Область техники, к которой относится изобретение
Настоящее изобретение относится к области каталитической химии, а именно к композитному носителю для катализатора, способу получения композитного носителя, катализатору дегидрирования, основанному на этом композитном носителе, способу получения катализатора и к способу дегидрирования газообразных углеводородов с использованием такого катализатора в режиме подвижного слоя.
Уровень техники
Легкие олефины (этилен, пропилен и бутен) являются ценными компонентами нефтехимической промышленности. Особенно активно такие олефины используются в полимерной отрасли. Изделия, содержащие полипропилен, полиэтилен, а также сополимеры пропилена, этилена и бутена, обладают хорошими эксплуатационными характеристиками и используются повсеместно, например, в производстве кабелей, бытовой техники, труб и т.д.
Одним из распространенных способов получения легких олефинов в промышленности является дегидрирование газообразных С2-С4-углеводородов в присутствии катализаторов. В качестве катализаторов дегидрирования широко используются гетерогенные катализаторы, содержащие в качестве активного компонента элемент VIII группы Периодической системы Д.И. Менделеева (далее Периодической системы). Активный компонент наносят на носитель, представляющий собой оксид алюминия, алюмосиликат или иной синтетический носитель. Помимо активного компонента, на носитель наносят другие компоненты, которые не являются активными в каталитическом процессе дегидрирования, однако позволяют улучшить эксплуатационные характеристики катализатора, в частности, срок эксплуатации катализатора. Одними из таких компонентов являются элементы IV группы Периодической системы, например, олово. Такие компоненты, как известно из уровня техники, в т.ч. из документов US3998900 и US3909451, препятствуют образованию крупных платиновых наночастиц в ходе эксплуатации. Введение дополнительных компонентов в состав катализатора позволяет сохранять высокую дисперсность активного компонента на поверхности соответствующего носителя и, следовательно, сохранять высокую активность катализатора во времени. Кроме элементов IV группы на носитель наносят другие компоненты: щелочные металлы, щелочноземельные металлы, лантаноиды, галогены и т.д.
Типичным катализатором дегидрирования является катализатор, описанный в WO2010076928, в котором в качестве активного компонента используют платину. Катализатор также содержит: один или несколько вспомогательных металлов, выбранных из группы, состоящей из олова, германия, галлия, индия, цинка и марганца, щелочной металл или щелочноземельный металл и галогеновый компонент, нанесенные на носитель, представляющий собой тета-оксид алюминия. Катализатор получают последовательной пропиткой носителя раствором вспомогательного металла (например, водорастворимым соединением олова), платиносодержащим раствором и затем раствором щелочного или щелочноземельного металла, при этом после каждой стадии пропитки носителя следуют стадии сушки и прокаливания.
Недостатком данного способа получения катализатора дегидрирования газообразных углеводородов является сложность его получения. На каждой стадии нанесения используются хлористоводородная и азотная кислоты, однако целесообразность их использования не указана. Кроме того, после каждой стадии нанесения проводят сушку в общей сложности в течение 18 часов и прокаливание в течение 3 часов, что является энергозатратным процессом. Следует отметить, что низкая температура прокаливания катализатора после нанесения компонента, выбранного из группы, состоящей из олова, германия, галлия, индия, цинка и марганца, вызывает понижение каталитической активности катализатора в процессе дегидрирования. Конверсия пропана при температуре 620°C и соотношении пропана к водороду 1:1 составляет около 35% после 100 минут проведения реакции и продолжает падать с течением времени, не достигая стабильного уровня.
Из уровня техники известны катализаторы дегидрирования, в которых используют композитный носитель, содержащий олово. Например, в документе WO2010069548 раскрыт платиносодержащий катализатор, в котором носитель состоит, по меньшей мере, из одного оксида элемента главной (A) или побочной (B) подгруппы II-IV группы Периодической системы или их смеси и дополнительный компонент, включающий оксид элемента главной подгруппы IV группы Периодической системы, который добавляют в процессе формования носителя. Описанный носитель получают одним из следующих способов:
1) по меньшей мере, один оксид элемента главной или побочной подгруппы II-IV группы Периодической системы или их смесь и дополнительный компонент, включающий оксид элемента главной подгруппы IV группы Периодической системы, размельчают, смешивают с вяжущим веществом и проводят формование (например, спекание, гранулирование, таблетирование, зернение, экструзию);
2) водный или спиртовой раствор, по меньшей мере, одного элемента главной или побочной подгруппы II-IV группы Периодической системы или составленный из них смешанный раствор и водный или спиртовой раствор дополнительного компонента, включающего соединение элемента главной подгруппы IV группы Периодической системы, при необходимости смешивают с деионизированной водой, нейтрализуют и осаждают. После осаждения полученный материал фильтруют, сушат и формуют подходящим способом.
Дегидрирование пропана с использованием описанного в WO2010069548 катализатора проводится в присутствии паров воды, что делает процесс технологически более сложным по причине необходимости дополнительной стадии отделения паров воды и осушки продуктов реакции перед разделением полученного олефина и непрореагировавшей части пропана.
Также композитный носитель для катализатора дегидрирования газообразных углеводородов известен из US6281160. Носитель получают гидролизом раствора изопропилата алюминия Al(OiPr)3 в изопропаноле с использованием раствора водорастворимого соединения германия, олова, свинца, рения, галлия, индия или таллия, с последующей термообработкой при 450°С. На носитель из раствора водорастворимого соединения наносят активный компонент, представляющий собой элемент VIII группы Периодической системы, предшественник катализатора сушат, прокаливают, а затем наносят другие вспомогательные элементы, например, щелочные металлы и галоген.
Недостатком данного носителя является его высокая ресурсоемкость вследствие использования изопропилата алюминия в качестве исходного компонента для получения носителя, который является весьма редким и дорогостоящим сырьем.
Таким образом, из уровня техники не известен простой и умеренный по ресурсоемкости способ получения катализатора дегидрирования газообразных углеводородов, который характеризовался бы высокими селективностью и активностью, а также имел высокую устойчивость к коксообразованию. Кроме того, так как катализатор дегидрирования используют в режиме подвижного слоя (пневматического подъема), важным является получение катализатора не только с высокой активностью, но и устойчивого к истиранию, т.е. способного сохранять форму при воздействии различных и значительных ударных и сдвиговых нагрузок.
Раскрытие Изобретения
Задачей настоящего изобретения является разработка катализатора дегидрирования газообразных углеводородов с высокой устойчивостью к коксообразованию, высокими активностью и селективностью.
Техническим результатом настоящего изобретения является увеличение срока эксплуатации катализатора дегидрирования газообразных углеводородов с сохранением эффективности и активности катализатора во времени за счет высокой устойчивости катализатора к коксообразованию и устойчивости к истиранию.
Техническим результатом настоящего изобретения также является сокращение числа производственных стадий получения активного и стабильного катализатора дегидрирования газообразных углеводородов.
Дополнительным техническим результатом является возможность увеличения срока эксплуатации катализатора с сохранением высокой активности и селективности катализатора за счет проведения стадии оксихлорирования.
Задача настоящего изобретения решается, и технический результат достигается за счет обеспечения носителя, в состав которого элемент IV группы Периодической системы включается посредством введения в золь оксида алюминия водорастворимых соединений элемента IV группы на этапе приготовления носителя, а именно на стадии пептизации с дальнейшим формованием сферических гранул капельным методом, позволяющим обеспечить достаточную сферичность гранул и их необходимую механическую прочность, их отверждением в щелочной среде, последующей просушкой, и прокаливанием с получением композитного носителя.
Не желая связывать себя какой-либо теорией, авторы изобретения полагают, что наличие в составе носителя элемента IV группы, введенного в носитель на стадии пептизации, позволяет обеспечить равномерное распределение элемента IV группы по объему носителя и, соответственно, его постоянную концентрацию на поверхности катализатора, которая не будет изменяться в ходе эксплуатации катализатора. Постоянная концентрация элемента IV группы на поверхности катализатора, свою очередь, позволяет сохранять активность катализатора за счет сохранения распределения более 90% активного компонента (элемента VIII группы) в приповерхностном слое с дисперсностью активного компонента в диапазоне 60-70%.
Кроме того, авторами было обнаружено, что проведение стадии оксихлорирования при многоцикличном процессе дегидрирования позволяет длительное время сохранять высокие значения селективности и активности катализатора, содержащего композитный носитель.
Описание фигур
Фиг. 1 иллюстрирует результаты тестирования катализаторов A, B, C, полученных на алюмоксидном и композитном носителях.
Фиг. 2 иллюстрирует результаты тестирования катализаторов А, D, G с разным содержанием олова в реакции дегидрирования пропана с и без стадии оксихлорирования.
