Изобретение относится к области синтеза органических соединений, более конкретно - к получению действующих веществ для лекарственных средств, в частности - к способу получения толбутамида (1-(4-метилфенилсульфонил)-3-бутилмочевины (I), регистрационный номер CAS [64-77-7]). Эта сульфонилмочевина, разработанная в 1956 г., относится к синтетическим гипогликемическим средствам первого поколения и используется для лечения сахарного диабета II типа средней степени тяжести (см., напр., Dictionary of Drugs: Chemical Data, Structures, and Bibliographies. Edited by J. Elks and C.R. Ganellin, 1st Ed., Springer New York, NY, 1990, p. 1214; патенты GB №808071A, US №2968158А).
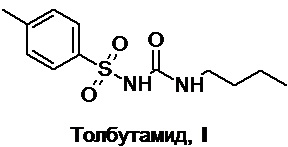
Известные методы синтеза толбутамида в большинстве случаев сводятся к двум его ключевым предшественникам, первый из которых - производное 1-(Х-сульфонил)-4-метилбензола [Ts-X (Ts = 4-Me-Ph-SO2-), где Х = NH2 (тозиламид), NHNa(K) (соли тозиламида), NCO (тозилизоцианат), NHC(O)Cl (тозилкарбамоилхлорид), NHCO2R (R = Me, Et и др.) (тозилкарбаматы), NHC(O)NH2 (тозилмочевина) и др.] (А), среди которых наиболее часто используется тозиламид и его соли, а также тозилмочевина, а второй - бутиламин (BuNH2) и его производные [Bu-Y, где Y = NCO (бутилизоцианат), NHCO2Ph (фенилкарбамат бутиламина), CHO (N-бутилформамид) и др.] (Б), среди которых наиболее часто используется сам бутиламин и бутилизоцианат (Схема 1). На заключительной стадии синтеза осуществляют формирование сульфонилмочевинного фрагмента толбутамида, для чего используют реакцию одной из возможных пар соответствующих предшественников А и Б между собой в подходящих условиях; при этом в синтезе обязательно требуется предварительная трансформация NH2-группы тозиламида и (или) бутиламина в один из подходящих для сборки сульфонилмочевинного фрагмента заместителей типа X или Y, что автоматически означает, что все известные подходы к структуре толбутамида требуют не менее двух синтетических стадий (Схема 1).
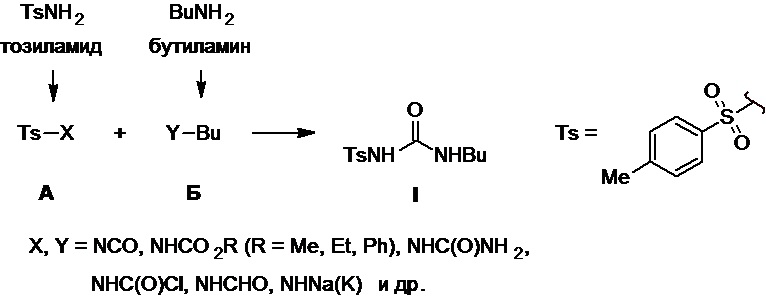
Схема 1.
Общие принципы получения толбутамида были раскрыты его разработчиками еще 70 лет назад (см., напр., патенты GB №808071A, US №2968158А, DE №974062C) и, среди прочего, включали трансформацию тозиламида с помощью метил- или этилхлорформиата, гидроксида/карбоната натрия или калия, фосгена и др. в соответствующие карбаматы, его соли с щелочными металлами, тозилизоцианат или тозилкарбамоилхлорид (Схема 2), что наглядно иллюстрирует предварительную подготовку NH2-группы тозиламида к созданию сульфонилмочевинного фрагмента целевой молекулы, упомянутую выше (Схема 1). Далее эти промежуточные соединения используют в реакции с бутиламином или бутилизоцианатом в подходящих условиях с получением толбутамида с выходом 70-80%.
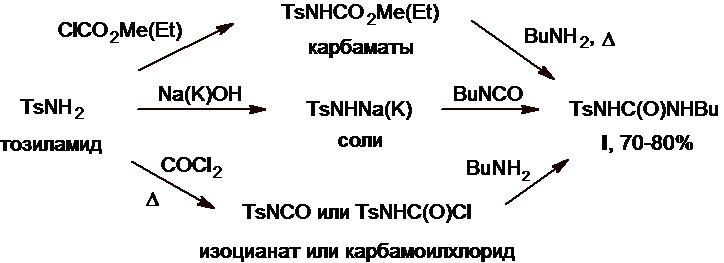
Схема 2.
Тогда же была описана реакция хлорамина Т с N-бутилформамидом, осуществляемая в воде в присутствии основания (патент DE №1066575B), которая приводит к получению целевого продукта с выходом 53% после перекристаллизации (Схема 3).
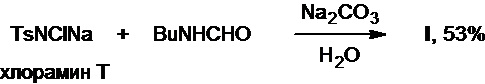
Схема 3.
В дальнейшем в рамках Схемы 2 было предложено множество вариантов оптимизации того или иного синтетического подхода к структуре целевого продукта, в целом позволивших облегчить трансфер лабораторных методов его синтеза в промышленную практику, в частности, за счет повышения безопасности процесса, упрощения способов его осуществления, снижения количества отходов и времени протекания реакций, облегчения выделения и очистки сырого продукта после реакции, отказа от использования нестабильных соединений в качестве сырья и полупродуктов и др. (см., напр., C.R. Sagandira et al., Tetrahedron, 2021, 96, 132378).
Так, с целью такой оптимизации рядом авторов была исследована наиболее простая схема синтеза толбутамида, включающая использование тозиламида или его солей с щелочными металлами в реакции с коммерчески доступным бутилизоцианатом (Схема 4). При этом основной фокус работ в этом направлении был сосредоточен на осуществлении этой реакции с применением современных методов синтетической органической химии, таких как гомогенный или гетерогенный катализ; реакции в потоке (проточный синтез); механосинтез с использованием эффективных шаровых мельниц, ультразвука, микроволнового излучения и т.п. (см., напр., V. Strukil, Beilstein J. Org. Chem., 2017, 13, 1828-1849; L. Gonnet et al., Angew. Chem. Int. Ed., 2022, 61(13), e202115030; L. Gonnet et al., ChemRxiv, 2021, 1-14; D. Tan et al., Chem. Comm., 2014, 50(40), 5248-5250; J. Cervello et al., Synthesis, 1990, 3, 221-222; H. Irie et al., J. Chem. Soc., Perkin Trans. 1, 1989, 7, 1209-1210; A. Dongarwar et al., World J. Pharm. Pharm. Sci. (WJPPS), 2020, 9(5), 1485-1492; патент JPH №01224356A).
