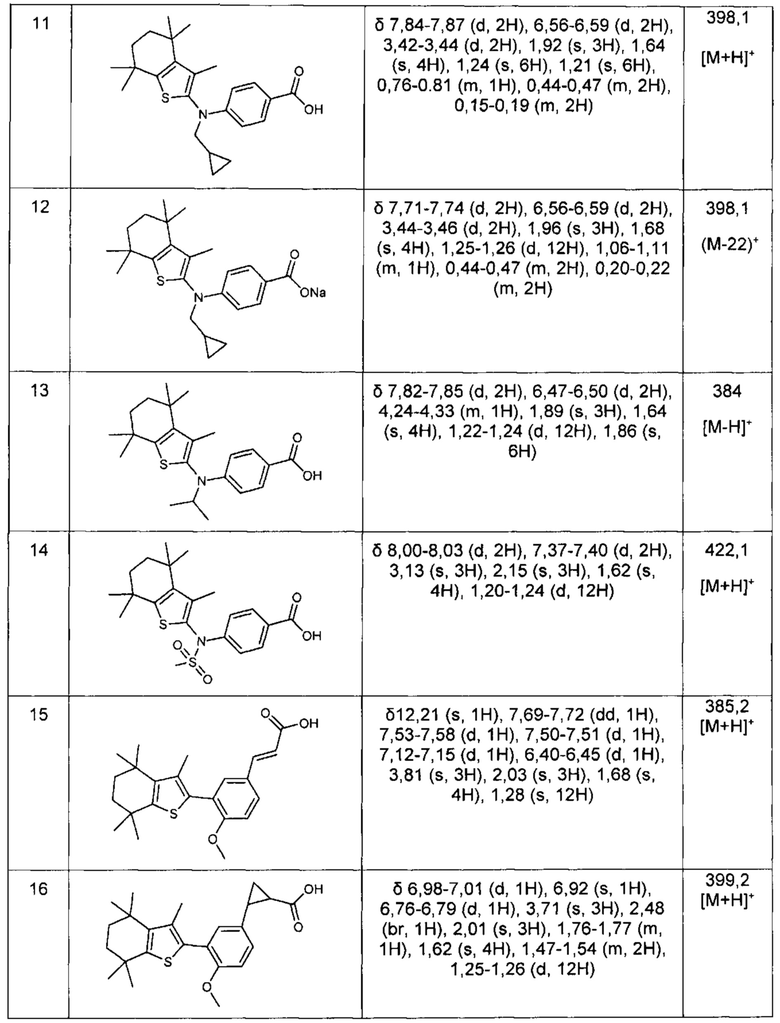
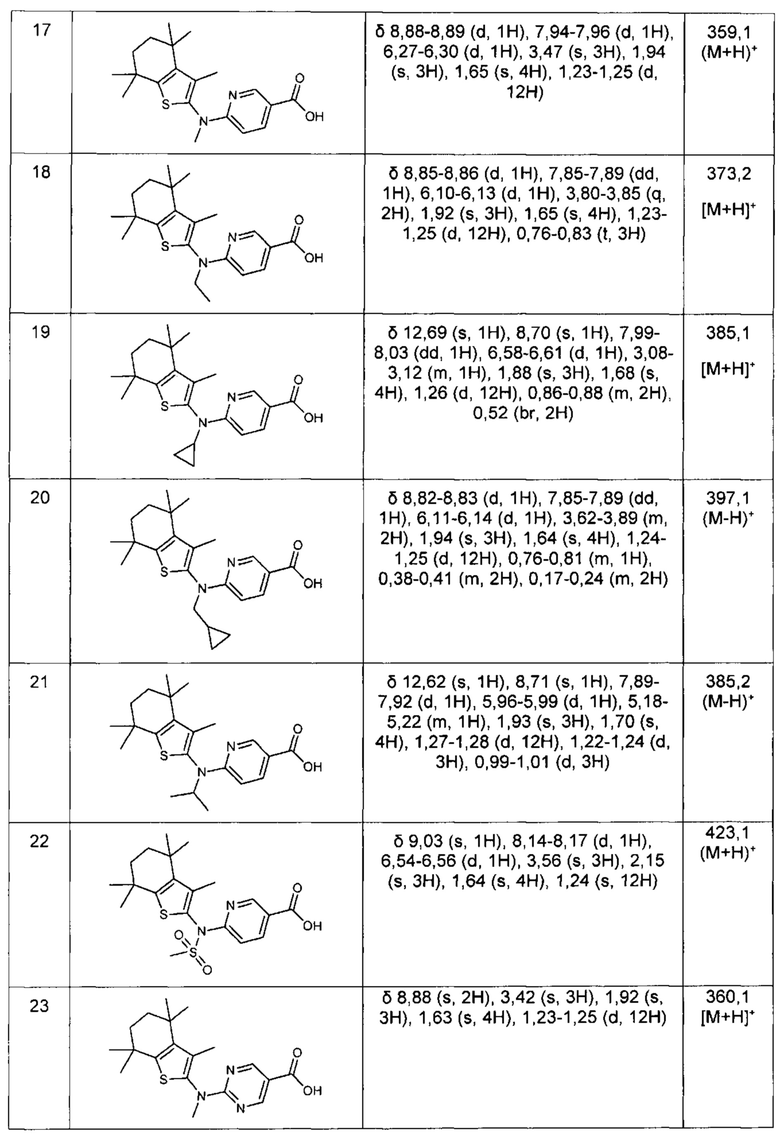
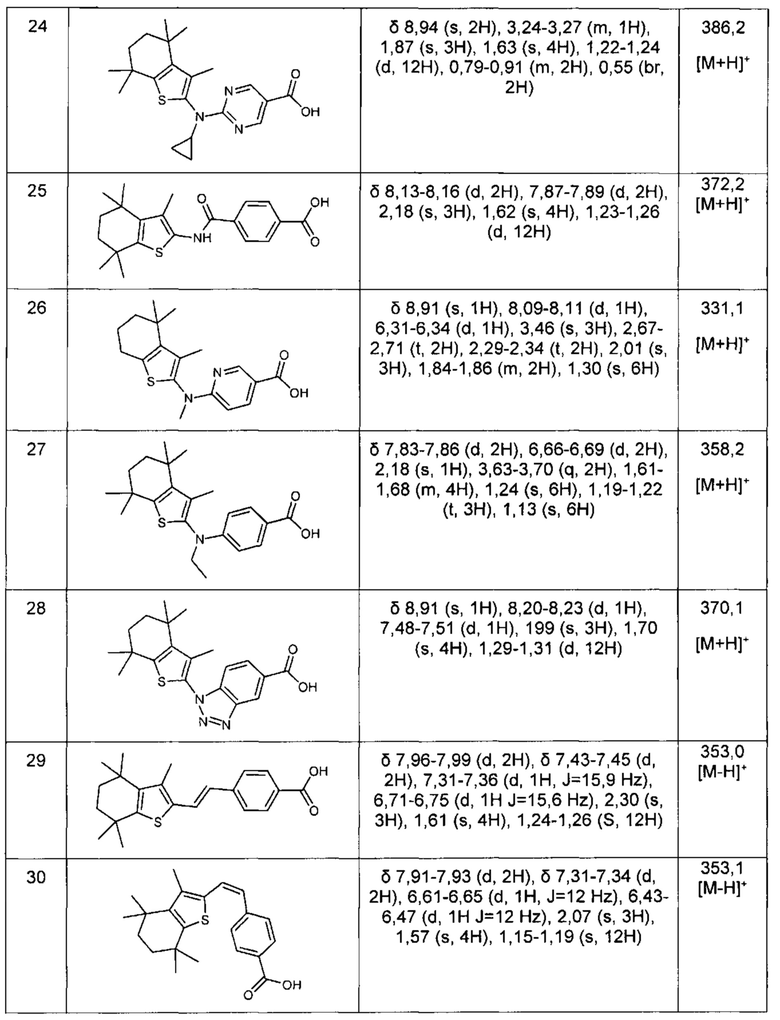
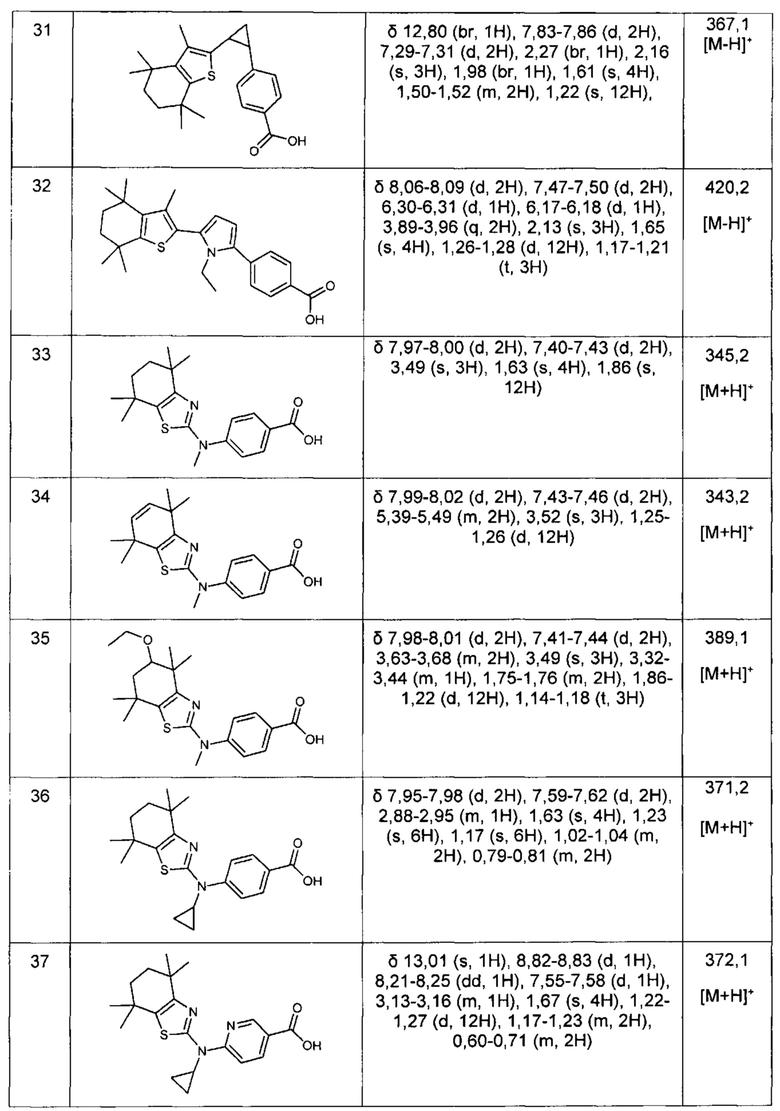
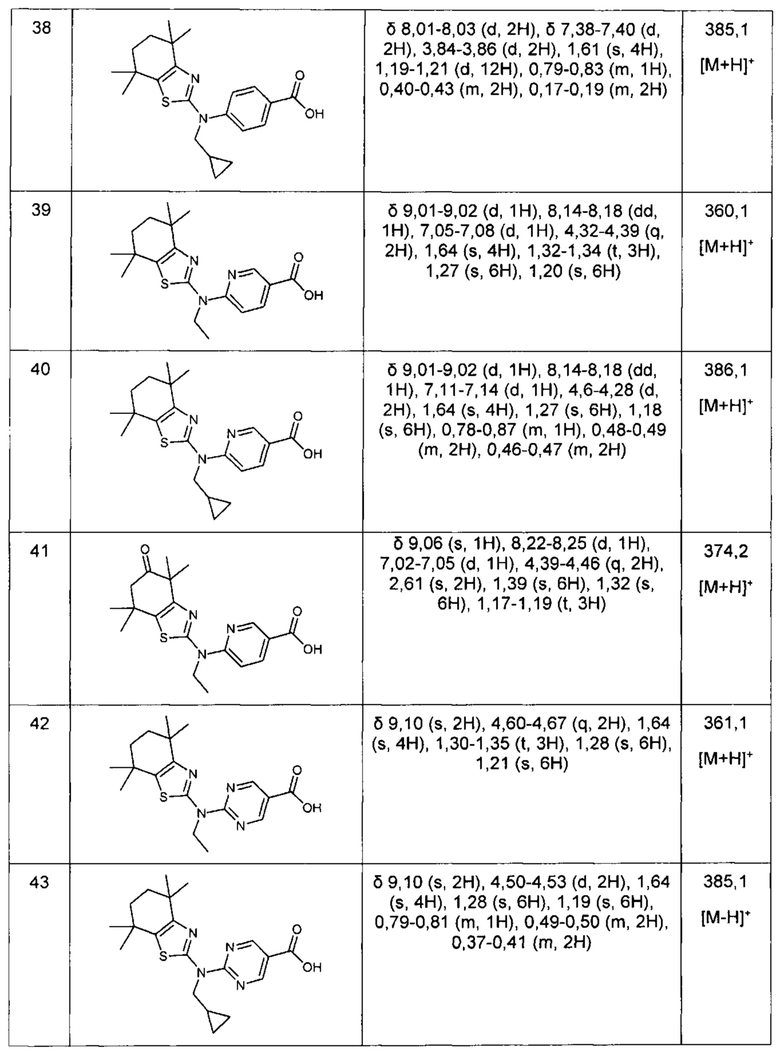
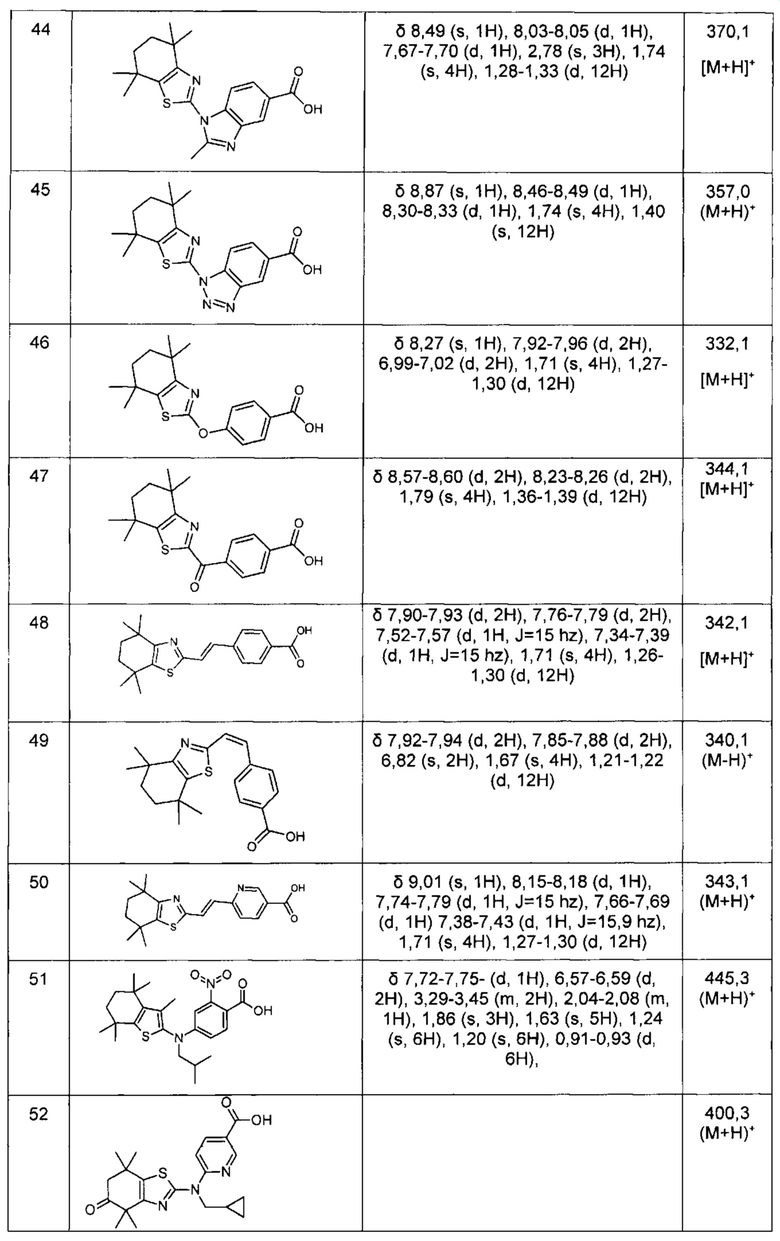
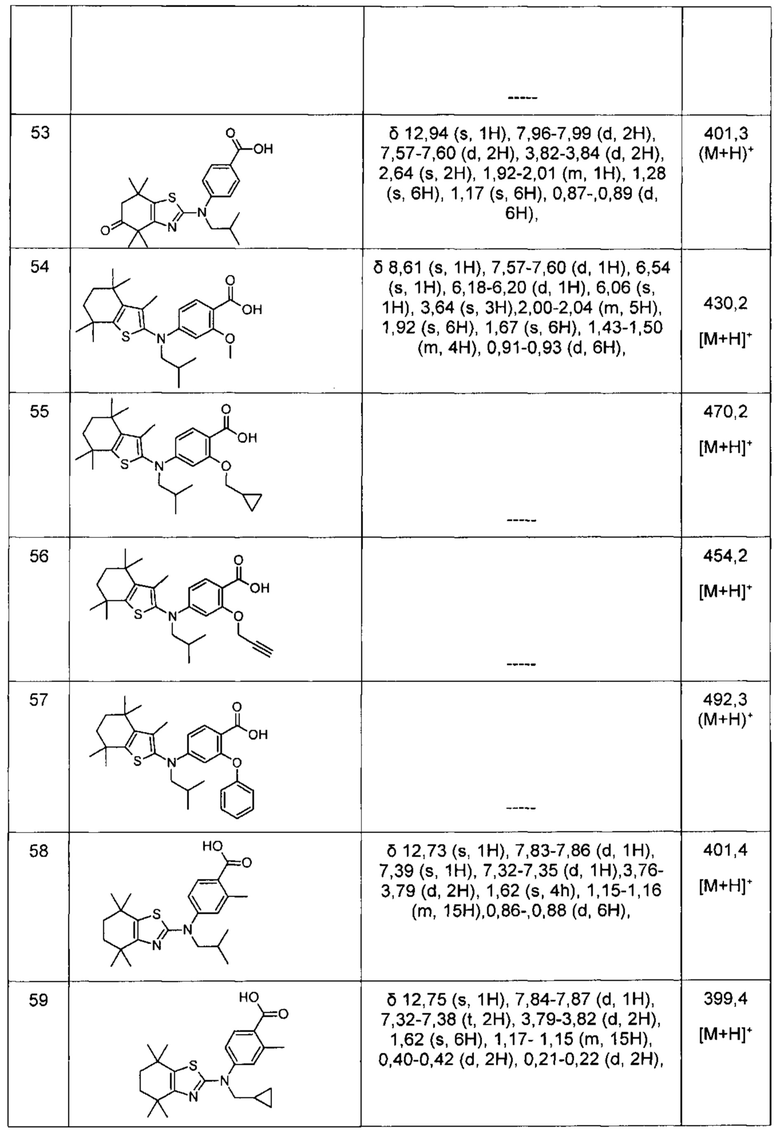
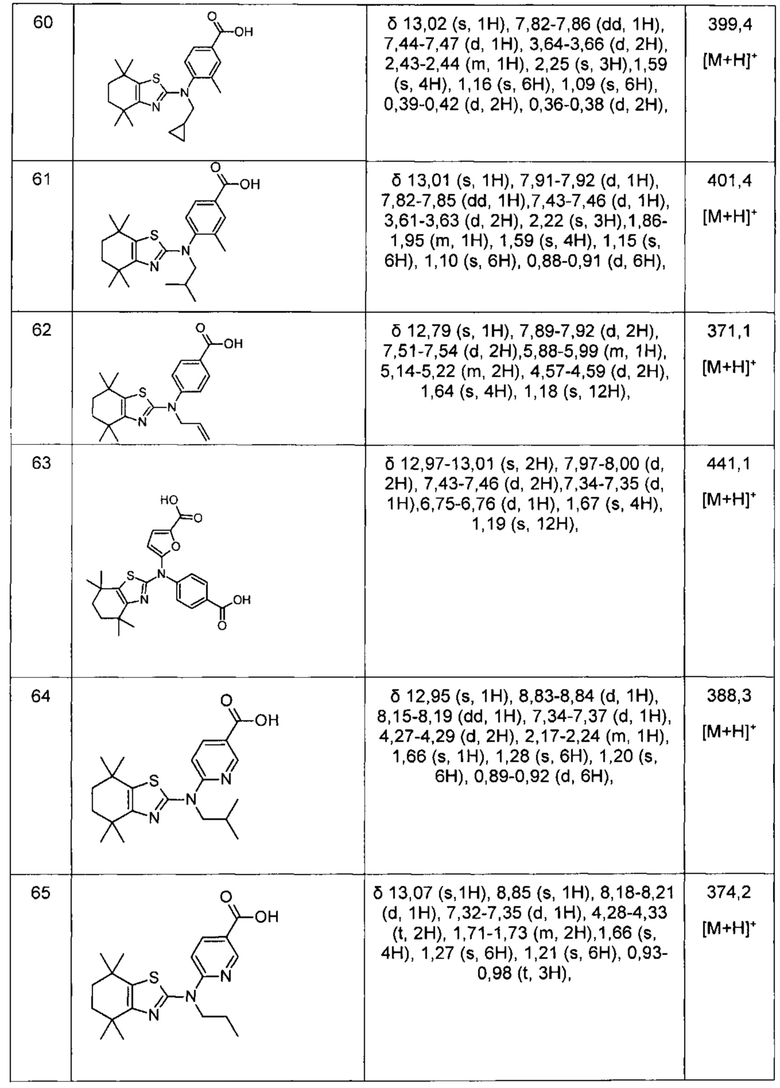
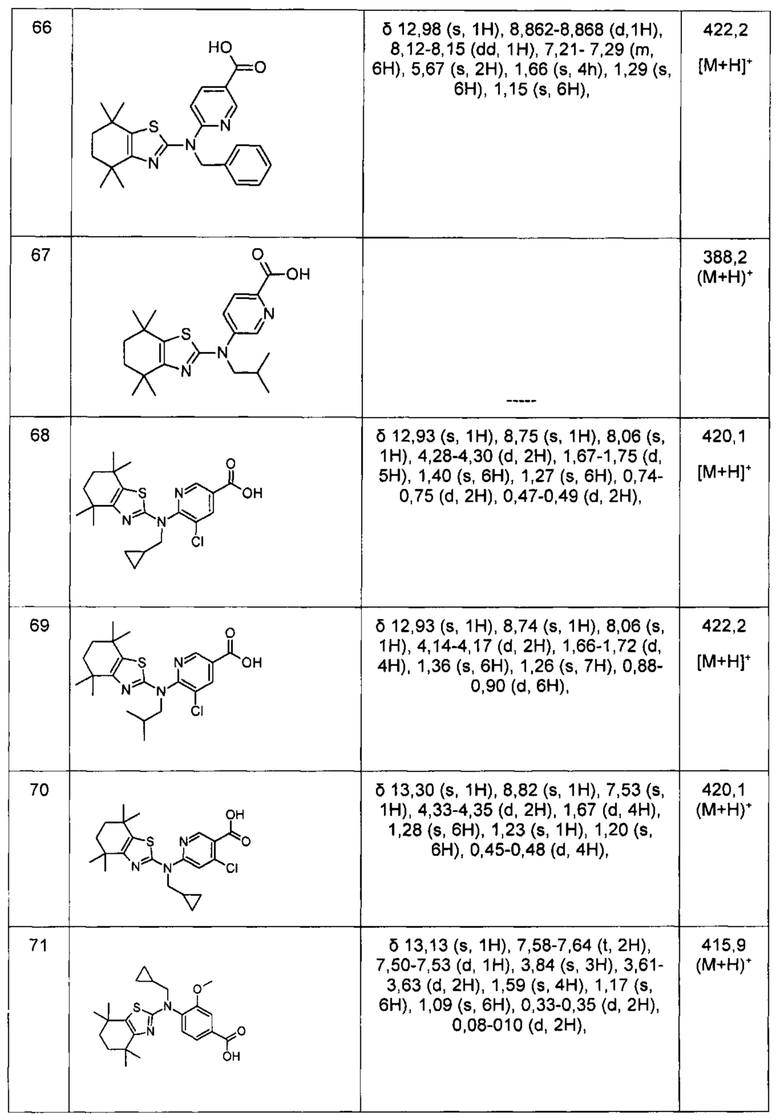
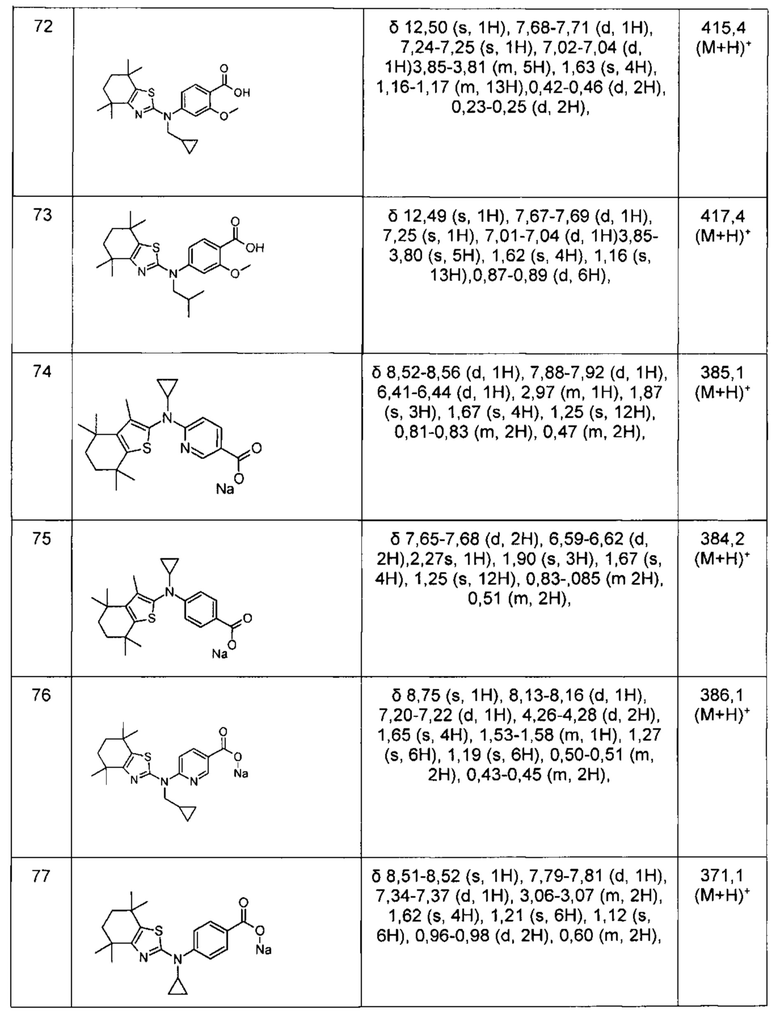
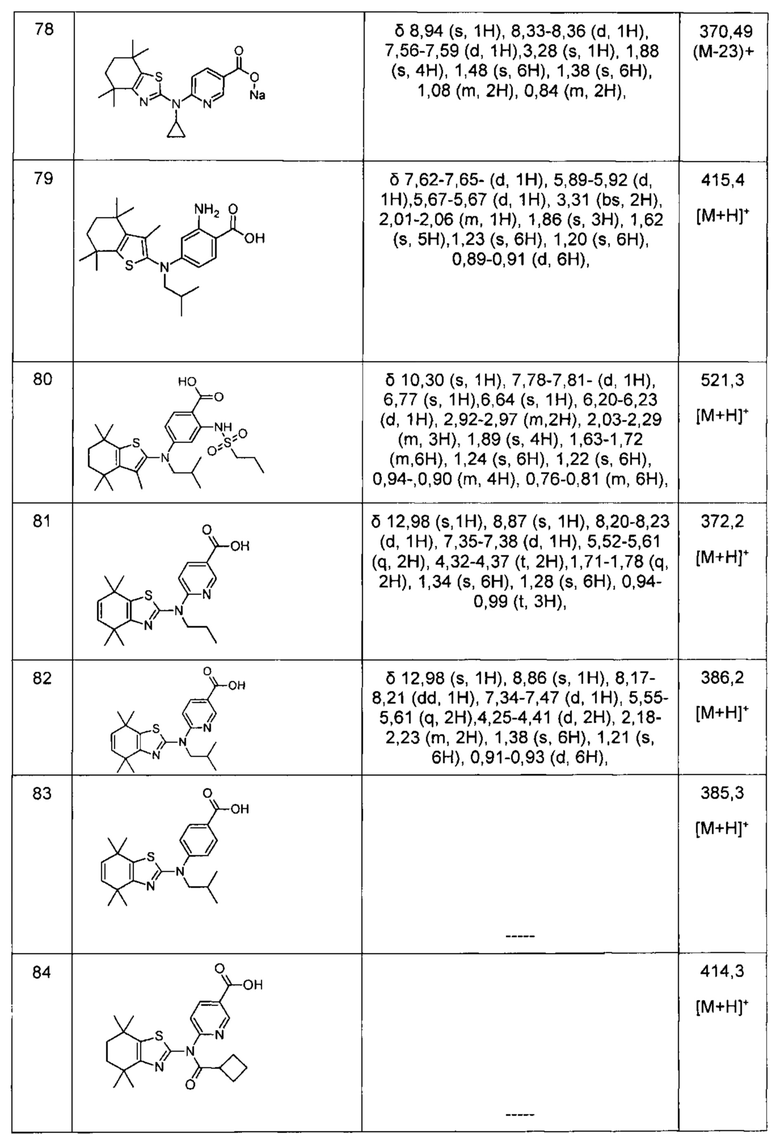
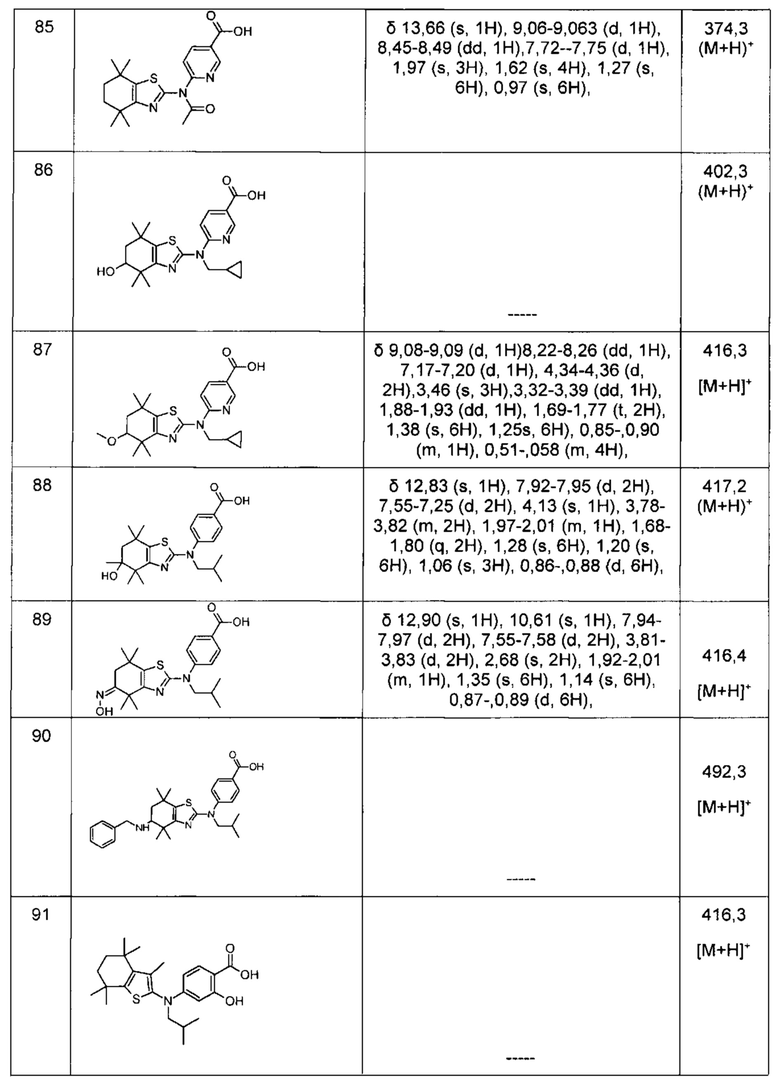
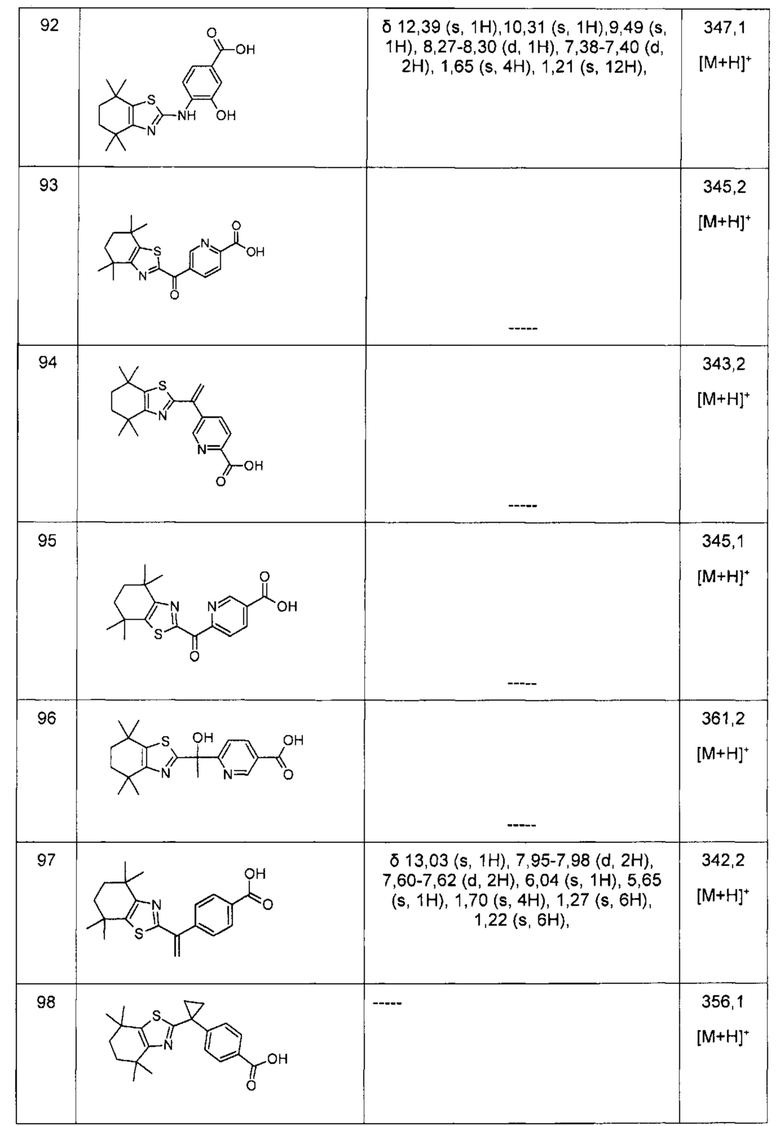
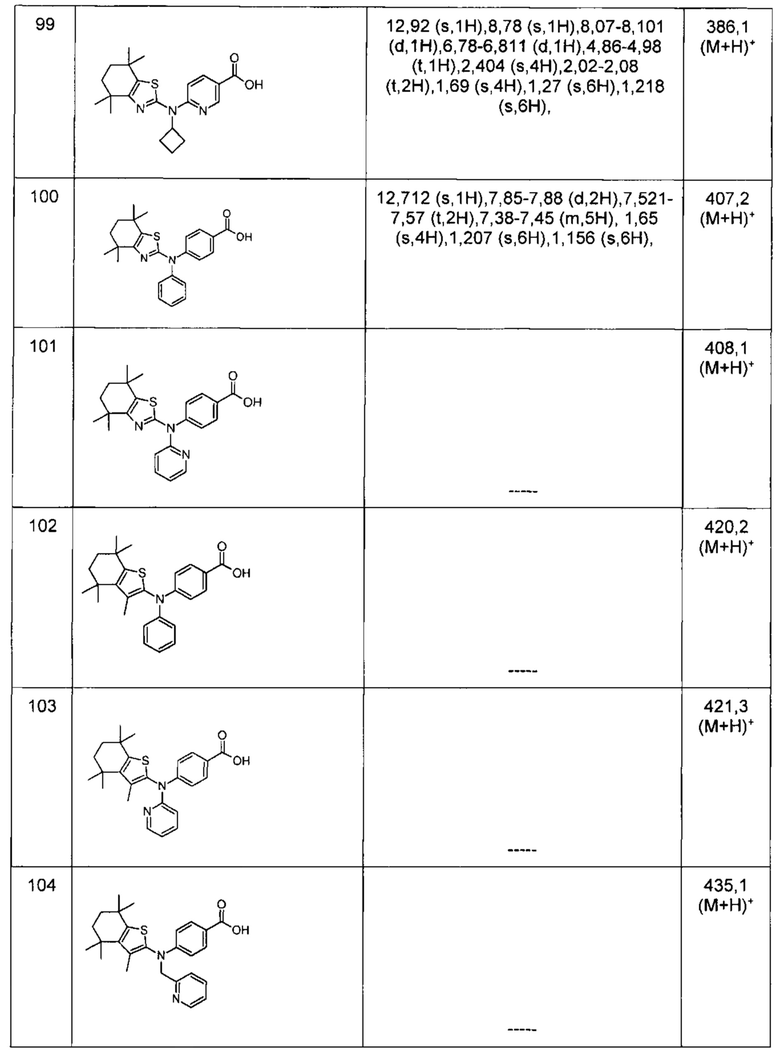
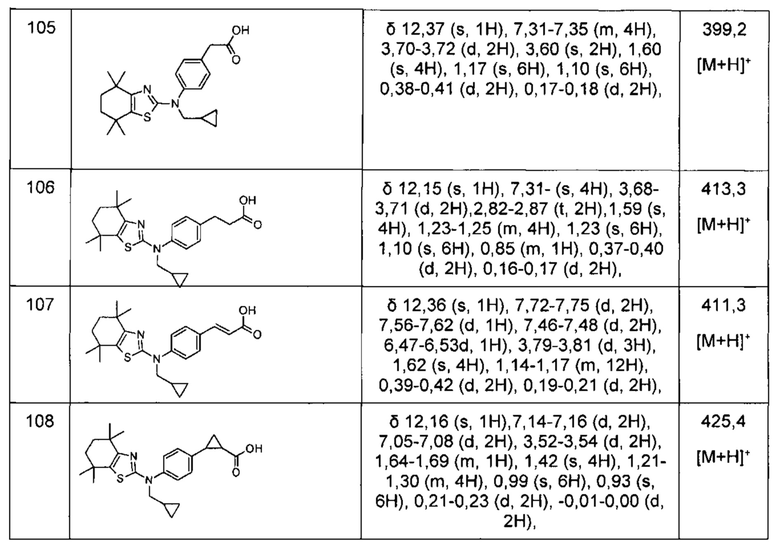
Биологическая активность
Способность соединений согласно настоящему изобретению действовать в качестве агонистов RXR и, таким образом, регулировать с повышением экспрессию генов в ряде органов, таких как печень, скелетные мышцы и жировая ткань, была исследована следующим образом.
Данные экспрессии генов печени (клетки HepG2):
Клетки HepG2 (источник АТСС, номер по каталогу НВ-8065) высевали в среду MEM (6,9 мМ глюкозы), содержащую 10% FBS (фетальная бычья сыворотка), и культивировали в течение 36 часов. После культивирования в течение 36 часов клетки обрабатывали исследуемым агонистом RXRa в количестве 1 мкМ в свежей среде MEM (6,9 мМ глюкозы и 10% FBS - фетальной бычьей сыворотки). После обработки в течение 24 часов сливали супернатант и к клеткам добавляли реактив Tri-reagent (Sigma, St. Louis, МО, США). Общую РНК извлекали из клеток HepG2 с применением реактива Tri-reagent (Sigma, St. Louis, МО, США), с последующей экстракцией хлороформом и осаждением изопропиловым спиртом. Комплиментарную ДНК синтезировали обратной транскрипцией (ABI, Foster City, СА, США). Уровень экспрессии генов измеряли для АсоХ1, АроА1, АроС3, THRSP, Sult2A1, SREBP-1c, CYP3A4 с использованием последовательности праймеров, известной в предшествующем уровне техники. Комплиментарную ДНК амплифицировали с использованием SYBR Green PCR Master Mix (Eurogenetic, Бельгия).