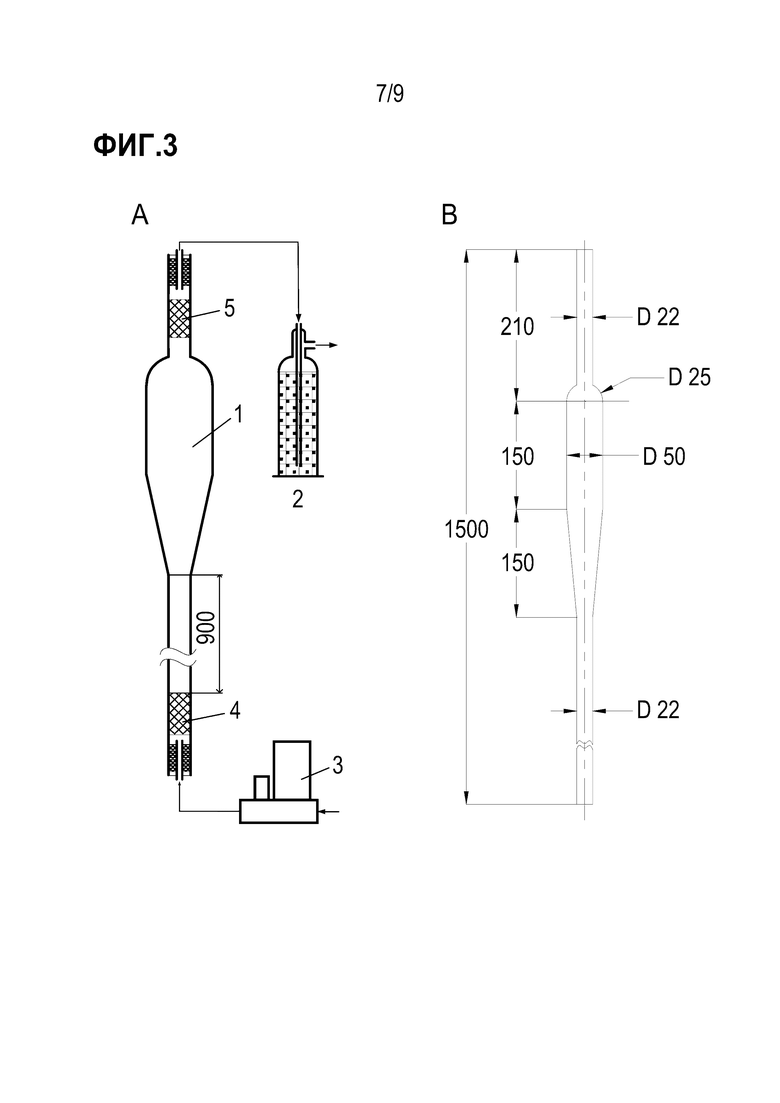
Фиг. 3 иллюстрирует схему установки для испытания показателя истираемости носителя: 1 - стеклянная трубка, 2 – абсорбер, 3 – регулятор расхода газа, 4,5 - сетчатый пыж.
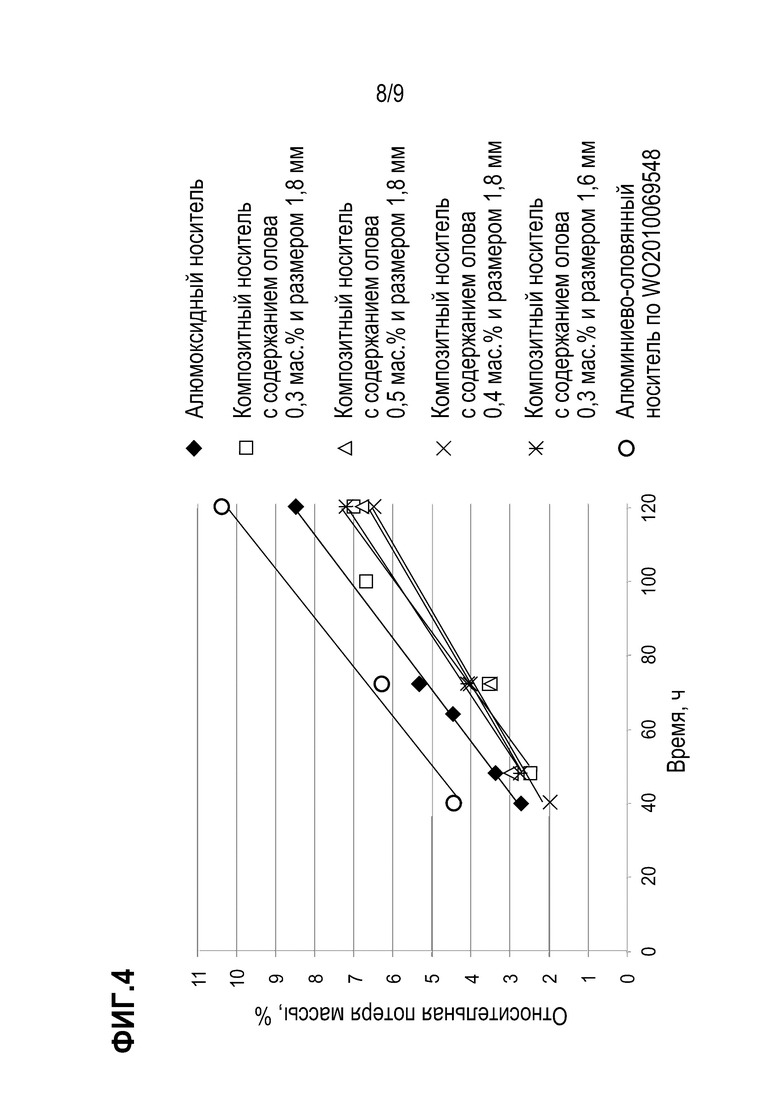
Фиг. 4 иллюстрирует результаты определения показателя истираемости носителя.
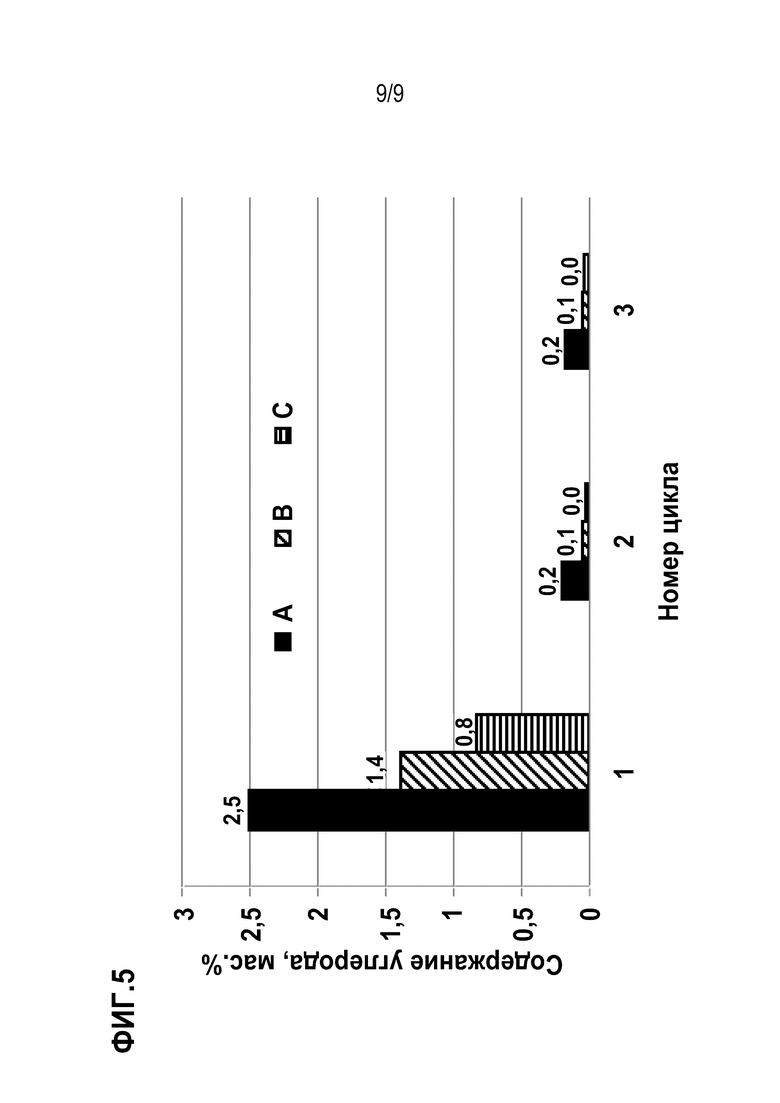
Фиг. 5 иллюстрирует результаты определения коксообразования на катализаторах А, В, С.
Подробное описание изобретения
Далее приводится подробное описание различных аспектов и вариантов реализации настоящего изобретения.
В одном своем аспекте настоящее изобретение относится к способу получения композитного носителя для катализатора, включающему следующие стадии:
a) получение псевдозоля, содержащего водорастворимое соединение элемента IV группы Периодической системы и гидроксид алюминия;
b) выдерживание псевдозоля в течение от 0,5 до 12 часов;
с) формование гранул предшественника носителя;
d) промывка полученных гранул предшественника носителя;
e) сушка и прокаливание промытых гранул предшественника носителя с получением носителя.
Получение псевдозоля можно проводить согласно любой известной из уровня техники методики. Неограничивающим примером такой методики может служить способ получения псевдозоля, описанный в источнике P. Ravindra et al. «Synthesis and characterization of millimetric gamma alumina spherical particles by oil drop granulation method», J. Porous. Mater. 19 (2012) 807-817.
После стадии получения, псевдозоль выдерживают на воздухе в течение от 0,5 до 12 часов, предпочтительно от 0,5 до 4 часов, более предпочтительно от 1 до 2 часов. Выдерживание проводят при комнатной температуре, где под комнатной температурой в соответствии с настоящим изобретением следует считать температуру (20±10)°С. Определение такой температуры, как комнатной известно, в частности, из ГОСТ 9454-78 (пункт 3.3), а также нормативного документа «Государственная фармакопея РФ XII издания часть 1» (применительно к температуре хранения лекарственных средств).
Подходящими для формования гранул предшественника носителя являются методики, описанные в следующих документах: «Synthesis and characterization of millimetric gamma alumina spherical particles by oil drop granulation method» P. Ravindra et al., J. Porous. Mater. 19 (2012) 807-817; «Parametric investigation of g-alumina granule preparation via the oil-drop route» H. Atashi et al., Adv. Powder Technol. 28 (2017) 1356-1371; Ismagilov, Z. R. New technology for production of spherical alumina supports for fluidized bed combustion/Z. R. Ismagilov, R. A. Shkrabina, N. A. Koryabkina // Catalysis Today. 1999. - Т. 47, № 1-4. С. 51-71; Preparation of Strong Alumina Supports for Fluidized Bed Catalysts. Volume 63/M. N. Shepeleva [и др.]. - Elsevier, 1991. - С. 583-590; Wang, Z.-M. Sol-Gel Synthesis of Pure and Copper Oxide Coated Mesoporous Alumina Granular Particles/Z.-M. Wang, Y. S. Lin // Journal of Catalysis. - 1998. - Т. 174, № 1. - С. 43-51; Yang, Z. Sol-Gel Synthesis of Silicalite/γ-Alumina Granules/Z. Yang, Y. S. Lin // Industrial & Engineering Chemistry Research. - 2000. - Т. 39, № 12. - С. 4944-4948; Buelna, G. Sol-gel-derived mesoporous γ-alumina granules/G. Buelna, Y. S. Lin // Microporous and Mesoporous Materials. - 1999. - Т. 30, № 2. - С. 359-369; Structural and Mechanical Properties of Nanostructured Granular Alumina Catalysts/G. Buelna [и др.] // Industrial & Engineering Chemistry Research. 2003. - Т. 42, № 3. - С. 442-447. Данные методики приводятся в качестве примера и не ограничивают настоящее изобретения.