Следует отметить, что те же самые принципы были использованы и для другой пары возможных исходных соединений в синтезе толбутамида, а именно - тозилизоцианата и бутиламина (Схема 4), хотя, вследствие ограниченной стабильности первого, они пока не нашли широкого применения.

Схема 4.
Работы в этом направлении привели к тому, что удалось, например, а) в присутствии каталитических количеств хлорида меди(I), триэтиламина, эфирата трехфтористого бора и др. отказаться от использования в этом превращении солей тозиламида с их заменой на сам тозиламид, что понизило стадийность процесса и сделало его более эффективным; б) частично или полностью отказаться от использования органического растворителя (нитрометана, N,N-диметилформамида (ДМФ), тетрагидрофурана (ТГФ) и др.), что облегчило выделение целевого продукта и сократило количество отходов процесса; в) повысить выход толбутамида до 95%; г) снизить экономические затраты для осуществления процесса.
Другим важным методом, используемым в промышленном производстве толбутамида, является трансформация тозилмочевины (Схема 5), доступной из тозиламида и мочевины (см., напр., патенты SU №128015А1, SU №190891A1, SU №204329А1, DD №267631A3, PL №146468B1, CS №259750B1, JPS №49248A, JPS №4886845A, DE №2023350А1, DE №2053740А1, DE №2053741А1; Рубцов М.В., Байчиков А.Г. Синтетические химико-фармацевтические препараты (справочник). М.: Медицина, 1971, с. 140). В этом случае целевое соединение получают из тозилмочевины двумя способами: а) вводят ее реакцию с бутиламином или его солями с соляной или серной кислотой при нагревании или б) первоначально действием метанола и серной кислоты превращают ее в метилкарбамат тозиламида, который далее нагревают с бутиламином. Несмотря на то, что при использовании этого метода для получения толбутамида из тозиламида требуется 2-3 синтетические стадии, данный процесс его производства характеризуется дешевизной и стабильностью исходного сырья и полупродуктов, при этом устойчиво и воспроизводимо приводит к конечному результату с выходом продукта, составляющим 52-87% в зависимости от условий его проведения.
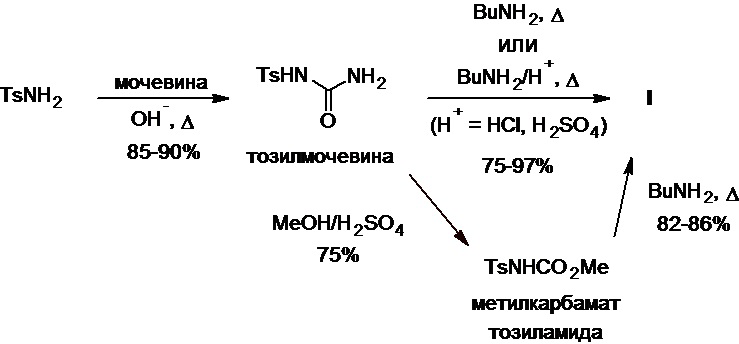
Схема 5.
Другой метод синтеза толбутамида (F. Saczewski et al., Green Chem., 2006, 8(7), 647-656) основан на реакции тозиламида с дифенилкарбонатом в присутствии 4-диметиламинопиридина (4-DMAP), которая приводит к образованию промежуточного стабильного цвиттер-ионного соединения (Схема 6), выступающего в роли синтетического эквивалента нестабильного тозилизоцианата (Схема 4). Это вещество выделяют в чистом виде и используют далее в реакции с бутиламином, которая приводит к получению целевого продукта с выходом 49% в расчете на исходный тозиламид, при этом вся синтетическая последовательность может быть реализована в мягких условиях.
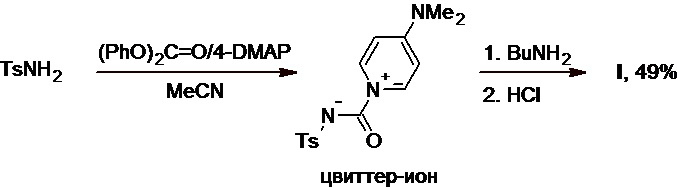
Схема 6.
Известна (патент DE №2213602A1) реакция тозиламида с N-бутиламидом трихлоруксусной кислоты в присутствии карбоната калия при нагревании (Схема 7). Несмотря на кажущуюся простоту процесса, выход целевого продукта составляет лишь 34%.
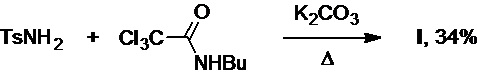
Схема 7.
В патенте WO №1993010086A1 предложен способ получения толбутамида, основанный на первоначальном превращении тозиламида в (тозилиминойод)бензол, который далее вводят в реакцию с монооксидом углерода (CO) в присутствии каталитических количеств комплекса PdCl2(PhCN)2, что приводит к генерированию в растворе тозилизоцианата, взаимодействие которого с бутиламином позволяет получать целевой продукт с выходом 60% в расчете на тозиламид (Схема 8).
 Схема 8.
Схема 8.
Известен метод синтеза толбутамида, основанный на взаимодействии натриевой соли тозиламида с диэтилпирокарбонатом при нагревании (патент NL №6603398А). В ходе этого процесса после подкисления уксусной кислотой или хлороводородом в растворе промежуточно генерируется этилкарбамат тозиламида, который далее вступает в реакцию с бутиламином с образованием целевого продукта с выходом 78% (Схема 9).
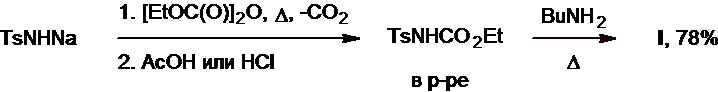
Схема 9.
В патенте DE №2256979C3 представлен способ получения толбутамида из тозилимино-алкил-N-бутилкарбаматов (алкил = метил, этил, пропил), доступных в несколько стадий из тозиламида. Эти соединения вступают в реакцию с пирролидином или пиперидином при комнатной температуре с образованием целевого продукта, выход которого не указан (Схема 10).