Данные экспрессии генов мышцы (клетки С2С12):
Клетки С2С12 (клетки миобластов, источник АТСС, номер по каталогу: CRL-1772) были высеяны в DMEM среде (25 мМ глюкозы), содержащей 10% FBS и (в день 0) были культивированы, через 24 ч сменили среду (1 день). После смены среды обработанные клетки были помещены в дифференцирующую среду (25 мМ глюкозы и 2% FBS) (с 3-его по 7-ой день). Дифференцированные клетки (мышечные волокна) обрабатывали 1 мкМ исследуемого агониста RXRa. После обработки в течение 24 часов сливали супернатант и к клеткам добавляли реактив Tri-reagent (Sigma, St. Louis, МО, США). Общую РНК экстрагировали из клеток С2С12 с использованием Tri-реагента (Sigma, St. Louis, МО, США), с последующей экстракцией хлороформом и осаждением изопропиловым спиртом. Комплиментарную ДНК синтезировали методом обратной транскрипции (ABI, Foster City, СА, США). Уровень экспрессии генов был измерен для СРТ1, UCP3, АВСА1, SREBP-1c, PDK4 с использованием последовательности праймеров, известной в предшествующем уровне техники. Комплиментарную ДНК амплифицировали с использованием SYBR Green PCR Master Mix (Eurogenetic, Бельгия).
Данные экспрессии генов жировых клеток (клетки 3Т3 L1):
Клетки 3T3L1 (пре-адипоциты, источник АТСС, номер по каталогу CL-173) высевали в DMEM средах (25 мМ глюкозы), содержащих 10% BCS и (в этот же день, день 0) культивировали. Через 24 часа была произведена смена среды (день 1). Через 48 ч после смены среды клетки обрабатывали агонистом RXRa и дифференцирующей средой (25 мМ глюкозы, 10% FBS, 500 мкМ IBMX, 1 мкМ дексаметазона и 100 нМ инсулина) (с 3-его по 5-й день). До терминации клетки (адипоциты) поддерживали в поддерживающих средах (25 мм глюкозы, 10% FBS, 100 нМ инсулина). Супернатант сливали, к клеткам добавляли реактив Tri-reagent (Sigma, St. Louis, МО, США). Общую РНК экстрагировали из клеток с использованием реактива Tri-reagent (Sigma, St. Louis, МО, США), с последующей экстракцией хлороформом и осаждением изопропиловым спиртом. Комплиментарную ДНК синтезировали методом обратной транскрипции (ABI, Foster City, СА, США). Уровень экспрессии генов был измерен для FABP4, SCD1, РСК1, UCP1, АВСА1, CPT1b, PPARg, SREBP-1c с использованием последовательности праймеров, известной в предшествующем уровне техники. Комплиментарную ДНК амплифицировали с использованием SYBR Green PCR Master Mix (Eurogenetic, Бельгия).
Биологическая активность
Определение уровня трансактивации для оценки активности
Клетки НЕK-293 (АТСС) высевали за день до трансфекции. Для оценки ЕС50 действия изоформ RXR 2 мкг hRXR а, β или γ сверхэкспресирующего вектора (OriGENE, США), 1 мкг плазмиды, экспрессирующей люциферазу светлячка под воздействием RARE компонента (RARE-Luc) и 25 нг вектора люциферазы Рениллы (QIAGEN, США) ко-трансфицируют с использованием реагента липофектамина. После 24 ч трансфекции клетки обрабатывали различными концентрациями агонистов RXR (100 нМ, 300 нМ и 1 мкМ) в течение 24 ч с последующей оценкой активности люциферазы с использованием системы анализа двойной люциферазы (Dual Luciferase Reporter Assay System (Promega). Активность люциферазы нормализовали до активности люциферазы Рениллы. Действие соединений RXR было выражено относительно LG268, известного агониста RXR.
Были получены следующие результаты:
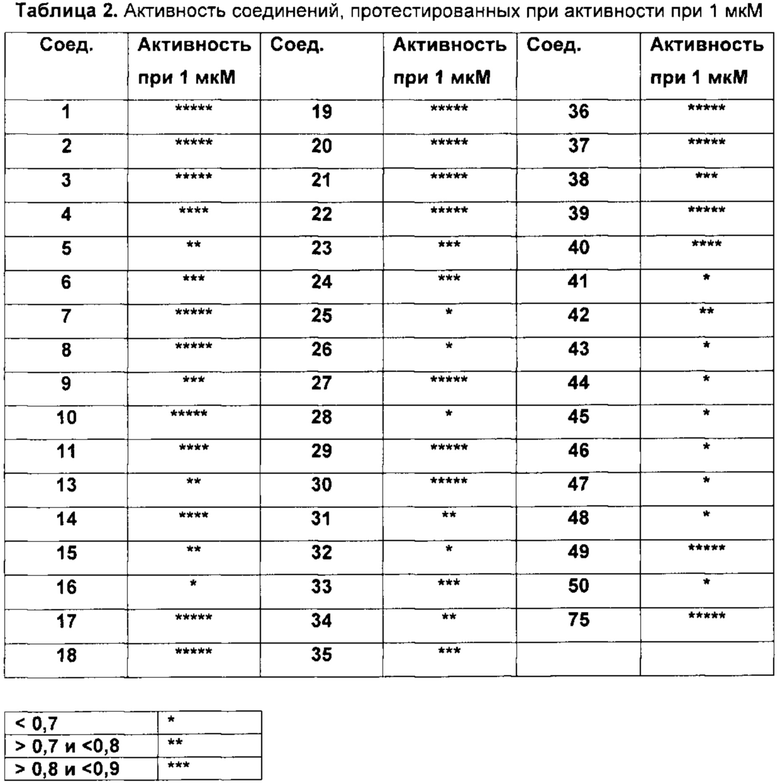
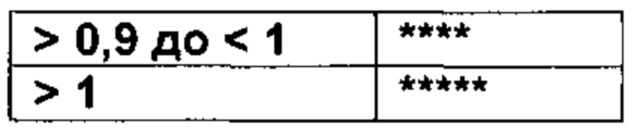
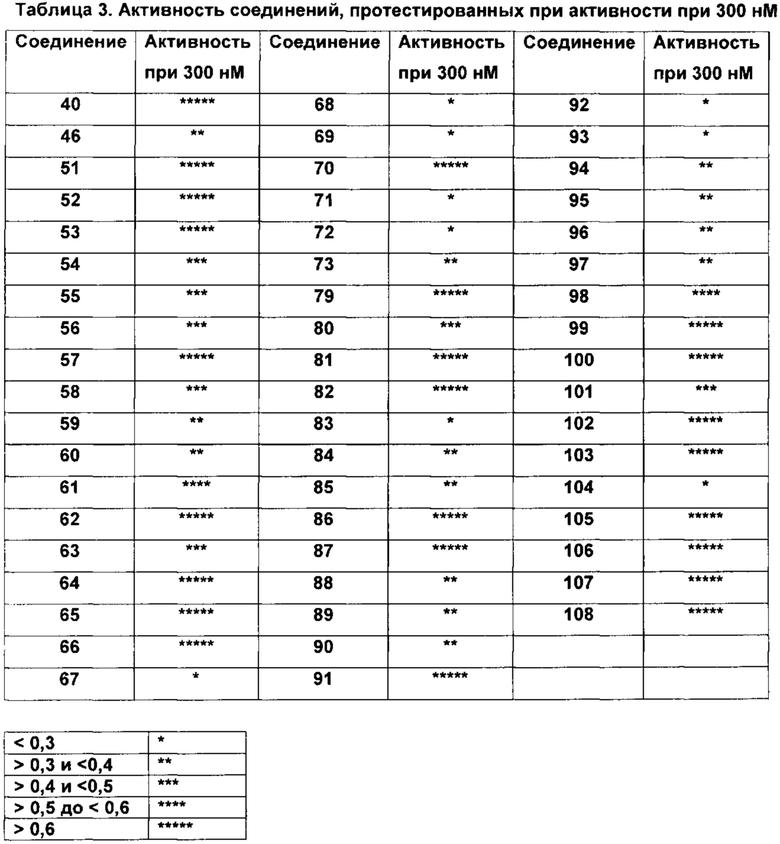
Данные исследований in vitro для лечения болезни Альцгеймера
Проводили несколько анализов in vitro для изучения влияния соединения 40 на клетки N2A и астроциты. Было проведено несколько измерений, чтобы продемонстрировать его использование в лечении болезни Альцгеймера. Болезнь Альцгеймера (БА) представляет собой хроническое, прогрессирующее, нейродегенеративное заболевание. Широкий спектр анти-амилоидных и нейропротекторных терапевтических средств изучается на основе гипотезы, что белок амилоид бета (Аβ) играет ключевую роль в начале заболевания и его прогрессировании, и что вторичные последствия, включая тау-гиперфосфорилирование и формирование нейрофибриллярных клубочков, окисление и воспаление вносят свой вклад в течение болезни. Вмешательство в такие процессы с применением средств, которые снижают выработку амилоида, уменьшают его накопление или увеличивают выведение, остановит каскад событий, включающих патогенез БА. Снижение тау-гиперфосфорилирования, ограничение окисления и эксайтотоксичности и контролирование воспаления может быть полезной стратегией борьбы с болезнью.
С целью изучения влияния соединения 40 на болезнь Альцгеймера культивировали нейронные клетки мыши (клетки N2A) и обрабатывали 320 мкМ холестерина отдельно виде или с исследуемым соединением в течение 24 ч. Затем холестерин удаляли и лечение продолжали в течение 24 ч. Содержание холестерина в клетках измеряли с помощью биохимических методов по отношению к общему содержанию белка. Для исследования экспрессии генов, клетки обрабатывали 320 мкМ холестерина отдельно или с исследуемым соединением в течение 48 ч. Получили РНК, затем преобразовали в кДНК и измерили экспрессию генов. Было продемонстрировано, что соединение 40 уменьшает накопление холестерина (Фиг. 1А), снижает окислительный стресс (уровни ROS) (Фиг. 1В), снижает апоптоз (активность каспазы) (Фиг. 1С) и увеличивает отток холестерина (экспрессию генов АВСА1 и ABCG1) (Фиг. 1D). Такие клеточные изменения приводят к уменьшению образования бляшек и улучшению здоровья нервных клеток.
Кроме того, культивированные астроциты были обработаны форбол-12-миристат-13-ацетатом (РМА) и липополисахаридами (LPS) отдельно или с исследуемым соединением в течение 48 часов с целью измерения уровней ROS. Для оценки влияния на маркеры воспаления, астроциты обрабатывали РМА и выдерживали с исследуемым соединением в течение 48 часов. Данные из астроцитов показали потенциал соединения 40 для уменьшения как оксидантного стресса (уровни ROS) (Фиг. 2А), так и воспаления (lL6 секреция и экспрессия, экспрессия lL1b, СОХ2 и МСР1) (Фиг. 2В, С, D, Е и F). Поскольку соединение 40 способно уменьшить воспаление, авторы настоящего изобретения полагают, что оно будет значительно влиять на ремиелинизацию нейронов и, следовательно, указанное соединение может быть рекомендовано для лечения рассеянного склероза.
Влияние агониста RXR на клеточные линии рака поджелудочной железы
Рак характеризуется неконтролируемым ростом клеток. Любое средство, которое может уменьшить клеточный рост, будет иметь высокий терапевтический потенциал. Таким образом, оценивали влияние агониста RXR соединения 40 на клетки MlA РаСа-2. Клетки MIA PaCa-2 (АТСС) высевали в 6-луночные планшеты (20000 клеток на лунку) в среде DMEM, содержащей 25 мМ глюкозы, 10% FBS, 2,5% лошадиной сыворотки в присутствии или в отсутствие различных концентраций соединения 40, как указано. Клетки собирали на 2-й день, 4-й день и 6-й день, и измеряли количество клеток. Клетки MIA РаСа-2 выращивали с или без различных концентраций соединения 40 (0,0, 0,1, 0,3, 1,0, 3,0 и 10.0 мкМ) и рост клеток измеряли по количеству клеток в различные моменты времени (день 2, 4 и 6). Необработанные клетки MIA РаСа-2 показали экспоненциальную скорость роста клеток, которая была понижена при использовании соединения 40 для лечения. Даже 0,1 мкМ соединения 40 давало значительное снижение роста клеток, которое было дополнительно снижено увеличением концентрации соединения 40 (Фиг. 3А).
Влияние соединения 40 на пролиферацию клеток далее оценивали путем анализа инкорпорирования BrdU (бромдезоксиуридина). Клетки MIA РаСа-2 выращивали в течение 4 дней в присутствии или в отсутствие соединения 40 (3 мкМ). После обработки контрольным носителем или соединением 40 (3 мкМ) клетки подвергали воздействию бромдезоксиуридина. Инкорпорирование BrdU измеряли с использованием коммерчески доступного набора (Roche) в соответствии с протоколом производителя. Инкорпорирование BrdU было значительно снижено в случае обработки соединением 40, что указывает на его эффективность для снижения роста клеток (Фиг. 3В). Сверхэкспрессия RXRa также уменьшила инкорпорирование BrdU, что предполагает ведущую роль метаболического пути RXR в снижении роста раковых клеток. Лечение соединением 40 дополнительно уменьшило инкорпорирование BrdU в MIA РаСа-2 панкреатической линии раковых клеток, сверхэкспресирующей RXRa. Следовательно, полученные данные указывают на то, что сигнальный путь RXR, регулируемый соединением 40, уменьшает рост клеток в панкреатических раковых клетках.
Для оценки воздействия соединения 40 на уровни ROS клетки подвергали воздействию индикаторной пробы ROS (DCFH-DA) в течение 1 часа. Увеличение флуоресценции деоксикоформицина, индуцированное ROS, оценивали при возбуждении 485 нм и излучении 528 нм. Уровни ROS были нормированы до уровней клеточной ДНК, которые были измерены с использованием флуоресценции бис-бензамида при возбуждении 360 нм и излучении 460 нм. Уровни ROS представлены в % от контрольных. Лечение соединением 40 в течение 4 дней привело к значительному увеличению уровня стресса в клетках MIA РаСа-2, что было измерено увеличением уровней активных форм кислорода (ROS) (Фиг. 4А). Избыточная экспрессия RXRa дополнительно увеличила уровни ROS. Следовательно, RXR сигнальный путь, опосредованный соединением 40, способствовал повышению уровней ROS в панкреатических раковых клетках.
Для того, чтобы оценить, привел ли этот повышенный стресс к апоптозу, измеряли активность каспазы-3 и гибель клеток. Активность каспазы-3 измеряли через ее способность расщеплять свой субстрат (Ac-DEVD-R110). Повышенная активность каспазы-3 привела бы к увеличению выпуска люминесцентной R110, которая была измерена при возбуждении 485 нм и излучении 530 нм. Активность каспазы-3 (в виде выделяющихся нано-молей R110) нормирована к общему клеточному белку. Активность каспазы-3, измеренная в терминальной стадии апоптоза, была значительно увеличена обработкой соединением 40 и дополнительно усилена сверхэкспрессией RXRa (Фиг. 4В). Аналогичным образом, увеличение гибели клеток, измеренная по увеличению фрагментации ДНК, также наблюдалась в линии клеток MIA РаСа-2, обработанной соединением 40, и сверхэкспрессия RXRa еще больше увеличила гибель клеток. Гибель клеток измеряли по повышенным уровням ДНК-фрагментов в соответствии с протоколом производителя (Roche) и нормализовали к общей клеточной ДНК. Данные представлены в виде % от контрольных. Однако избыточная экспрессия отдельно RXRa без соединения 40 не привела к увеличению гибели клеток (Фиг. 4С).
Совместно, такие данные указывают на то, что агонист RXR, соединение 40, увеличивает апоптоз в MIA РаСа-2 раковых панкреатических клетках.
Влияние агониста RXR на линии клеток рака груди
Культура MCF7 линии клеток рака груди в присутствии соединения 40 (3 мкМ) показала снижение роста клеток (Фиг. 5А). Это влияние, оказываемое соединением 40, поддерживали в течение длительного времени, как показывают данные, полученные в течение 15 пересевов. Кроме того, инкорпорирование 5'-бромо-2'-дезоксиуридина (BrdU) также значительно снижается в клетках MCF7, обработанных соединением 40 (Фиг. 5В). Снижение пролиферации клеток сопровождалось значительным увеличением клеточного стресса, как показали возросшие уровни активных форм кислорода (ROS) (Фиг. 5С). В заключение, повышение уровня апоптоза, как было измерено по увеличению фрагментации ДНК, было также увеличено в клетках MCF7 культивированием их с соединением 40 (Фиг. 5D). В заключение можно отметить, что данные указывают на то, что соединение 40 обладает активностью, позволяющей уменьшить рост клеток рака молочной железы и индуцировать в них апоптоз.
Влияние агониста RXR на линии лейкозных клеток
Прогрессирование промиелоцитарного лейкоза может быть аннулировано их дифференцировкой. Следовательно, агент, вызывающий дифференцировку, может быть терапевтически значимым. Для того, чтобы оценить дифференцирующее способности соединения 40, в качестве модели заболевания in vitro использовали HL-60 промиелоцитарную линию лейкозных клеток. Клетки линии HL-60 культивировали в течение 72 часов либо в виде контрольной группы, либо с обработкой, как указано. После культивирования в течение 72 часов клетки подвергали воздействию раствора нитро-тетразолия (NBT), содержащего форболмиристатацетат (РМА), в течение 1 часа при температуре 37°С. У дифференцированные клетки увеличился потенциал окислительной реакции, что уменьшает NBT. Уровни восстановленного NBT были измерены по поглощению при длине волны 540 нм. Дифференцировка HL-60 клеток была значительно увеличена путем обработки соединением 40 (Фиг. 6). Аналогичным образом, транс-ретиноевая кислота (ATRA), известный дифференцирующий агент, использующийся для терапии, также стимулирует дифференциацию в HL-60 клетках. Интересно отметить, что лечение соединением 40 с ATRA оказало аддитивное действие на дифференциацию. В заключение можно отметить, что соединение 40 может вызвать дифференцировку промиелоцитарной линии лейкозных клеток и оказывает аддитивное действие с ATRA.
Экспериментальные данные in vivo
I. Оценка влияния агониста RXR на мышах с алиментарным ожирением
Были выбраны шестинедельные самцы мыши C57BL/6J, акклиматизированы в течение 1 недели. Животные содержались либо на корме (10% ккал из жиров), либо на диете с высоким содержанием жиров (HFD) (D12492; 60% ккал из жиров), Research Diets, Inc. New Jearsey, США). Животных содержали в полипропиленовых клетках при температуре 23±1°С при влажности 60±10% и циклах день-ночь по 12 часов и со свободным доступом к корму и воде (вволю, ad libitum). Через 11 недель после вмешательства в питание, животных, которых содержали на диете с высоким содержанием жиров (HFD), отнесли к экспериментальным группам с отбором на основе массы тела, AUC глюкозы во время ОПТ (глюкозотолерантного теста), уровня глюкозы в крови натощак и после еды и уровня триглицеридов натощак (TG). Мышей контрольной группы без ожирения (n=10) содержали на обычном корме, в то время как группа, в которой проводилось лечение (n=10), и HFD контрольная группа (n=10) находились на диете с высоким содержанием жиров в течение всего экспериментального периода. Животные из группы, в которой проводилось лечение, получали соединение 40 в дозе 5-20 мг на кг массы тела один раз в день перорально в виде суспензии в 1% МС (метилцеллюлоза) в качестве носителя. Мышам контрольной группы без ожирения и мышам HFD контрольной группы вводили носитель.
Вес тела записывался еженедельно, а потребление корма - ежедневно. Кровь, собранную из хвостовой вены, исследовали для оценки глюкозы и TG с использованием полоски Co-agucheck TG. Мышам, которые не потребляли пищи в течение 6 часов, проводили глюкозотолерантный тест пероральным введением глюкозы в дозе 2 г на кг после 9 недель лечения. Было обнаружено, что соединение 40 значительно уменьшает гиперинсулинемию и улучшает утилизацию глюкозы (Фиг. 7). Также было обнаружено, что соединение 40 улучшает индекс инсулинорезистентности (HOMA-IR), что указывает на улучшение чувствительности организма в целом к инсулину (Фиг. 8).
Измерение термогенеза: Температуру тела измеряли в конце периода лечения с использованием ректального зонда. Для экспериментального холодового воздействия мышей размещали в клетках по отдельности и переносили в холодную окружающую среду с температурой 4°С. Температуру измеряли каждые 15 мин в течение 75 мин и затем температуру окружающей среды повышали до комнатной, при этом продолжали измерять ректальную температуру в течение еще 20 минут с 10-минутным интервалом. После 10 недель лечения из ретроорбитального венозного сплетения брали кровь для биохимических измерений. Затем животных умерщвляли и вскрывали, немедленно вырезали печень, взвешивали и оценивали печеночные триглицериды (TG). Были выделены и взвешены различные жировые депо. Было отмечено, что соединение 40 значительно снижает увеличение веса тела с уменьшением веса различных жировых депо (Фиг. 12). Кроме того, соединение 40 также значительно повышает нелихорадочный термогенез и уровень экспрессии UCP1 в паховых белых жировых тканях, что выявлялось по бурому окрашиванию белой жировой ткани (WAT) (Фиг. 13).
Животных забивали после конечного измерения натощак уровня глицерина, свободных жирных кислот, холестерина, ЛПНП. Было обнаружено, что лечение с помощью соединения 40 значительно снижает уровень TG натощак без увеличения накопления эктопического жира (Фиг. 9). Кроме того, было отмечено, что соединение 40 значительно снижает уровень холестерина в крови после еды (Фиг. 10). Кроме того, после лечения было замечено значительное снижение уровня циркулирующих свободных жирных кислот и глицерина (Фиг. 11).
Упражнение на выносливость или упражнение на беговой дорожке (ТМТ): Упражнение на выносливость проводилось на моторной беговой дорожке при интенсивности от низкой до умеренной при 5-20 см/сек в качестве максимальной скорости бега с интенсивностью шока в 0,2 мА и с наклоном в один градус. Животных подвергали ТМТ-тесту до истощения, для всех мышей в разных группах измеряли пройденное расстояние (см), время, проведенное в шоковой зоне (сек), количество шоковых ударов, общее время (мин).
Синергетический эффект соединения 40 и упражнений на метаболический профиль у мышей с алиментарным ожирением (DIO)
Были выбраны шестинедельные самцы мыши C57BL/6J, акклиматизированы в течение 1 недели. Животные содержались либо на корме (10% ккал из жиров), либо на диете с высоким содержанием жиров (HFD) (D12492; 60% ккал из жиров), Research Diets, Inc. New Jearsey, США). Через 11 недель после вмешательства в питание, животных, которых содержали на диете с высоким содержанием жиров (HFD), отнесли к экспериментальным группам с отбором на основе массы тела, AUC глюкозы во время ОПТ (глюкозотолерантного теста), уровня глюкозы в крови натощак и после еды и уровня триглицеридов натощак (TG). Мышей контрольной группы без ожирения (n=10) содержали на обычном корме, в то время как группа, в которой проводилось лечение (n=10), и HFD контрольная группа (n=10), с упражнениями и без упражнений, находились на диете с высоким содержанием жиров в течение всего экспериментального периода. Животные из группы, в которой проводилось лечение, получали соединение 40 в дозе 10 мг на кг массы тела один раз в день перорально в виде суспензии в 1% МС (метилцеллюлоза) в качестве носителя. Мышам контрольной группы без ожирения и мышам HFD контрольной группы вводили носитель. Тренировка на выносливость проводилось на моторной беговой дорожке при интенсивности от низкой до умеренной при 5-20 см/сек в качестве максимальной скорости бега с интенсивностью шока в 0,2 мА и с наклоном в один градус. Животные были приучены к этой процедуре 5 дней в неделю, для всех мышей в разных группах измеряли пройденное расстояние (см), время, проведенное в шоковой зоне (сек), количество шоковых ударов, общее время (мин). Вес тела записывался еженедельно, а потребление корма - ежедневно. Кровь, собранную из хвостовой вены, исследовали для оценки глюкозы и TG. Мышам, которые не потребляли пищи в течение 6 часов, проводили глюкозотолерантный тест пероральным введением глюкозы в дозе 2 г на кг после 4 и 8 недель лечения. Затем животных умерщвляли и вскрывали, немедленно вырезали печень, взвешивали и оценивали печеночные триглицериды (TG). Были выделены и взвешены различные жировые депо.
Было отмечено, что во время лечения соединение 40 значительно улучшается выносливость мышц (Фиг. 14). Кроме того, в группе, в которой проводилось лечение, наблюдалось значительное улучшение в уровнях глюкозы натощак и уровнях ПТТГ (Фиг. 29). Кроме того, в группе, в которой проводилось лечение, отмечалось улучшение в весе тела и уровне TG натощак (Фиг. 30).
II. Оценка влияния агониста RXR на мышах ob/ob
Самцы ob/ob и мыши без ожирения были доставлены из лабораторий Harlan, акклиматизированы в течение недели, содержались на стандартной лабораторной диете. Контрольным мышам без ожирения и мышам ob/ob в начале исследования было 16 недель от роду, мыши были безвыборочным методом поделены на контрольную группу и группу для лечения (соединение 40, в дозе 3, 10 30 мг/кг, 2 раза в день, перорально), с отбором на основе массы тела, глюкозы после еды и AUC глюкозы во время ОПТ (глюкозотолерантного теста). Лечение продолжалось в течение 4-х недель. Еженедельно контролировали уровень глюкозы и триглицеридов натощак (надрез хвоста). В конце 4-й недели животных забивали, затем проводили конечные измерения натощак уровня глицерина, свободных жирных кислот, холестерина, ЛПНП. Было обнаружено, что у животных из группы, в которой проводили лечение, одновременно присутствовали раннее начало и устойчивый контроль гипергликемии, вызванной приемом пищи (Фиг. 15). Кроме того, было обнаружено, что лечение с помощью соединение 40 значительно уменьшило общее содержание холестерина, главным образом за счет снижения холестерола липопротеинов низкой плотности (ЛПНП) (Фиг. 16).
III. Оценка влияния агониста RXR на мышах db/db
Самцы db/db и мыши без ожирения были доставлены из лабораторий Jackson, США, акклиматизированы в течение недели, содержались на стандартной лабораторной диете. Мыши без ожирения и мыши db/db на начало исследования были 8 недель от роду; животные были безвыборочным методом на контрольную группу и группу для лечения (соединение 40, в дозе 2,5, 5, 10 15 мг/кг, 2 раза в день, перорально), с отбором на основе массы тела, глюкозы после еды и AUC глюкозы во время ОГТТ (глюкозотолерантного теста). Лечение продолжалось в течение 8 недель. Еженедельно контролировали уровень глюкозы и триглицеридов натощак (надрез хвоста). В конце 8 недели лечения были измерены уровни глюкозы и инсулина HbA1c через различные промежутки времени. Образцы крови, собранные методом надреза хвоста, были подвергнуты анализу в конце 7 недели лечения, с целью измерения HbA1c на анализаторе DCA Vantage (Siemens).
Оценка 24 ч профиля глюкозы: в конце 7 недели лечения методом надреза хвоста отбирали образцы крови после еды, измеряли уровень глюкозы после еды через каждые 2 часа времени до 8 часов, а затем с интервалом в 4 часа.
Было обнаружено, что лечение соединением 40 значительно снижало уровень глюкозы в крови после приема пищи (Фиг. 17). Лечение также значительно снижало перемещение глюкозы в прандиальной фазе (Фиг. 18). Было также обнаружено, что лечение соединением 40 привело к резкому и дозозависимому снижению уровня HbA1c (Фиг. 19).
IV. Оценка влияния агониста RXR на самцах крыс Zucker с ожирением
Пятинедельные самцы крыс Zucker с ожирением от Charles River и соответствующие по возрасту контрольные крысы содержались при температуре 22±3°С и относительной влажности 50-70%, при циклах день-ночь по 12 часов с искусственными люминесцентными лампами. Животных содержали в группе по 3 в стандартных полипропиленовых клетках с верхней решеткой из нержавеющей стали, оснащенных оборудованием для гранулированных продуктов питания и питьевой водой в бутылке. Животных содержали на нормальной сбалансированной диете (Provomi), Bisleri, коммерчески доступная минеральная питьевая вода ad libitum. Контрольные крысы без ожирения и самцы XDF крыс были безвыборочным методом поделены на контрольную группу и группу для лечения (соединение 40, в дозе 10 мг/кг, 2 раза в день, перорально) - по массе тела, уровню глюкозы и глюкозы после еды. Лечение продолжалось в течение 8 недель. Еженедельно контролировали уровень глюкозы и триглицеридов натощак (надрез хвоста). В конце 8 недели лечения были измерены уровни глюкозы и инсулина HbA1c через различные промежутки времени. В образцах крови, собранных методом надреза хвоста, определялся уровень HbA1 на DCA Vantage (Siemens). Было обнаружено, что лечение с помощью соединения 40 значительно увеличивает активность сукцинатдегидрогеназы (СДГ), что указывает на улучшение окисления глюкозы в мышцах. Кроме того, лечение продемонстрировало раннее начало и устойчивый контроль гипергликемии после приема пищи, а также показало значительный контроль уровня глюкозы натощак (Фиг. 20). Также было обнаружено, что лечение значительно снижает гиперинсулинемию и улучшает распад глюкозы (Фиг. 21). Лечение также показало существенное улучшение уровня HbA1c (Фиг. 22). При измерении триглицеридов натощак во время лечения и после него было обнаружено, что соединение 40 не вызывает гипертриглицеридемии. Кроме того, оно значительно снижает уровень триглицеридов (TG) натощак (Фиг. 23). Также было обнаружено, что соединение 40 способствует уменьшению уровня С-реактивного белка (CRP), что указывает на потенциальные выгоды для сердечно-сосудистой системы (Фиг. 24). Также отмечалось существенное улучшение сердечной функции (Фиг. 25), а также улучшение уровней биомаркеров сердечно сосудистого риска (Фиг. 26).
Измерение скорости нервной проводимости: Скорость нервной проводимости была измерена с помощью электронного стимулятора, подключенного к AD Power Lab 8/30, фирмы AD instruments. Животных на короткое время анестезировали и подсоединяли к ним три различных электрода (+ve, -ve и нейтральный) от BioAmp. +ve электрод был размещен на передней стороне безымянного пальца на задней лапе, -ve электрод был помещен непосредственно над анодом на расстоянии 10 мм, нейтральный электрод был помещен в области живота. Напряжение 5 мВ с длительностью импульса 0,001 сек было представлено на двух разных регионах на расстоянии 1 см на седалищном нерве. Нервная проводимость измерялась с использованием программного обеспечения LabChart software в пределах видимости панели просмотра. Задержки в двух различных точках были измерены с помощью установки курсора мыши на начало пиков. Скорость нервной проводимости (NCV) была измерена с использованием следующих формул
NCV (м/с) = Задержка в точке В (мс) - Задержка в точке А (мс)
Расстояние между точками А и В (мм).
Значительное улучшение в NCV было отмечено в группе, в которой проводилось лечение (Фиг. 27).
Измерение параметров гемодинамики с помощью аппарата измерения давления Millar MPVS:
В конце 22 недели эксперимента у животного измерили гемодинамические параметры с использованием системы Millar MPVS, на короткое время животное анестезировали с использованием уретана и все хирургические процедуры проводили на тепловой пластине, как яремную вену, так и сонную артерию канюлировали, при этом яремную вену использовали для солевой калибровки, в то время как через сонную артерию в левый желудочек был введен крысиный катетер Millar для записи контура объемного давления. Кровь отбирали пункцией из хвостовой вены с помощью укола иглой в предварительно заполненные этилендиаминтетрауксусной кислотой пробирки, перемешивали и помещали сразу в лед, проводили центрифугирование при 10000 оборотах в минуту в течение 10 минут с использованием холодной центрифуги. Плазму отделяли и исследовали для оценки ANP, BNP, остеопротегерина с помощью набора USCN, а также мочевины в сыворотке крови и BUN с использованием набора Robonik. У больных животных, пролеченных соединением 40, наблюдалось существенное снижение уровня BUN в крови и креатинина в сыворотке крови (Фиг. 28).
V. Оценка влияния агониста RXR на гипертрофию сердца у крыс, индуцированную изопреналином
Четырнадцатинедельные самцы крыс линии Wistar были размещены при температуре, поддерживаемой при 22±3°С, относительной влажности 50-70%, при 12-ти часовых циклах день-ночь с искусственными люминесцентными лампами. Животные были акклиматизированы в течение семи дней. На восьмой день животные были распределены безвыборочным методом в зависимости от их веса тела на девять групп животных. Животные были распределены на следующие группы: G1: контроль среды, G2: контроль изопреналином (5 мг/кг, подкожно два раза в неделю) и G3: изопреналин (5 мг/кг, подкожно два раза в неделю) + соединение 40 (10 мг/кг, перорально раз в день). Лечение с помощью соединение 40 (10 мкг) для группы G3 было начато на одну неделю ранее, чем введение изопреналина, и далее в течение одиннадцати недель продолжали лечение соединением 40 и изопреналином. У животных измеряли суточное потребление корма и еженедельный вес тела на протяжении всего исследования. В конце экспериментального периода (11 неделя) пять животных из каждой группы были подвергнуты кардиоваскулярной оценке гемодинамики катетером Millar с использованием интерфейса Millar MPVS, соединенного с многоканальным универсальным усилителем PowerLab AD instruments. Измерялись различные параметры: частота сердечных сокращений, систолическая работа, систолический объем крови, сердечный выброс, систолическое давление левого желудочка, диастолическое давление левого желудочка, диастолический объем левого желудочка. Сразу же после того, как были получены гемодинамические оценки, у всех животных собрали кровь посмертно, далее последовало извлечение сердец. Все выделенные образцы сыворотки были использованы для оценки нескольких кардиомаркеров, таких как ANP, BNP, CRP, LDH, CKN, СКМВ и SGOT. Собранные сердца были обследованы на предмет гистологических изменений и экспрессии генов. Все полученные численные данные были выражены в виде средних величин ± SEM, с применением однофакторного дисперсионного анализа (ANOVA) и критерия Даннетта, использовали программное обеспечение Graph pad prism. В группе, в которой проводилось лечение, наблюдалось существенное уменьшение абсолютного веса сердца (Фиг. 31). Наблюдалось значительное снижение толщины стенки желудочка и перегородки, что означает, что соединение 40 является потенциальным средством для лечения желудочковой гипертрофии и сердечной недостаточности (Фиг. 32). У животных в группе, в которой проводилось лечение, наблюдалось значительное улучшение систолической работы (SW) и систолического объема крови (SV) (Фиг. 33), улучшение сердечного выброса, частоты сердечных сокращений и доли выброса (Фиг. 34), улучшение наполнения желудочков (Фиг. 35) и (Фиг. 36), что показывает, что соединение 40 может играть существенную роль в улучшении сердечной функции.
VI. Оценка влияния агониста RXR на гиперлипидемию у хомяков, вызванную диетой с высоким содержанием холестерина
Самцы сирийских хомяков от 8 до 10 месяцев от роду были размещены в полипропиленовых клетках, снабженных стерильной рисовой шелухой в качестве подстилки. Животных содержали при температуре 22±3°С, относительной влажности 50-70%, с 15-18 циклами смены воздуха с 12 часовым циклом день-ночь. Животные были обеспечены кормом для грызунов (ad libitum), поставляемым лабораториями Harlan для нормальных контрольных животных, а экспериментальные группы получали корм с высоким содержанием холестерина, который был подготовлен самостоятельно, для чего в сбалансированную диету подмешивали 11,5% кокосового масла, 11,5% кукурузного масла, 0,5% холестерина и 0,25% дезоксихолата. Была предоставлена вода Bisleri (ad libitum). Животные были распределены безвыборочным методом на основании массы тела и разделены на контрольную и экспериментальную группы. Животным группы, в которой проводилось лечение, вводили соединение 40 в дозе 10 мг на кг веса в течение 8-12 недель. Потребление корма измерялось ежедневно, а вес тела, уровень общего холестерина и триглицеридов измерялись еженедельно. У экспериментальных животных наблюдалось значительное снижение общего холестерина, не-ЛПВП холестерина и ЛПНП-С (Фиг. 37).