Формованные гранулы предшественника носителя промывают органическим растворителем, предпочтительно спиртом, например, метиловым, этиловым и/или пропиловым спиртом, кетоном, например, ацетоном, простым эфиром, например, диметиловым, диэтиловым и/или метил-этиловыми эфирами, предпочтительно, формованные гранулы предшественника носителя промывают спиртом, наиболее предпочтительным спиртом является метанол. Промывание формованных гранул предшественника носителя проводят 2-5 раз, предпочтительно 3, в течение от 10 до 240 минут, предпочтительно от 30 до 120 минут, наиболее предпочтительно от 30 до 60 минут.
После промывки полученные гранулы предшественника носителя сушат и прокаливают любым известным из уровня техники способом, выбор которого остается за специалистом в данной области техники. Авторы настоящего изобретения рекомендуют проводить сушку при температуре 100°С в течение от 12 до 36 часов, предпочтительно в течение от 24 до 30 часов, прокаливание при температуре 1000°С в течение от 60 до 240 минут, предпочтительно от 100 до 110 минут, с использованием, например, ступенчатого или линейного нагрева печи со скоростью нагрева от 1 до 20°С в минуту, предпочтительно от 5 до 10°С в минуту.
В одном из вариантов изобретения носитель для катализатора получают следующим способом:
В стеклянную емкость с дистиллированной водой вносят заданное количество хлорида олова и перемешивают до полного растворения соли (концентрация хлорида олова - 3,0-3,3 мг/мл).
В раствор помещают гидроксид алюминия и добавляют при перемешивании раствор азотной кислоты с концентрацией 3,0-3,5 М в количестве достаточном для обеспечения массовой доли оксида алюминия в полученном псевдозоле от 25 до 35 мас.%, предпочтительно от 27 до 33 мас.%.
Приготовленный псевдозоль выдерживают на воздухе в течение 60-120 минут. В полученную пасту добавляют дистиллированную воду в количестве достаточном для обеспечения массовой доли оксида алюминия в полученной суспензии от 18 до 25 мас.%, предпочтительно от 20 до 24 мас.%, интенсивно перемешивают до получения однородной суспензии и выдерживают на воздухе в течение 60-120 минут.
Пластифицированную массу формуют методом капельной формовки над слоем углеводорода (предпочтительно н-ундекан) высотой 0,3-0,4 см, предпочтительно 0,3 см, предпочтительно в водном растворе аммиака с концентрацией 20-25 мас.% с необязательным добавлением ПАВ.
Формованные гранулы предшественника носителя трижды промывают метанолом методом рециркуляции в течение 30-60 минут.
Промытые гранулы предшественника носителя сушат при 100°С в течение 12-36 часов, предпочтительно 24-30 часов, с использованием ступенчатого или линейного нагрева печи в токе азота или на воздухе, предпочтительно в токе азота.
Просушенные гранулы предшественника носителя прокаливают при 1000°С в течение 60-120 минут, предпочтительно 100-110 минут с использованием ступенчатого или линейного нагрева печи.
Дополнительно композитный носитель для катализатора перед нанесением активного компонента может быть обработан кислотой. Например, обработку проводят соляной кислотой с концентрацией 0,01-3 М в течение 10-120 минут. После обработки кислотой носитель прокаливают в диапазоне температур 100-1000°С в течение 1-5 часов с применением линейного или ступенчатого нагревания.
В одном из предпочтительных вариантов осуществления носитель, получаемый способом согласно изобретению, используется при изготовлении катализатора.
В еще одном из предпочтительных вариантов осуществления носитель, получаемый способом согласно изобретению, используется при изготовлении катализатора дегидрирования газообразных углеводородов.
В еще одном из предпочтительных вариантов осуществления носитель, получаемый способом согласно изобретению, содержит от 0,2 до 0,6 мас.% элемента IV группы Периодической системы и характеризуется относительной потерей массы спустя 120 часов в режиме подвижного слоя не более 7,5%.
В одном своем аспекте настоящее изобретение относится к композитному носителю для катализатора, полученному способом согласно изобретению.
В одном своем аспекте настоящее изобретение относится к композитному носителю для катализатора дегидрирования газообразных углеводородов, включающему оксид алюминия и оксид элемента IV группы Периодической таблицы, содержащий от 0,2 до 0,6 мас.% элемента IV группы Периодической системы, характеризующемуся относительной потерей массы спустя 120 часов в режиме подвижного слоя не более 7,5%.
В качестве элемента IV группы Периодической системы могут быть использованы следующие элементы: олово, германий, свинец или их смеси. Предпочтительно используют олово. Помимо элементов IV группы Периодической системы в носителе согласно настоящему изобретению также могут присутствовать другие элементы, известные из уровня техники как улучшающие эксплуатационные характеристики катализатора элементы, например, галлий, индий, цинк, марганец или их смеси.
При получении носителя в качестве источника элемента IV группы Периодической системы используют его водорастворимое соединение. Например: бромид олова, хлорид олова (II), хлорид олова (IV), пентагидрат хлорида олова (IV), тетраэтоксид германия, хлорид германия (IV), нитрат свинца, ацетат свинца, хлорат свинца и т.д.
Содержание элемента IV группы Периодической системы в носителе составляет от 0,2 до 0,6 мас.%, предпочтительно от 0,25 до 0,55 мас.%, наиболее предпочтительно от 0,3 до 0,4 мас.%.
В еще одном своем аспекте настоящее изобретение относится к катализатору дегидрирования газообразных углеводородов
Катализатор согласно настоящему изобретению содержит в расчете на общую массу катализатора:
- от 0,1 до 4,5 мас.% элемента VIII группы Периодической системы;
- от 0,1 до 6,0 мас.% элемента I и/или II группы Периодической системы;
- от 0,1 до 6,0 мас.% галогена;
- носитель согласно настоящему изобретению.
В некоторых вариантах осуществления катализатор по изобретению содержит от 0,15 до 2 мас.%, предпочтительно от 0,15 до 0,8 мас.% элемента VIII группы Периодической системы в расчете на общую массу катализатора.
В некоторых вариантах осуществления содержание элемента I и/или II группы Периодической системы в катализаторе согласно изобретению может составлять от 0,2 до 3 мас.%, предпочтительно от 0,5 до 2 мас.%.
В некоторых вариантах осуществления содержание галогена в катализаторе согласно изобретению может составлять от 0,2 до 3 мас.%, предпочтительно от 0,2 до 2 мас.%.
Предпочтительно катализатор по изобретению содержит в расчете на общую массу катализатора:
- от 0,15 до 2 мас.% элемента VIII группы Периодической системы;
- от 0,2 до 3 мас.% элемента I и/или II группы Периодической системы;
- от 0,2 до 3 мас.% галогена;
- носитель - остальное, где носитель содержит оксид алюминия и от 0,25 до 0,55 мас.% элемента IV группы Периодической системы.
Наиболее предпочтительно катализатор по изобретению содержит в расчете на общую массу катализатора:
- от 0,15 до 0,8 мас.% элемента VIII группы Периодической системы;
- от 0,5 до 2 мас.% элемента I и/или II группы Периодической системы;
- от 0,2 до 2 мас.% галогена;
- носитель - остальное, где носитель содержит оксид алюминия и от 0,3 до 0,4 мас.% элемента IV группы Периодической системы.
Катализатор дегидрирования также может содержать иные компоненты, позволяющие дополнительно увеличить срок эксплуатации, активность или селективность катализатора дегидрирования. Примерами таких добавок служат, но не ограничиваются ими: рений, галлий, церий, лантан, европий, индий, фосфор, никель, железо, вольфрам, молибден, цинк, кадмий и другие. Каталитически эффективные количества этих компонентов, как правило, составляют от 0,01 до 5 мас.%, их можно вводить в катализатор любым подходящим способом во время или после получения катализатора, отдельно или в смеси.
В качестве элемента VIII группы Периодической системы могут быть использованы следующие элементы: платина, палладий, рутений, родий, иридий, осмий или их смеси. В катализаторе элемент VIII группы может присутствовать как соединение, такое как оксид, сульфид, галогенид, или оксигалогенид, в химическом соединении с одним или более других ингредиентов композита или как элементарный металл. Предпочтительным элементом VIII группы является платина.
В качестве элементов I и II группы Периодической системы могут быть использованы следующие элементы: кальций, калий, натрий, магний, литий, стронций, барий, бериллий или их смеси. Предпочтительным элементом является калий.
В качестве галогена используют элемент VII группы Периодической таблицы, такие как хлор, бром, предпочтительно используют хлор.
В еще одном своем аспекте настоящее изобретение относится к способу получения катализатора дегидрирования газообразных углеводородов включающему следующие стадии:
a) получение носителя способом согласно изобретению;
b) обработка носителя кислотой;
c) прокаливание обработанного кислотой носителя;
d) нанесение на носитель, полученный на стадии с) элемента VIII группы Периодической системы;
e) сушка и прокаливание предшественника катализатора, полученного на стадии d);
f) нанесение на предшественник катализатора, полученный на стадии e) элементов I и/или II группы Периодической системы;
g) сушка и прокаливание предшественника катализатора, полученного на стадии f);
h) нанесение на предшественник катализатора, полученный на стадии g) галогена; и
i) сушка и прокаливание предшественника катализатора, полученного на стадии h).