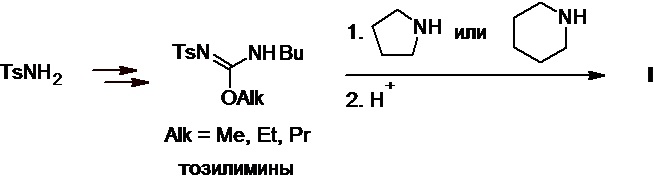
Схема 10.
Наконец, тозиламид также служит исходным сырьем в синтезе тозилимино-1,3-оксотиолана (Схема 11), раскрытие которого под действием бутиламина протекает гладко с образованием целевого толбутамида с выходом 96% (S. Suzue et al., Chem. Pharm. Bull., 1969, 17(8), 1535-1540).
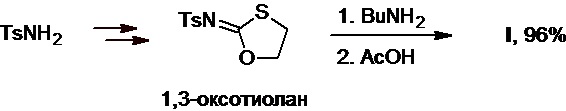
Схема 11.
Помимо приведенных выше примеров эффективного использования тозиламида в качестве исходного сырья для получения толбутамида, в последнее время появились работы, в которых представлены примеры успешного применения бутиламина в тех же целях.
Так, первоначальное превращение бутиламина в соответствующий фенилкарбамат действием фенилхлорформиата или дифенилкарбоната с его последующей реакцией с тозиламидом в присутствии сильных органических оснований (1,8-диазабицикло[5.4.0]ундец-7-ен (DBU), триэтиламин, трибутиламин и др.) приводит к получению толбутамида с выходом 83-97% (Схема 12). Эта двухстадийная синтетическая последовательность превращений оказалась настолько проста в исполнении, устойчива и универсальна, что легла в основу новой поточной методологии получения толбутамида, основанной на использовании системы проточных реакторов, что позволяет быстро получать целевой продукт напрямую из исходных компонентов (бутиламин, фенилхлорформиат, органическое основание, тозиламид), минуя стадию выделения и очистки промежуточного фенилкарбамата (см., напр., D.K. Tanwar et al., Org. Biomol. Chem., 2017, 15(23), 4992-4999; C.R. Sagandira et al., Synthesis, 2022, 54(5), 1365-1374; патент WO №2023017474A1). В настоящее время данный метод получения толбутамида является сугубо лабораторным, так как требует сложного и дорогостоящего аппаратурного оформления.
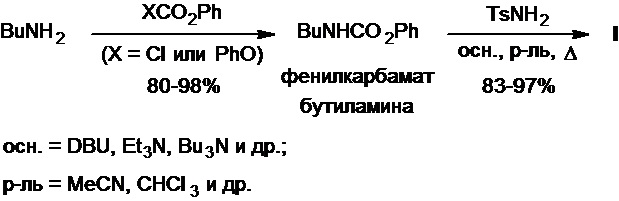
Схема 12.
Рядом авторов было показано, что взаимодействие бутиламина с углекислым газом в присутствии DBU в качестве сильного органического основания приводит к генерированию в растворе соли N-бутилкарбаминовой кислоты, которая вступает в реакцию с 1-хлорбензотриазолом (BtCl) в присутствии трифенилфосфина с образованием активированной Bt-мочевины (Схема 13). Реакция последней со смесью тозиламида с карбонатом калия или с натриевой солью тозиламида при нагревании приводит к получению толбутамида с выходом 44-53% в расчете на исходный бутиламин (R. Hunter et al., Synlett, 2011, 16, 2335-2338; I. Butula et al., Croat. Chem. Acta, 1979, 52(1), 47-49).
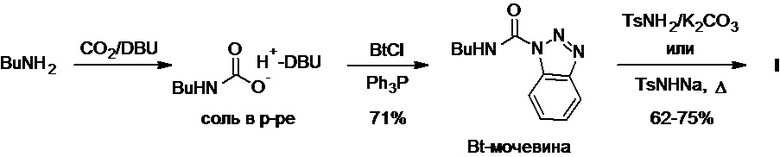
Схема 13.
Известно, что в присутствии элементарной серы и каталитических количеств селена бутиламин взаимодействует с монооксидом углерода, при этом промежуточный продукт этой реакции карбонилирования может быть проалкилирован йодметаном с образованием соответствующего N-бутил-S-метилтиокарбамата (Схема 14). Последний вступает в реакцию с тозиламидом в присутствии DBU в качестве основания при нагревании с образованием толбутамида с выходом 54% в расчете на исходный бутиламин (T. Mizuno et al., Synth. Commun., 2000, 30(17), 3081-3089).

Схема 14.
В патенте DE №2124742А1 показано, что раскрытие цикла этилен- или пропиленкарбоната бутиламином при нагревании приводит к образованию соответствующих гидроксиэтил-N-бутилкарбаматов (Схема 15). Последние вступают в реакцию с натриевой солью тозиламида, что приводит к получению толбутамида с выходом 18-40% в расчете на исходный бутиламин.
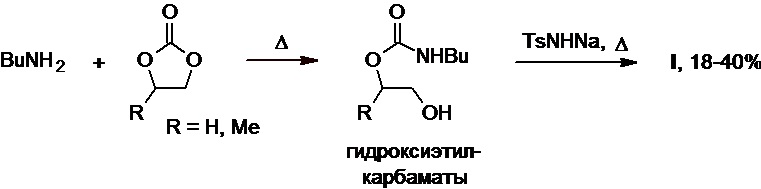
Схема 15.
Известно, что легко доступные в одну стадию из бутиламина 1,3-дибутилмочевина и бутилмочевина (Схема 16) также являются удобным исходным сырьем для получения толбутамида (R.J. Tull et al., J. Chem. Soc. C, 1967, 8, 701-702; патенты FR №1558886А, DE №2232761A1). Взаимодействие первой из них с натриевой солью тозиламида при нагревании приводит к целевому продукту с выходом 93%. С другой стороны, бутилмочевину можно превратить в толбутамид с выходом 78-83% двумя различными способами: а) путем ее последовательной обработки гидридом натрия и тозилхлоридом (TsCl) при нагревании или б) путем ее обработки тозилхлоридом в расплаве смеси карбонатов аммония и калия при высокой температуре.