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ РЕТИНОИДЫ ДЛЯ ЛЕЧЕНИЯ ЭМФИЗЕМЫ | 2001 |
|
RU2282616C2 |
| ИНГИБИТОРЫ РЕПЛИКАЦИИ ВИЧ | 2011 |
|
RU2564445C2 |
| ПРОИЗВОДНЫЕ И СПОСОБЫ ЛЕЧЕНИЯ ИНФЕКЦИЙ ГЕПАТИТА В | 2015 |
|
RU2742305C2 |
| Новые 3-индол замещенные производные, фармацевтические композиции и способы применения | 2016 |
|
RU2672252C1 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНОВ | 2011 |
|
RU2554353C2 |
| ПИРИДОНОВЫЕ И АЗАПИРИДОНОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПРИМЕНЕНИЯ | 2011 |
|
RU2617405C2 |
| СОЕДИНЕНИЯ 2-(2-ОКСОИНДОЛИН-3-ИЛИДЕН)МЕТИЛ-5-(2-ГИДРОКСИ-3-МОРФОЛИН-4-ИЛПРОПИЛ)-6,7 ДИГИДРО-1-Н-ПИРРОЛ[3,2-С]ПИРИДИН-4(5Н)-ОНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2008 |
|
RU2472792C2 |
| ПРОИЗВОДНЫЕ АМИНОКУМАРАНА ИЛИ ИХ СОЛИ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, КОМПОЗИЦИЯ ДЛЯ ИНГИБИРОВАНИЯ ОБРАЗОВАНИЯ ЛИПОПЕРОКСИДА | 1992 |
|
RU2087473C1 |
| ПИРАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ СОЕДИНЕНИЙ-АНТАГОНИСТОВ H4 | 2019 |
|
RU2821516C2 |
| ПИРРОЛОПИРАЗИНОВЫЕ ИНГИБИТОРЫ КИНАЗЫ | 2012 |
|
RU2673064C2 |
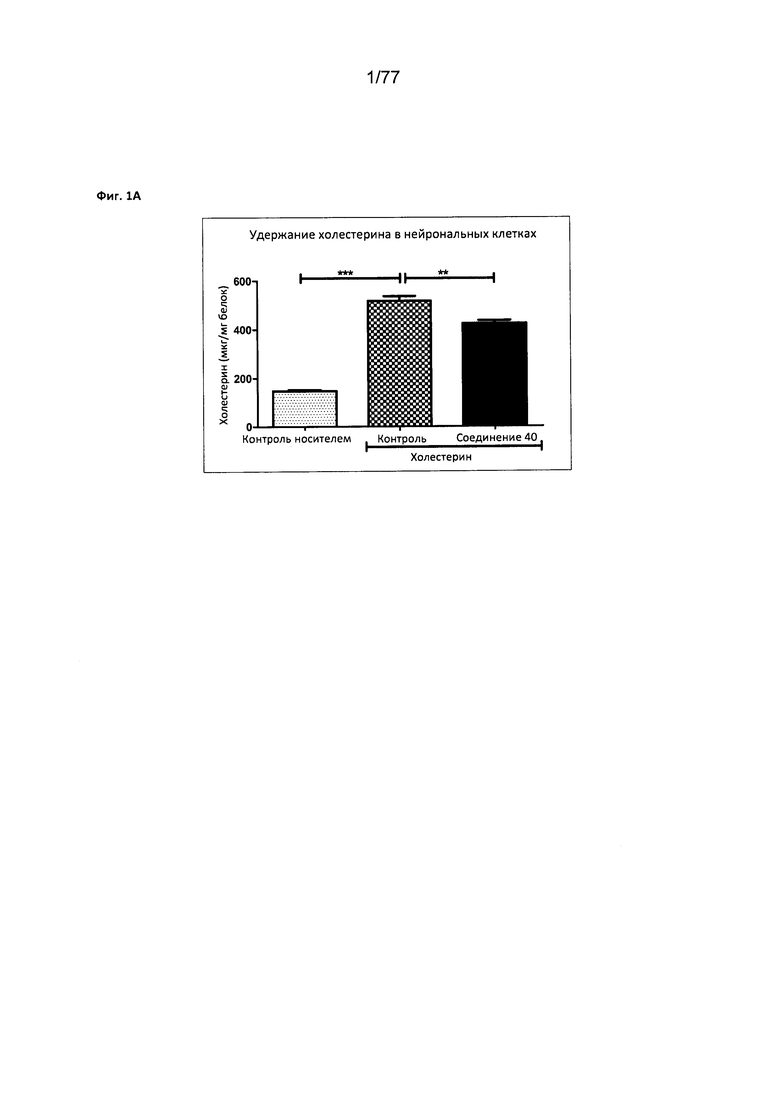
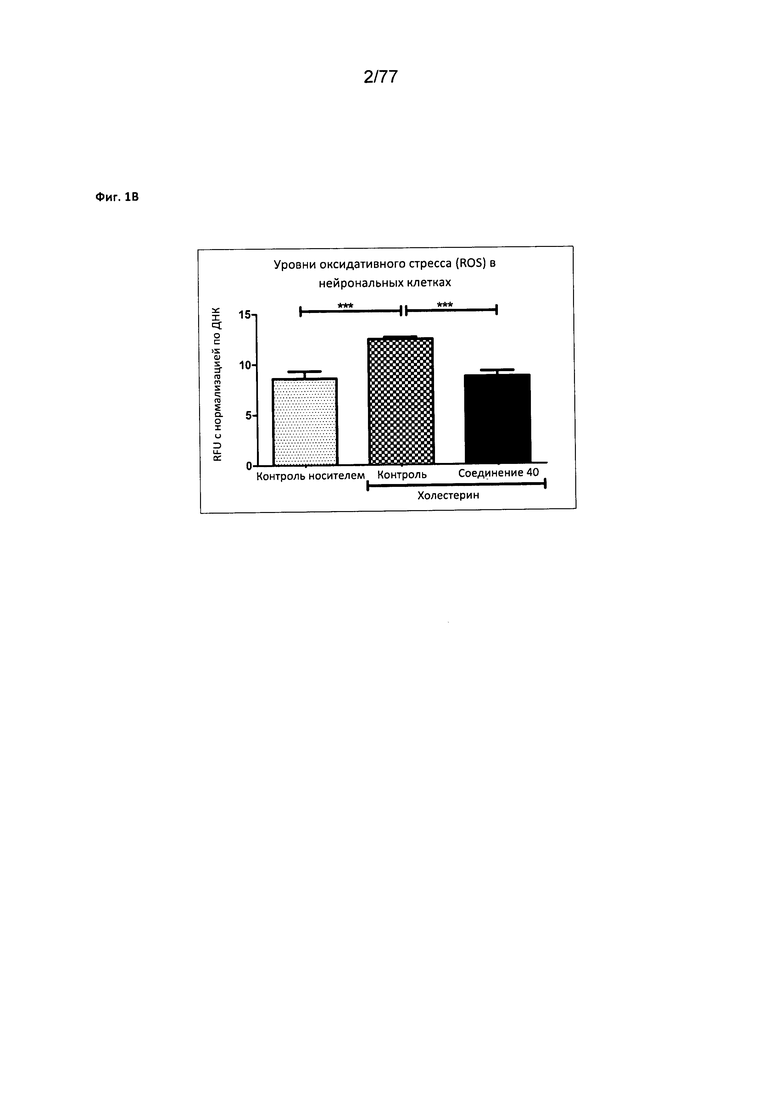
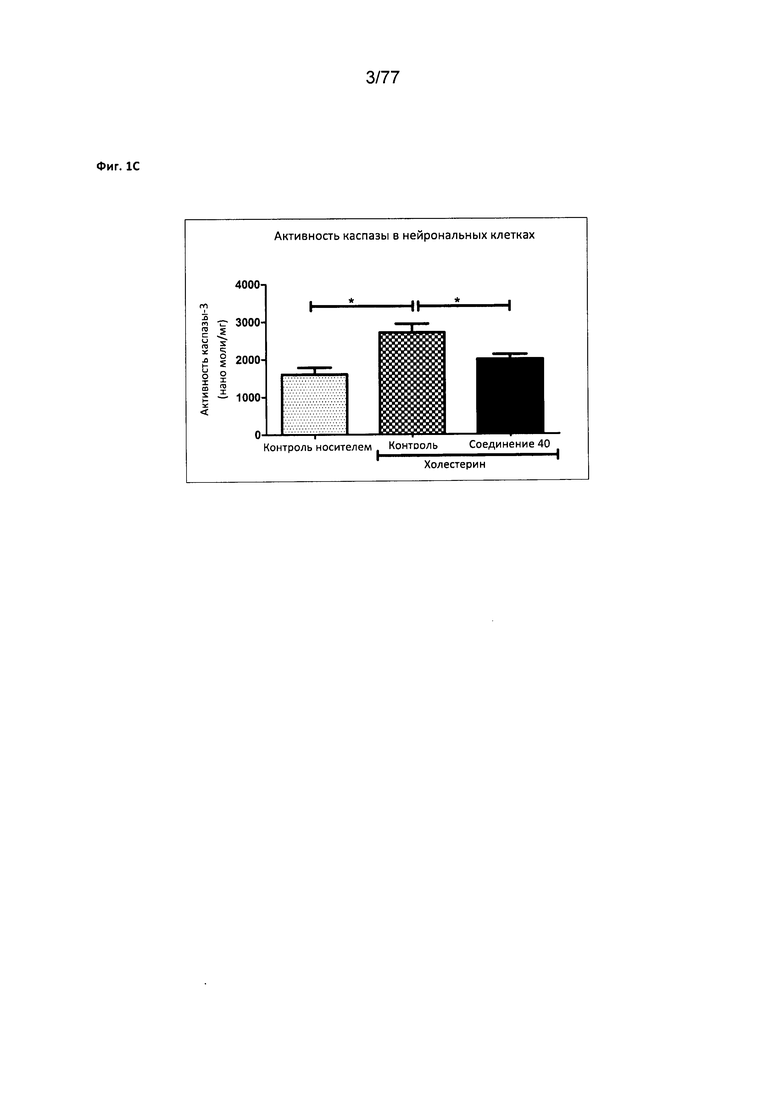
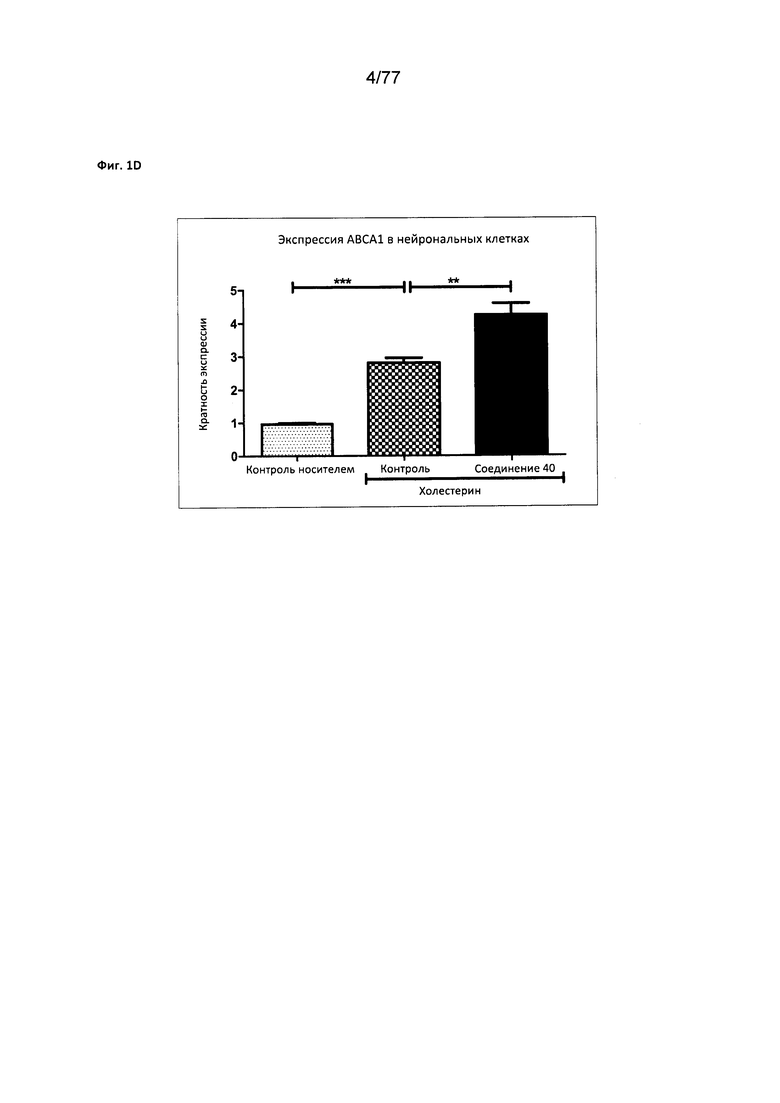