В некоторых вариантах осуществления обработку носителя проводят раствором кислоты.
В еще одном своем аспекте настоящее изобретение относится к способу получения катализатора дегидрирования газообразных углеводородов включающему следующие стадии:
a) получение носителя способом согласно изобретению;
b) нанесение на носитель, полученный на стадии а) элемента VIII группы Периодической системы;
c) сушка и прокаливание предшественника катализатора, полученного на стадии b);
d) нанесение на предшественник катализатора, полученный на стадии с) элементов I и/или II группы Периодической системы;
e) сушка и прокаливание предшественника катализатора, полученного на стадии d);
f) нанесение на предшественник катализатора, полученный на стадии e) галогена; и
g) сушка и прокаливание предшественника катализатора, полученного на стадии f).
Элемент VIII группы Периодической системы может быть нанесен на носитель любым известным из уровня техники способом, например: соосаждением, ионным обменом или пропиткой. Например, как указано в «Handbook of Heterogeneous Catalysis», Ed. G.Ertl, H.Knozinger, J.Weitkamp. - VCH A Wiley company, Vol.1, 2008, p. 469-484 Предпочтительный способ включает использование водорастворимого соединения элемента VIII группы для относительно равномерной пропитки носителя. В качестве водорастворимого соединения используют, например: хлороплатиновую, хлороиридивую, хлоропалладиевую, платинобромистоводородную кислоты, трихлорид платины, комплексы металлов платиновой группы, включающие: хлороплатинат аммония, гидрат тетрахлорида платины и т.д. Предпочтительно используют соединения платины, наиболее предпочтительно используют хлороплатиновую кислоту. Предпочтительно пропитывать носитель непосредственно после стадии прокалки для уменьшения риска вымывания активного компонента с поверхности.
Предпочтительно толщина слоя осаждения элемента VIII группы от внешней границы гранулы катализатора к ее центру составляет не менее 25% радиуса гранулы катализатора.
Элементы I и/или II группы Периодической системы и галоген могут быть нанесены пропиткой по влагоемкости, а также пропиткой из избытка растворителя, как это описано, например, в следующих документах: Studies in surface science and catalysis 1997. V.111, P. 191-198. Cristina L. Padro, Sergio R. de Miguel, Alberto A. Castro and Osvaldo A. Scelza, Fuel Processing Technology 109 (2013) 118-123, Farnaz Tahriri Zangeneh, Fuel Processing Technology 111 (2013) 94-104, Yiwei Zhang. Для полноты введения компонентов метод пропитки по влагоемкости является предпочтительным. Предпочтительно элементы I и/или II группы Периодической системы и галоген вводятся следующим образом:
В колбу роторного испарителя приливают растворы водорастворимых соединений элементов I и/или II групп Периодической системы и галогена, предпочтительно используют водорастворимое соединение, которое одновременно содержит элементы I и/или II групп и галогена, например, хлорид калия. Образец носителя с нанесенным активным компонентом (элементом VIII группы Периодической системы) пересыпают в колбу и встряхивают в течение 1-5 минут.
Колбу подсоединяют к роторному испарителю и перемешивают содержимое в течение 1-2 часов со скоростью вращения 20 об/мин.
Пропитанные гранулы сушат на воздухе при 100°С в течение 3-10 часов с использованием ступенчатого или линейного нагрева печи.
Просушенные гранулы прокаливают при 550°С в течение 2-4 часов с использованием ступенчатого или линейного нагрева печи.
Получают катализатор по существу сферической формы со средним диаметром от приблизительно 0,2 до приблизительно 10 мм, предпочтительно от приблизительно 0,5 до приблизительно 5 мм, наиболее предпочтительно от приблизительно 1 до приблизительно 3 мм. Термин "по существу сферический" означает, что большинство частиц катализатора имеют сферическую форму с незначительными отклонениями, где под «большинством» подразумевают по меньшей мере более 80% частиц, предпочтительно более 90% частиц, более предпочтительно более 95% частиц.
В еще одном своем аспекте настоящее изобретение относится к способу дегидрирования газообразных углеводородов в режиме подвижного слоя с использованием катализатора согласно настоящему изобретению.
В предпочтительном варианте осуществления способ дегидрирования предусматривает дегидрирование газообразных (С2-С4) углеводородов при температуре от 580 до 650°С, предпочтительно 630°С. В еще одном предпочтительном варианте осуществления способа дегидрирования согласно настоящему изобретению отношение компонентов реакционной смеси Н2:С2-С4 составляет от 0,2 до 0,8, предпочтительно 0,5. Давление в реакторе может составлять от 1 до 8 атм, предпочтительно от 1 до 6 атм. В еще одном предпочтительном варианте осуществления способ дегидрирования согласно настоящему изобретению предусматривает использование катализатора дегидрирования более одного цикла.
В еще одном предпочтительном варианте осуществления способ дегидрирования согласно настоящему изобретению предусматривает периодическое проведение стадии оксихлорирования катализатора. Оксихлорирование проводят согласно известным из уровня техники методикам. Частными примерами оксихлорирования являются, но не ограничиваются ими способы, описанные в следующих документах: US5880050, CN105797788, CN107866237, WO2017151361. Предпочтительно оксихлорирование проводят при температуре от 500 до 550°C, более предпочтительно от 510 до 540°C, наиболее предпочтительно от 520 до 530°C, в токе кислород-хлор-аргоновой (азотной) смеси. Предпочтительно концентрация хлора составляет от 0,7 до 1,1 об.%, более предпочтительно от 0,9 до 1,0 об.%. Предпочтительно концентрация кислорода составляет от 1,2 до 1,8 об.%, более предпочтительно от 1,2 до 1,4 об. %. Объемный расход смеси и продолжительность процедуры оксихлорирования выбираются таким образом, чтобы отношение массы поданного хлора к массе обрабатываемого катализатора составляла от 0,005 до 0,01, предпочтительно от 0,006 до 0,009, более предпочтительно от 0,007 до 0,008 при обработке неподвижного катализатора, либо с обеспечением подачи хлора на уровне от 0,5 до 1 мас.% от кратности циркуляции катализатора при обработке движущегося катализатора, предпочтительно от 0,6 до 0,9 мас.%, более предпочтительно от 0,7 до 0,08 мас.%.
Осуществление изобретения
Данное изобретение более конкретно описано в приведенных ниже примерах, которые приведены исключительно для иллюстрации настоящего изобретения и не ограничивают его.
Пример 1. Сравнительный: Получение катализатора А (состав:0,3% Pt - 0,2% Sn - 1,0% K - 1,0% Cl/Al2O3).
Олово наносили на носитель, представляющий собой оксид алюминия, методом пропитки из раствора хлорида олова (II) SnCl2 в 1М хлористоводородной кислоте HCl. 10 грамм носителя (Ɵ-Al2O3) пропитывали раствором, содержащим 5 мл SnCl2 с концентрацией олова 4.10 мг/мл и 2.8 мл дистиллированной воды. Влажные гранулы выдерживали на воздухе при комнатной температуре в течение ночи, сушили в сушильном шкафу при 60°C в течение 1 часа и при 120°C в течение 2 часов. Гранулы прокаливали при 950°C в течение 2 часов. 10,0 г высушенного и прокаленного модифицированного носителя, содержащего оксид алюминия и оксид олова, пропитывали раствором, содержащим 0,965 мл раствора платинохлористоводородной кислоты H2PtCl6 с концентрацией Pt 31,87 мг/мл и 6,84 мл дистиллированной воды. Влажные гранулы выдерживали на воздухе при комнатной температуре в течение 2 часов, сушили в сушильном шкафу при 60°C в течение 1 часа и при 120°С в течение 2 часов. Pt-содержащий катализатор помещали в реактор и пропитывали раствором, содержащим 4,1 мл раствора хлорида калия KCl с концентрацией K равной 25,0 мл/мл в 3,7 мл дистиллированной воды. Влажные гранулы выдерживали на воздухе при комнатной температуре в течение 2 часов, затем в сушильном шкафу при 60°C в течение 1 часа и при 120°C в течение 2 часов. Гранулы прокаливали в муфельной печи при 550°C в течение 2 часов. Подъем температуры до заданных значений осуществляли со скоростью 10°C/мин.
Полученный катализатор восстанавливали следующим способом: систему продували аргоном со скоростью 50 см3/мин в течение 20 мин, затем в реактор вместо аргона подавали водород со скоростью 50 см3/мин. Реактор в токе водорода нагрели со скоростью 15°C/мин до 550°C и выдерживали при этой температуре в течение 2 часов.
Пример 2: Получение катализатора B (состав 0,3% Pt - 1,0% K - 1,0% Cl/ Sn(0,4%)-Al2O3).
Композитный носитель был получен методом смешения растворимого соединения олова SnCl2·2H2O с водной суспензией гидроксида алюминия (высокодисперсного псевдобемита). В водный раствор 0,44 г хлорида олова (марка Sigma Aldrich, 98% CAS 10025-69-1), содержащий 0,002 моль ионов олова добавляли гидроксид алюминия (марка Pural SB, «Sasol», Германия) массой 21,6 г, содержащем 0,277 моль ионов алюминия, соответствующем количеству ионов олова в растворе и необходимом для получения конечного носителя с расчетным содержанием олова 0,4% мас..При этом раствор интенсивно перемешивали. При постоянном перемешивании во взвесь добавляли водный раствор азотной кислоты с концентрацией 3,1 моль/л в количестве, достаточном для обеспечения массовой доли оксида алюминия в полученном псевдозоле 30 мас.%. для получения однородной суспензии, которую перемешивали в течение 15 минут. Приготовленный псевдозоль выдерживали на воздухе в течение 30 минут при комнатной температуре (20оС). В полученную гелеобразную пасту добавляли дистиллированную воду в количестве достаточном для обеспечения массовой доли оксида алюминия в полученной суспензии 22 мас.%, и интенсивно перемешивали до получения однородной суспензии. Формование гранул носителя проводили методом капельного формования: полученную суспензию через фильеры заданного размера прикапывали в 20% водный раствор аммиака через слой углеводорода (ундекана), толщина которого составляла 0,3 см. Полученные гранулы сушили и прокаливали поэтапно: 10 ч при 60ºC, 1 ч при 100°C, 1 ч при 400°C и 1 час при 1050ºC при скорости нагрева 15°C/мин для получения в составе носителя оксида алюминия тета-модификации. Нанесение платины, калия, хлора и последующее восстановление катализатора проводили по той же методике, что и в Примере 1.