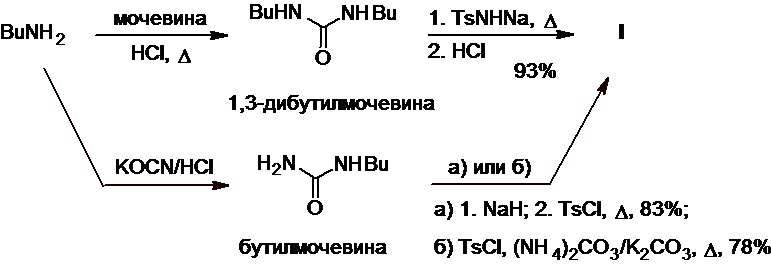
Схема 16.
Известен одностадийный метод синтеза толбутамида из тозилхлорида, азида натрия, монооксида углерода и бутиламина в присутствии ацетата палладия(II) в качестве катализатора (J. Zhao et al., Angew. Chem. Int. Ed., 2016, 55(18), 5545-5549). В ходе процесса происходит первоначальное генерирование в растворе тозилазида с его последующим карбонилированием в присутствии палладиевого катализатора с генерированием тозилизоцианата, который уже и вступает в реакцию с бутиламином, приводя к получению целевого продукта с выходом 90% в расчете на исходный тозилхлорид (Схема 17).
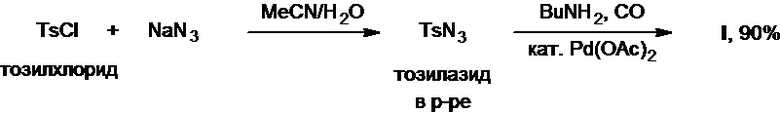
Схема 17.
В патентах FR №1588266А и GB №1185013А показано, что 4-толуолсульфиновая кислота, ее натриевая соль или хлорангидрид могут выступать в роли исходного сырья в синтезе толбутамида. Взаимодействие этих соединений с 1-гидрокси-3-бутилмочевиной в присутствии полифосфорной кислоты или тионилхлорида при нагревании приводит к получению целевого соединения с выходом до 50% (Схема 18).
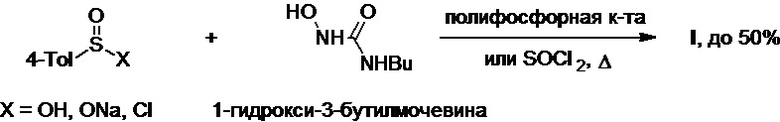
Схема 18.
Также известен метод получения толбутамида (патент US №3281412А), основанный на реакции окисления 1-(4-толуолсульфинил)-3-бутилмочевины в мягких условиях под действием перманганата калия в водном растворе гидроксида натрия, приводящей к образованию целевого продукта с выходом 87% (Схема 19).
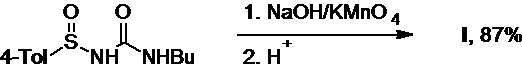
Схема 19.
Рядом авторов показано, что сернистый аналог толбутамида, 1-тозил-3-бутилтиомочевина, может выступать удобным исходным сырьем в его производстве (патент JPS №4940463B1; T. Kodama et al., Yuki Gosei Kagaku Kyokaishi (J. Synthetic Org. Chem., Japan), 1967, 25(6), 498-502). Окисление этой тиомочевины хлоритом или гипохлоритом натрия, диоксидом селена или его смесью с водным раствором гидроксида натрия при нагревании приводит к получению целевого продукта с выходом 85-96% (Схема 20).
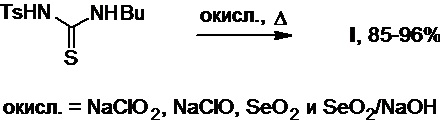
Схема 20.
Также было найдено (патент JPS №4929184B1; T. Kodama et al., Yuki Gosei Kagaku Kyokaishi (J. Synthetic Org. Chem., Japan), 1968, 26(8), 674-679), что в результате обработки вышеупомянутой тиомочевины хлором или тионилхлоридом при нагревании можно получить и выделить в чистом виде соответствующий замещенный дисульфид, который в присутствии окислителей (хлорит или гипохлорит натрия, пероксид водорода, оксид ртути(II) и др.) превращается в целевой продукт с выходом, достигающим 96% (Схема 21).
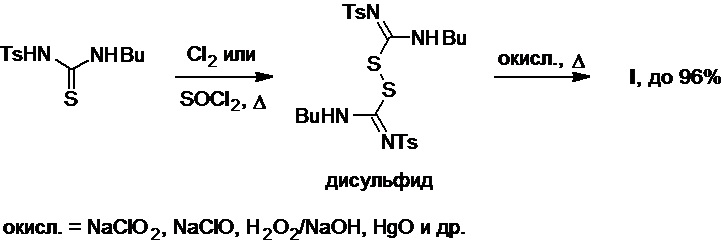
Схема 21.
Наконец, исчерпывающее хлорирование этой тиомочевины хлором или хлоридом фосфора(V) при нагревании приводит к образованию соответствующего замещенного хлорформамидина (Схема 22), который при обработке водным раствором гидроксида натрия или калия превращается в толбутамид с выходом до 97% (T. Kodama et al., Yuki Gosei Kagaku Kyokaishi (J. Synthetic Org. Chem., Japan), 1967, 25(6), 493-497).
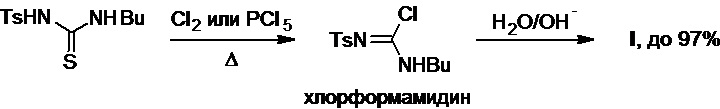
Схема 22.
Суммируя вышесказанное, большинство известных методов получения толбутамида (Схемы 2-22) характеризуются следующими технологическими недостатками: а) многостадийностью (как правило, не менее двух синтетических стадий); б) наличием стадий, подразумевающих работу с нестабильными, опасными и токсичными исходными соединениями, полупродуктами и (или) реагентами (напр., фосгеном, низшими хлорформиатами, изоцианатами, монооксидом углерода, азидом натрия, хлоридом фосфора(V), хлором, соединениями селена(IV) или йода(III) и др.), осуществление которых требует соблюдения специальных мер промышленной безопасности и специальных условий проведения процесса; в) использованием в большинстве случаев повышенных температур, иногда достигающих 160-170°С, пониженного или повышенного давления; г) низкой или умеренной конверсией исходного сырья и связанными с этим сложностями при выделении и очистке целевого продукта до надлежащего качества; д) большим количеством технологических операций вследствие многостадийности процессов. В целом, все эти факторы негативно сказываются на безопасности, масштабируемости, эффективности и технико-экономических показателях описанных выше методов.