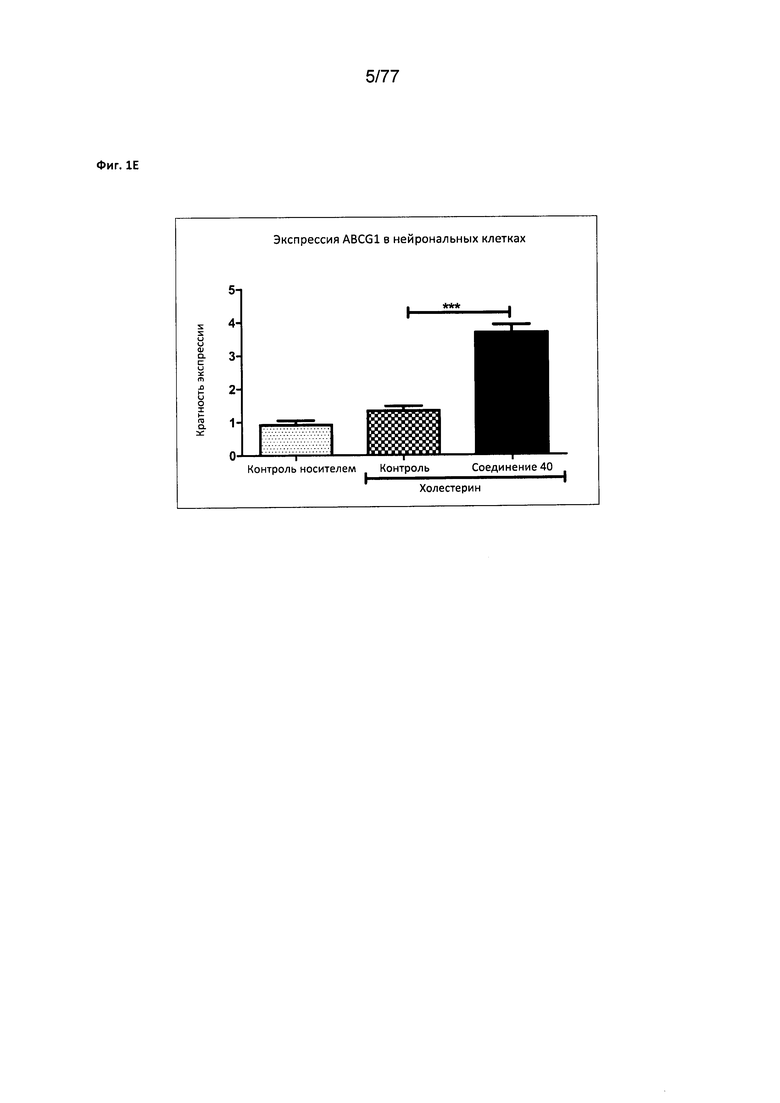
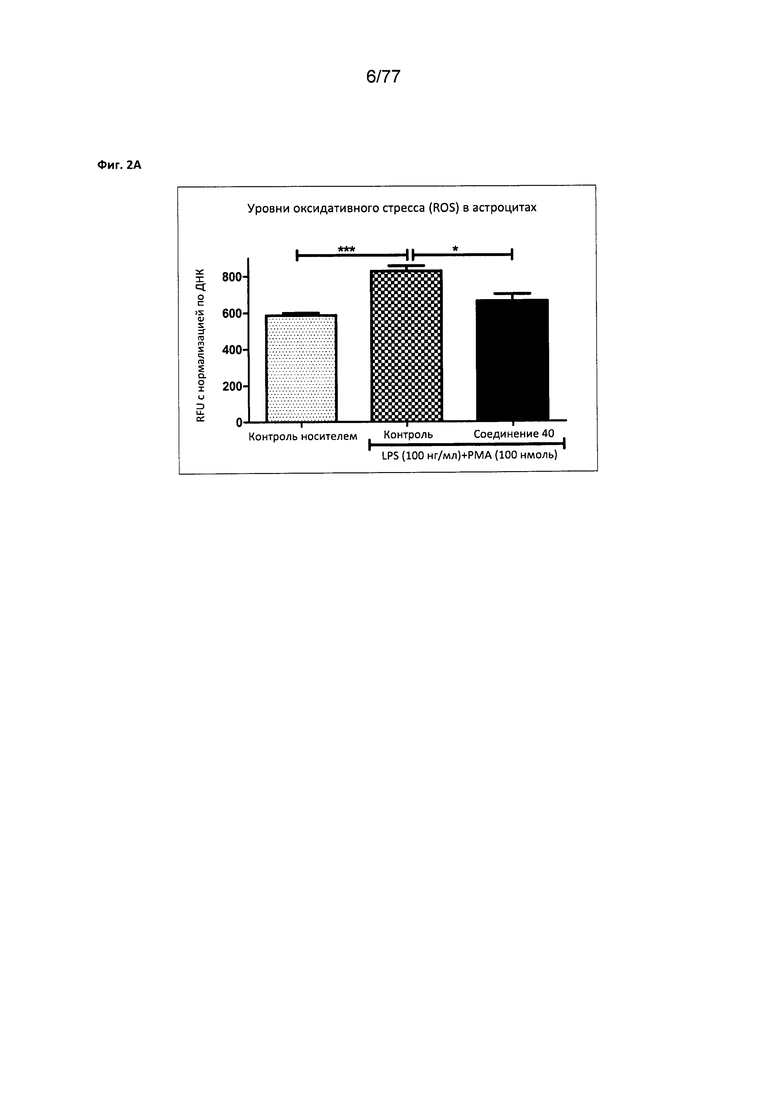
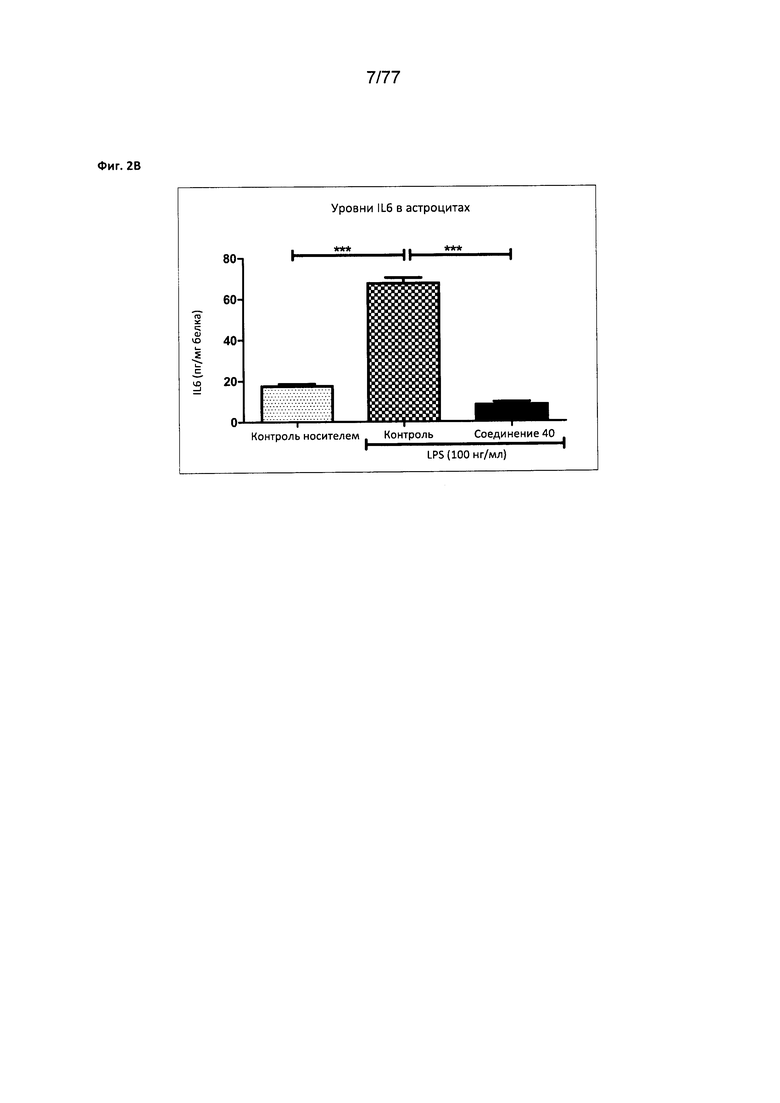
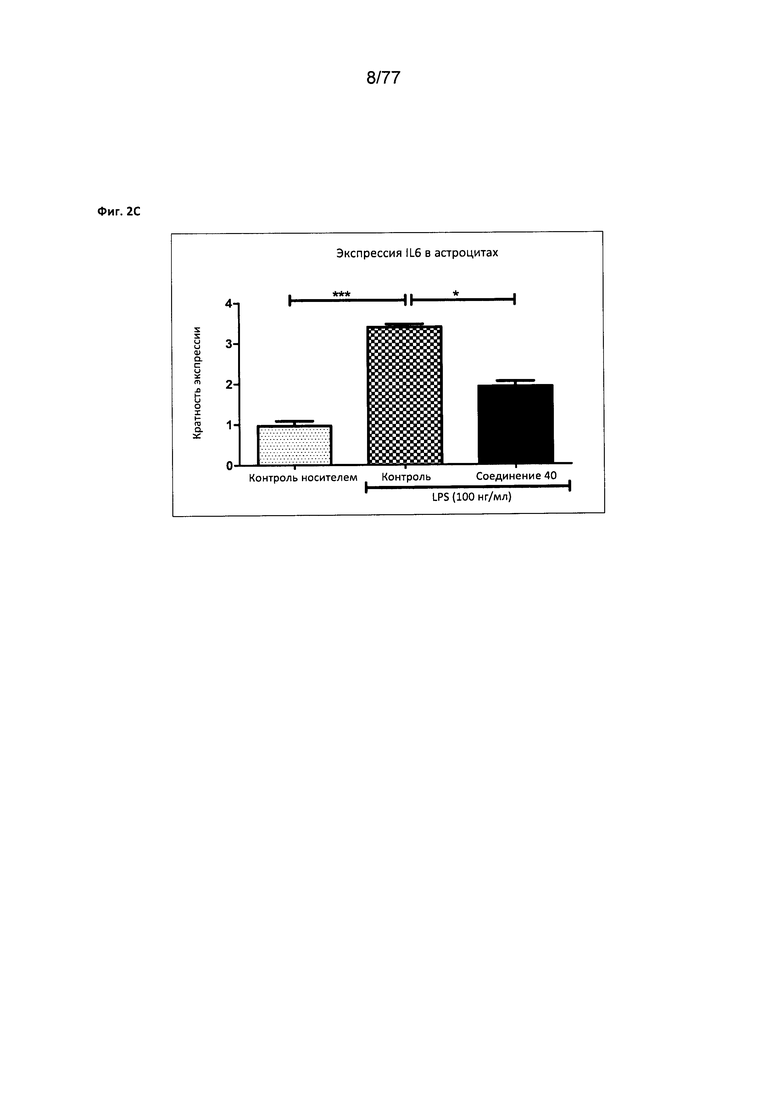
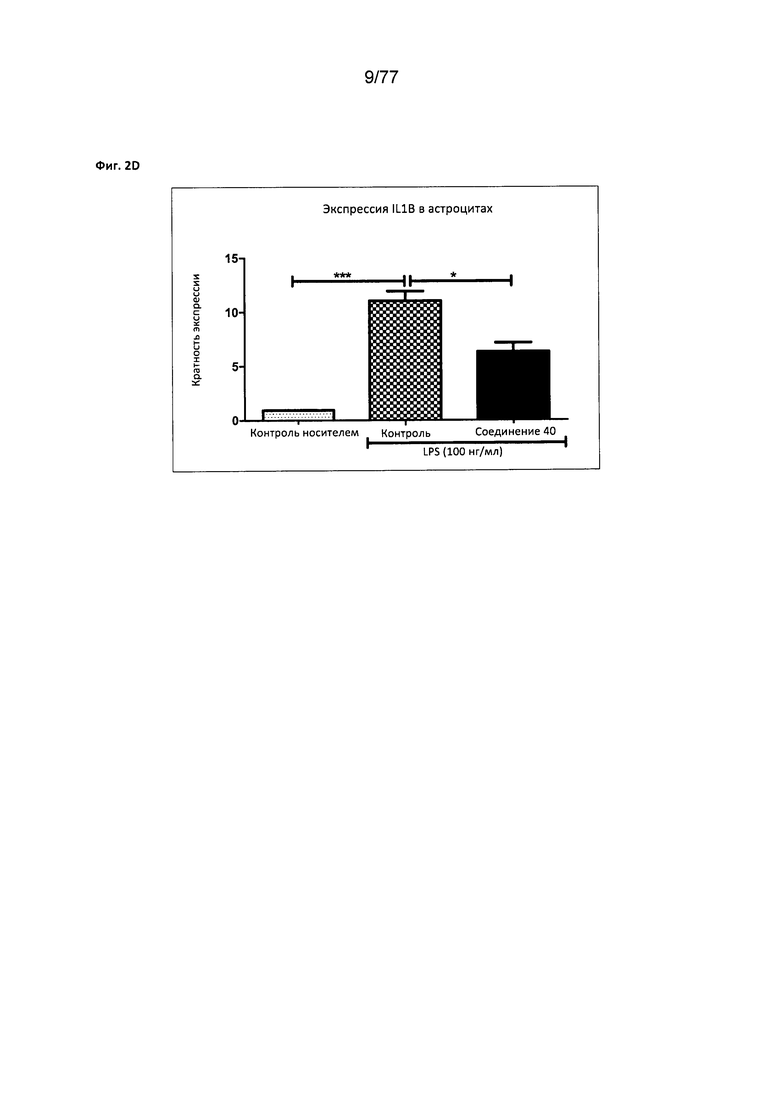
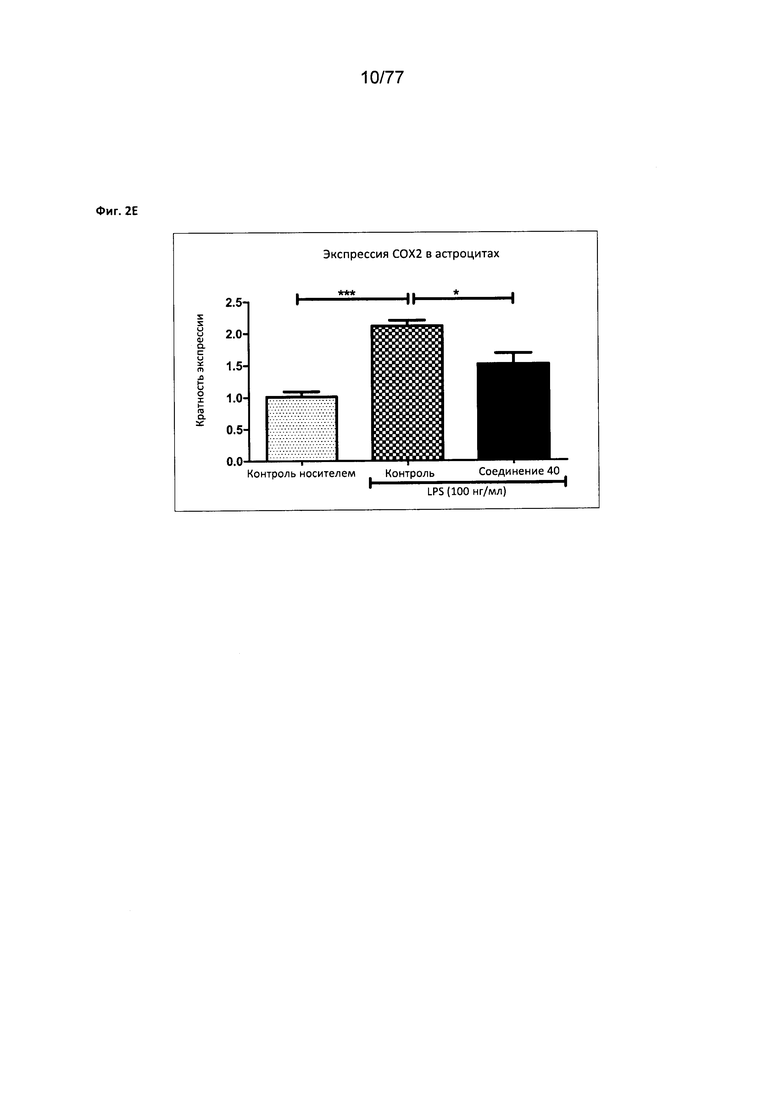
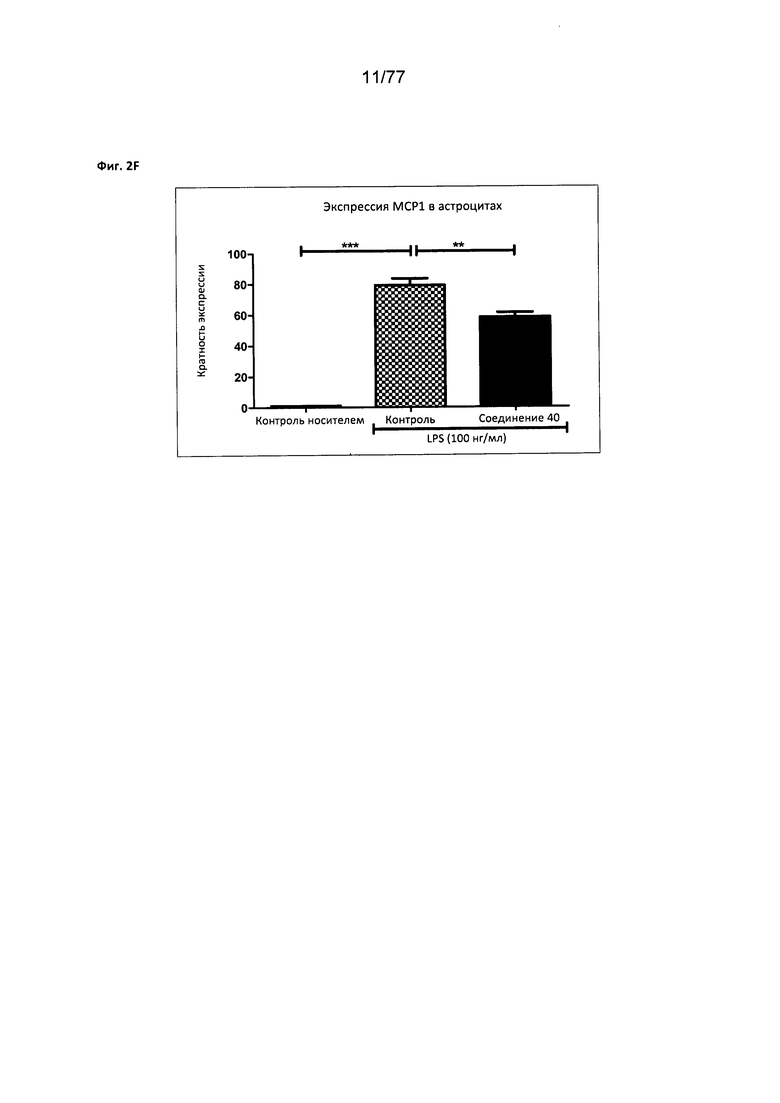
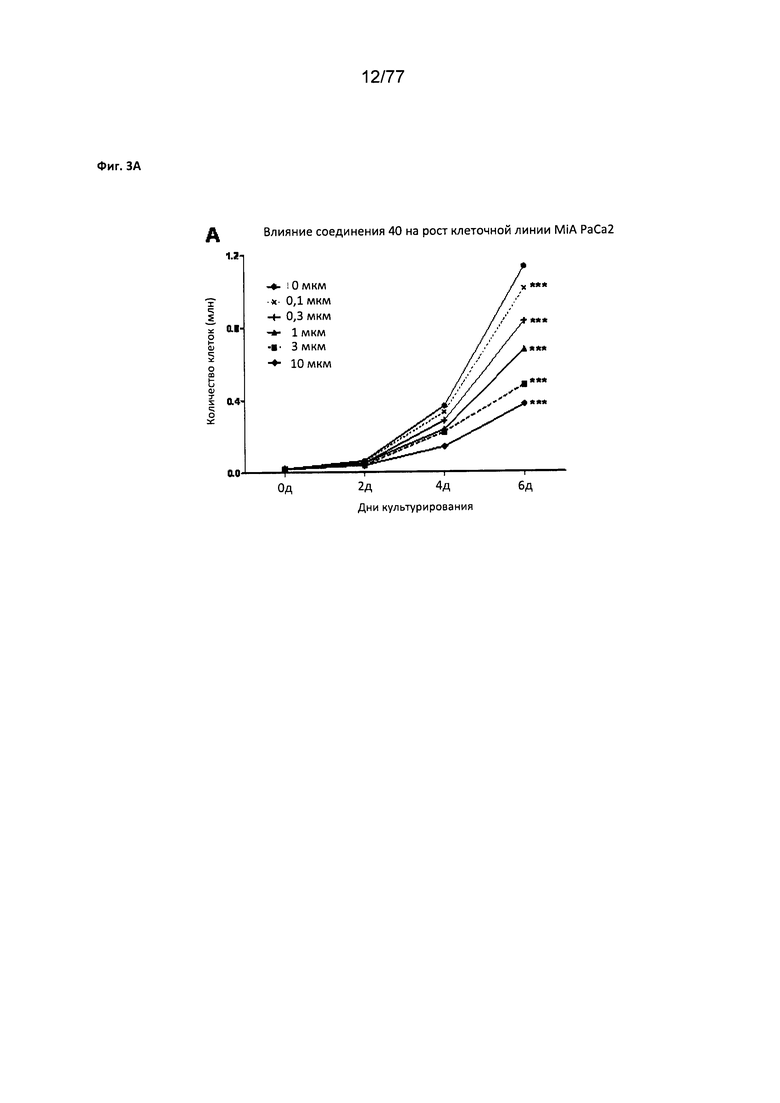
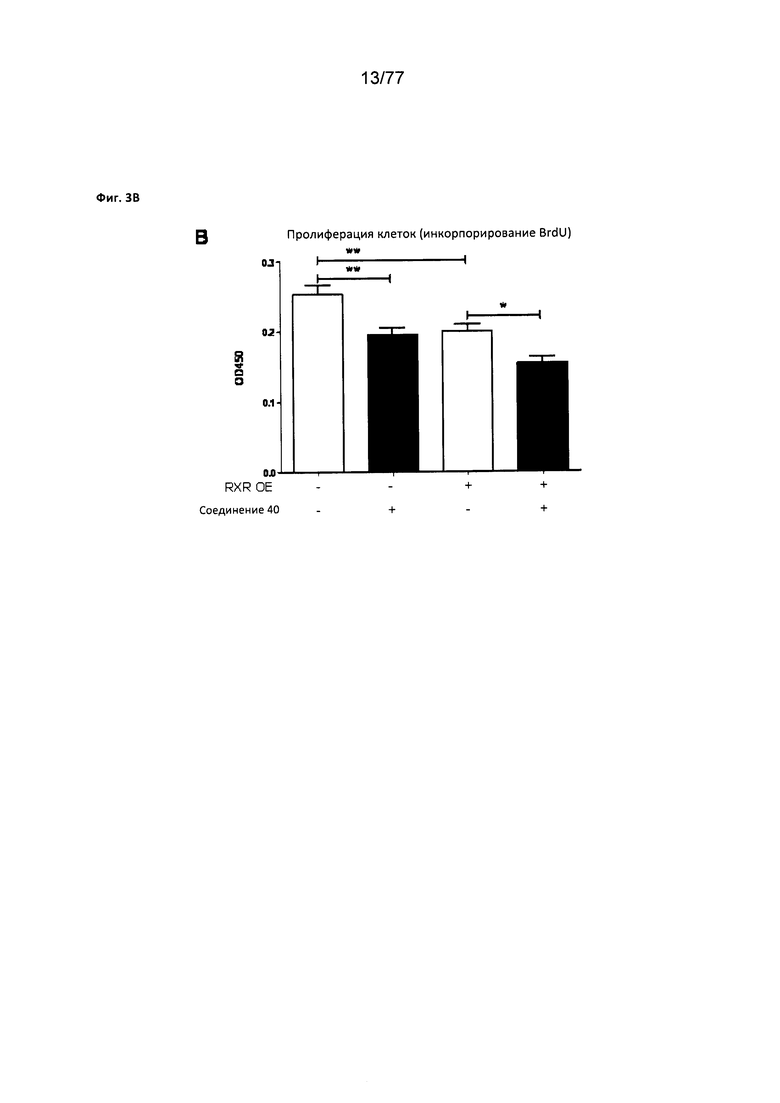
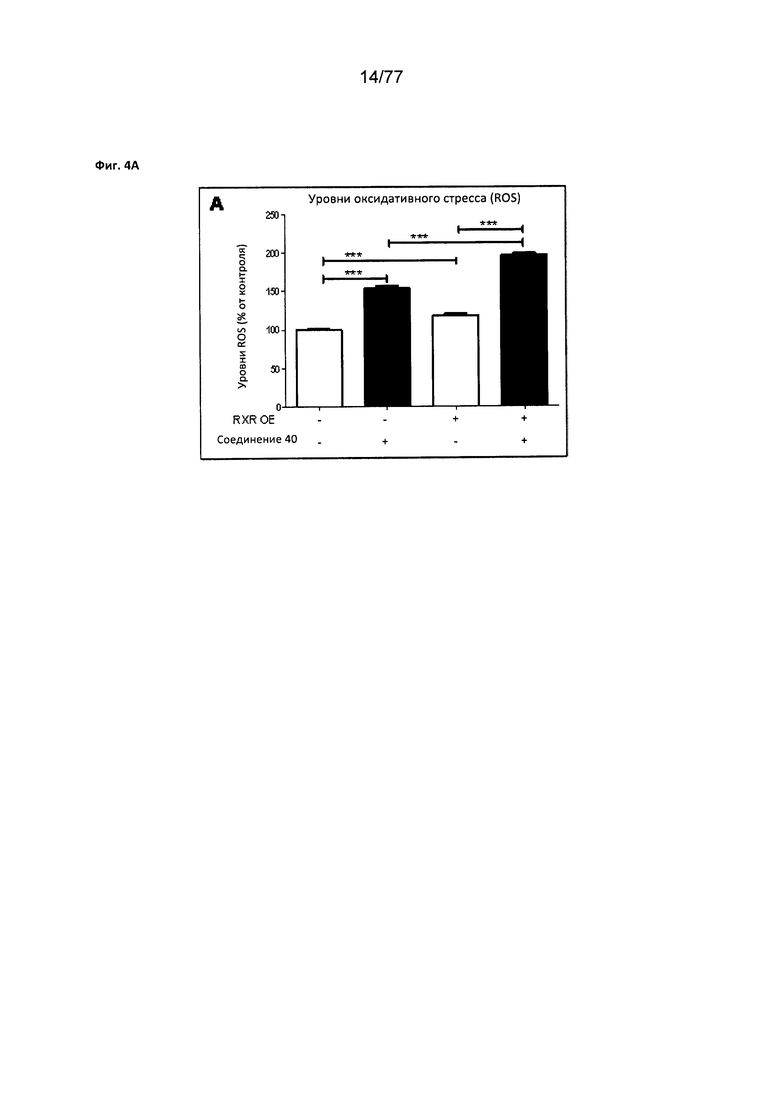
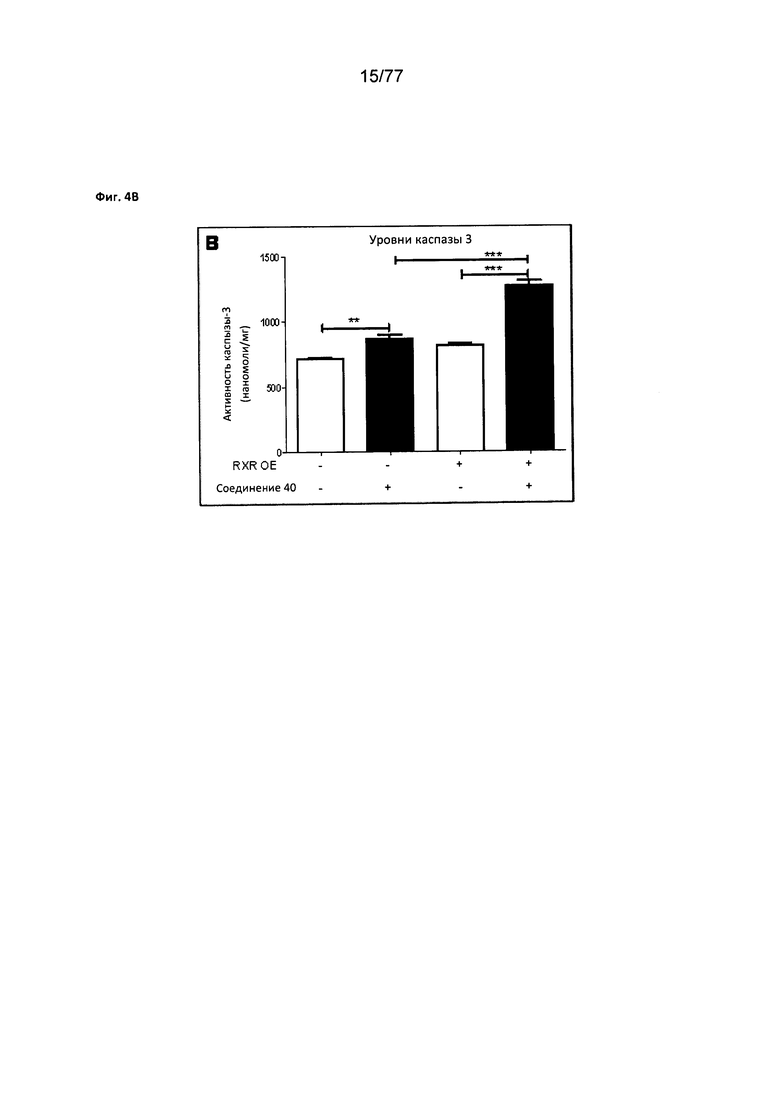
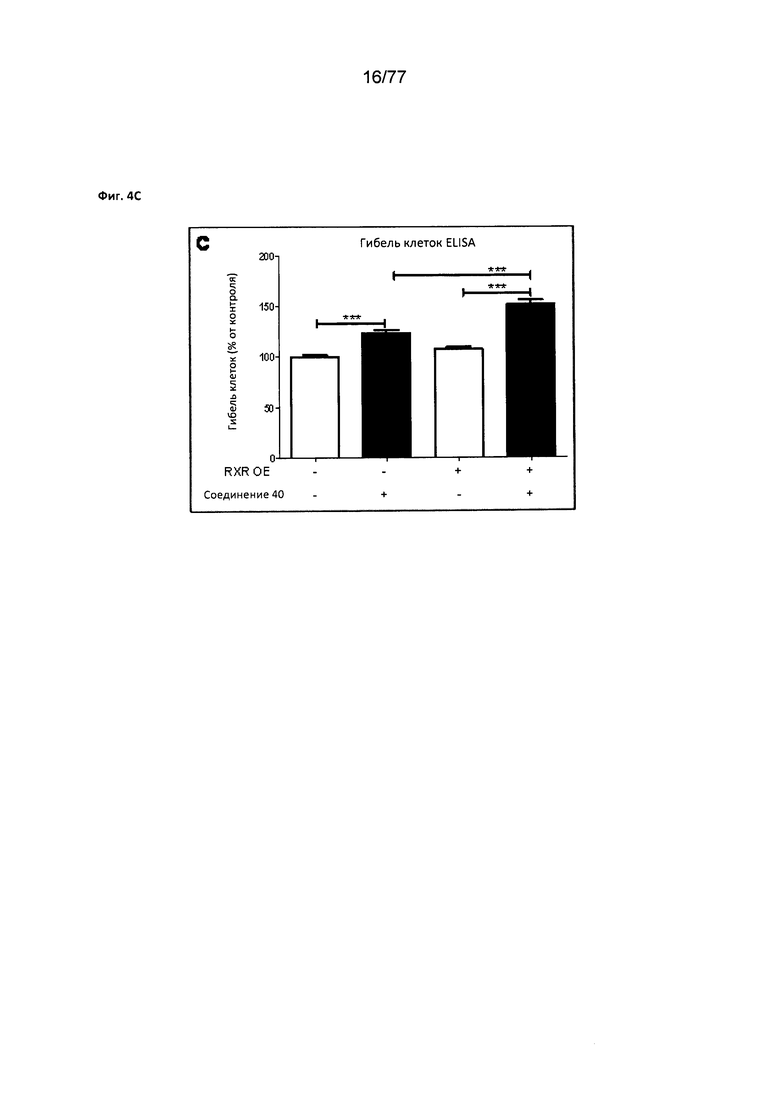
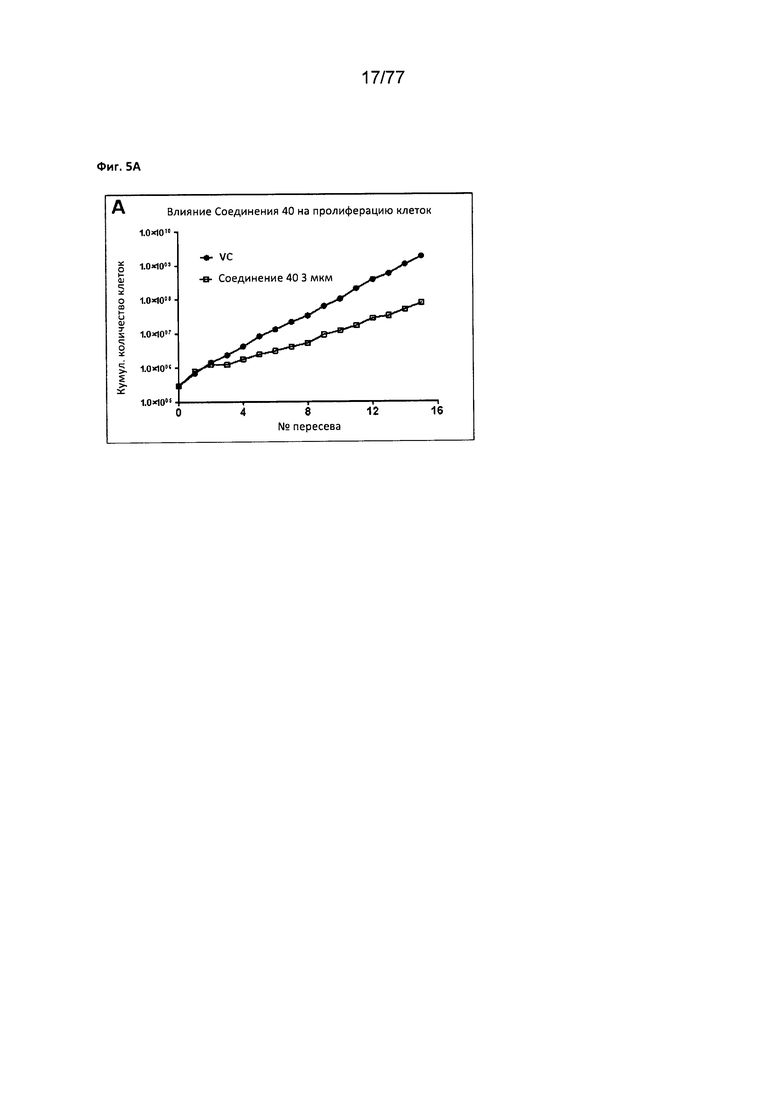
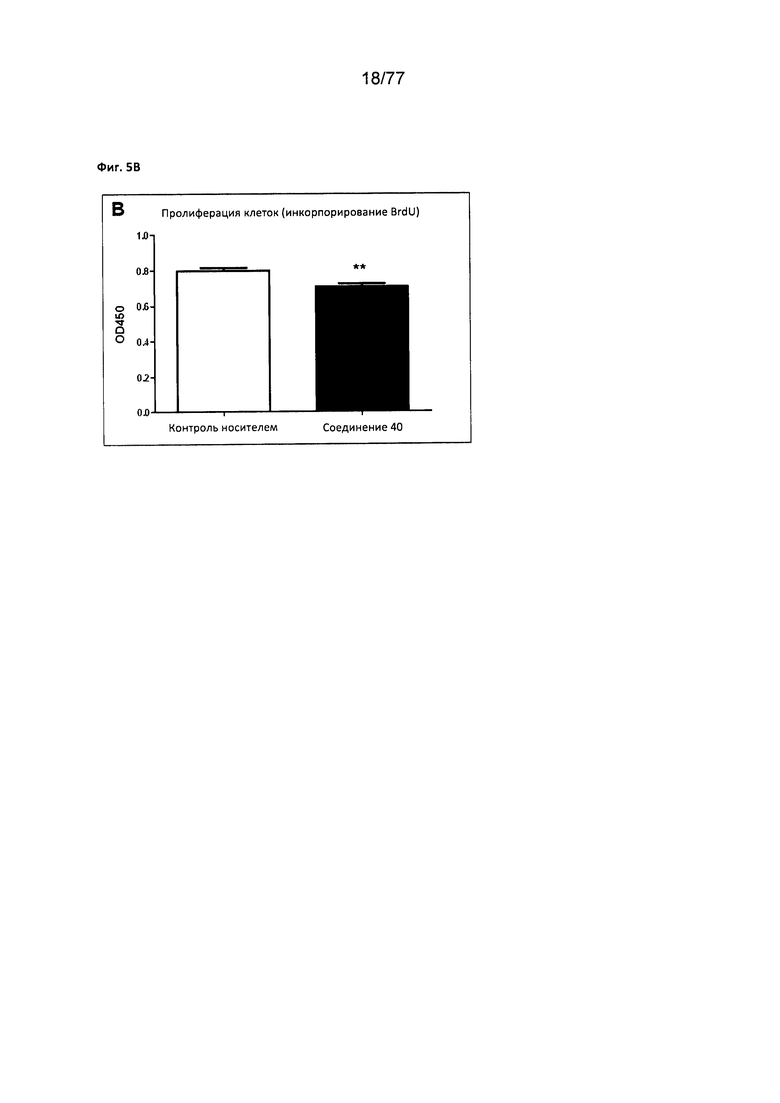
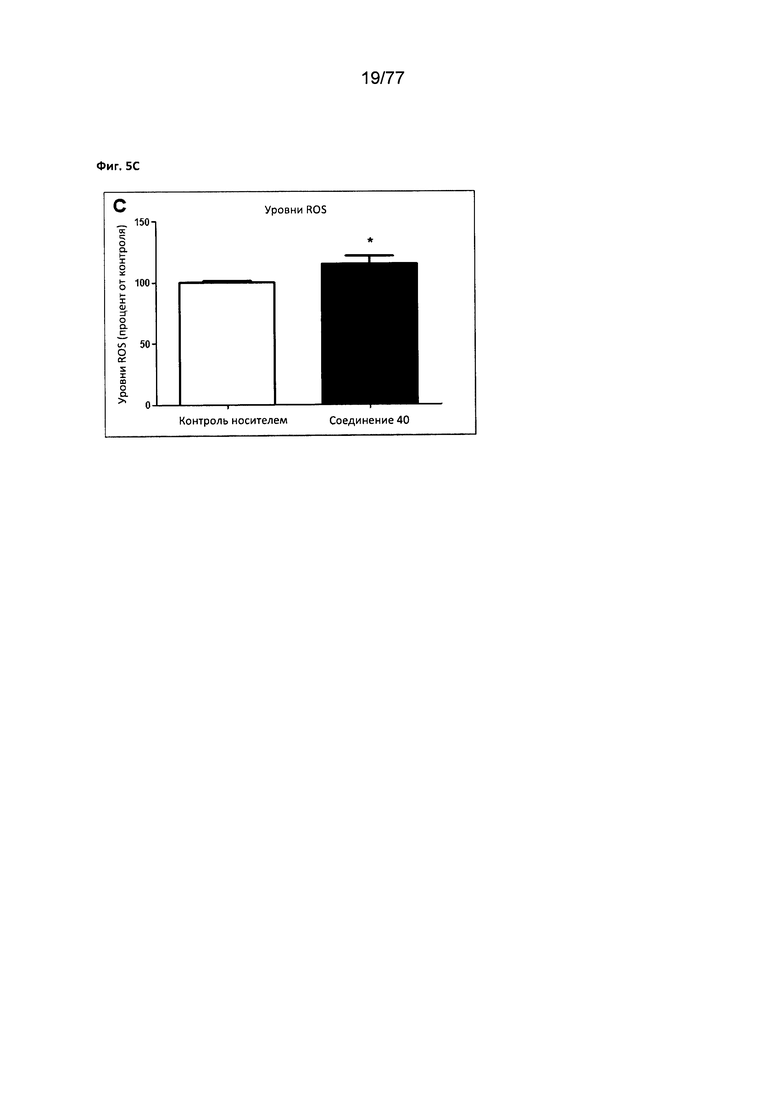
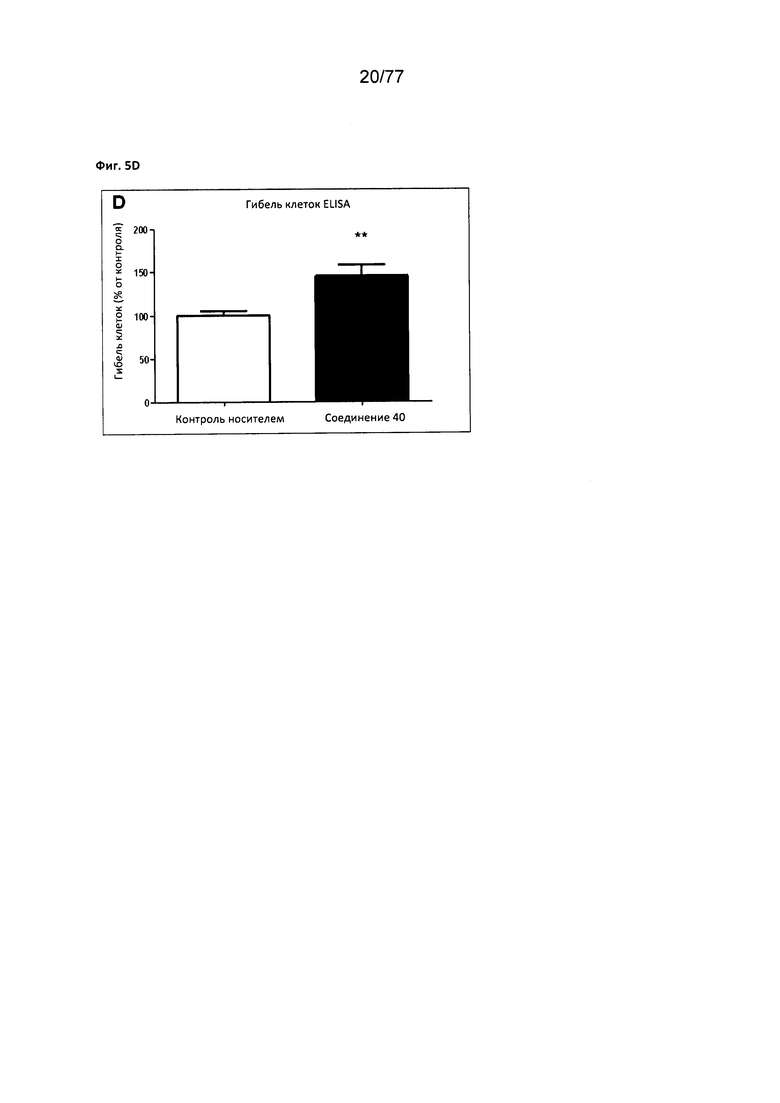
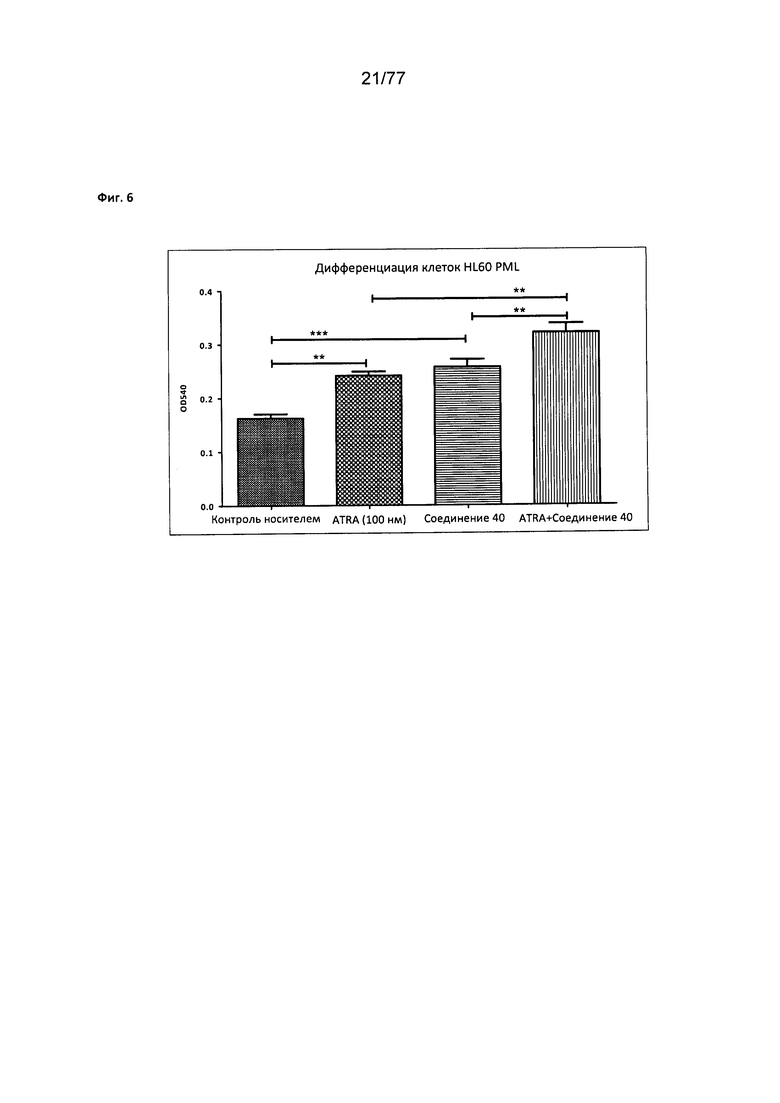
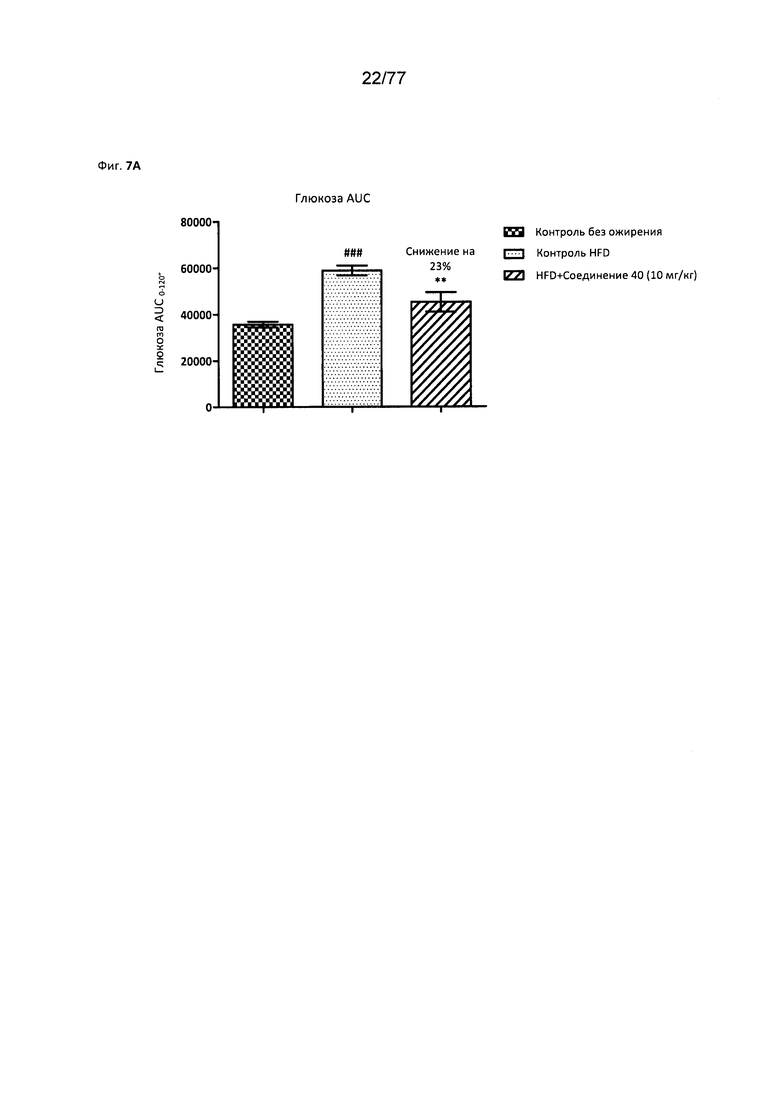
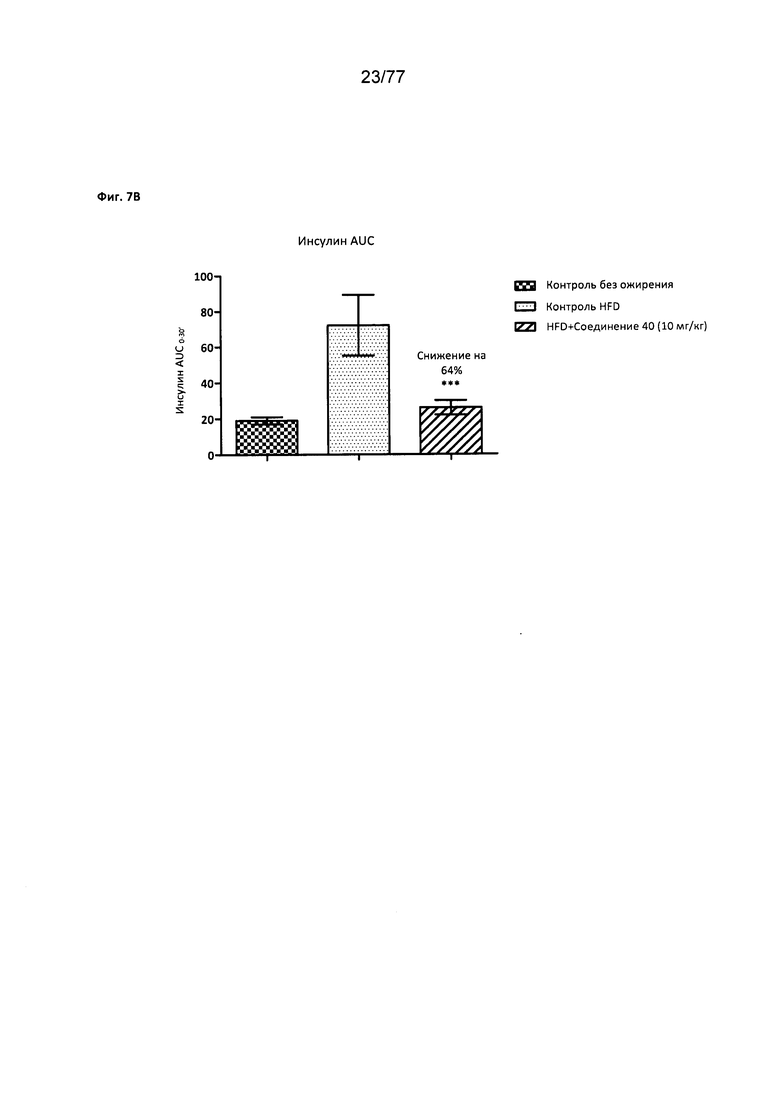
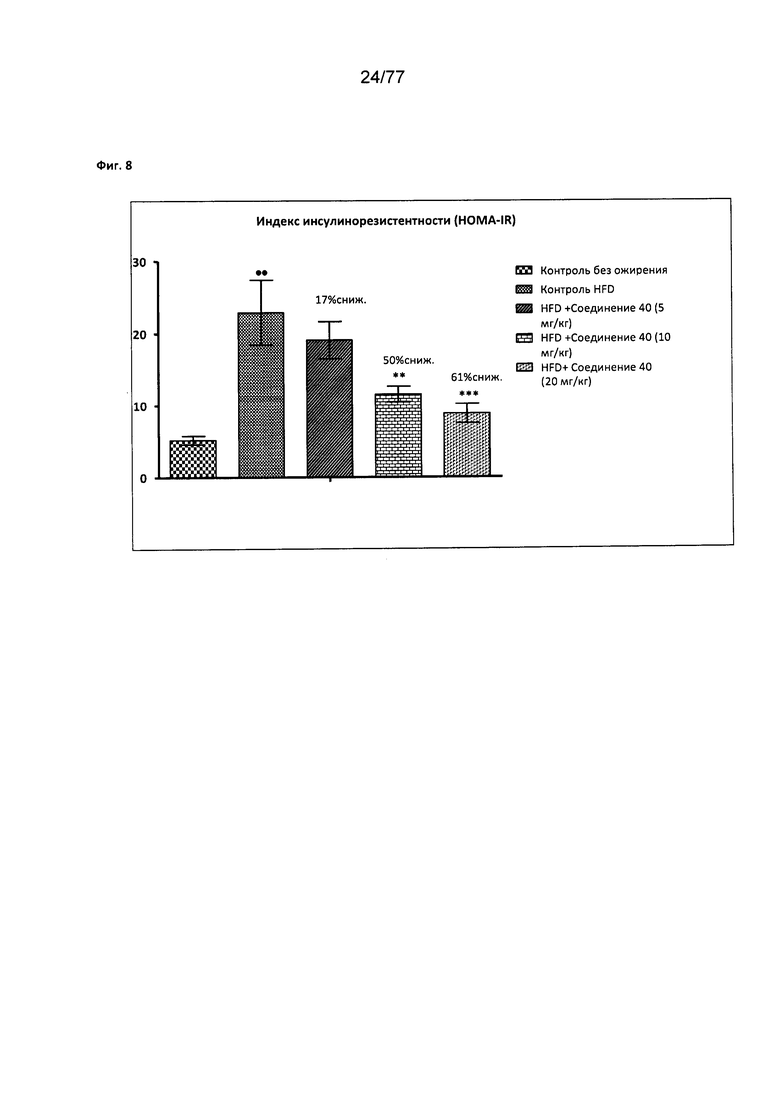
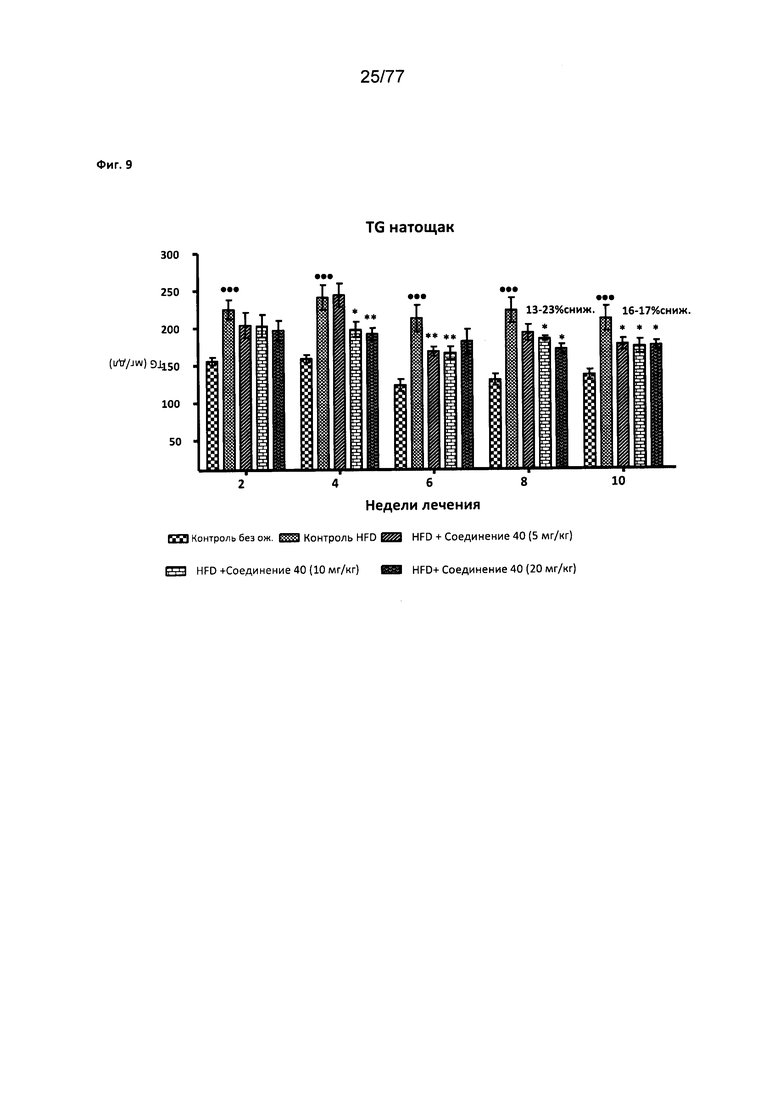
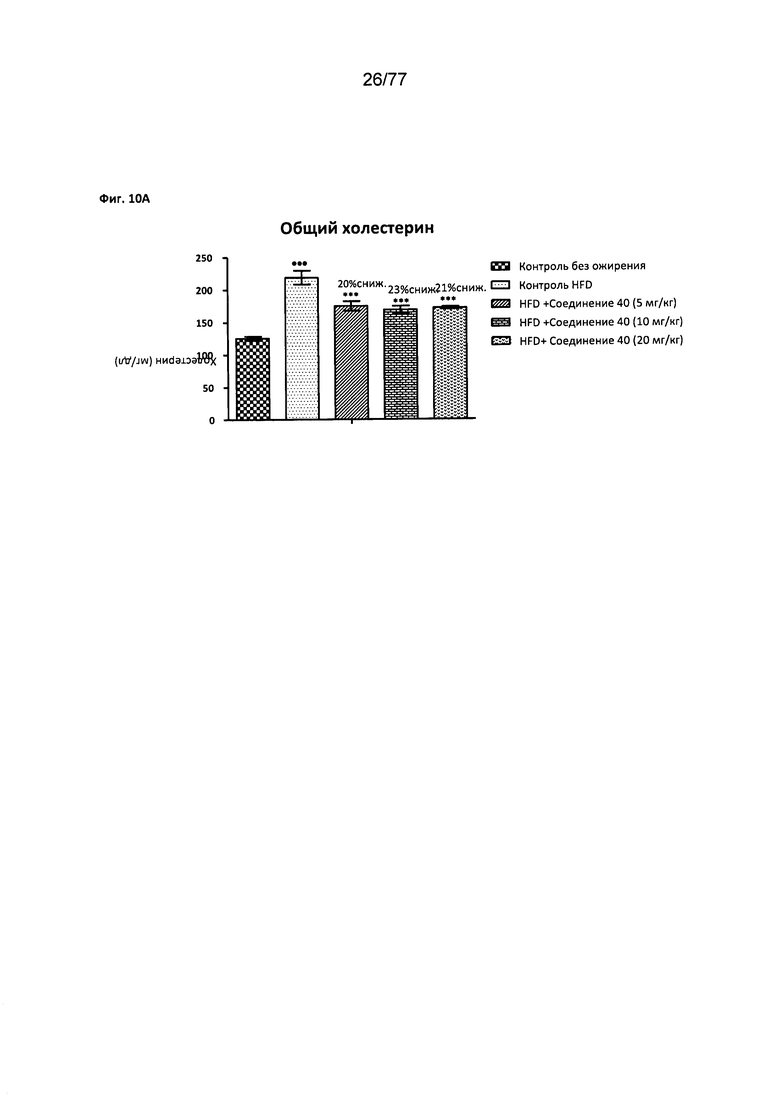
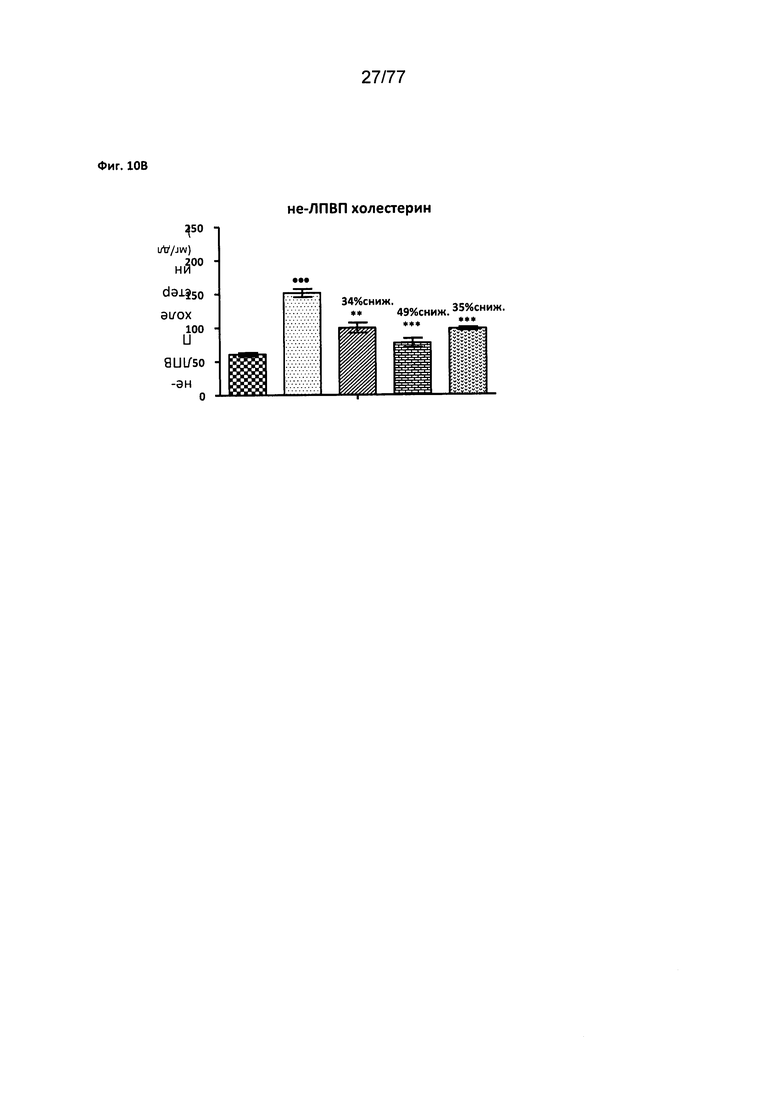
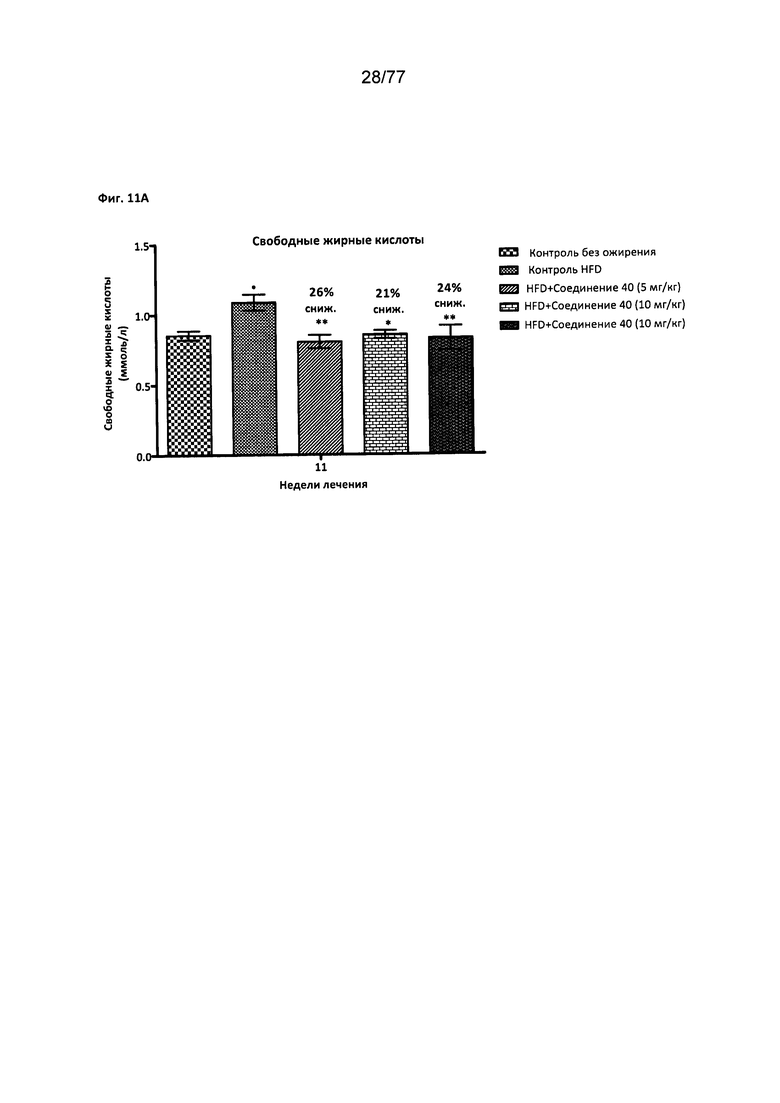
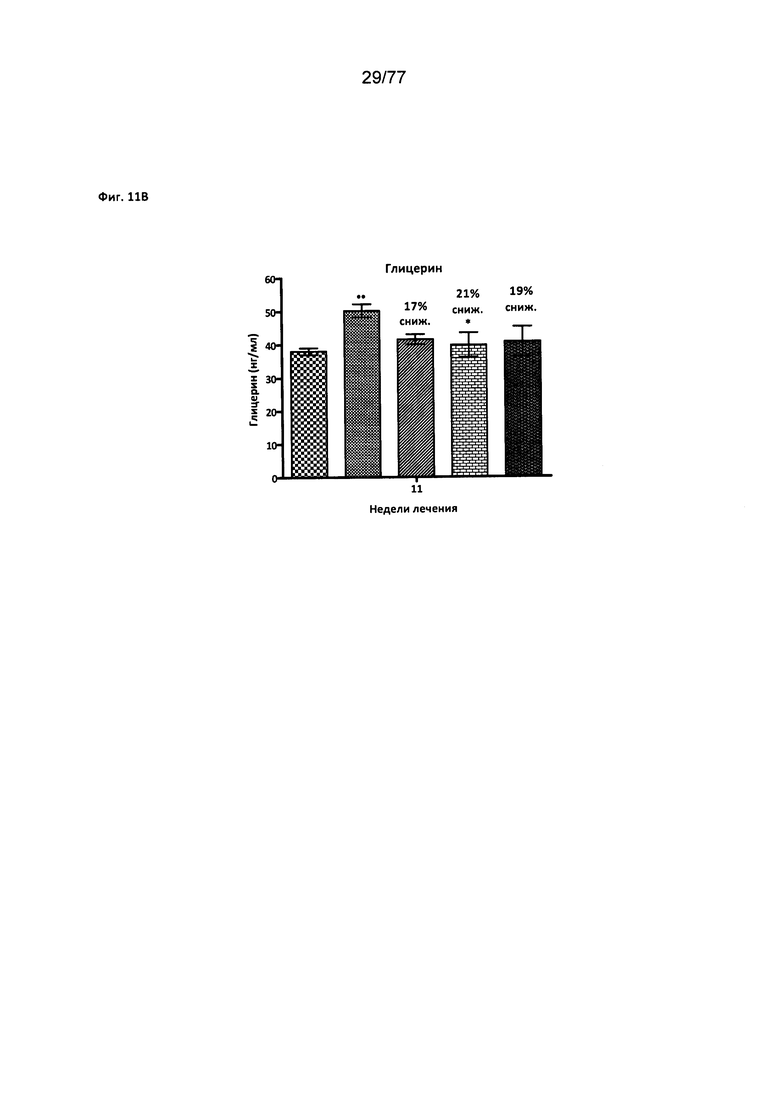
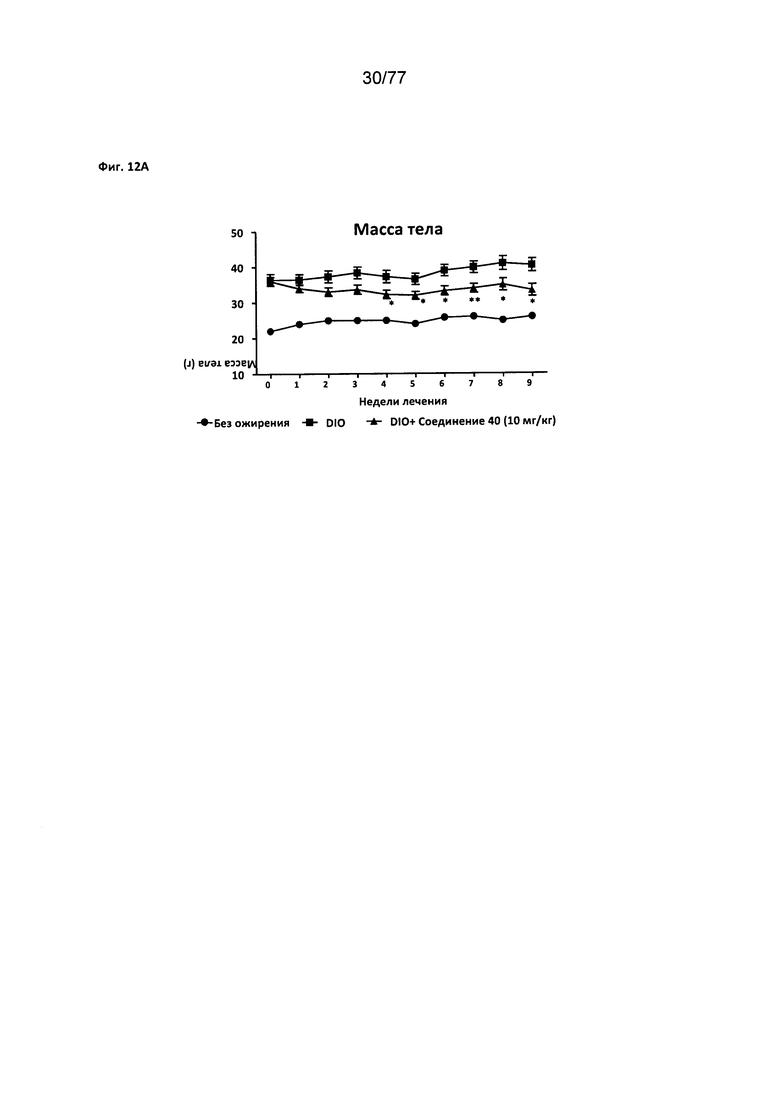
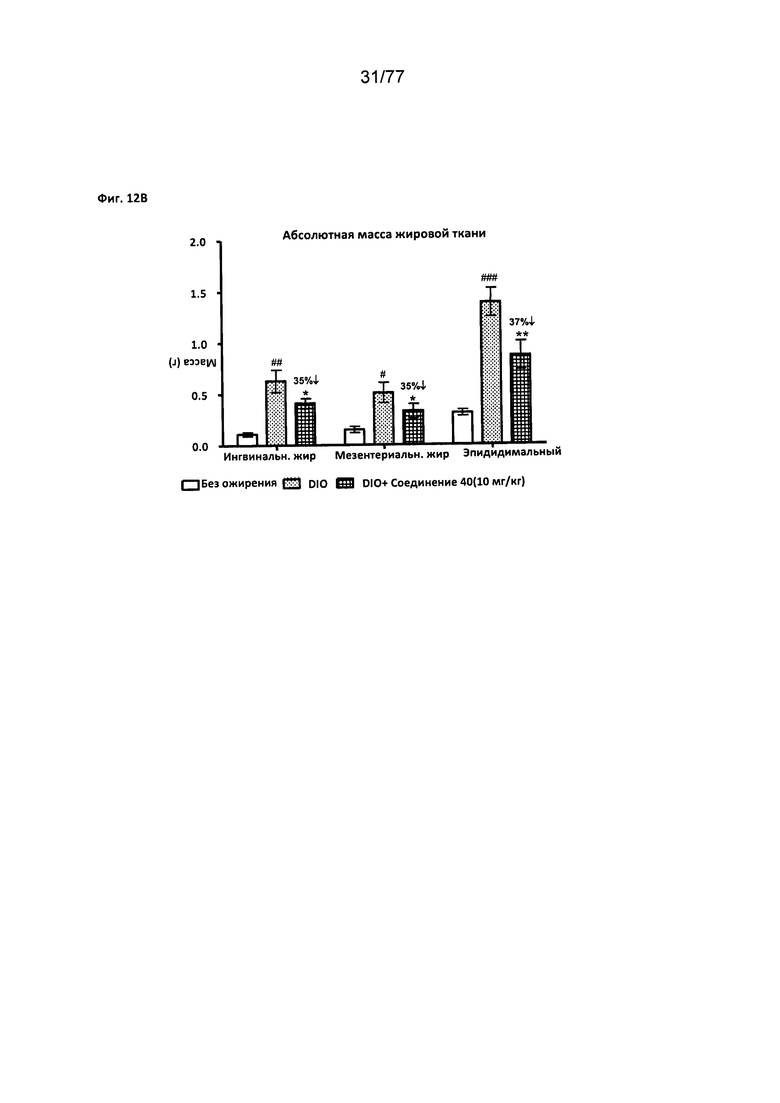
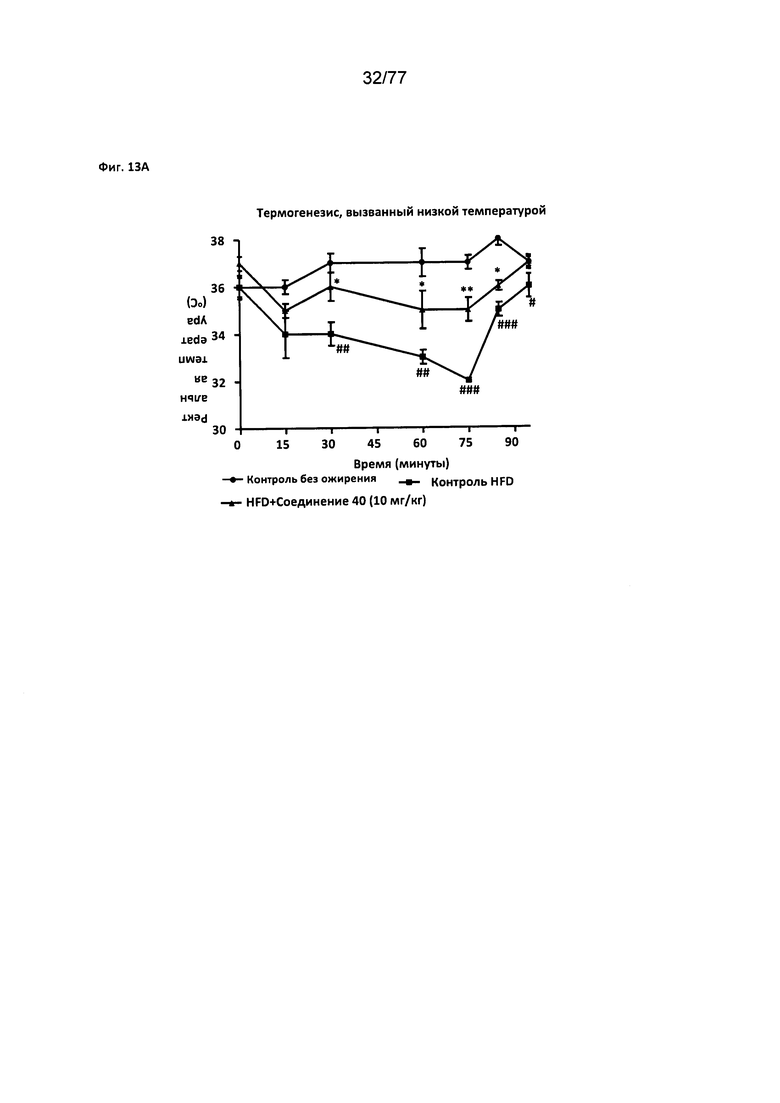
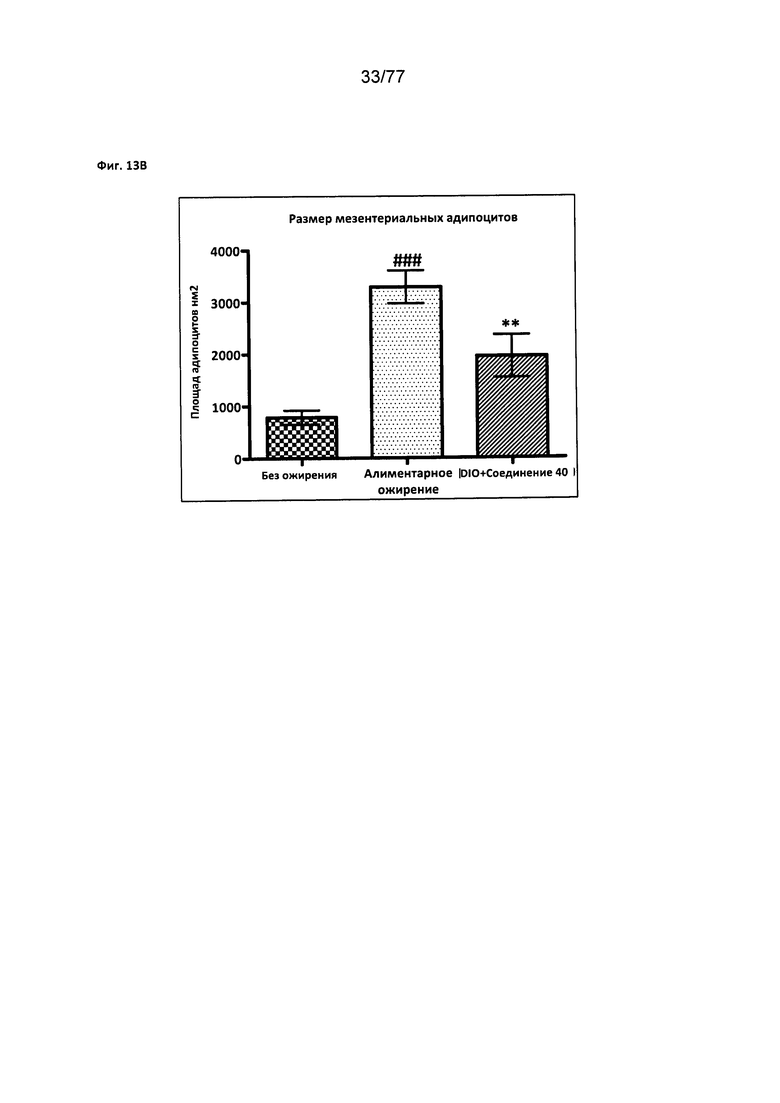
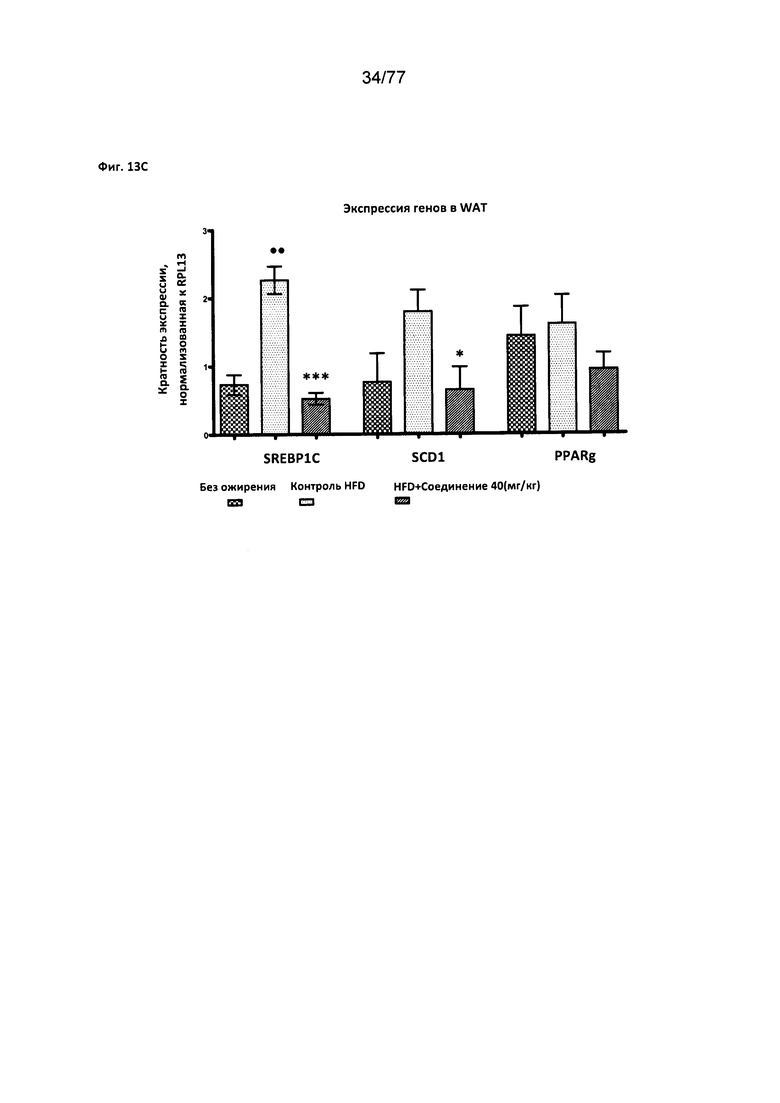
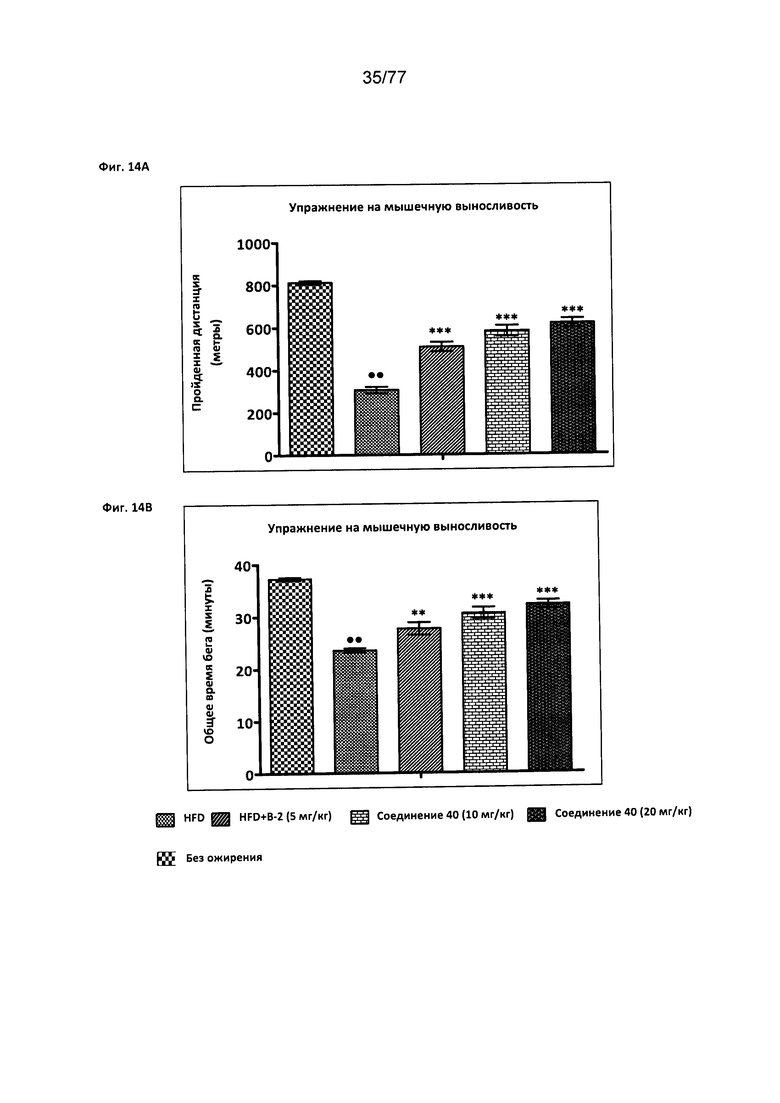