Пример 3: Получение катализатора C (состав 0,3% Pt - 1,0% K - 1,0% Cl/ Sn(0.4%)-Al2O3).
Катализатор C отличается от катализатора Примера 2 тем, что композитный носитель был обработан водным раствором соляной кислоты с концентрацией 0,5-1 моль/л и прокален при 950°C, как и в случае с катализатором Примера 1, после стадии нанесения олова. Нанесение платины, калия, хлора и последующее восстановление катализатора проводили по той же методике, что и в Примере 1.
Примеры 4, 5 иллюстрируют применение полученных катализаторов в реакции дегидрирования пропана (ДГП).
Пример 4: Испытание катализаторов дегидрирования пропана.
Полученные катализаторы A, B и C исследовали в реакции дегидрирования пропана. Выполнение исследований по сравнительному тестированию катализаторов в реакции дегидрирования пропана проводили в соответствии со следующей методикой. На начальном этапе реактор нагревали со скоростью 20°C/мин до 110°C и выдерживали при этой температуре в течение 1 часа. Катализатор восстанавливали в токе водорода при нагревании со скоростью 15°C/мин до 650°C и выдерживали при этой температуре в течение 2 часов. Реакцию дегидрирования пропана проводили при 650°C в течение 2 часов при следующем режиме подачи газов: C3H8-30 см3/мин, H2-6,0 см3/мин. Результаты каталитической активности катализаторов в дегидрировании пропана представлены на Фиг. 1 в виде конверсии пропана и селективности по пропилену.
Конверсия пропана на всех катализаторах в первом цикле составляет 60-63% и не падает с течением времени. Селективность по пропилену совпадает для катализатора А и катализатора С и составляет после 20 минут эксперимента около 87-88%, тогда как в случае катализатора B селективность несколько ниже и достигает 85%. Можно сказать, что метод введения олова в носитель не влияет на каталитическую активность катализатора, основным определяющим фактором является поверхностное содержание олова, которое для катализаторов одинаково.
Пример 5: Испытание катализаторов дегидрирования пропана после регенерации в двух циклах дегидрирования пропана.
Полученные катализаторы A, B и C исследовали в реакции дегидрирования пропана после регенерации катализатора (Фиг. 1). Для каждого образца предусматривается 3 последовательных цикла испытаний, включающих стадии восстановления нанесенного металла, непосредственно процесса дегидрирования пропана и регенерации поверхности катализатора. Стадии сушки, восстановления, реакции дегидрирования совпадали с описанными стадиями в примере 4. Катализатор охлаждали до температуры 480°C в токе аргона, после чего проводили выжигание кокса. В ток аргона (60,5 см3/мин) вводили 1 об.% кислорода и нагревали реактор со скоростью 10°C/мин до 650°С. Удаление кокса проводили в течение 4 часов. Полученный регенерированный катализатор продували инертным газом. Второй и третий цикл дегидрирования пропана проводили таким же образом. В третьем цикле отсутствовали стадии регенерации и продувки катализатора инертным газом.
Во втором цикле селективность по пропилену не меняется, сохраняется такая же тенденция, как и в первом цикле проведения реакции дегидрирования пропана. В третьем цикле все катализаторы проявляют одинаковую селективность в пределах 87-90% (Фиг. 1). Зависимость конверсии пропана от катализатора более сложная. Во втором и третьем цикле в ходе реакции наблюдается снижение конверсии пропана на всех катализаторах, однако для катализатора, полученного на композитном носителе по изобретению, снижение конверсии более медленное, катализатор остается активным дольше.
Важной характеристикой катализаторов является их склонность к коксобразованию. Этот показатель оценивается из количества углерода, выжигаемого с поверхности катализатора после проведения процесса дегидрирования пропана в ходе стадии отжига. В таблице 1 представлены данные для образцов А, В и С. Содержание кокса в образцах, синтезированных на основе носителя с внедренным оловом, с учетом погрешности измерений сопоставимо, хотя для катализатора В после первого цикла испытаний это значение немного выше, чем для других. Тем не менее, весьма существенна разница между образцами В и С, полученных на композитном носителе, и образцом А, полученном на алюмоксидном носителе: после первого цикла содержание кокса в последнем в 1,8-3 раза выше, чем в катализаторах, полученных с использованием композитных носителей.
Таблица 1. Содержание углерода в катализаторах А, В и С, образованного в ходе трех циклов испытаний в ДГП.
Исходя из вышесказанного, можно заключить, что использование в качестве носителя композитного шарика с внедренным оловом позволяет получать катализатор дегидрирования пропана, по своей устойчивости к коксообразованию превосходящий катализатор на основе носителя с нанесенным оловом.
Пример 6: Получение катализатора D (состав 0,34% Pt - 1,0% K - 1,0% Cl/ Sn(0,3%)-Al2O3).
Катализатор D получали согласно методике получения катализатора по Примеру 2 на основе носителя Al2O3 с содержанием внедренного олова 0,3 мас.%. Композитный носитель был получен методом смешения растворимого соединения олова SnCl2·2H2O с водной суспензией гидроксида алюминия (высокодисперсного псевдобемита). В водный раствор хлорида олова (марка Sigma Aldrich, 98% CAS 10025-69-1), содержащий заданное количество ионов олова добавляли гидроксид алюминия (марка Pural SB, «Sasol», Германия) в количестве соответствующем количеству ионов олова в растворе и необходимом для получения конечного носителя с заданным содержанием олова. При этом раствор интенсивно перемешивали. При постоянном перемешивании во взвесь добавляли водный раствор азотной кислоты с концентрацией 3,1 моль/л для получения однородной суспензии, которую перемешивали в течение 15 минут. Приготовленный псевдозоль выдерживали на воздухе в течение 30 минут. В полученную гелеобразную пасту добавляли дистиллированную воду и интенсивно перемешивали до получения однородной суспензии. Формование гранул носителя проводили методом капельного формования: полученную суспензию через фильеры заданного размера прикапывали в 20%-ный водный раствор аммиака через слой углеводорода (ундекана), толщина которого составляла 0,3 см. Полученные гранулы сушили и прокаливали поэтапно: 10 часов при 60ºC, 1 час при 100ºC, 1 час при 400ºC и 1 час при 1050ºC при скорости нагрева 15ºC/мин для получения в составе носителя оксида алюминия тета-модификации. Нанесение платины, калия, хлора и последующее восстановление катализатора проводили по той же методике, что и в Примере 1, за исключением количества платинохлористоводородной кислоты H2PtCl6: 10,0 г высушенного и прокаленного композитного носителя, содержащего оксид алюминия и оксид олова пропитывали раствором, содержащим 1,092 мл раствора платинохлористоводородной кислоты с концентрацией Pt 31,87 мг/мл и 5,91 мл дистиллированной воды.
Пример 7: Получение катализатора E (состав 0,34% Pt - 1,0% K - 1,0% Cl/ Sn(0,4%)-Al2O3).
Катализатор Е получен согласно методике получения катализатора Примера 6 на основе носителя Al2O3 с содержанием внедренного олова 0,4 мас.%. Композитный носитель был получен методом смешения растворимого соединения олова SnCl2·2H2O с водной суспензией гидроксида алюминия (высокодисперсного псевдобемита). В водный раствор хлорида олова (марка Sigma Aldrich, 98% CAS 10025-69-1), содержащий заданное количество ионов олова добавляли гидроксид алюминия (марка Pural SB, «Sasol», Германия) в количестве соответствующем количеству ионов олова в растворе и необходимом для получения конечного носителя с заданным содержанием олова. При этом раствор интенсивно перемешивали. При постоянном перемешивании во взвесь добавляли водный раствор азотной кислоты с концентрацией 3,1 моль/л для получения однородной суспензии, которую перемешивали в течение 15 минут. Приготовленный псевдозоль выдерживали на воздухе в течение 30 минут. В полученную гелеобразную пасту добавляли дистиллированную воду и интенсивно перемешивали до получения однородной суспензии. Формование гранул носителя проводили методом капельного формования: полученную суспензию через фильеры заданного размера прикапывали в 20-% водный раствор аммиака через слой углеводорода (ундекана), толщина которого составляла 0,3 см. Полученные гранулы сушили и прокаливали поэтапно: 10 часов при 60ºC, 1 час при 100ºC, 1 час при 400ºC и 1 час при 1050ºC при скорости нагрева 15ºC/мин для получения в составе носителя оксида алюминия тета-модификации. Нанесение платины, калия, хлора и последующее восстановление катализатора проводили по той же методике, что и в Примере 1, за исключением количества платинохлористоводородной кислоты H2PtCl6 10,0 г высушенного и прокаленного композитного носителя, содержащего оксид алюминия и оксид олова пропитывали раствором, содержащим 1,092 мл раствора платинохлористоводородной кислоты с концентрацией Pt 31,87 мг/мл и 5,91 мл дистиллированной воды.
Пример 8: Получение катализатора G (состав 0,34% Pt - 1,0% K - 1,0% Cl/ Sn(0,5%)-Al2O3).