Необходимо также отметить, что в синтезе тозиламида, являющегося ключевым исходным сырьем или предшественником в синтезе такого сырья при промышленном производстве толбутамида, используется доступный тозилхлорид. Таким образом, непосредственное применение тозилхлорида в синтезе толбутамида в качестве исходного сырья приведет к автоматическому снижению стадийности процесса как минимум на одну стадию, и как следствие, его упрощению и удешевлению.
На текущий момент лишь в одном из многочисленных известных методов получения толбутамида рассматривается возможность прямого использования тозилхлорида в качестве исходного сырья (Схема 17), при этом еще в двух примерах его используют в качестве реагента (Схема 16). Несмотря на кажущуюся простоту одностадийного подхода, представленного на Схеме 17, и его общую эффективность, к его недостаткам при переходе на уровень перспективного промышленного внедрения можно отнести а) использование в нем опасных и токсичных азида натрия и газообразного монооксида углерода; б) генерирование в ходе процесса нестабильного тозилазида; в) использование в процессе пониженного давления; г) сложности при обработке реакционной массы после реакции для отделения отработанного палладиевого катализатора и выделения целевого продукта (напр., экстракция продукта органическим растворителем после реакции с последующей его очисткой на силикагеле). Все это приведет к сложности аппаратурного оформления процесса, большому количеству требуемых технологических операций и необходимости в специальных мерах безопасности.
На основании приведенного анализа доступных опубликованных данных очевидно, что проблема поиска новых безопасных и хорошо масштабируемых способов получения толбутамида, позволяющих получать его из доступного сырья с использованием минимального количества синтетических стадий, в ходе которых будет исключено использование в синтезе повышенной или пониженной температуры или давления, а также нестабильных, опасных и токсичных исходных соединений, полупродуктов и (или) реагентов, является актуальной.
Задачей предлагаемого технического решения является улучшение технико-экономических показателей технологического процесса производства толбутамида за счет применения новой технологии его синтеза исходя из тозилхлорида, позволяющей в одну стадию получать целевой продукт высокого качества с хорошим выходом.
Техническим результатом является а) улучшение технико-экономических показателей технологического процесса производства толбутамида за счет понижения его стадийности до одной стадии и исключения использования в нем нестабильных, опасных и токсичных исходных соединений, полупродуктов и (или) реагентов, агрессивных коррозионных сред, повышенной температуры или давления и др.; б) повышение безопасности и общей эффективности процесса; в) снижение количества технологических операций и, как следствие, упрощение масштабируемости процесса; г) получение целевого продукта высокого качества с выходом до 81%.
Технический результат достигается за счет использования способа получения толбутамида, заключающегося во взаимодействии тозилхлорида с цианатом натрия или калия в присутствии органического основания в среде полярного органического растворителя при комнатной температуре в атмосфере аргона или азота или на воздухе без доступа влаги с последующим введением в образующуюся реакционную смесь бутиламина или его гидрохлорида с добавкой неорганической соли или без таковой с дальнейшей обработкой содержимого реактора соляной или уксусной кислотой и водой с последующим выделением и очисткой целевого продукта.
В качестве органического основания используют пиридин, 2-пиколин, 3-пиколин, 4-пиколин или 2,6-лутидин; в качестве полярного органического растворителя используют ацетонитрил, пропионитрил или их смеси; в качестве неорганической соли используют карбонаты натрия, калия или кальция или гидрокарбонаты натрия или калия.
В ходе оптимизации условий проведения процесса предпочтительным оказалось использование: а) от 1,1 до 1,3 мол. экв. тозилхлорида по отношению к бутиламину или его гидрохлориду; б) от 2,0 до 2,5 мол. экв. цианата натрия или цианата калия по отношению к бутиламину или его гидрохлориду; в) от 2,5 до 3,5 мол. экв. органического основания по отношению к бутиламину; г) от 3,5 до 5,0 мол. экв. органического основания по отношению к гидрохлориду бутиламина; д) от 2,0 до 3,0 мол. экв. органического основания по отношению к гидрохлориду бутиламина в случае добавки неорганической соли в количестве от 0,1 до 1,0 мол. экв. по отношению к гидрохлориду бутиламина.
Изобретение иллюстрируется следующими примерами (подходы А-В, примеры 1-15):
А) Получение толбутамида c использованием бутиламина (Схема 23, примеры 1-5):
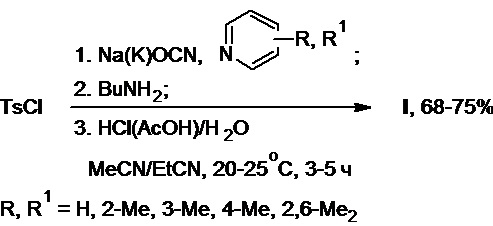
Схема 23.
Пример 1.
В реактор синтеза в атмосфере азота помещают тозилхлорид (18,6 г, 0,10 моль), цианат натрия (12,1 г, 0,19 моль) и ацетонитрил. К полученной суспензии при интенсивном перемешивании добавляют 2-пиколин (20,7 г, 0,22 моль), после чего содержимое реактора перемешивают при температуре 20-25°С в течение 2-3 ч и добавляют к нему бутиламин (6,5 г, 0,09 моль). Осуществляют выдержку реакционной массы в течение 1-2 ч (ТСХ-контроль). Далее полученную смесь подкисляют 70%-ным водным раствором уксусной кислоты до pH 2-3 и разбавляют водой. Образовавшийся осадок продукта отфильтровывают, промывают на фильтре водой и высушивают на воздухе. Получают 18,0 г (75% в расчете на бутиламин) толбутамида в виде белого порошка с т.пл. 127,5-129,0°С и чистотой не менее 98,5% (по данным ВЭЖХ). Структура полученного продукта подтверждена на основании комплекса спектральных данных: 1Н ЯМР-спектр (500 МГц, CDCl3, δ, м.д.): 9,39 (с, 1Н, NH), 7,80 (д, 2Н, J = 7,7 Гц, CH(аром.)), 7,30 (д, 2Н, J = 7,7 Гц, CH(аром.)), 6,55 (с, 1Н, NH), 3,27-3,17 (м, 2Н, CH2N), 2,44 (с, 3Н, CH3), 1,51-1,42 (м, 2Н, CH2), 1,33-1,23 (м, 2Н, CH2), 0,89 (т, 3H, J = 7,0 Гц, СН3); ИК-спектр (в порошке, ν, см-1): 3332 ср, 3093 ср, 2954 ср, 2929 ср, 2872 ср, 1701 сл, 1655 с, 1598 ср, 1541 с, 1465 ср, 1447 ср, 1345 ср, 1245 ср, 1160 с, 1120 ср, 1091 с, 1044 ср, 1004 ср, 887 с, 842 ср, 814 ср; УФ-спектр: λмакс = 228 нм (с = 1,0 мг/100 мл MeOH); масс-спектр (APCI): [M+H]+: 271,1.