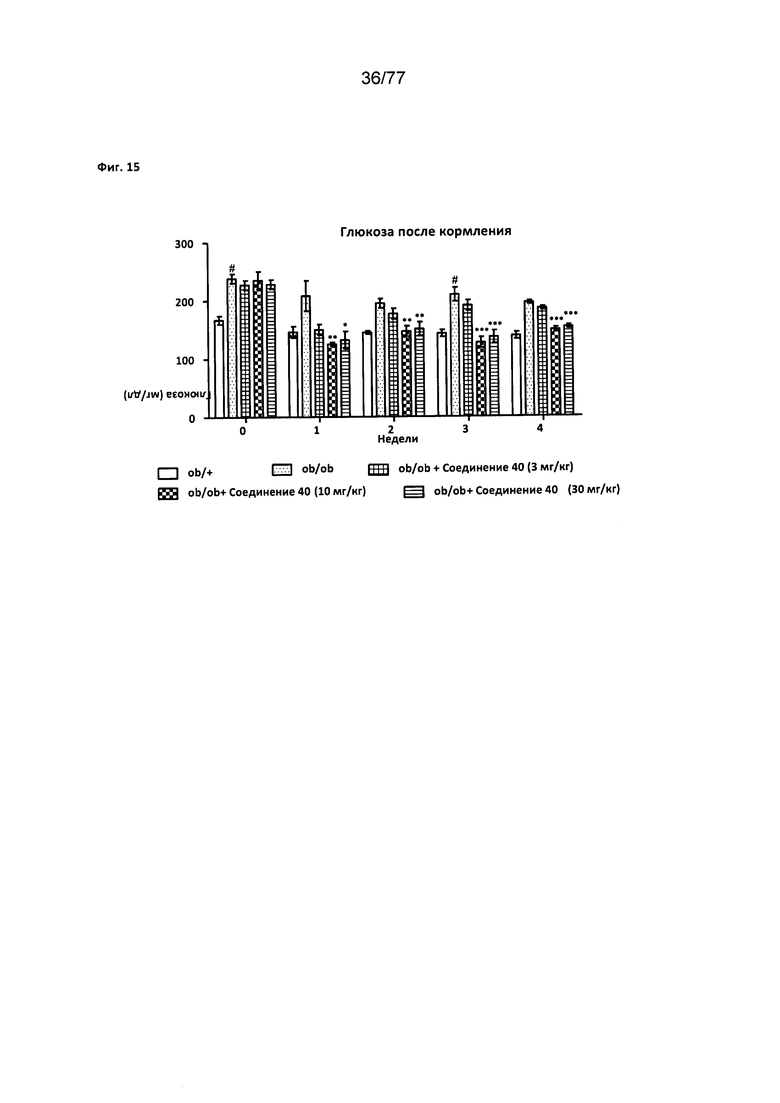
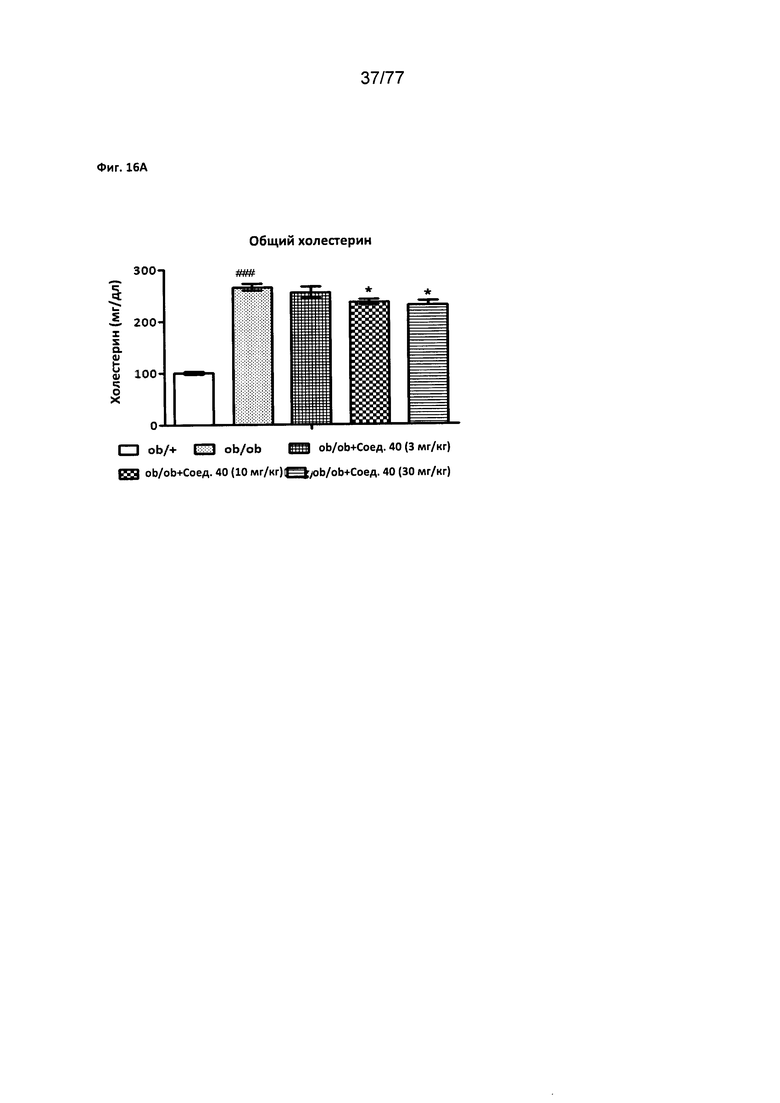
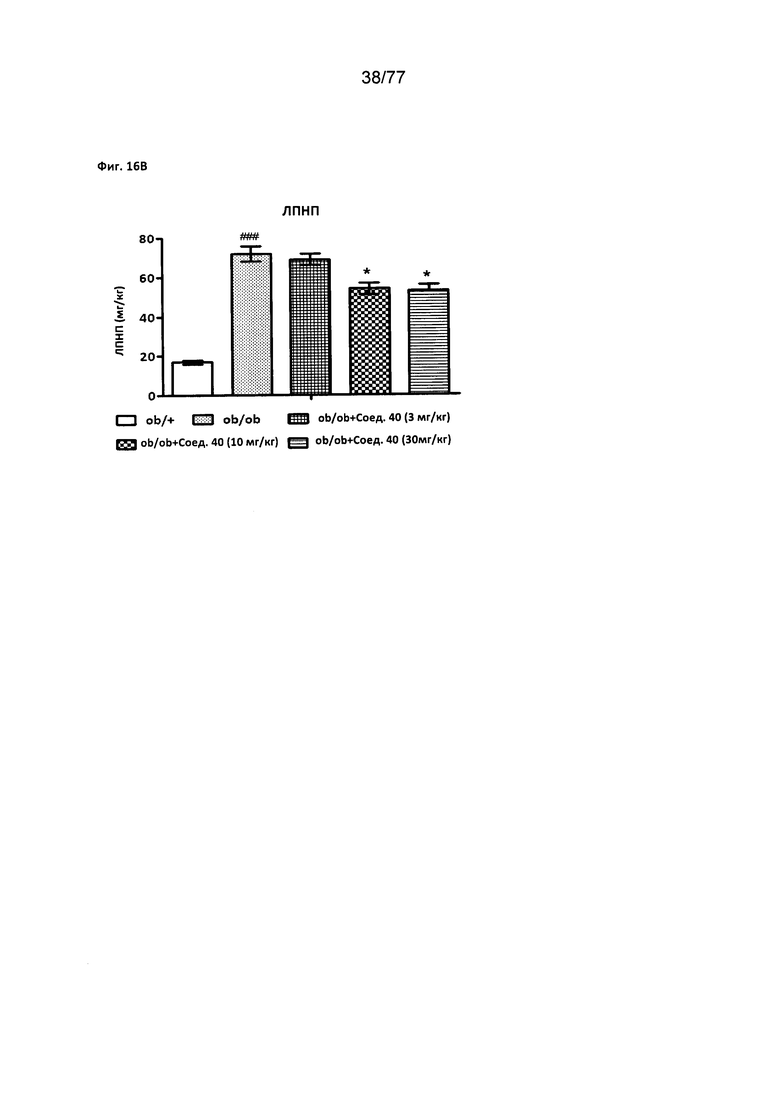
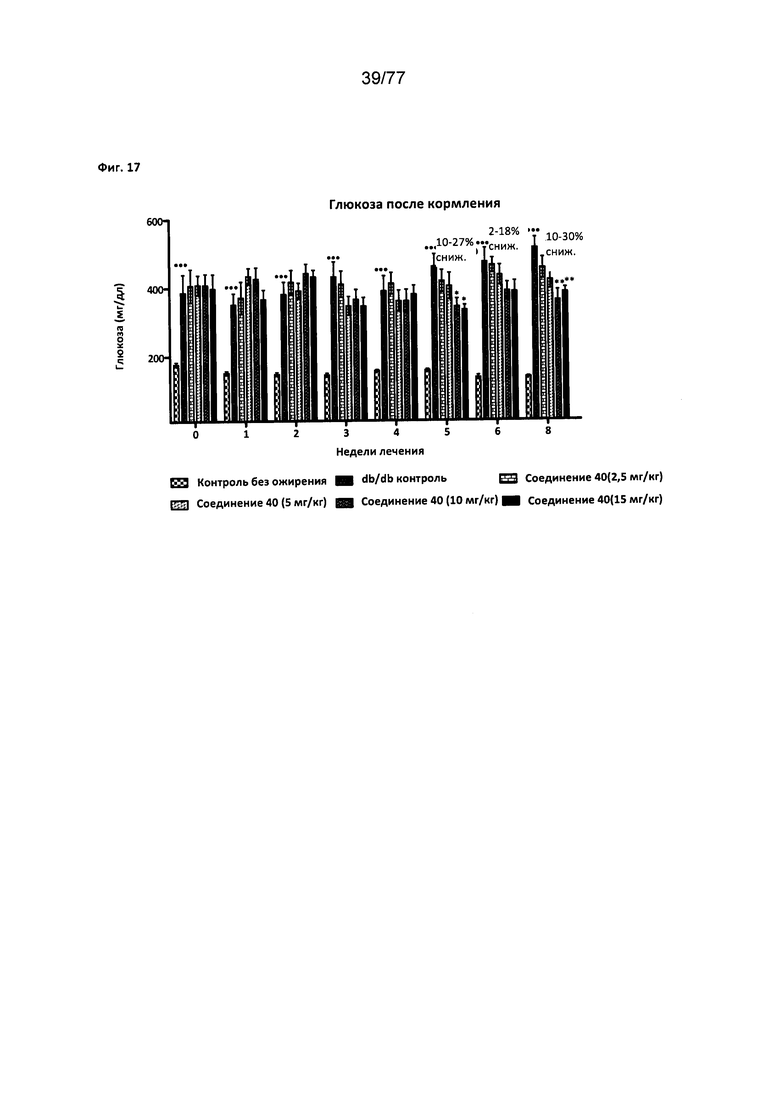
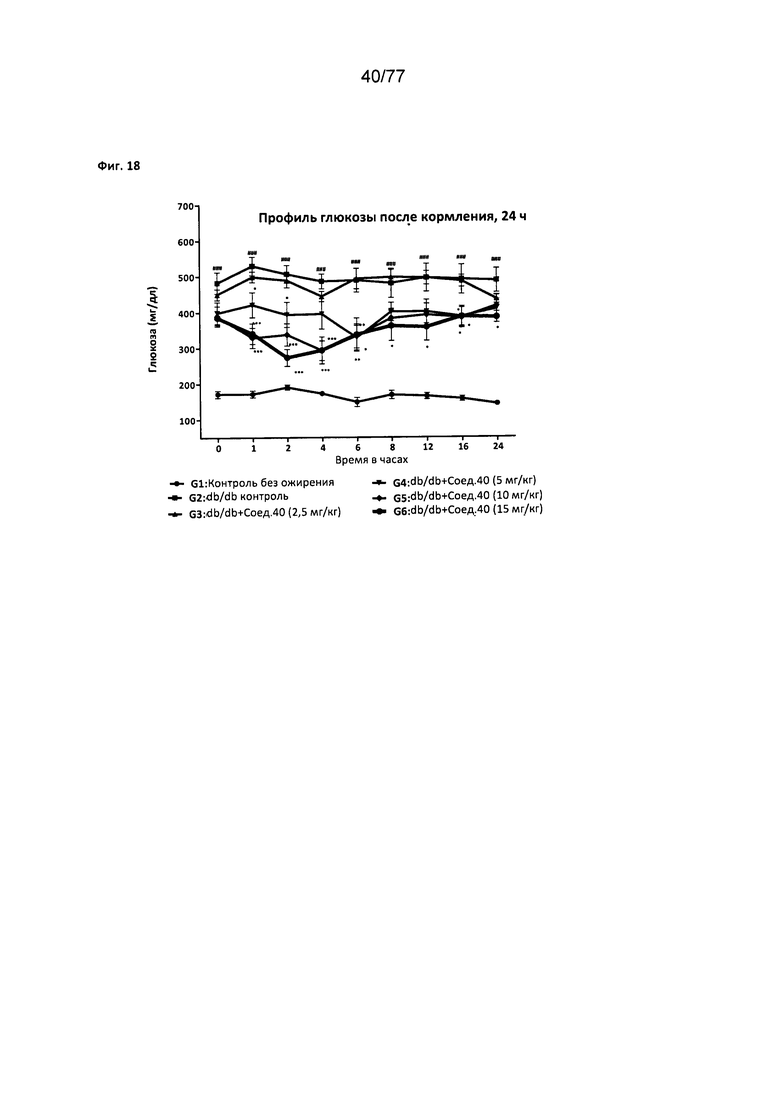
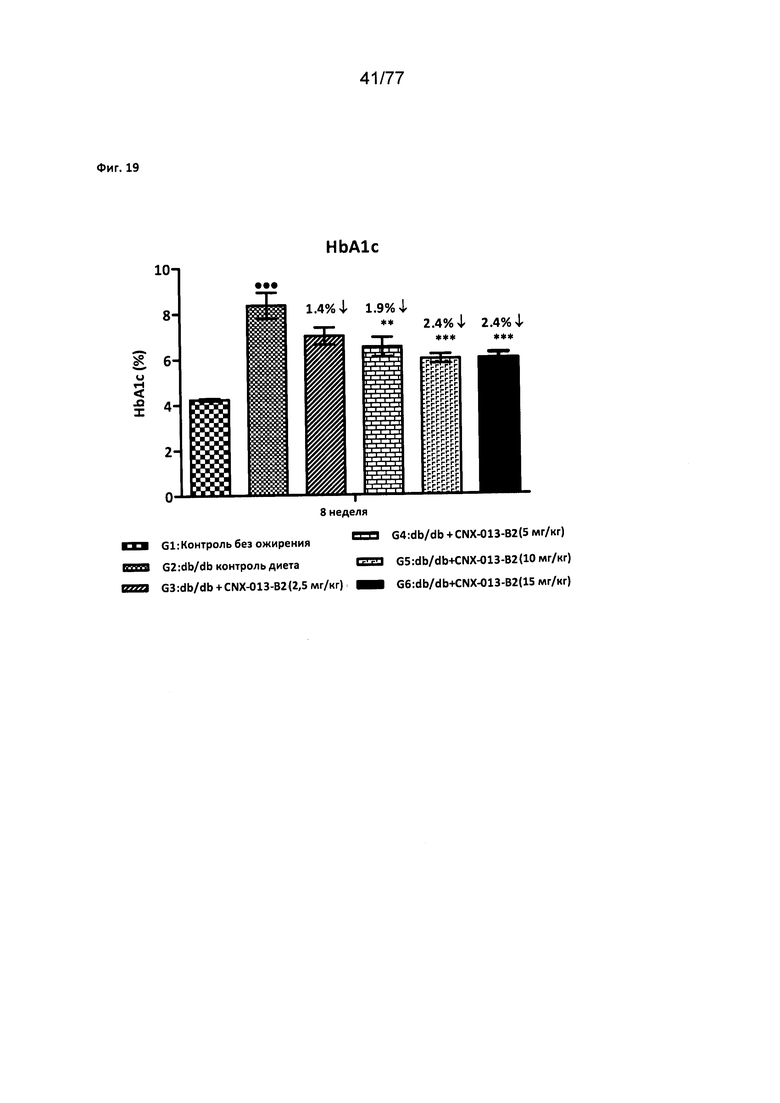
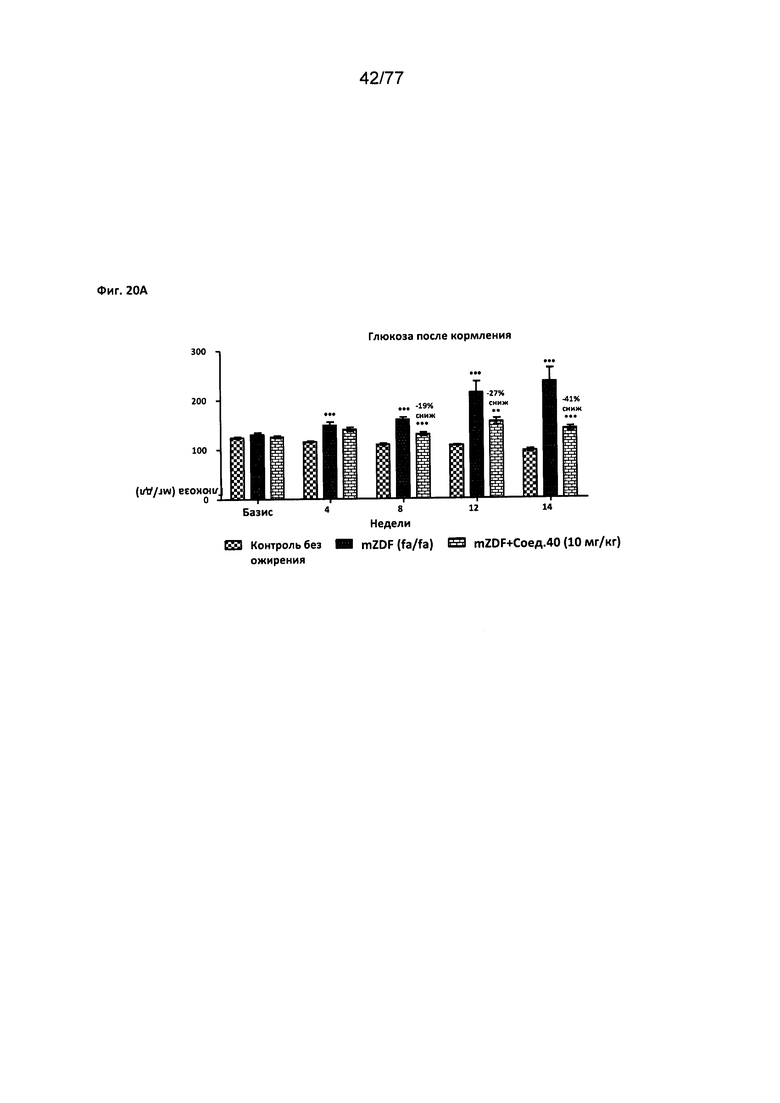
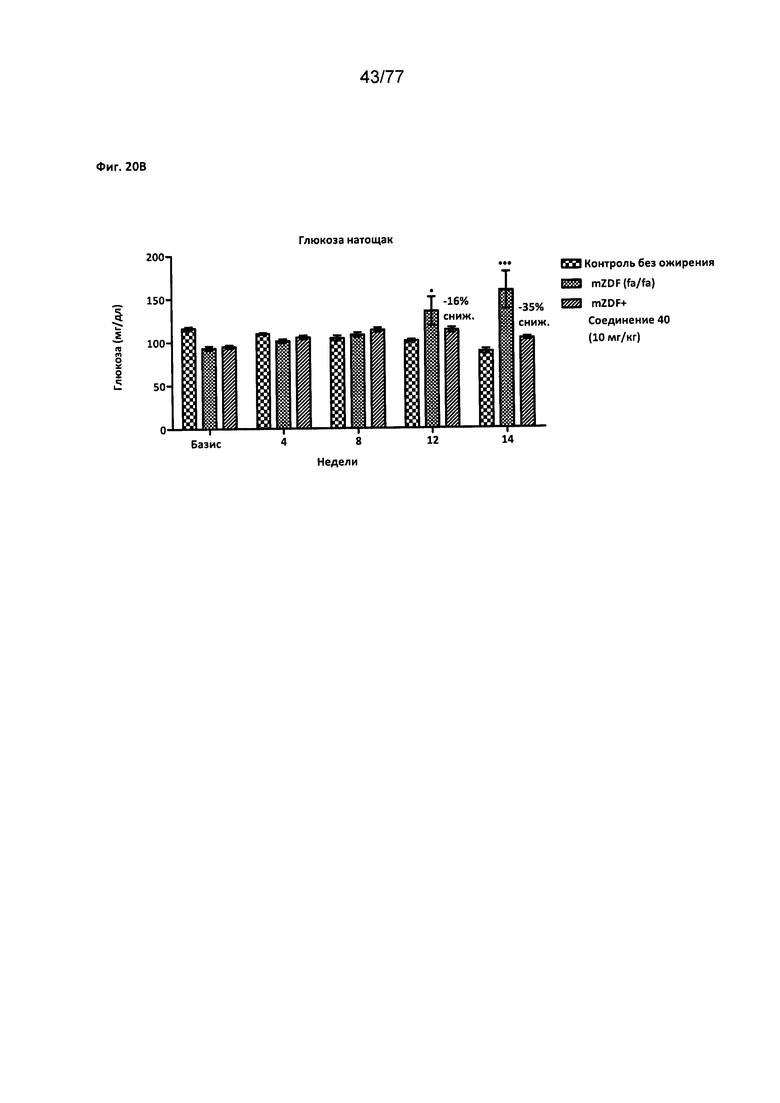
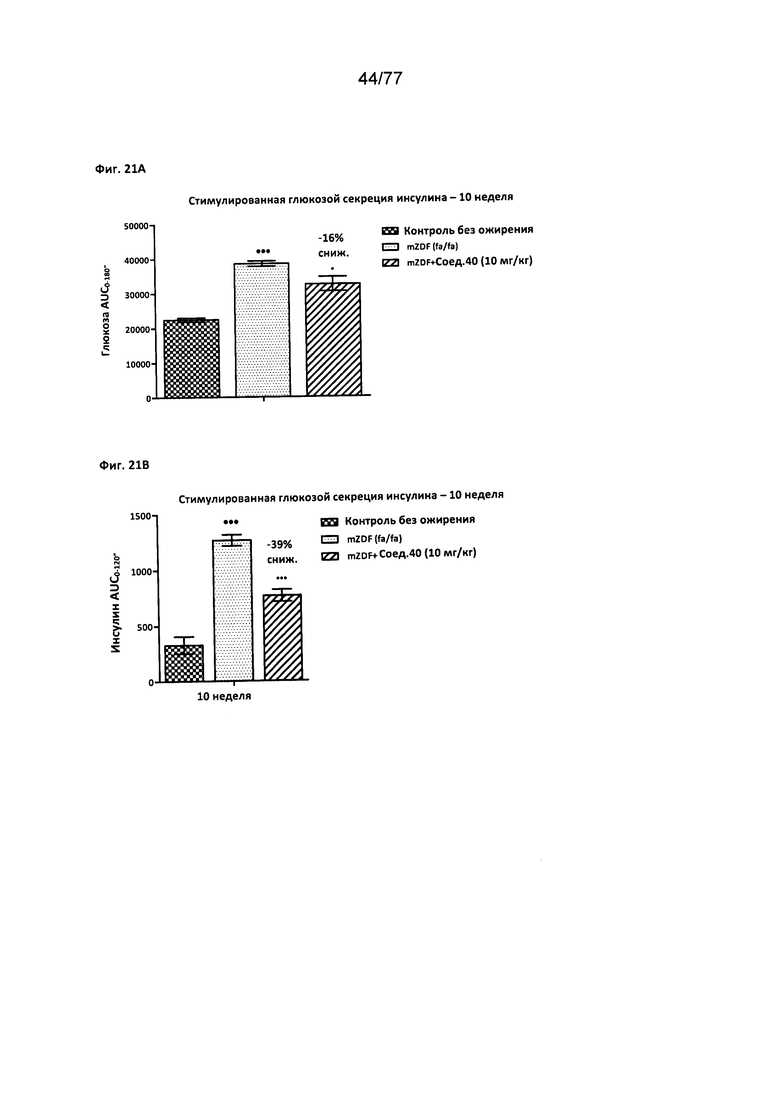
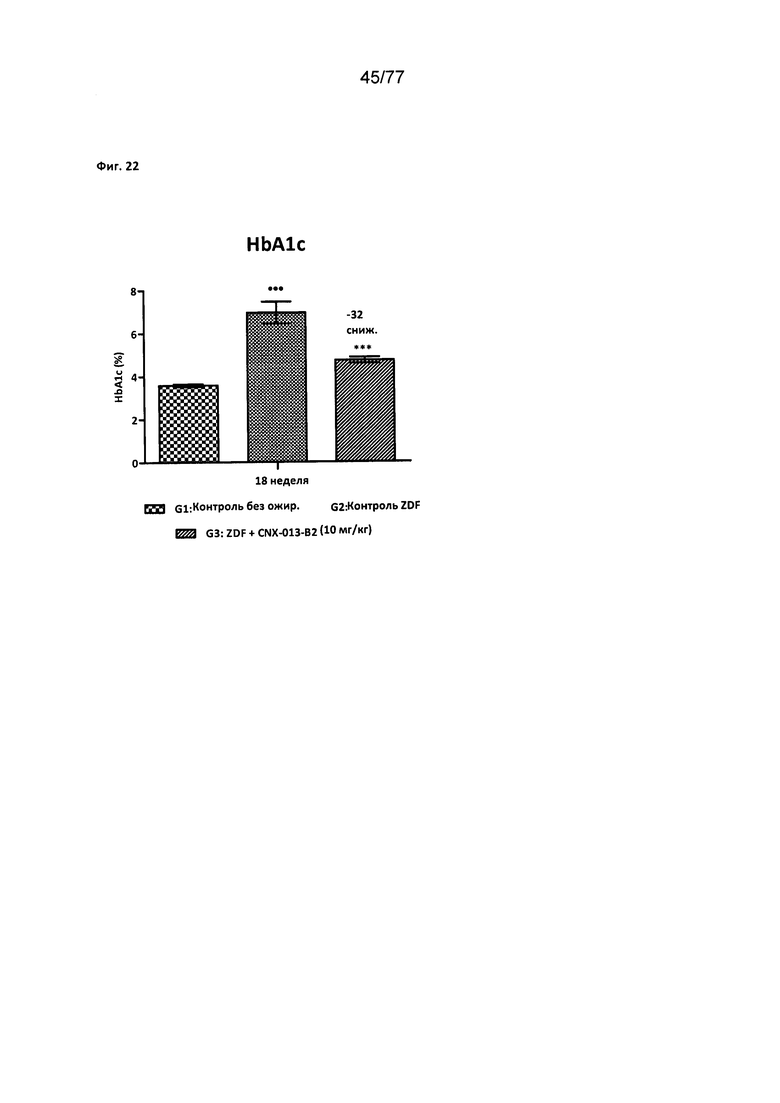
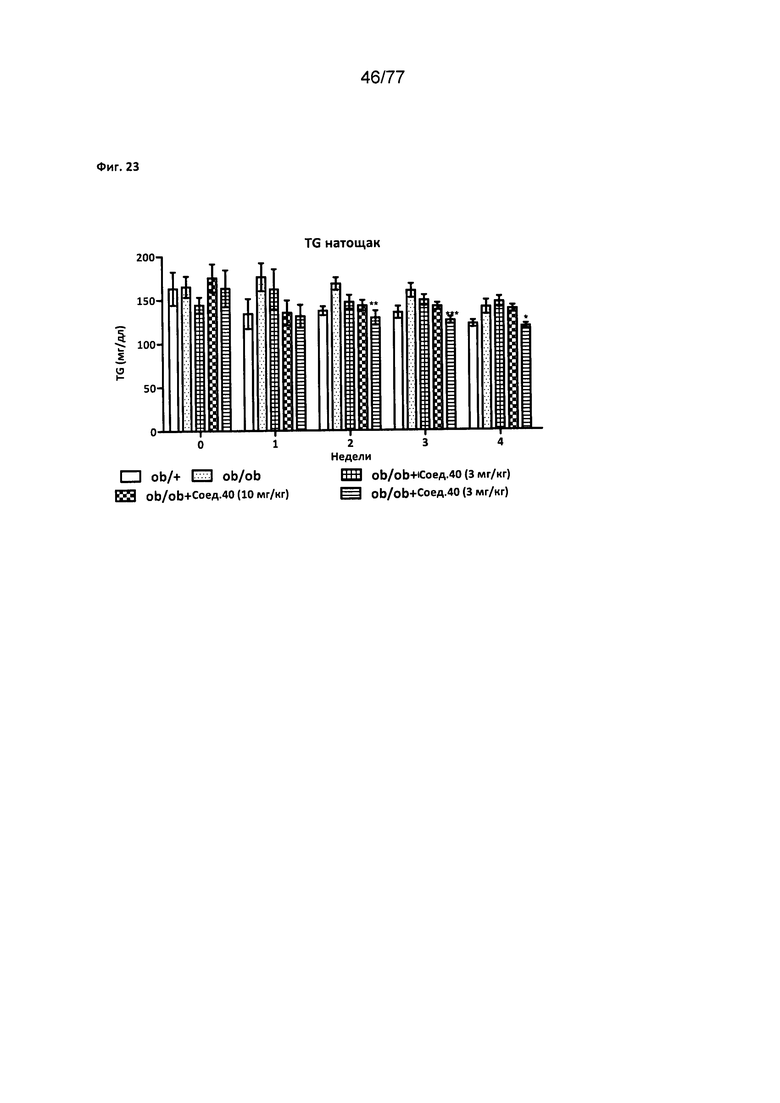
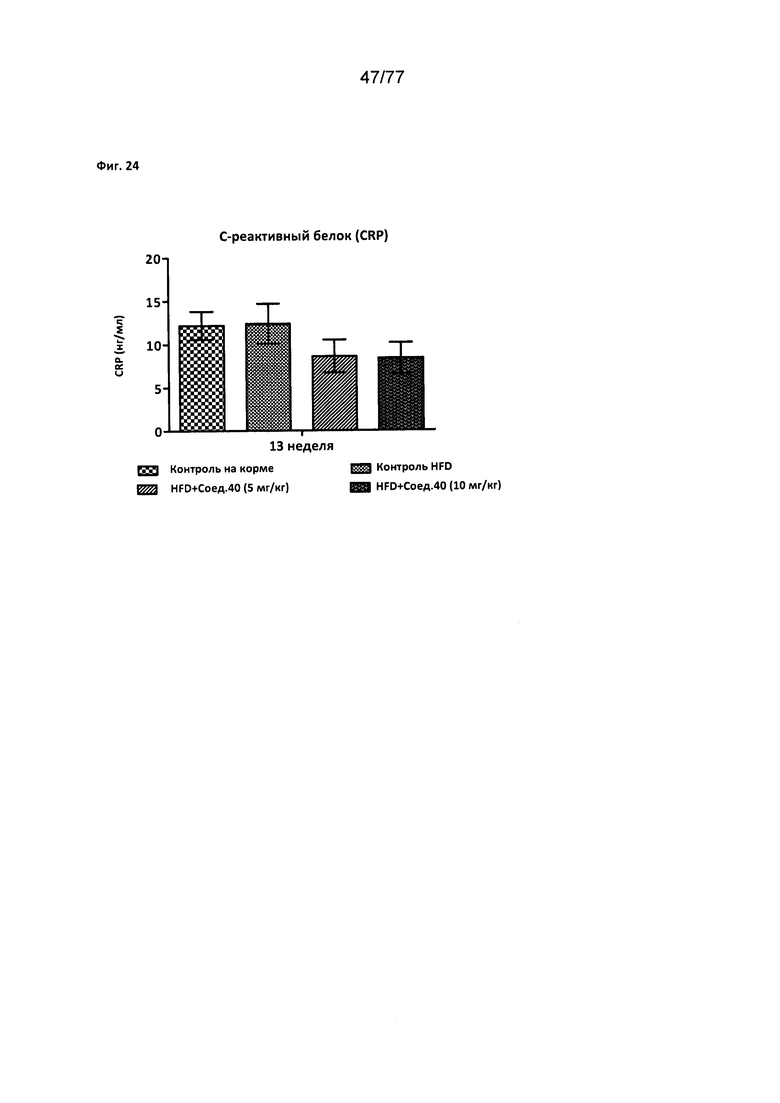
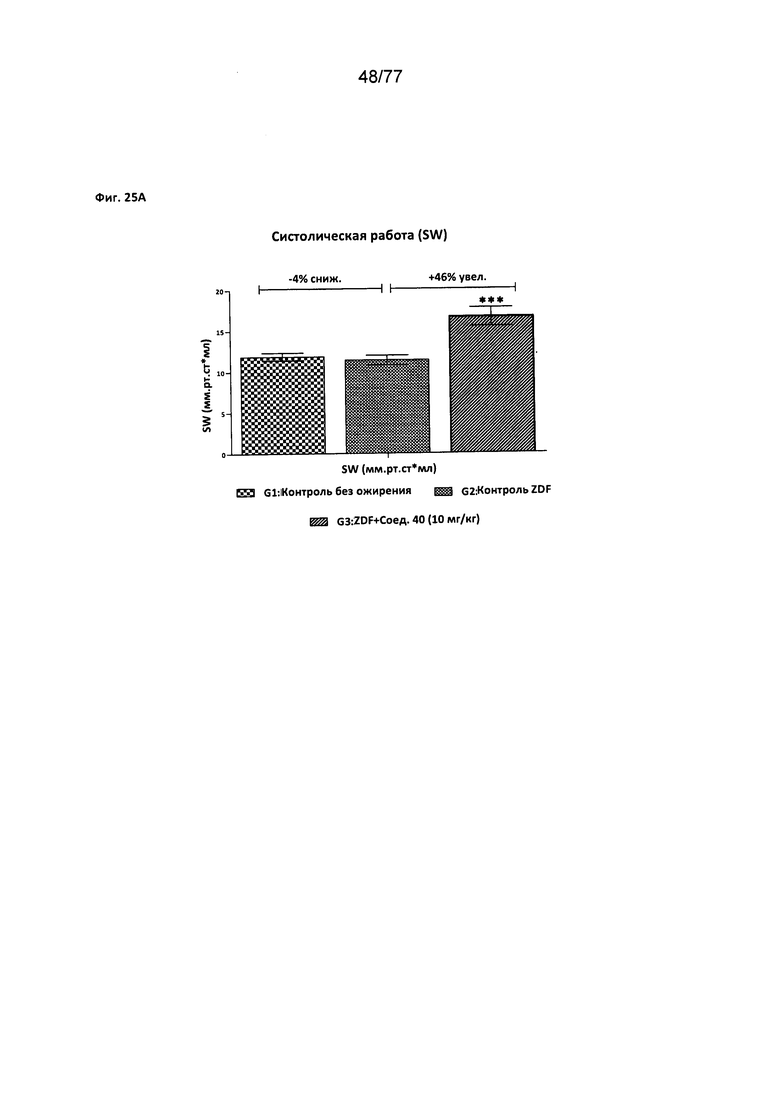
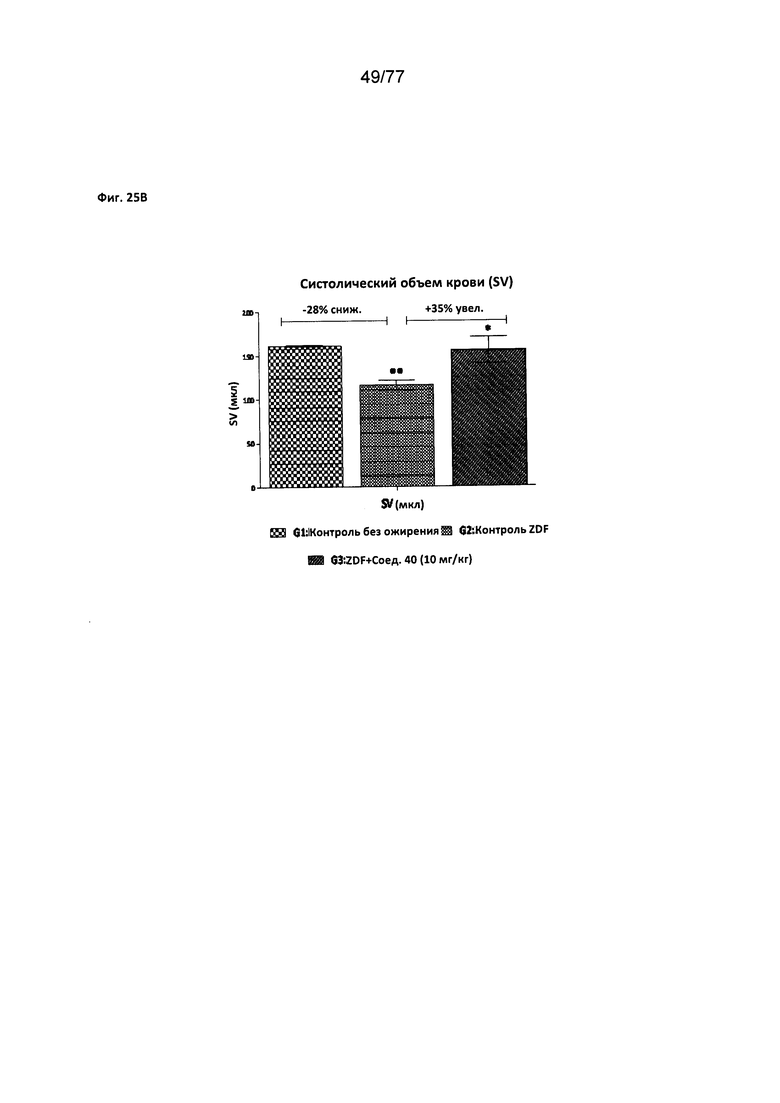
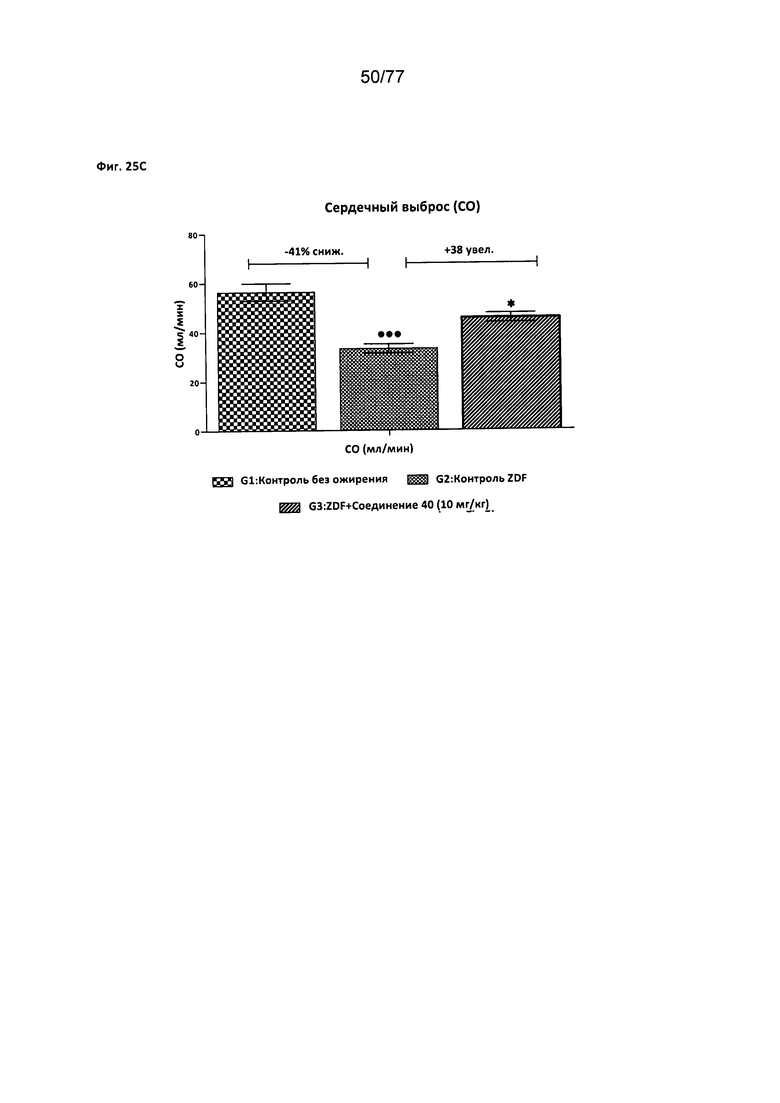
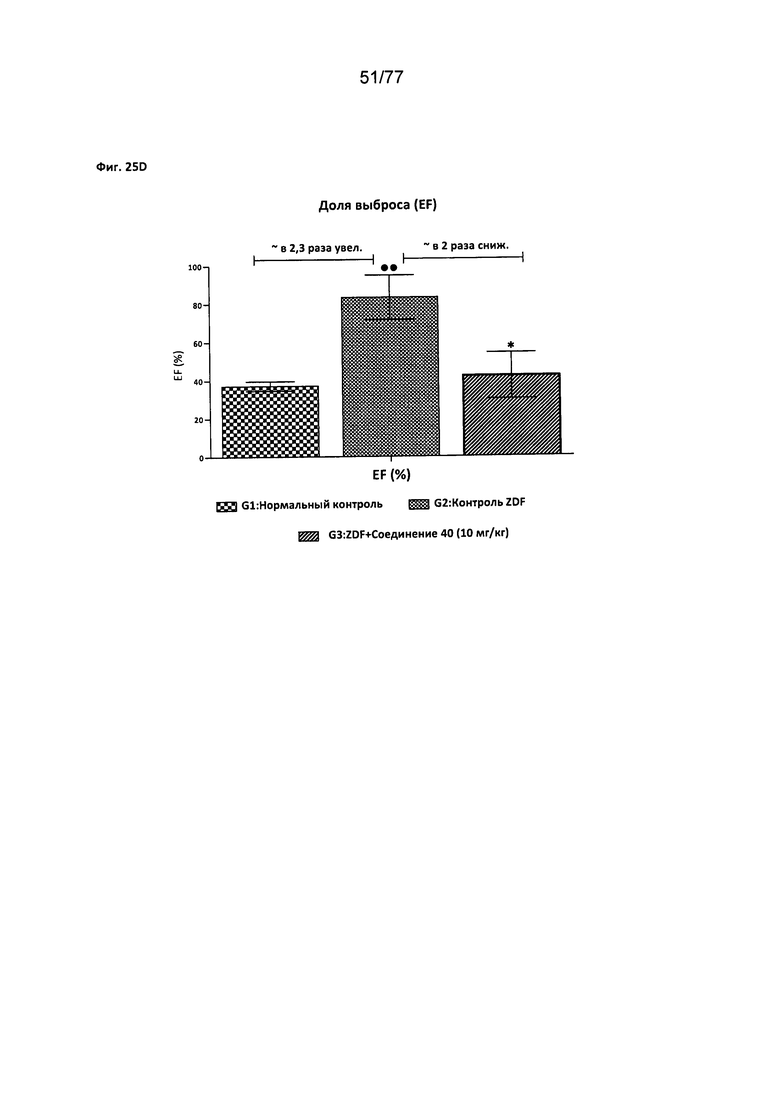
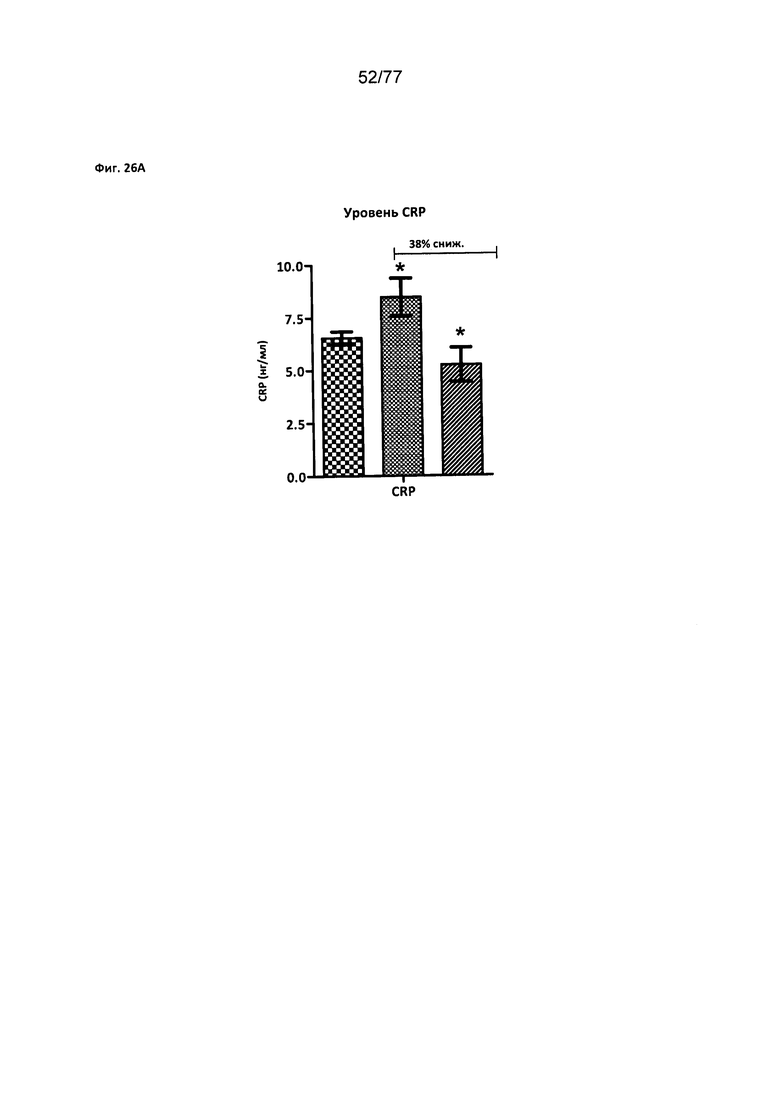
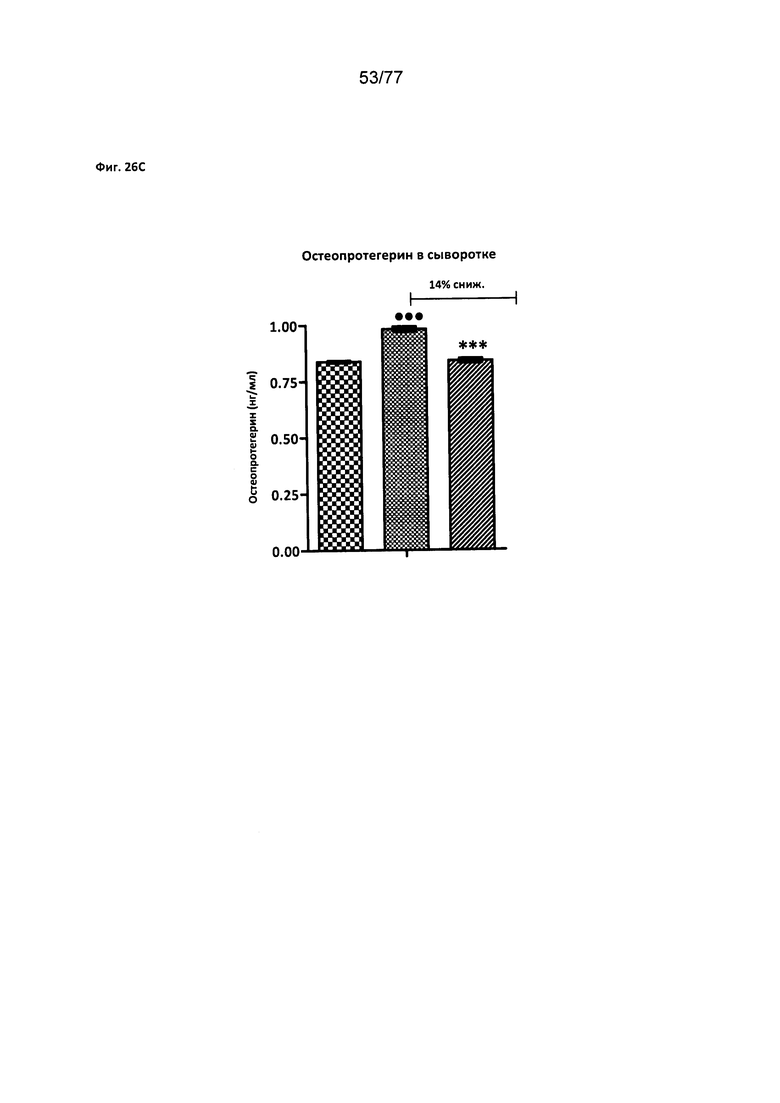
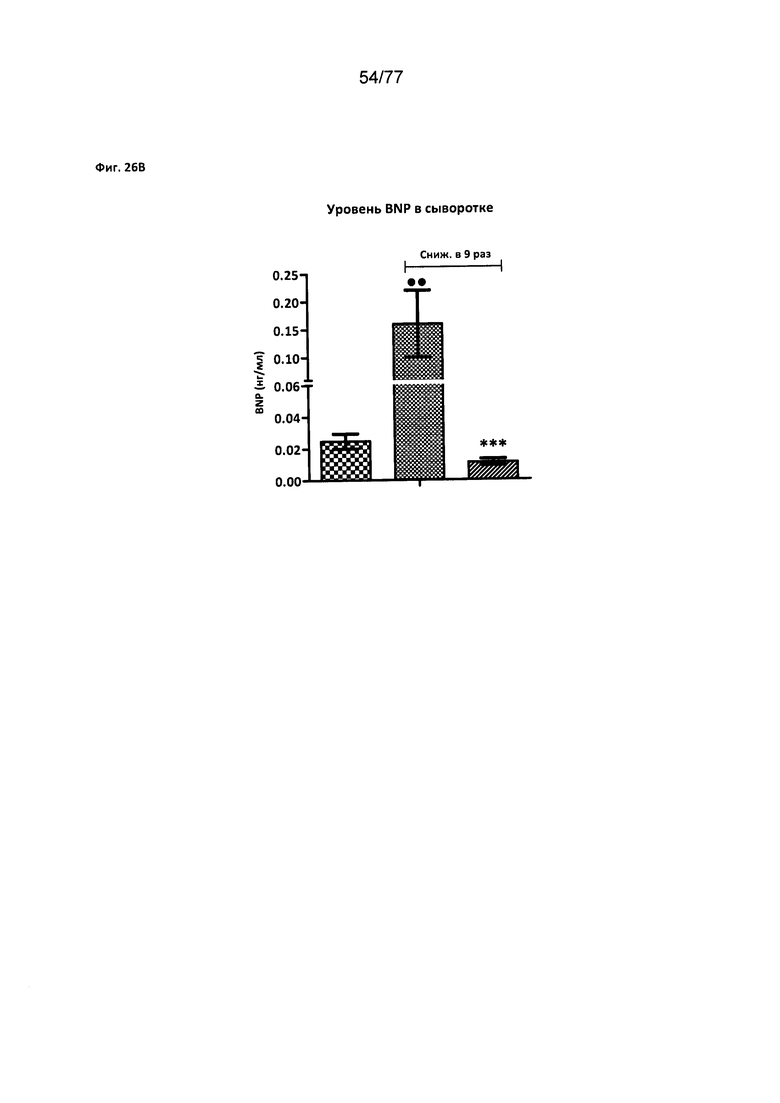
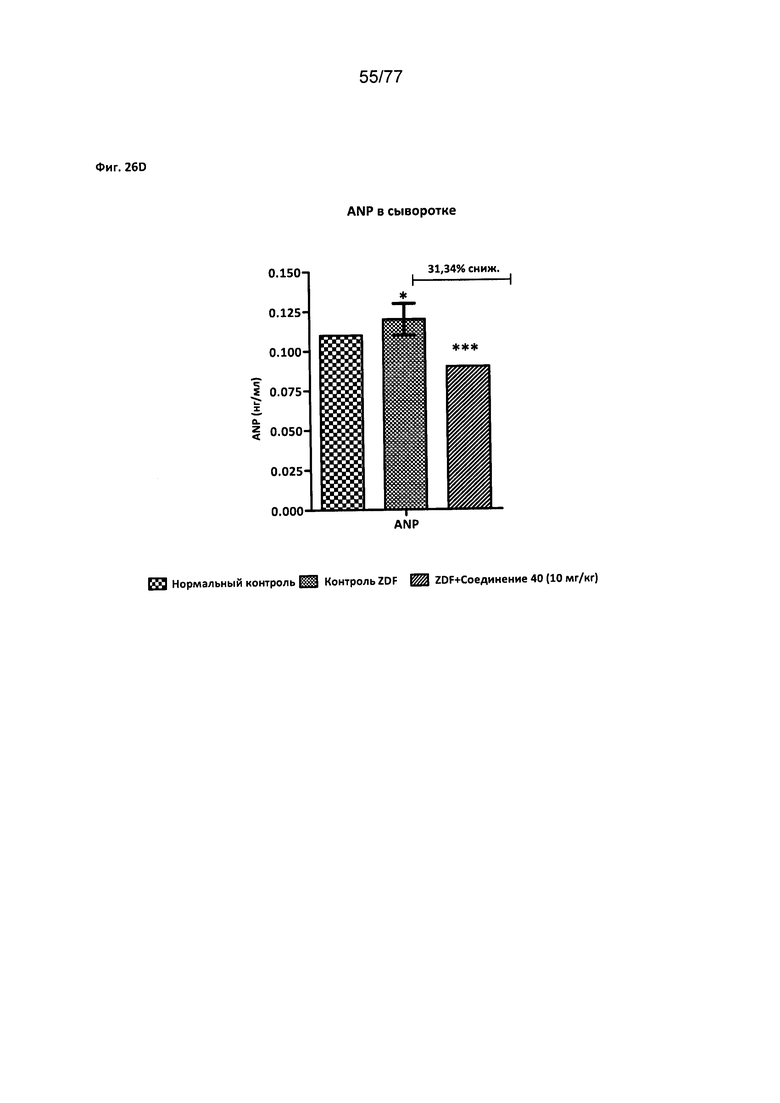
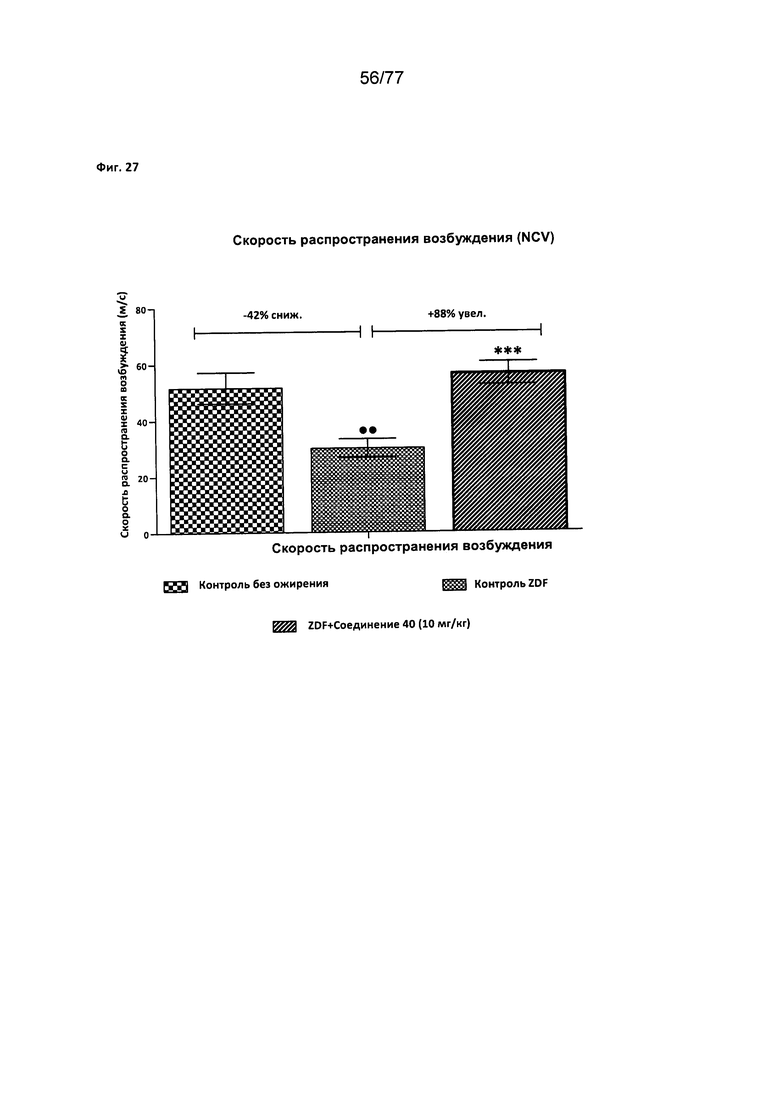
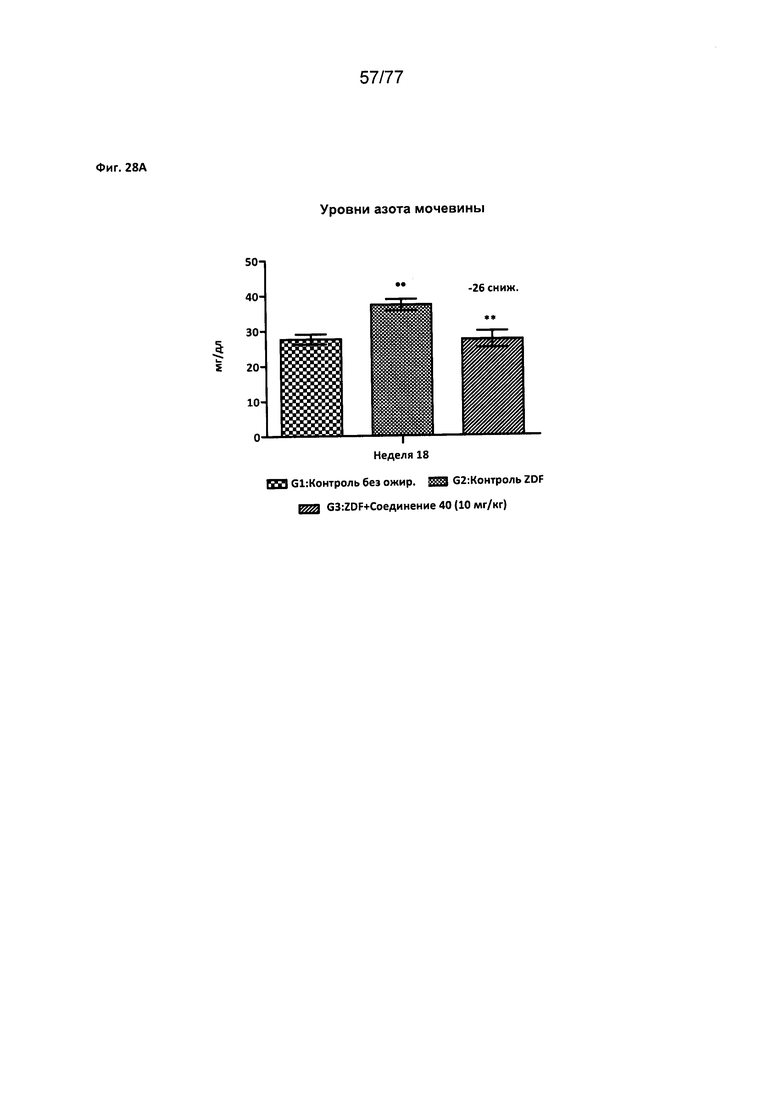
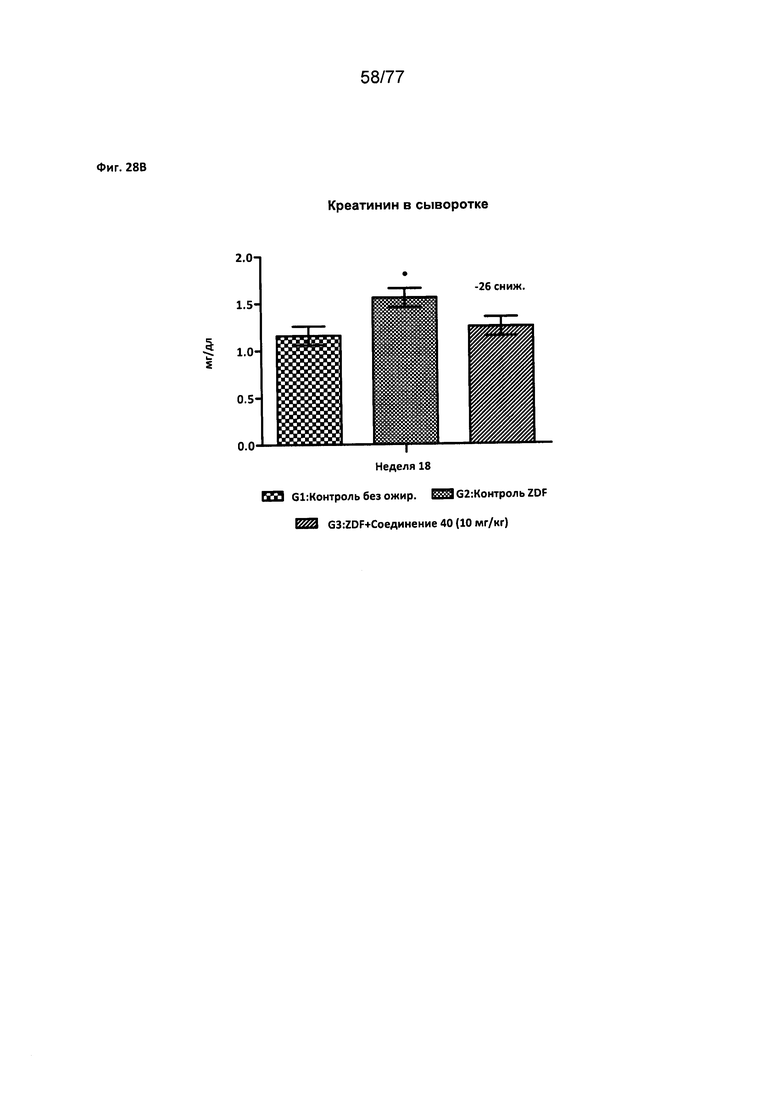
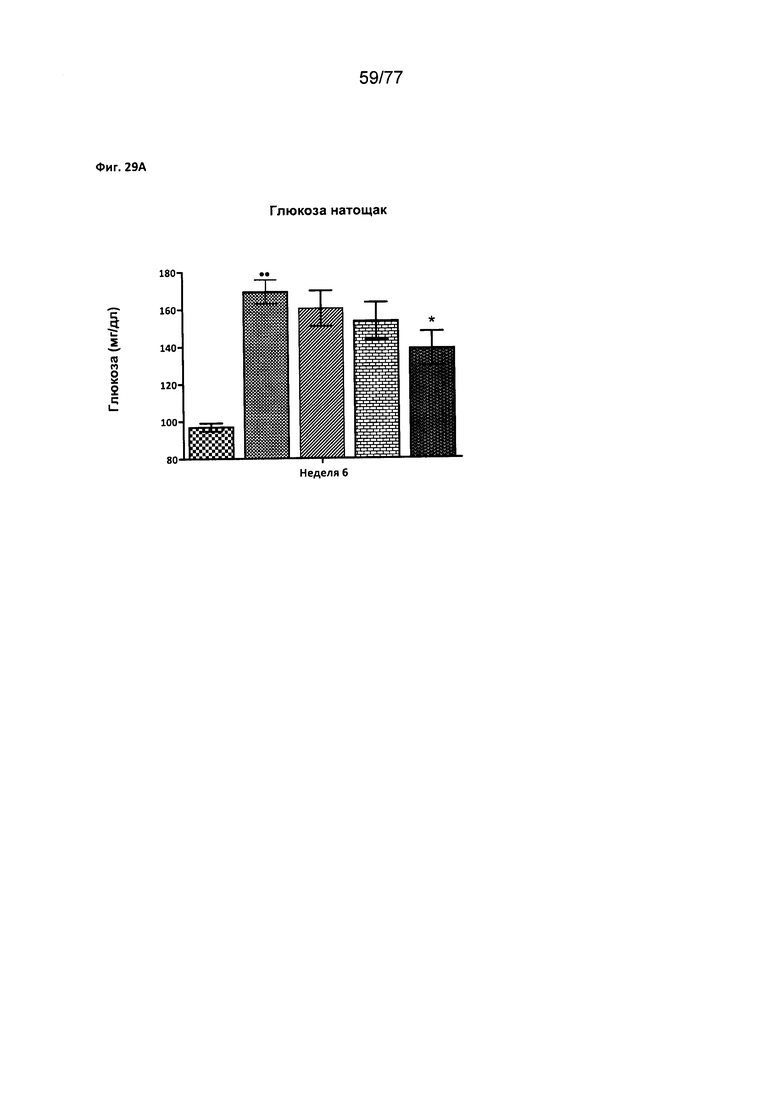
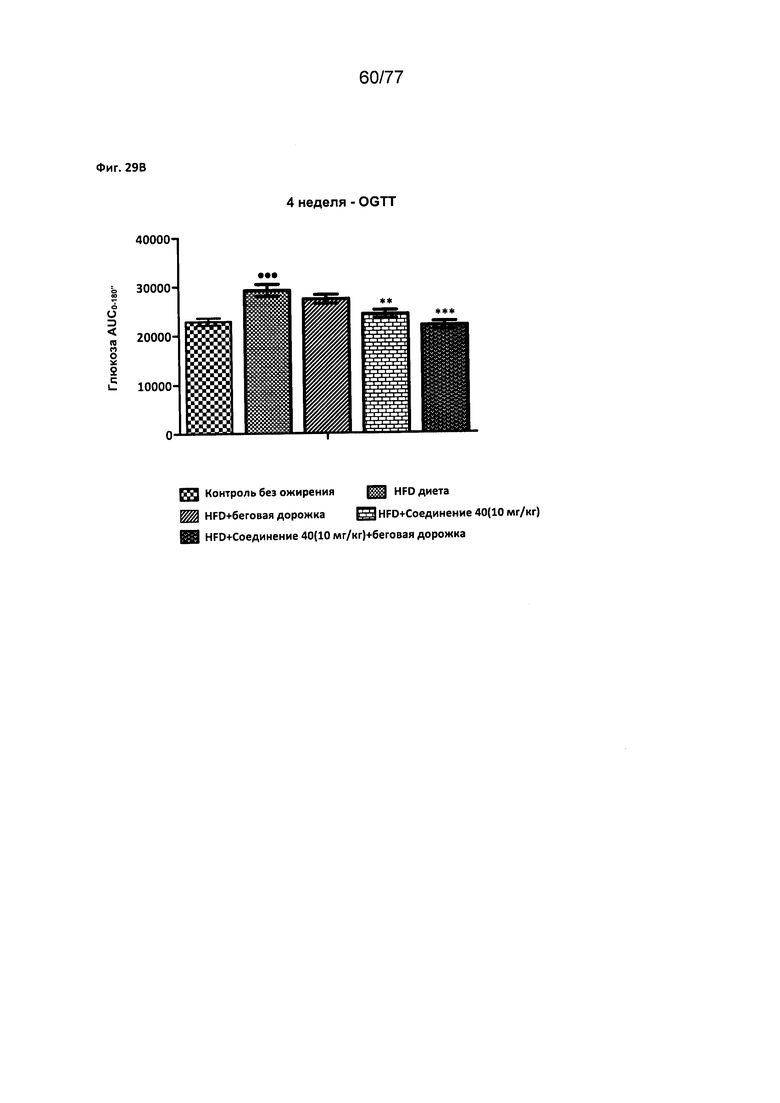
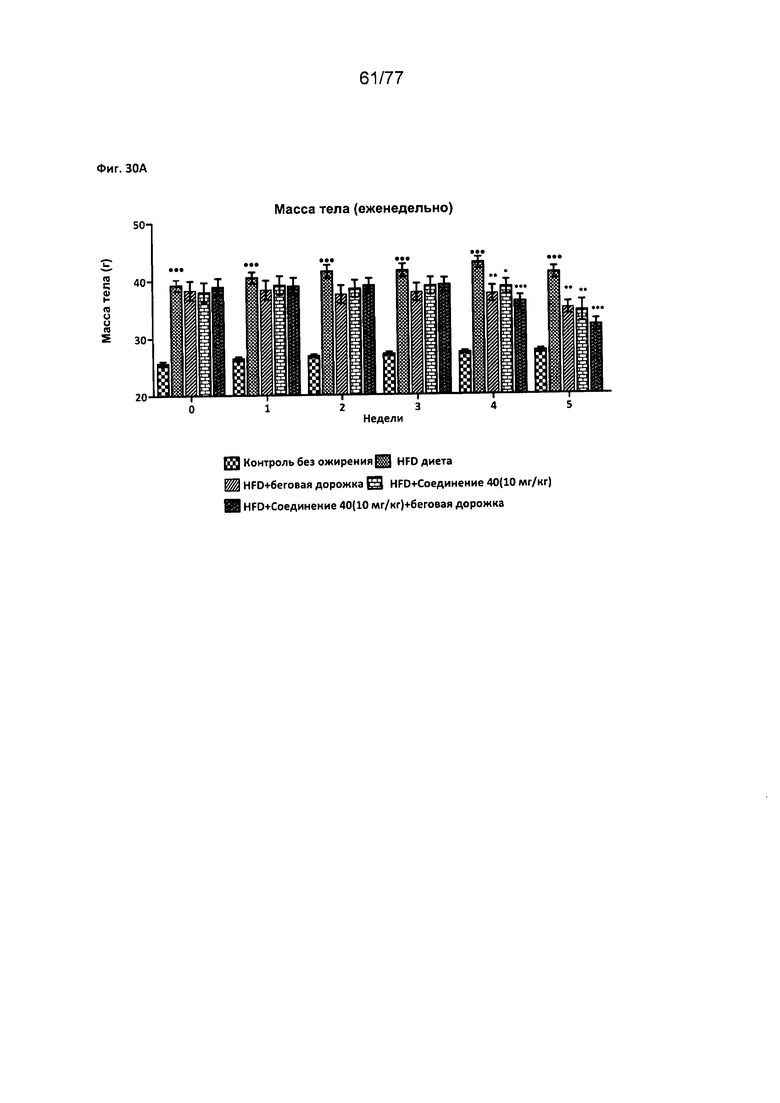
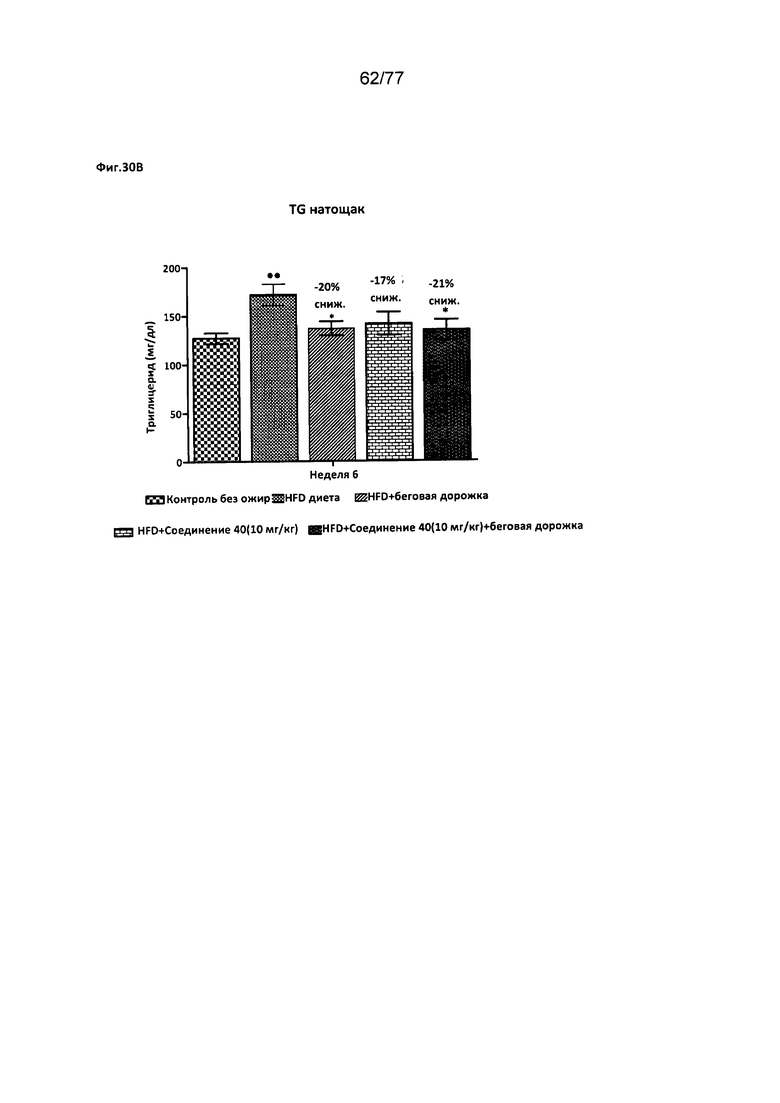
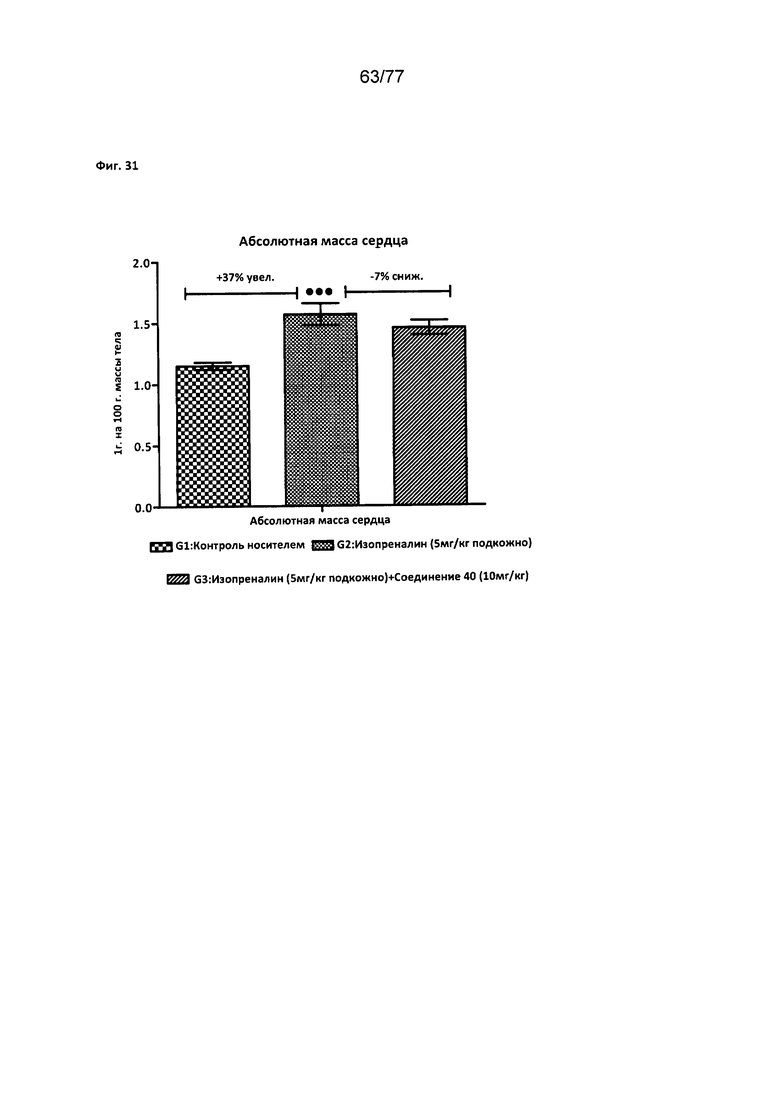
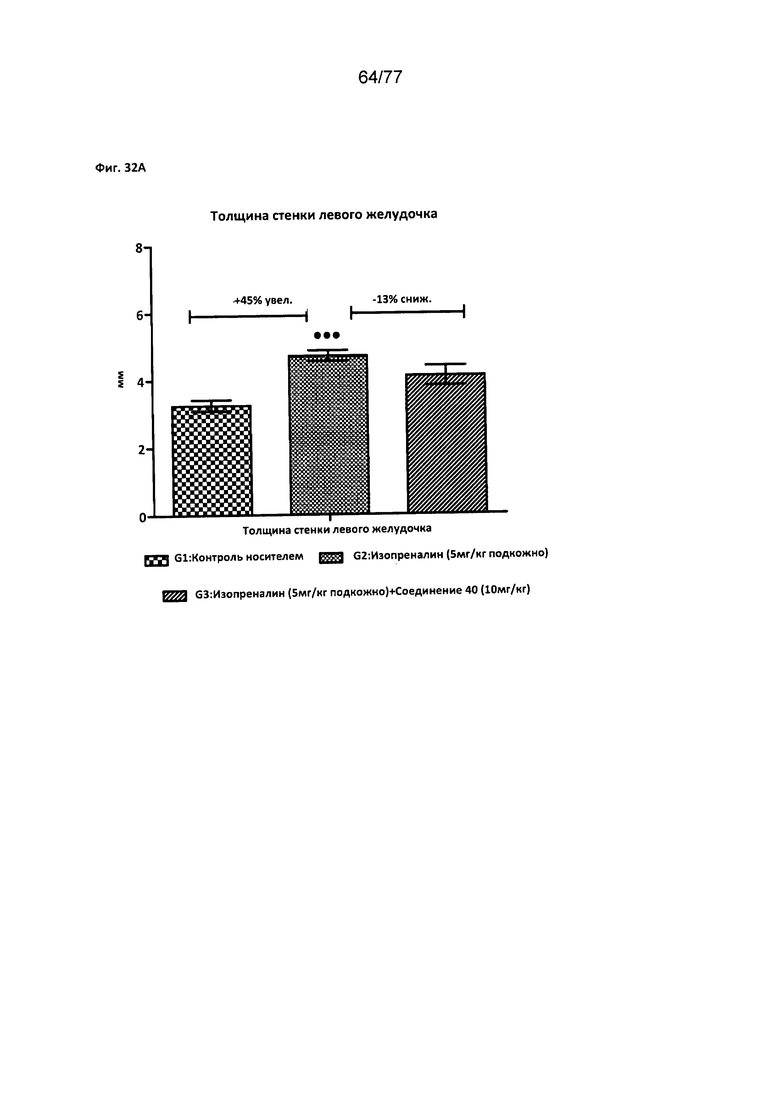
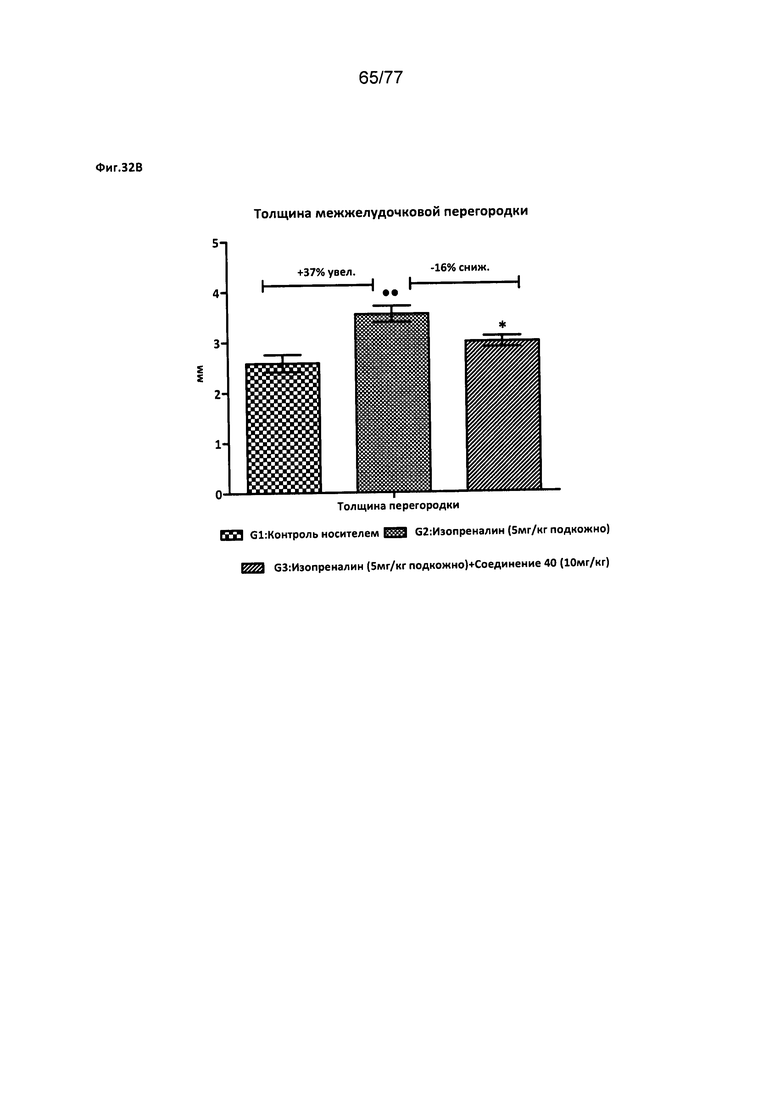
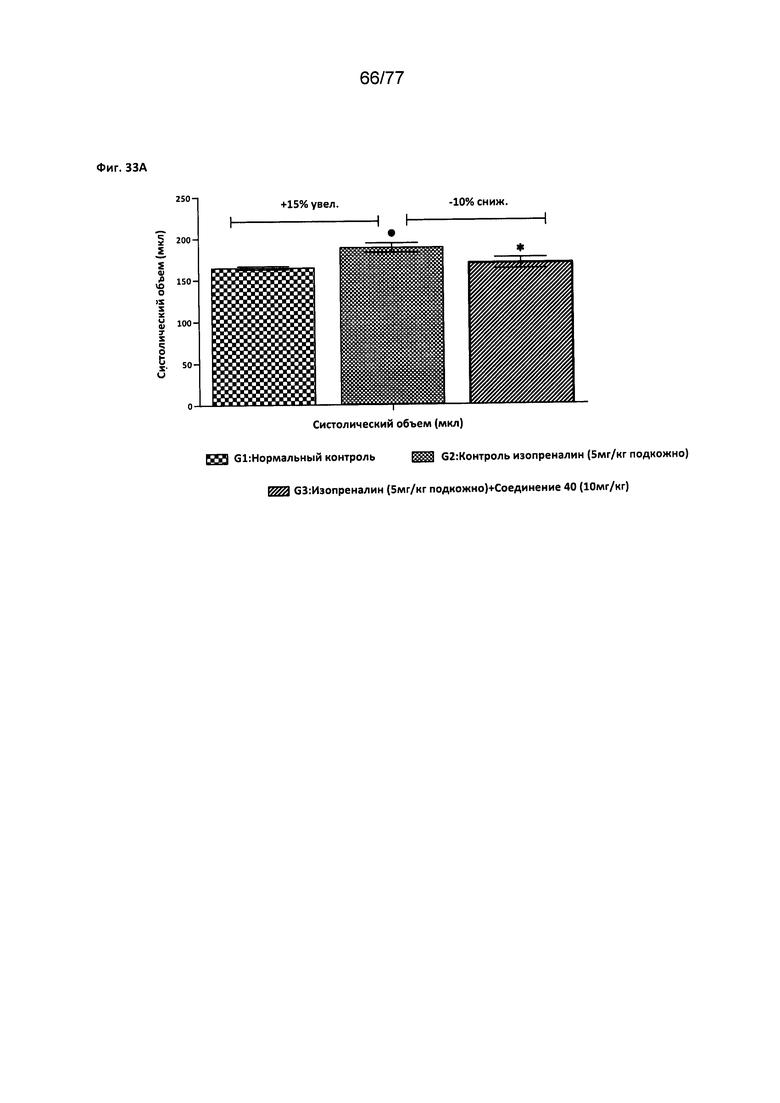