Композитный носитель был получен методом смешения растворимого соединения олова SnCl2·2H2O с водной суспензией гидроксида алюминия (высокодисперсного псевдобемита). В водный раствор хлорида олова (марка Sigma Aldrich, 98% CAS 10025-69-1), содержащий заданное количество ионов олова добавляли гидроксид алюминия (марка Pural SB, «Sasol», Германия) в количестве соответствующем количеству ионов олова в растворе и необходимом для получения конечного носителя с заданным содержанием олова. При этом раствор интенсивно перемешивали. При постоянном перемешивании во взвесь добавляли водный раствор азотной кислоты с концентрацией 3,1 моль/л для получения однородной суспензии, которую перемешивали в течение 15 минут. Приготовленный псевдозоль выдерживали на воздухе в течение 30 минут. В полученную гелеобразную пасту добавляли дистиллированную воду и интенсивно перемешивали до получения однородной суспензии.. Формование гранул носителя проводили методом капельного формования: полученную суспензию через фильеры заданного размера прикапывали в 20% водный раствор аммиака через слой углеводорода (ундекана), толщина которого составляла 0,3 см. Полученные гранулы сушили и прокаливали поэтапно: 10 часов при 60°C, 1 час при 100°C, 1 час при 400°C и 1 час при 1050ºC при скорости нагрева 15°C/мин для получения в составе носителя оксида алюминия тета-модификации.
Катализатор G получен согласно методике получения катализатора Примера 6 на основе носителя Al2O3 с содержанием внедренного олова 0,5 мас.%.
Пример 9 иллюстрирует применение полученных катализаторов D, E и G в реакции дегидрирования пропана с применением стадии оксихлорирования.
Пример 9: Испытание катализаторов дегидрирования пропана в ходе 3-10 циклов с использованием стадии оксихлорирования.
Исследование каталитических свойств представленных катализаторов D, E и G в реакции дегидрирования пропана проводили в проточном кварцевом реакторе с неподвижным слоем катализатора массой 1,6-3 г при удельном расходе пропана на единицу массы катализатора от 1,1 л⋅г(кат)-1⋅ч-1 до 1,7 л⋅г(кат)-1⋅ч-1 и отношении H2: С2-С4 в составе исходной сырьевой смеси от 0,2 до 0,7. Тестирование проводили в циклическом режиме с последовательным чередованием следующих стадий:
1. Сушка катализаторов при 110-240°C 1,0-1,5 часов в токе азота или водорода с объемной скоростью подачи 1,8-11,5 л/ч;
2. Восстановление катализаторов в течение 1-3 часов при 550-650°C в токе водорода 1,8-3 л/ч;
3. Проведение реакции дегидрирования при 590-650°C в течение 2-4 часов в токе сырьевой смеси H2 с углеводородом C2-C4 2-9 л/час;
4. Выжигание кокса при 550-650°C в течение 2-4 часов в токе аргон/азот-воздушной смеси с расходом 3,8-30 л/ч с содержанием кислорода в составе смеси 0,5-2 об.%;
5. Оксихлорирование в течение 1-3 часов при 500-550°C в токе кислород-хлор-аргоновой (азотной) смеси 5-6 л/ч с содержанием хлора и кислорода в составе смеси 0,5-1 и 1-2 об.% соответственно;
6. Последовательное повторение стадий 2-5 для реализации последующих циклов;
7. Охлаждение катализаторов до комнатной температуры в токе аргона (азота) 5-12 л/ч.
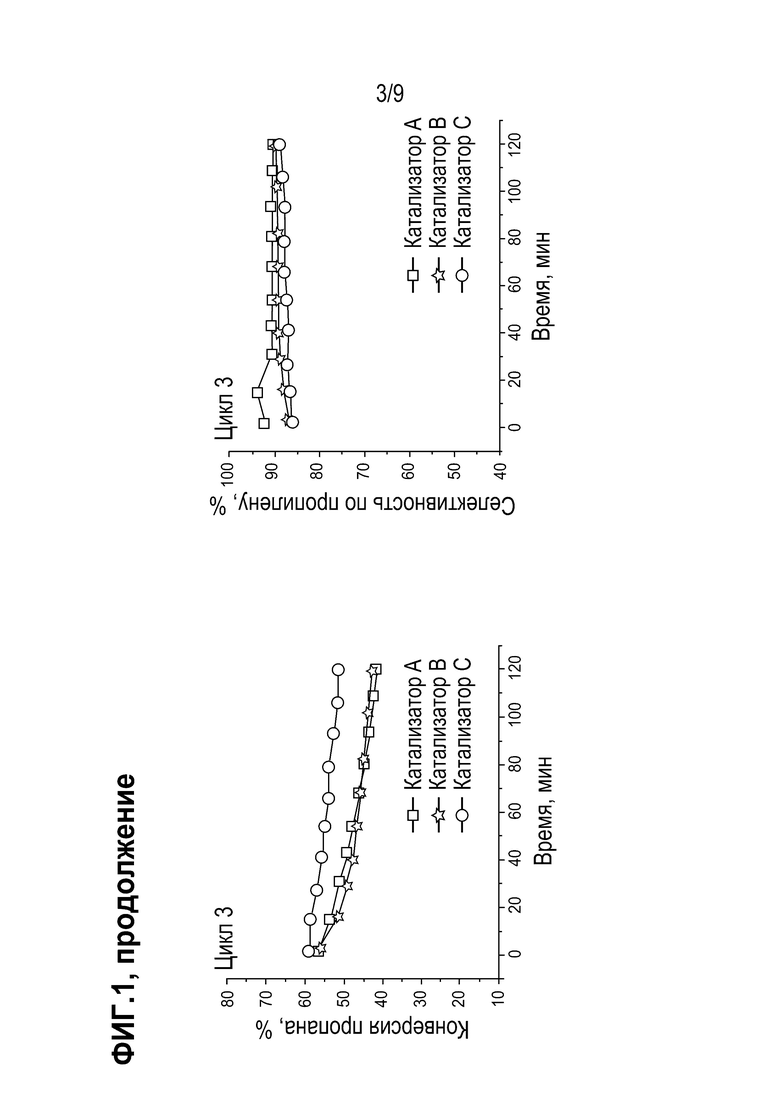
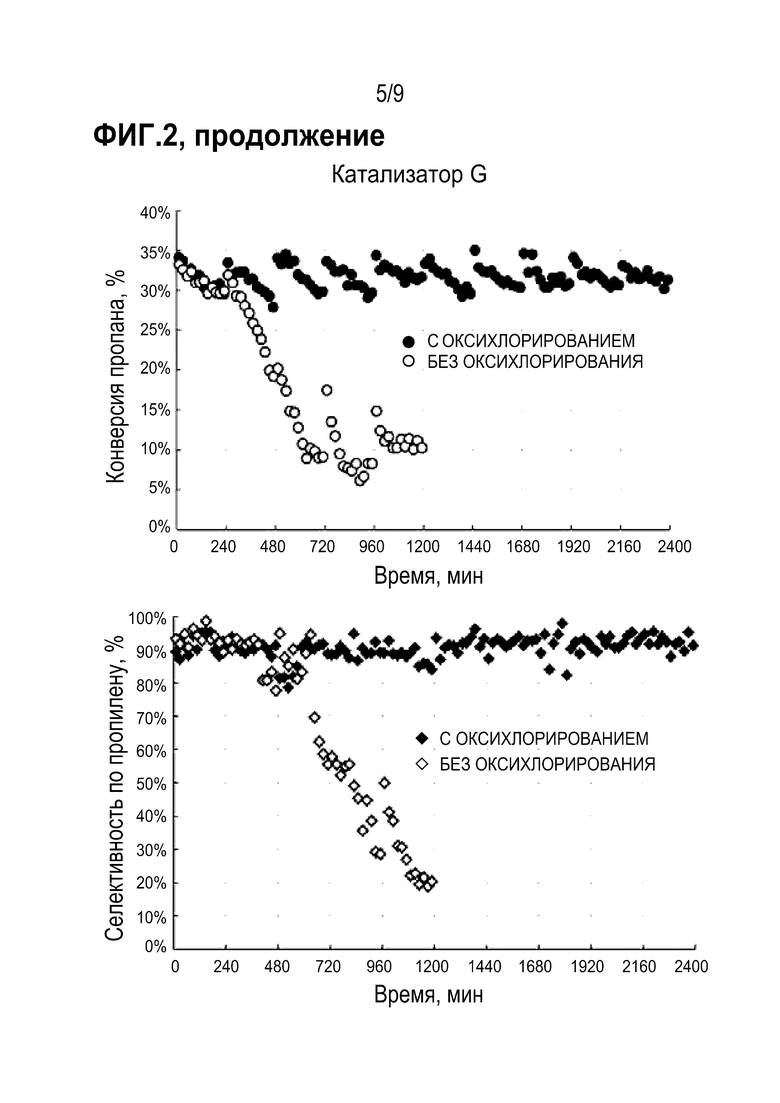
Результаты испытания катализаторов в реакции дегидрирования пропана (конверсия пропана и селективность образования пропилена) представлены на Фиг 2.
Из приведенных данных видно, что при проведении испытаний со стадией оксихлорирования образцы катализаторов D и E демонстрируют стабильные значения конверсии пропана в течение десяти циклов. При этом катализаторы D и Е более стабильны по сравнению с катализатором А, для которого наблюдается заметное снижение конверсии пропана в ходе каждого цикла с восстановлением исходных показателей после регенерации.
Пример 10: Испытание устойчивости к истиранию сферических алюмооксидных и оловянно-алюмооксидных носителей катализаторов дегидрирования пропана.
Образцы сферических носителей: алюмооксидного носителя, полученного согласно описанному в WO2010069548 варианту 2, и композитного оловянно-алюмооксидного с содержанием олова 0,3-0,5 мас.% испытывают на устойчивость к истиранию в движущемся слое с использованием установки, включающей в себя стеклянную трубку 1 переменного сечения, абсорбер 2 и регулятор расхода газа (3). Схема установки и чертеж стеклянной трубки представлены на Фиг 3.