В зависимости от характера дальнейшего использования толбутамида его дополнительная очистка может быть выполнена путем перекристаллизации полученного образца из низших спиртов, ацетона, их смесей с водой или этилацетата с получением толбутамида с чистотой 99,5-99,8% (по данным ВЭЖХ).
Пример 2.
В реактор синтеза на воздухе без доступа влаги помещают тозилхлорид (27,1 г, 0,14 моль), цианат калия (20,4 г, 0,25 моль) и пропионитрил. К полученной суспензии при интенсивном перемешивании добавляют пиридин (26,0 г, 0,33 моль), после чего содержимое реактора перемешивают при температуре 20-25°С в течение 2-3 ч и добавляют к нему бутиламин (8,0 г, 0,11 моль). Осуществляют выдержку реакционной массы в течение 1-2 ч (ТСХ-контроль). Далее полученную смесь подкисляют конц. соляной кислотой до pH 2-3 и разбавляют водой. Образовавшийся осадок продукта отфильтровывают, промывают на фильтре водой и высушивают на воздухе. Получают 20,7 г (70% в расчете на бутиламин) толбутамида в виде белого порошка.
Пример 3.
В реактор синтеза в атмосфере аргона помещают тозилхлорид (15,6 г, 0,08 моль), цианат натрия (10,7 г, 0,16 моль) и смесь ацетонитрила с пропионитрилом (50/50 об.%). К полученной суспензии при интенсивном перемешивании добавляют 2,6-лутидин (25,6 г, 0,24 моль), после чего содержимое реактора перемешивают при температуре 20-25°С в течение 2-3 ч и добавляют к нему бутиламин (5,0 г, 0,07 моль). Осуществляют выдержку реакционной массы в течение 1-2 ч (ТСХ-контроль). Далее полученную смесь подкисляют 10%-ным водным раствором соляной кислоты до pH 2-3 и разбавляют водой. Образовавшийся осадок продукта отфильтровывают, промывают на фильтре водой и высушивают на воздухе. Получают 12,6 г (68% в расчете на бутиламин) толбутамида в виде белого порошка.
Пример 4.
В реактор синтеза в атмосфере азота помещают тозилхлорид (28,7 г, 0,15 моль), цианат калия (22,2 г, 0,27 моль) и смесь ацетонитрила с пропионитрилом (20/80 об.%). К полученной суспензии при интенсивном перемешивании добавляют 3-пиколин (34,4 г, 0,37 моль), после чего содержимое реактора перемешивают при температуре 20-25°С в течение 2-3 ч и добавляют к нему бутиламин (10,0 г, 0,14 моль). Осуществляют выдержку реакционной массы в течение 1-2 ч (ТСХ-контроль). Далее полученную смесь подкисляют ледяной уксусной кислотой до pH 2-3 и разбавляют водой. Образовавшийся осадок продукта отфильтровывают, промывают на фильтре водой и высушивают на воздухе. Получают 26,2 г (71% в расчете на бутиламин) толбутамида в виде белого порошка.
Пример 5.
В реактор синтеза в атмосфере аргона помещают тозилхлорид (46,9 г, 0,25 моль), цианат натрия (33,3 г, 0,51 моль) и смесь ацетонитрила с пропионитрилом (70/30 об.%). К полученной суспензии при интенсивном перемешивании добавляют 4-пиколин (47,8 г, 0,51 моль), после чего содержимое реактора перемешивают при температуре 20-25°С в течение 2-3 ч и добавляют к нему бутиламин (15,0 г, 0,21 моль). Осуществляют выдержку реакционной массы в течение 1-2 ч (ТСХ-контроль). Далее полученную смесь подкисляют конц. соляной кислотой до pH 2-3 и разбавляют водой. Образовавшийся осадок продукта отфильтровывают, промывают на фильтре водой и высушивают на воздухе. Получают 40,5 г (73% в расчете на бутиламин) толбутамида в виде белого порошка.
Б) Получение толбутамида c использованием гидрохлорида бутиламина (Схема 24, примеры 6-10):
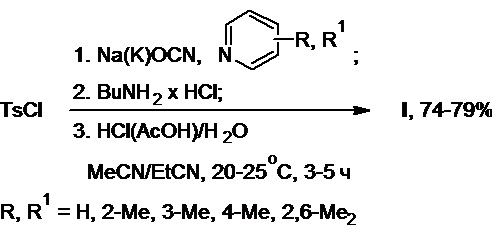
Схема 24.
Пример 6.
В реактор синтеза в атмосфере азота помещают тозилхлорид (19,1 г, 0,10 моль), цианат натрия (12,5 г, 0,19 моль) и ацетонитрил. К полученной суспензии при интенсивном перемешивании добавляют 3-пиколин (29,7 г, 0,32 моль), после чего содержимое реактора перемешивают при температуре 20-25°С в течение 2-3 ч и добавляют к нему гидрохлорид бутиламина (10,0 г, 0,09 моль). Осуществляют выдержку реакционной массы в течение 1-2 ч (ТСХ-контроль). Далее полученную смесь подкисляют 20%-ным водным раствором соляной кислоты до pH 2-3 и разбавляют водой. Образовавшийся осадок продукта отфильтровывают, промывают на фильтре водой и высушивают на воздухе. Получают 19,0 г (77% в расчете на гидрохлорид бутиламина) толбутамида в виде белого порошка. Спектральные и аналитические характеристики полученного продукта полностью идентичны таковым, представленным в примере 1.
Пример 7.
В реактор синтеза на воздухе без доступа влаги помещают тозилхлорид (33,9 г, 0,18 моль), цианат калия (25,5 г, 0,31 моль) и пропионитрил. К полученной суспензии при интенсивном перемешивании добавляют пиридин (43,3 г, 0,55 моль), после чего содержимое реактора перемешивают при температуре 20-25°С в течение 2-3 ч и добавляют к нему гидрохлорид бутиламина (15,0 г, 0,14 моль). Осуществляют выдержку реакционной массы в течение 1-2 ч (ТСХ-контроль). Далее полученную смесь подкисляют ледяной уксусной кислотой до pH 2-3 и разбавляют водой. Образовавшийся осадок продукта отфильтровывают, промывают на фильтре водой и высушивают на воздухе. Получают 27,4 г (74% в расчете на гидрохлорид бутиламина) толбутамида в виде белого порошка.