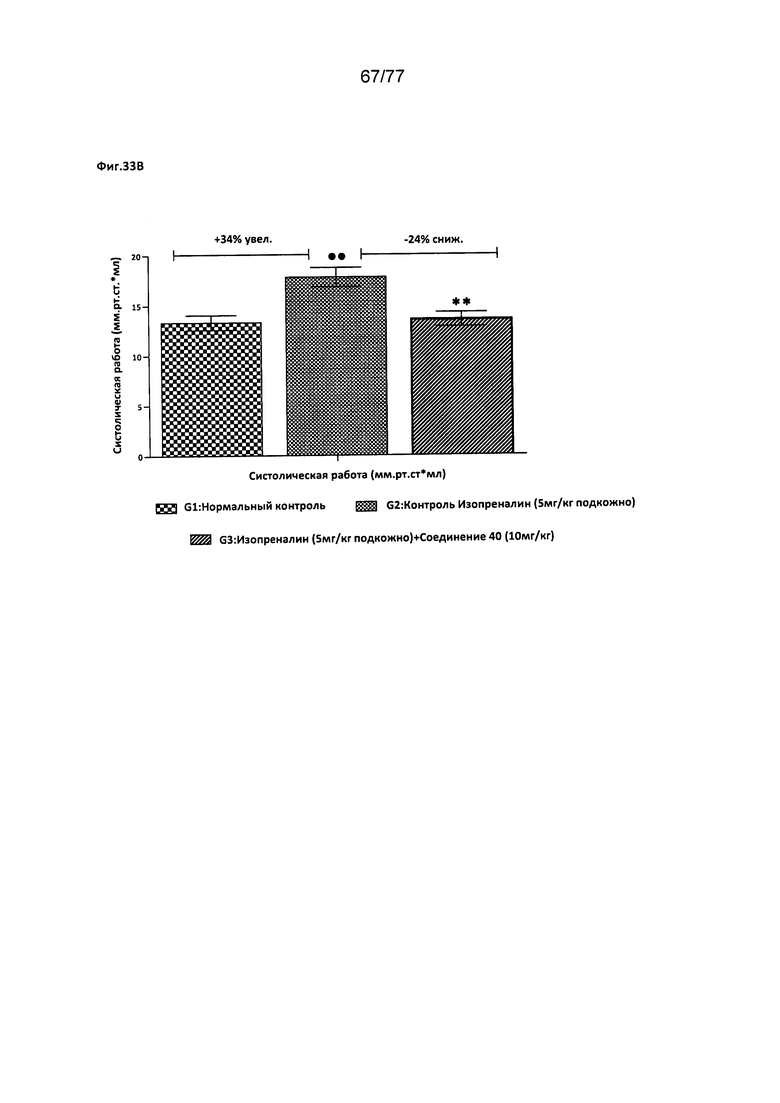
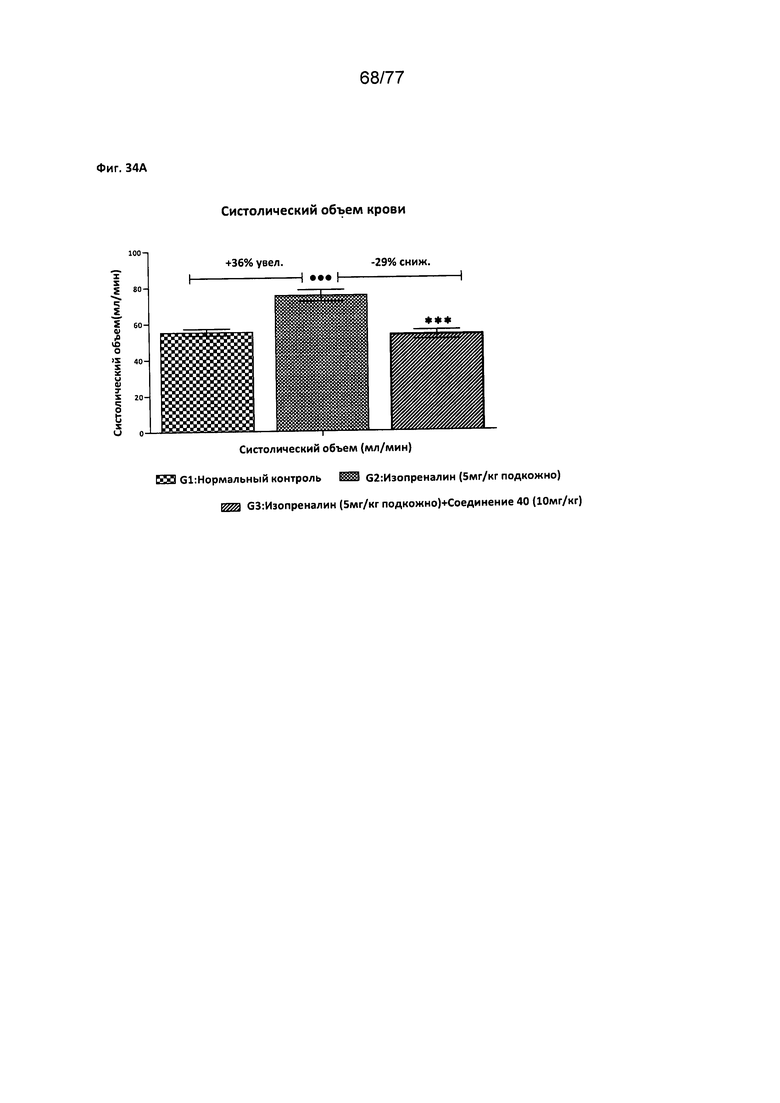
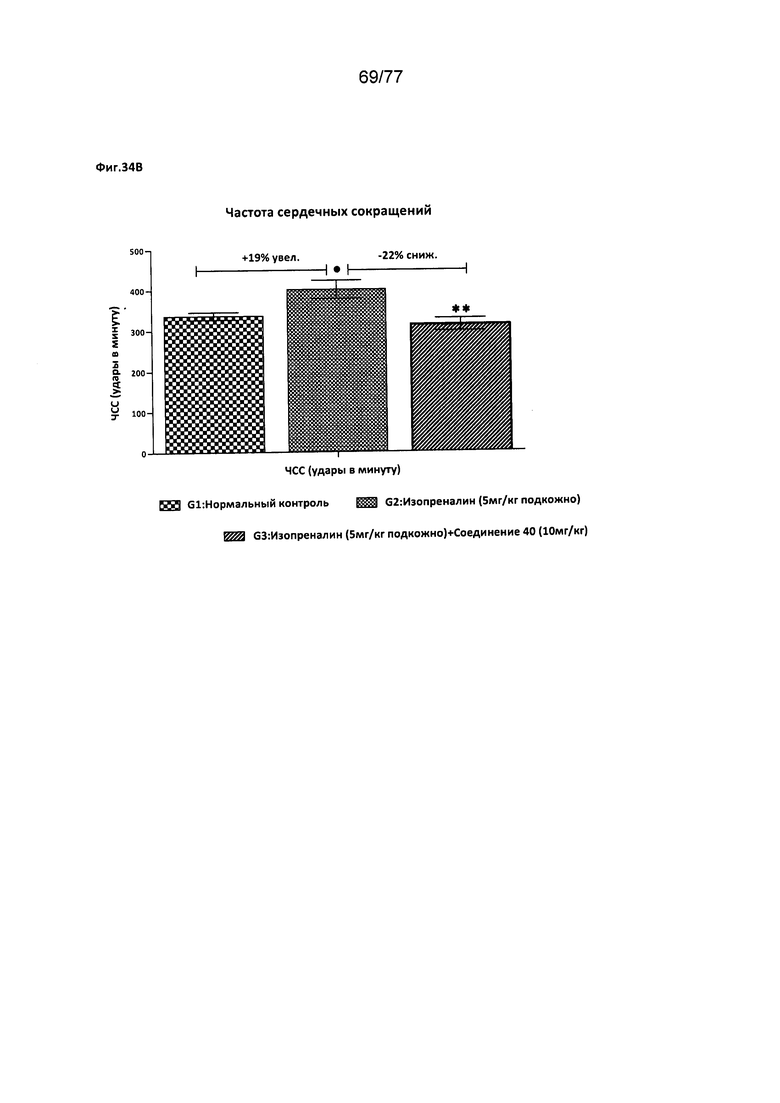
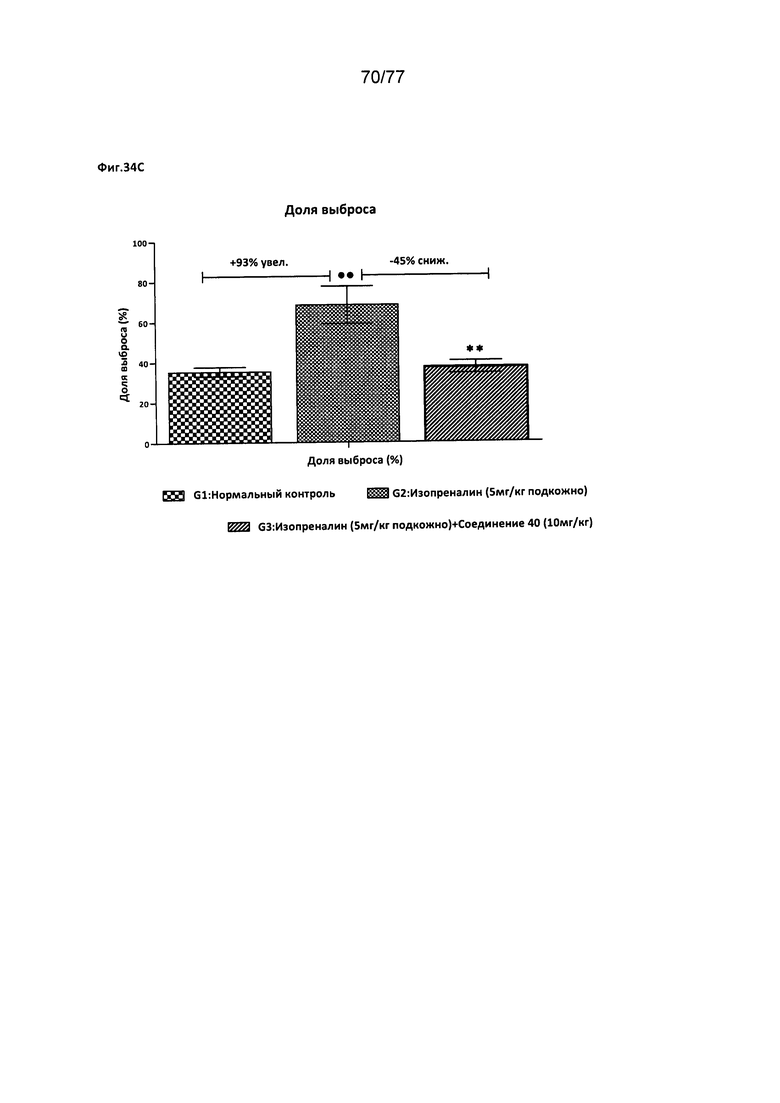
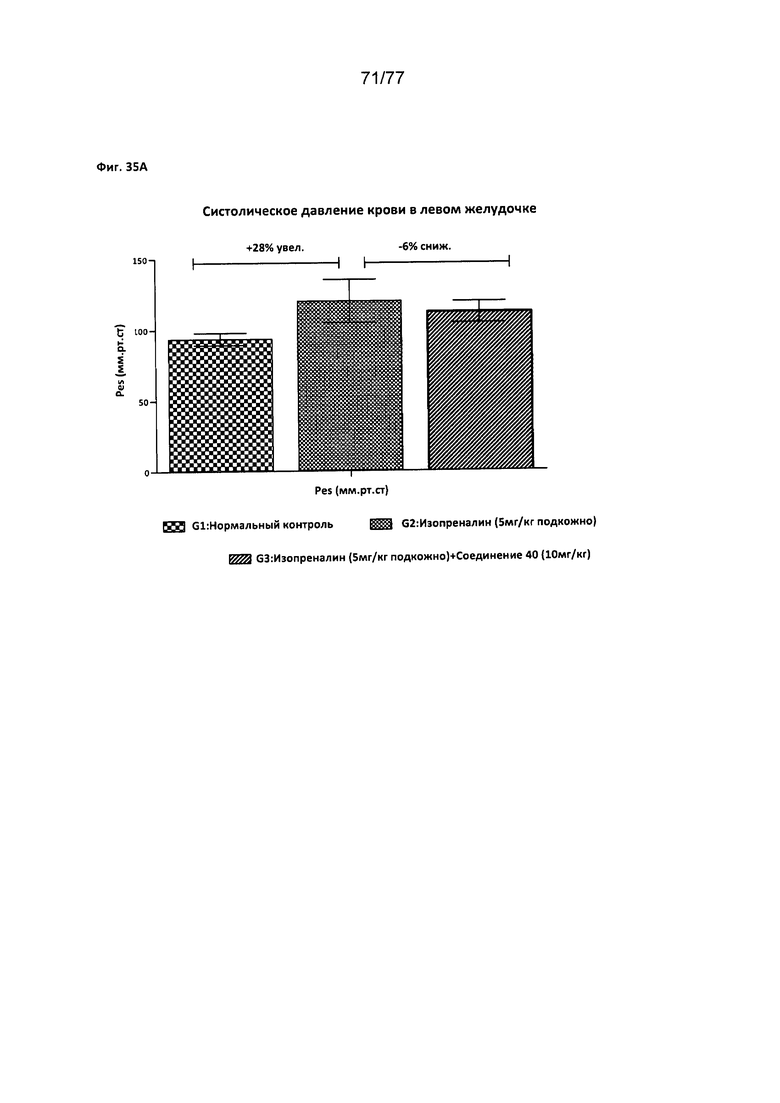
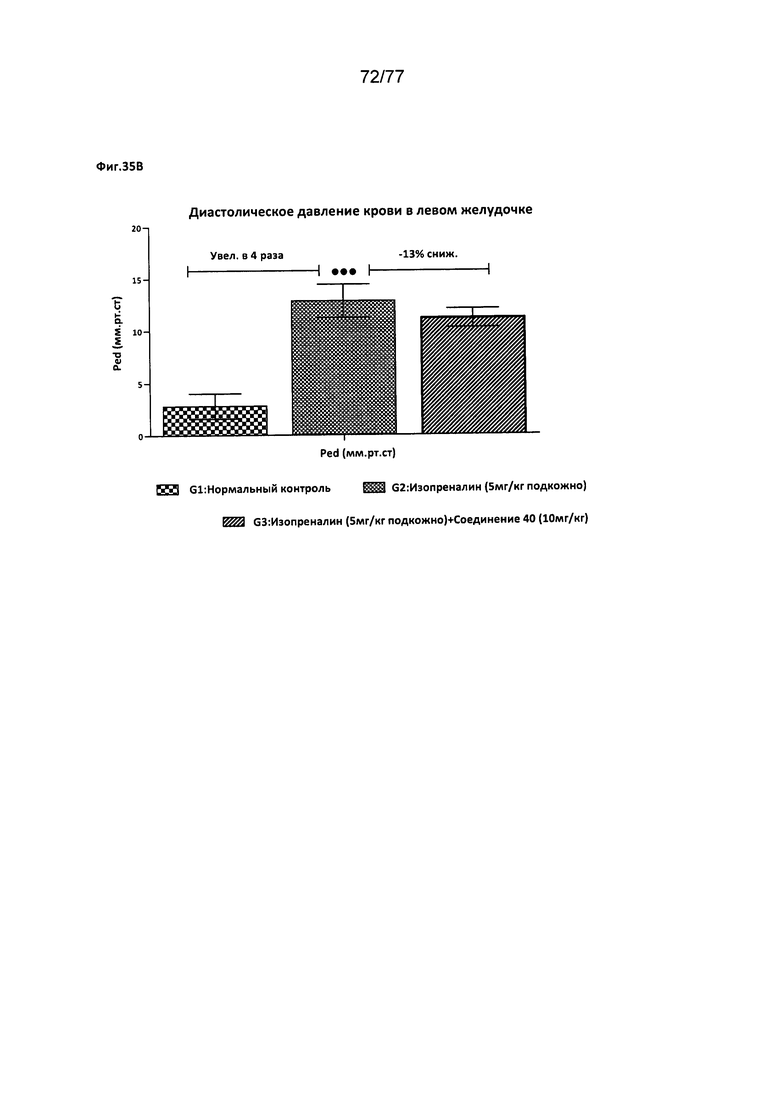
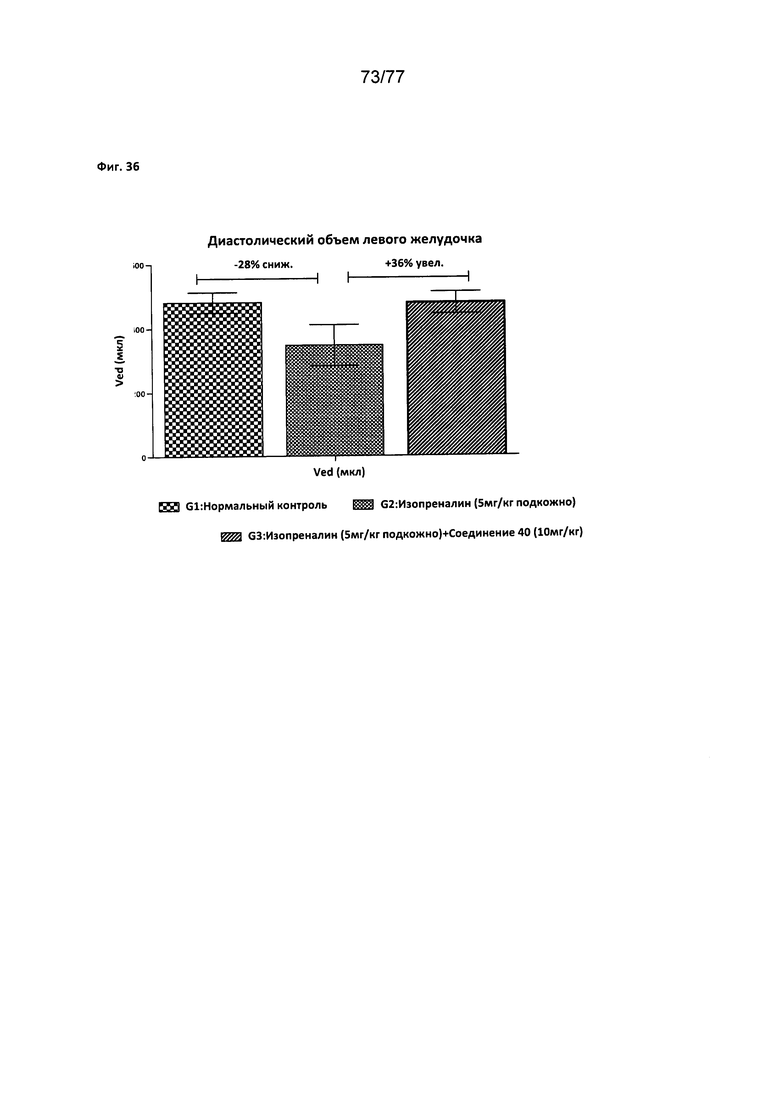
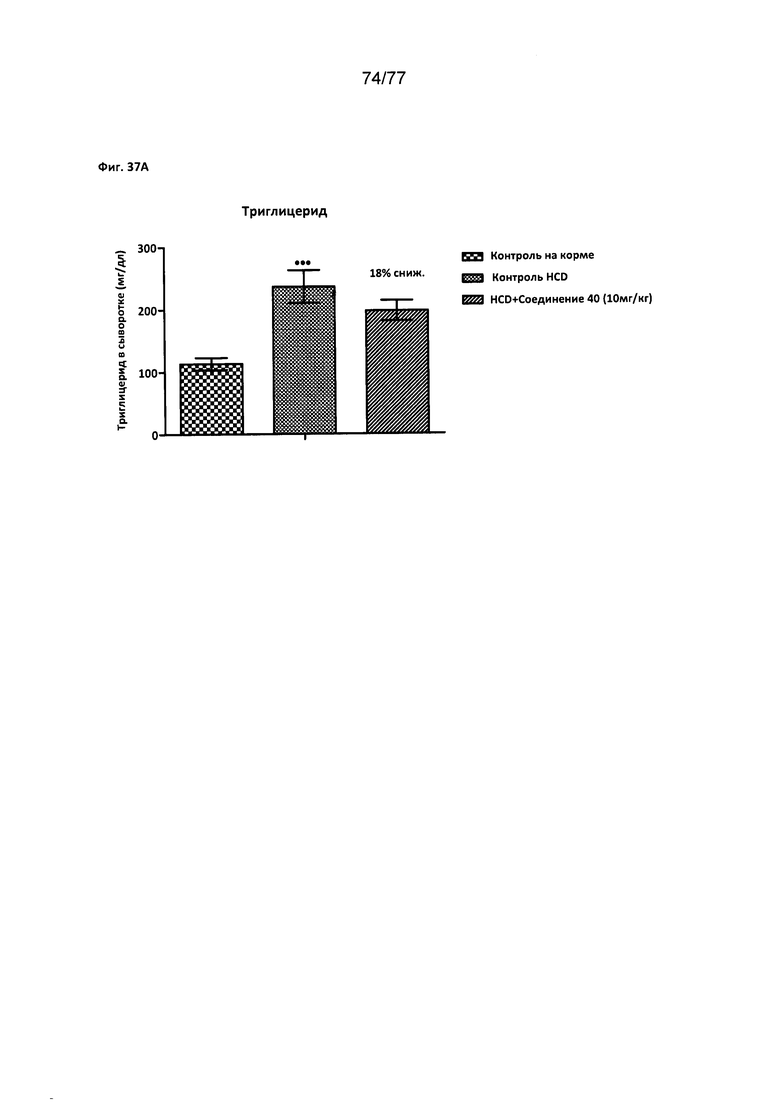
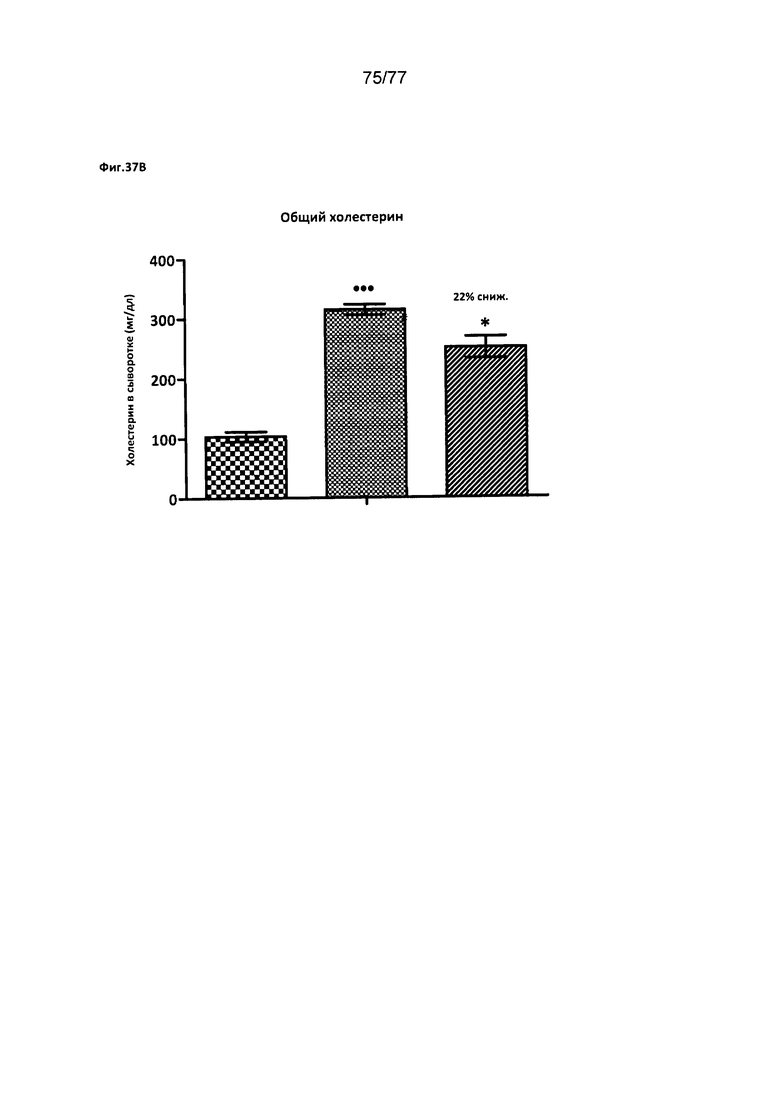
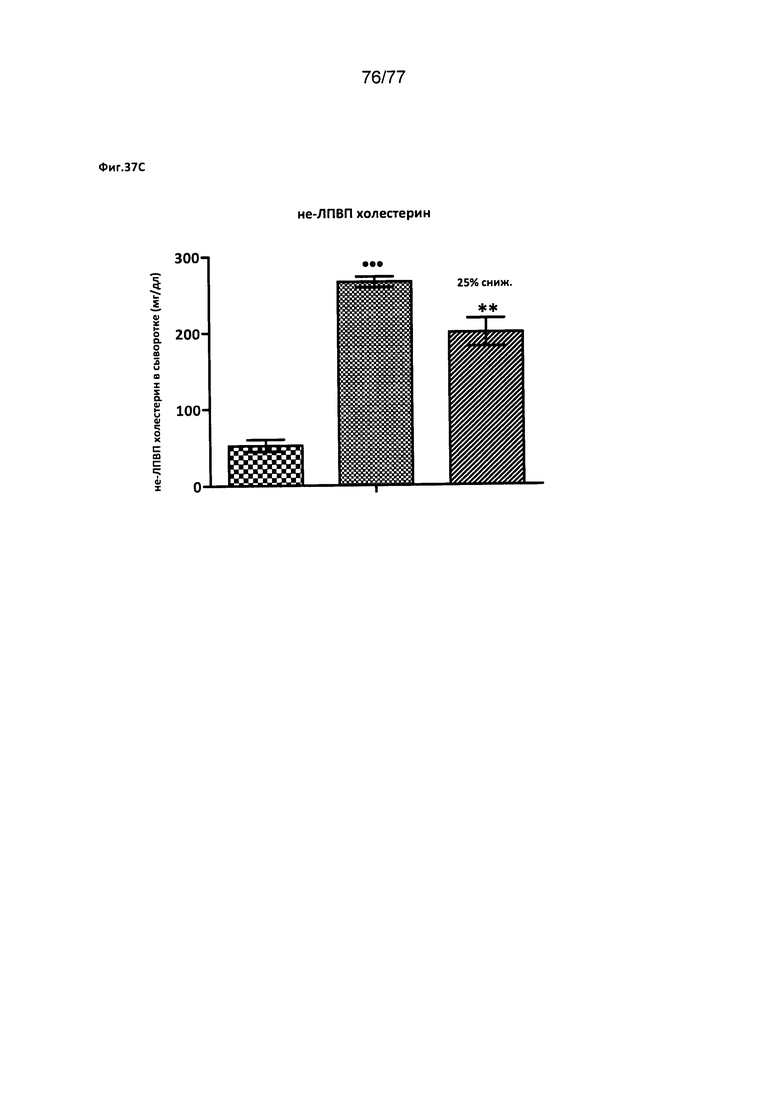
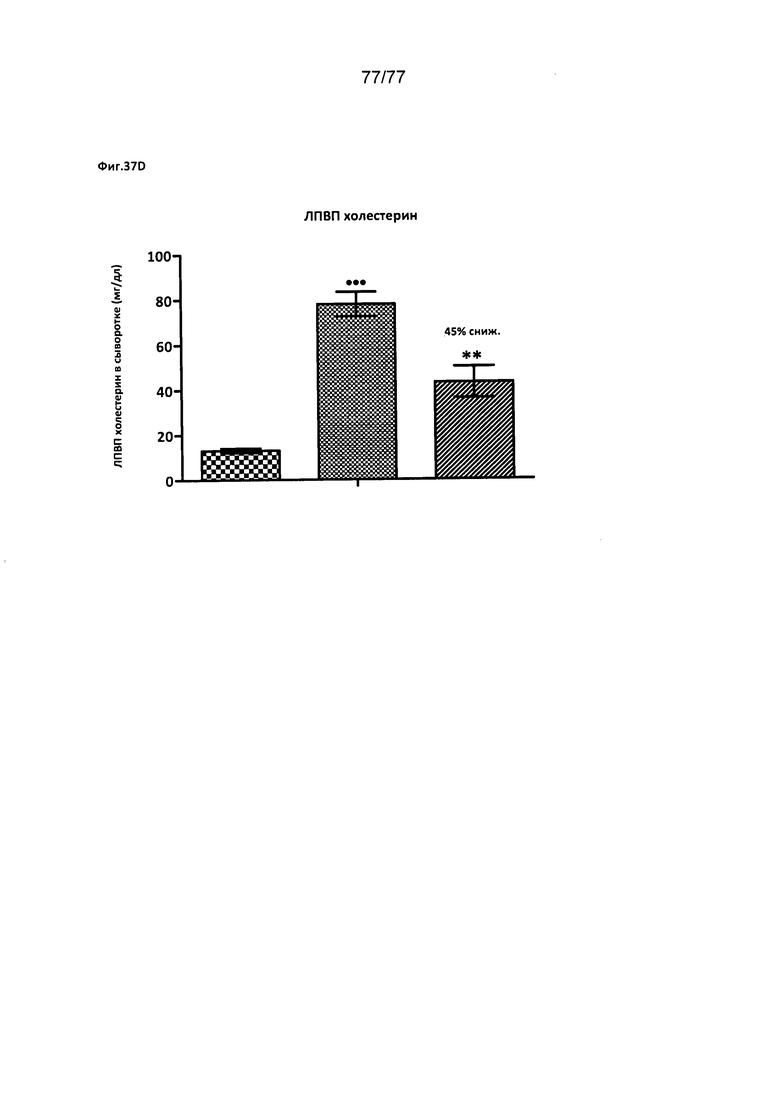
Изобретение относится к соединению формулы (I):
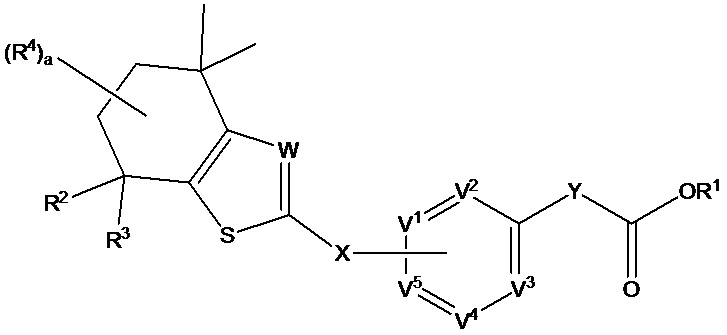
Формула (I)
где R1 представляет собой Н; R2 и R3 независимо выбраны из группы, состоящей из Н и метила; каждый R4 независимо выбран из группы, состоящей из H, OH, C1-C12алкила, или два R4 при одном и том же атоме углерода совместно образуют заместитель =O или группу формулы =NOH, или два R4 при соседних атомах углерода совместно образуют двойную связь; а представляет собой целое число, выбранное из группы, состоящей из 0 и 1; где Х выбран из группы, состоящей из связи, -O-, -S-, -NR10-, NR10C(=O)-, -C(=R11)- и - (CR12R13)b-; где R10 выбран из группы, состоящей из Н, С1-С3алкила, С3-C5циклоалкила, C1алкила, дополнительно замещенного C3циклоалкилом, C5гетероарил-С1алкила, где гетероатом выбран из N, SO3H, SO2R5, COR5; где R11 выбран из