Стеклянная трубка включает в себя следующие отделы:
Ввод азота через патрубок и его распределение на сетчатом пыже 4 (размер ячейки 1 мм);
Рабочая часть с внутренним диаметром 18 мм и длиной 900 мм;
Коническое расширение до внутреннего диаметра 46 мм для улавливания сферического носителя и предотвращения его уноса из трубки.
Дополнительно на выходе из трубки установлен сетчатый пыж 5 с размером ячейки 1 мм.
Подачу азота осуществляют в нижнюю часть трубки через регулятор расхода газа с диапазоном подачи 200-1600 л/час. На выходе из трубки газ проходит через барботер, заполненный водой, для улавливания частиц пыли.
Испытание устойчивости к истиранию носителей проводили в соответствии с представленной ниже процедурой:
1. Испытуемый образец массой 50 г прокаливали в муфельной печи при температуре 370°С в течение 3-х часов для удаления сорбированной влаги.
2. После остывания в печи до температуры 150°С образец помещали в эксикатор для остывания до комнатной температуры.
3. Остывший образец взвешивали с точностью до 0,01 г.
4. Помещали испытуемый образец в стеклянную трубку 1. Для этого в верхней части стеклянной трубки отсоединяли трубку отвода азота, извлекали сетчатый пыж 5 и засыпали образец в трубку через воронку.
5. Устанавливали пыж 5 и отводящую трубку на место.
6. Устанавливали расход азота 1000-1200 л/час на регуляторе расхода газа. Убеждались в том, что сферы испытуемого образца начали двигаться порциями вверх по трубке с постепенным осыпанием вниз. Также контролировали, чтобы не происходил проскок сфер через коническую часть трубки. При необходимости следует изменить расход газа на входе в трубку.
7. Проводили испытание в течение установленного промежутка времени: от 20 до 120 часов. Продолжительность испытания может быть изменена исходя из свойств конкретного образца.
8. По истечении времени испытания прекращали подачу азота в трубку.
9. Отсоединяли трубку ввода газа в нижней части стеклянной трубки 1, извлекали резиновый уплотнитель с политетрафторэтиленовой лентой.
10. Подставляли под нижний конец трубки большой полиэтиленовый пакет (гриппер) и извлекали сетчатый пыж 4 пинцетом с тем, чтобы испытанный носитель высыпался в пакет. При этом важно не потерять ни одной сферы испытуемого образца.
11. Убирали пакет с собранным материалом.
12. При необходимости сбора пыли для анализа к нижнему концу трубки прикрепляли скотчем маленький гриппер и постукиваниями по верхней части трубки ссыпают пыль в пакет. При необходимости можно продуть трубку сверху вниз азотом.
13. Отсоединяли гриппер с собранным образцом пыли.
14. Продували трубку азотом для удаления остатков пыли. При этом для удаления частиц пыли, осевшей на стенках в расширенной части трубки следует использовать ершик. Рекомендуемый расход азота для продувки - 1200 л/час, продолжительность продувки составляет порядка 5 минут.
15. Извлеченный образец прокаливали в муфельной печи при температуре 370°С в течение трех часов для удаления сорбированной влаги.
16. После остывания в печи до температуры 150°С образец помещали в эксикатор для остывания до комнатной температуры.
17. Остывший образец взвешивали с точностью 0,01 г.
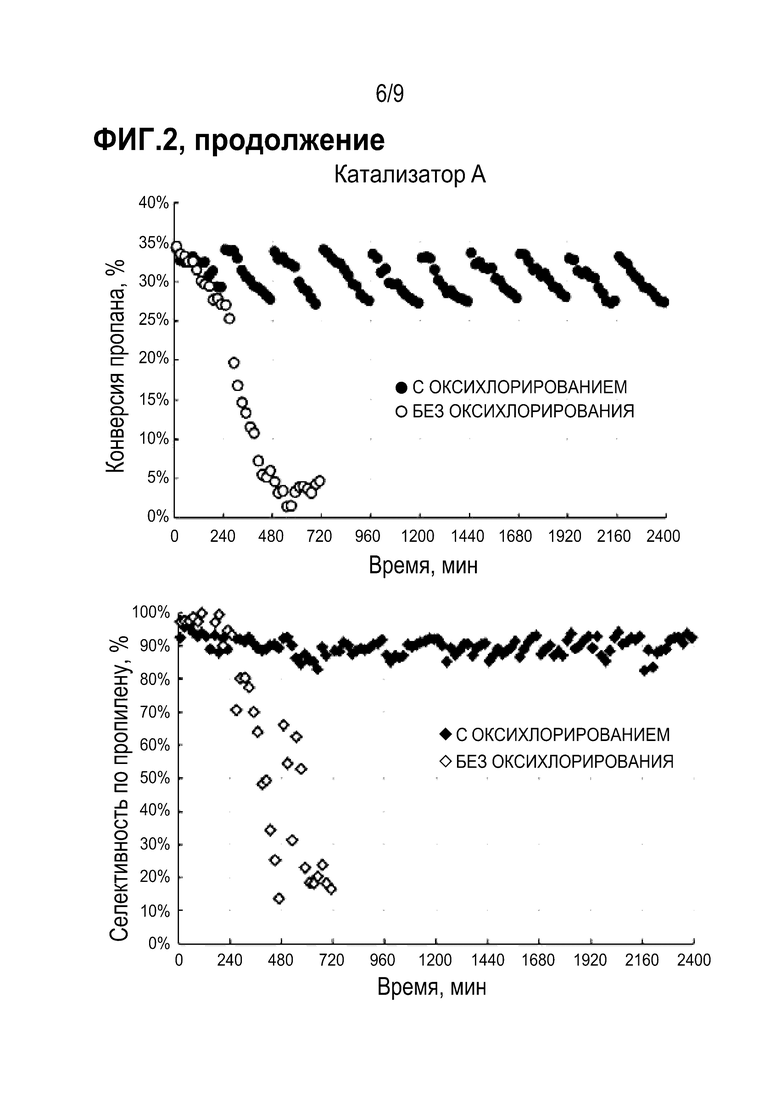
Относительная потеря массы исследованного образца рассчитывается по формуле:
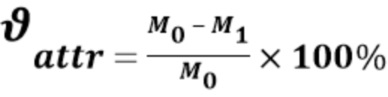 (1),
(1),
где  - относительная потеря массы,
- относительная потеря массы,
M 0 - исходная масса образца,
M 1 - конечная масса образца,
Результаты сравнительного испытания устойчивости к истиранию носителей представлены на Фиг 4, результаты испытания устойчивости к истиранию носителей в течение 120 часов представлены в Таблице 2 .
Таблица 2. Значение относительной потери массы носителя при истирании в течение 120 часов.
Из результатов проведенных испытаний видно, что алюмооксидный сферический носитель характеризуется на 0,5-1% большей потерей массы образца, чем образцы сферического композитного оловянно-алюмооксидного носителя, при одинаковом времени истирания. Наибольшей потерей массы обладает носитель, полученный согласно варианту 2 WO2010069548. При этом данный результат достигается во всем заявленном диапазоне содержания олова. С увеличением содержания олова истираемость возрастает лишь незначительно при соблюдении заявленных пределов.
Таким образом, предлагаемый катализатор на основе заявляемого носителя, обладает высокой активностью и селективностью, хорошей устойчивостью к истиранию.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРА ДЛЯ ДЕГИДРИРОВАНИЯ УГЛЕВОДОРОДОВ | 1994 |
|
RU2132731C1 |
| Катализатор риформинга бензиновых фракций и способ его получения | 2021 |
|
RU2767882C1 |
| СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРОВ ДЛЯ ДЕГИДРИРОВАНИЯ УГЛЕВОДОРОДОВ | 1994 |
|
RU2128551C1 |
| Катализаторы на основе металлов платиновой группы на носителях из оксида алюминия | 2023 |
|
RU2823764C1 |
| КАТАЛИЗАТОР И СПОСОБ | 2010 |
|
RU2565757C2 |
| КАТАЛИТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРЕВРАЩЕНИЯ АЛКАНОВ В АЛКЕНЫ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2019 |
|
RU2705574C1 |
| ВЫСОКОАКТИВНЫЙ КАТАЛИЗАТОР ДЕГИДРИРОВАНИЯ АЛКАНОВ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2019 |
|
RU2724902C1 |
| КОМПОЗИЦИЯ КАТАЛИЗАТОРА ДЕГИДРИРОВАНИЯ | 2020 |
|
RU2809169C2 |
| ЦЕОЛИТНЫЙ КАТАЛИЗАТОР И СПОСОБ ДЕГИДРИРОВАНИЯ ПРОПАНА С ЕГО ИСПОЛЬЗОВАНИЕМ | 2024 |
|
RU2840849C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОЛЕФИНОВЫХ УГЛЕВОДОРОДОВ | 1997 |
|
RU2127242C1 |



Изобретение относится к области каталитической химии. Описан способ получения композитного носителя для катализатора, содержащего элемент IV группы Периодической системы в количестве от 0,2 до 0,6 мас.%, включающий следующие стадии: получение псевдозоля, содержащего водорастворимое соединение элемента IV группы Периодической системы и гидроксид алюминия; выдерживание псевдозоля в течение от 0,5 до 12 часов; формование гранул предшественника носителя; промывку гранул предшественника носителя; сушку и прокаливание промытых гранул предшественника носителя с получением носителя. Описаны композитные носители для катализатора дегидрирования газообразных углеводородов, полученные описанным выше способом. Описан катализатор дегидрирования газообразных углеводородов содержащий: от 0,1 до 4,5 мас.% элемента VIII группы Периодической системы; от 0,1 до 6,0 мас.% элемента I и/или II группы Периодической системы; от 0,1 до 6,0 мас.% галогена; носитель, полученный описанным выше способом. Описан способ получения катализатора дегидрирования газообразных углеводородов, включающий стадии: a) получение носителя описанным выше способом, b) обработку носителя кислотой; c) прокаливание обработанного кислотой носителя; d) нанесение на носитель, полученный на стадии с), элемента VIII группы Периодической системы; e) сушку и прокаливание предшественника катализатора, полученного на стадии d); f) нанесение на предшественник катализатора, полученный на стадии e), элементов I и/или II группы Периодической системы; g) сушку и прокаливание предшественника катализатора, полученного на стадии f), h) нанесение на предшественник катализатора, полученный на стадии g), галогена; i) сушкуи прокаливание предшественника катализатора, полученного на стадии h). Описан способ получения катализатора дегидрирования газообразных углеводородов, включающий следующие стадии: a) получение носителя описанным выше способом; b) нанесение на носитель, полученный на стадии а), элемента VIII группы Периодической системы; c) сушку и прокаливание предшественника катализатора, полученного на стадии b); d) нанесение на предшественник катализатора, полученный на стадии с), элементов I и/или II группы Периодической системы; e) сушку и прокаливание предшественника катализатора, полученного на стадии d); f) нанесение на предшественник катализатора, полученный на стадии e), галогена; g) сушку и прокаливание предшественника катализатора, полученного на стадии f). Описан способ дегидрирования газообразных углеводородов в режиме подвижного слоя с использованием описанношго выше катализатора. Технический результат - увеличение срока эксплуатации катализатора дегидрирования газообразных углеводородов с сохранением эффективности и активности катализатора во времени, сокращение числа производственных стадий получения активного и стабильного катализатора дегидрирования газообразных углеводородов. 7 н. и 39 з.п. ф-лы, 5 ил., 2 табл., 10 пр.