Пример 8.
В реактор синтеза в атмосфере аргона помещают тозилхлорид (31,3 г, 0,16 моль), цианат натрия (21,4 г, 0,33 моль) и смесь ацетонитрила с пропионитрилом (80/20 об.%). К полученной суспензии при интенсивном перемешивании добавляют 2-пиколин (63,7 г, 0,68 моль), после чего содержимое реактора перемешивают при температуре 20-25°С в течение 2-3 ч и добавляют к нему гидрохлорид бутиламина (15,0 г, 0,14 моль). Осуществляют выдержку реакционной массы в течение 1-2 ч (ТСХ-контроль). Далее полученную смесь подкисляют конц. соляной кислотой до pH 2-3 и разбавляют водой. Образовавшийся осадок продукта отфильтровывают, промывают на фильтре водой и высушивают на воздухе. Получают 29,2 г (79% в расчете на гидрохлорид бутиламина) толбутамида в виде белого порошка.
Пример 9.
В реактор синтеза в атмосфере азота помещают тозилхлорид (14,4 г, 0,08 моль), цианат калия (11,1 г, 0,14 моль) и смесь ацетонитрила с пропионитрилом (50/50 об.%). К полученной суспензии при интенсивном перемешивании добавляют 2,6-лутидин (25,7 г, 0,24 моль), после чего содержимое реактора перемешивают при температуре 20-25°С в течение 2-3 ч и добавляют к нему гидрохлорид бутиламина (7,5 г, 0,07 моль). Осуществляют выдержку реакционной массы в течение 1-2 ч (ТСХ-контроль). Далее полученную смесь подкисляют 50%-ным водным раствором уксусной кислоты до pH 2-3 и разбавляют водой. Образовавшийся осадок продукта отфильтровывают, промывают на фильтре водой и высушивают на воздухе. Получают 14,4 г (78% в расчете на гидрохлорид бутиламина) толбутамида в виде белого порошка.
Пример 10.
В реактор синтеза в атмосфере аргона помещают тозилхлорид (20,9 г, 0,11 моль), цианат натрия (14,8 г, 0,23 моль) и ацетонитрил. К полученной суспензии при интенсивном перемешивании добавляют 4-пиколин (38,2 г, 0,41 моль), после чего содержимое реактора перемешивают при температуре 20-25°С в течение 2-3 ч и добавляют к нему гидрохлорид бутиламина (10,0 г, 0,09 моль). Осуществляют выдержку реакционной массы в течение 1-2 ч (ТСХ-контроль). Далее полученную смесь подкисляют 10%-ным водным раствором соляной кислоты до pH 2-3 и разбавляют водой. Образовавшийся осадок продукта отфильтровывают, промывают на фильтре водой и высушивают на воздухе. Получают 19,5 г (79% в расчете на гидрохлорид бутиламина) толбутамида в виде белого порошка.
В) Получение толбутамида c использованием гидрохлорида бутиламина в присутствии неорганических солей (Схема 25, примеры 11-15):
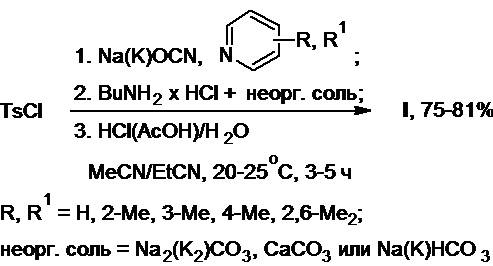
Схема 25.
Пример 11.
В реактор синтеза в атмосфере азота помещают тозилхлорид (19,1 г, 0,10 моль), цианат калия (15,5 г, 0,19 моль) и пропионитрил. К полученной суспензии при интенсивном перемешивании добавляют пиридин (14,4 г, 0,18 моль), после чего содержимое реактора перемешивают при температуре 20-25°С в течение 2-3 ч и добавляют к нему гидрохлорид бутиламина (10,0 г, 0,09 моль) и карбонат натрия (4,8 г, 0,05 моль). Осуществляют выдержку реакционной массы в течение 1-2 ч (ТСХ-контроль). Далее полученную смесь подкисляют конц. соляной кислотой до pH 2-3 и разбавляют водой. Образовавшийся осадок продукта отфильтровывают, промывают на фильтре водой и высушивают на воздухе. Получают 20,0 г (81% в расчете на гидрохлорид бутиламина) толбутамида в виде белого порошка. Спектральные и аналитические характеристики полученного продукта полностью идентичны таковым, представленным в примере 1.
Пример 12.
В реактор синтеза на воздухе без доступа влаги помещают тозилхлорид (33,9 г, 0,18 моль), цианат натрия (20,5 г, 0,31 моль) и ацетонитрил. К полученной суспензии при интенсивном перемешивании добавляют 3-пиколин (31,9 г, 0,34 моль), после чего содержимое реактора перемешивают при температуре 20-25°С в течение 2-3 ч и добавляют к нему гидрохлорид бутиламина (15,0 г, 0,14 моль) и карбонат кальция (9,6 г, 0,10 моль). Осуществляют выдержку реакционной массы в течение 1-2 ч (ТСХ-контроль). Далее полученную смесь подкисляют ледяной уксусной кислотой до pH 2-3 и разбавляют водой. Образовавшийся осадок продукта отфильтровывают, промывают на фильтре водой и высушивают на воздухе. Получают 27,7 г (75% в расчете на гидрохлорид бутиламина) толбутамида в виде белого порошка.
Пример 13.
В реактор синтеза в атмосфере аргона помещают тозилхлорид (15,7 г, 0,08 моль), цианат натрия (10,7 г, 0,16 моль) и смесь ацетонитрила с пропионитрилом (60/40 об.%). К полученной суспензии при интенсивном перемешивании добавляют 4-пиколин (19,1 г, 0,21 моль), после чего содержимое реактора перемешивают при температуре 20-25°С в течение 2-3 ч и добавляют к нему гидрохлорид бутиламина (7,5 г, 0,07 моль) и гидрокарбонат калия (6,9 г, 0,07 моль). Осуществляют выдержку реакционной массы в течение 1-2 ч (ТСХ-контроль). Далее полученную смесь подкисляют 20%-ным водным раствором соляной кислоты до pH 2-3 и разбавляют водой. Образовавшийся осадок продукта отфильтровывают, промывают на фильтре водой и высушивают на воздухе. Получают 14,2 г (77% в расчете на гидрохлорид бутиламина) толбутамида в виде белого порошка.