группы, состоящей из О, NR14, NOR14 и CR14R15, где каждый из R14 и R15 независимо выбран из группы, состоящей из Н и С1-С3алкила; каждый R12 и R13 независимо выбран из группы, состоящей из Н, С1-С3алкила, или R12 и R13 при одном и том же атоме углерода совместно образуют циклическую группу; b представляет собой целое число, выбранное из группы, состоящей из 0, 1 и 2; V2, V3 и V4 каждый независимо выбран из группы, состоящей из N и CR6; каждый R6 независимо выбран из группы, состоящей из H, галогена, OH, C1-C3алкила, необязательно замещенного C1-C3алкилокси, где указанный необязательный заместитель выбран из циклопропила, циано, фенокси или NR5SO2R5; где каждый R5 независимо выбран из группы, состоящей из Н, С3циклоалкила; V1 и V5 выбраны из группы, состоящей из N, CR7 и CR8, таким образом, что один из V1 и V5 представляет собой CR8, а другой представляет собой N или CR7; R7 представляет собой H; R8 представляет собой связь с X, Y представляет собой связь; W выбран из группы, состоящей из N и CR9; R9 выбран из Н и С1-С6алкила; или фармацевтически приемлемым солям указанного соединения, которые являются агонистами RXR. А также к фармацевтической композиции, применению и способам лечения путем активации RXR. 10 н. и 24 з.п. ф-лы, 37 ил., 1 табл., 108 пр.