1. Способ получения композитного носителя для катализатора, содержащего элемент IV группы Периодической системы в количестве от 0,2 до 0,6 мас.%, включающий следующие стадии:
- получение псевдозоля, содержащего водорастворимое соединение элемента IV группы Периодической системы и гидроксид алюминия;
- выдерживание псевдозоля в течение от 0,5 до 12 часов;
- формование гранул предшественника носителя;
- промывка гранул предшественника носителя;
- сушка и прокаливание промытых гранул предшественника носителя с получением носителя.
2. Способ по п.1, в котором выдерживание псевдозоля проводят в течение от 0,5 до 4 часов.
3. Способ по п.2, в котором выдерживание псевдозоля проводят в течение от 1 до 2 часов.
4. Способ по п.1, в котором выдерживание псевдозоля проводят при комнатной температуре.
5. Способ по п.1, в котором формование гранул предшественника носителя проводят методом капельной формовки.
6. Способ по п.1, в котором формование гранул предшественника носителя проводят в среде углеводорода.
7. Способ по п.6, в котором формование гранул предшественника носителя проводят в среде ундекана.
8. Способ по п.1, в котором гранулы предшественника носителя промывают органическим растворителем.
9. Способ по п.8, в котором в качестве органического растворителя используют спирт, простой эфир и/или кетон.
10. Способ по п.9, в котором в качестве органического растворителя используют метиловый, этиловый и/или пропиловый спирт.
11. Способ по п.10, в котором гранулы предшественника носителя промывают метанолом.
12. Способ по п.1, в котором носитель сушат при температуре 100°С в течение от 12 до 36 часов.
13. Способ по п.12, в котором носитель сушат при температуре 100°С в течение от 24 до 30 часов.
14. Способ по п.1, в котором носитель прокаливают при температуре 1000°С в течение 60-240 минут.
15. Способ по п.14, в котором носитель прокаливают при температуре 1000°С в течение 100-110 минут.
16. Способ по п.1, в котором носитель предназначен для катализатора дегидрирования углеводородов.
17. Способ по п.1, в котором носитель содержит от 0,2 до 0,6 мас.% элемента IV группы Периодической системы и характеризуется относительной потерей массы спустя 120 часов в режиме подвижного слоя не более 7,5%.
18. Композитный носитель для катализатора дегидрирования газообразных углеводородов, полученный способом по любому из пп. 1-17, включающий оксид алюминия и оксид элемента IV группы Периодической таблицы, при этом содержание элемента IV группы Периодической системы составляет от 0,2 до 0,6 мас.%, отличающийся тем, что он характеризуется относительной потерей массы спустя 120 часов в режиме подвижного слоя не более 7,5%.
19. Композитный носитель по п.18, в котором элемент IV группы Периодической системы представляет собой олово, германий, свинец.
20. Композитный носитель по п.19, в котором элементом IV группы Периодической системы является олово.
21. Композитный носитель по п.18, содержащий от 0,25 до 0,55 мас.% элемента IV группы Периодической системы.
22. Композитный носитель по п.21, содержащий от 0,3 до 0,4 мас.% элемента IV группы Периодической системы.
23. Композитный носитель по п.18, дополнительно содержащий галлий, индий, цинк, марганец или их смеси.
24. Композитный носитель для катализатора дегидрирования газообразных углеводородов, полученный способом по любому из пп.1-17, включающий оксид алюминия и оксид элемента IV группы Периодической таблицы, при этом содержание элемента IV группы Периодической системы составляет от 0,2 до 0,6 мас.%.
25. Катализатор дегидрирования газообразных углеводородов содержащий:
от 0,1 до 4,5 мас.% элемента VIII группы Периодической системы;
от 0,1 до 6,0 мас.% элемента I и/или II группы Периодической системы;
от 0,1 до 6,0 мас.% галогена;
носитель по любому из пп.18-24.
26. Катализатор по п.25, в котором носитель в качестве элемента IV группы Периодической системы содержит олово, германий, свинец.
27. Катализатор по п.26, в котором носитель в качестве элемента IV группы Периодической системы содержит олово.
28. Катализатор по п.25, в котором носитель дополнительно содержит галлий, индий, цинк, марганец или их смеси.
29. Катализатор по п.25, в котором носитель содержит от 0,2 до 0,6 мас.% элемента IV группы Периодической системы.
30. Катализатор по п.29, в котором носитель содержит от 0,25 до 0,55 мас.% элемента IV группы Периодической системы.
31. Катализатор по п.25, в котором в качестве элемента VIII группы Периодической системы используют платину, палладий, рутений, родий, иридий, осмий или их смеси.
32. Катализатор по п.31, в котором в качестве элемента VIII группы Периодической системы используют платину.
33. Катализатор по п.25, который содержит от 0,15 до 2 мас.% элемента VIII группы Периодической системы, предпочтительно от 0,15 до 0,8 мас.%.
34. Катализатор по п.25, в котором в качестве элементов I и II группы Периодической системы используют кальций, калий, натрий, магний, литий, стронций, барий, бериллий или их смеси.
35. Катализатор по п.34, в котором в качестве элемента I используют калий.
36. Катализатор по п.25, который содержит от 0,2 до 3 мас.% элемента I или II групп Периодической системы, предпочтительно от 0,5 до 2 мас.%.
37. Катализатор по п.25, в котором в качестве галогена используют хлор.
38. Катализатор по п.25, который содержит от 0,2 до 3 мас.% галогена, предпочтительно от 0,2 до 2 мас.%.
39. Способ получения катализатора дегидрирования газообразных углеводородов, включающий следующие стадии:
a) получение носителя способом по любому из пп.1-17;
b) обработка носителя кислотой;
c) прокаливание обработанного кислотой носителя;
d) нанесение на носитель, полученный на стадии с), элемента VIII группы Периодической системы;
e) сушка и прокаливание предшественника катализатора, полученного на стадии d);
f) нанесение на предшественник катализатора, полученный на стадии e), элементов I и/или II группы Периодической системы;
g) сушка и прокаливание предшественника катализатора, полученного на стадии f);
h) нанесение на предшественник катализатора, полученный на стадии g), галогена; и
i) сушка и прокаливание предшественника катализатора, полученного на стадии h).
40. Способ по п.39, в котором обработку носителя проводят раствором кислоты.
41. Способ получения катализатора дегидрирования газообразных углеводородов, включающий следующие стадии:
a) получение носителя способом по любому из пп.1-17;
b) нанесение на носитель, полученный на стадии а), элемента VIII группы Периодической системы;
c) сушка и прокаливание предшественника катализатора, полученного на стадии b);
d) нанесение на предшественник катализатора, полученный на стадии с), элементов I и/или II группы Периодической системы;
e) сушка и прокаливание предшественника катализатора, полученного на стадии d);
f) нанесение на предшественник катализатора, полученный на стадии e), галогена; и
g) сушка и прокаливание предшественника катализатора, полученного на стадии f).
42. Способ дегидрирования газообразных углеводородов в режиме подвижного слоя с использованием катализатора по любому из пп.25-38.
43. Способ дегидрирования по п.42, в котором дегидрирование проводят при температуре от 580 до 650°С.
44. Способ дегидрирования по любому из пп.42-43, в котором отношение компонентов реакционной смеси Н2:С2-С4 составляет от 0,2 до 0,8.
45. Способ дегидрирования по любому из пп.42-44, в котором катализатор дегидрирования используется более одного цикла.
46. Способ дегидрирования по п.45, в котором периодически проводят стадии оксихлорирования катализатора дегидрирования.
| CN 105214657 A, 20.03.2018 | |||
| Способ получения катализатора гидроочистки нефтяного сырья | 1987 |
|
SU1424863A1 |
| CN 102451766 A, 16.05.2012 | |||
| СПОСОБ ПОЛУЧЕНИЯ ПСЕВДОЗОЛЯ ГИДРОКСИДА АЛЮМИНИЯ | 1990 |
|
SU1771174A1 |
| CN 104445317 В, 14.09.2016 | |||
| HEIBAZ.K | |||
| et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Способ получения олифы или массы для приготовления лаков | 1913 |
|
SU507A1 |
| Прибор для измерения угла наклона | 1921 |
|
SU253A1 |
| KIRSZENSZEJN P | |||
| et al | |||
| "Effect | |||