Пример 14.
В реактор синтеза в атмосфере азота помещают тозилхлорид (28,7 г, 0,15 моль), цианат калия (22,2 г, 0,27 моль) и смесь ацетонитрила с пропионитрилом (20/80 об.%). К полученной суспензии при интенсивном перемешивании добавляют 2,6-лутидин (33,7 г, 0,31 моль), после чего содержимое реактора перемешивают при температуре 20-25°С в течение 2-3 ч и добавляют к нему гидрохлорид бутиламина (15,0 г, 0,14 моль) и карбонат калия (3,8 г, 0,03 моль). Осуществляют выдержку реакционной массы в течение 1-2 ч (ТСХ-контроль). Далее полученную смесь подкисляют 70%-ным водным раствором уксусной кислоты до pH 2-3 и разбавляют водой. Образовавшийся осадок продукта отфильтровывают, промывают на фильтре водой и высушивают на воздухе. Получают 30,1 г (81% в расчете на гидрохлорид бутиламина) толбутамида в виде белого порошка.
Пример 15.
В реактор синтеза в атмосфере аргона помещают тозилхлорид (20,9 г, 0,11 моль), цианат натрия (14,8 г, 0,23 моль) и пропионитрил. К полученной суспензии при интенсивном перемешивании добавляют 2-пиколин (22,9 г, 0,25 моль), после чего содержимое реактора перемешивают при температуре 20-25°С в течение 2-3 ч и добавляют к нему гидрохлорид бутиламина (10,0 г, 0,09 моль) и гидрокарбонат натрия (6,1 г, 0,07 моль). Осуществляют выдержку реакционной массы в течение 1-2 ч (ТСХ-контроль). Далее полученную смесь подкисляют конц. соляной кислотой до pH 2-3 и разбавляют водой. Образовавшийся осадок продукта отфильтровывают, промывают на фильтре водой и высушивают на воздухе. Получают 19,5 г (79% в расчете на гидрохлорид бутиламина) толбутамида в виде белого порошка.
В результате использования предлагаемого способа получения толбутамида удается а) улучшить технико-экономические показатели технологического процесса его производства за счет понижения стадийности процесса до одной стадии и исключения использования в нем нестабильных, опасных и токсичных исходных соединений, полупродуктов и (или) реагентов, агрессивных коррозионных сред, повышенной температуры или давления и др.; б) повысить безопасность и общую эффективность процесса; в) снизить количество технологических операций и, как следствие, упростить масштабируемость процесса; г) получить целевой продукт высокого качества с выходом до 81%.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения гликлазида | 2021 |
|
RU2754708C1 |
| Способ получения метил-N-(3-гидроксифенил)карбамата | 2023 |
|
RU2812527C1 |
| Способ получения этил-N-(3-гидроксифенил)карбамата | 2023 |
|
RU2805745C1 |
| Способ получения фенмедифама | 2023 |
|
RU2813459C1 |
| Способ получения моносульфурон-метила | 2024 |
|
RU2834839C1 |
| Способ получения R-N-[[3-[(диметиламино)карбонил]пиридин-2-ил]сульфонил]карбаматов, в которых заместителем R является метил или этил | 2023 |
|
RU2816572C1 |
| Способ получения моносульфурона | 2024 |
|
RU2834000C1 |
| Способ получения пиразосульфурон-этила | 2024 |
|
RU2828085C1 |
| Способ получения никосульфурона | 2023 |
|
RU2807708C1 |
| Способ получения десмедифама | 2023 |
|
RU2810479C1 |
Настоящее изобретение относится к способу получения толбутамида, который относится к синтетическим гипогликемическим средствам первого поколения и используется для лечения сахарного диабета II типа средней степени тяжести. Способ заключается во взаимодействии тозилхлорида с цианатом натрия или калия в присутствии органического основания в среде полярного органического растворителя при комнатной температуре в атмосфере аргона или азота или на воздухе без доступа влаги. Далее в образующуюся реакционную смесь вводят смесь бутиламина или его гидрохлорида с добавкой неорганической соли или без таковой с дальнейшей обработкой содержимого реактора соляной или уксусной кислотой и водой, с последующим выделением и очисткой целевого продукта. Технический результат - улучшение технико-экономических показателей технологического процесса, повышение безопасности и общей эффективности процесса, снижение количества технологических операций и получение целевого продукта высокого качества с выходом до 81%. 3 з.п. ф-лы, 15 пр.
1. Способ получения толбутамида, заключающийся во взаимодействии тозилхлорида с цианатом натрия или калия в присутствии органического основания в среде полярного органического растворителя при комнатной температуре в атмосфере аргона или азота или на воздухе без доступа влаги с последующим введением в образующуюся реакционную смесь бутиламина или его гидрохлорида с добавкой неорганической соли или без таковой с дальнейшей обработкой содержимого реактора соляной или уксусной кислотой и водой с последующим выделением и очисткой целевого продукта.
2. Способ по п. 1, характеризующийся тем, что в качестве органического основания используют пиридин, 2-пиколин, 3-пиколин, 4-пиколин или 2,6-лутидин.
3. Способ по п. 1, характеризующийся тем, что в качестве полярного органического растворителя используют ацетонитрил, пропионитрил или их смеси.
4. Способ по п. 1, характеризующийся тем, что в качестве неорганической соли используют карбонаты натрия, калия или кальция или гидрокарбонаты натрия или калия.
| Jin Zhao et al | |||
| Product-Derived Bimetallic Palladium Complex Catalyzes Direct Carbonylation of Sulfonylazides | |||
| Angew Chem | |||
| Int | |||
| Ed., 2016, 55(18), 5545-5549 | |||
| Davin Tan et al | |||
| Mechanosynthesis of pharmaceutically relevant sulfonyl-(thio)ureas | |||
| Chem | |||
| Commun., 2014, 50(40), 5248-5250 | |||
| WO 1993010086 A1, 27.05.1993 | |||
| СПОСОБ ПОЛУЧЕНИЯ №- | 0 |
|
SU204329A1 |
| Способ получения замещенной арилсульфонилмочевины | 1972 |
|
SU503513A3 |