1. Cоединение формулы (I):
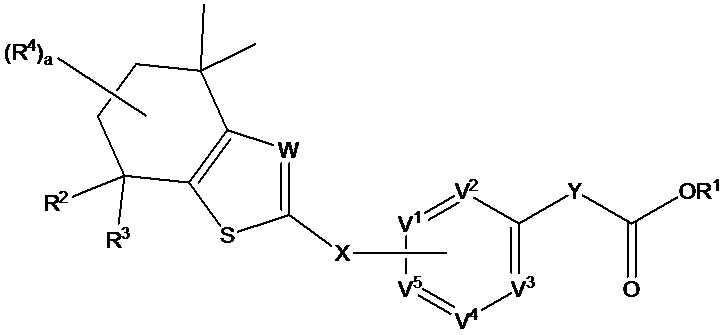
Формула (I)
где
R1 представляет собой Н;
R2 и R3 независимо выбраны из группы, состоящей из Н и метила;
каждый R4 независимо выбран из группы, состоящей из H, OH, C1-C12алкила, или два R4 при одном и том же атоме углерода совместно образуют заместитель =O или группу формулы =NOH, или два R4 при соседних атомах углерода совместно образуют двойную связь;
а представляет собой целое число, выбранное из группы, состоящей из 0 и 1;
где Х выбран из группы, состоящей из связи, -O-, -S-, -NR10-, NR10C(=O)-, -C(=R11)- и - (CR12R13)b-;
где R10 выбран из группы, состоящей из Н, С1-С3алкила, С3-C5циклоалкила, C1алкила, дополнительно замещенного C3циклоалкилом, C5гетероарил-С1алкила, где гетероатом выбран из N, SO3H, SO2R5, COR5;
где R11 выбран из группы, состоящей из О, NR14, NOR14 и CR14R15, где каждый из R14 и R15 независимо выбран из группы, состоящей из Н и С1-С3алкила;
каждый R12 и R13 независимо выбран из группы, состоящей из Н, С1-С3алкила, или R12 и R13 при одном и том же атоме углерода совместно образуют циклическую группу;
b представляет собой целое число, выбранное из группы, состоящей из 0, 1 и 2;
V2, V3 и V4 каждый независимо выбран из группы, состоящей из N и CR6;
каждый R6 независимо выбран из группы, состоящей из H, галогена, OH, C1-C3алкила, необязательно замещенного C1-C3алкилокси, где указанный необязательный заместитель выбран из циклопропила, циано, фенокси или NR5SO2R5;
где каждый R5 независимо выбран из группы, состоящей из Н, С3циклоалкила;
V1 и V5 выбраны из группы, состоящей из N, CR7 и CR8, таким образом, что один из V1 и V5 представляет собой CR8, а другой представляет собой N или CR7;
R7 представляет собой H;
R8 представляет собой связь с X,
Y представляет собой связь;
W выбран из группы, состоящей из N и CR9;
R9 выбран из Н и С1-С 6алкила;
или фармацевтически приемлемая соль указанного соединения.
2. Соединение по п. 1, отличающееся тем, что а представляет собой 0.
3. Соединение по п. 1 или 2, отличающееся тем, что R2 и R3 представляют собой метил.
4. Соединение по любому из пп. 1-3, отличающееся тем, что W представляет собой N.
5. Соединение по любому из пп. 1-3, отличающееся тем, что W представляет собой CR9.
6. Соединение по п. 5, отличающееся тем, что R9 представляет собой метил.
7. Соединение по п. 1, отличающееся тем, что X представляет собой -NR10-.
8. Соединение по п. 7, отличающееся тем, что R10 представляет собой Н.
9. Соединение по любому из пп. 1-8, отличающееся тем, что V2 представляет собой CR6.
10. Соединение по любому из пп. 1-9, отличающееся тем, что V3 представляет собой CR6.
11. Соединение по любому из пп. 1-10, отличающееся тем, что V5 представляет собой CR8.
12. Соединение по любому из пп. 1-11, отличающееся тем, что V4 представляет собой CR6.
13. Соединение по любому из пп. 1-11, отличающееся тем, что V4 представляет собой N.
14. Соединение по любому из пп. 1-13, отличающееся тем, что V1 представляет собой CR6.
15. Соединение по любому из пп. 1-13, отличающееся тем, что V1 представляет собой N.
16. Соединение по любому из предшествующих пунктов, отличающееся тем, что каждый R6 независимо выбран из группы, состоящей из Н, галогена, ОН и C1-C3алкила.
17. Соединение по п. 1, выбранное из группы, состоящей из:

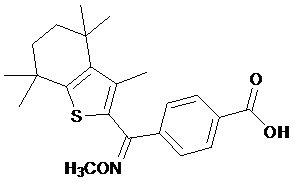
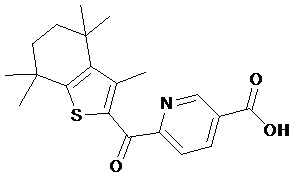
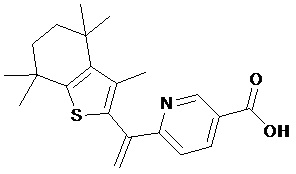
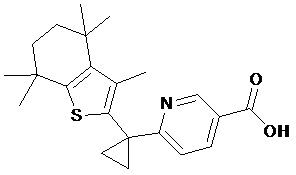
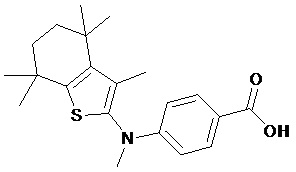
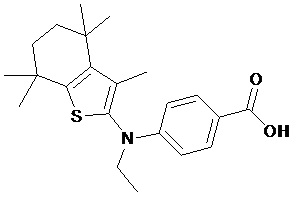
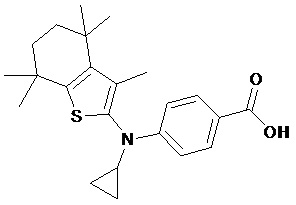
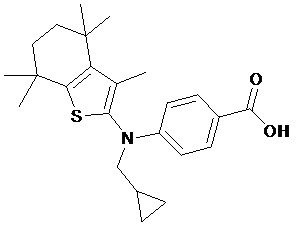
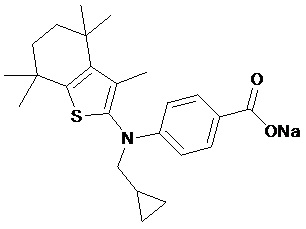
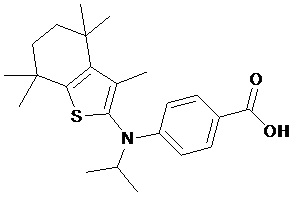
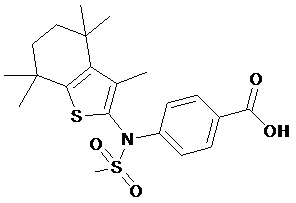
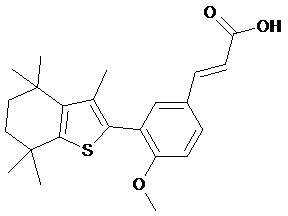
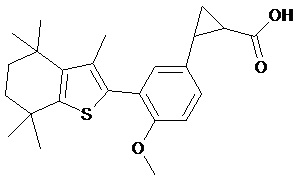
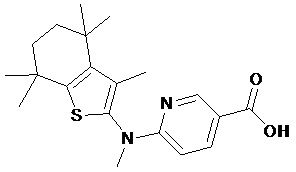
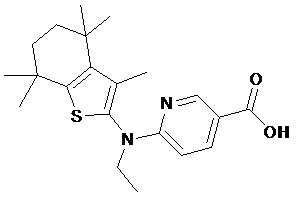
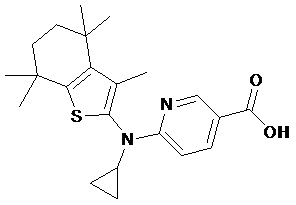
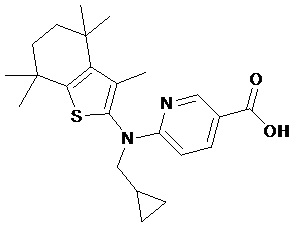
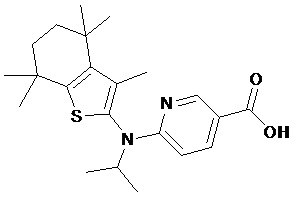
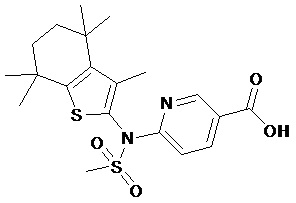
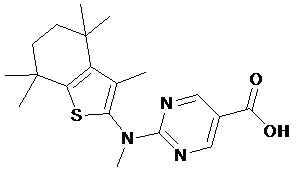
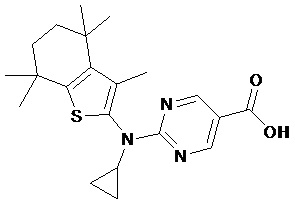
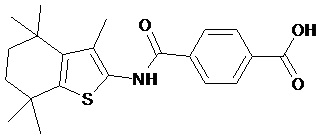
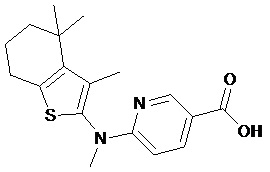
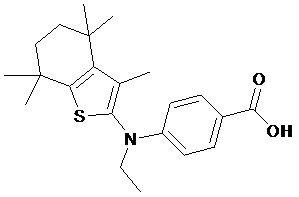
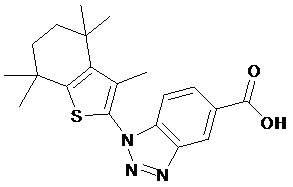
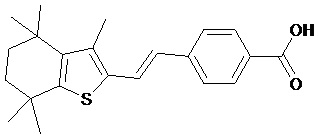
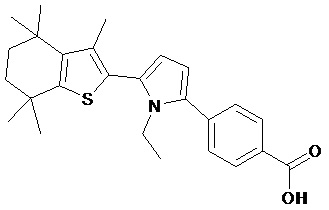
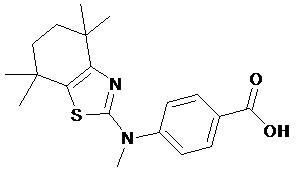
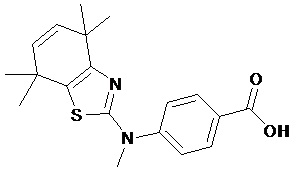
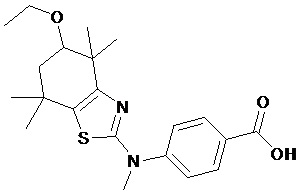
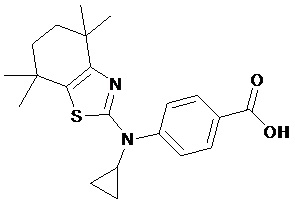
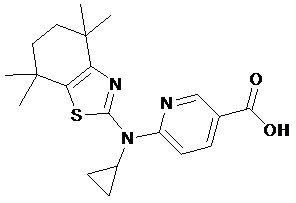
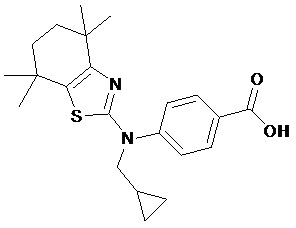
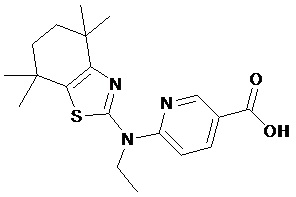
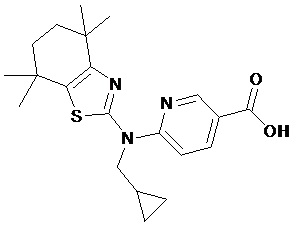
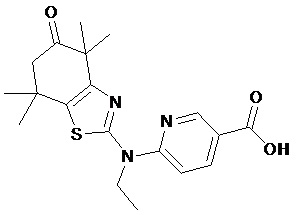
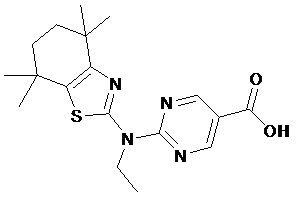
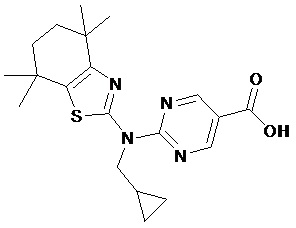
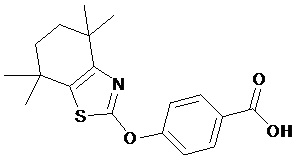
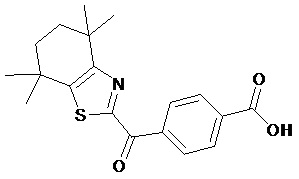
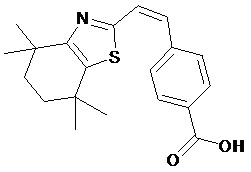
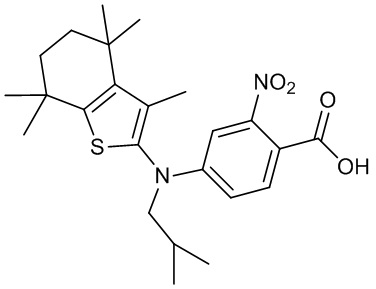
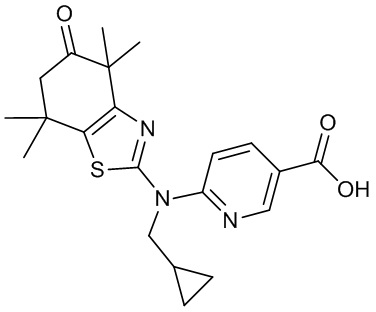
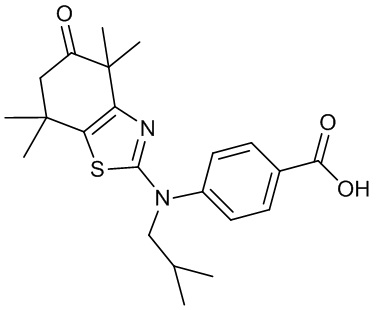
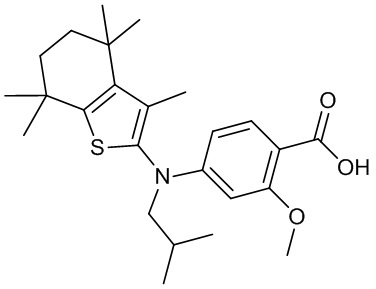
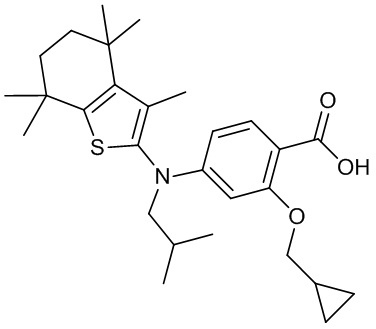
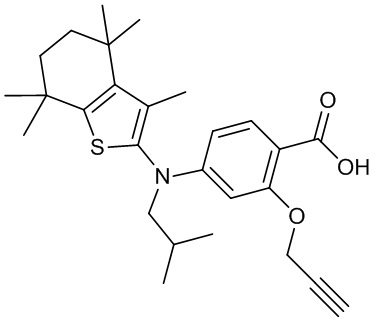
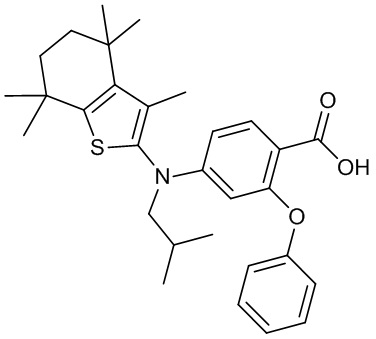
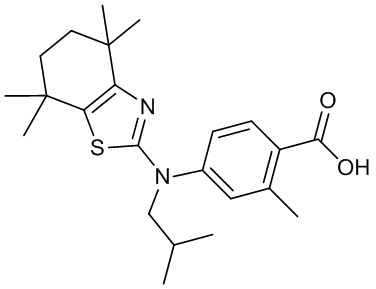
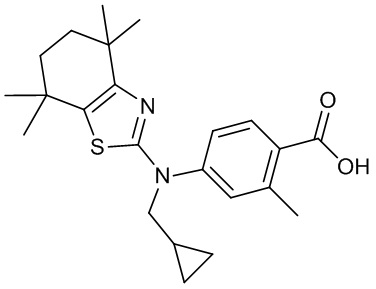
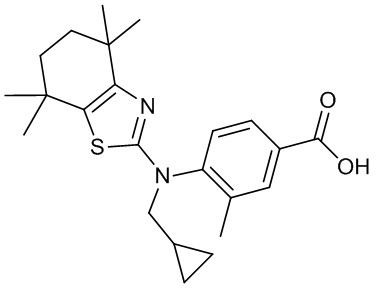
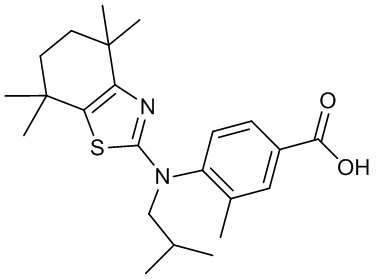
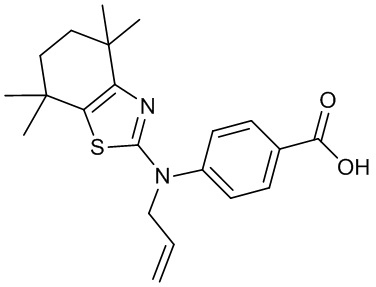
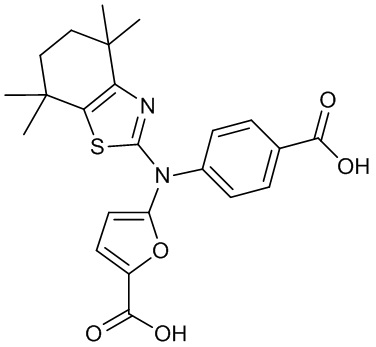
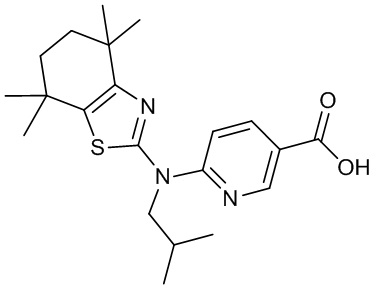
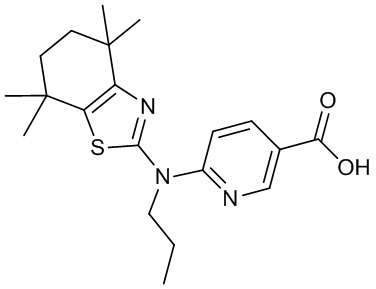
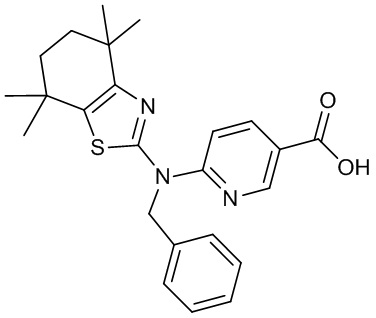
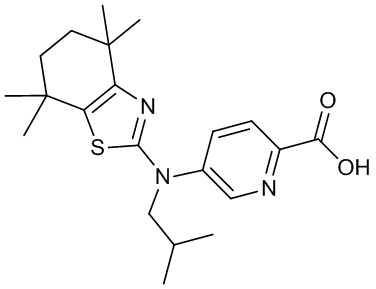
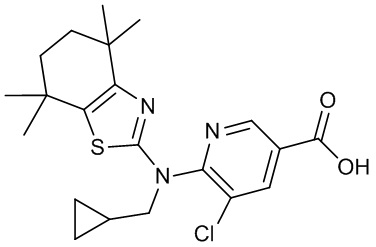
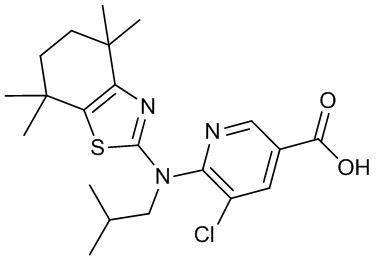
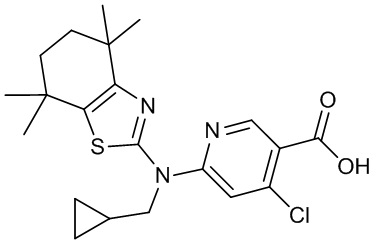
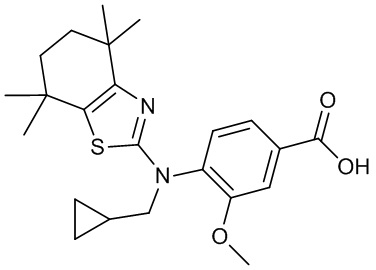
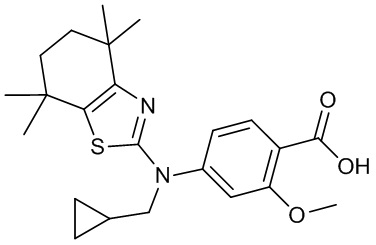
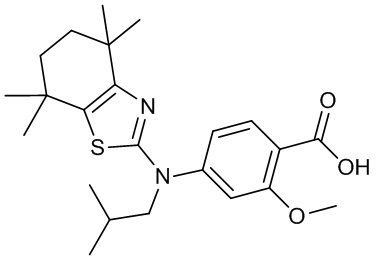
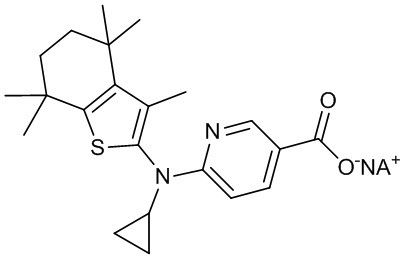
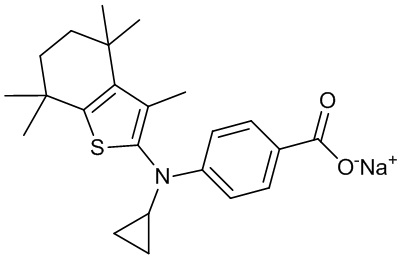
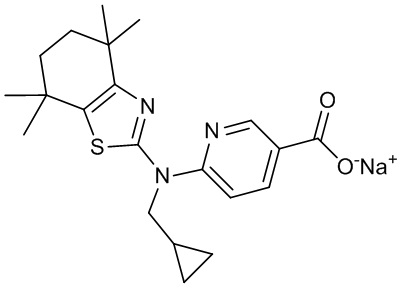
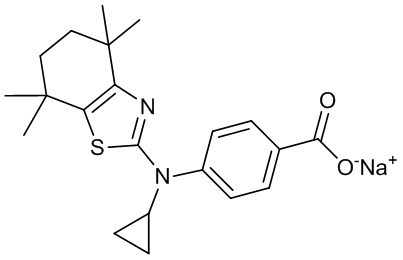
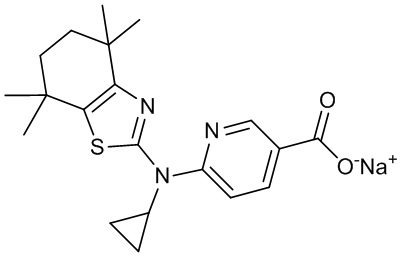
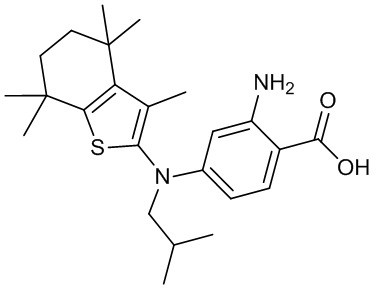
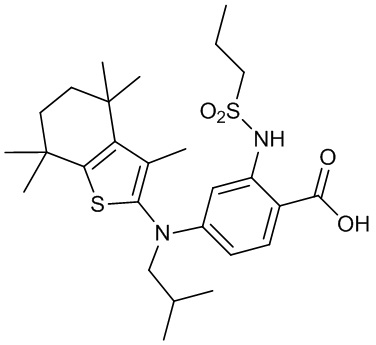
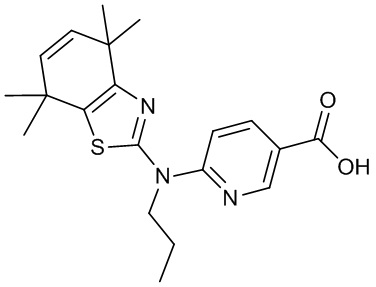
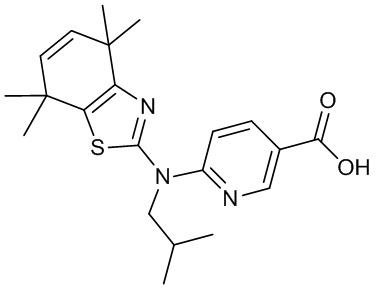
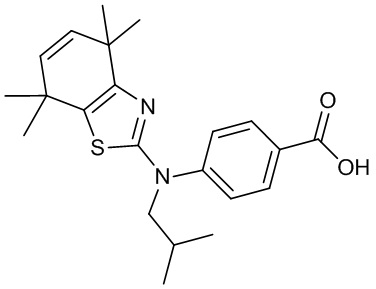
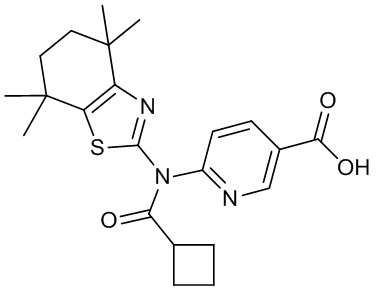
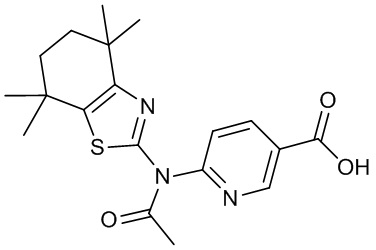
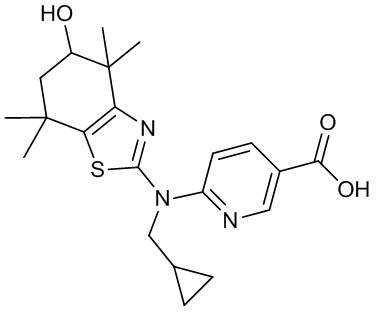
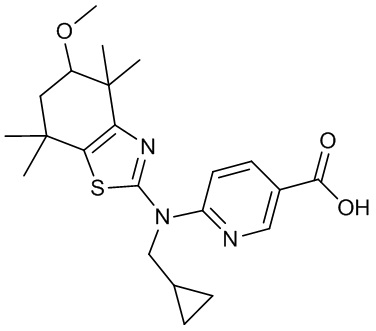
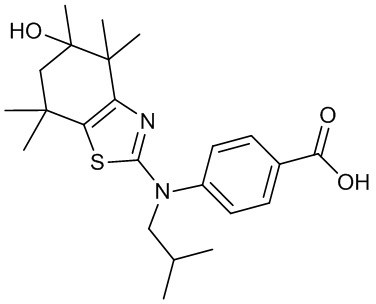
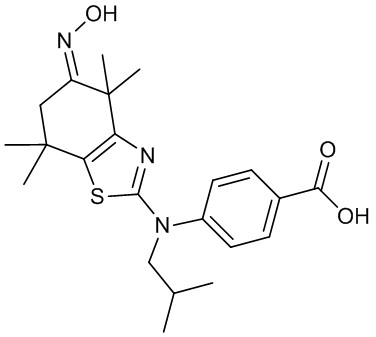
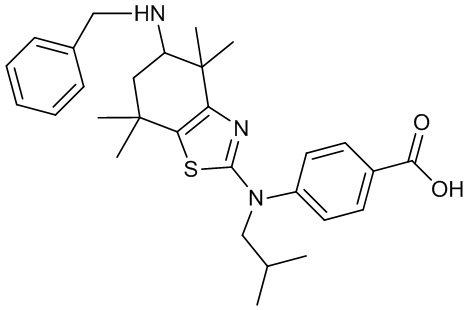
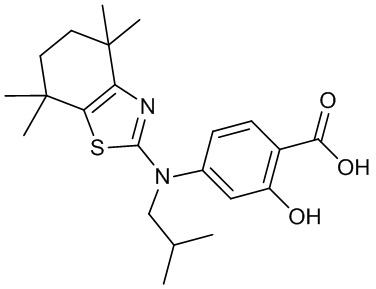
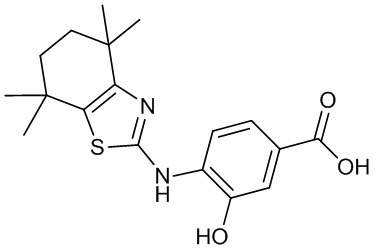
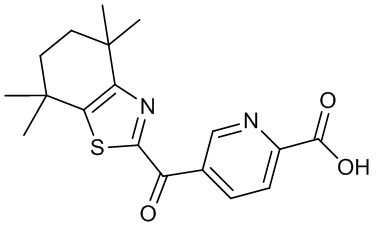
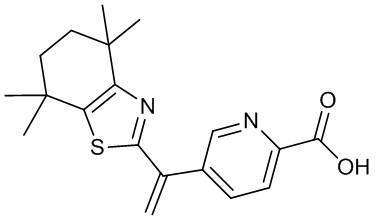
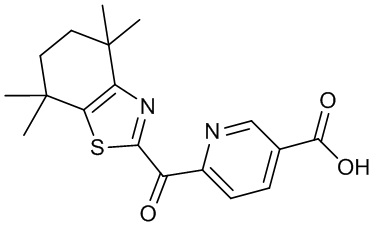
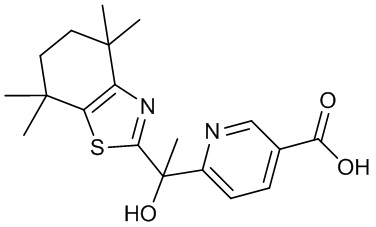
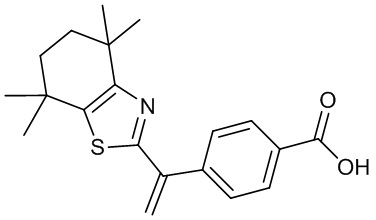
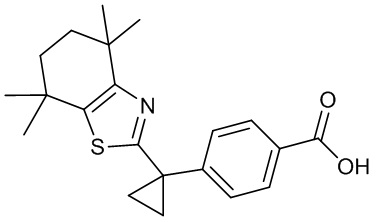
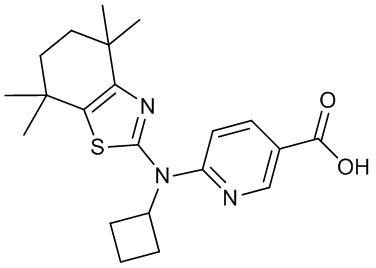
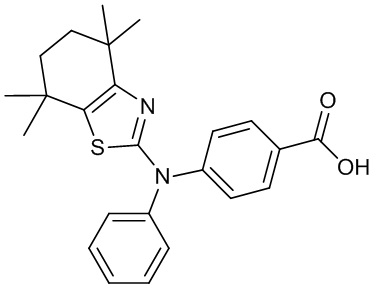
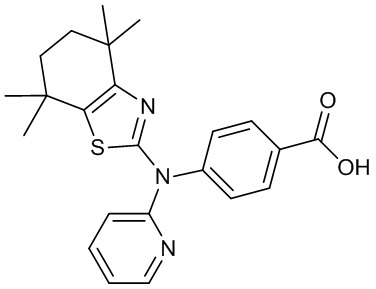
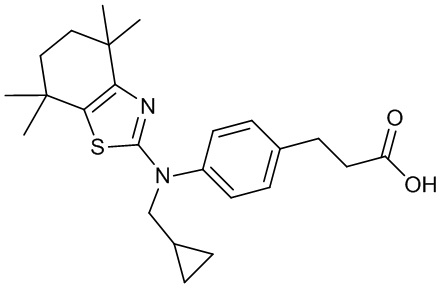
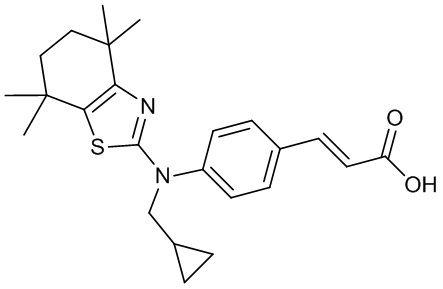
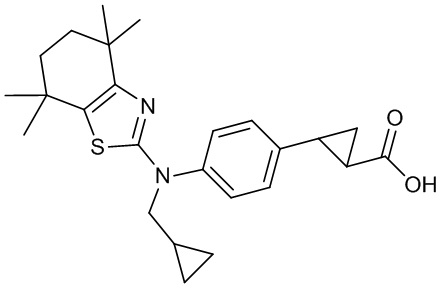
или фармацевтически приемлемая соль или пролекарство указанного соединения.
18. Фармацевтическая композиция для лечения состояния, поддающегося лечению путем активации RXR, содержащая эффективное количество соединения по любому из пп. 1-17 и фармацевтически приемлемые разбавитель, наполнитель или носитель.
19. Способ профилактики или лечения состояния у млекопитающего, где указанное состояние поддается профилактике или лечению путем активации RXR, включающий введение эффективного количества соединения по любому из пп. 1-17.
20. Способ по п. 19, отличающийся тем, что указанное состояние представляет собой диабет.
21. Способ по п. 20, отличающийся тем, что указанное состояние представляет собой диабет II типа.
22. Способ по п. 19, отличающийся тем, что указанное состояние представляет собой ожирение.
23. Способ по п. 19, отличающийся тем, что указанное состояние представляет собой сердечно-сосудистое заболевание.
24. Способ увеличения мышечной выносливости у млекопитающего, включающий введение эффективного количества соединения по любому из пп. 1-17.
25. Способ улучшения сердечной функции у млекопитающего, включающий введение эффективного количества соединения по любому из пп. 1-17.
26. Способ достижения эффекта, миметического физическим упражнениям, у млекопитающего, включающий введение эффективного количества соединения по любому из пп. 1-17.
27. Применение соединения по любому из пп. 1-17 для изготовления лекарственного средства, где указанное лекарственное средство предназначено для лечения состояния, поддающегося профилактике или лечению путем активации RXR.
28. Применение по п. 27, отличающееся тем, что указанное состояние представляет собой диабет.
29. Применение по п. 28, отличающееся тем, что указанное состояние представляет собой диабет II типа.
30. Применение по п. 27, отличающееся тем, что указанное состояние представляет собой ожирение.
31. Применение по п. 27, отличающееся тем, что указанное состояние представляет собой сердечно-сосудистое заболевание.
32. Применение соединения по любому из пп. 1-17 для изготовления лекарственного средства для увеличения мышечной выносливости у млекопитающего.
33. Применение соединения по любому из пп. 1-17 для изготовления лекарственного средства для улучшения сердечной функции у млекопитающего.
34. Применение соединения по любому из пп. 1-17 для изготовления лекарственного средства для достижения эффекта, миметического физическим упражнениям, у млекопитающего.
| Буровой станок для подземного бурения | 1953 |
|
SU98591A1 |
| WO 2008030618 A1, 13.03.2008 | |||
| Bonchev, D., Seitz, W | |||
| A., Mountain, C | |||
| F., & Balaban, A | |||
| T | |||
| Прибор для охлаждения жидкостей в зимнее время | 1921 |
|
SU1994A1 |
| Modeling the anticarcinogenic action of retinoids by making use of the OASIS method | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Inhibition of the induction of ornithine decarboxylase by arotinoids | |||
| Journal of Medicinal Chemistry, 37(15), 2300-2307 | |||

